WO2016015676A1 - 吡啶取代的2-氨基吡啶类蛋白激酶抑制剂 - Google Patents
吡啶取代的2-氨基吡啶类蛋白激酶抑制剂 Download PDFInfo
- Publication number
- WO2016015676A1 WO2016015676A1 PCT/CN2015/085727 CN2015085727W WO2016015676A1 WO 2016015676 A1 WO2016015676 A1 WO 2016015676A1 CN 2015085727 W CN2015085727 W CN 2015085727W WO 2016015676 A1 WO2016015676 A1 WO 2016015676A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- acid
- compound
- formula
- pharmaceutically acceptable
- salt
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- PMTPFBWHUOWTNN-UHFFFAOYSA-N COc1ccnc(Cl)c1 Chemical compound COc1ccnc(Cl)c1 PMTPFBWHUOWTNN-UHFFFAOYSA-N 0.000 description 1
- RSWNZSUTNZTHOI-NSHDSACASA-N C[C@@H](CN(CC1)C(OC(C)(C)C)=O)N1c(nc1)cc(OC)c1Br Chemical compound C[C@@H](CN(CC1)C(OC(C)(C)C)=O)N1c(nc1)cc(OC)c1Br RSWNZSUTNZTHOI-NSHDSACASA-N 0.000 description 1
- RVJJEIWGPMKJKV-LBPRGKRZSA-N C[C@@H](CN(CC1)C(OC(C)(C)C)=O)N1c1cc(OC)ccn1 Chemical compound C[C@@H](CN(CC1)C(OC(C)(C)C)=O)N1c1cc(OC)ccn1 RVJJEIWGPMKJKV-LBPRGKRZSA-N 0.000 description 1
- KXJMJPMGZHFONJ-DLBZAZTESA-N C[C@H](c(c(Cl)c(cc1)F)c1Cl)Oc1c(N)ncc(-c(c(OC)c2)cnc2N(CC2)[C@@H](C)CN2C(OC(C)(C)C)=O)c1 Chemical compound C[C@H](c(c(Cl)c(cc1)F)c1Cl)Oc1c(N)ncc(-c(c(OC)c2)cnc2N(CC2)[C@@H](C)CN2C(OC(C)(C)C)=O)c1 KXJMJPMGZHFONJ-DLBZAZTESA-N 0.000 description 1
- BVGDAZBTIVRTGO-UONOGXRCSA-N C[C@H](c(c(Cl)ccc1F)c1Cl)Oc1c(N)ncc(-c(c(OC)c2)cnc2N2[C@@H](C)CNCC2)c1 Chemical compound C[C@H](c(c(Cl)ccc1F)c1Cl)Oc1c(N)ncc(-c(c(OC)c2)cnc2N2[C@@H](C)CNCC2)c1 BVGDAZBTIVRTGO-UONOGXRCSA-N 0.000 description 1
- GIXNNVOMGTZNCH-SNVBAGLBSA-N C[C@H](c(c(Cl)ccc1F)c1Cl)Oc1c(N)ncc(B2OC(C)(C)C(C)(C)O2)c1 Chemical compound C[C@H](c(c(Cl)ccc1F)c1Cl)Oc1c(N)ncc(B2OC(C)(C)C(C)(C)O2)c1 GIXNNVOMGTZNCH-SNVBAGLBSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/73—Unsubstituted amino or imino radicals
Definitions
- the present invention is in the field of medicinal chemistry, and in particular relates to salts of pharmaceutically acceptable acids of pyridine substituted 2-aminopyridine derivatives.
- Anaplastic lymphoma kinase is a receptor tyrosine kinase that is a member of the insulin receptor superfamily and plays an important role in tumor cell growth and development.
- the ALK gene can be fused to a variety of protein genes to express ALK proteins, as well as mutations, amplifications, and the like.
- ALK gene can be encoded by the fusion gene with the EML4 gene to produce ALK, thereby promoting lung cancer cell growth.
- EML4-ALK fusion is caused by short arm insertion of chromosome 2, and various variant types have been found so far. All of these fusion genes have been tested for biological function, and their expression products are a chimeric tyrosine kinase. Since 2007, they have gradually appeared in NSCLC related research reports.
- EML4-ALK fusion gene The discovery of the EML4-ALK fusion gene and the unique effects of ALK inhibitors in its subpopulations allow NSCLC to be divided into different subtypes depending on the molecular pathogenesis, such as EGFR mutant, KRAS mutant, EML4-ALK. Gene fusion type and the like. In patients with general non-small cell lung cancer, the positive rate of EML4-ALK fusion gene is low, about 3% to 7%. The EML4-ALK fusion gene is mainly found in non-smoker patients with lung adenocarcinoma.
- ALK kinase inhibitors are used to prevent, alleviate and/or treat cancers mediated by protein kinases (eg, ALK), such as ALK-positive non-small cell lung cancer (NSCLC) and the like.
- ALK protein kinases
- NSCLC ALK-positive non-small cell lung cancer
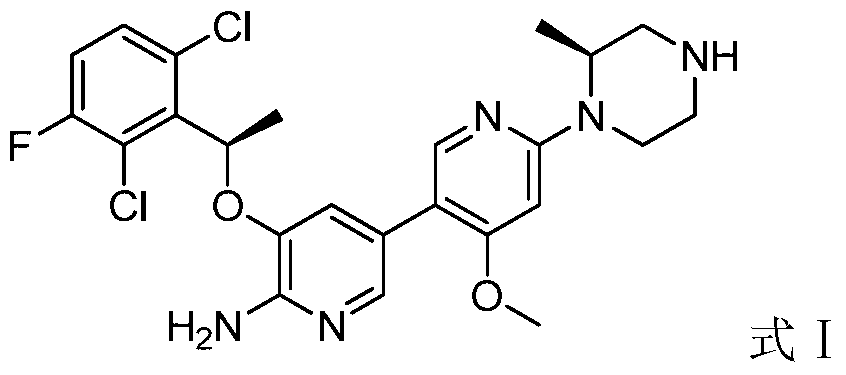
- the application provides 5-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-4'-methoxy-6'-((S)-
- a pharmaceutically acceptable acid salt of 2-methylpiperazin-1-yl)-3,3'-bipyridin-6-amine (having the structure of formula I below, hereinafter referred to as a compound of formula I).
- the application provides a method of preparing a salt of a pharmaceutically acceptable acid of the compound of Formula I, which comprises reacting the compound of Formula I with the pharmaceutically acceptable acid.
- the application provides a pharmaceutical composition
- a pharmaceutical composition comprising a therapeutically effective amount of a pharmaceutically acceptable acid salt of a compound of formula I and a pharmaceutically acceptable carrier, excipient or diluent.
- the application provides the use of a pharmaceutically acceptable acid salt of a compound of formula I or a pharmaceutical composition thereof for the manufacture of a medicament for the treatment and/or prevention of a protein kinase-associated disease.
- the application provides a method for treating and/or preventing a protein kinase-associated disease comprising administering a pharmaceutically acceptable acid salt of a compound of formula I or a pharmaceutical composition thereof to a mammal in need thereof
- the animal preferably a human, is administered.
- the application provides a pharmaceutically acceptable acid salt or a pharmaceutical composition thereof for use in the treatment and/or prevention of a protein kinase-associated disease.
- One embodiment or “implementation” or “in another embodiment” as referred to throughout this specification Or “in certain embodiments” is meant to include in a particular embodiment a particular reference element, structure, or feature that is associated with the embodiment.
- the appearances of the phrase “in one embodiment” or “in an embodiment” or “in another embodiment” or “in some embodiments” are not necessarily all referring to the same embodiment.
- the particular elements, structures, or characteristics may be combined in any suitable manner in one or more embodiments.
- a reaction including a “catalyst” includes a catalyst, or two or more catalysts.
- the term “or” is generally used in its meaning including “and/or” unless it is specifically defined otherwise.
- the application provides 5-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-4'-methoxy-6'-((S)-
- a pharmaceutically acceptable acid salt of 2-methylpiperazin-1-yl)-3,3'-bipyridin-6-amine (having the structure of formula I below, hereinafter referred to as a compound of formula I).
- the pharmaceutically acceptable acid comprises a mineral acid and an organic acid.
- examples of the inorganic acid include, but are not limited to, sulfuric acid, carbonic acid, nitric acid, hydrochloric acid, hydrobromic acid, hydroiodic acid, phosphoric acid, and metaphosphoric acid; and the organic acid includes an aliphatic organic acid and an aromatic organic acid, examples thereof Including but not limited to trifluoroacetic acid, lactic acid, fumaric acid, mandelic acid, glycolic acid, toluenesulfonic acid (eg p-toluenesulfonic acid, o-toluenesulfonic acid), citric acid, methanesulfonic acid, formic acid, acetic acid, benzoic acid, phenylacetic acid , malonic acid, cinnamic acid, malic acid, maleic acid, tartaric acid, oxalic acid, fumaric acid, acrylic acid, crotonic acid,
- the pharmaceutically acceptable acid is selected from the group consisting of sulfuric acid, hydrochloric acid, phosphoric acid, p-toluenesulfonic acid, citric acid, methanesulfonic acid, malic acid, maleic acid, tartaric acid, and fumaric acid.
- the pharmaceutically acceptable acid may be in an equimolar ratio (ie a molar ratio of the compound of the formula I to the acid of 1:1) or a different molar ratio
- the molar ratio of the compound of formula I to the pharmaceutically acceptable acid in the salt can be from 1:0.5 to 4, or from 1:1-3.
- the molar ratio of the compound of Formula I to the pharmaceutically acceptable acid is 1:0.5, 1:1, 1:1.5, 1:2, 1:2.5, 1:3, 1:3.5, or 1:4. .
- the pharmaceutically acceptable acid salt of the compound of Formula I provided herein is selected from the group consisting of the following salts.
- the molar ratio of the compound of formula I to hydrochloric acid is 1:0.5, 1:1, 1:1.5, 1 : 2, 1:2.5, 1:3, 1:3.5 or 1:4; in one embodiment, the molar ratio of the compound of the formula I to hydrochloric acid is 1:0.5; in one embodiment, the molar ratio of the compound of the formula I to hydrochloric acid Ratio 1:1; in one embodiment, the molar ratio of the compound of formula I to hydrochloric acid is 1:1.5; in one embodiment, the molar ratio of the compound of formula I to hydrochloric acid is 1:2; in one embodiment, the compound of formula I The molar ratio to hydrochloric acid is 1:2.5;
- the molar ratio of the compound of formula I to sulfuric acid is 1:0.5, 1:1, 1:1.5, 1: 2, 1:2.5, 1:3, 1:3.5 or 1:4; in one embodiment, the molar ratio of the compound of the formula I to sulfuric acid is 1:0.5; in one embodiment, the molar ratio of the compound of the formula I to the sulfuric acid 1:1; In one embodiment, the molar ratio of the compound of formula I to sulfuric acid is 1:1.5; in one embodiment, the molar ratio of the compound of formula I to sulfuric acid is 1:2; in one embodiment, the compound of formula I is The molar ratio of sulfuric acid is 1:2.5; in one embodiment, the molar ratio of the compound of
- the molar ratio of the compound of formula i and citric acid 1: 0.5, 1: 1, 1: 1.5 , 1:2, 1:2.5, 1:3, 1:3.5 or 1:4; in one embodiment, the molar ratio of the compound of formula I to citric acid is 1:0.5; in one embodiment, the compound of formula I is The molar ratio of citric acid is 1:1; in one embodiment, the molar ratio of the compound of formula I to citric acid is 1:1.5; in one embodiment, the molar ratio of the compound of formula I to citric acid is 1:2; in one implementation In the scheme, the molar ratio of the compound of formula I to citric acid is 1:2.5; in one embodiment, the molar ratio of the compound of formula I to
- the molar ratio of the compound of formula i and malic acid 1: 0.5, 1: 1, 1: 1.5 , 1:2, 1:2.5, 1:3, 1:3.5 or 1:4; in one embodiment, the molar ratio of the compound of the formula I to malic acid is 1:0.5; in one embodiment, the compound of the formula I
- the molar ratio of malic acid is 1:1; in one embodiment, the molar ratio of the compound of formula I to malic acid is 1:1.5; in one embodiment, the molar ratio of the compound of formula I to malic acid is 1:2; in one implementation In the scheme, the molar ratio of the compound of the formula I to malic acid is 1:2.5; in one embodiment, the molar ratio of the compound of
- the compound of formula i and maleic acid molar ratio of 1: 0.5, 1: 1,1 : 1.5, 1:2, 1:2.5, 1:3, 1:3.5 or 1:4; in one embodiment, the molar ratio of the compound of the formula I to maleic acid is 1:0.5; in one embodiment, The molar ratio of the compound I to the maleic acid is 1:1; in one embodiment, the molar ratio of the compound of the formula I to maleic acid is 1:1.5; in one embodiment, the molar ratio of the compound of the formula I to maleic acid is 1 In one embodiment, the molar ratio of the compound of formula I to maleic acid is 1:2.5; in one embodiment, the molar ratio of the compound of formula I to maleic acid
- the molar ratio of the compound of formula I to fumaric acid is 1:0.5; in one embodiment, the molar ratio of the compound of formula I to fumaric acid is 1:1; in one embodiment, the compound of formula I is The molar ratio of fumaric acid is 1:1.5; in one embodiment, the molar ratio of the compound of formula I to fumaric acid is 1:2; in one embodiment, the molar ratio of the compound of formula I to fumaric acid is 1:2.5; In one embodiment, the molar ratio of the compound of formula I to fumaric acid is 1:3.
- the present application provides 5-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-4'-methoxy-6'-((S)
- a process for the preparation of a pharmaceutically acceptable acid salt of 2-methylpiperazin-1-yl)-3,3'-bipyridyl-6-amine which comprises; 5-((R)-1-( 2,6-Dichloro-3-fluorophenyl)ethoxy)-4'-methoxy-6'-((S)-2-methylpiperazin-1-yl)-3,3'- Bipyridyl-6-amine (having the structure of formula I below, hereinafter referred to as a compound of formula I) is reacted with the pharmaceutically acceptable acid.
- the pharmaceutically acceptable acid comprises a mineral acid and an organic acid.
- the inorganic acid include, but are not limited to, sulfuric acid, carbonic acid, nitric acid, hydrochloric acid, hydrobromic acid, hydroiodic acid, phosphoric acid, and metaphosphoric acid.
- the organic acid includes an aliphatic organic acid and an aromatic organic acid, and examples thereof include, but are not limited to, trifluoroacetic acid, lactic acid, fumaric acid, mandelic acid, glycolic acid, toluenesulfonic acid (for example, p-toluenesulfonic acid, o-toluenesulfonic acid).
- citric acid, methanesulfonic acid formic acid, acetic acid, benzoic acid, phenylacetic acid, malonic acid, cinnamic acid, malic acid, maleic acid, tartaric acid, oxalic acid, fumaric acid, acrylic acid, crotonic acid, oleic acid and linoleic acid acid.
- the pharmaceutically acceptable acid is selected from the group consisting of sulfuric acid, hydrochloric acid, and phosphorus. Acid, p-toluenesulfonic acid, citric acid, methanesulfonic acid, malic acid, maleic acid, tartaric acid and fumaric acid.
- the compound of Formula I in the preparation of the pharmaceutically acceptable acid salt of the compound of Formula I, can be dissolved in a suitable single solvent or mixed solvent containing the corresponding acid.
- the reaction is carried out to obtain the salt, or the compound of the formula I is dissolved in a suitable single solvent or a mixed solvent, and then the corresponding acid is added thereto to carry out a reaction to obtain the salt.
- the pharmaceutically acceptable acid in the method of preparing a pharmaceutically acceptable acid salt of the compound of Formula I, can be in an equimolar ratio (ie, the mole of the compound of Formula I and the acid) The ratio is 1:1) or reacts with the compound of formula I in different molar ratios, depending on whether the acid is a monobasic or polybasic acid, and the desired salt.
- the molar ratio of the compound of formula I to the acid can be from 1:0.5 to 4, or from 1:1-3.
- the pharmaceutically acceptable acid salt of the compound of formula I can be prepared by adjusting the amount of the pharmaceutically acceptable acid, for example, using a stoichiometric amount or an excess of a pharmaceutically acceptable acid relative to the compound of formula I.
- the molar ratio of the compound of Formula I to the pharmaceutically acceptable acid is 1:0.5, 1:1, 1:1.5, 1:2, 1:2.5, 1 :3, 1:3.5 or 1:4.
- the reaction temperature may range from 0 ° C to the boiling point of the solvent system, preferably from 0 ° C to 50 ° C, more preferably It is preferably 0 ° C to 35 ° C.
- the reaction in the method of preparing a pharmaceutically acceptable acid salt of the compound of Formula I, can be carried out under the protection of an inert gas such as nitrogen.
- the reaction time can be from 0.5 to 5 h.
- suitable solvents include ethers, alcohols, ketones, nitriles, esters, alkanes, halogenated alkanes, and any combination thereof.
- suitable solvents include ethers, alcohols, ketones, nitriles, esters, alkanes, halogenated alkanes, and any combination thereof. Examples thereof include, but are not limited to, tetrahydrofuran, dioxane, diisopropyl ether, diethyl ether, methanol, ethanol, isopropanol, acetone, acetonitrile, ethyl acetate, ethyl formate, hexane, dichloromethane, chloroform and Any combination of them.
- the application provides a pharmaceutical composition
- a pharmaceutical composition comprising a therapeutically effective amount of a pharmaceutically acceptable acid salt of a compound of formula I and a pharmaceutically acceptable carrier, excipient or diluent.
- the "pharmaceutically acceptable carrier and excipient” means an inert substance which facilitates administration of the active ingredient, including but not limited to the acceptable use by the State Food and Drug Administration for human or animal (eg livestock). Any of the glidants, sweeteners, diluents, preservatives, dyes/colorants, flavor enhancers, surfactants, wetting agents, dispersants, disintegrants, suspending agents, stabilizers, isotonic agents Agent, solvent or emulsifier.
- Non-limiting examples of such carriers and excipients include, but are not limited to, calcium carbonate, calcium phosphate, various sugars and various types of starch, cellulose derivatives, gelatin, vegetable oils, and polyethylene glycols.
- the pharmaceutical composition of the present invention can be formulated into solid, semi-solid, liquid or gaseous preparations such as tablets, pills, capsules Agents, powders, granules, ointments, emulsions, suspensions, solutions, suppositories, injections, inhalants, gels, microspheres and aerosols, and the like.
- Typical routes of administration of the pharmaceutical compositions of the invention include, but are not limited to, oral, rectal, transmucosal, enteral, or topical, transdermal, inhalation, parenteral, sublingual, intravaginal, intranasal, intraocular, peritoneal Internal, intramuscular, subcutaneous, intravenous administration.
- a preferred route of administration is oral administration.
- the application provides a method of modulating protein kinase activity comprising contacting the protein kinase with a salt of a pharmaceutically acceptable acid of the compound of Formula I.
- the protein kinase is selected from the group consisting of ALK.
- the protein kinase comprises a mutated kinase, wherein the mutated kinase is selected from the group consisting of a mutated ALK kinase.
- the application provides the use of a pharmaceutically acceptable acid salt of a compound of formula I or a pharmaceutical composition thereof for the manufacture of a medicament for the treatment and/or prophylaxis of a disease, wherein the disease is with a protein kinase (eg, ALK) activity-related diseases, such as abnormal cell proliferation, wherein abnormal cell proliferation includes cancer.
- a protein kinase eg, ALK
- the present application also provides the use of a pharmaceutically acceptable acid salt of a compound of Formula I, or a pharmaceutical composition thereof, for the manufacture of a medicament for the treatment and/or prevention of a disease mediated by ALK .
- ALK-mediated diseases include, but are not limited to, ALK-positive non-small cell lung cancer, anaplastic large cell lymphoma, inflammatory myofibroblastic tumor, nasopharyngeal carcinoma, breast cancer, colorectal cancer, diffuse large B-cell lymphoid Tumors, systemic histiocytosis, neuroblastoma, and the like, preferably include ALK-positive non-small cell lung cancer.
- the present application also provides a method of treating and/or preventing a disease in a mammal (e.g., a human), wherein the disease is a disease associated with protein kinase (e.g., ALK) activity, the method comprising administering to a mammal (e.g., a human) A therapeutically effective amount of a pharmaceutically acceptable acid salt of the compound of formula I or a pharmaceutical composition thereof.
- a mammal e.g., a human
- a therapeutically effective amount of a pharmaceutically acceptable acid salt of the compound of formula I or a pharmaceutical composition thereof
- the protein kinase is selected from the group consisting of ALK.
- the protein kinase comprises a mutated kinase, wherein the mutated kinase is selected from the group consisting of a mutated ALK kinase.
- the present application also provides methods of treating and/or preventing a disease in a mammal (eg, a human), wherein the disease is a disease associated with ALK activity, the method comprising administering to a mammal (eg, a human) A therapeutically effective amount of a pharmaceutically acceptable acid salt of the compound of formula I or a pharmaceutical composition thereof.
- the diseases associated with ALK activity include, but are not limited to, ALK-positive non-small cell lung cancer, anaplastic large cell lymphoma, inflammatory myofibroblastic tumor, nasopharyngeal carcinoma, breast cancer, colorectal cancer, diffuse large B-cell lymphoid Tumors, systemic histiocytosis, neuroblastoma, and the like, preferably include ALK-positive non-small cell lung cancer.
- the present application also provides a pharmaceutically acceptable acid salt of the compound of formula I for use in modulating protein kinase activity or for treating and/or preventing a disease associated with protein kinase activity in a mammal (eg, a human) or Their pharmaceutical compositions.
- the protein kinase is preferably ALK.
- the protein kinase comprises a mutated kinase, wherein the mutated kinase is selected from the group consisting of a mutated ALK kinase.
- the present application also provides a pharmaceutically acceptable acid of the compound of Formula I for use in modulating ALK activity or for treating and/or preventing a disease associated with ALK activity in a mammal (eg, a human) Salts or their pharmaceutical compositions.
- the diseases associated with ALK activity include, but are not limited to, ALK-positive non-small cell lung cancer, anaplastic large cell lymphoma, inflammatory myofibroblastic tumor, nasopharyngeal carcinoma, breast cancer, colorectal cancer, diffuse large B-cell lymphoid Tumors, systemic histiocytosis, neuroblastoma, and the like, preferably include ALK-positive non-small cell lung cancer.
- Step 1 Dissolve (S)-1-(2,6-dichloro-3-fluorophenyl)ethanol (20.9 g, 0.10 mol) in 200 mL of anhydrous tetrahydrofuran, and sequentially add 3-hydroxy-2 under nitrogen atmosphere. -Nitropyridine (16.0 g, 0.11 mol) and triphenylphosphine (40.0 g, 0.15 mol), and the reaction mixture was stirred at room temperature for 1 hour. After cooling to 0 ° C, diisopropyl azodicarboxylate (40 mL, 0.15 mol) was added dropwise, and after stirring, stirring was continued at 0 ° C for 12 hours. The solvent was evaporated to give an oily crystal. 20.2 g), yield: 61%.
- Step 2 (R)-3-(1-(2,6-Dichloro-3-fluorophenyl)ethoxy)-2-nitropyridine (20.0 g, 60 mmol) at 0 ° C with stirring
- To the ethanol solution (300 mL) 15 mL of 2M hydrochloric acid and reduced iron powder (27 g, 480 mmol) were added. After the addition, the mixture was heated to reflux for 12 hours. After cooling to room temperature, filtration and concentration of the filtrate, (R)-3-(1-(2,6-dichloro-3-fluorophenyl)ethoxy)-2-aminopyridine (17.0 g) was obtained. Directly used in the next step, yield: 94%. MS m/z [ESI]: 301.0 [M + 1].
- Step 3 To a solution of (R)-3-(1-(2,6-dichloro-3-fluorophenyl)ethoxy)-2-aminopyridine (15.0 g, 50 mmol) at 0 ° C. Bromosuccinimide (10 g, 56 mmol) was added portionwise in acetonitrile (200 mL). The addition was carried out at 0 ° C for 1 hour. Then, the solvent was evaporated, the methylene chloride was added, and the organic layer was washed with saturated aqueous sodium hydrogen sulfate, and dried over anhydrous sodium sulfate. -(2,6-Dichloro-3-fluorophenyl)ethoxy)-5-bromo-2-aminopyridine (9.88 g), yield: 52%. MS m/z [ESI]: 380.9 [M + 1].
- Step 4 (R)-3-(1-(2,6-Dichloro-3-fluorophenyl)ethoxy)-5-bromo-2-aminopyridine (7.6 g, 20 mmol), boronic acid Which alcohol ester (7.56g, 30mmol), Pd(dppf)Cl 2 (732mg, 1mmol) and anhydrous potassium acetate (4.90g, 50mmol) were added to dry dioxane (200mL), replaced with nitrogen at 100 ° C Reaction for 4 hours.
- Step 1 2-Chloro-4-methoxypyridine (2.58 g, 18 mmol), (S)-3-methyl-1-tert-butoxycarbonylpiperazine (5.4 g, 27 mmol), Pd 2 (dba) 3 (824 mg, 0.9 mmol), BINAP (1.12 g, 1.8 mmol) and potassium tert-butoxide (5.01 g, 45 mmol) were added to dry toluene (200 mL) and refluxed for 16 hr under nitrogen atmosphere. Then, the reaction mixture was cooled to room temperature, filtered, and concentrated under reduced pressure. MS m/z [ESI]: 308.2 [M + 1].
- Step 2 To a solution of (S)-4-(4-methoxypyridin-2-yl)-3-methylpiperazine-1-carboxylic acid tert-butyl ester (2.46 g, 8 mmol). Bromosuccinimide (1.57 g, 8.8 mmol) was added portionwise in acetonitrile (50 mL). The reaction was carried out for 2 hours at room temperature. The solvent was evaporated under reduced pressure. dichloromethane was evaporated, evaporated, evaporated, evaporated, evaporated. 75%. MS m/z [ESI]: 386.1 [M + 1].
- Step 1 tert-Butyl (S)-4-(5-bromo-4-methoxypyridin-2-yl)-3-methylpiperazine-1-carboxylate (106 mg, 0.275 mmol), (R) )-3-(1-(2,6-Dichloro-3-fluorophenyl)ethoxy)-5-(4,4,5,5-tetramethyl-1,3,2-dioxa Cyclopentane-2-yl)-2-aminopyridine (140 mg, 0.33 mmol), tetrakis(triphenylphosphine)palladium (32 mg, 0.0275 mmol) and cesium carbonate (179 mg, 0.55 mmol) were added to dioxane ( 10 mL) and water (1.5 mL) were replaced with nitrogen and reacted at 100 ° C overnight.
- Step 2 To 5-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-4'-methoxy-6'-(( Add a solution of S)-2-methyl-4-tert-butoxycarbonylpiperazin-1-yl)-3,3'-bipyridin-6-amine (67 mg, 0.11 mmol) in dichloromethane (10 mL) The fluoroacetic acid (1 mL) was stirred for 1 hour, then the pH of the reaction mixture was adjusted to more than 13 with 10 mol/L sodium hydroxide solution, then extracted with dichloromethane, and the organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated.
- the inhibitory activity of IC 50 uses the indicators to represent, i.e., IC 50 activity of ALK kinase, is inhibited by 50% of the compound.
- the homogeneous activity time-resolved fluorescence (HTRF, Cisbio) method was used to establish and optimize the kinase activity detection platform of ALK (purchased from Millipore) for the determination of compound activity.
- HEPES buffer Prepare 50ml 0.05M HEPES buffer with 1M HEPES buffer (Invitrogen, Catalog No. 15630-080), take 1M HEPES buffer 2.5ml, add appropriate amount of distilled water (ddH 2 O), with NaOH The pH was adjusted to 7.0 and finally ddH 2 O (re-distilled water) was added to 50 ml.
- Test compound a compound of formula I
- the compound was diluted 3 times with 100% DMSO starting from 1 mM, 4 ⁇ l of each concentration was added to 96 ⁇ l of reaction buffer, 2.5 ⁇ l was added to a 384-well plate (OptiPlate-384, PerkinElmer), and then added. 5 ⁇ l of the kinase was mixed by centrifugation, and then 2.5 ⁇ l of ATP was added to the TK peptide mixture (the final concentration of ATP was Km) to initiate the reaction.
- the compound inhibits generation of ALK enzyme IC 50: IC 50 values calculated using compound GraFit6.
- IC above compound of formula I 50 measured value of 1.96nM. This data indicates that the compound of formula I has excellent ALK inhibitory activity.
- Table 1 lists the ALK kinase inhibitory activity of the compound of formula I against the mutation.
- ALK kinases in which L1196M and G1269S mutations are commercially available are commercially available.
- the experimental method was as follows: nude mice were subcutaneously inoculated with human non-small cell lung cancer NCI-H2228 cells, and after the tumors were grown to 80-200 mm 3 , the animals were randomly divided into groups (D0) and administered, and the dosage and administration schedule were specifically See Table 2. The tumor volume was measured twice a week, the rats were weighed, and data were recorded. The tumor volume (V) is calculated as:
- V 1/2 ⁇ a ⁇ b 2 , where a and b represent length and width, respectively.
- T/C(%) (TT 0 )/(CC 0 ) ⁇ 100, where T and C are the tumor volumes at the end of the experiment, respectively; T 0 and C 0 are the tumor volumes at the beginning of the experiment, respectively.
- T/C (%) (TT 0 ) / T 0 ⁇ 100, where T is the tumor volume at the end of the experiment; T 0 is the tumor volume at the beginning of the experiment.
- the tumor inhibition rate (%) 100-T/C (%), partial regression means that the tumor becomes small but does not disappear, and complete regression means that the tumor disappears.
- solvent distilled water containing 0.1% Tween-80
- treatment group was prepared with distilled water containing 0.1% Tween-80.
- D0-20 indicates that continuous administration was performed once a day for 21 days from 0 days (D0); D0-13 indicates that administration was continued once every day for 14 days from 0 days.
- n is the number of mice, the number of experimental mice is 12 for the control group, and n for the treatment group.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description








Claims (25)
- 式Ⅰ化合物的药学上可接受酸的盐,其中式Ⅰ化合物具有如下结构:

- 如权利要求1所述的式Ⅰ化合物的药学上可接受酸的盐,其中所述药学上可接受酸包括无机酸和有机酸。
- 如权利要求1所述的式Ⅰ化合物的药学上可接受酸的盐,其中所述药学上可接受酸选自硫酸、碳酸、硝酸、盐酸、氢溴酸、氢碘酸、磷酸、偏磷酸、三氟乙酸、乳酸、延胡索酸、扁桃酸、乙醇酸、对甲苯磺酸、邻甲苯磺酸、柠檬酸、甲磺酸、甲酸、乙酸、苯甲酸、苯乙酸、丙二酸、肉桂酸、苹果酸、马来酸、酒石酸、草酸、富马酸、丙烯酸、巴豆酸、油酸和亚油酸。
- 如权利要求1所述的式Ⅰ化合物的药学上可接受酸的盐,其选自:5-((R)-1-(2,6-二氯-3-氟苯基)乙氧基)-4'-甲氧基-6'-((S)-2-甲基哌嗪-1-基)-3,3'-联吡啶-6-胺盐酸盐,5-((R)-1-(2,6-二氯-3-氟苯基)乙氧基)-4'-甲氧基-6'-((S)-2-甲基哌嗪-1-基)-3,3'-联吡啶-6-胺硫酸盐,5-((R)-1-(2,6-二氯-3-氟苯基)乙氧基)-4'-甲氧基-6'-((S)-2-甲基哌嗪-1-基)-3,3'-联吡啶-6-胺柠檬酸盐,5-((R)-1-(2,6-二氯-3-氟苯基)乙氧基)-4'-甲氧基-6'-((S)-2-甲基哌嗪-1-基)-3,3'-联吡啶-6-胺苹果酸盐,5-((R)-1-(2,6-二氯-3-氟苯基)乙氧基)-4'-甲氧基-6'-((S)-2-甲基哌嗪-1-基)-3,3'-联吡啶-6-胺马来酸盐,5-((R)-1-(2,6-二氯-3-氟苯基)乙氧基)-4'-甲氧基-6'-((S)-2-甲基哌嗪-1-基)-3,3'-联吡啶-6-胺酒石酸盐,和5-((R)-1-(2,6-二氯-3-氟苯基)乙氧基)-4'-甲氧基-6'-((S)-2-甲基哌嗪-1-基)-3,3'-联吡啶-6-胺富马酸盐。
- 制备权利要求1-4中任一项所述的式Ⅰ化合物的药学上可接受酸的盐的方法,其包括将 所述式Ⅰ化合物和所述药学上可接受酸反应。
- 如权利要求5所述的制备方法,其中所述药学上可接受酸选自硫酸、碳酸、硝酸、盐酸、氢溴酸、氢碘酸、磷酸、偏磷酸、三氟乙酸、乳酸、延胡索酸、扁桃酸、乙醇酸、对甲苯磺酸、邻甲苯磺酸、柠檬酸、甲磺酸、甲酸、乙酸、苯甲酸、苯乙酸、丙二酸、肉桂酸、苹果酸、马来酸、酒石酸、草酸、富马酸、丙烯酸、巴豆酸、油酸和亚油酸,优选盐酸、硫酸、柠檬酸、苹果酸、马来酸、酒石酸和富马酸。
- 如权利要求5或6所述的制备方法,其中通过将所述式Ⅰ化合物溶于含有相应的药学上可接受酸的单一溶剂或混合溶剂中来进行反应以获得所述盐,或者将所述式Ⅰ化合物溶于单一溶剂或混合溶剂中,随后向其中加入相应的药学上可接受酸来进行反应以获得所述盐。
- 如权利要求5-7中任一项所述的制备方法,其中所述式Ⅰ化合物与所述药学上可接受酸的摩尔比可为1:0.5-4或1:1-3,例如1:0.5、1:1、1:1.5、1:2、1:2.5、1:3、1:3.5或1:4。
- 如权利要求5-8中任一项所述的制备方法,其中实施反应的温度可为0℃至溶剂系统的沸点,优选0℃-50℃,更优选0℃-35℃。
- 如权利要求5-9中任一项所述的制备方法,其中所述反应在诸如氮气氛的惰性气氛中实施。
- 如权利要求5-10中任一项所述的制备方法,其中所述反应的时间为0.5-5h。
- 如权利要求7-11中任一项所述的制备方法,其中所述溶剂选自四氢呋喃、二氧六环、异丙醚、乙醚、甲醇、乙醇、异丙醇、丙酮、乙腈、乙酸乙酯、甲酸乙酯、己烷、二氯甲烷、三氯甲烷及它们的任意组合。
- 药物组合物,其含有治疗有效量的权利要求1-4中任一项所述的式Ⅰ化合物的药学上可接受酸的盐和药学上可接受的载体、赋形剂或稀释剂。
- 权利要求1-4中任一项所述的式Ⅰ化合物的药学上可接受酸的盐或权利要求13所述的药物组合物在制备用于治疗和/或预防与蛋白激酶相关的疾病的药物中的用途。
- 权利要求1-4中任一项所述的式Ⅰ化合物的药学上可接受酸的盐或权利要求13所述的药物组合物在制备用于治疗和/或预防与突变的蛋白激酶相关的疾病的药物中的用途。
- 权利要求1-4中任一项所述的式Ⅰ化合物的药学上可接受酸的盐或权利要求13所述的药物组合物在制备用于治疗和/或预防由ALK介导的疾病的药物中的用途。
- 如权利要求16所述的用途,其中所述由ALK介导的疾病包括ALK阳性的非小细胞肺癌、间变性大细胞淋巴瘤、炎性肌纤维母细胞瘤、鼻咽癌、乳腺癌、结直肠癌、弥漫大B 细胞淋巴瘤、全身组织细胞增生症和神经母细胞瘤。
- 用于治疗和/或预防与蛋白激酶相关的疾病的方法,其包括将权利要求1-4中任一项所述的式Ⅰ化合物的药学上可接受酸的盐或权利要求13所述的药物组合物对有需要的哺乳动物、优选人进行给药。
- 用于治疗和/或预防与突变的蛋白激酶相关的疾病的方法,其包括将权利要求1-4中任一项所述的式Ⅰ化合物的药学上可接受酸的盐或权利要求13所述的药物组合物对有需要的哺乳动物、优选人进行给药。
- 用于治疗和/或预防由ALK介导的疾病的方法,其包括将权利要求1-4中任一项所述的式Ⅰ化合物的药学上可接受酸的盐或权利要求13所述的药物组合物对有需要的哺乳动物、优选人进行给药。
- 如权利要求20所述的方法,其中所述由ALK介导的疾病包括ALK阳性的非小细胞肺癌、间变性大细胞淋巴瘤、炎性肌纤维母细胞瘤、鼻咽癌、乳腺癌、结直肠癌、弥漫大B细胞淋巴瘤、全身组织细胞增生症和神经母细胞瘤。
- 用于治疗和/或预防与蛋白激酶相关的疾病的权利要求1-4中任一项所述的式Ⅰ化合物的药学上可接受酸的盐或权利要求13所述的药物组合物。
- 用于治疗和/或预防与突变的蛋白激酶相关的疾病的权利要求1-4中任一项所述的式Ⅰ化合物的药学上可接受酸的盐或权利要求13所述的药物组合物。
- 用于治疗和/或预防由ALK介导的疾病的权利要求1-4中任一项所述的式Ⅰ化合物的药学上可接受酸的盐或权利要求13所述的药物组合物。
- 如权利要求24所述的式Ⅰ化合物的药学上可接受酸的盐或药物组合物,其中所述由ALK介导的疾病包括ALK阳性的非小细胞肺癌、间变性大细胞淋巴瘤、炎性肌纤维母细胞瘤、鼻咽癌、乳腺癌、结直肠癌、弥漫大B细胞淋巴瘤、全身组织细胞增生症和神经母细胞瘤。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP15826817.7A EP3176160B1 (en) | 2014-07-31 | 2015-07-31 | Pyridine-substituted 2-aminopyridine protein kinase inhibitors |
| ES15826817T ES2762641T3 (es) | 2014-07-31 | 2015-07-31 | Inhibidores de proteína quinasa de 2-aminopiridina sustituida con piridina |
| JP2017505496A JP6581180B2 (ja) | 2014-07-31 | 2015-07-31 | ピリジン置換の2−アミノピリジン類タンパク質キナーゼ阻害剤 |
| CN201580039739.6A CN106536510B (zh) | 2014-07-31 | 2015-07-31 | 吡啶取代的2-氨基吡啶类蛋白激酶抑制剂 |
| US15/329,046 US9981946B2 (en) | 2014-07-31 | 2015-07-31 | Pyridine-substituted 2-aminopyridine protein kinase inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201410371344 | 2014-07-31 | ||
| CN201410371344.3 | 2014-07-31 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016015676A1 true WO2016015676A1 (zh) | 2016-02-04 |
Family
ID=55216786
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2015/085727 Ceased WO2016015676A1 (zh) | 2014-07-31 | 2015-07-31 | 吡啶取代的2-氨基吡啶类蛋白激酶抑制剂 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US9981946B2 (zh) |
| EP (1) | EP3176160B1 (zh) |
| JP (1) | JP6581180B2 (zh) |
| CN (1) | CN106536510B (zh) |
| ES (1) | ES2762641T3 (zh) |
| WO (1) | WO2016015676A1 (zh) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110283161A (zh) * | 2015-07-30 | 2019-09-27 | 正大天晴药业集团股份有限公司 | 吡啶取代的2-氨基吡啶类蛋白激酶抑制剂的结晶 |
| WO2021018310A1 (zh) * | 2019-08-01 | 2021-02-04 | 正大天晴药业集团股份有限公司 | 用于治疗非小细胞肺癌的氨基吡啶衍生物 |
| WO2021219137A1 (zh) * | 2020-04-30 | 2021-11-04 | 正大天晴药业集团股份有限公司 | 用于治疗met基因异常疾病的氨基吡啶衍生物 |
| CN113754579A (zh) * | 2020-06-01 | 2021-12-07 | 正大天晴药业集团股份有限公司 | 一种吡啶衍生物的制备方法 |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7270633B2 (ja) * | 2018-03-14 | 2023-05-10 | チア タイ ティエンチン ファーマシューティカル グループ カンパニー リミテッド | 上咽頭がん治療用キノリン誘導体 |
| CN112294813A (zh) * | 2019-07-30 | 2021-02-02 | 正大天晴药业集团股份有限公司 | 喹啉衍生物在治疗脊索瘤中的用途 |
| CN112336726A (zh) * | 2019-08-09 | 2021-02-09 | 正大天晴药业集团股份有限公司 | 治疗结直肠癌的联用药物组合物 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006021886A1 (en) * | 2004-08-26 | 2006-03-02 | Pfizer Inc. | Aminoheteroaryl compounds as protein tyrosine kinase inhibitors |
| CN103965168A (zh) * | 2013-02-02 | 2014-08-06 | 正大天晴药业集团股份有限公司 | 芳基、杂芳基取代的2-氨基吡啶类蛋白激酶抑制剂 |
| CN104650049A (zh) * | 2013-08-28 | 2015-05-27 | 广东东阳光药业有限公司 | 取代的吡啶化合物及其使用方法和用途 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BR122020017756B1 (pt) | 2004-08-26 | 2022-02-15 | Pfizer Inc | Uso de compostos de aminoeteroarila enantiomericamente puros na preparação de um medicamento para o tratamento de crescimento celular anormal em um mamífero |
| WO2009099982A1 (en) | 2008-02-04 | 2009-08-13 | Osi Pharmaceuticals, Inc. | 2-aminopyridine kinase inhibitors |
| EP2566858A2 (en) | 2010-05-04 | 2013-03-13 | Pfizer Inc. | Heterocyclic derivatives as alk inhibitors |
| MX2013009551A (es) | 2011-02-24 | 2013-09-06 | Jiangsu Hanson Pharmaceutical Co Ltd | Compuestos que contienen fosforo como inhibidores de proteina cinasa. |
| CN102718745A (zh) | 2011-03-30 | 2012-10-10 | 中国科学院上海药物研究所 | 新型胺基吡啶类化合物、其制备方法、包含此类化合物的药物组合物及其用途 |
| AU2014211856C1 (en) | 2013-02-02 | 2018-04-12 | Centaurus Biopharma Co., Ltd. | Substituted 2-aminopyridine protein kinase inhibitor |
| US10385038B2 (en) * | 2015-07-30 | 2019-08-20 | Chia Tai Tianqing Pharmaceutical Group Co., Ltd. | Pyridine substituted 2-aminopyridine protein kinase inhibitor crystal |
-
2015
- 2015-07-31 WO PCT/CN2015/085727 patent/WO2016015676A1/zh not_active Ceased
- 2015-07-31 JP JP2017505496A patent/JP6581180B2/ja active Active
- 2015-07-31 EP EP15826817.7A patent/EP3176160B1/en active Active
- 2015-07-31 CN CN201580039739.6A patent/CN106536510B/zh active Active
- 2015-07-31 US US15/329,046 patent/US9981946B2/en active Active
- 2015-07-31 ES ES15826817T patent/ES2762641T3/es active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006021886A1 (en) * | 2004-08-26 | 2006-03-02 | Pfizer Inc. | Aminoheteroaryl compounds as protein tyrosine kinase inhibitors |
| CN103965168A (zh) * | 2013-02-02 | 2014-08-06 | 正大天晴药业集团股份有限公司 | 芳基、杂芳基取代的2-氨基吡啶类蛋白激酶抑制剂 |
| CN104650049A (zh) * | 2013-08-28 | 2015-05-27 | 广东东阳光药业有限公司 | 取代的吡啶化合物及其使用方法和用途 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3176160A4 * |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110283161A (zh) * | 2015-07-30 | 2019-09-27 | 正大天晴药业集团股份有限公司 | 吡啶取代的2-氨基吡啶类蛋白激酶抑制剂的结晶 |
| CN110330483A (zh) * | 2015-07-30 | 2019-10-15 | 正大天晴药业集团股份有限公司 | 吡啶取代的2-氨基吡啶类蛋白激酶抑制剂的结晶 |
| CN110283161B (zh) * | 2015-07-30 | 2021-09-03 | 正大天晴药业集团股份有限公司 | 吡啶取代的2-氨基吡啶类蛋白激酶抑制剂的结晶 |
| WO2021018310A1 (zh) * | 2019-08-01 | 2021-02-04 | 正大天晴药业集团股份有限公司 | 用于治疗非小细胞肺癌的氨基吡啶衍生物 |
| WO2021219137A1 (zh) * | 2020-04-30 | 2021-11-04 | 正大天晴药业集团股份有限公司 | 用于治疗met基因异常疾病的氨基吡啶衍生物 |
| CN115484955A (zh) * | 2020-04-30 | 2022-12-16 | 正大天晴药业集团股份有限公司 | 用于治疗met基因异常疾病的氨基吡啶衍生物 |
| CN113754579A (zh) * | 2020-06-01 | 2021-12-07 | 正大天晴药业集团股份有限公司 | 一种吡啶衍生物的制备方法 |
| CN113754579B (zh) * | 2020-06-01 | 2025-08-01 | 正大天晴药业集团股份有限公司 | 一种吡啶衍生物的制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3176160B1 (en) | 2019-09-18 |
| US9981946B2 (en) | 2018-05-29 |
| JP6581180B2 (ja) | 2019-09-25 |
| EP3176160A1 (en) | 2017-06-07 |
| JP2017521475A (ja) | 2017-08-03 |
| CN106536510B (zh) | 2019-04-26 |
| ES2762641T3 (es) | 2020-05-25 |
| CN106536510A (zh) | 2017-03-22 |
| US20170226084A1 (en) | 2017-08-10 |
| EP3176160A4 (en) | 2017-12-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10064848B2 (en) | Pyridic ketone derivatives, method of preparing same, and pharmaceutical application thereof | |
| WO2016015676A1 (zh) | 吡啶取代的2-氨基吡啶类蛋白激酶抑制剂 | |
| EP2952510B1 (en) | Substituted 2-aminopyridine protein kinase inhibitor | |
| EP2799437B1 (en) | Quinoline and cinnoline derivatives and use thereof | |
| CN102548987B (zh) | 作为激酶抑制剂的氟取代化合物及其使用方法 | |
| AU2019218187B2 (en) | Dioxinoquinoline compounds, preparation method and uses thereof | |
| KR101301533B1 (ko) | 암세포 성장 억제 효과를 갖는 신규 피리미딘 유도체 | |
| US20140350050A1 (en) | Pyridine compounds as inhibitors of kinase | |
| JP6513294B2 (ja) | ピリジン置換の2−アミノピリジン類タンパク質キナーゼ阻害剤の結晶 | |
| KR102685187B1 (ko) | Alk 및/또는 egfr 돌연변이 키나제 억제 효과를 나타내는 화합물 및 이의 의약 용도 | |
| JP2010111702A (ja) | 複素環化合物、その製造法および用途 | |
| US11407760B2 (en) | Dioxinoquinoline compounds, preparation method and uses thereof | |
| CN106146468B (zh) | 吡啶酮类蛋白激酶抑制剂 | |
| KR20190003242A (ko) | 변이 상피세포성장인자 수용체 키나아제 저해제로서 융합 피리미딘 유도체 | |
| CN112442105A (zh) | 作为蛋白激酶抑制剂的新型嘧啶并[4,5-d]嘧啶-2-酮衍生物 | |
| CN114555597A (zh) | 异柠檬酸脱氢酶(idh)抑制剂 | |
| WO2019170086A1 (zh) | 一种酰基取代的噁嗪并喹唑啉类化合物、制备方法及其应用 | |
| WO2023046114A1 (zh) | 蝶啶酮衍生物及其应用 | |
| WO2023010354A1 (zh) | 一种具有egfr抑制活性的小分子化合物及其制备方法与应用 | |
| CN118059101A (zh) | 8-羟基喹啉衍生物的用途 | |
| CN120398871A (zh) | 用于预防或治疗癌症的新型杂环化合物和包括与DNA聚合酶θ抑制剂相同的杂环化合物的药物组合物 | |
| HK40042713B (zh) | 一种恶嗪并喹唑啉与恶嗪并喹啉类化合物及其制备方法和应用 | |
| HK40042713A (zh) | 一种恶嗪并喹唑啉与恶嗪并喹啉类化合物及其制备方法和应用 | |
| HK1210774B (zh) | 取代的2-氨基吡啶类蛋白激酶抑制剂 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15826817 Country of ref document: EP Kind code of ref document: A1 |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015826817 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2015826817 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15329046 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2017505496 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |