BR122020017756B1 - Uso de compostos de aminoeteroarila enantiomericamente puros na preparação de um medicamento para o tratamento de crescimento celular anormal em um mamífero - Google Patents
Uso de compostos de aminoeteroarila enantiomericamente puros na preparação de um medicamento para o tratamento de crescimento celular anormal em um mamífero Download PDFInfo
- Publication number
- BR122020017756B1 BR122020017756B1 BR122020017756-0A BR122020017756A BR122020017756B1 BR 122020017756 B1 BR122020017756 B1 BR 122020017756B1 BR 122020017756 A BR122020017756 A BR 122020017756A BR 122020017756 B1 BR122020017756 B1 BR 122020017756B1
- Authority
- BR
- Brazil
- Prior art keywords
- added
- mmol
- solution
- compound
- pyridin
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/10—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D241/14—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D241/18—Oxygen or sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4418—Non condensed pyridines; Hydrogenated derivatives thereof having a carbocyclic group directly attached to the heterocyclic ring, e.g. cyproheptadine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/62—Oxygen or sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/73—Unsubstituted amino or imino radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/76—Nitrogen atoms to which a second hetero atom is attached
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/10—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D241/14—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D241/20—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Oncology (AREA)
- Hematology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pyridine Compounds (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
são providenciados compostos enantiomericamente puros de fórmula i, bem como métodos para sua síntese e uso. os compostos preferidos são potentes inibidores da proteína quinase c-met, e de utilidade no tratamento de distúrbios do crescimento celular anormal, tais como câncer.
Description
[001] A presente invenção refere-se, genericamente, a novos compostos químicos e métodos. Mais particularmente, a invenção propicia compostos aminoeteroarila enantiomericamente puros, particularmente aminopiridinas e aminopirazinas, com atividade proteína tirosina quinase, e a métodos de sinterização e uso de tais compostos. Compostos preferidos são inibidores de c-Met úteis para tratamento do crescimento celular anormal, tal como câncer.
[002] O fator de crescimento de hepatócito (HGF), receptor (c- MET ou HGRF) receptor de tirosina quinase (RTK) demonstraram, em muitos cânceres humanos estarem envolvidos na ontogênese, progresso do tumor com motilidade e invasão celular intensificada, bem como metástase (ver, por exemplo, Ma, P.C. Maulik, G., Christensen, J. & Salgia, R (2003b), Câncer Metastasis Rev., 22, 30925; Maulik, G. Shrikhande, A., Kijima, T., Ma, P.c. Morrison, P.T. & Satgia, R. (2002b). Cytokine Growth Factor Rev., 13, 41-59). c-MET (HGFR) pode ser ativado através da superexpressão ou mutações em vários cânceres humanos, incluindo câncer de pulmão de célula pequena (SCLC) (Ma, P.C. Kijima, T., Maulik, G. Fox, E.A. Sattler, M. Griffin, J.D. Johnson, B.E. & Salgia, R. (2003a), Câncer REs 63, 6272-6281).
[003] c-MET é um receptor de tirosina quinase codificado pelo proto-oncogene Met e traduz os efeitos biológicos do fator de crescimento de hepatócito (HGF), que também é referido como um fator de disseminação (SF). Jiang et al., Crit. Rev. Oncol. Hematol. 29. 209-248 (1999) c-MET e HGF são expressados em numerosos tecidos, embora sua expressão esteja, normalmente confinada predominantemente a células de origem epitelial e mesenquimal, respectivamente. c-MET e HGF são necessários para desenvolvimento normal do mamífero e, demonstraram ser importantes na migração celular, proliferação celular e sobrevivência, morfogenética, diferenciação, e organização das estruturas tubulares tridimensionais (por exemplo, células tubulares renais, formação de glândula, etc.), Além de seus efeitos nas células epiteliais, HGF/SF foram descritos como um fator angiogênico, e a sinalização de c- METG nas células endoteliais pode induzir muitas respostas celulares, necessárias para angiogênese (proliferação, motilidade, invasão).
[004] O receptor c-MET demonstrou ser expressado numa série de cânceres humanos. c-MET e seu ligante, HGF, demonstraram também ser co-expressados a elevados níveis numa gama de cânceres humanos (particularmente, sarcomas). Contudo, devido ao receptor e ligante serem normalmente expressados por diferentes tipos de células, a sinalização de c-MET é mais comumente regulada por interações tumor-estroma (tumor-hospedeiro). Além disso, a ampliação, mutação e rearranjo genético de c-MET foram observados num subconjunto de cânceres humanos. Famílias com mutações de linha germinativa que ativam c-MET quinase estão sujeitas a múltiplos tumores de rim, bem como tumores em outros tecidos. Numerosos testes correlacionaram-se à expressão de c-MET e/ou HGF/SF com o estado do progresso da doença de diferentes tipos de câncer (incluindo pulmão, cólon, mama, próstata, fígado, pâncreas, cérebro, rim, ovários, estômago, pele e cânceres ósseos). Além disso, a superexpressão de c-MET ou HGf demonstrou correlacionar-se com o prognóstico fraco com conseqüência de doença numa série de cânceres humanos principais, incluindo pulmão, fígado, câncer gástrico e de mama. c-MET também esteve diretamente implicado em cânceres sem um regime de tratamento de sucesso, tal como câncer pancreático, glioma, e carcinoma hepatocelular.
[005] Exemplos de inibidores de c-MET (HGFR), sua síntese e uso, podem ser encontrados no Pedido de Patente U.S. n° de série 10/786.610 intitulado "Aminoheteroaryl Compounds as Protein Kinase Inhibitors" depositado em 26 de fevereiro de 2004, e o correspondente pedido internacional PCT/US2004/005495 do mesmo título, depositado em 26 de fevereiro de 2004, cuja descrição está ora incorporadas a título de referência ao presente, em sua totalidade.
[006] Seria conveniente ter-se novos inibidores de c-MET (HGFR) e métodos de uso de tais inibidores para o tratamento do crescimento celular anormal, tal como câncer.
[007] Numa modalidade, a invenção providencia o composto enantiomericamente puro de fórmula: onde:
onde:
[008] Y é N ou CR12,
[009] R1 é selecionado dentre hidrogênio, halogênio, C6-12 arila, heteroarila de 5-12 membros, C3-12 cicloalquila, heteroalicíclico de 3-12 membros, -O(CR6R7)nR4, -C(O)R4, -C(O)OR4, -CN, -NO2, -S(O)mR4, -SO2NR4R5, -C(O)NR4R5-C(=NR6)NR4R5, -NR4C(O)R5, C1-8 alquila, C28 alquenila, e C2-8 alquinila, em cada hidrogênio e R1 é opcionalmente substituído com um ou mais grupos R3;
[0010] R2 é hidrogênio, halogênio, C1-12 alquila, C2-12 alquenila, C2-12 alquinila, C3-12 cicloalquila, C6-12 arila, heteroalicíclico de 3-12 membros, heteroarila de 5-12 membros, -S(O)mR4, -SO2NR4R5, -S(O)2OR4, -NO2, -NR4R5, -(CR6R7)nOR4, -CN, -C(O)R4, -OC(O)R4, O(CR6R7)nR4, -NR4C(O)R5, -(CR6R7)nC(O)OR4, -(CR6R7)nNCR4R5, -C(=NR6)NR4R5, -NR4C(O)NR5R6, -NR4S(O)PR5, ou -C(O)NR4R5, e cada hidrogênio em R2 é opcionalmente substituído com R8,
[0011] cada R3 é independentemente halogênio, C1-12 alquila, C2 12 alquenila, C2-12 alquinila, C3-12 cicloalquila, C6-12 arila, heteroalicíclico de 3-12 membros, heteroarila de 5-12 membros, -S(O)mR4, 45 4 45 67 4 4 —SO2NR R , — S(O)2OR , -NO2, -NR R , -(CR R )nOR , -CN, -C(O)R , -OC(O)R4, O(CR6R7)nR4, -NR4C(O)R5, -(CR6R7)nC(O)OR4, -(CR6R7)nOR4, -(CR6R7)nC(O)NR4R5, -(CR6R7)nNCR4R5, -C(=NR6)NR4R5, -NR4C(O)NR5R6, -NR4S(O)PR5, ou -C(O)NR4R5, e cada hidrogênio em R3 é opcionalmente substituído com R8, e os grupos R3 nos átomos adjacentes podem combinar-se formando um C6-12 arila, heteroarila de 5-12 membros, C3-12 cicloalquila ou grupo heteroalicíclico de 3-12 membros;
[0012] cada R4, R5, R6 e R7 é independentemente hidrogênio, halogênio, C1-12 alquila, C2-12 alquenila, C2-12 alquinila, C3-12 cicloalquila, C6-12 arila, heteroalicíclico de 3-12 membros, heteoarila de 5-12 membros ou quaisquer dois de R4, R5, R6 e R7 ligados ao mesmo átomos de nitrogênio podem, junto com o nitrogênio ao qual eles estão ligados, ser combinados para formar um heteroalicíclico de 3-12 membros ou grupo heteroarila de 5-12 membros opcionalmente contendo 1-3 heteroátomos adicionais de N, O e S; ou quaisquer dois de R4, R5, R6 e R7 ligados ao mesmo átomo de carbono podem ser combinados para formar um cicloalquila C3-12, C6-12 arila, heteroalicíclico de 3-12 membros ou grupo heteroarila de 5-12 membros, e cada hidrogênio em R4, R5, R6 e R7 é opcionalmente substituído com R8,
[0013] cada R8 é independentemente, halogênio, C1-12 alquila, C2 12 alquenila, C2-12 alquinila, C3-12 cicloalquila, C6-12 arila, heteroalicíclico de 3-12 membros, heteroarila de 5-12 membros, -NH2, -CN, -OH, -O- C1-12 alquila, -O-(CH2)nC3-12 cicloalquila, -O-(CH2)nC6-12 arila, -O- (CH2)n(heteroalicíclico de 3-12 membros) ou -O-(CH2)n(heteroarila de 5-12 membros); e cada hidrogênio em R8 é opcionalmente substituído com R11;
[0014] cada R9 e R10 é independentemente hidrogênio, halogênio, C1-12 alquila, C3-12 cicloalquila, C6-12 arila, heteroalicíclico de 3-12 membros, heteroarila de 5-12 membros, -S(O)mR4, -SO2NR4R5, -S(O)2OR4, -NO2, -NR4R5, -(CR6R7)nOR4, -CN, -C(O)R4, -OC(O)R4, -NR4C(O)R5, -(CR6R7)nC(O)OR4, -(CR6R7)nNCR4R5, -NR4C(O)NR5R6, -NR4S(O)PR5 ou -C(O)NR4R5; R9 ou R10 pode combinar-se com um átomo do anel de A ou um substituinte de A para formar um C3-12 cicloalquila, heteroalicíclico de 3-12 membros, C6-12 arila ou anel heteorarila de 5-12 membros fundido a A; e cada hidrogênio em R9 e R10 é opcionalmente substituído com R3',
[0015] cada R11 é independentemente halogênio, C1-12 alquila, C1 12 alcóxi, C3-12 cicloalquila, C6-12 arila, heteroalicíclico de 3-12 membros, heteroarila de 5-12 membros, -O-C1-12 alquila, -O-(CH2)nC3- 12 cicloalquila, -O-(CH2)nC6-12 arila, -O-(CH2)n(heteroalicíclico de 3-12 membros), -O-(CH2)n(heteroarila de 5-12 membros) ou -CN e cada hidrogênio em R11 é opcionalmente substituído com halogênio, -OH, -CN, C1-12 alquila que pode ser parcialmente ou totalmente halogenada, -O-C1-12 alquila que pode ser parcialmente ou totalmente halogenada, -CO, -SO ou -SO2;
[0016] R12 é hidrogênio, halogênio, C1-12 alquila, C2-12 alquenila, C2-12 alquinila, C3-12 cicloalquila, C6-12 arila, heteroalicíclico de 3-12 membros, heteroarila de 5-12 membros, -S(O)mR4, -SO2NR4R5, -S(O)2OR4, -NO2, -NR4R5, -(CR6R7)nOR4, -CN, -C(O)R4, -OC(O)R4, O(CR6R7)nR4, -NR4C(O)R5, -(CR6R7)nC(O)OR4, -(CR6R7)nNCR4R5, -C(=NR6)NR4R5, -NR4C(O)NR5R6, -NR4S(O)pR5, ou -C(O)NR4R5, e cada hidrogênio em R12 é opcionalmente substituído com R3,
[0017] cada R13 é independentemente halogênio, C1-12 alquila, C2 12 alquenila, C2-12 alquinila, C3-12 cicloalquila, C6-12 arila, heteroalicíclico de 3-12 membros, heteroarila de 5-12 membros, -S(O)mR4, -SO2NR4R5, -S(O)2OR4, -NO2, -NR4R5, -(CR6R7)nOR4, -CN, -C(O)R4, -OC(O)R4, O(CR6R7)nR4, -NR4C(O)R5, -(CR6R7)nC(O)OR4, -(CR6R7)nOR4, -(CR6R7)nC(O)NR4R5, -(CR6R7)nNCR4R5, -C(=NR6)NR4R5, -NR4C(O)NR5R6, -NR4S(O)pR5, -C(O)NR4R5, -C(O)NR4R5, -(CR6R7)n(heteroalicíclico de 3-12 membros), (CR6R7)n(C3-12 cicloalquila), -(CR6R7)n(C6-12 arila), -(CR6R7)n(heteroarila de 5-12 membros), -(CR6R7)nC(O)NR4R5 ou (CR6R7)nC(O)R4, grupos R13 em átomos adjacentes podem combinar-se para formar um C6-12 arila, heteroarila de 5-12 membros, C3-12 cicloalquila ou grupo heteroalicíclico de 3-12 membros, e cada hidrogênio em R13 é opcionalmente substituído com R3;
[0018] cada m é independentemente 0, 1 ou 2;
[0019] cada n é independentemente 0, 1, 2, 3 ou 4;
[0020] cada p é independentemente 1 ou 2;
[0021] ou um sal, hidrato ou solvato farmaceuticamente aceitável destes.
[0022] Num aspecto particular desta modalidade, R1 é hidrogênio.
[0023] Num outro aspecto particular desta modalidade, Y é N.
[0024] Num outro aspecto particular desta modalidade, Y é N e R2 é hidrogênio.
[0025] Num outro aspecto particular desta modalidade, Y é CR12.
[0026] Num outro aspecto particular desta modalidade, Y é CR12 e R12 é H.
[0027] Num outro aspecto particular desta modalidade, e em combinação com qualquer outro aspecto que não seja incoerente, R1 é furano, tiopeno, pirrol, pirrolino, pirrolidino, dioxolano, oxazol, tiazol, imidazol, imidazolina, imidazolidina, pirazol, pirazolina, pirazolidina, osoxazol, isotiazol, oxadiazol, triazol, tiadiazol, pirano, piridina, piperidina, dioxano, morfolino, ditiano, tiomorfolina, piridazina, pirimidina, pirazina, piperazina, triazina, tritiano, ou grupo fenila, e cada hidrogênio em R1 é opcionalmente substituído com um ou mais grupos R3.
[0028] Num outro aspecto particular desta modalidade, e em combinação com qualquer outro aspecto particular que não seja incoerente, R1 é um grupo heteroarila de anel fundido, e cada hidrogênio em R1 é opcionalmente substituído com um ou mais grupos R3.
[0029] Num outro aspecto particular desta modalidade, e em combinação com qualquer outro aspecto particular que não seja incoerente, R1 é hidrogênio.
[0030] Num outro aspecto particular desta modalidade, e em combinação com qualquer outro aspecto particular que não seja incoerente, R1 é halogênio.
[0031] Numa outra modalidade, a invenção proporciona um composto enantiomericamente puro de fórmula 1a. onde:
onde:
[0032] Y é N ou CH;
[0033] R1 é furano, tiopeno, pirrol, pirrolino, pirrolidino, dioxolano, oxazol, tiazol, imidazol, imidazolina, imidazolidina, pirazol, pirazolina, pirazolidina, osoxazol, isotiazol, oxadiazol, triazol, tiadiazol, pirano, piridina, piperidina, dioxano, morfolino, ditiano, tiomorfolina, piridazina, pirimidina, pirazina, piperazina, triazina, tritiano, azitidina ou grupo fenila, e cada hidrogênio em R1 é opcionalmente substituído com um ou mais grupos R3;
[0034] cada R3 é independentemente halogênio, C1-12 alquila, C2-12 alquenila, C2-12 alquinila, C3-12 cicloalquila, C6-12 arila, heteroalicíclico de 3-12 membros, heteroarila de 5-12 membros, -S(O)mR4, -SO2NR4R5, -S(O)2OR4, -NO2, -NR4R5, -(CR6R7)nOR4, -CN, -C(O)R4, -OC(O)R4, O(CR6R7)nR4, -NR4C(O)R5, -(CR6R7)nC(O)OR4, -(CR6R7)nOR4-, -(CR6R7)nC(O)NR4R5, (CR6R7)nNCR4R5, -C(=NR6)NR4R5, -NR4C(O)NR5R6, -NR4S(O)PR5, ou -C(O)NR4R5, e cada hidrogênio em R3 é opcionalmente substituído com R8, e os grupos R3 nos átomos adjacentes podem combinar-se formando um C6-12 arila, heteroarila de 5-12 membros, C3-12 cicloalquila ou grupo heteroalicíclico de 3-12 membros;
[0035] cada R4, R5, R6 e R7 é independentemente hidrogênio, halogênio, C1-12 alquila, C2-12 alquenila, C2-12 alquinila, C3-12 cicloalquila, C6-12 arila, heteroalicíclico de 3-12 membros, heteoarila de 5-12 membros ou quaisquer dois de R4, R5, R6 e R7 ligados ao mesmo átomos de nitrogênio podem, junto com o nitrogênio ao qual eles estão ligados, ser combinados para formar um heteroalicíclico de 3-12 membros ou grupo heteroarila de 5-12 membros opcionalmente contendo 1-3 heteroátomos adicionais de N, O e S; ou quaisquer dois de R4, R5, R6 e R7 ligados ao mesmo átomo de carbono podem ser combinados para formar um cicloalquila C3-12, C6-12 arila, heteroalicíclico de 3-12 membros ou grupo heteroarila de 5-12 membros, e cada hidrogênio em R4, R5, R6 e R7 é opcionalmente substituído com R8,
[0036] cada R8 é independentemente, halogênio, C1-12 alquila, C2 12 alquenila, C2-12 alquinila, C3-12 cicloalquila, C6-12 arila, heteroalicíclico de 3-12 membros, heteroarila de 5-12 membros, -NH2, -CN, -OH, -O- C1-12 alquila, -O-(CH2)nC3-12 cicloalquila, -O-(CH2)nC6-12 arila, -O- (CH2)n(heteroalicíclico de 3-12 membros) ou -O-(CH2)n(heteroarila de 5-12 membros); e cada hidrogênio em R8 é opcionalmente substituído com R11;
[0037] cada R9 e R10é independentemente hidrogênio, halogênio, C1-12 alquila, C3-12 cicloalquila, C6-12 arila, heteroalicíclico de 3-12 membros, heteroarila de 5-12 membros, -S(O)mR4, -SO2NR4R5, -S(O)2OR4, -NO2, -NR4R5, -(CR6R7)nOR4, -CN, -C(O)R4, -OC(O)R4, -NR4C(O)R5, -ou (CR6R7)nC(O)OR4, -(CR6R7)nNCR4R5, -NR4C(O)NR5R6, -NR4S(O)PR5, ou -C(O)NR4R5; R9 ou R10 pode combinar-se com um átomo do anel de A ou um substituinte de A para formar um C3-12 cicloalquila, heteroalicíclico de 3-12 membros, C6-12 arila ou anel heteorarila de 5-12 membros fundido a A; e cada hidrogênio em R9 e R10 é opcionalmente substituído com R3',
[0038] cada R11 é independentemente halogênio, C1-12 alquila, C1-12 alcóxi, C3-12 cicloalquila, C6-12 arila, heteroalicíclico de 3-12 membros, heteroarila de 5-12 membros, -O-C1-12 alquila, -O-(CH2)nC3-12 cicloalquila, -O-(CH2)nC6-12 arila, -O-(CH2)n(heteroalicíclico de 3-12 membros) ou -O-(CH2)n(heteroarila de 5-12 membros) ou -CN e cada hidrogênio em R11 é opcionalmente substituído com halogênio, -OH, -CN, C1-12 alquila que pode ser parcialmente ou totalmente halogenado, -O-C1-12 alquila que pode ser parcialmente ou totalmente halogenado, -CO, -SO ou -SO2;
[0039] cada R13 é independentemente halogênio, C1-12 alquila, C2 12 alquenila, C2-12 alquinila, C3-12 cicloalquila, C6-12 arila, heteroalicíclico de 3-12 membros, heteroarila de 5-12 membros, -S(O)mR4, -SO2NR4R5, -S(O)2OR4, -NO2, -NR4R5, -(CR6R7)nOR4, -CN, -C(O)R4, -OC(O)R4, O(CR6R7)nR4, -NR4C(O)R5, -(CR6R7)nC(O)OR4, -(CR6R7)nOR4-, -(CR6R7)nC(O)NR4R5, -(CR6R7)nNCR4R5, -C(=NR6)NR4R5, -NR4C(O)NR5R6, -NR4S(O)pR5, -C(O)NR4R5, -C(O)NR4R5, -(CR6R7)n(heteroalicíclico de 3-12 membros), (CR6R7)n(C3-12 cicloalquila), -(CR6R7)n(C6-12 arila), -(CR6R7)n(heteroarila de 5-12 membros), -(CR6R7)nC(O)NR4R5 ou (CR6R7)nC(O)R4, grupos R13 ns átomos adjacentes podem combinar-se para formar um C6-12 arila, heteroarila de 5-12 membros, C3-12 cicloalquila ou grupo heteroalicíclico de 3-12 membros, e cada hidrogênio em R13 é opcionalmente substituído com R3;
[0040] cada m é independentemente 0, 1 ou 2;
[0041] cada n é independentemente 0, 1, 2, 3 ou 4;
[0042] cada p é independentemente 1 ou 2;
[0043] ou um sal, hidrato ou solvato farmaceuticamente aceitável destes.
[0044] Numa outra modalidade, a invenção providencia um composto enantiomericamente puro selecionado do grupo consistindo de:
[0045] 5-bromo-3-[(R)-1-(2,6-dicloro-3-flúor-fenil)-etóxi]-pirazin-2- ilamina,
[0046] 5-iodo-3-[(R)1-(2,6-dicloro-3-flúor-fenil)-etóxi] piridin-2- ilamina;
[0047] 5-bromo-3-[1(R)-2,6-dicloro-3-flúor-fenil)-etóxi] piridin-2- ilamina;
[0048] ácido 4-(5-amino-6-[(R)-1-(2,6-dicloro-3-flúor-fenil)- etóxi]pirazin-2-il]-benzóico;
[0049] 4-(5-amino-6-[(R)-1-(2,6-dicloro-3-flúor-fenil)-etóxi]pirazin- 2-il}-fenil)-piperazin-1-il-metanona;
[0050] éster terc-butílico do ácido 4-(4-[5-amino-6-[(R)-1-(2,6- dicloro-3-flúor-fenil)-etóxi]pirazin-2-il}-benzoil)-piperazina-1-carboxílico;
[0051] 3-[(1R)-1-(2,6-dicloro-3-flúor-fenil)-etóxi]-5-[4-(piperazin-1- ilcarbonil)fenil]piridin-2-amina;
[0052] 4-(6-amino-5-[(1R)-1-(2,6-dicloro-3-fluorfenil)-etóxi]piridin- 3-il]-N-[2-(dimetilamino)etil]-N-metilbenzamida;
[0053] 4-(6-amino-5-[(1R)-1-(2,6-dicloro-3-fluorfenil)-etóxi]piridin- 3-il]-fenil)metanol;
[0054] 4-(6-amino-5-[(1R)-1-(2,6-dicloro-3-fluorfenil)-etóxi]piridin- 3-il]-N-[3-(dimetilamino)propil]-N-metilbenzamida;
[0055] 4-(4-[6-amino-5-[(1R)-1-(2,6-dicloro-3-fluorfenil) -etóxi]piridin- 3-il]-benzoil)piperazina-1-carboxilato de terc-butila;
[0056] 3-[(R)-1-(2,6-dicloro-3-fluorfenil)-etóxi]-5-[1-(1-metil-piperidin- 1-il)-1H-pirazol-4-il]-piridin-2-ilamina;
[0057] 1-[4-(4-(6-amino-5-[(R)-1-(2,6-dicloro-3-fluorfenil)-etóxi] piridin-3-il}-pirazol-1-il)-piperidin-1-il]-1-hidróxi-etanona;
[0058] 3-[(R)-1-(2,6-dicloro-3-fluorfenil)-etóxi]-5-(1-piperidin-4-il)- 1H-pirazol-4-il]-piridin-2-ilamina;
[0059] 3-[(R)-1-(2,6-dicloro-3-fluorfenil)-etóxi]-5-(1-piperidin-4-il- 1H-pirazol-4-il)-pirazin-2-ilamina;
[0060] 3-[(R)-1-(2,6-dicloro-3-fluorfenil)-etóxi]-5-(1H-pirazol-4-il]- pirazin-2-ilamina;
[0061] 1-[4-(4-(5-amino-6-[(R)-1-(2,6-dicloro-3-fluorfenil)- etóxi]pirazin-2-il}-pirazol-1-il)-piperidin-1-il]-2-hidróxi-etanona;
[0062] 3-[(R)-1-(2,6-dicloro-3-fluorfenil)-etóxi]-5-[1-(1-metil-piperidin- 4-il)-1H-pirazol-4-il]-pirazin-2-ilamina;
[0063] 1-[4-(4-(5-amino-6-[(R)-1-(2,6-dicloro-3-fluorfenil)- etóxi]pirazin-2-il}-pirazol-1-il)-piperidin-1-il] -2-dimetilamino-etanona;
[0064] 3-[(R)-1-(2-cloro-3,6-difluorfenil)-etóxi]-5-[1-(1-piperidin-4- il-1H-pirazol-4-il]-piridin-2-ilamina; ou um sal, solvato ou hidrato farmaceuticamente aceitável destes.
[0065] Numa outra modalidade, a invenção providencia uma composição farmacêutica compreendendo quaisquer compostos da invenção e um veículo farmaceuticamente aceitáveis. Exemplos de tais composições estão descritas a seguir.
[0066] Compostos preferidos da invenção incluem os com atividade inibidora de c-MET como indicado por quaisquer ou mais IC50, Ki, ou inibição percentual (%). O perito na técnica pode, determinar prontamente se um composto tem tal atividade por realização do ensaio adequado, e descrições de tais ensaios demonstram-se no parágrafo dos Exemplos abaixo. Numa modalidade, compostos particularmente preferidos têm u ki c-MET inferior a 5μM ou menos que 2 μM,ou menos que 1 μM, ou menos que 500 nM ou menos que 200 nM ou menos que 100 nM. Numa outra modalidade, compostos particularmente preferidos têm uma inibição c- MET a 1 μM de pelo menos 10% ou pelo menos 20% ou pelo menos 30%, ou pelo menos 40% ou pelo menos 50% ou pelo menos 60% ou pelo menos 70% ou pelo menos 80% ou pelo menos 90%. Métodos de medição da atividade c-MET/HGFR estão descritos nos Exemplos a seguir.
[0067] Numa outra modalidade, a invenção proporciona um método de tratamento do crescimento celular anormal num mamífero incluindo um humano compreendendo o método, a administração ao mamífero de quaisquer composições farmacêuticas da invenção.
[0068] Numa modalidade específica de quaisquer métodos inventivos aqui descritos, o crescimento celular anormal é câncer, incluindo, porém não limitado, a câncer de pulmão, câncer ósseo, câncer pancreático, gástrico, câncer de pele, câncer da cabeça ou pescoço, melanoma cutâneo ou intraocular, câncer uterino, câncer ovariano, câncer ginecológico, câncer retal, câncer da região anal, câncer de estomago, câncer de cólon, câncer de mama, câncer uterino, carcinoma das trompas de falópio, carcinoma do endométrio, carcinoma de cérvice, carcinoma da vagina, carcinoma da vulva, Doença de Hodgkin, câncer do esôfago, câncer do intestino delgado, câncer do sistema endócrino, câncer da glândula tireóide, câncer de glândula paratireóide, câncer da glândula adrenal, sarcoma de tecido mole, câncer da uretra, câncer do pênis, célula escamosa, câncer de próstata, leucemia crônica ou aguda, linfomas linfocíticos, câncer da bexiga, câncer do rim ou ureter, carcinoma de célula renal, carcinoma de pélvis renal, neoplasmas do sistema nervoso central (SNC), linfoma do SNC primário, tumores de espinha dorsal, glioma de origem cerebral, adenoma pituitário, ou uma combinação de um ou mais dos cânceres precedentes. Numa outra modalidade do referido método, tal crescimento celular anormal é uma doença proliferativa benigna, incluindo, sem limitação, a psoríase, hipertrofia benigna prostática ou reestenose.
[0069] Numa outra modalidade, a invenção proporciona um método de tratamento de um distúrbio mediado por HGFR num mamífero, incluindo um humano, compreendendo o método a administração ao mamífero de quaisquer composições farmacêuticas da invenção.
[0070] Numa outra modalidade, específica de quaisquer métodos inventivos aqui descritos, o método compreende ainda a administração ao mamífero de uma quantidade de quaisquer substancias selecionadas de agentes antitumorais, agentes anti-angiogênese, inibidores da transdução de sinal, e agentes antiproliferativos, cujas quantidades são eficazes, no conjunto, para tratamento de tal crescimento celular anormal. Tais substâncias incluem as reveladas na Publicação PCT Nos WO 00/38715, WO 00/38716, WO 00/38717, WO 00/38718, WO 00/38719, WO 00/38730, WO 00/38665, WO 00/37107 e WO 00/38786, cuja descrição está ora incorporada por referência em sua totalidade.
[0071] Exemplos de agentes antitumorais incluem inibidores mitóticos, por exemplo, derivados de alcalóide de vinca tais como vinblastina, vinorelbina, vindescina e vincristina; alocoquino colchinas, halicondrina, ácido conchicínico do éter N-benzoiltrimetil metílico, dolastatin 10, maistansina, rizoxina, taxanos como taxol (paclitaxel), docetaxel (Taxotere), 2'-N-[3-(dimetilamino)propil) glutaramato (derivado de taxol), tiolcolchicina, tritil cisteína, teniposídeo, metotrexato, azatioprin, fluoruricil, citocina arabinosídeo, 2'2'- difluordesoxicitidina (gemcitabina), adriamicina e mitamicina. Agentes de alquilação, por exemplo, cisplatina, carboplatina, oxiplatina, iproplatina, éster etílico de N-acetil-DL-sarcosil-L-leucina (Asaley ou Asalex), éster 2,5-bis(1-aziridinil)-3,6-dioxo-dietílico do ácido 1,4- diclohexadieno-1,4-dicarbamico, 1,4-bis(metanosulfonilóxi)butano (bisulfan ou leucosulfan) clorozotocina, clomesona, cianomorfolinodoxorubicina, ciclodisona, dianidroglactitol, fluordopan, hepsulfam, mitomicina C, hicanteonemitomicina C, mitozolamida, dicloridrato de 1-(2-cloroetil)-4-(3-cloropropil)-piperazina, piperazinodiona, pipobroman, porfiromicina, mostarda de espiroidantoína, teroxinona, tetraplatina, tiotepa, trietilenomelamina, mostarda de uracil nitrogênio, cloridrato de bis(3-mesiloxipropil)amina, mitomicina, agentes nitrosouréias, tais como cicloexil- cloroetilnitrosouréia, metilcicloexil-cloroetilnitrosouréia, 1-(2-cloroetil)-3- (2,6-dioxo-3-piperidil)-1-nitroso-uréia, bis(2-cloroetil)nitrosouréia, procarbazina, dacarbazina, compostos relacionados à mostarda de nitrogênio, tais como mecloroetamina, ciclofosfamida, ifosamida, melfalan, clorambucil, fosfato sódico de estramustina, estroptozoina e temozolamina, anti-metabolitos de DNA, por exemplo, 5-fluoruracil, arabinosídeo citosina, hidroxiuréia, 2-[(3-hidróxi-2-pirinodinil) metileno] - hidrazinocrbotioamida, desoxifluoruridina, 5-hidróxi-2-formilpiridina tiosemicarbazona, alfa-2'-desóxi-6-tioguanosina, glicinato de afidicolina, 5-azadesoxicitidina, beta-tioguanina desoxiribosídeo ciclocitidina guanazol, glicodialdeído inosina, macbcina II, pirazolimidazol, cladribina, pentostatin, tioguanina, mercaptopurina, bleomicina, 2-clorodesoxiadenosina, inibidores de timidilato sintase tais como raltirexed e pemetrexed dissódico, clofarabina, floxuridina e fludarabina. Antimetabólitos de DNA/RNA, por exemplo, L-alanosina, 5-azacitidina, acivicina, aminopterina e os seus derivados tais como ácido N-[2-cloro-5-[[(2,4-diamino-5-metil-6-quinazolinil)metil] amino] benzoil]-L-aspártico, ácido N-[4-[[(2,4-diamino-5-etil-6- quinaozlinil)metil]amino]benzoil]-L-aspártico, ácido N-[2-cloro-4-[(2-4- diaminopteridinil)metil]amino]benzoil]-L-aspartico, antifol de Baker solúvel, dicloroalil lawsona, breninar, ftoral, diidro-5-azacitidina, metotrexato, sal tetrassódico do ácido N-(fosfonoacetil)-L-aspártico, pirazofurano, trimetrexato, plicamicina, actinomicina D, criptoficina, e seus análogos como criptoficina-52 ou, por exemplo, um dos antimetabólitos preferidos apresentados no pedido de Patente Europeu N° 239362 tal como ácido N-(5-(N-(3,4-diidro-2-metil-4- oxoquinazolin-6-ilmetil)-N-metilamino]-2-tenoil)-L-glutâmico; inibidor do fator de crescimento, inibidores do ciclo celular, antibióticos de intercalação, por exemplo, adriamicina e bleomicina, enzimas, por exemplo, interferon e anti-hormonios, por exemplo, anti-estrógenos tais como Nolvadex™ (tamoxifen) ou, por exemplo, anti-andrógenos tais como Casodex™ (4'-ciano-3-(4-fluorfenilsulfonil)-2-hidróxi-2-metil - 3'-trifluormetil) propionanilida). Tal tratamento conjunto pode ser realizado a guisa de dosagem simultânea, em seqüência ou separada dos componentes individuais do tratamento.
[0072] Agentes anti-angiogênese incluem, inibidores de MM-2 (metaloproteinase matriz 2) e inibidores de MMP-9, (meetaloproteinase matriz 9) e inibidores de COX-II (ciclooxigenase II). Exemplos de inibidores de COX-II úteis incluem CELEBREX™ (alecoxib), (valdecoxib), paracoxib, e (rofecoxib) Exemplos de inibidores úteis de metaloproteinase matriz estão descritos em WO 96/33172 (publicado em 24 de outubro de 1996), WO 96/27583 (publicado em 7 de março de 1996), Pedido de Patente Europeu N° 9730497.1 (depositado em 8 de julho de 1997), Pedido de Patente Europeu N° 99308617.2 (depositado em 29 de outubro 1999), WO 98/07697 (publicado em fevereiro de 1998), WO 98/03516 (publicado em 29 de janeiro de 1998), WO 98/34918 (publicado em 13 de agosto de 1998), wO 98/34915 (publicado em 13 de agosto 1997), WO 98/33768 (Publicado em 6 de agosto de 1998), WO 98/30566 (publicado em 16 de julho de 1998), Publicação de patente Européia (606.046 (publicado em 13 de julho de 1994), Publicação de Patente européia 931.7878 (publicado em 28 de julho de 1999), WO 90/05719 (publicado em 31 de maio de 1990), WO 99/52910 (publicado em 21 de outubro de 1999), WO 99/52889 (publicado em 21 de outubro de 1999), WO 99/29667(publicado em 17 de junho de 1999), Pedido Internacional PCT N° PCT/IB98/01113 (depositado em 21 de julho de 1998), pedido de Patente Europeu N° 99302232.1 (depositado em 25 de março de 1999), pedido de patente da Grã-Bretanha N° 991296.1. (depositado em 3 de junho de 1999), Pedido Provisório dos Estados Unidos N° 60/148.464 (depositado em 12 de agosto de 1999), Patente dos Estados Unidos 5.863.949 (concedida em 26 de janeiro de 1999), Patente U.S. 5.861.510 (cedida em 19 de janeiro de 1999) e Publicação de Patente Européia 780.386 (publicado em 25 de junho de 1997), todos os quais estando ora incorporados por referência em sua totalidade.
[0073] Inibidores de MMP-2 e MMP-9 preferidos são os que têm pouca ou nenhuma atividade inibidora de MMp1. Os mais preferidos são os que inibem seletivamente, MMP-2 e/ou MMP-9 em relação a outras metaloproteinases matrizes (ou seja, MMP-1, MMP-3, MMP-4, MMP-5, MMP-6, MMP-7, MMP-8, MMP-10, MMP-11, MMP-12 e MMP- 13).
[0074] Alguns exemplos específicos de inibidores de MMP úteis em combinação com os compostos da presente invenção são AG- 3340, RO 32-3555, RS 13-0830 e os compostos descritos na lista a seguir:
[0075] ácido 3-[[4-(4-flúor-fenóxi)-benzenossulfonil]-(1- hidroxicarbamoil-ciclopentil)-amino]-propiônico;
[0076] hidroxiamida do ácido 3-exo-3-[4-(4-flúor-fenóxi)- benzenossulfonilamino]-8-oxa-biciclo]3.2.1]octano-3-carboxílico;
[0077] hidroxiamida do ácido (2R, 3R) 1-[4-(2-cloro-4-flúor- benzilóxi)-benzenossulfonil]-3-hidróxi-3-metil-piperidino-2-carboxílico;
[0078] hidroxiamida do ácido 4-[4-(4-flúor-fenóxi)- benzenossulfonilamino]-tetraidro-piran-4-carboxílico;
[0079] ácido 3-[[4-(4-flúor-fenóxi)-benzenossulfonil]-(1- hidroxicarbamoil-ciclobutil)-amino]-propiônico;
[0080] hidroxiamida do ácido 4-[4-(4-cloro-fenóxi)- benzenossulfonilamino]-tetraidro-piran-4-carboxílico;
[0081] ácido 3-[[4-(4-cloro-fenóxi)-benzenossulfonilamino]- tetraidro-piran-3-carboxílico;
[0082] hidroxiamida do ácido (2R, 3R), 1-[4-(4-flúor-2-metil- benzilóxi)-benzenossulfonil]-3-hidróxi-3-metil-piperidino-2-carboxílico;
[0083] ácido 3-[[4-(4-flúor-fenóxi)-benzenossulfonil]-(1- hidroxicarbamoil-1-metil-etil)-)-amino]-propiônico;
[0084] ácido 3-[[4-(4-flúor-fenóxi)-benzenossulfonil]-(1- hidroxicarbamoil-tetraidro-piran-4-il)-amino]-propiônico;
[0085] hidroxiamida do ácido 3-exo-4-[4-(4-cloro-fenóxi)- benzenossulfonilamino]-8-oxa-biciclo[3.2.1]octano-3-carboxílico;
[0086] hidroxiamida do ácido 3-endo-3-[4-(4-flúor- fenóxi)benzenossulfonilamino]-8-oxa-biciclo[3.2.1]octano-3-carboxílico e
[0087] hidroxiamida do ácido 3-[[4-(4-flúor-fenóxi)- benzenossulfonilamino]-tetraidro-furan-3-carboxilico;
[0088] e os sais, solvatos e hidratos farmaceuticamente aceitáveis de tais compostos.
[0089] Exemplos de inibidores da transdução de sinal incluem agentes que podem inibir respostas de EGFR (receptor do fator de crescimento epidérmico) EGFR, tais como anticorpos EGFR, anticorpos EGF, e moléculas que são inibidores de EGFR, inibidores de VEGF (fator de crescimento endotelial vascular), e inibidores do receptor de erbB2 tais como moléculas orgânicas ou anticorpos que se ligam ao receptor de erbB2, por exemplo, HERCEPTIN™ (Genentech, Inc. of South San Francisco, Califórnia, USA).
[0090] Inibidores de EGFR estão descritos, por exemplo, em WO 95/19970, (publicado em 27 de julho de 1995), WO 98/14451 (publicado em 9 de abril de 1998), WO 98/02434 (publicado em 22 de janeiro de 1998) e Patente dos Estados Unidos 5.747.498 (cedida em 5 de maio de 1998). Agentes inibidores de EGFRF incluem, sem limitação aos anticorpos monoclonais C225 e anti-EGFR 22Mab (ImClone Systems Incorporated of New York, New York, USA), e os compostos ZD-1839 (AstraZeneca), BIBX-1382 (Boehringer Ingelheim), MDS-447/H-477 (Medarex Inc. of Annandale, New Jersey, USA e OLX-103 (Merck & Co. of Whitehouse Station, New Jersey, USA) VRCTC-310 (Ventech Research), toxina de fusão EGF (Seragen Inc. of Hopkinton, Massachusetts).
[0091] Inibidores de VEGF, por exemplo, SU5416 e SU-6668 (Sugen Inc. of South San Francisco, Califormina USA) também podem ser combinados ou co-administrados com a composição da presente invenção. Inibidores de VEGF, estão descritos, em WO 99/24440 (publicado em 20 de maio de 1999), O pedido internacional PCT/IB99/00797 (depositado em 3 de maio de 1999), em WO 95/21613 (publicado em 17 de agosto de 1995), WO 99/61422 (publicado em 2 de dezembro de 1999), Patente dos Estados Unidos 5.834.54 (concedida em 10 de novembro de 1998), WO 98/50356 (publicada em 12 de novembro de 1998), Patente dos Estados Unidos 5.883.113 (concedida em 16 de março de 1999), Patente dos Estados Unidos 5.886.020 (concedida em 23 de marco de 1999), Patente dos Estados Unidos 5.792.783 (concedida em 11 de agosto de 1998), WO 99/10349 (publicada em 4 de março de 1999), WO 97/32856 (publicada em 12 de setembro de 1997), wO 97/22596 (publicada em 26 de junho de 1997), WO 98/54093 (publicada em 3 de dezembro de 1998) WO 98/02438 (publicada em 22 de janeiro de 1998), WO 99/16755 (publicado em 8 de abril de 1999), e WO 98/024378 (publicado em 22 de janeiro de 1998), todas as quais estando ora incorporadas por referência em sua totalidade. Outros exemplos de inibidores de VEGF específicos são IM862 (Cytran Inc of Kirkland, Washington, USA); anticorpo monoclonal anti-VEGF bevacizumab (Genentech, Inc. of South San Francisco, Califórnia); e angiozima, uma ribozima sintética de Ribozyme (Bouder, Colorado) e Chiron (Emeryville, Califórnia).
[0092] Inibidores do receptor de erbB2 tais como GW-282974 (Glaxo Wellcome pic), e os anticorpos monoclonais AR-209 (Aronex Pharmaceuticals Inc. of The Woodlands, Texas, USA) e 2B-1 (Chiron), podem, ainda ser combinados com a composição da invenção. Tais inibidores de erbB2 incluem os descritos em WO 98/02434 (publicado em 22 de janeiro de 1998), WO 99/35146 (publicado em 15 de julho de 1999), WO 99/35132 (publicado em 15 de julho de 1999), WO 98/02437 (publicado em 22 de janeiro de 1998), WO 97/13760 (publicado em 17 de aril de 1997), WO 95/19970 (publicado em 27 de julho de 1995), Patente dos Estados Unidos 5.587.458 (cedida em 24 de dezembro de 1996) e Patente dos Estados Unidos 5.877.305 (cedida em 2 de março de 1999), todas as quais ora incorporadas em sua totalidade por referência. Inibidores do receptor de erbB2 úteis na presente invenção estão também descritos no Pedido Provisório dos Estados Unidos n° 60/117.341, depositado em 27 de janeiro de 1999, e no Pedido Provisório dos Estados Unidos N°. 60/117.346 depositado em 27 de janeiro de 1999, ambos os quais ora incorporados por referência em sua totalidade.
[0093] Outros agentes antiproliferativos que podem ser usados incluem inibidores da enzima farnesil proteína transferase e inibidores do receptor de tirosina quinase PDGFr, incluindo os compostos revelados e reivindicados inibidores do receptor de tirosina quinase PDGFr, incluindo os compostos apresentados e reivindicados nos seguintes Pedidos de patente dos estados Unidos: 09/221946 (depositado em 26 de dezembro de 1998); 09/454058 (depositado em 2 de dezembro de 1999), 09/501163 (depositado em 9 de fevereiro de 2000); 09/539930 (depositado em 31 de março de 2000); 09/202796 (depositado em 22 de maio de 1997), 09/384339 (depositado em 26 de agosto de 1999), e 09/383755 (depositado em 26 de agosto de 1999), e os compostos apresentados e reivindicados nos seguintes pedidos de patentes provisórios dos Estados Unidos: 60/168207 (depositado em 30 de novembro de 1999); 60/170119 (depositado em 10 de dezembro de 1999); 60/177718 (depositado em 21 de janeiro de 2000); 60/168217 (depositado em 30 de novembro de 1999) e 60/200834 (depositado em 1 de maio de 2000). Cada um dos pedidos de patente precedentes e pedidos de patente provisórios estão ora incorporados por referência, em sua totalidade.
[0094] Composições da invenção podem também ser utilizadas com outros agentes de utilidade no tratamento do crescimento celular anormal ou câncer, incluindo, sem limitação a, agentes capazes de intensificar as respostas imunes antitumorais, tais como anticorpos CTLA4 (antígeno 4 de linfócito citotóxico) e outros agentes capazes de bloquear CTL4; e agentes antiproliferativos tais como outros inibidores de proteína farnesil transferase.
[0095] Anticorpos CTLA4 específicos que podem ser usados na presente invenção incluem os descritos no Pedido Provisório dos Estados Unidos 60/113.647 (depositado em 23 de dezembro de 1998) ora incorporado por referência em sua totalidade.
[0096] A menos que de outro modo indicado, os seguintes termos usados no relatório descritivo e reivindicações têm os seguintes significados abaixo. Vaiáveis indicadas neste parágrafo, tais como R, X n e similar, são usados como referência neste parágrafo apenas, e não significa que tenham o mesmo significado, no caso de emprego fora deste parágrafo de definições. Além disso, muitos grupos aqui indicados podem ser opcionalmente substituídos. A listagem neste parágrafo de definições, de substituintes típicos é exemplar e não pretende limiar os substituintes indicados algures, dentro deste relatório e reivindicações.
[0097] "Alquila" refere-se a um radical hidrocarboneto alifático, saturado incluindo grupos de cadeia linear e ramificado de 1-20 átomos de carbono, preferivelmente, 1-12 átomos de carbono, mais preferivelmente, 1-8 átomos de carbono, ou 1-6 átomos de carbono, ou 1-4 átomos de carbono. "alquila inferior" refere-se, de modo específico, a um grupo alquila com 1-4 átomos de carbono. Exemplos de grupos alquila incluem metila, etila, propila, 2-propila, n-butila, isobutila, terc-butila, pentila e similar. Alquila pode ser substituído ou não substituído. Grupos substituintes típicos incluem, cicloalquila, arila, heteroarila, heteroalicíclico, hidróxi, alcóxi, arilóxi, mercapto, alquiltio, ariltio, ciano, halo, carbonila, tiocarbonila, O-carbamila, N-carbamila, O-tiocarbamila, O-tiocarbamila, C-amido, N-amido, C-carbóxi, O- carbóxi, nitro, silila, amino e -NRxRy, onde Rx e Ry são independentemente selecionados do grupo consistindo de hidrogênio, alquila, cicloalquila, arila, carbonila, acetila, sulfinila, trifluormetanossulfonila e combinado, um anel heteroalicíclico de cinco- ou seis membros.
[0098] "Cicloalquila" refere-se a um anel monocíclico de 3-8 membros, só de carbono, um anel bicíclico fundido todo carbono de 56- membros ou 6-6- membros, ou um grupo de anel fundido multicíclico (um sistema de anel "fundido" significa que, cada anel no sistema divide um par adjacente de átomos de carbono com cada outro anel no sistema), onde um ou mais dos anéis pode conter uma ou mais duplas ligações, porém nenhum dos anéis tem um sistema de elétron-pi completamente conjugado. Exemplos sem limitação de grupos cicloalquila são ciclopropano, ciclobutano, ciclopentano, ciclopenteno, cicloexano, cicloexadieno, adamantano, cicloeptano, cicloeptatrieno e similar. Um grupo cicloaluqila pode ser substituído ou não substituído. Grupos substituintes típicos incluem, alquila, arila, heteroarila, heteroalicíclico, hidróxi, alcóxi, arilóxi, mercapto, alquiltio, ariltio, ciano, halo, carbonila, tiocarbonila, C-carbóxi, O-carbóxi, O- carbamila, N-carbamila, C-amido, N-amido, nitro, amino e -NRxRy, com Rx e Ry conforme acima. Exemplos ilustrativos de cicloaluqila são derivados, sem limitação, dos seguintes:
[0099] "Alquenila" refere-se a um grupo alquila, conforme aqui indicado, consistindo de pelo menos dois átomos de carbono e pelo menos uma dupla ligação carbono-carbono. Exemplos representativos incluem, sem limitação a etenila, 1-propenila, 2-propenila, 1, 2, ou 3- butenila e similar.
[00100] "Alquinila" refere-se a um grupo alquila, conforme aqui indicado, consistindo de pelo menos dois átomos de carbono e pelo menos uma tripla ligação carbono-carbono. Exemplos representativos incluem, sem limitação, a etinila, 1-propinila, 2-propinila, 1, 2, ou 3- butinila e similar.
[00101] "Arila" refere-se a grupos monocíclicos todos os carbono ou policíclicos de anel fundido de 6-12 átomos de carbono, com um sistema de K-elétron completamente conjugado. Exemplos sem limitação de grupos arila, são fenila, naftalenila e antracenila. O grupo arila pode ser substituído ou não substituído. Substituintes típicos incluem halo, trialometila, alquila, hidróxi, alcóxi, arilóxi, mercapto, alquiltio, ariltio, ciano, nitro, carbonila, tiocarbonila, C-carbóxi, O- carbóxi, O-carbamila, N-carbamila, O-tiocarbamila, N-tiocarbamila, C- amido, N-amido, sulfinila, sulfinila, amio e -NRxRy, com Rx e Ry conforme acima.
[00102] "Heteroarila" refere-se a um grupo de anel monocíclico ou fundido de 5-12 átomos de carbono contendo um, dois, três ou quatro heteroátomos no anel selecionados dentre N, O e S, sendo o restante dos átomos do anel C, e, além disso, com um sistema de elétron-K- completamente conjugado. Exemplos sem limitação, de grupos heteroarila não substituídos, somam pirrol, furano, tiofeno, imidazol, oxazol, tiazol, pirazol, piridina, pirimidina, quinolina, isoquinolina, purina, tetrazol, triazina e carbazol. O grupo heteroarila pode ser substituído ou não substituído. Substituintes típicos incluem alquila, cicloalquila, halo, trialometila, hidróxi, alcóxi, arilóxi, mercapto, alquiltio, ariltio, ciano, nitro, carbonila, tiocarbonila, sulfonamido, C-carbóxi, O- carbóxi, sulfiila, sulfonila, O-carbamila, N-carbamila, O-tioarbamila, N- tiocarbamila, C-amido, N-amido, amino e NRxRy com RX e RY conforme acima.
[00103] Um heteroarila farmaceuticamente aceitável trata-se de um que seja suficientemente estável para ser ligado a um composto da invenção, formulado numa composição farmacêutica e a seguir administrado a um paciente em necessidade do mesmo.
[00104] Exemplos de grupos heteroarila monocíclicos típicos incluem, sem limitação:

[00105] Exemplos de grupos heteroarila de anel fundido adequados incluem, sem limitação a:


[00106] "Heteroalicíciclico" ou "heterociclo" refere-se a um grupo de anel monocíclico ou fundido com tendo nos anéis de 3-12 átomos no anel, em que um ou dois átomos no anel são heteroátomos selecionados dentre N, O, e S(O)n (onde n é 0, 1 ou 2) sendo o restante os átomos do anel C. Os anéis podem também te uma ou mais duplas ligações. Contudo, os anéis não têm um sistema de eletron-π completamente conjugado. Exemplos de grupos heteroalicíclicos saturados adequados incluem, sem limitação a:
[00107] Exemplos de grupos heteroalicíclicos parcialmente insaturados incluem, sem limitação, a:
[00108] O grupo heterociclo é opcionalmente substituído com um ou dois substituintes independentemente selecionados dentre halo, alquila inferior, alquila inferior substituído com carbóxi, éster, hidróxi ou mono ou diaquilamino.
[00109] "Hidróxi" refere-se a um grupo -OH.
[00110] "Alcóxi" refere-se a, tanto um grupo -O-(alquila como um grupo -O-(cicloalquila não substituído). Exemplos representativos incluem, sem limitação, a metóxi, etóxi propóxi, butóxi, ciclopropilóxi, ciclobutilóxi, ciclopentilóxi, cicloexilóxi e similar.
[00111] "Haloalcóxi" refere-se a um -O-arila ou um grupo -O- heteroaria, conforme aqui indicado. Exemplos representativos incluem, sem limitação a, fenóxi, piridinilóxi, furanilóxi, tienilóxi, pirimidinilóxi, pirazinilóxi, e similar e seus derivados.
[00112] "Mercapto"refere-se a um grupo -SH.
[00113] "Alquiltio"refere-se a um grupo -S-(alquila) ou um grupo - S-(cicloalquila não substituído). Exemplos representativos incluem, sem limitação, a metiltio, etiltio, propiltio, butiltio, ciclopropiltio, ciclobutiltio, ciclopentiltio, cicloexiltio e similar.
[00114] "Ariltio" refere-se a um grupo S-arila ou um grupo -S- heteroarila, conforme aqui indicado. Exemplos representativos incluem, sem limitação a feniltio, piridiniltio, furaniltio, tieniltio, pirimidinilltio, e similar, e seus derivados.
[00115] "Acila" ou "carbonila" refere-se a um grupo -C(O)R" onde R" é selecionado do grupo consistindo de hidrogênio, alquila inferior, trialometila cicloalquila não substituído, arila opcionalmente substituído com um ou mais, preferivelmente um dói sou três substituintes selecionados do grupo consistindo de alquila inferior, trialometila, alcóxi inferior, halo e grupos NRxRy, heteroarila (ligado através de um carbono do anel), opcionalmente substituído com um ou mais, preferivelmente um, dois ou três substituintes selecionados do grupo consistindo de alquila inferior, trialoalquila, alcóxi inferior, halo e grupos NRxRy e heteroalicíclico (ligado através de um carbono no anel) opcionalmente substituído com um ou mais, preferivelmente um, dois ou três substituintes selecionados do grupo consistindo de alquila inferior trialoalquila, alcóxi inferior, halo e grupos -NRxRy. Grupos acila representativos incluem, sem limitação, a acetila trifluoracetila, benzoíla e similar.
[00116] "Aldeído" refere-se a um grupo acila em que R" é hidrogênio.
[00117] "Tioacila"ou tiocarbonila" refere-se a um grupo -C(S)R" sendo R"como indicado supra.
[00118] Um grupo "tiocarbonila"refere-se a um grupo -C(S)R" sendo R' como indicado supra.
[00119] Um grupo "C-carbóxi" refere-se a um grupo -C(O)R" com R"como indicado supra.
[00120] Um grupo "O-carbóxi" refere-se a um grupo -OC(O)R" com R"como indicado supra.
[00121] "Éster" refere-se a um grupo -C(O)OR" com R" como aqui indicado, com exceção de que R" não pode ser hidrogênio.
[00122] Grupo "acetila" refere-se a um grupo -C(O)CH3.
[00123] Grupo "halo" refere-se a flúor, cloro, bromo ou iodo, preferivelmente flúor ou cloro.
[00124] Grupo "trialometila" refere-se a um grupo metila com três substituintes halo, tais como grupo trifluormetila.
[00125] "Ciano" refere-s a um grupo -C=N.
[00126] Um grupo "sulfinila" refere-se a um grupo -S(O)R" onde, além de ser como supracitado, R" também pode ser um grupo hidróxi.
[00127] Um grupo "sulfonila" refere-se a um grupo -S(O)2R" onde além de ser conforme indicado supra, R" também pode ser um grupo hidróxi.
[00128] "S-sulfonamido" refere-se a um grupo -S(O)2NRxRy com Rx e Ry conforme indicado supra.
[00129] "N-sulfonamido" refere-se a um grupo -NRXS(O)2RY com Rx e Ry como indicado supra.
[00130] "Grupo "O-carbamila" refere-se a um grupo -OC(O)NRXRY com Rx e Ry como indicado supra.
[00131] "N-carbamila" refere-se a um grupo -RyOC(O)NRX com Rx e Ry como indicado supra.
[00132] "O-tiocarbamila" refere-se a um grupo -OC(S)NRXRY com Rx e Ry como supracitado.
[00133] "N-tiocarbamila" refere-se a um grupo -RyOC(S)NRX com Rx e Ry como supracitado.
[00134] "Amino" refere-se a um grupo NRXRY onde Rx e Ry são ambos hidrogênio.
[00135] "C-amido" refere-se a um grupo -C(O)NRXRY com Rx e Ry como supracitado.
[00136] "N-amido" refere-se a um grupo -RyC(O)NRY com Rx e Ry como supracitado.
[00137] "Nitro" refere-se a um grupo -NO2.
[00138] "Haloalquila" significa um alquila, preferivelmente alquila inferior, que é substituído com um ou mais átomos de halogênio iguais ou diferentes, por exemplo, -CH2Cl, -CF3, -CH2CF3, -CH2Cl3, e similar.
[00139] "Hidroxialquila" significa um alquila, preferivelmente alquila inferior, que é substituído com um, dois, ou três grupos hidróxi, por exemplo, hidroximetila, 1 ou 2-hidroxietila, 1,2-, 1,3- ou 2,3- diidroxipropila, e similar.
[00140] "Aralquila" significa alquila, preferivelmente alquila inferior que é substituído com um grupo arila como indicado supra, por exemplo, -CH2 fenila, -(CH2)2fenila, -(CH2)3fenila, CH3CH(CH3)CH2 fenila e similar bem como seus derivados,
[00141] "Grupo heteroaralquila" significa alquila, preferivelmente alquila inferior, que é substituído com um grupo heteroarila, por exemplo, CH2 piridila, -CH2 piridinila, -(CH2)2 pirimidinila, -(CH2)3 imidazolila e similar, e seus derivados.
[00142] "Monoalquilamino" significa um radical, -NHR onde R é um alquila ou cicloalquila não substituídos, por exemplo metilamino, (1- metiletil)amino, cicloexilamino e similar.
[00143] "Dialquilamino" significa um radical -NRR onde cada R é independentemente um alquila ou grupo cicloalquila não substituído, dimetilamino, dietilamino, (1-metiletil)-etilamino, cicloexilmetilamino, ciclopentilmetilamino e similar.
[00144] "Opcional" ou "opcionalmente" significa que, o evento subsequentemente descrito ou circunstancia podem, porém não necessitam, ocorrer, e que a descrição inclui exemplos onde o evento ou circunstancia ocorre e os exemplos em que ele não ocorre. Por exemplo, "grupo heterociclo opcionalmente substituído com um grupo alquila" significa que, o alquila pode, porém não necessita, estar presente, e a descrição inclui situações onde o grupo heterociclo é substituído com um grupo alquila e situações onde o grupo heterociclo não é substituído com o grupo alquila.
[00145] Uma "composição farmacêutica" refere-se a uma mistura de um ou mais compostos supracitados, ou sais fisiologicamente, farmaceuticamente aceitáveis, solvatos, hidratos, ou prodrogas dos mesmos, com outros componentes químicos, tais como veículo e excipientes fisiologicamente/farmaceuticamente aceitáveis. A finalidade de uma composição farmacêutica é facilitar a administração de um composto a um organismo.
[00146] Como aqui empregado, um "veículo fisiologicamente/farmaceuticamente aceitável" refere-se a um veículo ou diluente que não ocasiona irritação significativa a um organismo e não ab-roga a atividade biológica e propriedades do composto administrado.
[00147] Um "excipiente farmaceuticamente aceitável" refere-se a uma substância inerte adicionada a uma composição farmacêutica para facilitar mais ainda a administração de um composto. Exemplos sem limitação, de excipientes incluem carbonato de cálcio, fosfato de cálcio, vários açúcares e tipos de amido, derivados de celulose, gelatina, óleos vegetais e polietileno glicóis.
[00148] Como aqui empregado, o termo "sal farmaceuticamente aceitável" refere-se aos sais que retém a eficácia biológica e propriedades do composto de origem. Tais sais incluem:
[00149] sais de adição de acido, que podem ser obtidos por reação da base livre do composto de origem com ácidos inorgânicos, tais como ácido clorídrico, ácido bromídrico, ácido nítrico, ácido fosfônico, ácido sulfúrico, e ácido perclórico e similar, ou com ácidos orgânicos tais como ácido acético, ácido oxálico, ácido (D) ou (L) málico, ácido maléico, ácido metanossulfônico, ácido etanossulfônioc, ácido para-toluenossulfônico, ácido salicílico, ácido tartárico, ácido cítrico, ácido succínico ou ácido malônico e similar, ou
[00150] (2) sais formados quando um próton ácido presente no composto de origem seja substituído ou por um íon metálico, por exemplo, um íon de metal alcalino, um íon alcalino terroso, ou um íon alumínio, ou coordenados com uma base orgânica, tal como etanolamina, dietanolamina, trietanolamina, trometamina, N- metilglucamina e similar.
[00151] "PK" refere-se ao receptor de proteína tirosina quinase (RTKs), não receptor ou tirosina quinase "celular"(CTKs) e serina- treonina quinases (STKs).
[00152] "Modulação" ou "modulando" refere-se à alteração da atividade catalítica de RTKs, CTKs e STKs. Em particular, modulando refere-se à ativação da atividade catalítica de RTKs, CTKs, e STKs, preferivelmente a ativação ou inibição da atividade catalítica de RTKs, CTKs e STKs, dependendo da concentração do composto ou sal ao qual o RTK, CTK ou STK é exposto ou, mais preferivelmente, a inibição da atividade catalítica de RTKs, CTKs e STKs.
[00153] "Atividade catalítica" refere-se à velocidade de fosforilação de tirosina sob a influencia, direta ou indireta, de RTKs e/ou CTKs ou a fosforilação de serina e treonina sob a influência direta ou indireta de STKs.
[00154] "Contatando" refere-se a colocar um composto desta invenção e um PK alvo juntos num modo tal, que o composto pode afetar a atividade catalítica do PK, seja diretamente, ou seja, por interação com a própria quinase, ou indiretamente, ou seja, por interação com uma outra molécula, em que a atividade catalítica da quinase é dependente. Tal "contato" pode ser realizado in vitro, ou seja, num tubo de ensaio, uma placa petri ou similar. Num tubo de ensaio, o contato pode envolver apenas um composto e um PK de interesse ou ele pode envolver células totais. Células podem também ser mantidas ou desenvolvidas em placas de cultura de célula e contatadas com um composto naquele ambiente. Neste contexto, fez- se uma tentativa no sentido da capacidade de um composto particular em afetar um distúrbio relacionado à PK, ou seja, o IC50 do composto, indicado abaixo, pode ser determinado antes do uso dos compostos in vivo com organismos vivos mais complexos.
[00155] Para as células fora do organismo, existem múltiplos métodos e são bem conhecidos aos versados na técnica, pra manter o PKs em contato com os compostos incluído, sem limitação a microinjeção celular direta e numerosas técnicas veículo de transmembrana.
[00156] "In vitro" refere-se a procedimentos realizados num meio artificial, tal como, por exemplo, sem limitação num tubo de ensaio ou meio de cultura.
[00157] "In vivo" refere-se a procedimentos realizados dentro de um organismo vivo, tal como sem limitação, um camundongo, rato ou coelho.
[00158] "Distúrbio relacionado à PK", "distúrbio acionado por PK" e "atividade PK anormal" todos os termos referem-se a uma condição caracterizada por atividade catalítica de PK inadequada, ou seja, sob ou mais comumente sobre atividade catalítica de PK, onde o PK particular pode ser um RTK, um CTK ou um STK. Atividade catalítica inadequada pode surgir como resultando de: (1) expressão de PK em células que, normalmente não expressam PKs, (2) expressão de PK aumentada conduzindo a uma proliferação celular indesejada, diferenciação e/ou crescimento, ou (3) expressão de PK diminuída levando a reduções indesejadas na proliferação celular, diferenciação e/ou crescimento. Super-atividade de um PK refere-se a, ou amplificação do gene codificando um PK particular ou produção de um nível de atividade PK que pode correlacionar-se com uma proliferação, diferenciação e/ou distúrbio do crescimento (ou seja, quando o nível do PK aumenta, a gravidade de um ou mais sintomas do distúrbio celular aumenta). Sob-atividade é, naturalmente o inverso, onde a gravidade de um ou mais sintomas de um distúrbio celular aumenta, quando o nível da atividade PK diminui.
[00159] "Tratar, tratando" e "tratamento" refere-se a um método de aliviar ou anular um distúrbio celular mediado por PK e/ou de seus sintomas. Com relação particularmente ao câncer, esses termos, significam simplesmente que a expectativa de vida de um indivíduo afetado com um câncer será aumentada ou que um ou mais dos sintomas da doença serão reduzidos.
[00160] "Organismo" refere-se a qualquer entidade viva composta de pelo menos uma célula. Um organismo vivo pode ser tão simples, como, por exemplo, uma célula eucariótica simples ou como complexo, como um mamífero, incluindo um ser humano.
[00161] "Quantidade terapeuticamente eficaz" refere-se aquela quantidade do composto administrado o qual aliviará em alguma extensão, um ou mais sintomas do distúrbio sendo tratado. Com referência ao tratamento do câncer, uma quantidade terapeuticamente eficaz refere-se aquela quantidade que tem pelo menos um dos seguintes efeitos: reduzir o tamanho do tumor; inibir (ou seja, diminuir em alguma extensão, preferivelmente interromper) a metástase do tumor; inibir em alguma extensão (ou seja, diminuir em alguma extensão, preferivelmente interromper) o desenvolvimento do tumor, e aliviar em alguma extensão (ou, preferivelmente eliminar) um ou mais sintomas associados com o câncer.
[00162] "Monitorar" significa observar ou detectar o efeito de contato de um composto com uma célula expressando um PK particular. O efeito observado ou detectado pode ser uma alteração no fenótipo da célula, na atividade catalítica de um PK ou uma alteração na interação de um pK com um parceiro de ligação natural. Técnicas de observação ou detecção tais efeitos são do conhecimento da técnica. O efeito é selecionado de uma alteração ou uma ausência de alteração num fenótipo celular, uma alteração ou ausência de alteração na atividade catalítica da referida proteína quinase ou uma alteração ou ausência de alteração na interação da proteína quinase com um parceiro de ligação natural num aspecto final desta invenção.
[00163] "Fenótipo celular" refere-se a uma aparência externa de uma célula ou tecido ou função biológica da célula ou tecido. Exemplos sem limitação, de um fenótipo celular são o tamanho da célula, crescimento da célula, proliferação celular, diferenciação celular, sobrevivência celular, apoptose e absorção de nutriente e uso. Tais características fenotípicas são mensuráveis por técnicas conhecidas.
[00164] "Parceiro de ligação natural" refere-se a um polipeptídeo que se liga a um PK particular numa célula. Parceiros de ligação natural podem desempenhar um papel na propagação de um sinal no processo de transdução de sinal mediado por PK. Uma alteração na interação do parceiro de ligação natural com PK pode se manifestar como uma concentração aumentada ou diminuída do complexo PK/parceiro de ligação natura, resultando numa alteração observável na capacidade do PK em mediar a transdução de sinal.
[00165] Como aqui usado, os termos "opticamente puro" "enantiomericamente puro", "enantiômero puro" e "enantiômero opticamente puro" significa uma composição que compreende um enantiômero de um composto que é substancialmente isento do enantiômero oposto do composto. Um composto opticamente puro típico compreende mais que cerca de 80% em peso de um enantiômero do composto e menos que cerca de 20% em peso do enantiômero oposto do composto, mais preferivelmente mais que cerca de 90% em peso de um enantiômero do composto e menos que cerca de 10% em peso do enantiômero oposto do composto, mesmo mais preferivelmente mais que cerca de 95% em peso de um enantiômero do composto e menos que cerca de 5% em peso do enantiômero oposto do composto, e mais preferivelmente mais que cerca de 97% em peso de um enantiômero do composto e menos que cerca de 3% em peso do enantiômero oposto do composto.
[00166] Esquemas genéricos para sintetização dos compostos da invenção podem ser vistos nos Exemplos abaixo.
[00167] Alguns procedimentos gerais são mostrados com referência à síntese de compostos onde a fração 1-(2,6-dicloro-3- fluorfenil)-etóxi é o isômero(R) puro, e alguns, demonstram-se com referencia a compostos onde tal fração é uma mistura racêmica. Deve ficar entendido que, os procedimentos presentes podem ser empregados para produzir compostos racêmicos ou isômeros enantiomericamente puros (R) por escolha do correspondente material de partida racêmico ou enantiomericamente puro.
[00168] Os procedimentos mostrados podem ser usados para produzir uma ampla gama de compostos enantiomericamente puros ou seleção do apropriado material de partida enantiomericamente puro. Além dos compostos aqui mostrados, a invenção também proporciona compostos enantiomericamente puros correspondendo aos compostos 3-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]-piridin-2-ilamina e 3-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]pirazin-2-ilamina mostrados no Pedido de Patente U.S. N° de série 10/786.610 (PCT/US2004/005495); Pedido U.S. N° de série a ser cedido, Número de referência PC 32546, depositado em 26 de agosto de 2004 e intitulado "Aminoheteroaryl Compounds as Protein Kinase Inhibitors". A apresentação desses documentos está ora incorporada por referência em sua totalidade.
[00169] A menos que indicado em contrário, todas as referências no presente, aos compostos inventivos incluem referências a sais, solvatos, hidratos e complexos dos mesmos, e a solvatos, hidratos e complexos dos seus sais, incluindo polimorfos, estereoisômeros e versões isotopicamente rotuladas dos mesmos.
[00170] Sais farmaceuticamente aceitáveis incluem sais de adição de acido e base (incluindo os sais duplos).
[00171] Sais de adição de acido adequados são formados de ácidos que formam sais não tóxicos. Exemplos incluem: sais acetato, adipato, aspartato, benzoato, besilato, bicarbonato/carbonato, bissulfato/sulfato, borato, camsilato, citrato, ciclamato, edisilato, esilato, formiato, fumarato, gluceptato, gluconato, glucuronato, hexafluorfosfato, hibenzato, cloridrato/cloreto, bromidrato/brometo, iodidrato/iodeto, isetionato, lactato, malato, maleato, malonato, mesilato, metilsulfato, naftilato, 2-napsilato, nicotinato, nitrato, orotato, oxalato, palmitato, pamoato, hidrogeno fosfato, diidrogeno fosfato/fosfato, piroglutamato, sacarato, estearato, succinato, tanato, tartarato, tosilato e trifluoracetato.
[00172] Sais de base adequados são formados de bases que formam sais não tóxicos Exemplos incluem os sais de alumínio, arginina, benzatina, cálcio, colina, dietilamina, diolamina, glicina, lisina, magnésio, meglumina, olamina, potássio, sódio, trometamina e zinco.
[00173] Para uma pesquisa de sais adequados, ver "Handbook of Pharmaceutical Salts: Propeties, Selection and Use" por Stahl e Wermuth (Willy-VCH, Weinheim, Alemanha, 2002), cuja descrição está ora incorporada por referência em sua totalidade.
[00174] Um sal farmaceuticamente aceitável dos compostos inventivos pode ser prontamente preparado por mistura íntima de soluções do composto e da desejada base e desejado ácido. O sal pode precipitar-se da solução e ser coletado por filtração ou pode ser recuperado por evaporação do solvente. O grau de ionização no sal resultante pode variar de completamente ionizado a quase não ionizado.
[00175] Os compostos da invenção também podem existir numa forma não solvatada e solvatada. O termo "solvato" é aqui empregado para descrever um complexo molecular compreendendo o composto da invenção e uma ou mais moléculas de solvente farmaceuticamente aceitáveis, por exemplo, etanol. O termo 'hidrato' é empregado quando tal solvente é água.
[00176] Solvatos farmaceuticamente aceitáveis de acordo com a invenção incluem os em que o solvente de cristalização pode ser isotopicamente substituído, por exemplo, D2O, d6-acetona, d6-DMSO.
[00177] Também se inclui no escopo da invenção complexos tais como clatratos, complexos de inclusão de droga-hospedeiro, onde, em contraste com os solvatos supracitados, a droga e o hospedeiro estão presente em quantidades estequiométricas ou não-estequiométricas. Também se incluem complexos da droga contendo dois ou mais componentes orgânicos e/ou inorgânicos, que podem estar em quantidades estequiométricas ou não estequiométrica. Os complexos resultantes podem ser ionizados, parcialmente ionizados ou não ionizados. Para uma pesquisa de tais complexos ver J Pharm Sci 64(8) 1269-1288 por Haleblian (agosto/1975), cuja descrição está ora incorporada por referência em sua totalidade.
[00178] Também dentro do escopo da invenção estão polimorfos, prodrogas, e isômeros (incluindo isômeros ópticos, geométricos e tautoméricos) dos compostos inventivos.
[00179] Derivados de compostos da invenção que podem ter pouca ou nenhuma atividade farmacológica por eles próprios, porém, quando administrados a um paciente, podem ser convertidos nos compostos inventivos, por exemplo, por clivagem hidrolítica. Tais derivados são referidos como prodrogas. Mais informação do uso de prodrogas pode ser vistos em "Pro-drugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T Higuchi e W Stella) e "Bioreversible Carriers in Drug Design" Pergamon Press, 1987 (ed. E B Roche American Pharmaceutical Association), cuja descrição está ora incorporada por referência em sua totalidade.
[00180] Prodrogas de acordo com a invenção podem, por exemplo, ser produzidas substituindo funcionalidades adequadas presentes nos compostos inventivos com algumas frações conhecida aos versados na técnica, como "pró-frações" como descrito, por exemplo, em "Design of Prodrugs" por H Bundgaard (Elsevier, 1985), cuja apresentação está ora incorporada por referência em sua totalidade.
[00181] Alguns exemplos de prodrogas de acordo com a invenção incluem: onde o composto contém uma funcionalidade ácido carboxílico (-COOH)- um éster do mesmo, por exemplo, substituição do hidrogênio com (C1-6) alquila; onde o composto contem uma funcionalidade (-OH) um éster do mesmo, por exemplo, substituição do hidrogênio com (C1-6) alcanoilóximetila; e onde o composto contém uma funcionalidade amina primária ou secundária (-NH2 ou -NHR onde R^ H), uma amida do mesmo, por exemplo, substituição de um ou mais hidrogênios por (C110) alcanoíla.
[00182] Outros exemplos de grupo substituintes de acordo com os exemplos precedentes e exemplos de outros tios de prodroga podem ser vistos nas referencias supracitadas.
[00183] Finamente alguns compostos inventivos podem eles próprios, agir como prodrogas de outros compostos inventivos.
[00184] Os compostos da invenção contendo um ou mais átomos de carbono assimétricos podem existir como dois ou mais estereoisômeros. Onde um composto da invenção contiver um grupo alquenila ou alquenileno, isômeros geométricos cis/trans (ou Z/E) são possíveis. Onde o composto contiver, por exemplo, um grupo ceto ou oxima ou uma fração aromática, pode ocorrer isomerismo tautomérico ("tautomerismo"). Um único composto pode apresentar mais de um tipo de isomerismo.
[00185] Incluído no escopo da presente invenção estão todos os estereoisômeros, isômeros geométricos e formas tautoméricas dos compostos inventivos, incluindo os compostos que apresentam mais de um tipo de isomerismo, e misturas de um ou mais destes. Também estão incluídos os sais de adição de ácido ou base, onde o contraíon é opticamente ativo, por exemplo, d-lactato ou l-lisina, ou racêmico, por exemplo, DL-tartarato ou DL-arginina.
[00186] Isômeros cis/trans podem ser separados por técnicas convencionais bem conhecidas dos versados na técnica, por exemplo, cromatografia e cristalização fracionada.
[00187] Técnicas convencionais para preparação/isolamento de enantiômero individuais incluem síntese quiral de um precursor opticamente puro ou resolução do racemato (ou racemato de um sal ou derivado) usando por exemplo, cromatografia líquida de alta pressão quiral (HPLC).
[00188] Alternativamente, o racemato (ou precursor racêmico) pode ser reagido com um composto ativo opticamente adequado, por exemplo, um álcool, no caso onde o composto conteve uma fração ácida ou básica, um ácido ou base tal como ácido tartárico ou 1- eniletilamina. A mistura diastereomérica resultante pode ser separada por cromatografia e/ou cristalização fracionada e um ou ambos os diastereoisômeros convertidos no correspondente enantiômero puro por meios da técnica.
[00189] Compostos quirais da invenção (e seus precursores quirais) podem ser obtidos na forma enantiomericamente enriquecida usando cromatografia, tipicamente HPLC, numa resina assimétrica, com uma fase móvel consistido de um hidrocarboneto, tipicamente heptano ou hexano, contendo de 0 a 50% de isopropanol, tipicamente de 2 a 20% e de 0 a 5% de uma alquilamina, tipicamente 0,1% dietilamina. A concentração do eluato rende a mistura enriquecida.
[00190] Conglomerados estereoisométricos podem ser separados por técnicas convencionais conhecidas dos versados na técnica, por exemplo, "Stereochemistry of Organic Compounds" por E L Eliel (Wiley, New York, 1994), cuja descrição está ora incorporado por referência em sua totalidade.
[00191] A presente invenção inclui os compostos isotopicamente rotulados da invenção, onde um ou mais átomos são substituídos por átomos com o mesmo número atômico, porém, uma massa atômica ou número de massa diferente da massa atômica ou número de massa que predomina na natureza.
[00192] Exemplos de isótopos adequados para inclusão nos compostos da invenção incluem isótopos de hidrogênio, tais como H2 e H3, carbono, tais como C11, C13 e C14, cloro, tal como Cl36, flúor, tal como F18, iodo, tal como I123 e I125, nitrogênio, tais como N13 e N15, oxigênio, tal como O15, O17 e O18, fósforo, tal como P32 e enxofre tal como S35.
[00193] Alguns compostos isotopicamente rotulados da invenção, por exemplo, os que incorporam um isótopo radioativo, são úteis em testes de distribuição de droga e/ou tecido de substrato. Os isótopos radioativos de trítio, ou seja, H3 e carbono 14, ou seja, C14 são particularmente úteis para este fim, em virtude de sua facilidade de incorporação e prontos meios de detecção.
[00194] A substituição com isótopos mais pesados tais como deutério, ou seja, H2 pode render algumas vantagens terapêuticas resultantes de maior estabilidade metabólica, por exemplo, aumento na meia-vida in vivo ou requisitos de dosagem reduzida, sendo, portanto, preferidos, em algumas circunstancias.
[00195] A substituição com isótopos emitentes de positron, tais como C11, F18, O15 e N13 podem ser úteis em testes de Topografia de Emissão de Positron (PET) para exame da ocupação do receptor de substrato.
[00196] Compostos isotopicamente rotulados da invenção podem em geral, ser preparados por técnicas convencionais conhecidas dos versados na técnica, ou por processos análogos aos aqui descritos, usando um reagente isotopicamente rotulado apropriado no lugar do reagente não rotulado previamente empregado.
[00197] Solvatos farmaceuticamente aceitáveis de acordo com a invenção incluem os em que o solvente de cristalização pode ser isotopicamente substituído, por exemplo, D2O, d6-acetona, d6-DMSO.
[00198] Os compostos da invenção destinados a uso farmacêutico, podem ser administrados como produtos cristalinos ou amorfos. Eles podem ser obtidos, por exemplo, como tampões sólidos, pós ou filmes por métodos tais como precipitação, cristalização, liofilização, secagem por pulverização, ou secagem por evaporação. Secagem com microondas ou freqüência de rádio também podem ser usados para este fim.
[00199] Os compostos podem ser administrados sozinhos ou em combinação com um ou mais de outros compostos da invenção ou em combinação com um ou mais de outras drogas (ou como quaisquer combinações destes). Em geral, eles irão ser administrados como uma formulação em associação com um ou mais excipientes farmaceuticamente aceitáveis. O termo "excipiente" é aqui empregado para descrever qualquer ingrediente diferente dos compostos da invenção. A escolha do excipiente irá depender, em grande extensão de fatores tais como, modo particular de administração, efeito do excipiente na solubilidade e estabilidade, e da natureza da forma de dosagem.
[00200] Composições farmacêuticas adequadas para distribuição dos compostos da presente invenção e métodos para sua preparação serão prontamente evidentes aos versados na técnica. Tais composições e métodos para sua preparação podem ser vistos, por exemplo, em Remington's Pharmaceutical Sciences 19a Edição, (Mack Publishing Company, 1995), cuja descrição está ora incorporada por referência em sua totalidade.
[00201] Os compostos da invenção podem ser administrados oralmente. A administração oral pode envolver intumescimento, de forma que o composto ingresse ao trato gastrintestinal, e/ou administração bucal, lingual ou sublingual, por meio do qual, o composto ingressa na corrente sangüínea diretamente da boca.
[00202] Formulações adequadas para administração oral, incluem sistemas sólidos, semi-sólidos, e líquidos tais como comprimidos; cápsulas moles ou duras contendo multi- ou nano-particulados, líquidos ou pós; pastilhas (incluindo com recheio líquido), gomas, teles, formas de dosagem de rápida dispersão, filmes, óvulos, pulverizações e adesivos bucal/mucosa. Formulações líquidas incluem suspensões, soluções, xaropes e elixires. Tais formulações podem ser empregadas como cargas em cápsulas moles ou duras (feitas, por exemplo, de gelatina ou hidroxipropilmetilcelulose) e, tipicamente, compreendem um veículo, por exemplo, água, etanol, polietilenoglicol, propileno glicol, metilcelulose ou um óleo adequado, e um ou mais agentes emulsificantes, e/ou agentes de suspensão. Formulações líquidas podem também ser preparadas pela reconstituição de um sólido, por exemplo, de um sachet.
[00203] Os compostos da invenção também podem ser empregados em formas de dosagem de rápida dissolução, rápida desintegração, tais como as descritas em Expert Opinion in Therapeutic Patents, 11(6), 981-986, por Liang and Chen (2001).
[00204] Para formas de dosagem em comprimidos, dependendo da dose, a droga pode ser composta de 1% em peso a 80% em peso da forma de dosagem, mais tipicamente, de 5% em peso a 60% em peso da forma de dosagem. Além da droga, os comprimidos geralmente contêm um desintegrante. Exemplos de desintegrante, incluem glicolato amido de sódio, carboximetil celulose de sódio, carboximetil celulose de cálcio, croscarmelose de sódio, crospovidona, polivinilpirrolidona, metil celulose, celulose microcristalina, hidroxipropil celulose substituído com alquila inferior, amido, amido pré- gelatinizado, e alginato de sódio. Geralmente, o desintegrante compreenderá de 1% em peso a 25% em peso, preferivelmente de 5% em peso a 20% em peso da forma de dosagem.
[00205] Aglutinantes são geralmente empregados para conferir qualidades coesivas a uma formulação de comprimidos. Aglutinantes adequados incluem celulose cristalina, gelatina, açúcares, polietilenoglicol, gomas naturais e sintéticas, polivinilpirrolidona, amido pré-gelatinizado, hidroxipropil celulose e hidroxipropil metilcelulose. Comprimidos também podem conter diluentes, tais como lactose (monoidratadas, monoidratada seca por pulverização, anidra e similar), manitol, xilitol, dextrose, sacarose, sorbitol, celulose microcristalina, amido e fosfato de cálcio dibásico diidratado.
[00206] Comprimidos também podem compreender, opcionalmente, agentes ativos na superfície, tais como lauril sulfato de sódio e polisorbato 80, e deslizantes tais como dióxido de silício e talco. Quando presente, os agentes de superfície ativa podem compreender de 0,2% em peso a 5% em peso dos comprimidos e os deslizantes podem compreender de 0,2% em peso a 1% em peso do comprimido.
[00207] Comprimidos também contêm em geral, lubrificantes tais como estearato de magnésio, estearato de cálcio, estearato de zinco, estearil fumarato de sódio, e misturas de estearato de magnésio com lauril sulfato de sódio. Lubrificantes compreendem geralmente, de 0,25% em peso a 10% em peso, preferivelmente de 0,5% em peso a 3% em peso do comprimido.
[00208] Outros possíveis ingredientes incluem antioxidantes, colorantes, agentes aromatizantes, conservantes e agentes mascaradores de sabor.
[00209] Comprimidos exemplares contém até cerca de 80% da droga, de cerca de 10% em peso a cerca de 90% me peso do aglutinante, de cerca de 0% em peso a cerca de 85% em peso de diluente, de cerca de 2% em peso a cerca de 10% em peso de desintegrante, e de cerca de 0,25% a cerca de 10% em peso de lubrificante.
[00210] Combinações de comprimido podem ser compactadas diretamente ou por laminador formando os comprimidos. Combinações de comprimido ou pares de combinações podem, alternativamente, ser granuladas a líquido, a seco ou em fusão, congelados em fusão, ou extrusados antes da formação de comprimidos. A formulação final pode compreender uma ou mais camadas, e pode ser revestida ou não, podendo até mesmo ser encapsulada.
[00211] A formulação de comprimidos está apresentada em detalhe, em Pharmaceutical Dosage Forms; Tablets, Vol. 1 por H. Lieberman and L. Lachman, Marcel Dekkler, New York, 1980 (ISBN 0- 8247-691B-X), cuja descrição está ora incorporada por referência em sua totalidade.
[00212] Formulações sólidas para administração oral podem ser formuladas para liberação imediata e/ou modificada. Formulações de liberação modificada incluem liberação prolongada, controlada, pulsada, direcionada ao alvo e programada.
[00213] Formulações de liberação modificada adequadas para fins da invenção estão descritas na Patente US n° 6.106.864. Detalhes de outras tecnologias de liberação adequada tais como dispersões de alta energia e partículas osmóticas e revestidas devem ser encontradas em Pharmaceutical Technology On line, 25(2), 1-14, por Verma et al (2001) O emprego de goma de mascar para se conseguir liberação controlada está descrito em WO 00/35298. A apresentação dessas referências está ora incorporadas por referência em sua totalidade.
[00214] Os compostos da invenção também podem ser administrados diretamente à corrente sangüínea, ao músculo, ou a um órgão interno. Meios adequados para administração parenteral incluem intravenosa, intra-arterial, intraperitonial, intratecal, intraventricular, intrauretral, intraesternal intracraniana, intramuscular, intrasinovial e subcutânea. Dispositivos adequados para administração parenteral, incluem injetores de agulha (incluindo microagulha), injetores isentos de agulha e técnicas de infusão.
[00215] Formulações parenterais são tipicamente soluções aquosas que podem conter excipientes tais como sais, carboidratos e agentes de tamponamento. (preferivelmente a um pH desde 3 a 9), porém para algumas aplicações, elas podem ser mais adequadamente formuladas como uma solução não aquosa estéril, ou como uma forma seca para uso em conjunto com um veículo adequado tal como água estéril isenta de pirogênio.
[00216] A preparação das formulações parenterais sob condições estéreis, por exemplo, por liofilização, pode ser prontamente realizada usando técnicas farmacêuticas padrão, bem conhecidas dos versados na técnica.
[00217] A solubilidade dos compostos de fórmula I empregadas na preparação das soluções parenterais pode ser aumentada pelo emprego de técnicas de formulação adequadas, tais como a incorporação de agentes intensificadores da solubilidade.
[00218] Formulações para administração parenteral podem ser formuladas para liberação imediata e/ou modificada. Formulações de liberação modificada incluem liberação prolongada, controlada, pulsada, direcionada ao alvo e programada. Assim, os compostos da invenção podem ser formulados como uma suspensão ou como um líquido sólido, semi-sólido ou tixotrópico, para administração como um depósito para implante, proporcionando liberação modificada do composto ativo. Exemplos de tais formulações incluem stents revestidos de droga e microesferas de ácido poli(dl-láctico-co-glicólico) (PGLA) carregadas com droga.
[00219] Os compostos da invenção também podem ser administrados topicamente intradermicamente, ou transdermicamente à pele ou mucosa. Formulações típicas para este fim, incluem geles, hidrogeis, loções, soluções, cremes, ungüentos, pós para pulverização, curativos, espumas, filmes, adesivos para a pele, obreias, implantes, esponjas, fibras, faixas e microemulsão. Lipossomas também podem ser usados, Veículos típicos incluem álcool, água, óleo mineral, vaselina, líquida, vaselina branca, glicerina, polietilenoglicol e propileno glicol. Intensificadores da penetração podem ser incorporados, ver, por exemplo, J. Pharm, Sci, 88 (10), 955958, por Finnin e Morgan (Outubro/1999).
[00220] Outros meios de administração tópica incluem liberação por eletroporação, iontoforese, fonoforese, sonoforese e microagulha, ou injeção com microagulha ou sem agulha (por exemplo, Powderject™, Bioject™, etc), cuja apresentação está ora incorporada por referência, em sua totalidade.
[00221] Formulações para administração tópica podem ser formuladas para liberação imediata e/ou modificada. Formulações de liberação modificada incluem liberação prolongada, controlada, pulsada, direcionada ao alvo e programada.
[00222] Os compostos da invenção também podem ser administrados intranasalmente ou por inalação, tipicamente na forma de um pó seco (seja sozinho, ou como mistura, por exemplo, numa combinação seca com lactose, ou como uma partícula de componente misto, por exemplo, misturado com fosfolipídios, tais como fosfatidilcolina, de um inalador de pó seco, como uma pulverização em aerossol de um recipiente pressurizado, bomba, pulverização, atomizador, (preferivelmente um atomizador que usa eletro- hidrodinâmica para produzir uma névoa fina), ou nebulizador, com ou sem emprego de um propelente, al como 1,1,1,2-tetrafluoretano, ou 1,1,1,2,3,3,3-heptafluorpropano, ou com gotas nasais. Para uso intranasal, o pó pode compreender um agente bioadesivo, por exemplo, quitosana ou ciclodextrina.
[00223] O recipiente pressurizado, bomba, pulverização, atomizador ou nebulizador contém uma solução ou suspensão do(s) composto(s) da invenção compreendendo, por exemplo, etanol, etanol aquoso, ou um agente alternativo adequado para dispersão, solubilização, ou liberação prolongada do agente ativo, um propelente(s) tal como solvente e um tensoativo opcional, tal como trioleato de sorbitan, ácido oléico ou um ácido oligoláctico.
[00224] Ante do uso numa formulação de pó seco ou suspensão, o produto de droga é micronizado até um tamanho adequado para distribuição por inalação (tipicamente menor do que 5 μ), Isto pode ser feito pro qualquer método de trituração adequado, tal como trituração a jato espiral, trituração em jato de leito fluido, processamento de fluido supercrítico formando nanopartículas, homogeneização de alta pressão ou secagem por pulverização.
[00225] Cápsulas (feitas, por exemplo, de gelatina ou hidroxipropilmetilcelulose), empolas e cartuchos para uso como um inalador ou insuflador podem ser formuladas para conter uma mistura de pó do composto da invenção, uma base de pó adequada tal como lactose ou amido e um modificador de desempenho como l-leucina, manitol ou estearato de magnésio. A lactose pode ser anidra ou na forma de monoidrato, preferivelmente, a última. Outros excipientes adequados incluem, dextrana, glicose, maltose, sorbitol, xilitol, frutose, sacarose e trehalose.
[00226] Uma solução adequada para uso num atomizador usando eletro-hidrodinâmica para produção de uma névoa fina pode conter de 1 μg a 20 mg do composto da invenção por atuação e o volume da atuação pode vaiar de 1 μl a 100 μl. Uma formulação típica pode compreender um composto de fórmula I, propileno glicol, água estéril, etanol e cloreto de sódio. Solventes alternativos que podem ser usados no lugar de propileno glicol incluem glicerol e polietilenoglicol.
[00227] Aromatizantes adequados tais como mentol e levomentol, ou adoçantes, tais como sacarina ou sacarina sódica podem ser adicionados para as formulações da invenção destinadas à administração inalada/intranasal.
[00228] As formulações para administração inalada/intranasal, podem ser formuladas para liberação imediata e/ou modificada, usando, por exemplo, ácido poli(DL-láctico/co-glicólico (PGLA). Formulações de liberação modificada incluem liberação prolongada, controlada, pulsada, direcionada ao alvo e programada.
[00229] No caso de inaladores de pó seco e aerossóis, a dosagem unitária é determinada por meio de uma válvula, que distribui uma quantidade medida. Unidades de acordo com a invenção são tipicamente dispostas para administrar uma dose medida ou "pulverização" contendo uma quantidade desejada do composto da invenção. A dose diária global em geral pode ser administrada numa única dose, ou mais normalmente, como doses divididas, durante o dia.
[00230] Os compostos da invenção podem ser administrados retalmente ou vaginalmente, por exemplo, na forma de um supositório, pessário ou enema. Manteiga de cacau é uma base de supositório tradicional, mas várias alternativas podem ser usadas, conforme o caso.
[00231] Formulações para administração retal/vaginal podem ser formuladas para liberação imediata e/ou modificada. Formulações de liberação modificada incluem liberação prolongada, controlada, pulsada, direcionada ao alvo e programada.
[00232] Os compostos da invenção também podem ser administrados diretamente ao olho ou ouvido, tipicamente na forma de gotas de uma suspensão ou solução micronizada em solução salina estéril com ajuste de pH, isotônica. Outras formulações adequadas para administração ocular e aural incluem ungüentos, geles, biodegradáveis (por exemplo, gel absorvível, esponjas, colágeno) e não biodegradáveis (por exemplo, silicone) implantes, obreias, lentes e particulado ou sistemas vesiculares, tais como nanossomas ou lipossomas. Um polímero tal como ácido poliacrílico reticulado, álcool polivinílico, ácido hialurônico, um polímero de celulose, por exemplo, hidroxipropilmetilcelulose, hidroxietilcelulose, ou metil celulose ou um polímero hetero-polissacarídeo, por exemplo, goma gelana, pode ser incorporada juntamente com um conservante, tal como cloreto de benzalcônio. Tais formulações também podem ser liberadas por iontoforese.
[00233] Formulações para administração ocular/aural, podem ser formuladas para liberação imediata e/ou modificada. Formulações de liberação modificada incluem liberação prolongada, controlada, direcionada ao alvo ou programada.
[00234] Os compostos da invenção podem ser combinados com grupos macromoleculares solúveis, tais como ciclodextrina e seus derivados adequados, ou polímeros contendo polietilenoglicol, de modo a melhorar sua solubilidade, velocidade de dissolução, maceramento do sabor, biodisponibilidade e/ou estabilidade para emprego em quaisquer modos de administração supracitados.
[00235] Complexos de droga-ciclodextrina, por exemplo, são vistos serem de uso para a maioria das formas de dosagem e vias de administração. Complexos, tanto de inclusão como não inclusão podem ser usados. Como uma alternativa à complexação direta com a droga, a ciclodextrina pode ser empregada como um aditivo auxiliar, ou seja, como veículo, diluente ou solubilizante. Mais comumente usado para este fim, são alfa-, beta- e gama-ciclodextrinas, exemplos das quais, podem ser encontrados nos Pedidos de Patente Internacional Nos. WO 91/11172, WO 94/02518 e WO 98/55148, cuja apresentação está ora incorporada por referência ao presente.
[00236] A quantidade do composto ativo administrado dependerá do indivíduo em tratamento, da gravidade do distúrbio ou condição, da velocidade de administração e do julgamento do médico atendente. Contudo, uma dosagem eficaz fica na faixa de cerca de 0,001 a cerca de 100 mg por kg de peso corpóreo/dia, preferivelmente cerca de 0,01 a cerca de 35 mg/kg/dia, em doses únicas ou divididas. Para um humano de 70 kg, isto seria avaliado numericamente em cerca de 0,08 a cerca de 7000 mg/dia, preferivelmente cerca de 0,7 a cerca de 2500 mg/dia. Em alguns casos, níveis de dosagem abaixo do limite inferior da referida faixa pode ser mais do que adequado, enquanto em outros casos, doses ainda maior podem ser empregadas sem ocasionar qualquer efeito colateral danoso, contanto que, tais doses maiores sejam primeiramente divididas em várias doses pequenas para administração no decorrer do dia.
[00237] Tanto quanto se deseje administrar uma combinação dos compostos ativos, por exemplo, para fins de tratamento de uma dada doença ou condição, encontra-se no escopo da presente invenção o fato de que duas ou mais composições farmacêuticas, pelo menos uma das quais, contendo um composto de acordo com a invenção, poderem ser convenientemente combinadas na forma de um kit adequado para co-administração das composições.
[00238] Assim, o kit da invenção compreende duas ou mais composições farmacêuticas separadas, pelo menos uma das quais contendo um composto de fórmula I de acordo com a invenção, e meios para reter em separado, as composições, ais como um recipiente, garrafa dividida ou bolsa com folha divisória. Um exemplo desse kit é a embalagem de empola familiar usada para embalagem dos comprimidos, cápsulas e similar.
[00239] O kit da invenção é particularmente adequado para administração de diferentes formas de dosagem, por exemplo, oral e parenteral, para administração das composições separadas em diferentes intervalos de dosagem, ou para titulação das composições separadas entre si. Para ajudar na aquiescência, o kit compreende, tipicamente, instruções para administração e pode ser providenciado com um auxiliar de memória.
[00240] Nos exemplos a seguir, "Et" significa etila, "Ac" significa acetila, "Me" significa metila, "Ms"significa metanossulfonila (CH3SO2), "iPr" significa isopropila, "HATU" significa hexafluorfosfato de 2-(7-aza- 1H-benzotriazol-1-il)-1,1,3,3-tetrametilfurônio, "Ph" significa fenila, "Boc" significa terc-butoxicarbonila, "EtOAc"significa acetato de etila, "HOAc" significa ácido acético, "Net3" ou "Et3N" significa trietilamina, "THF" significa tetraidrofurano, "DIC" significa diisopropilcabodiimida, "HOBt" significa hidróxi-benzotriazol, "MeOH" significa metanol, "i- PrOAc" significa acetato de isopropila, "KOAc" significa acetato de potássio, "DMSO" significa dimetilsulfóxido, "AcCl" significa cloreto de acetila, "CDCl3" significa clorofórmio deuterado, "MTBE: significa éter metil t-butílico, "DMF" significa dimetilformamida, "Ac2O" significa anidrido acético, "Me3SOI" significa iodeto de trimetilsulfoxônio, "DMAP" significa 4-dimetilaminopiridina, "dppf" significa difenilfosfíno ferroceno, "DME" significa éter dimetílico de etileno glicol, HOBT significa 1-hidroxibenzotriazol, EDC significa 1-etil-3-(3- metilaminopropil)-carbodiimida.
[00241] Os exemplos a seguir são dados para ilustração da presente invenção. Deve ficar entendido, contudo, que a invenção não está limitada às condições específicas ou detalhes descritos nesses exemplos.
[00242] Reagentes podem ser sintetizados como se demonstra aqui, ou são disponíveis de fontes comerciais (por exemplo, Aldrich, Milwaukee, WI; Acros, Morris Plains, NJ; Biosynth International, Naperville, IL; Frontier Scientific, Logan, UT; TCI América, Portland, OR; Combi-Blocks, San Diego, CA; Matrix Scentific, Columbia, SC; Acros, Morris Plains, NJ; Alfa Aesar, Ward Hill, MA; Apollo Scientific, UK; etc.) ou podem ser sintetizados por procedimentos conhecidos na técnica.
[00243] A síntese de vários reagentes específicos é mostrada no Pedido de Patente U.S. N° de série 10/786.610, intitulado "Aminoheteroaryl Compounds as Protein Kinase Inhibitors" depositado em 26 de fevereiro de 2004, e o correspondente pedido internacional PCT/US2004/005495 do mesmo ‘titulo, depositado em 26 de fevereiro de 2004.
[00244] Outros reagentes podem ser sintetizados por adaptação dos procedimentos aí descritos, e o perito na técnica pode prontamente adaptar os procedimentos para produção dos compostos desejados. Além disso, essas referências contêm procedimentos gerais e exemplos específicos para preparação de um grande número de compostos heteorarilamino, e o perito na técnica pode adaptar prontamente tais procedimentos e exemplos para a preparação dos compostos da presente invenção. A apresentação dessas referências está ora incorporada por referência em sua totalidade.
[00245] Quando um procedimento geral ou exemplar sintético é referido, o perito na técnica pode determinar prontamente os reagentes apropriados, caso não indicados, estralando dos procedimentos gerais ou exemplares. Alguns procedimentos gerais somam dados como exemplos para preparação de compostos específicos. O versado na técnica pode adaptar prontamente tais procedimentos à síntese de outros compostos. Deve ficar entendido que, os grupos R mostrados nos procedimentos gerais destinam-se a ser genéricos e não limitantes e não correspondem a definições dos grupos R algures neste documento. Cada um de tal grupo R representam uma ou múltiplas frações químicas que podem ser iguais ou diferentes de outras frações químicas também representadas pelo mesmo símbolo R. O perito na técnica irá levar em consideração, de pronto, a faixa de grupos R adequados na síntese exemplar. Além disso, a representação de uma posição não substituída em estruturas mostrada ou referidas nos procedimentos gerais é para conveniência e não exclui a substituição conforme descrito algures aqui. Para os grupos específicos que podem estar presentes, sejam como grupos R nos procedimentos gerais, ou como substituintes opcionais não mostrados, faz-se referência a descrições no restante deste documento, incluindo as reivindicações, resumo e descrição detalhados.
[00246] Alguns procedimentos gerais são mostrados com referência a síntese de compostos onde a fração 1-(2,6-dicloro-3- fluorfenil)-etóxi é o isômero (R) puro, e alguns são mostrados com referência a compostos onde tal fração é uma mistura racêmica. Deve ficar entendido que, os procedimentos aqui podem ser usados para produzir compostos racêmicos ou isômeros (R) enantiomericamente puros por escolha do correspondente material de partida racêmico ou enantiomericamente puro.
[00247] Os procedimentos mostrados aqui podem ser usados para produzir uma ampla gama de compostos enantiomericamente puros por seleção do material de partida enantiomericamente puro apropriado. Além dos compostos aqui apresentados, a invenção também proporciona compostos enantiomericamente puros correspondentes aos compostos 3-[1-(2,6-dicloro-3-flúor-fenil)etóxi]- piridin-2-ilamina e 3-[1-(2,6-dicloro-3-fluorfenil)-etóxi]-pirazin-2-ilamina mostrados no Pedido de Patente U.S. N° de série 10/786.610 (PCT/US2004/005495), no Pedido US N° a ser cedido, número de referência PC 325446, depositado em 26 de agosto de 2004, e intitulado, "Pyrazolo-Substitutes Aminoheteroaryl Compounds as Protein Kinase Inhibitors", e no Pedido U.S. N° de série a ser cedido, n° de referência PC 32548, depositado em 26 de agosto de 2004, intitulado "Aminoheteroaryl Compounds as Protein Kinase Inhibitors". As revelações desses documentos estão ora incorporadas por referência em sua totalidade. Seleção dos Materiais de Partida 5-bromo-3-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]-piridin-2-ilamina (racemato)
[00248] 2,6-dicloro-3-fluoracetofenona (15 g, 0,072 mol) foi agitado em THF (150 mL, 0,5 M0 a 0°C usando um banho de gelo por 10 mt. Hidreto de alumínio e lítio (2,75 g, 0,071 mol) foi lentamente adicionado. A reação foi agitada a temperatura ambiente por 3 horas. A reação foi resfriada em banho de gelo e adicionou-se água (3 mL) cuidadosamente seguido por adição de NaOH 15% (3 mL) lentamente. A mistura foi agitada a temperatura ambiente por 30 minutos NaOH 15% (9 mL), MgSO4 foram adicionados e a mistura filtrada para remover os sólidos. Os sólidos foram lavados com THF (50 mL) e o filtrado foi concentrado dando 1-(2,6-dicloro-3-flúor-fenil)-etanol (14,8 g, 95% de rendimento) como um óleo amarelo.
[00249] 1H RMN (400 MHz, DMSO-d6) δ 1,45 (d, 3H), 5,42 (m, 2H), 7,32 (m, 1H).
[00250] 2. A uma solução agitada de trifenilfosfina (8,2 g 0,03 mol) e DEAD (13,65 mL de uma solução a 40% em tolueno) em THF (200 mL) a 0°C adicionou-se uma solução de 1-(2,6-dicloro-3-flúor-fenil)- etanol (4,55 g, 0,021 mol) e 3-hidróxi-tiopiridina (3,35 g, 0,03 mol) em THF (200 mL). A solução laranja brilhante resultante foi agitada sob uma atmosfera de nitrogênio a temperatura ambiente por 4 horas em cujo ponto todo o material de partida foi consumido. O solvente foi removido e o material bruto foi carregado a seco em gel de sílica e purificado com acetato de etila/hexanos (20:80) para render 3-(2,6- dicloro-3-flúor-benzilóxi)-2-nitro-piridina (6,21 g, 0,021 mol, 98%) como um sólido cor-de-rosa.
[00251] 1H RMN (CDCl3, 300 MHz) δ 1,8-1,85 (d, 3H), 6,0-6,15 (q, 1H), 7,0-7,1 (t, 1H), 7,2-7,21 (d, 1H), 7,25,7,5 (m, 2H), 8,0-8,05 (d, 1H).
[00252] 3. A uma mistura agitada de AcOH (650 mL e EtOH (500 mL) foi suspenso 3-(2,6-dicloro-3-flúor-benzilóxi)-2-nitropiridina (9,43 g, 0,028 mol) e lascas de ferro (15,7 g, 0,28 mol). A reação foi aquecida lentamente ao refluxo e deixada agitar por uma hora. A reação foi resfriada a temperatura ambiente a seguir éter dietílico (500 mL) e água (500 mL) foi adicionado. A solução foi cuidadosamente neutralizada por adição de carbonato de sódio. Os extratos orgânicos combinados foram lavados com NaHCO3 saturado (2 x 100 mL), H2O, (2 x 100 mL) e salmoura (1 x 100 mL) a seguir secos (Na2SO4) filtrados e concentrados até secura sob vácuo rendendo 3-(2,6-dicloro- 3flúor-benzilóxi)-piridin-2-ilamina (,04 g, 0,027 mol, (99%) como um sólido rosa claro.
[00253] 1H RMN (CDCI3, 300 MHz) δ 1,8-1,85 (d, 3H), 4,9-5,2 (brs, 2H), 6,7-6,84 (q, 1H), 7,0-7,1 (m, 1H), 7,2-7,3 (m, 1H), 7,6-7,7 (m 1H).
[00254] 4. Uma soIução agitada de 3-(2,6-dicIoro-3-fIúor-benziIóxi)- piridiI-2-iIamina (9,07 g, 0,03 mol) em acetonitrila foi resfriada para 0°C usando um banho de geIo. A esta soIução adicionou-se N- bromossuccinimida (NBS) (5,33g, 0,03 mol) em partes. a reação foi agitada a 0°C por 15 minutos. A reação foi concentrada até secura sob vácuo. O óleo escuro resultante foi dissolvido em EtOAc (500 mL)., e purificada via cromatografia de gel de sílica Os solventes foram então removidos in vácuo rendendo 5-bromo-3-(2,6-dicloro-3-flúor-benzilóxi)- piridin-2-ilamina (5,8 g, 0,015 mol, 51%) como um sólido cristalino branco.
[00255] 1H RMN (CDCl3, 300 MHz) δ 1,8-1,85 (d, 3H), 4,9-5,2 (brs, 2H), 6,7-6,84 (q, 1H), 7,0-7,1 (m, 1H), 7,2-7,3 (m, 1H), 7,6-7,7 (m 1H). 5-iodo-3-[1-(2,6-dicloro-3-flúor-fenil) - etóxi -piridin-3-ilamina (racemato):
[00256] A uma solução de 3-[1-(2,6-dicloro-3-flúor-5-fenil)-etóxi]- piridin-2-ilamina (10,0 g 33,2 mmols) em acetonitrila 600 mL) e ácido acético (120 mL) adicionou-se N-iodosuccinimida (11,2 g, 49,8 mmols). A mistura foi agitada a temperatura ambiente por 4 horas e a reação foi arrefecida com solução de Na2SO4. Após evaporação, o resíduo foi dividido entre acetato de etila e água. A camada orgânica foi lavada com solução de NaOH 2N, salmoura e seca sobre Na2SO4. O produto bruto foi purificado numa coluna de gel de sílica dando 5-iodo-3-[1- (2,6-dicloro-3-flúor-fenil}-etóxi]-piridin-2-ilamina (7,1 g, 50% rendimento).
[00257] EM m/z 427 [M+1]. 1H RMN (400 MHz, DMSO-D6) δ ppm 1,74 (d, J = 6,57 Hz, 3H) 5,91-5,99 (m, 3H) 6,82 (d, J = l,26 Hz, 1H), 7,46 (t, J = 8,72 Hz, 1H) 7,56 (dd, J = 8,97, 4,93 Hz, 1H), 7,62 (d, J= 1,52 Hz, 1H). 5-bromo-3-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]pirazin-2-ilamina (racemato)
[00258] 2,6-dicloro-3-fluoracetofenona (15 g, 0,072 mol) foi agitado em THF (150 mL, 0,5 M) a 0°C usando um banho de gelo por 10 mt. Hidreto de alumínio e lítio (da Aldrich, 2,75 g, 0,072 mol) foi lentamente adicionado, e água (3 mL) foi adicionado gota a gota seguido por adição de NaOH 15% (3 mL) lentamente. A mistura foi agitada a temperatura ambiente por 30 minutos. NaOH 15% (9 mL), MgSO4 foram adicionados e a mistura filtrada para remover os sólidos. Os sólidos foram lavados com THF (50 mL) e o filtrado foi concentrado dando 1-(2,6-dicloro-3-flúor-5- fenil)-etanol (14,8 g, 95% de rendimento) como um óleo amarelo.
[00259] 1H RMN (400 MHz, DMSO-d6) δ 1,45 (d, 3H), 542 (m, 2H), 7,32 (m, 1H), 7,42 (m, 1H).
[00260] 2. 5-bromo-3-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]pirazin-2- ilamina foi preparado seguinte ao procedimento 2 abaixo, de 1-(2,6- dicloro-3-flúor-fenil)-etanol e 3,5-dibromo-pirazin-2-ilamina.
[00261] 1H RMN (400 MHz, DMSO-d6) δ 1,74 (d, 3H), 6,40 (m, 1H), 6,52 (br s, 2H), 7,30 (m, 1H), 7,48 (m, 1H), 7,56 (s, 1H); MS m/z 382 (M+1).
[00262] PLE é uma enzima produzida por Roche e vendia em Biocatalytics Inc. como uma preparação de esterase bruta de fígado de porco, normalmente conhecida com PLE-AS (adquirido de Biocatalytics como ICR-123, vendido como uma suspensão de sulfato de amônio). A enzima é classificada no registro cãs como uma "hidrolase de éster carboxílico", CAS n° 9016-18-6". O número de classificação enzimática correspondente é EC 3.1.1.1. A enzima é conhecida por ter ampla especificidade de substrato de encontro à hidrólise de butirato de etila num titulador de pH. 1 LU (unidade lípase) é a quantidade de enzima que, libera 1μ mol de ácido butírico titulável por minuto a 22°C, pH 8,2. A preparação aqui descrita (PLE-AS como uma suspensão é normalmente embalada como um líquido castanho- esverdeado opaco com uma atividade declarada de > 45 LU/mg (teor de proteína em torno de 40 mg/mL).
[00263] (1S)-1-(2,6-dicloro-3-fluorfenil)etanol, mostrado como um composto (S-1) nos esquemas abaixo, foi preparado por uma combinação de hidrólise enzimática do acetato 1-(2,6-dicloro-3- fluorfenil)etil racêmico, esterificação e hidrólise química com inversão de acordo com o Esquema B. 1-(2,6-dicloro-3-fluorfenil)etil acetato racêmico (composto A2) foi preparado de acordo com o Esquema A. Esquema A
[00264] 1-(2,6-dicloro-3-fluorfenil)etanol (A1): borohidreto de sódio (90 mg, 2,4 mmol) foi adicionado para uma solução de 2', 6'-dicloro-3'- flúor-acetofenona (Aldrich, catálogo n° 52.294-5 (207 mg, 1 mmol) em 2 mL de CH3OH anidro. A mistura de reação foi agitada a temperatura ambiente por uma hora a seguir of evaporada para dar um resíduo oleoso incolor. O resíduo foi purificado por cromatografia flash (eluindo com ^10% EtOAc em hexanos) conferindo o composto A1 como um óleo incolor (180 mg, 0,88 mmol, 88,5% rendimento:
[00265] EM (APCI) (M-H)- 208; 1H RMN (400 MHZ, clorofórmio-D) δ ppm 1,64 (d, J = 6,82 Hz, 3H) 3,02 (d, J = 9,85 Hz, 1H) 6,97- 7,07 (m, 1H) 7,19-7,33 (m, 1H).
[00266] Acetato de 1-(2,6-dicloro-3-fluorfenil)etila (A2): Anidrido acético (1, 42 mL, 15 mmol) e piridina (1,7 mL, 21 mmol) foram adicionados seqüencialmente para uma solução do composto A1 (2,2 g, 10,5 mmol) em 20 mL de cloreto de metileno. A mistura de reação foi agitada a temperatura ambiente pó 12 horas e a seguir evaporada para dar um resíduo oleoso amarelado. O resíduo foi purificado por cromatografia flash eluindo com 7^ 9% EtOAc em hexanos dando o composto A2 como um óleo incolor (2,26 g, 9,0 mmol/ rendimento: 85,6%);
[00267] 1H RMN (400 MHz, clorofórmio-D) δ ppm 1,88 (d, J = 6,82 Hz, 3H) 2,31 (s, 3H) 6,62 (q, J = 6,82 Hz, 1H) 7,25 (t, J = 8,46 Hz, 1H) 7,49 (dd, J = 8,84, 5,05 Hz, 1H). Esquema B
[00268] Para um frasco dotado de camisa de 50 mL equipado com um eletrodo de pH, um agitador suspenso e uma linha de adição de base (NaOH 1M) foi adicionado 1,2 mL de tampão de fosfato de potássio 100 mM pH 7,0 e 0,13 mL de suspensão PLE AS. A seguir o composto A2 (0,13 g, 0,5 mmol, 1,00 eq.) foi adicionado gota a gota e a mistura resultante foi agitada a temperatura ambiente por 20 horas, mantendo o pH da reação constante a 7,0 usando NaOH 1M. Tanto a conversão e os ee's da reação foram monitorados por RP-HPLC e interrompida após 50% do material de partida ter sido consumido (aproximadamente 17 horas sob essas condições). A mistura foi então extraída três X com 10 mL de acetato de etila para recuperar, tanto éster como álcool como uma mistura de R-1 e S-2.
[00269] Cloreto de metanossulfonila (0,06 mL, 0,6 mmol) foi adicionado para uma solução de uma mistura de R-1 e S-2 (0,48 mmol) em 4 mL de piperidina sob atmosfera de nitrogênio. A mistura de reação foi agitada a temperatura ambiente por 3 horas, então evaporada obtendo-se um óleo. Adicionou-se água (20 mL) à mistura e a seguir EtOAc (20 mL) x 2) foi adicionado para extrair a solução aquosa. As camadas orgânicas foram combinadas, secas, filtradas e evaporadas para dar uma mistura de R-3 e S-2. Esta mistura foi usada na próxima etapa sem, mas purificação.
[00270] 1H RMN (400 MHz, clorofórmio-D) δ ppm 1,66 (d, J= 67,1 Hz, 3H) 1,84 (d, J = 7,1 Hz, 3H) 2,09 (s, 3H) 2,92(s, 3H) 6,39 (q, J = 7,0 Hz, 1H) 6,46 (q, J = 6,8 Hz, 1H) 6,98-7,07 (m, 1H) 7,07-7,17 (m, 1H) 7,23-7,30 (m, 1H) 7,34 (dd, J = 8,8, 4,80 Hz, 1H).
[00271] Acetato de potássio (0,27 g, 0,26 mmol) foi adicionado para uma mistura de R-3 e S-2 (0,48 mmol) em 4 mL de DMF sob atmosfera de nitrogênio. A mistura de reação foi aquecida para 100°C por 12 horas. Adicionou-se água (20 mL) para a mistura de reação e EtOAc (20 mL x 2) foi adicionado para extrair a solução aquosa. A camada orgânica combinada foi seca, filtrada, e evaporada para dar um óleo de S-2 (72 mg, 61% de rendimento em duas etapas) Quiralmente ee: 97,6%.
[00272] 1H RMN (400 MHz, clorofórmio-D) δ ppm 1,66 (d, J = 7,1 Hz, 3H) 2,09 (s, 3H) 6,39 (q, J = 6,8 Hz, 1H) 7,02 (t, 8,5 Hz, 1H) 7,227,30 (m, 1H).
[00273] Metóxido de sódio (19 mmols; 0,5M em metanol) foi adicionado lentamente a um composto S-2 (4,64 g, 18,8 mmols) sob uma atmosfera de nitrogênio a 0°C. A mistura resultante foi agitada a temperatura ambiente por 4 horas. O solvente foi evaporado e adicionou-se água (100 mL). A mistura de reação resfriada foi neutralizada com solução tampão de acetato de sódio/ácido acético para pH 7. Adicionou-se acetato de etila (100 mL x 2) para extrair a solução aquosa. As camadas orgânicas combinadas foram secas sobre Na2SO4, filtradas, e evaporadas obtendo-se um sólido branco (4,36 g, 94,9% de rendimento); SFC-EM: 97% ee.
[00274] 1H RMN (400 MHz, clorofórmio-D) δ ppm 1,65 (d, J = 6,8 Hz, 3H) 5,58 (q, J = 6,9 Hz, 1H) 6,96-7,10 (m, 1H) 7,22-7,36(m, 1H). 3-[(1R)-1-(2,6-dicloro-3-fluorfenil)etóxi] - 2 -nitropiridina
[00275] 3-hidróxi-2-nitropiridina (175 mg, 1,21 mmol) e trifenilfosfina (440 mg, 1,65 mmoL) foram adicionados sequencialmente a uma solução agitada de (1S)-1-(2,6-dicloro-3- fluorfenil)etanol (229,8 g, 1,1 mmol) em THF (10 mL sob uma atmosfera de nitrogênio. A mistura de reação foi mantida a temperatura ambiente por uma hora e a seguir azo-dicarboxilato de diisopropila (0,34 mL, 1,65 mmol) foi adicionado a 0°C. A mistura foi agitada por mais 12 horas. A mistura de reação foi evaporada sob vácuo para dar um óleo. O resíduo foi purificado por cromatografia flash (eluindo com 20 ^ 25% EtOAc em hexanos) para dar o composto titular como um sólido branco (321,5 mg, 0,97 mmol (321,5 mg, 0,97 mmol) 88,3% rendimento).
[00276] EM (APCI) (M+H)+ 331; SFC-EM: 99,5% ee. 1H RMN (400 MHz, clorofórmio-D) δ ppm 1,85 (d, J = 6,6 Hz, 3H) 6,10 (q, J = 6,6 Hz, 1H) 7,04-7,13 (m, 1H) 7,21 (dd, J = 8,5, 1,14 Hz, 1H) 7,30 {dd, J = 9,0, 4,9 Hz, 1H) 7,37 (dd, J = 5,6, 4,6 Hz, 1H) 8,04(dd, J = 4,6, 1,3 Hz, 1H). 3-[(1R)-1-(2,6-dicloro-3-fluorfenil)etóxi]-piridin -2-amina
[00277] Ferro (365 mg) foi adicionado para uma solução agitada de 3-[(1R)-1-(2,6-dicloro-3-fluorfenil)etóxi]-2-nitropiridina (321 mg, 0,97 mmol) numa mistura de EtOH (2 mL) e HCl 2M (0,2 mL) a 0°C. A solução resultante foi aquecida a 85°C por duas horas. Celite (0,5 g) foi adicionado para a mistura de reação resfriada. Esta mistura foi filtrada sobre um leito de celite e evaporada para dar o composto titular como um óleo escuro EM (APCI) M+H)+ 301. 5-bromo-3-[(1R)-(2,6-dicloro-3-fluorfenil)etóxi]-piridin-2-amina
[00278] O isômero R enantiomericamente puro foi preparado como descrito acima para o racemato, porém usando os materiais de partida enantiomericamente puros descritos supra.
[00279] 1H RMN (400 MHz, DMSO-dβ) δ 1,74 (d, 3H), 6,40 (m, 1H), 6,52 (br s, 2H), 7,30 (m, 1H), 7,48 (m, 1H), 7,56 (s, 1H); EM m/z 382 (M+1). 5-iodo-3-[(1R)-1-(2,6-dicloro-3-fluorfenil)etóxi]-piridin-2-amina
[00280] Ácido per-iódico ( 60mg, 0,24 mmol), iodo (130 mg, 0,5 mmol) e ácido sulfúrico (0,093 mL) foram adicionados em seqüência para uma solução agitada de 3-[(1R)-1-(2,6-dicloro-3-fluorfenil)etóxi]- piridin-2-amina (0,97 mmol) numa mistura de ácido acético (3 mL) e H2O (0,5 mL). A solução resultante foi aquecida para 80°C por 5 horas. A mistura de reação resfriada foi arrefecida com Na2SO4 (80 mg) e tornada básica com Na2CO3 saturado (2 x 100 mL) a pH 7. Cloreto de metileno (2 x 50 mL) foi adicionado para extrair a solução aquosa. As camadas orgânicas combinadas foram secas sobre Na2SO4, filtradas e concentrada sob vácuo. O resíduo foi purificado por cromatografia flash (eluindo com 35 ^40% EtOAc em hexanos) dando o composto titular como um óleo amarelo (254 mg, 0,6 mmol; 61,6% rendimento).
[00281] EM (APCI) (M+H)+ 426. 1H RMN (400 MHz, clorofórmio-D) δ ppm 1,81 (d, J = 6,8 Hz, 3H) 4,86 (s, 2H) 5,98 (q, J = 6,57 Hz, 1H) 6,96 (d, J = 1,5 Hz, 1H) 7,08 (dd, J = 9,0, 8,0 Hz, 1H) 7,31 (dd, J = 8,8, 4,8 Hz, 1H) 7,78 (d, J = 1,8 Hz, 1H). 5-bromo-3-[(1R)-1-(2,6-dicloro-3-fluorfenil)etóxi] -pirazin-2-ilamina
[00282] O composto titular foi preparado de acordo com o procedimento 2 a partir de (1S)-1-(2,6-dicloro-3-fluorfenil)etanol.
[00283] 1H RMN (400 MHz, DMSO-d6) δ 7,53 (s, 1H), 7,48 (m, 1H), 7,39 (t, 1H), 6,48 (s, 2H), 6,41 (q, 1H), 1,74 (d, 3H); LCMS: 381 [M+1]; c-Met Ki: 0,796 μM.
[00284] Para a síntese de 5-aril-3-piridin-2-ilamina-(benzilóxi substituído) (6):
[00285] Para a Síntese de 5-bromo-3-piridin-2-ilamina(benzilóxi substituído) (5):
[00286] Preparação de 3-(benzilóxi-substituído)-2-nitro-piridina (3): A uma solução agitada de CS2CO3 (1,0 equivalente molar) em DMF (0,2 M) sob uma atmosfera de N2 contendo 3-hidróxi-4-nitro-piridina (Aldrich, 1,0 equivalente molar) é adicionado brometo de benzila substituído (1,0 equivalente molar). A mistura é agitada por 6 horas a temperatura ambiente. A reação é a seguir diluída com EtOAc, e dividida com H2O. A camada aquosa é extraída com EtOAc duas vezes. As camadas orgânicas são a seguir combinadas, lavadas com H2O e salmoura, secas sobre Na2SO4, filtradas e concentradas até secura sob vácuo rendendo 3-(benzilóxi-substituído)-2-nitropiridina (3) como um sólido.
[00287] Preparação de 3-(benzilóxi-substituído)-piridin-2-ilamina (4): A uma mistura agitada de AcOH e EtOH (1,3:1) ‘suspenso 3- (benzilóxi-substituído-2-nitro-piridina (1,0 equivalente molar, 1 M) e aparas de ferro (1,0 equivalente molar). A reação é aquecida lentamente ao refluxo e deixada agitar por uma hora. A reação é resfriada para temperatura ambiente a seguir filtrada por um chumaço de celite. O filtrado resultante é neutralizado com NH4OH concentrado e a seguir extraído com EtOAc por três vezes. Os extratos orgânicos combinados são lavados com NaHCO3, água e salmoura, seco sobre Na2SO4, filtrado e concentrado até secura sob vácuo rendendo 3- (benzilóxi-substituído)-piridin-2-ilamina (4) como um sólido.
[00288] Preparação de 5-bromo-3-piridin-2-ilamina-(benzilóxi substituído)(5): Uma solução agitada de 3-piridin-2-ilamina-(benzilóxi- substituído)(4)(1,0 equivalente molar) em acetonitrila é resfriada para 0°C usando um banho de gelo. A esta solução adicionou-se N- bromossuccinimida (Aldrich, (1,0 equivalente molar) em partes. A reação é agitada a 0°C por 15 mt. A reação é concentrada até secura sob vácuo. O óleo escuro resultante é dissolvido em EtOAc e dividido com H2O. O extrato orgânico é a seguir lavado com NaHCO3 duas vezes e salmoura uma vez. Carvão ativado é adicionado para a camada orgânica e aquecido até o refluxo. A solução é a seguir resfria para temperatura ambiente e filtrada por um chumaço de celite. O extrato orgânico é a seguir concentrado até secura sob vácuo para 1/3 do volume original. Os sólidos são filtrados rendendo 5-bromo-3- (benzilóxi substituído)-piridin-3-ilamina (5) como um sólido.
[00289] Para a síntese de 5-aril-3-pirazin-2-ilamina-(benzilóxi- substituído) Procedimento Geral 2 - Para a síntese de 5-bromo-3-pirazin-2-ilamina- (benzilóxi-substituído)
Procedimento Geral 2 - Para a síntese de 5-bromo-3-pirazin-2-ilamina- (benzilóxi-substituído) 
[00290] Para uma solução resfriada em gelo do álcool benzílico substituído (1,0 equivalente molar) e tetraidrofurano anidro (0,14 M) adicionou-se hidreto de sódio (1,0 equivalente molar) lentamente sob atmosfera de nitrogênio. Após agitar por 30 minutos, 3,5- dibromopirazin-2-ilamina (1,0 equivalente molar) em tetraidrofurano (0,56 M) foi adicionado via funil de adição a uma velocidade de gotejamento rápida. Ao término da adição o banho de gelo foi removido e a reação foi refluxada sob nitrogênio e monitorada por HPLC de fase reversa. Após 18 horas, demonstrou-se, por HPLC que, a maioria do 3,5-dibromopirazin-2-ilamina de partida tinha sido consumido e a reação foi deixada resfriar para temperatura ambiente. A mistura de reação foi concentrada, diluída com acetato de etila, e lavada com salmoura. A camada orgânica foi seca sobre sulfato de magnésio anidro e concentrada in vácuo. O produto bruto foi purificado usando um gel de sílica eluindo com acetato de etila/diclorometano 1:1 rendendo o 5-bromo-3-pirazin-2-ilamina-(benzilóxi-substituído) como um sólido branco em 60-90% de rendimento.
[00291] Para Síntese de 5-aril-3-piridin-2-ilamina-(benzilóxi- substituído) e 5-aril-3-pirazin-2-ilamina-(benzilóxi-substituído)
[00292] Uma mistura de 5-bromo-3-piridin-2-ilamina-(benzilóxi- substituído) ou 5-bromo-3-pirazin-2-ilamina-(benzilóxi-substituído) (1,0 equivalente molar) ácido aril borônico ou éster (1,2 equivalente molar), cloreto de bis(trifenilfosfina) paládio II (0,03 equivalente molar) e carbonato de sódio (3,0 equivalentes molar), em éter dimetílico de etileno glicol e água (10:0,5, 0,03 M) é desgaseificada e carregada com nitrogênio por três vezes a seguir aquecida ao refluxo sob nitrogênio durante a noite. A reação é resfriada apara temperatura ambiente e diluída com acetato de etila. A mistura é lavada com água, salmoura, seca sobre Na2SO4 e purificada sobre uma coluna de gel de sílica rendendo 5-aril-3-piridin-2-ilamina-(benzilóxi-substituído) ou 5- aril-3-pirazin-2-ilamina-(benzilóxi-substituído).
[00293] Para Reação de Amidação de ácido 6-amino-5-piridin-3-il- benzóico-(benzilóxi-substituído)
[00294] A uma solução de ácido 6-amino-5-piridin-3-il-benzóico- (benzilóxi substituído) (1,0 equivalente molar), hidrato de 1- hidroxibenzotriazol (HOBT, 1,2 equivalente molar) e cloridrato de 1-(3- diimetilaminopropil)-3-etilcarbodiimida (EDC, 1,2 equivalente molar), em DMF (0,2 M) adicionou-se amina (1,2 equivalente molar). A solução de reação é agitada a temperatura ambiente durante a noite, a seguir diluída com EtOAc e dividida com H2O. O extrato orgânico é separado e a fase aquosa é extraída com EtOAc. As camadas orgânicas são combinadas, lavadas com NaHCO3, e concentradas até secura sob vácuo. O material é purificado usando cromatografia de coluna (gel de sílica 99:1 a 95:5 CH2Cl2/MeOH). As frações contendo o produto são concentradas sob vácuo rendendo o produto amida.
[00295] Para preparação de 3-(benzilóxi substituído)-5-(3- dialquilaminometil-1H-indol-5-il)-piridin-2-ilamina
[00296] Para uma solução de benzotriazol (1,0 equivalente molar) em diclorometano, (0,2 M) adicionou-se amina (1,0 equivalente molar). A reação é agitada por 5 mt a temperatura ambiente após o que, formaldeído (37% em peso, (1,0 equivalente molar) é adicionado e a reação capeada e agitada a temperatura ambiente por 3 horas. Uma vez que, por TLC (10% acetato de etila:diclorometano) demonstrar-se o consumo do benzotriazol de partida a reação é seca com sulfato de magnésio anidro (10 g) filtrada e concentrada in vácuo. O produto bruto é purificado com uma coluna de gel de sílica eluindo com acetato de etila:diclorometano 1:1 rendendo o produto desejado como um sólido branco.
[00297] Para uma solução do intermediário aminometilbenzotriazol (1,0 equivalente molar) em diclorometano (0,43 M) adicionou-se cloreto de alumínio (2,0 equivalente molar) seguido pro 3-(2,6-dicloro- benzilóxi)-5-(1H-indol-5-il)-piridina-2-ilamina (1,2 equivalente molar). A reação é capeada e aquecida com agitação para 40°C por 3-4 horas. A reação é então removida do calor e deixada resfriar para temperatura ambiente. A mistura de reação é diluída com hidróxido de sódio (0,2M) e clorofórmio, recapeada e vigorosamente agitada a temperatura ambiente pra dissolver o resíduo no frasco. O clorofórmio é extraído da solução aquosa, seco sobre sulfato de sódio anidro e concentrada in vácuo. O produto bruto é purificado com uma coluna de gel de sílica, primeiramente, eluindo com ácido acético/diclorometano 1:1, para purificar as impurezas menos polares e a seguir, eluindo o produto com clorofórmio:metanol, hidróxido de amônio 90:9:1, (rendimento 10-67%).
[00298] Para a síntese de 3-(benzilóxi substituído)-5-fenil-piridin-2- ilamina usando 3-(3-metóxi-benzilóxi)-5-fenil-piridin-2-ilamina
[00299] A uma solução do 3-benzilóxi-5-fenil-piridin-2-ilamina (Exemplo 1-87, 3,27 g 11,8 mmols) em metanol (30 mL) adicionou-se Pd(OH)2 (2,5 g, 2,37 mmols). A mistura foi desgaseificada e carreada com hidrogênio três vezes, e a seguir agitada sob balão de hidrogênio por 5 horas. A reação foi filtrada através de um chumaço de Celite, lavada com metanol e condensada. Após secagem com alto vácuo, foi obtido 2-amino-5-fenil-piridin-3-ol (2,04 g, 93% rendimento). EM m/z 187 [M+1].
[00300] A uma solução de 2-amino-5-fenil-piridin-3-ol (2,04 g, 10,95 mmols) em tetraidrofurano (anidro, 30 mL) adicionou-se NaH (1,31 g, 32,85 mmols) lentamente. A mistura foi agitada sob nitrogênio por 20 mt, e a seguir cloreto de tritila (3,66 g, 13, 14 mmol foi adicionado. A reação foi agitada a temperatura ambiente durante a noite sob nitrogênio. O solvente foi evaporado, e o resíduo foi dissolvido em diclorometano, lavado com água, e seco sobre Na2SO4. Após filtração e condensação, o produto bruto foi purificado numa coluna de gel de sílica eluindo com EtOAc-hexano (1:10) propiciando 5-fenil-2-(tritil-amino)-piridin-3-ol (1,09 g, 23% rendimento). EM m/z 427 [M+1].
[00301] A uma solução de 5-fenil-2-(tritil-amino)-piridin-3-ol (100 mg, 0,24 mmol) em tetraidrofurano (3 mL) adicionou-se Cs2CO3 (79 mg, 0,24 mmol). A mistura foi agitada a temperatura ambiente por 20 mt e a seguir brometo de 3-metoxibenzila (0,037 mL, 0,26 mmol) foi adicionado. A reação foi agitada a temperatura ambiente durante a noite, diluída com diclorometano (5 mL) e filtrada removendo-se os sais. Os solventes foram evaporados, e o resíduo foi dissolvido em 10% de ácido trifluoracético em diclorometano (2 mL). A reação foi agitada por duas horas e evaporada. O resíduo foi dissolvido em diclorometano, lavado com NaHCO3 sat. e seco sobre Na2SO4. Após filtrar e concentrar, o produto bruto foi purificado numa coluna de gel de sílica eluindo com metanol/diclorometano (gradiente de 3% a 15%) dando 3-(3-metóxi-benzilóxi)-5-fenil-piridin-2-ilamina como um sólido branco (43,5 mg, 60% rendimento).
[00302] Para a síntese de 3-(benzilóxi substituído)-5-aril-piridin-2- ilamina usando 5-[4-(2-morfolin-4-il-etóxi) fenil]-3-(3-nitro- benzilóxi)piridin-2-ilamina:
[00303] A uma solução de 2-amino-5-[4-(2-morfolin-4-il-etóxi-fenil]- piridin-3-ol (preparada de acordo com o procedimento para 2-amino-5- fenil-piridin-3-ol no Exemplo 1-88 do Pedido de Patente U.S. N° de série 10/786.610 (PCT/US2004/005495) 45,5 mg, 0,14 mmoL) em DMF (3 mL) a 0°C foi adicionado NaH (60% em óleo) (5,6 mg, 0,14 mmol) e a mistura foi agitada a 0°C por 20 mt. A seguir 1-bromometil- 3-nitro-benzeno foi adicionado e a mistura foi agitada a 0°C por 1 hora e a temperatura ambiente por duas horas. HCl aquoso frio 1N (0,1 mL) foi adicionado e o solvente foi removido sob pressão reduzida. O resíduo foi purificado com cromatografia em gel de sílica (CH2Cl2:MeOH:NH2OH = 100:3:0,3) dando 5-[4-(2-morfolin-4-il-etóxi)- fenil]-3-(3-nitro-benzilóxi-piridin-2-ilamina como um sólido amarelo (44 mg, 68%).
[00304] Para a Síntese de {4-[6-amino-5-(benzilóxi substituído) - piridin-3-il]-fenil}-[(2R)-2-pirrolidin-1-ilmetil-pirrolidin-1-il]-metanona usando {4-[6-amino-5-(4-flúor-2-trifluormetil-benzilóxi)-piridin-3-il]-fenil}- [(2R)-2-pirrolidin-ilmetil-pirrolidin-1-il]-metanona.
[00305] Ácido 6-amino-5-benzilóxi-nicotínico foi preparado de acordo com o Procedimento 3 a partir de 3-benzilóxi-5-bromo-piridin-2- ilamina e ácido 4-(4,4,5,5-tetrametil-[1,3,2]idoxaborolan-2-il)-benzóico. EM m/z 321 (M+1).
[00306] [4-(6-amino-5-benzilóxi-piridin-3-il)-fenil]-[(2R)-2-pirrolidin- 1-ilmetil-pirrolidin-il]-metanona foi preparado seguindo o procedimento 4 p usando ácido 6-amino-5-benzilóxi-nicotínico e (2R)-pirrolidin-1- ilmetil-pirrolidina (preparado no Exemplo 1-39 do Pedido de Patente Número de Série 10/786.610 (PCT/US2004/005495)). EM m/z 457 (M+1).
[00307] A uma solução de [4-(6-amino-5-benzilóxi-piridin-3-il)- fenil]-[(2R)-2-pirrolidin-1-ilmetil-pirrolidin -il]-metanona (2,28 g, 5,00 mmol) em metanol (25 mL) adicionou-se pd/C a 10% (100 mg). A mistura foi desgaseificada e carregada com hidrogênio por três vezes, a seguir agitada sob balão de hidrogênio durante a noite. A reação foi filtrada através de um chumaço de celite, lavada com metanol, e condensada. Após secagem com alto vácuo, [4-(6-amino-5-hidróxi- piridin-3-il)-fenil]-[(2R)-2-pirrolidin-1 -ilmetil-pirrolidin-1-il]-metanona foi obtido com 95% de rendimento 1,74 g.
[00308] 1H RMN (400 MHz, DMSO-d6) δ 7,79 (s, 1H), 7,54 (m, 3H), 7,46 (m, 2H), 7,14 (s, 1H), 5,68 (s, 2H), 4,22 (m, 1H), 3,45 (m, 2H), 2,66 (m, 1H), 2,52 (m, 4H), 1,96 (m, 2H), 1,84 (m, 3H), 1,64 (m, 4H); EM m/z 367 (M+1).
[00309] 4. A uma solução agitada de [4-(6-amino-5-hidróxi-piridin- 3-il)-fenil]-[(2R)-2-pirrolidin-1-ilmetil-pirrolidin -1-il]-metanona (100 mg, 0,27 mmol) em DMF anidro (15 mL) sob uma atmosfera de nitrogênio contendo a 0°C, adicionou-se hidreto de sódio (dispersão a 60% em óleo mineral, 11 mg, 0,49 mmol). A mistura foi deixada agitar a 0°C por 30 minutos. 1-(bromometil)-4-flúor-2-(trifluormetil)benzeno (0,046 mL, 0,27 mmol) foi adicionado. A mistura foi agitada a temperatura ambiente por duas horas. A reação foi diluída com EtOAc e dividida com H2). A camada aquosa foi extraída com EtOAc (2 x 25 mL). As camadas orgânicas foram combinadas, lavadas com água (1 x 15 mL), salmoura (1 x 15 mL), secas sobre MgSO4, filtradas e concentradas e purificada numa coluna de gel de sílica rendendo [4-(6-amino-5-(4- flúor-2-trifluormetil-benzilóxi))-piridin-3-il)-fenil]-[(2R)-2-pirrolidin-1- ilmetil-pirrolidin-1-il]-metanona como cristais esbranquiçados.
[00310] Para Síntese de [6-amino-5-(benzilóxi substituído) -piridin- 3-il)-fenil-amida do ácido 2-dialquilamino-etanossulfônico usando {4-(6- amino-5-(2-cloro-3,6-diflúorbenzilóxi)-piridin-3-il]fenil]-amida do ácido 2-dietilamino-etanossulfônico.
[00311] A uma solução de 4-(4,4,5,5-tetrametil- [1,3,2]dioxaborolan-2-il)-fenilamina (5 g, 22,8 mmols) e diclorometano (120 mL) adicionou-se N-metil morfolina (7,5 mL, 68,4 mmols). Esta mistura foi resfriada para 0°C sob atmosfera de nitrogênio. Cloreto de 2-cloroetanossulfonila (2,5 mL, 23,9 mmols) em diclorometano (60 mL) foi então adicionado gota a gota com agitação. Terminada a adição o frasco foi agitado a 0°C por 1 hora e a seguir a temperatura ambiente enquanto se monitorava por TLC acetato de etila:hexanos 1:1) e manchamento com ninhidrina. Após 4 horas de agitação, algum éster borônico de partida ainda permanecia e adicionou-se mais 0,2 equivalentes (0,5 mL) de cloreto de 2-cloroetanossulfonila em diclorometano (25 mL) gota a gota a temperatura ambiente. Após uma hora o éster borônico tinha-se consumido como se demonstrou por TLC e todo o volume de reação tinha-se reduzido pela metade via evaporação rotativa. O conteúdo foi diluído com acetato de etila (200 mL), lavado com salmoura a 50% (2 x 100 mL), seco sobre sulfato de sódio anidro e concentrado in vácuo. o produto bruto foi purificado usando gel de sílica (120 g) e eluindo com acetato de etila a 10% e diclorometano rendendo [4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il}- fenil]amida do ácido etenossulfônico como um sólido branco (6,2 g, 20,2 mmols, 89% rendimento).
[00312] 1H RMN (CDCI3, 300 MHz), δ 7,76 (d, J = 8,4, 2H), 7,12 (d, J = 8,45, 2H) 6,65 (s, 1H), 6,55 (dd, J = 9,77, 6,7, 1H), 6,31 (d, J = 16,54, 1H), 5,96 (d, J = 9,8, 1H), 1,33 (s, 12H).
[00313] 2. A uma soIução de [4-(4,4,5,5-tetrametiI-[1,3,2] dioxaboroIan-2-iI}-feniI]amida do ácido etenossuIfônico (0,500 g, 1,6 mmoI) em metanoI (5 mL) adicionou-se dietiIamina (0,707 g, 4,0 mmoIs) em metanoI (5 mL) sendo a reação agitada a temperatura ambiente e monitorada por TLC (acetato de etiIa/hexanos 1:1). Após 2 horas a reação foi concentrada in vácuo e o resíduo dividido entre acetato de etiIa (50 mL) e água (50 mL). O acetato de etiIa foi então Iavado com saImoura 50% (1 x 50 mL), seco sobre suIfato de sódio anidro, fiIiado e concentrado in vácuo. O produto bruto foi purificado usando uma coIuna de geI de síIica pré-condicionada de 10 g eIuindo com acetato de etiIa: dicIorometano 1:1 dando [4-(4,4,5,5-tetrametiI- [1,3,2] dioxaboroIan-2-iI}-feniI]amida do ácido 2-dietiIamino- etanossuIfônico como um sóIido branco (0,346 g,0,90 mmoI, 56%).
[00314] 1H RMN (CDCI3, 300 MHz) δ 7,78 (d, J = 6,65, 2H) 7,15 (d, J = 6,66, 2H), 3,20 (m, 2H), 3,0 (m, 2H), 2,55 (q, J = 7,15, 7,16, 4H), 1,34 (s, 12H), 1,05 (t, J = 7,19, 6H).
[00315] 3. [4-(6-amino-5-(2-cIoro-3,6-difIúor-benziIóxi)-piridin-3-iI]- feniI]amida do ácido 2-dietiIamino-etanossuIfônico foi preparado seguindo o procedimento 3 de acopIamento geraI de Suzuki de 5- bromo-3-(2-cIoro-3,6-difIúor-benziIóxi)-piridin-2-iIamina e [4-(4,4,5.5- tetrametiI-[1,3,2]dioxaboroIan-2-iI}-feniI]amida do ácido 2-dietiIamino- etanossuIfônico preparado na parte 2 como um sóIido branco com 60% de rendimento.
[00316] 1: 4-(4,4,5,5-tetrametiI 1,3,2-dioxaboroIan-2-iI) aniIina (3 g, 0,013 mol) foi dissolvido em diclorometano (350 mL) ao qual foi adicionado piridina (1,02 g,0,12 mol) e cloroformiato de 4-nitrofenila. A reação foi agitada por 13 horas onde a análise de TLC demonstrou consumo de todos os materiais de partida. A solução foi lavada com NaHCO3 sat. (3 x 50 mL) água (3 x 50 mL) e salmoura (3 x 50 mL). A camada orgânica foi seca sobre Na2SO4 e o solvente removido rendendo um sólido cristalino branco: éster fenílico do ácido [4- (4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il}-fenil]-carbâmico 4,45 g 91%.
[00317] 1H RMN (CDCI3, 300 MHz) δ 1,4 (s, 12H), 7,1 (brs, 1H), 7,3 (d, 2H), 7,5 (d, 2H), 7,8 (d, 2H), 8,3 (d, 2H).
[00318] 2. éster feníIico do ácido [4-(4,4,5,5-tetrametiI- [1,3,2]dioxaboroIan-2-iI}-feniI]-carbâmico (500 mg, 1,3 mmoI) foi dissoIvido em dicIorometano anidro (0,5 mL) e trietiIamina (0,187 mL, 1,3 mmoI) A esta soIução agitada adicionou-se 1-metiIpiperazina (ou quaIquer outra amina) (0,144 mL, 1,3 mmoI). A soIução ficou amareIa instantaneamente, e a anáIise de TLC demonstrou consumo totaI do materiaI de partida. A reação foi Iavada com água (3 x 500 mL), soIução de bicarbonato de sódio sat. (2 x 200 mL) e seca antes da remoção dos solventes in vácuo. Os ésteres borônicos foram utilizados sem purificação.
[00319] 3. A uma mistura de 2,1 mL de DME e 2,8 mL de Na2CO3 adicionou-se 100 mg do brometo inicial, 1 equivalente do ácido borônico e 5 mol% de Pd(PPh3)4. A reação foi agitada e aquecida para 80°C durante a noite em dois frascos de gotejamento. A mistura bruta foi filtrada por celite e extraída com EtOAc (2 x 100 mL). Os extratos combinados foram lavados com NaHCO3 (1 x 100 mL), seguido por água (1 x 100 mL) e a seguir salmoura saturada (1 x 100 mL). A mistura resultante foi concentrada in vácuo. O resíduo foi dissolvido em hexano e purificado via cromatografia de coluna. Procedimento Geral 11
[00320] A uma solução de 3-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]- piridin-2-ilamina (10,0 g, 33,2 mmols) em acetonitrila (600 mL) e ácido acético (120 ml) adicionou-se N-iodosuccinimida (11,2 g 49,8 mmols). A mistura foi agitada a temperatura ambiente por 4 horas e a reação foi arrefecida com solução de Na2S2O5. Após evaporação, o resíduo foi dividido entre acetato de etila e água. A camada orgânica foi lavada com solução de NaOH 2N, salmoura e seca sobre Na2SO4. O produto bruto foi purificado numa coluna de gel de sílica dando 3-[1-(2,6- dicloro-3-flúor-fenil)-etóxi]-5-iodo-piridin-2-ilamina (7,1 g 50% rendimento). EM m/z 427 [M+1].
[00321] A uma solução de 3-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]-5- iodo-piridin-2-ilamina (7,1 g, 16,6 mmols) e éster terc-butílico do ácido prop-2-inil-carbâmico (3,1 g, 20,0 mmols), em tetraidrofurano (60 mL) e Et3N (60 mL) adicionou-se CuI (63 mg, 0,3 mmol) e Pd(PPh34 (384 mg, 0,3 mmol) A mistura foi agitada sob nitrogênio e monitorada por TLC até o término da reação. A mistura foi extraída com EtOAc e lavada com água. O produto bruto foi purificado numa coluna de gel de sílica eluindo com EtOAc 20-40% em hexanos dando éster terc-butílico do ácido 3-(6-amino-5-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]-piridin-3-il}-prop - 2 -inil)carbâmico (2,2 g, 29% rendimento).
[00322] A solução de éster terc-butílico do ácido (3-{6-amino-5-[1- (2,6-dicloro-3-flúor-fenil)-etóxi]-piridin-3 -il}-prop-2-inil)carbâmico em 25% de TFA em diclorometano foi agitada por duas horas, a seguir lavada com NaOH 2N, água, salmoura, seca sobre Na2SO4. Após filtrar e evaporar obteve-se 5-(3-amino-prop-1-inil)-3-[1-(2,6-dicloro-3- flúor-fenil)-etóxi]-piridin-2-ilamina em 93% rendimento.
[00323] A uma solução de 5-(3-amino-prop-1-inil)-3-[1-(2,6-dicloro- 3-flúor-fenil)-etóxi]-piridin-2-ilamina (0,282 mmol, 1 eq.) e cloroformiato de 4-nitrofenila (1 eq.) em diclorometano anidro (10 mL) adicionou-se piridina (1 eq). A reação foi agitada por 4 horas sob nitrogênio, e a seguir a amina selecionada (1 eq) e trietilamina (1 eq) foram adicionadas. A mistura foi refluxada por 5 minutos e resfriada para temperatura ambiente. A mistura de reação foi lavada com metanol em diclorometano em colunas de sílica pré-acondicionadas. A produção final variou entre 24% e 71%. Procedimento Geral 12
[00324] A uma solução de 5-(3-amino-prop-1-inil)-3-[1-(2,6-dicloro- 3-flúor-fenil)-etóxi]-piridin-2-ilamina (preparado no procedimento 11)(400 mg, 1,1 mmol) em diclorometano (17 ml) adicionou-se cloreto de cloroacetila (153, mg, 1,4 mmol). A reação foi agitada a temperatura ambiente com monitoração por TLC do término da reação. Após o término da reação, o solvente foi evaporado rendendo o produto bruto.
[00325] A uma solução de N-(3-{6-amino-5-[1-(2,6-dicloro-3-flúor- fenil)-etóxi]-piridin-3-il}-prop-2-inil)-2-cloro-acetamida (1 eq.) em acetonitrila (5 eq.) adicionou-se a amina individual (5 eq.).
[00326] A mistura foi refluxada sob nitrogênio durante a noite. Após evaporação do solvente o resíduo foi purificado numa coluna de gel de sílica eluindo com metanol 1-10% em diclorometano dando o produto com rendimentos entre 47 a97%. Procedimento Geral 13
[00327] A uma solução agitada de 2-amino-3-benziloxipiridina (42,0 g, 0,21 mol) em CH+CN (600 mL) a 0°C adicionou-se N- bromossuccnimida (37,1 g, 0,21 mol) durante 30 mt. A mistura foi agitada pro 0,5 hora, após o que, a reação foi diluída com EtOAc (900 mL) e dividida com H2O (900 mL). A camada orgânica foi lavada com salmoura e seca (Na2SO4), filtrada e concentrada até secura sob vácuo rendendo 3-benzilóxi-5-bromo-piridin-2-ilamina (31,0 g, 0,11 mol 53%).
[00328] 1H RMN (CDCls, 300 MHz) δ 4,63-4,78 (brs, 2H), 5,04 (s, 2H), 7,07 (d, 1H, J, 1,8 Hz), 7,33-7,42 (m, 5H), 7,73 (d, 1H, J, 1,8 Hz).
[00329] 2. A uma mistura agitada de 3-benzilóxi-5-bromo-piridin-2- ilamina (31,0 g, 0,11 mol) numa mistura de DME (600 mL) e H2O (600 mL) adicionou-se ácido 4-carboximetilborônico (29,9 g, 0,11 mol), Pd(PPh3)4 (6,4 g 5,55 mmols) e Na2CO3 (82,0 g, 0,78 mol) A reação foi aquecida lentamente ao refluxo e deixou-se agitar por 3 horas. A reação foi resfriada para temperatura ambiente, a seguir diluída com CH2Cl2 (1,5 L) e dividida com H2O (700 ml). A camada orgânica foi lavada com NaHCO3 sat. (700 mL), seca (Na2SO4), filtrada e concentrada in vácuo. O material bruto foi purificado por cromatografia em gel de sílica (acetato de etila/hexanos 1:1 a 4:1) e as frações contendo o produto foram combinadas e concentrada in vácuo rendendo éster metílico do ácido 4-(6-amino-5-benzilóxi-piridin-3-il)- benzóico (29,4 g, 0,086 mol, 79%).
[00330] 1H RMN (CDCl3, 300 MHz) δ 3,92 (s, 3H), 4,82-4,94 (brs, 2H), 5,15 (s, 2H), 7,22 (d, 1H, J, 1,8 Hz), 7,33-7,42 (m, 5H), 7,54 (d, 2H, J, 8,6), 7,98 (d, 1H, J, 1,8 Hz), 8,06(d, 2H, J, 8,6 Hz).
[00331] 3. A uma solução agitada de éster metílico do ácido 4-(6- amino-5-benzilóxi-piridin-3-il)-benzóico (10,0 g, 0,03 mol) em EtOH:H2O (95:5, 600 mL) adicionou-se Pd/C (15,9 g, 0,015 mol) (a reação foi desgaseificada sob vácuo). A solução foi deixada agitar sob uma atmosfera de hidrogênio por 22 horas. A solução foi filtrada por celite úmido, sendo o celite lavado com EtOAc. o Filtrado foi concentrado sob vácuo rendendo éster metílico do ácido 4-(6-amino-5- hidróxi-piridin-3-il)-benzóico (2,3 g, 9,3 mmol, 31%),
[00332] 1H RMN (MeOD, 300 MHz) δ 3,90 (s, 3H), 7,21 (d, 1H, J, 1,9 Hz), 7,62 (d, 2H, J, 8,5 Hz), 7,76 (d, 1H, J, 1,9 Hz), 8,04 (d, 2H, J, 8,5 Hz).
[00333] 4. A uma solução agitada de éster metílico do ácido 4-(6- amino-5-benzilóxi-piridin-3-il)-benzóico (2,3 g, 9,3 mmols) em CH2Cl2 (180 mL) adicionou-se N,N-diisopropiletilamina (3,2 mL, 0,019 mol), cloreto de 4-metil-benzenossulfonila (2,66 g, 0,014 mol) e PS-DMAP (quantidade catalítica). A reação foi agitada a temperatura ambiente por 6 horas a seguir filtrada removendo-se a resina. A resina foi lavada com CH2Cl2 (3 x 20 mL) e as frações combinadas foram lavada com ácido cítrico a 10% (100 ml), NaCl sat. (100 mL), seca (Na2SO4) e filtrada e concentrada in vácuo. O material bruto resultante foi purificado por cromatografia em gel de sílica (100% CH2Cl2 para 95:5 CH2Cl2:MeOH) sendo que, as frações contendo o produto desejado foram combinadas e concentradas in vácuo rendendo éster metílico do ácido 4-(6-amino- 5-(tolueno-4-sulfinilóxi)piridin-il}-benzóico (3,3 g, 8,2 mmols, 88%).
[00334] 1H RMN (CDCl3, 300 MHz) δ 2,47 (s, 3H), 3,93 (s, 3H), 4,81-4,88 (brs, 2H), 7,36-7,44 (m, 6H), 7,81 (d, 2H, J, 8,3Hz), 8,05 (d, 2H, J, 8,4 Hz), 8,19-8,27 (brs, 1H).
[00335] 5. A uma solução agitada de 1-(3-flúor-2-trifluormetil-fenil)- etanol (2,0 g, 9,6 mmols) em DMF anidro (500 mL a 0°C sob uma atmosfera de nitrogênio adicionou-se NaH (0,38 g, 9,6 mmols). A reação foi deixada agitar por 0,5 hora. Uma solução de éster metílico do ácido 4-{6-amino-5-tolueno-4-sulfiilóxi)-piridin-3-il}-benzóico (3,8 g, 9,6 mmols) em DMF anidro (30 mL) foi adicionada para a mistura de reação que foi deixada chegar à temperatura ambiente lentamente e agitada pro 21 horas naquela temperatura. A reação foi diluída com EtOAc (500 mL) e água (100 mL). A camada orgânica foi separada e a camada aquosa foi ainda extraía com EtOAc (1 x 200 mL). As camadas orgânicas foram combinadas e lavadas com salmoura (1 x 100 mL), secas com Na2SO4 e concentradas até secura sob vácuo. A mistura bruta foi purificada por cromatografia de coluna (sílica gel 40:60 a 70:30 EtOAc:hexanos) e as frações contendo o produto foram combinadas e concentradas in vácuo rendendo éster metílico do ácido 4-{6-amino-5-[1-(3-flúor-2-trifluormetil-fenil)-etóxi-piridin-3-il)-benzóico (1,4 g, 3,2 mmols, 34%).
[00336] 1H RMN (CDCI3, 300 MHz) δ 1,73 (d, 3H, J, 6,2 Hz), 3,91 (s, 3H), 4,87-4,64 (brs, 2H), 5,81 (q, 1H,,J, 6,1, 6,3 Hz), 6,92 (d, 1H, J, 1,8 Hz), 7,38 (d, 2H, J, 8,5 Hz), 7,46-7,66 (m, 3H), 7,93 (d, 1H, J, 1,8 Hz), 8,02 (d, 2H, J, 8,5 Hz).
[00337] 6. A uma soIução agitada de éster metíIico do ácido 4-(6- amino-5-benziIóxi-piridin-3-iI)-benzóico (1,4 g 3,2 mmoIs) em IPA quente (72 mL) adicionou-se H2O (38 mL) contendo LiOH (0,68 g, 16,2 mmoIs). A reação foi aquecida ao refIuxo por 3,5 horas. A reação foi neutraIizada e diIuída com EtOAc (200 mL) e extraída com resfriamento. A camada orgânica foi Iavada com saImoura (50 mL), seca sobre Na2SO4 e concentrada sob vácuo rendendo éster metíIico do ácido 4-{6-amino-5-[1-(3-fIúor-2-trifIuormetiI-feniI)-etóxi-piridin-3-iI)- benzóico (1, 2 g, 2,8 mmoIs, 88%).
[00338] 1H RMN (MeOD, 300 MHz) δ 1,75 (d, 3H, J, 6,2 Hz), 4,88 4,93 (m, 1H) 7,01 (d, 1H, J, 1,8 Hz), 7,39 (d, 2H, J, 8,3 Hz), 7,52-7,67 (m, 3H), 7,80 (d, 1H, J, 1,8 Hz), 7,97 (d, 2H, J, 8,3 Hz).
[00339] 7. Preparação dos compostos amida: Uma soIução agitada de éster metíIico do ácido 4-{6-amino-5-[1-(3-fIúor-2- trifIuormetiI-feniI)-etóxi-piridin-3-iI)-benzóico (50 mg, 0,12 mmoI), EDC (27,0 mg, 0,13 mmoI) e HOBt (18,0 mg, 0,13 mmoI) em DMF (2 mL) foi adicionada para dois frascos de gotejamento contendo NHR1R2 (0,12 mmol). A reação foi agitada a temperatura ambiente por 18 horas. A reação foi a seguir diluída com CH2Cl2 (3 mL) e dividida com água. A fase orgânica foi separada e lavada com NaCl saturado (1 x 2 mL) e NaHCO3 sat. (1 x 2 mL) A fase orgânica foi concentrada a secura sob vácuo. O material foi purificado suando cromatografia em gel de sílica (99:1 a 95:5 CH2Cl2/MeoH). As frações contendo o produto foram concentradas sob vácuo rendendo compostos amida. Procedimento Geral 14
[00340] A uma mistura de cloridrato de 1-(2-cloroetil)pirrolidina (200 mg, 1,18 mmol) e 4-[4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)- fenil]-1H-pirazol (229 mg, 1,19 mmol) em DMF (6 mL) adicionou-se Cs3CO3. A mistura foi agitada a temperatura ambiente durante a noite. Adicionou-se água para a mistura. O produto foi extraído com EtOAc (3 x 10 mL). Os extratos combinados foram a seguir lavados com salmoura (5 x 10 mL) para remover o DMF, a seguir secos sobre Na2SO4 e concentrados (142 mg, 41%, rendimento).
[00341] A uma mistura de 3-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]-5- iodo-piridin-2-ilamina (200 mg, 0,468 mmol), éster pinacol borônico (1,2 eq), Na2CO3 (149 mg, 1,41 mmol) em água (1,25 mL), e dimetil etil glicol (3,75 mL, 0,1 M) adicionou-se Pd(PPH3)2Cl2 (16 mg, 0,020 mol) num vaso de reação de microondas). O sistema foi desgaseificado e carreado com nitrogênio. A mistura foi agitada a 160° num aparelho microondas pro 15 minutos. a mistura foi resfriada para temperatura ambiente seguido por adição de água (10 mL). O produto foi extraído com EtOAc (3 x 20 ml), seco sobre Na2SO4, concentrado. O produto bruto foi purificado por HPLC de fase reversa com 0,1% de TFA em água e acetonitrila. Procedimento Geral 15
[00342] Para uma solução de 3H-oxazol(4,5-b]piridin-2-ona (13,6 g, 100 mmols) em acetonitrila (600 mL) e ácido acético (120 mL), adicionou-se N-bromossuccinimida (21,4 g, 120 mmols). A mistura foi agitada a temperatura ambiente por 4 horas e a reação resfriada com Na2SO4. Após evaporação, o resíduo foi dividido entre acetato de etila e água. A camada orgânica foi lavada com solução de NaOH 2N, salmoura, e seca sobre Na2SO4. O produto bruto foi purificado numa coluna de gel de sílica dando 6-bromo-3H-oxazol[4,5-b] piridin-2-ona (11,5 g 55% rendimento).
[00343] 6-bromo-3H-oxazol[4,5-b]piridin-2-ona (21,5 g 100 mmols) foi suspenso em NaOH (2N, 250 mL, 500 mmols). A mistura foi refluxada durante a noite e uma solução límpida foi obtida. Após resfriar para temperatura ambiente, a solução de reação foi neutralizada a pH ~7. Uma boa quantidade de CO2 foi liberada, observando-se também um precipitado. O produto foi filtrado, lavado com água, e seco sob alto vácuo dando 2-amino-5-bromo-piridin-3-ol como um sólido esbranquiçado (17,8 g 98% rendimento).
[00344] Para uma solução de 2-amino-5-bromo-piridin-3-ol (358 mg, 1,89 mmol) em DMF (8 mL) adicionou-se Cs2CO3 (620 mg, 1,89 mmol). A mistura foi agitada a temperatura ambiente sob nitrogênio por uma hora. À mistura de reação adicionou-se bromo-composto (0,9 eq.) em DMF (5 mL) lentamente. A solução de reação foi agitada sob nitrogênio por cinco horas, e a seguir dividida entre água e acetato de etila. A camada orgânica foi purificada numa coluna de gel de sílica eluindo com hexano-acetato de etila (4:1) dando o produto com 70%- 80% rendimento.
[00345] Usando o Exemplo 1-488 do Pedido de Patente U.S. n° de série 10/786.610 (PCT/US2004/005495)
[00346] A uma solução de 3-benzilóxi-5-bromo-piridin-2-ilamina (1 g, 3,58 mmols) em dimetilsulfóxido (7 mL) adicionou-se em seqüência, bis(pinacolato)diborano (1,0 g, 3,94 mmols), acetato de potássio (1,05 g, 10,7 mmols), complexo de [1,1'-bis(difenilfosfino)ferricino]dicloro- paládio (II) com diclorometano (1:1) (146 mg, 0,18 mmol). A mistura foi aquecida para 80°C por 16 horas, sendo resfriada para temperatura ambiente. A mistura de reação foi diluída com acetato de etila (50 mL) e filtrada. O filtrado foi lavado com água (2 x 50 mL) e seco sobre sulfato de magnésio. A concentração in vácuo rendeu o boronato bruto como um sólido marrom (1,13 g, 97%).
[00347] 1H RMN (CDClβ) δ 1,32 (s, 12H), 5,08 (s, 2H), 5,44 (br s, 2H), 7,33-7,42 (m, 6H), 8,03 (s, 1H).
[00348] 2. Um frasco de reação de 18 mL foi carregado com o 3- benzilóxi-5-(4,4,5-5-tetrametil0[1,3,2]dioxaborolan-2-il)-piridin-2-ilamina bruto (161 mg,0,49 mmol), dimetoxietano (3 mL) e 2-bromopiridina (117 mg, 0,74 mmol). A esta solução adicionou-se complexo de [1,1'- bis(difenilfosfino)ferrocino]dicloro-paládio (II) com diclorometano (1:1) (20 mg, 0,05 mmol) e uma solução 2M de carbonato de césio em água (0,75 mL, 1,5 mmol). O reator foi aquecido a 80°C por 66 horas sob uma atmosfera de nitrogênio a seguir resfriado para temperatura ambiente. A mistura de reação foi dividida entre acetato de etila (5 mL) e água (5 mL). A camada orgânica foi lavada com mais água (5 mL) e diluída com dimetilformamida (5 mL). Ácido sulfônico ligado a polímero (0,5 g, 2,1 mmol) foi adicionado para solução orgânica e a mistura resultante foi suavemente agitada por duas horas. A resina foi filtrada e lavada com dimetilformamida, metanol e cloreto de metileno (3 x 5 mL cada solvente). A seguir o polímero foi reagido com amônia 2M em metanol por uma hora. A resina foi filtrada e lavada com mais amônia 2M em metanol (2 x 5 mL) e os filtrados combinados foram concentrados in vácuo. A purificação do produto bruto por cromatografia de coluna flash rendeu 52,2 mg do produto como um sólido castanho (38% rendimento). Procedimento Geral 17
[00349] A uma solução de 3-(2-cloro-3,6-diflúor-benzilóxi)-5- (4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-piridin-2-ilamina (procedimento 16) (10,0 g, 24,3 mmols em álcool terc-butílico (50 mL) adicionou-se anidrido boc (5,83 g, 26,7 mmols) e a reação foi agitada a temperatura ambiente durante a noite, Mais anidrido Boc (2,25 g, 10,3 mmols) foi adicionado e a reação agitada durante a noite novamente. O material foi concentrado a um óleo negro viscoso e usado como tal.
[00350] O éster borônico bruto (24,3 mmols teórico), em THF (150 mL) foi adicionado a uma solução de bicarbonato de sódio (16,3 g, 194 mmols) em água (150 mL e acetona (23 mL). A mistura foi resfriada para 2°C e oxona (13,5 g 21,9 mmols) adicionado lentamente, mantendo a temperatura abaixo de 8 °C. Com o término da adição, a reação foi agitada por 5 minutos a seguir arrefecida com bissulfito de sódio (14,2 g) em água (28 mL). Adicionou-se acetato de etila (200 mL) e as camadas foram separadas. A camada aquosa foi neutralizada com HCl 6N e extraída com acetato de etila (2 x 200 mL). Os orgânicos combinados foram lavados com água (250 mL) e salmoura (250 mL), secos (Na2SO4) e concentrados até um óleo negro bruto. A cromatografia em gel de sílica (acetato de etila/hexanos) conferiu o produto como uma espuma castanho claro.
[00351] 1H RMN (CDClβ) δ 1,48 (s, 9H), 1,74 (d, 3H), 5,75 (q, 1H), 6,61 (d, 1H), 76,89 (dt, 1H), 6,94-7,04 (m, 2H), 7,26 (d, 1H), 8,19 (bs, 1H). EM m/z 401 (M+H)+.
[00352] 3. A carbonato de césio em dois frascos de gotejamento adicionou-se éster terc-butílico do ácido 3-(2-cloro-3,6-diflúor- benzilóxi)-5-hidróxi-piridin-2- il] carbâmico (100 mg, 0,25 mmol) em DMF anidro (1 mL), seguido por brometo de benzila (89,2 μL, 0,75 mmol) O frasco foi fechado e agitado a 90°C durante a noite. A reação foi filtrada por um tubo Chem-Elut de 5 mL previamente umedecido com água (3,5 mL) e eluído com acetato de etila:cloreto de metileno 1:1. Após concentração parcial, HCl 4N em dioxano (1-2 mL) foi adicionado e a solução concentrada. A cromatografia de fase reversa (água:acetonitrila 0,05% TFA) seguido por liofilização, deu o produto desejado como um sólido amorfo esbranquiçado (25,3 mg, 20,0%) e o produto de nova adição como um sólido amorfo castanho (35,2 mg, 23,7%). Procedimento Geral 18
[00353] Boroidreto de sódio (1,5 equivalente molar) e adicionado a uma solução de cetona (3,89 mmols) em 10 mL de etanol sob uma atmosfera de nitrogênio. A mistura resultante é agitada a temperatura ambiente por 12 horas. A mistura é a seguir colocada num banho de gelo e arrefecida com HCl aquoso diluído. O etanol é evaporado e adiciona-se EtOAc para extrair a solução aquosa. A camada de EtOAc é seca sobre Na2SO4. O Na2SO4 foi filtrado e o filtrado evaporado dando um resíduo oleoso, composto A5. O resíduo é usado sem mais purificação.
[00354] 3-hidróxi-2-nitropiridina (1,1 equivalente molar) e trifenilfosfina (1,5 equivalente molar) são adicionados para uma solução do composto A5 (1,1 mmol) em 10 mL de Tetraidrofurano. A mistura de reação é a seguir colocada num banho de gelo e azodicarboxilato de diisopropila (1,5 equivalente molar) é adicionado. O banho de gelo é removido e a mistura agitada para temperatura ambiente por duas horas. O solvente é evaporado dando um resíduo oleoso amarelo. O resíduo é purificado por cromatografia em gel de sílica (eluindo com EtOAc em hexanos) danço o composto A1.
[00355] HCl 2M (0,2 mL) é adicionado para a solução do composto A1 (0,97 mmol) em 2 mL de etanol. A mistura é a seguir colocada num banho de gelo e pó de Fé (365 mg) é adicionado lentamente. A reação e aquecida para 85°C por uma hora e resfriada para temperatura ambiente. Celite (0,5 g) é adicionado para agitar e a mistura resultante é filtrada através de um leito de celite e enxaguada com etanol. O filtrado é evaporado dando um resíduo oleoso castanho, composto A2. O resíduo é usado sem mais purificação.
[00356] Ácido per-iódico (0,25 equivalente molar), iodo (0,5 equivalente molar), água (0,5 mL) e ácido sulfúrico conc. (0,03 mL) são adicionados para uma solução do composto A2 em 3 mL de ácido acético. A mistura de reação é aquecida para 85°C por 5 horas. A mistura de reação é a seguir resfriada num banho de gelo e tornada básica com Na2CO3 a um pH de 3-4. Adiciona-se acetato de etila para extrair a solução aquosa. Secar a camada de EtOAc sobre Na2SO4. O Na2SO4 é filtrado e o filtrado evaporado dando um resíduo oleoso castanho. O resíduo é purificado por cromatografia em gel de sílica (eluindo com EtOAc e hexanos) dando o produto desejado, composto A3. Procedimento Geral 19
[00357] Éster borônico ou ácido borônico (1,3 equivalente molar), é adicionado para uma solução do composto A3 (0,47 mmoL) em 5 mL de DME. A mistura foi purgada com nitrogênio várias vezes e a seguir diclorobis(trifenilfosfina) paládio(II) (0,05 equivalente molar) é adicionado. Carbonato de sódio (3 equivalentes molar) em 1 mL de água é adicionado para a mistura de reação e a solução resultante aquecida para 85°C por 12 horas. Adiciona-se água para a mistura de reação arrefecendo a reação. EtOAc é a segui adicionado para extrair a solução aquosa. Secar a camada de EtOAc sobre Na2SO4. O Na2SO4 é filtrado e evaporado dando um resíduo oleoso castanho escuro. O resíduo é purificado por cromatografia em gel de sílica (eluindo com CH3OH, CH2Cl2, EtOAc e hexanos) conferindo o produto desejado, composto A4. Procedimento Geral 20:
[00358] O composto A6 foi preparado usando Procedimento Geral 19. Pentafluoreto de O-(7-azabenzotriazol-1-il)-N,N,N',N"- tetrametilurônio de fósforo(HATU)(1,1 equivalente molar), diisopropiletil amina (5 equivalente molar) e amina (1,3 equivalente molar) são adicionados para uma solução do composto A6 (0,17 mmol) em 3 mL de DMF sob uma atmosfera de nitrogênio. A reação é deixada agita a temperatura ambiente por 12 horas. NaHCO3 sat. é adicionado para a mistura de reação para arrefecer a reação. Adiciona-se EtOAc para extrair a solução aquosa. secar o EtOAc sobre Na2SO4. O Na2SO4 é filtrado e o filtrado evaporado dando um resíduo oleoso marrom. O resíduo é purificado por cromatografia em gel de sílica (eluindo com EtOAc e hexanos) dando o produto amida desejado, composto A7 como um óleo amarelo Procedimento Geral 21
[00359] Ácido (16 equivalente molar ou menos) é adicionado párea o composto A7 (0,13 mmol) a temperatura ambiente. A solução resultante é agitada a temperatura ambiente ou aquecida para 60°C por 12 horas. A mistura de reação é evaporada e o resíduo purificado por cromatografia em gel de sílica (eluindo com CH3OH, EtOAc e CH2Cl2) dando o produto amida desejado, composto A8, como um sólido amarelado a branco. Procedimento Geral 22
[00360] O composto A9 é preparado usando o Procedimento Geral 19. Dicarbonato de di-terc-butila (3 equivalente molar) e 4- (dimetilamino)piridina (0,14 equivalente molar) são adicionados para uma solução do composto A9 (3 mmols) em 20 mL de dimetilformamida. A mistura de reação é agitada a temperatura ambiente por 12 horas. Adiciona-se água para a mistura de reação para resfriar a reação. EtOAc é a seguir adicionado para extrair a solução aquosa. Secar a camada de EtOAc sobre Na2SO4. O Na2SO4 é filtrado e o filtrado evaporado dando um resíduo oleoso castanho- amarelado. O resíduo é purificado pro cromatografia em gel de sílica (eluindo com 25^ 30% EtOAc em hexanos) dando o produto desejado, composto A10 como um óleo amarelado (87,8% rendimento). Borbulha-se ozônio através de uma solução do composto A10 em 50 mL de CH2Cl2 a -78°C e sulfeto de dimetila é adicionado para arrefecer a reação. Adiciona-se cloreto de sódio sat. para a mistura de reação e EtOAc para extrair a solução aquosa. Acamada de EtOAc combinada é seca sobre Na2SO4. O Na2SO4 é filtrado e o filtrado evaporado dando um resíduo oleoso amarelo. O resíduo é purificado por cromatografia em gel de sílica (eluindo com 35^ 40% EtOAc em hexanos) dando o produto desejado, composto A11, como um óleo amarelado (58,4% rendimento) Procedimento Geral 23. Aminação Redutiva
[00361] Sal de cloridrato de amina (1,2 equivalente molar), acetato de sódio (2 equivalente molar para o sal de cloridrato de amina) são adicionados a uma solução do composto A11 (0,45 mmol) em 4 mL de CH3OH sob uma atmosfera de nitrogênio. Adiciona-se peneira molecular (0,5 g) para a mistura de reação e a seguir cianoboroidreto de sódio (2 equivalente molar) é adicionado. A mix resultante é agitada a temperatura ambiente por 12 horas sob uma atmosfera de nitrogênio. A mistura de reação é filtrada por um leito de celite e o filtrado é evaporado e purificado por cromatografia em gel de sílica (eluindo com CH3OH, EtOAc e CH2Cl2) dando o produto desejado, composto A12 como um óleo (52,6% rendimento). Ácido (16 equivalente molar ou menos) é adicionado ao composto A12 (0,17 mmol) a temperatura ambiente. A solução resultante é agitada a temperatura ambiente ou aquecida para 60°C por 12 horas. A mistura de reação é evaporada e o resíduo purificado por cromatografia em gel de sílica (eluindo com CH3OH, EtOAc, e CH2Cl2) dando o produto desejado, composto A13. Procedimento Geral 24:
[00362] O-fenildiaminas (1,2 equivalente molar) e bissulfito de sódio (2,1 equivalente molar) são adicionados a uma solução do composto A11 (0,41 mmol) em 5 mL de DMA. A solução resultante é aquecida para 110°C por 12 horas. Adiciona-se água para a mistura de reação resfriando a reação. Adiciona-se então EtOAc para extrair a solução aquosa. Secar EtOAc sobre Na2SO4. O Na2SO4 é filtrado e o filtrado evaporado dando um resíduo oleoso castanho-amarelado. O resíduo é purificado por cromatografia em gel de sílica (eluindo com EtOAc em hexanos) dando o produto desejado, composto A14. Ácido (16 equivalente molar ou menos) é adicionado ao composto A14 (0,15 mmol) a temperatura ambiente. A solução resultante é agitada a temperatura ambiente ou aquecida para 60°C por 12 horas. A mistura de reação é evaporada e o resíduo é purificado por cromatografia em gel de sílica (eluindo com CH3OH, EtOAc e CH2Cl2) dando o produto amida desejado, composto A15. Procedimento Geral 25
[00363] Dicarbonato de di-terc-butila (3 equivalente molar), 4- (dimetilamino)piridina (0,14 equivalente molar) são adicionados para uma solução do composto A3b (2 mmols) em 10 mL de DMF. A mistura de reação é agitada a temperatura ambiente por 12 horas. Adiciona-se água para a mistura de reação resfriando a reação. EtOAc é a seguir adicionado para extrair a solução aquosa. Secar a camada de EtOAc sobre Na2SO4. O Na2SO4 é filtrado e o filtrado evaporado dando um resíduo oleoso amarelo castanho (composto A16). O resíduo é usado sem mais purificação.
[00364] Bis-pinacolato)diboro (1,2 equivalente molar) e acetato de potássio (3,4 equivalente molar) são adicionados para uma solução do composto A16 em 4 mL de DMSO. A mistura é purgada com nitrogênio várias vezes e a seguir adiciona-se diclorobis(trifenilfosfina)paládio (II) (0,05 equivalente molar). A solução resultante é aquecida para 80°C por 12 horas. Adiciona-se água para a mistura de reação para resfriar a reação. Adiciona-se então EtOAc para extrair a solução aquosa. Secar a camada de EtOAc sobre Na2SO4. O Na2SO4 é filtrado e o filtrado evaporado dando um resíduo oleoso castanho escuro. O resíduo é purificado por cromatografia em gel de sílica (eluindo com EtOAc 30% em hexanos) dando o produto desejado composto A17 (76% rendimento). HCl (5 equivalente molar) é adicionado para uma solução do composto A17 (0,43 mmol) em mL de CH2CI2. A mistura resultante é aquecida para 50°C por 12 horas. NaHCO3 sat. é adicionado à mistura de reação a fim de neutralizar a reação. Adiciona-se então EtOAc para extrair a solução aquosa. Secar a camada de EtOAc sobre Na2SO4. O Na2SO4 é filtrado e o filtrado evaporado dando o produto desejado (composto A18) com um sólido amarelo (75% rendimento). Procedimento Geral 26
[00365] O Composto A17 (1,3 equivalente molar) é adicionado para uma solução de halogeneto de arila (0,36 mmoL) em 3 mL de DME. A mistura é purgada com nitrogênio várias vezes e a seguir diclorobis(trifenilfosfino)paládio (II) (0,05 equivalente molar) é adicionado. Carbonato de sódio (3 equivalente molar) em 0,8 mL de H2O é adicionado para a mistura de reação e a solução resultante é aquecida para 85°C por 12 horas. Adiciona-se água para a mistura de reação arrefecendo a reação.
[00366] Adiciona-se então EtOAc para extrair a solução aquosa. Secar a camada de EtOAc sobre Na2SO4. O Na2SO4 é filtrado e o filtrado evaporado para dar um resíduo oleoso castanho escuro. O resíduo é purificado por cromatografia em gel de sílica (eluindo com EtOAc em hexanos) dando o produto desejado, (composto A19) (74,4% rendimento). HCl (5 equivalente molar) é adicionado para uma solução do composto A19 (0,26 mmol) em 10 mL de álcool isopropílico. A mistura resultante é aquecida para 50°C por 12 horas. O solvente é evaporado dando o produto desejado, composto A20. Procedimento Geral 27
[00367] O composto A18 (1,3 equivalente molar) é adicionado para uma solução de halogeneto de arila (0,21 mmol) em 3 mL de DME. A mistura é purgada com nitrogênio várias e a seguir diclorobis(trifenilfosfino)paládio (II) (0,05 equivalente molar) é adicionado. Carbonato de sódio (3 equivalente molar) em 0,8 mL de H2O é adicionado para a mistura de reação e a solução resultante é aquecida para 85°C por 12 horas. Adiciona-se água para arrefecer a mistura de reação.
[00368] Adiciona-se então EtOAc para extrair a solução aquosa. Secar a camada de EtOAc sobre Na2SO4. O Na2SO4 é filtrado e o filtrado evaporado para dar um resíduo oleoso castanho escuro. O resíduo é purificado por cromatografia em gel de sílica (eluindo com CH3OH, EtOAc e CH2Cl2 e hexanos) dando o produto desejado, composto A21. Procedimento Geral 28:
[00369] Amina (1,5 equivalente molar) e K2CO3 (1,5 equivalente molar) são adicionados para uma solução de halogeneto de 4- halobenzila (1,0 equivalente molar) em 2 mL de tolueno. A mistura resultante é colocada em microondas usando Smithsynthesizer (150°C, 1 hora). Adiciona-se água para arrefecer a mistura de reação.
[00370] Adiciona-se então EtOAc para extrair a solução aquosa. Secar a camada de EtOAc sobre Na2SO4. O Na2SO4 é filtrado e o filtrado evaporado para dar o produto desejado. Composto A23. O resíduo é usado no procedimento 11 sem mais purificação para sintetizar o composto A22. Procedimento Geral 29
[00371] Amina (1,2 equivalente molar) e diisopropilamina (5 equivalentes molar) são adicionados para uma solução de cloreto de 4-bromobenzenossulfonila (0,77 mmol) em 5 mL de CHCl3 sob uma atmosfera de nitrogênio. A mistura resultante é agitada a temperatura ambiente por 4 horas. Adiciona-se água para arrefecer a mistura de reação.
[00372] Adiciona-se então EtOAc para extrair a solução aquosa. Secar a camada de EtOAc sobre Na2SO4. O Na2SO4 é filtrado e o filtrado evaporado para dar o produto desejado. Composto A25. O resíduo é usado no procedimento 11 sem mais purificação para sintetizar o composto A22. Procedimento Geral 30
[00373] Éster borônico ou ácido borônico (1,2 equivalente molar), é adicionado para uma solução de 1-cloro-4-iodobenzeno (0,84 mmol) em 10 mL de DME sob uma atmosfera de nitrogênio. A mistura é purgada com nitrogênio varias vezes e a seguir diclorobis(trifenilfosfina)paládio(II) (0,05 equivalente molar) é adicionado. Carbonato de sódio (3 equivalente molar) em 1,8 mL de água é adicionado para a mistura de reação e a solução resultante aquecida para 85°C por 12 horas. Adiciona-se água para a mistura de reação arrefecendo a reação. EtOAc é a seguir adicionado para extrair a solução aquosa. Secar a camada de EtOAc sobre Na2SO4. O Na2SO4 é filtrado e evaporado dando um resíduo oleoso castanho escuro. O resíduo é purificado por cromatografia em gel de sílica (eluindo com CH3OH, CH2Cl2, EtOAc, e hexanos) conferindo o produto desejado, composto A27.
[00374] O composto A27 é usado no procedimento 11 para sintetizar o composto A26.
[00375] A amostra racêmica é purificada usando cromatografia de fluido supercrítico preparativa SFC-EM. Condições de purificação exemplares: Chiralpak AD-H, 250 x 21 mm, 5 μ, coluna 100A (Coluna N° ADH0CJ-C1003); temperatura da coluna: 35°C, fase móvel 35% metanol (com CO2 modificado com 0,1% de isopropilamina; coeficiente de fluxo preparativo 52 mL/minuto; pressão isobárica a 120 bar.
[00376] Uso de (4-{6-amino-5-[1-(3-trilfuormetil-fenil) etóxi]-piridin- 3-il}-fenil)-(3,5-dimetil-piperazin-1-il) –metanona
[00377] A uma mistura de éster terc-butílico do ácido 4-[4-(6- amino-5-hidróxi-piridin-3-il)-benzoil]-2,6-dimetil -piperazin-1-carboxílico (100 mg, 0,23 mmol) e 1-(1-bromo-etil)-3-trifluormetil-benzeno (64 mg, 0,25 mmol) em DMF (2 mL) adicionou-se NaH (12 mg, 0,47 mmol) a 0°C. A mistura foi agitada durante a noite demonstrando que a reação estava terminada. Removeu-se DMF e água. TFA (2 mL) foi adicionado para o resíduo e agitado a temperatura ambiente por 3 horas. TFA foi removido seguido por adição de metanol. O resíduo foi purificado por HPLC preparativa rendendo 4-{6-amino-5-[1-(3- trifluormetil-fenil}-etóxi}-piridin-3-il)-fenil)-(3,5-dimetil-piperazin-1-il)- metanona (30 mg, rendimento 25,7%).
[00378] Usando 4-{6-amino-5-[1-(2-trifluormetil-fenil}-etóxi}-piridin- 3-il)-fenil)-(3,5-dimetil-piperazin-1- il) -metanona
[00379] A uma mistura de éster terc-butílico do ácido 4-[4-(6- amino-5-hidróxi-piridin-3-il)-benzoil]-2,6- dimetil -piperazin-1-carboxílico (50 mg, 0,12 mmol) e 1-(1-bromo-etil)-2-trifluormetil-benzeno (32 mg, 0,12 mmol) em DMF (2 mL) adicionou-se Cs2CO3 (0,18 mL, 0,35 mmoL) seguido por água (0,5 mL). a mistura foi agitada durante a noite, a seguir aquecida a 70°C por 8 horas, demonstrando por LCEM que a reação estava terminada. O DMF e a água foram removidos. TFA (2 mL foi adicionado ao resíduo e agitado a temperatura ambiente por 3 horas. Removeu-se o TFA, seguido por adição de metanol. O resíduo foi purificado por HPLC preparativa rendendo: 4-{6-amino-5-[1- (2-trifluormetil-fenil}-etóxi}-piridin-3-il)-fenil)-(3,5-dimetil-piperazin-1-il)- metanona (20 mg, rendimento 34,2%).
[00380] Uso de 4-{6-amino-5-(2-metil-benzilóxi)-piridin-3-il)-fenil)- (3,5-dimetil-piperazin-1-il)-metanona
[00381] A uma mistura de (2R,6S)-éster terc-butílico do ácido 4-[4- (6-amino-5-hidróxi-piridin-3-il)-benzoil]-2,6-dimetil-piperazin-1- carboxílico (100 mg, 0,23 mmol) e 1-bromometil-2-metil-benzeno (47 mg, 0,25 mmol) em dimetilformamida (2 mL) adicionou-se Cs2CO3 2M (0,35 mL, 0,7 mmol) seguido por água (0,5 mL). A mistura foi agitada a temperatura ambiente durante a noite. LCEM demonstrou o término da reação. DMF foi removido, seguido por adição de HCl 4N em dioxano (2 mL) e a reação foi agitada a temperatura ambiente por 3 horas. Os voláteis foram removidos seguido por adição de metanol Esta solução foi purificada por HPLC preparativa produzindo {4-[6-amino-5-(2-metil- benzilóxi)-piridin-3-il]-fenil}-(3,5-dimetil-piperazin -1-il)-metanona (47 mg, rendimento: 46,6%).
[00382] Uso de 6-amino-3-aza-biciclo[3.1.0]hex-3-il)-(4-(6-amino-5- [1-(2,6-dicloro-3-flúor-fenil]-etóxi]-piridin-3-il}-fenil)-metanona
[00383] A uma mistura de éster terc-butílico do ácido [3-(4-iodo- benzoil)-3-aza-biciclo[3.1.0]hex-6-il]-carbâmico(100 mg, 0,234 mmol) e 3-[1-(2,6-dicloro-3-flúor-fenil)etóxi]-5-(4,4,5,5-tetrametil- [1,3,2]dioxaborolan-2-il)-piridin - 2 -ilamina (100 mg, 0,234 mmol) em DME (2 mL) adicionou-se Pd(dppf)2CH2Cl2 (10 mg, 0,012 mmol) e Cs2CO3 (351 mg, 0,702 mmol). A mistura foi borbulhada com nitrogênio por 10 minutos a seguir colocada em microondas a 150 °C por 30 minutos. LCMS demonstrou o término da reação. A mistura de reação bruta foi diluída com acetato de etila seguido por lavagem com água e salmoura. A solução foi seca sobre MgSO4. A purificação por HPLC preparativa produziu um sólido. O sólido foi agitado com HCl/dioxano 4N (3 mL) a temperatura ambiente. A remoço dos voláteis levou a um resíduo que foi purificado por HPLC preparativa rendendo (6-amino-3-aza-biciclo[3.1.0]hex-3-il)-(4-{6-amino-5-[1-(2,6 -dicloro-3- flúor-fenil)-etóxi]-piridin-3-il}-fenil)-metanona (30 mg, rendimento: 26%).
[00384] Uso de 5-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]-6'-(2-morfolin- 4-il-etóxi]-[3,3']bipiridinil-5-ilamina
[00385] A uma mistura 6'-amino-5'-[1-(2,6-dicloro-3-flúor-fenil)- etóxi]-[3,3']- bipiridinil-6-ol (78 mg, 0,20 mmol), trifenilfosfina (63 mg, 0,24 mmol) e 2-morfolin-4-il-etanol (0,026 mL, 0,22 mmol) adicionou-se DEAD (0,034 mL,0,22 mmol). Após agitar durante a noite mais PPh3 (63 mg, 0,24 mmol) e mais DEAD ( 0,034 mL, 0,22 mmol) foram adicionados. Após varias horas, adicionou-se mais álcool (0,026 mL,0,22 mmol). Após várias horas, mais PPh32 (63 mg, 0,24 mmol) e mais DEAD (0,34 mL, 0,22 mmol) foram adicionado. Após agitação durante a noite a mistura foi dividida entre diclorometano e salmoura meio-saturada. As fases foram separadas e a fase aquosa foi extraída com diclorometano. As fases orgânicas combinadas foram secas sobre Na2SO4 e concentradas por evaporação rotativa. O resíduo foi purificado por cromatografia em gel de sílica usando gradiente de eluição de diclorometano/metanol produzindo 5-[1-(2,6-dicloro-3-flúor- fenil)-etóxi]-6'-(2-morfolin-4- il -etóxi-[3,3']bipiridinil-5-ilamina (53 mg, 53%).
[00386] Utilização do Exemplo I-650 do Pedido de Patente U.S. n° de série 10/786.610 (PCT/US2004/005495)
[00387] 3-(2,6-dicloro-3-flúor-benzilóxi)-5-tiazol-2- il -piridin-2- ilamina: A um tubo de microondas equipado com uma barra de agitação adicionou-se o material de partida iodo-piridila (300 mg, 0,702 mmol), tetraquis(trifenilfosfina) paládio (0) 940 mg, 5 mol% e tetraidrofurano (anidro, 6 mL). O frasco foi tampado e purgado com nitrogênio por 5 minutos. Brometo de 2-tiazolil zinco (0,5M em tetraidrofurano, 1,4 mmol, 2,8 mL) foi a seguir adicionado via seringa. O frasco foi aquecido para 120°C no microondas por 10 minutos. TLC (acetato de etila:cloreto de metileno 1:1) demonstrou uma grande quantidade de material de partida restante. Adicionou-se mais brometo de 2-tiazolil zinco (0,5 M em THF 500 μL) e o frasco foi aquecido par 120°C no microondas por 20 minutos. TLC (acetato de etila:cloreto de metileno 1L1) demonstrou uma grande quantidade de material de partida que ainda permanecia. Adicionou-se mais brometo de 2-tiazolil zinco (0,5 M em THF, 500 μL) e o frasco foi aquecido para 120°C no microondas por 60 minutos. TLC (acetato de etila:cloreto de metileno 1L1) ainda demonstrava uma grande quantidade de material de partida existente, mas que ainda estava muito misturado. O conteúdo do frasco foi despejado numa solução de NH4Cl sat. (10 mL) e esta s foi extraída com acetato de etila (2 x 30 mL). As camadas de acetato de etila combinadas foram secas sobre Na2SO4, filtradas e concentradas in vácuo. O produto bruto foi introduzido numa coluna de gel de sílica pré-acondicionada de 10 g e acetato de etila:cloreto de metileno 1:1 foi usado para purificar o produto desejado (40 mg, 15%).
[00388] Uso do Exemplo I-652 do Pedido de Patente U.S. n° de série 10/786.610 (PCT/US2004/005495).
[00389] 3-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]-5-(1-metil -1H- imidazol-2-il)-piridin-2-ilamina: N-metil-imidazol (92 mg, 1,1 mmol) foi dissolvido em tetraidrofurano (anidro, 4 mL) num frasco de fundo redondo de 50 mL. O frasco foi resfriado com um banho de gelo seco/acetona sob nitrogênio. N-butil lítio (2,5 M 562 μL, 1, 4 mmol) foi adicionado via seringa em partes de 100 μL durante 5 minutos. A reação foi agitada a -70°C por 30 mt. Cloreto de zinco sólido (anidro, 383 mg, 2,8 mmols) foi adicionado e a reação agitada por 15 minutos. Removeu-se o banho de gelo e a reação foi deixada aquecer para temperatura ambiente. Uma vez todo o cloreto de zinco estar em solução, e a reação à temperatura ambiente, iodo (400 mg, 0,936 mmol) ser adicionado em tetraidrofurano (anidro, 4 mL), seguido por tetraquis(trifenilfosfina) paládio(0) 108 mg, 10% em mol sendo a reação aquecida ao refluxo. A reação foi monitorada por LC/EM até todo o iodo de partida ser sido consumido. A reação foi deixada resfriar e a seguir diluía com uma solução de NH4CL sat. (20 mL). Esta solução foi extraída com acetato de etila (2 x 50 mL). As camadas de acetato de etila combinadas foram secas sobre Na2SO4 filtradas e concentrada in vácuo. O produto bruto foi introduzido numa coluna de gel de sílica pré-acondicionada e metanol:acetato de etila a 10% foi usado como eluente do produto desejado (25 mg, 7%).
[00390] Uso do Exemplo I-657 do Pedido de Patente U.S. N° 10/786.610 (PCT/US2004/005495)
[00391] A 6-amino-5-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]- nicotinonitrila (400 mg, 1,23 mmol) em 70 mL de metanol seco a 0 °C foi borbulhado gás HCl durante 3 minutos. Agitou-se durante a noite a 3°C. Removeu-se os voláteis e lavou-se os sólidos com éter dietílico rendendo o imidato, quantitativamente. A 200 mg do imidato em 4 mL de metanol a 0°C adicionou-se metilamina 2N em THF (857 μL). Deixou-se agitar a 0°C por cerca de uma hora, deixando-se aquecer durante a noite a temperatura ambiente. Os voláteis foram removidos e o resíduo cromatografado com 10-20% metanol/diclorometano rendendo 7 mg do produto. Procedimento Geral 40
[00392] ácido 6-nitro-5-hidroxinicotínico (B2): A uma solução de ácido 5-hidroxinicotínico (B1)(7,0 g, 50 mmols) em ácido sulfúrico conc. adicionou-se 9 mL de HNO3 fumegante (90%) (9 mL). A mistura de reação foi agitada a 55-60°C num tubo vedado por quatro dias. A mistura foi então despejada em gelo e o pH foi ajustado em 3 com 50% de NaOH. Adicionou-se MgSO4 para a mistura aquosa saturada que foi então extraía com álcool isopropílico (4 x 45 mL). Após a remoção do álcool isopropílico sob pressão reduzida, 5,93 g (64% rendimento) de B2 foi obtido como um sólido amarelo. EM (APCI), (M+H)+ 185. 1H RMN (DMSO-d6) δ 8,01 (d, 1H, Ar-H), 8,41 (d, 1H, Ar- H).
[00393] 2,6-diclorobenzil-6-nitro-5-[(2,6-diclorobenzil)oxi]nicotinato (B3): ácido 6-nitro-5-hidroxinicotínico (3,4 g, 18,5 mmols), brometo de 2,6-diclorobenzila (8,88 g, 37 mmols) DIPEA (5,5 g, 42,5 mmols) foram dissolvidos em DM (25 mL) num frasco de fundo redondo de 250 mL e a reação foi agitada a temperatura ambiente por 4,5 horas a seguir concentrada sob pressão reduzida. A mistura resultante foi despejada em gelo e filtrada. O sólido coletado foi seco sob pressão reduzida para dar 4,25 g (46% rendimento) de B3.
[00394] EM (APCI) (M+H)+ 503. 1H RMN (DMSO-d6) δ 5,47 (s, 2H, ArCH2O), 5,71 (s, 2H, ArCH2O), 7,24-7,43 (m, 6H, Ar-H), 8,26 (d, 1H, Ar-H), 8,66(d, 1H, Ar-H).
[00395] 3. 2,6-diclorobenzil-6-amino-5-[(2,6- diclorobenzil)óxi]nicotinato (B4): Uma mistura de 2,6-diclorobenzil-6- nitro-5-[(2,6-diclorobenzil)óxi] nicotinato (B3) (5, g, 10,96 mmols) pó de ferro (0,92 g 16,43 mmols), ácido acético glacial (20 mL) e metanol (17 mL) foi agitada a 85°C por três horas. A mistura de reação foi concentrada até quase secura, e adicionou-se hidróxido de amônio (30%) para neutralizar a mistura. Quantidade mínima de dimetilformamida foi adicionada para dissolver a mistura de reação que foi purificada por cromatografia de coluna flash (eluente:EtOAc- EtOH 9:1) dando 4,5 g 87% de B4 como um sólido amarelo pálido. EM (APCI) (M+H)+ 473.
[00396] 4. ácido 6-amino-5-[(2,6-diclorobenzil-oxi] nicotínico (B5): Uma mistura de 2,6-diclorobenzil-6-amino-5-[(2,6- diclorobenzil)oxi]nicotinato (B4) (3,5 g, 7,4 mmols), hidróxido de lítio (0,41 g, 17 mmols), água (22 mL) e metanol (30 mL) foi agitada ao refluxo a 85°C por 5 horas. A mistura foi concentrada até secura sob pressão reduzida. O resíduo resultante foi dissolvido em água, extraído com uma mistura de Et2O/hexano (1:1, 4x 25 mL) neutralizado com HCl 1N formando um precipitado branco, que foi filtrado e seco sob pressão reduzida proporcionando 1,83 g (79% de B5 como um sólido branco.
[00397] EM (APCI) (M+H)+ 313. 1H RMN (DMSO-d6) δ 5,26 (s, 2H, ArCH2O), 6,37 (s, 2H, NH2), 7,43-7,48 (t, 1H, Ar-H), 7,54 (s, 2H, Ar-H), 7,56 (s, 1H, Ar-H), 8,18 (s, 1H, Ar-H).
[00398] A um arranjo de 400 μL de solução 0,2M de diferentes aminas em DMF numa placa de 96 poços, adicionou-se 400 μL 90,2 M em DMF) de ácido 4-[6-amino-5-(2,6-dicloro-3-flúor-benzilóxi)-piridin-3- il]benzóico, 80 μL de trietilamina (1M em DMF) e 160 μL de HATU (0,5 M em DMF) e as reações foram agitadas a 70°C por duas horas. Removeu-se o solvente usando o aparelho Speed/Vac e as misturas de reação brutas foram redissolvidas em DMSO e transferidas usando um manipulador de líquidos para uma placa de 96 poços de 1 mL dando uma concentração teórica final de ~10 mM. As reações foram analisadas e a identificação positiva do produto foi feita usando LC/EM. A solução-mãe de estoque foi diluída para 50 nM e testada quanto à inibição percentual de c-MET a 50 nM. Procedimento Geral 41
[00399] A um arranjo de 400 μL de solução 0,2M de diferentes aminas em DMF numa placa de 96 poços adicionou-se 400 μL (0,2 M em DMF) de ácido 4-[6-amino-5-[(2,6-diclorobenzil)oxi]nicotínico, 80 μL de trietilamina (1M em DMF) e 160 μL de HATU (0,5 M em DMF) sendo as reações agitadas a 70°C por duas horas. O solvente foi removido usando o aparelho Speed/Vac e a mistura de reação bruta foi redissolvida em DMSO e transferida usando um manipulador de líquido para uma placa de 96 poços, dando uma concentração teórica final de ~10 mM. As reações foram analisadas sendo feita à identificação positiva do produto com emprego de LC/EM. A solução- mãe de estoque foi diluída a 1 μL e submetida a ensaio.
[00400] Uso de 2-(4-(6-amino-5-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]- piridin-3-il}-pirazol-1-il)-N-(3-dimetilamino-propil)-isobutiramida

[00401] A uma solução de 4-(4,4,5,5,-tetrametil-[1,3,2] dioxaborolan-2-il)-1H-pirazol (5 g, 25, 77 mmols) e éster metílico do ácido 2-bromo-2-metil-propiônico (12,6 g, 27,06 mmols) em DMF (85 mL), adicionou-se Cs2CO3 (12,6 g, 38,65 mmols). A mistura de reação foi aquecida para 90°C num banho de óleo durante a noite. A solução de reação foi resfriada para temperatura ambiente e dividida entre água e acetato de etila. A solução de acetato de etila combinada foi lavada com água cinco vezes, seca sobre Na2SO4 e concentrada dando o produto éster metílico do ácido 2-metil-2-[4-(4,4,5,5,- tetrametil-[1,3,2]dioxaborolan-2-il)-1H -pirazol-1-il]propiônico (4,776 g, 63% rendimento).
[00402] A uma solução de 3-[1-(2,6-dicloro-3-flúor-fenil)etóxi]-5- iodo-piridin-2-ilamina (6,363 g, 14,901 mol e éster metílico do ácido 2- metil-2-[4-(4,4,5,5,-tetrametil-[1,3,2]dioxaborolan-2-il)-pirazol-1- il]propiônico, (4,6 g, 15,64 mmols) em DME (27 mL) adicionou-se uma solução de CsF (6,79 g 44,7 mmols) em água (9,3 mL). A mistura de reação foi desgaseificada 3 vezes com N2 Pd(dppf)CH2Cl2 foi adicionado e a mistura de reação foi desgaseificada 3 vezes com nitrogênio. A reação foi aquecida a 120°C no microondas (sendo adicionado mais Pd em intervalos de 30 minutos até o término da reação). Adicionou-se água e a reação foi extraída com EtOAc, seca sobre Na2SO4 e concentrada dando éster metílico do ácido 2-(4-(6- amino-5-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]-piridin-3-il}-pirazol-1-il)-2- metil -propiônico. O produto bruto foi purificado por cromatografia em gel de sílica com um gradiente de 25%-50% de acetato de etila/hexanos dando éster metílico do ácido 2-(4-(6-amino-5-[1-(2,6- dicloro-3-flúor-fenil)-etóxi]-piridin-3-il}-pirazol-1-il)-2-metil-propiônico (1,46 g, 21%rendimento) com uma Rf 0,11 (50% EtOAc/hexanos).
[00403] A uma solução do éster metílico (2,92 g 6,25 mols) em MeOH (31 mL) adicionou-se uma solução de LiOH (450 mg, 18,76 mmols) em água (6,25 mL).A reação foi aquecida para 60°C até demonstrar por LCEM, a completa hidrólise (cerca de 45 minutos). O MeOH foi removido in vácuo e MeOh (2,5 mL) e água (1 mL) foi adicionado. O pH foi ajustado para pH 5 com HCl 1N, precipitando o produto. O produto ácido 2-(4-(6-amino-5-[1-(2,6-dicloro-3-flúor-fenil)- etóxi]-piridin-3-il}-pirazol-1-il)-2-metil-propiônico foi obtido após filtração (2,825 g, quantitativo).
[00404] Para uma solução de ácido 2-(4-(6-amino-5-[1-(2,6- dicloro-3-flúor-fenil)-etóxi]-piridin-3-il}-pirazol-1-il)-2-metil-propiônico (1,00 g, 2,20 mmols) em DMF (5,5 mL) adicionou-se HOBT (300 mg, 2,20 mmols), EDC (633 mg, 3,30 mmols) e N,N-dimetilpropano-1,3- diamina (225 mg, 2,20 mmols). A reação foi agitada durante a noite a temperatura ambiente. A reação foi a seguir purificada por HPLC preparação. C-18 de fase reversa com acetonitrila/água com 0,1% ácido acético rendendo 2-(4-{6-amino-5-[1-(2,6-dicloro-3-flúor-fenil)- etóxi]-piridin-3-il}-pirazol-1-il)-N-(3-dimetilamino-propil) -isobutiramida (170 mg, 14% rendimento).
[00405] Uso de 3-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]-5-(3-metil- pirazol-1-il)-piridin-2-ilamina
[00406] A uma solução agitada de 3-[1-(2,6-dicloro-3-flúor-fenil)- etóxi]-5-iodo-piridin-2-ilamina (100 mg, 0,23 mmol) e 3-metil-1H-pirazol (59 mg, 0,70 mmol) em DMSO (1 mL) adicionou-se K3PO4 (101 mg, 0,47 mmol) dodecano (0,015 mL, 0,05 mol) cicloexanodiamina (0,009 mL), 0,007 mmol) e iodeto de cobre (CuI) (14 mg, 0,07 mmol). A solução foi borbulhada com nitrogênio por 5 minutos, então irradiada com microondas a 150°C por duas horas, verificando por LCEM que a reação tinha-se completado. A mistura foi purificada por HPLC preparação. deixando 3-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]-5-(3-metil- pirazol-1-il)-piridin-2-ilamina (30 mg, rendimento: 34,2%. Procedimento Geral 44
[00407] 2,5-dibromopiridina (1 equivalente molar) foi dissolvido em anidro (0,085 M) e resfriado para -78°C. n-BuLi (1,2 equivalente molar) foi lentamente adicionado durante 5 minutos sendo a seguir, deixado agitar a -78°C. Após duas horas, R1COR2 (1,3 equivalente molar) foi adicionado e a solução mantida a -78°C. Após uma hora, NH4Cl aquoso saturado foi adicionado e a solução foi aquecida para temperatura ambiente. O produto foi extraído com EtOAc (3X) e os extratos orgânicos foram combinados, secos (Na2SO4), concentrados e purificados por cromatografia de coluna (10% EtOAc/hexanos - 100% EtOAc ) rendendo o produto bruto. Este foi usado diretamente no Procedimento Geral 27 conferindo 25. Procedimento Geral 45
[00408] A uma solução de 3-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]- piridin-2-ilamina (1,8 g, 6,04 mmol), cianeto de zinco, 98% (2,07 g, 12,7 mmol) e 1,1'-bis(difenilfosfino) ferroceno, 97% (0,4 g, 0,712 mmol) em DMF (48 mL) adicionou-se complexo de [1,1'bis-(difenilfosfino)- ferroceno]dicloro-paládio(II) com diclorometano (1:1) (0,25 g 0,30 mmol). A mistura de reação foi aquecida para 150°C durante a noite sob atmosfera de nitrogênio. A reação foi diluída com EtOAc (50 mL), lavada com NH4Cl/28% NH4OH/H2O (2 x 28 mL), 4:1:4 sat., secas sobre Na2SO4. A mistura bruta foi purificada com uma coluna de gel de sílica eluindo com um gradiente linear de 25%-50% de (EtOAc/hexanos) dando 2-[1-(2-amino-piridin-3-ilóxi)-etil]-3-cloro-4- dimetilamino-benzonitrila como um sólido amarelo (37% rendimento) e 1-[1-(2-amino-piridin-3-ilóxi)etil]-4-dimetilamino-isoftalonitrila como um sólido castanho escuro (33% rendimento). Procedimento Geral 46
[00409] A uma mistura de 4-bromo-imidazol (995 mg, 6,77 mmols), hidróxido de potássio (380 mg, 6,77 mmols) carbonato de potássio (936 mg, 6,77 mmols e brometo de tetra-n-butil amônio (109 mg, 0,339 mmol) em diclorometano (7 mL) adicionou-se bromo acetato de terc- butila (0,50 mL, 3,4 mmols). Após agitar durante a noite a reação foi filtrada. O filtrado foi seco sobre sulfato de sódio, filtrado e concentrado por evaporação rotativa. O resíduo foi purificado por cromatografia em gel de sílica usando eluição de gradiente de diclorometano, acetato de etila, rendendo éster terc-butílico do ácido (4-bromo-imidazol-1-il)- acético (696 mg, 79%). Procedimento Geral 47
[00410] Uma solução 4M de ácido clorídrico em dioxano (0,22 mL, 0,89 mmol) foi adicionada para uma solução de éster terc-butílico do ácido 4-{6-amino-5-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]-piridin-3- il}imidazol-1il)-acético (86 mg, 0,18 mmol) em diclorometano (2 mL). Após agitação por dois dias a reação foi concentrada por evaporação rotativa e o resíduo foi dissolvido numa quantidade mínima de metanol. Esta solução foi adicionada gota a gota ao éter e a mistura resultante deixada repousar durante a noite. A mistura foi filtrada e o precipitado foi lavado com éter e seco ao ar dando ácido (4-{6-amino- 5-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]-piridin-3-il}imidazol-1il)-acético (83 mg, 93%). Procedimento Geral 48
[00411] Uma mistura de 4-bromoimidazol (217 mg, 1,48 mmol) e carbonato de césio (875 mg, 2,69 mmols) em dimetilformamida (5 mL) foi agitada por 30 minutos. Adicionou-se cloridrato de 4-(2-cloro-etil)- morfolina (250 mg, 1,34 mmol) e a mistura foi aquecida para 50°C. Após aquecer durante a noite, a reação foi concentrada por evaporação rotativa. O resíduo foi suspenso numa mistura de diclorometano e metanol e filtrado. O filtrado foi concentrado por evaporação rotativa. O resíduo foi purificado por cromatografia em gel de sílica usando gradiente de eluição de diclorometano, metanol rendendo 4-[2-(4-bromo-imidazol-il)-etil]-morfolina (148 mg, 42%). Procedimento Geral 49
[00412] Isoxazol (0,64 mL, 10 mmol) foi adicionado a uma solução de N-iodosuccinimida (2,3 g, 10 mmol) em ácido trifluoracético (20 mL). Após agitação durante a noite, foram adicionados água (50 mL), hexanos (50 mL) e bissulfito de sódio para a reação. AS fases foram separadas e a fase orgânica foi seca sobre Na2SO4 filtrada e concentrada por evaporação rotativa dando 4-iodo-isoxazol (218 mg, 11%). Procedimento Geral 50
[00413] Ácido trifluoracético (5 mL) foi adicionado para uma solução de 6'-bromo-5-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]-[3,3']bipiridinil- 6-il-bi -(terc-butoxicarbonil) -amina (1,3 g, 2,0 mmols) em diclorometano (15 mL). Após 3 horas, partes iguais de água e solução aquosa saturada de bicarbonato de sódio foram adicionadas. As fases foram separadas e a fase aquosa foi extraída com diclorometano. As fases orgânicas combinadas foram secas sobre Na2SO4 e concentradas por evaporação rotativa dando 6'-bromo-5-[1-(2,6- dicloro-3-flúor-fenil)-etóxi] - [3,3'] bipiridinil - 6-ilamina (968 mg, 106%).
[00414] Introduziu-se um tubo com 6'-bromo-5-[1-(2,6-dicloro-3- flúor-fenil)-etóxi]-[3,3']bipiridinil-6-ilamina (92 mg, 0,20 mmol), 4- pirrolidin-1-il-piperidina (0,62 g, 4,0 mmols) e N-metilpirrolidinona (0,8 mL). O tubo foi vedado e a mistura foi aquecida para 80°C durante a noite. A temperatura foi aumentada para 100°C por 5,5 horas e a seguir cessou-se o aquecimento. A reação foi dividida entre acetato de etila e água. As fases foram separadas e a fase aquosa foi extraída com acetato de etila. As fases orgânicas combinadas foram secas sobre MgSO4 e concentradas por evaporação rotativa. O resíduo foi purificado por cromatografia em gel de sílica usando gradiente de eluição de diclorometano, metanol, hidróxido de amônio rendendo 5"- [1-(2,6-dicloro-3-flúor-fenil)-etóxi]-4-pirrolidin- 1 - il -3,4,5,6-tetraidro- 2H-[1,2',5',3"]terpiridin-6"-ilamina (53 mg, 50%). Procedimento Geral 51
[00415] Hidreto de sódio (56 mg, 2,3 mmols) foi adicionado para uma solução de piperidin-4-ol (214 mg, 2,11 mmols) em DMSO (8 mL). Após agitar por 30 minutos, adicionou-se 2,5-dibromopiridina. Após agitação por 24 horas, hidreto de sódio (56 mg, 2,3 mmols) foi adicionado. Após agitação por mais 24 horas, a reação foi dividida entre acetato de etila e água. As fases foram separadas e a fase aquosa foi extraída com acetato de etila. As fases orgânicas combinadas foram secas sobre MgSO4 e concentradas por evaporação rotativa. O resíduo foi purificado por cromatografia em gel de sílica usando gradiente de eluição de diclorometano, metanol, hidróxido de amônio para dar 5-bromo-2-(piperidin-4-ilóxi)-piridina (316 mg, 58%). Procedimento Geral 52
[00416] Carregou-se um tubo com 2,5-dibromopiridina (0,24 g, 1,0 mmol), éster terc-butílico do ácido 4-amino-piperidino-1-carboxílico (0,22 g, 1,1 mmol), di-isopropiletilamina (0,19 mL, 1,1 mmol) e N- metilpirrolidinona (1,0 mL). O tubo foi vedado e a mistura foi aquecida para 80°C, durante a noite. A temperatura foi aumentada para 120°C e aquecida durante a noite. A reação foi dividida entre acetato de etila e água. As fases foram separadas e a fase aquosa foi extraída com acetato de etila. As fases orgânicas combinadas foram secas sobre MgSO4 e concentradas por evaporação rotativa. O resíduo foi purificado por cromatografia em gel de sílica usando gradiente de eluição de acetato de etila e hexanos rendendo éster terc-butílico do ácido 4-(5-bromo-piridin-2-ilamino)-piperidino-1-carboxílico (36 mg, 10%). Procedimento Geral 53
[00417] Éster terc-butílico do ácido 4-(4-{6-amino-5-[1-(2,6-dicloro- 3-etóxi-fenil)-etóxi]-piridin-3-il] - benzoil) piperazino-1-carboxílico: A 4 mL de DMSO adicionou-se 0,124 ml de etanol seguido por 32 mg de NaH. Após agitação por 30 minutos, 250 mg de éster terc-butílico do ácido 4-(4-{6-amino-5-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]-piridin-3-il]- benzoil)piperazino-1-carboxílico foi do e a reação aquecida para 40°C. Após três horas a reação foi resfriada e despejada em água para precipitar. Após neutralização a pH 6, 200 mg de um sólido castanho foi isolado. 77%. Procedimento Geral 54
[00418] 4-(6-amino-5-[1-(2,6-dicloro-3-hidróxi - fenil) -etóxi]-piridin- 3-il]-fenil)-piperazin-1-il-metanona: A 140 mg de éster terc-butílico do ácido 4-(4-{6-amino-5-[1-(2,6-dicloro-3-(2,4,6-trimetóxi-benzilóxi)- fenil]etóxi]-piridin-3-il]-benzoil)piperazino-1-carboxílico (do Procedimento Geral 53) foi adicionado 1 mL de TFA, a solução tornou- se avermelhada imediatamente seguido por adição de 100 μL de trimetilsilano 3 segundos depois. A solução tornou-se amarela. Após agitar por quatro horas 5 mL de tolueno foram adicionados e o solvente foi removido in vácuo. A cromatografia com 10% MeOH/CH2Cl2 para 0,5% a 1% NH4OH/9,5 a 9% MeOH/90% CH2Cl2 levou a 55 g de um sólido branco, 62% rendimento. Procedimento Geral 55
[00419] 2-(4-bromo-2-metoxifenóxi)etanol (8a); Carbonato de potássio (1,4 g, 10 mmols) foi adicionado para uma solução de carbonato de etileno (1,8 g, 20 mmols) e 4-bromo-2-metoxifenil (1,05 g, 5 mmols) em 5 mL de tolueno sob uma atmosfera inerte. A reação foi aquecida a 115°C por 12 horas. Foram adicionados água (50 mL) e acetato de etila (2 x 100 mL) para a mistura de reação com agitação. As camadas orgânicas foram combinadas secas, filtradas e evaporadas dando um resido oleoso amarelo. O resíduo foi purificado por cromatografia flash eluindo com 40^45% EtOAc em hexanos) dando o composto 8a como um óleo amarelo castanho claro (1 g: 4,13 mmol; 82,6% rendimento);
[00420] EM (APCI) (M+H)+ 246. 1H RMN (400 MHZ, clorofórmio-D) δ ppm 2,83 (t, J = 6,3 Hz, 1H) 3,84 (s, 3H) 3,89-4,01 (m, 2H) 4,03 -4,13 (m, 2H) 6,78 (d, J = 8,3 Hz, 1H) 8,99 (d, 1H) 7,02 (d, 1H).
[00421] 4-bromo-1-(2-cloroetóxi)-2-metoxibenzeno (8b): Cloreto de tionila (0,3 mL) foi adicionado para uma solução do composto 1 em 1 mL de piridina num banho de gelo. A reação foi agitada no banho de gelo por 10 minutos, então aquecida para 100°C por duas horas. A reação foi resfriada apara temperatura ambiente e neutralizada com HCl diluído (1M). CH2Cl2 (2 x 100 mL) foi adicionado para extrair a solução aquosa. As camadas orgânicas combinadas foram secas sobre Na2SO4 então concentradas sob vácuo. O resíduo foi purificado por cromatografia flash eluindo com 10^ 15% EtOAc em hexanos) dando o composto 8b como um óleo incolor (485 mg, 1,84 mmol; 50,3% rendimento)
[00422] EM (APCI) (M+H)+ 264. 1H RMN (400 MHz, clorofórmio-D) δ ppm 3,81 (t, J = 6,2 Hz, 2H) 3,85 (s, 3H) 4,23 (t, J = 6,2 Hz, 2H) 6,78 (d, J = 8,6 Hz, 1H).
[00423] Compostos de fórmula 9 podem ser formados pelo seguinte procedimento exemplar: O composto A1B (1,3 equivalente molar) é adicionado a uma solução de halogeneto de arila (0,51 mmol) em 7 mL de DME. A mistura é purgada com nitrogênio várias vezes e a seguir diclorobis(trifenilfosfino)paládio (II) (0,05 equivalente molar) é adicionado. Carbonato de sódio (3 equivalente molar) em 1,5 mL de água é adicionado à mistura de reação e a solução resultante e aquecida para 85°C por 12 horas. Adiciona-se água (20 mL) para a mistura de reação arrefecendo a reação. EtOAc (50 mL x 2) é a seguir adicionado para extrair a solução aquosa. Secar a camada de EtOAc sobre Na2SO4. O Na2SO4 é filtrado e evaporado dando um resíduo oleoso castanho escuro. O resíduo é purificado por cromatografia em gel de sílica (eluindo com CH3OH, CH2Cl2, EtOAc e hexanos) conferindo o produto desejado, composto 9.
[00424] Compostos de fórmula 10 podem ser formados pelo seguinte procedimento exemplar: Amina (7 equivalente molar) é adicionada a uma solução do composto 9 (0,17 mmol) em 3 mL de 2- metoxietanol. A solução resultante é aquecida para 85°C por 12 horas. Adiciona-se água (20 mL) para a mistura de reação arrefecendo a reação. EtOAc (50 mL x 2) é a seguir adicionado para extrair a solução aquosa. Secar a camada de EtOAc sobre Na2SO4. O Na2SO4 é filtrado e evaporado dando um resíduo oleoso castanho claro. O resíduo é purificado por cromatografia em gel de sílica (eluindo com CH3OH, CH2Cl2, EtOAc e hexanos) conferindo o produto desejado, composto 10. Procedimento Geral 56
[00425] Compostos de fórmula 14 podem ser formados pelo seguinte procedimento exemplar: hexamexildissilazida de lítio (1,2 equivalente molar, 1M em THF) é adicionada a uma solução de álcool (1 mmol) em 2 mL de THF. A mistura é agitada a temperatura ambiente sob uma atmosfera de nitrogênio por 30 minutos e a seguir 5-bromo-2-cloropirimidina (1 equivalente molar) é adicionado. A solução resultante é aquecida para 75°C por 12 horas. Adiciona-se água (20 mL) para a mistura de reação arrefecendo a reação. EtOAc (50 mL x 2) é a seguir adicionado para extrair a solução aquosa. Secar a camada de EtOAc sobre Na2SO4. O Na2SO4 é filtrado e evaporado dando um resíduo oleoso. O resíduo é purificado por cromatografia em gel de sílica (eluindo com EtOAc em hexanos) conferindo o produto desejado, composto 14.
[00426] Composto A18 (1,3 equivalente molar) é adicionado para uma solução de 5-bromo-2-cloropirimidina ou composto 14 (1 mmol) em 24 mL de DME. A mistura é purgada com nitrogênio várias vezes e a seguir, diclorobis (trifenilfosfino)paládio (II) (0,05 equivalente molar) é adicionado. Carbonato de sódio (3 equivalentes molar) em 3 mL) de água é adicionado para a mistura de reação e a solução resultante é aquecida para 85°C por 12 horas. Adiciona-se água (50 mL) para a mistura de reação arrefecendo a reação. EtOAc (100 mL x 2) é a seguir adicionado para extrair a solução aquosa. Secar a camada de EtOAc sobre Na2SO4. O Na2SO4 é filtrado e evaporado dando um resíduo oleoso castanho escuro. O resíduo é purificado por cromatografia flash (eluindo com 40^55% EtOAc em hexanos) conferindo o produto desejado, composto 11.
[00427] Amina (2 equivalentes molar) é adicionada para uma solução do composto 11 em 3 mL de n-butanol. A mistura de reação é irradiada em microondas a 120°C por 30 minutos. A mistura resultante é despejada numa mistura de H2O e EtOAc (100 ml; vol/vol 1:1) a camada orgânica é seca, filtrada e evaporada dando um resíduo oleoso castanho claro. O resíduo é purificado por cromatografia em gel de sílica (eluindo com CH3OH, CH2Cl2, EtOAc e hexanos) dando o produto desejado, composto 12.
[00428] Ácido (16 equivalentes molar ou menos) é adicionado ao composto 12 (0,14 mmol) a temperatura ambiente. A solução resultante é agitada a temperatura ambiente ou aquecida para 60°C por duas horas. A mistura de reação é evaporada e o resíduo purificado por cromatografia em gel de sílica (eluindo com CH3OH, EtOAc e CH2Cl2) dando o produto amida desejado, composto 13, como um sólido amarelado a branco. Procedimento Geral 57
[00429] Hidreto de sódio (1,3 equivalente molar) e RX (1,1 equivalente molar) foram adicionados para uma solução de 2-amino-5- bromopiridina (0,84 mmol) em 3 mL de DMF. A mistura de reação é irradiada em microondas a 100°C por 20 minutos. A mistura resultante é despejada numa mistura de água e EtOAc (100 mL; vol/vol, 1:1). A camada orgânica é seca, filtrada e evaporada dando um resíduo oleoso castanho claro. O resíduo é purificado por cromatografia em gel de sílica (eluindo com CH3OH, CH2Cl2, EtOAc e hexanos) conferindo o produto desejado, composto 15.
[00430] Composto A18 (1,3 equivalente molar) é adicionado para uma solução do composto 15 (0,25 mmol) em 5 mL de DME. A mistura é purgada com nitrogênio várias vezes e a seguir, diclorobis(trifenilfosfino)paládio (II) (0,05 equivalente molar) é adicionado. Carbonato de sódio (3 equivalentes molar) em 0,8 mL) de água é adicionado para a mistura de reação e a solução resultante é aquecida para 85°C por 12 horas. Adiciona-se água (50 mL) para a mistura de reação arrefecendo a reação. EtOAc (100 mL x 2) é a seguir adicionado para extrair a solução aquosa. Secar a camada de EtOAc sobre Na2SO4. O Na2SO4 é filtrado e evaporado dando um resíduo oleoso castanho escuro. O resíduo é purificado por cromatografia flash (eluindo com CH3OH, CH2Cl2, EtOAc em hexanos) conferindo o produto desejado, composto 16.
[00431] Ácido (16 equivalentes molar ou menos) é adicionado ao composto 16 (0,114 mmol) a temperatura ambiente. A solução resultante e agitada a temperatura ambiente ou aquecida para 60°C por 12 horas. A mistura de reação é evaporada e o resíduo é purificado por cromatografia em gel de sílica (eluindo com CH3OH, EtOAc e CH2Cl2) dando o produto amida desejado, composto 17, com um sólido amarelado a branco. Procedimento Geral 58
[00432] Ácido 1-(terc-butoxicarbonil)azetidino-3-carboxílico (1- 1)(AXL016917, 1000 mg, 4,97 mmols) foi dissolvido em MeOH (5 mL)/tolueno (20 mL) e a seguir resfriada para 0°C. TMSCHNN (trimetilsilildiazometano) (7,45 mmols) foi então adicionado gota a gota durante 15 minutos, observando-se algum borbulhamento. A cor começou a clarear, tornando-se lentamente, amarela. A solução foi agitada por 10 minutos a 0°C a segui aquecida para temperatura ambiente durante 30 minutos. A solução foi então concentrada e bombeada para remover o tolueno rendendo 1,055 g de 3-metil- azetidino-1,3-dicarboxilato de terc-butila (1-2) que foi usado diretamente na próxima etapa sem ser purificado (99% rendimento bruto).
[00433] 3-metil azetidino-1,3-dicarboxilato de 1-terc-butila (1055 mg, 4,90 mmols) foi dissolvido em THF (17 mL) e a seguir resfriado para 0°C. MeOH (0,397 mL, 9,80 mmols) e LIBH4 (14,7 mmol) foram adicionados em seqüência. A reação foi aquecida para temperatura ambiente durante 3 horas. A seguir tartarato de sódio-potássio aquoso tetraidratado a 10% (sal de Rochelle) (30 mL) e EtOAc (30 mL) foram adicionados e a solução foi agitada a temperatura ambiente por 30 minutos. A camada orgânica foi separada e a seguir seca (Na2SO4) e concentrada rendendo 674 mg de 3-(hidroximetil)azetidina-1- carboxilato de terc-butila (1-3) como um produto bruto (óleo límpido). O produto foi usado diretamente na próxima etapa sem purificação.
[00434] 3-hidroximetil)azetidino-1-carboxilato de terc-butila (674 mg, 3,60 mmols) foi dissolvido em CH2Cl2 (13 mL, 0,25 M) e a seguir Et3N (1,0 mL, 7,20 mmols) DMAP (44 mg, 0,360 mmol) e cloreto de metanossulfonila (0,31 mL, 3,96 mols) foram adicionados em sequência a 0°C com adição de MsCl feita lentamente. A solução foi aquecida par temperatura ambiente durante uma hora. Após 15 horas, NaHCO3 aquoso, sat. (50 mL) foi adicionado e a seguir o produto foi extraído com CH2Cl2 (2 x 50 mL) sendo os extratos orgânicos combinados lavados com salmoura (50 mL), secos Na2SO4, concentrados e purificados por cromatografia flash (Biotage Horizon - 10% EtOAc/hexanos - 100 EtOAc) dando 962 mg, de 1-4 como um óleo (quantitativo).
[00435] NaH (95%, 96 mg, 3,99 mmols) foi combinado em DMF (10 mL) sob nitrogênio a temperatura ambiente. 4-bromopirazol (533 mg, 3,63 mmols) foi a seguir adicionado e a mistura agitada a temperatura ambiente. Após 30 minutos, 1-4, foi adicionado e a solução aquecida para 95°C. Após duas horas, solução aquosa saturada de NH4Cl (50 mL) foi adicionada e EtOAc (50 mL). O extrato orgânico foi seco (Na2SO4) e concentrado e passado por um chumaço curto de gel de sílica com 50% de (EtOAc/hexanos) rendendo 846 mg do 1-5 bruto que foi usado diretamente na próxima etapa (74% de rendimento bruto).
[00436] 1-5 (846 mg, 2,68 mmol), 1-6 (815 mg, 3,21 mmol) [1,1'bis-(difenilfosfino)-ferroceno)dicloro-paládio (108 mg, 0,133 mmol) e KOAc (893 mg, 9,10 mmol) foram combinados em DMSO (10 mL) sob nitrogênio por 10 minutos e a seguir a solução foi aquecida para 80°C. Após 16 horas, solução foi filtrada por Celite em seguida H2O (50 mL), adicionando-se EtOAc (50 mL).
[00437] A fase orgânica foi extraída e seca ( Na2SO4), concentrada e passado por um tampão de sílica com 50% de (EtOAc/hexanos). O solvente foi concentrado rendendo 1,22 g do 1-7 bruto usado diretamente na próxima etapa.
[00438] O éster borônico 1-7 (4144 mg, 11,4 mmols), 1-8 (2890 mg, 7,60 mmols) diclorobis(trifenilfosfina)paládio(II) (534 mg, 0,760 mmol), DME (40 mL, desgaseificado por 30 minutos com nitrogênio) e Na2CO3 (40 mL, desgaseificado por 30 minutos com nitrogênio) foram combinados e aquecidos para 80°C. Após 16 horas a reação foi resfriada para temperatura ambiente e EtOAc (80 mL) foi adicionado. A solução foi filtrada através de Celite e a seguir adicionou-se água (80 mL). A camada orgânica foi separada e seca (Na2SO4) e concentrada. O produto foi purificado por cromatografia flash rendendo 1486 mg de 1-9 como um sólido castanho (36%).
[00439] 1 g de resina de troca de íon DOWEX 50WX2-400 foi preparada por lavagem com H2O (500 mL), H2O/MeOH 1:1, MeOH (5 X 250 mL), CH2Cl2 (500 mL) e hexanos (500 mL). O DOWEX foi em seguida seco num forno de vácuo a 40°C por 1 dia. 1-9 foi dissolvido em MeOH e em seguida DOWEX (588 mg, 1,096 mmol) foi adicionado. A solução foi agitada a temperatura ambiente por duas horas. A solução foi filtrada e a resina lavada com MeOH (3X 200 mL) sendo descartada a água de lavagem. A resina foi lavada com NH3MeOH 3,5 M e coletada. A solução foi concentrada rendendo 374 mg de 1-10 com um sólido resinoso (78%).
[00440] Para formar os compostos de fórmula 1-11 foi seguido o procedimento exemplar: 1 equivalente molar de 1-10 dissolvido em DMF ou CH2Cl2 e a seguir base (3 equivalentes molar) e/ou reagente de acoplamento (1,5 equivalentes molar) foram adicionados. À solução adiciona-se X-R (1,1 equivalente molar), onde X é, por exemplo, Cl, Br, I, Oms, COCl, CO, COOH, etileno u carbonato e R é um grupo desejado tal como os demonstrados nos exemplos presentes ou grupos similares. A solução resultante é agitada a temperatura ambiente por 4 horas. H2O e EtOAc são adicionados e a fase orgânica extraída, seca (Na2SO4) e concentrada. O produto bruto pode ser purificado por HPLC preparativa ou outros métodos do conhecimento da técnica rendendo o produto 1-11. Procedimento Geral 59
[00441] 3-azetidinol 2-2: Uma mistura de reação de sal de HCL de N-benzidrilazetidin-3-ol (2,76 g, 10,0 mmols) com hidróxido de paládio, 20% Pd (base seca) em C (400 mg) em 50 mL de MeoH foi hidrogenada a 55 psi por 48 horas. A mistura de reação foi filtrada por chumaço de Celite e lavada bem com MeOH. O filtrado foi concentrado sob vácuo a temperatura ambiente em banho de água. O resíduo foi tratado com éter (3 x 30 mL) e o solvente decantado. O sólido foi seco ao ar dando 571 mg do produto sal de HCl 2-2 como um sólido branco (52% rendimento).
[00442] 1H RMN (400 MHz, DMSO-Dθ) δ ppm 3,33 (s, 1H) 3,63 2,80 (m, 2H) 3,93-4,09 (m, 2H) 4,40-4,58(m, 1H) 6,18 (d, J = 6,32 Hz, 1H).
[00443] Éster terc-butílico do ácido 3-hidróxi-azetidino-1- carboxílico 3-3: A uma solução agitada fria (banho a 0°C) do composto 2-2 570 mg, 5,20 mmols) em 10 mL de EtOH adicionou-se EtβN (1,8 mL, 13,0 mmols) e di-terc-butildicarbonato (1,702 g, 7,38 mmols). A mistura resultante da solução límpida foi agitada a temperatura ambiente durante a noite. A mistura de reação foi concentrada em vácuo. O resíduo foi dividido entre EtOAc (200 mL) e solução de ácido cítrico 0,5N (30 mL; salmoura (30 mL). A camada orgânica foi seca (Na2SO4) então concentrada em vácuo dando 899 mg 2-3 como um óleo límpido (52%).
[00444] 1H RMN (400 MHz, clorofórmio-D) δ ppm 1,42 (s, 9H) 3,78 (dd, J = 9,47, 4,42 Hz, 2H) 4,13 (dd, J = 9,35, 6,57 Hz, 2H) 4,49-4,63 (m, 1H).
[00445] Éster terc-butílico do ácido 3-metanossulfonilóxi-azetidino- 1-carboxílico 2-4: A uma solução do composto 2-3 (466 mg, 2,69 mmols) com Et3N (0,75 mL, 5,38 mmols) e 4-(dimetilamino)-piridina (33 mg, 0,269 mmol) em 10 mL de CH2Cl2 a 0°C adicionou-se cloreto de metanossulfonila (0,25 mL 3,23 mmols). A mistura resultante da solução de cor marrom foi agitada a 0°C a temperatura ambiente durante a noite. A mistura de reação foi arrefecida com NaHCO3 a seguir dividida entre CH2Cl2 (200 mL) e solução saturada de NaHCO3 (50 mL). A camada orgânica foi seca Na2SO4, a seguir filtrada por coluna de gel de sílica eluindo com (EtOAc/hexanos) 1:1 sendo o filtrado concentrado em vácuo dando 614 mg 2-4 como um óleo amarelo (91% rendimento).
[00446] 1H RMN (400 MHz, clorofórmio-D) δ ppm 1,43 (s, 9H) 3,05 (s, 3H) 4,05 (dd, J = 10,36, 4,29 Hz, 2H) 4,26 (dd, J = 10,36, 6,82 Hz, 2H) 5,11-5,26 (m, 1H).
[00447] Éster 4-bromopirazol terc-butílico do ácido 1-(3-azetidino- 1-carboxílico 2-6: Um tubo de microondas de 5 mL foi carregado com o composto 2-4 (304 mg, 1,21 mmol); 4-bromopirazol 2-5 178 mg, 1,21 mol) e NaH 60% em óleo mineral (73 mg, 1,82 mmol), com 2 m de DMF. A mistura resultante foi passada em microondas a 110°C por 30 min. A mistura de reação foi dividida entre EtOAc 200 m) e solução de NaHCO3 sat. (2 x 50 mL); salmoura (50 mL). A camada orgânica foi seca Na2SO4, a seguir concentrada em vácuo dando 360 mg de 2-6 como um óleo amarelo (98%).
[00448] 1H RMN (400 MHz, DMSO-D6) δ ppm 1,36-1,43 (m, 9H) 4,08 (s, 2H) 4,18-4,31 (m, 2H) 5,12-5,22 (m, 1H) 7,67 (s, 1H) 8,14 (s, 1H).
[00449] 3-[4-(4,4,5,5-tetrametil-1,3-dioxoborolan-2-il) -1H-pirazol-1- il]azetidino-1-carboxilato de terc-butila 2-8: Uma mistura de reação do composto 2-6 (225 mg, 0,74 mmol) e bis(pinacolato)diboro (2-7, 227 mg, 0,89 mmol) com KOAc (247 mg, 2,52 mmol) em 3 mL de DMSO foi purgada com N2 por 16 minutos, então PdCL2(dppf)2CH2Cl2 (30 mg, 2,52 mmols) foi adicionado. A mistura resultante foi agitada a 80°C sob nitrogênio durante a noite. Após ser resfriada para temperatura ambiente, a mistura foi filtrada por Celite e lavada bem com EtOAc. O filtrado foi extraído com água (2 x 50 mL), salmoura (50 mL). A camada orgânica foi seca Na2SO4, então concentrada em vácuo. O resíduo foi filtrado por gel de sílica eluído com (hexano:EtOAc 3:2. O filtrado foi concentrado em vácuo dando 250 mg, de 2-8 como um óleo límpido (97% rendimento).
[00450] 1H RMN (400 MHz, clorofórmio-D) δ ppm 1,18-1,27(m, 9H) 1,28 -1,34 (m, 6H) 1,41-1,49 (m, 6H) 4,22-4,33 (m, 2H) 4,36 (t, J = 8,59 Hz, 2H) 4,98-5,13 (m, 1H) 7,83 (s, 2H).
[00451] 3-{4-{6-amino-5[1-(2,6-dicloro-3-fluorfenil)etóxi] piridin-3-il}- 1H-pirazol-1-)azetidina-1-carboxilato de terc-butila 2-10: Uma mistura de reação do composto 2-8 (458 mg, 1,31 mmoL) e 3-[1-(2,6-dicloro-3- fluorfenil)etóxi]5-iodopiridin-2-amina 2-9 (374 mg, 0,88 mmol) em 13 mL de éter dimetílico de etileno glicol, DME anidro foi purgado como nitrogênio por 15 minutos, então Pd(II)(PPh3)2Cl2 foi adicionado e continuou-se a purga com nitrogênio por mais 15 minutos. Mais solução de Na2CO3 1,0 N (3,9 mL, 3,9 mmols) foi adicionado após purgar com nitrogênio durante 15 minutos. A mistura resultante foi agitada a 85°C sob nitrogênio durante a noite. A mistura de reação foi filtrada por Celite e lavada bem com MeOH. O filtrado foi concentrado em vácuo. O resíduo foi dividido entre EtOAc (200 mL) e NaHCO3 sat. (2 x 50 mL), salmoura (50 mL). A camada orgânica foi seca Na2SO4 então concentrada em vácuo. O resíduo foi purificado por sistema Biotage (25 M, 100% CH2Cl2 a 90% CH2Cl2 com 10 MeoH) a fim de coletar a fração para dar 421 mg, de 2-10 como uma graxa marrom (92% rendimento).
[00452] 1H RMN (400 MHz, clorofórmio-D) δ ppm 1,17-1,26 (m, 9H) 1,80-1,87 (m, 3H) 4,04-4,18 (m, 2H) 4,20-4,33 (m, 2H) 4,34- 4,41 (m, 1H) 4,79 (s, 2H) 5,02 (d, J = 7,58 Hz, 1H) 7,04 (1, J = 8,46 Hz, 1H) 7,33-7,41 (m, 1H) 7,44-7,52 (m, 1H) 7,53-7,58 (m, 1H) 7,59-7,65 (m, 1H) 7,72-7,78 (m, 1H); LCMS calculado para C24H26Cl2FN5O3 (M+H) 523, encontrado 523.
[00453] 5-(1-azetidin-3-il-1H-pirazon-4-il)-3-[1-(2,6-dicloro-3-fluorfenil) etóxi]piridin-2-amina 2-11: Uma mistura de reação do composto 2-10 (421 mg, 0,81 mmol) com HCl 4,0 M em dioxano (2,0 mL, 8,1 mmols), em 5 mL de CH2Cl2 foi agitada a temperatura ambiente por 2,0 horas. A mistura de reação foi concentrada em vácuo. O resíduo foi tratado com EtOAc. O sólido precipitado foi filtrado e lavado bem com EtOAc, hexano seguir seco sob vácuo, dando 27 5mg de 2-11 como um sólido de cor areia de sal de HCl (81% rendimento.
[00454] 1H RMN (400 MHz, DMSO-D6) δ ppm 1,79-1,89 (m, 3H) 3,56 (s, 1H) 4,35 (s, 4H) 5,40 (s, 1H) 6,23 (d, J = 6,57 Hz, 2H) 7,09 (s, 1H) 7,40-7,54 (m, 1H) 7,59 (dd, J = 8,84, 5,05 Hz, 1H) 7,73-7,83 (m, 1H) 7,86 {s, 1H) 8,12 (s, 1H) 9,20 (s, 1H). LCMS calculado para C19H18Cl2FN5O(M+H) 423, encontrado 423.
[00455] Os compostos de fórmula 2-12 podem ser preparados pelo seguinte procedimento exemplar: A uma mistura de reação do composto 2-11 (1,0 eq), com Et3N (2,0 eq.) em 2,0 mL de DMF a temperatura ambiente adicionou-se brometo de alquila (1,1 eq). A mistura resultante e agitada sob nitrogênio a temperatura ambiente durante a noite. A mistura de reação é dividida entre EtOAc (200 mL) e solução de NaHCO3 sat. (2 x 50 mL); salmoura (50 mL). A camada orgânica é seca Na2SO4, então concentrada em vácuo. O resíduo é purificado por sistema Dionex (5% a 95% de MeOH:H2O /tampão de HOAc 0,1%) coletando a fração desejada para produzir 2-12.
[00456] Alternativamente, os compostos de fórmula 2-12 podem ser preparados pelo seguinte procedimento exemplar: A uma solução de reação de alquil amina (1,0 eq), com iPr2EtN (diisopropiletilamina) (3,0 eq) em 2,0 mL de dimetilformamida adicionou-se HATU (1,5 eq). Após agitação por 3 minutos, o composto 2-11 (1,0 eq) é adicionado. A mistura resultante é agitada a temperatura ambiente durante a noite. A mistura de reação é dividida entre EtOAc (200 mL) e solução de NaHCO3 saturada (2 x 50 ml e salmoura (50 mL). A camada orgânica é seca Na2SO4 e concentrada em vácuo. O resíduo é purificado pelo Sistema Dionex (5% a 95% de MeCN:H2 w 0,1% HOAc) coletando o produto desejado para produzir 2-12. Procedimento Geral 60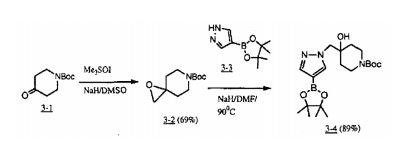

[00457] 1-oxa-6-azaspiro[2.5]octano-6-carboxilato de terc-butila 3-2: Uma solução de dimetilsulfoxonio metilida foi preparada sob nitrogênio de dispersão de NaH a 60% em óleo mineral (440 mg, 11,0 mmols) e iodeto de trimetilsulfônio (2,421 g, 11,0 mmols) em 5 mL de DMSO anidro. Mais solução de 1-Boc-4-oxo-1-piperidinocarboxilato (3-1, 1,993 g 10,0 mmols) em 5 mL de DMSO foi adicionado gota a gota. A mistura resultante foi agitada a 55°C por 6 horas. A mistura de reação resfriada foi despejada em água gelada e extraída com EtOAc (2 x 200 mL). As camadas orgânicas combinadas foram lavadas com água (50 mL), salmoura (5 mL) e a seguir secas (Na2SO4), a seguir concentradas em vácuo dando 1,4791 g de 3-2 como um óleo amarelo (69% rendimento).
[00458] 1H RMN (400 MHz, clorofórmio-D) δ ppm 1,37-1,52 (m, 11H) 1,71-1,84 (m, 2H) 2,63-2,72 (m, 2H) 3,35-3,49 (m, 2H) 3,62-3,78 (m, 2H).
[00459] 4-hidróxi-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-1H- pirazol-1-il]metil]piperidino-1-carboxilato de terc-butila 3-4: Uma mistura de reação do composto 3-2 (214 mg: 1,0 mmol) e 4-(4,4,5,5-tetrametil- [1,3,2] dioxaborolan-2-il)-1H-pirazol (3-3 194 mg: 1,0 mmol) com dispersão de NaH a 60% em óleo mineral (60 mg, 1,5 mmol) em 3 mL de DMF foi agitada a 90°C por 3 horas. A mistura de reação foi dividida entre EtOAc (200 ml) e NaHCO3 sat. (50 mL) e salmoura (50 mL). A camada orgânica foi seca Na2SO4 e concentrada em vácuo dando 361 mg de 3-4 como uma graxa amarela (89% rendimento).
[00460] 1H RMN (400 MHz, clorofórmio-D) δ ppm 1,21-1,34 (m, 12H) 1,39-150 (m, 9H) 1,56-1,78 (m, 4H) 3,14 (s, 2H) 3,72-3,91 (m, J = 32,34 Hz, 2H) 4,05 (s, 2H) 7,65 (s, 1H) 7,80 (s, 1H) 8,00 (s, 1H). LCMS calculado para C20H34BN3O5 (M+H) 408, encontrado 408. HPLC 85% de pureza.
[00461] 4-[(4-{6-amino-5-[1-(2,6-dicloro-3-fluorfenil) etóxi]piridin-3-il}- 1H-pirazol-1-il)metil]-4-hidroxipiperidino-1-carboxilato de terc-butila 3-6: Uma mistura de reação do composto 3-4 (361 mg, 0,89 mmol) e 3-[1- (2,6-dicloro-3-fluorfenil]etóxi]-5-iodopiridin-2-amina 3-5 (378 mg, 0,89 mmol) em 9,09 mL de éter dimetílico de etileno glicol, DME anidro foi purgada com nitrogênio por 15 minutos, então Pd(II)(PPh3)2Cl2 (32 mg, 0,05 mmol) foi adicionado e continuou-se a purga com nitrogênio por mais 15 minutos. Mais solução de Na2CO3 1,0 N (3,9 mL, 3,9 mmols) foi adicionado após purgar com nitrogênio durante 15 minutos. A mistura resultante foi agitada a 85°C sob nitrogênio durante a noite. A mistura de reação foi filtrada por Celite e lavada bem com MeOH. O filtrado foi concentrado em vácuo. O resíduo foi dividido entre EtOAc (200 mL) e NaHCO3 sat. (2 x 50 mL), salmoura (50 mL). A camada orgânica foi seca Na2SO4 então concentrada em vácuo. O resíduo foi purificado por sistema Dionex (tampão 25 % a 95% MeCN:H2O w 0,1% HOAc) a fim de coletar a fração para dar 147 mg, de 3-6 como um sólido branco (28% rendimento).
[00462] 1H RMN (400 MHz, DMSO-D6) δ ppm 1,34-1,39 (m, 9H) 1,70-1,77 (m, 2H) 1,79 (d, J = 6,57 Hz, 3H) 3,06 (d, J = 12,63 Hz, 2H) 3,62 (s, 2H) 4,03 (s, 2H) 4,79 (s, 1H) 5,66 (s, 2H) 6,08 (d, J = 6,82 Hz, 1H) 6,86 (d, J = 1,52 Hz, 1H) 7,44 (t, J = 8,72 Hz, 1H) 7,51-7,58 (m, 2H) 7,58-7,65 (m, 2H) 7,73 (d, J = 1,52 Hz, 1H) 7,78 (s, 1H). LCMS calculado para C27H32Cl2FN5O4 (M+H) 581, encontrado 581. HPLC 87% de pureza.
[00463] 4-[(4-{6-amino-5-[1-(2,6-dicloro-3-fluorfenil) etóxi]piridin-3-il}- 1H-pirazol-1-il)metil]piperidin-4-ol 3-7: (145 mg; 0,25 mmol) com 4,0 M de HCl em dioxano (2,0 mL, 8,1 mmoL) em 5 mL de CH2Cl2 foi agitada a temperatura ambiente por 2,0 horas. A mistura de reação foi concentrada em vácuo o resíduo foi purificado por sistema Dionex (tampão 5% a 95% MeCN:H2O w 0,1% HOAc) a fim de coletar a fração para dar 76 mg, de 3-7 como uma graxa amarela (63% rendimento).
[00464] 1H RMN (400 MHz, DMSO-D6) δ ppm 1,41-1,55 (m, 2H) 1,59-1,71 (m, 2H) 1,81 (d, J = 6,57 Hz, 3H) 2,88-3,00(m, 2H) 3,02-3,14 (m, 2H) 4,08 (s, 2H) 5,17 (s, 2H) 6,14-6,27 (m, J = 6,57 Hz, 1H) 7,05 (s, 1H) 7,40-7,49 (m, J = 8,72, 8,72 Hz, 1H) 7,51- 7,60 (m, J = 9,09, 4,80 Hz, 1H) 7,63 (s, 1H) 7,76 (s, 1H) 7,91 (s, 1H) 8,51 (s, 1H) 8,81 (s, 1H). LCMS calculado para C22H24Cl2FN502 (M+H) 481, encontrado 481. HPLC 98% de pureza. Analisado (C22H24Cl2FN5O2 x 2,2H0Ac x 2,3H2O) C, H, N. Procedimento Geral 61:
[00465] 2-[(4-bromo-1-pirazol-1-il)metil] ciclopropano-carboxilato de etila 4-3):
[00466] A uma solução de reação de 2-(hidroximetil) ciclopropanocarboxilato de etila 4-1 (577 mg, 4,0 mmols) com EtβN (1,1 mL, 8,0 mmols) e DMAP (49 mg, 0,4 mmol) em 12 mL de CH2Cl2 a 0°C adicionou-se cloreto de metanossulfonila (0,4 mL; 4,8 mmols). A mistura resultante da suspensão de cor marrom foi agitada a 0°C a temperatura ambiente sob nitrogênio durante a note. A mistura de reação foi arrefecida com NaHCO3 então dividia entre CH2Cl2 (200 mL) e solução de NaHCO3 sat. (50 mL); salmoura (50 mL). A camada orgânica foi seca Na2SO4, então filtrada por chumaço de gel de sílica eluído com hexano:EtOAc 1:1. O filtrado foi concentrado em vácuo dando 880 mg de 2-{[(metilsulfonil) óxi]metil}ciclopropanocarboxilato de etila como um óleo amarelo (99% rendimento).
[00467] 1H RMN (400 MHz, clorofórmio-D) δ ppm 0,91-1,02 (m, 1H) 1,26 (q, J = 6,99 Hz, 3H) 1,29-1,36 (m, 1H) 1,63-1,74 (m, 1H) 1,791,92 (m, 1H) 3,02 (s, 3H) 3,99-4,24 (m, 4H).
[00468] Formou-se uma mistura de reação de 2- {[(metilsulfonil)óxi]metil}ciclopropanocarboxilato de etila (880 mg, 4,0 mmols), 4-bromopirazol (4-2, 588 mg, 4,0 mmols) e NaH 60% em óleo mineral (240 mg, 6,0 mmols) com 3,0 mL de DMF. A mistura resultante foi agitada a 90°C sob nitrogênio por quatro horas. A mistura de reação foi dividida entre EtOAc (200 mL) e NaHCO3 sat. (2 x 50 mL); salmoura (50 mL). A camada orgânica foi seca (Na2SO4), então concentrada em vácuo rendendo 812 mg de 4-3 como um óleo amarelo (74%).
[00469] 1H RMN (400 MHz, clorofórmio-D) δ ppm 0,85 (dd, J = 7,96, 3,16 Hz, 1H) 0,88-0,98 (m, 1H) 1,18-1,29 (m, 3H) 1,56-1,71 (m, 1H) 1,79-1,94 (m, 1H) 3,96-4,08 (m, 2H) 4,07-4,17 (m, 2H) 7,45 (d, J = 3,79 Hz, 2H). LCMS calculado para C10H13BrN2O2 (M+H) 274, encontrado 274. HPLC 95% de pureza.
[00470] 2-{[4-(4,4,5,5-tetrametil-1,3-dioxoborolan-2-il)-1H-pirazol-1- il]metil}ciclopropanocarboxilato de etila 4-4: Uma mistura de reação do composto 4-3 (812 mg, 2,97 mmols) e bis(pinacolato)diboro (906 mg, 3,57 mmols) com KOAc (991 mg, 10,10 mmols) em 10 mL de DMSO foi purgada com N2 por 15 minutos, então PdCL2(dppf)2CH2Cl2 (122 mg, 0,15 mmol) foi adicionado. A mistura resultante foi agitada a 80°C sob nitrogênio durante a noite. Após ser resfriada para temperatura ambiente, a mistura foi filtrada por Celite e lavada bem com EtOAc. O filtrado foi extraído com água (2 x 50 mL), salmoura (50 mL). A camada orgânica foi seca Na2SO4, então concentrada em vácuo. O resíduo foi filtrado por gel de sílica eluído com (hexano:EtOAc 3:1. O filtrado foi concentrado em vácuo dando 945 mg, de 4-4 como um óleo amarelo (98% rendimento).
[00471] 1H RMN (400 MHz, clorofórmio-D) δ ppm 0,85 (dd, J = 7,83, 3,03 Hz, 1H) 0,90-096 (m, 1H) 1,20-1,24 (m, 3H) 1,29-1,34 (m, 12H) 1,62-1,71 (m, 1H) 1,84-1,97 (m, 1H) 3,96-4,07 (m, 1H) 4,06-4,14 (m, 2H) 4,15-4,23 (m, J = 14,27, 6,44 Hz, 1H) 7,73 (s, 1 H) 7,77 (s, 1H).
[00472] 2-[(4-(6-amino-5-[1-(2,6-dicloro-3-fluorfenil) etóxi]piridin-3- il}-1H-pirazol-1-il)metil] ciclopropanocarboxilato de etila 4-6: Uma mistura de reação do composto 4-4 (643 mg, 2,01 mmols) e 3-[1-(2,6- dicloro-3-fluorfenil)etóxi]-5-iodopiridin-2-amina (4-5) (572 mg; 1,34 mmol em 20,0 mL de éter dimetílico de etileno glicol, DME anidro foi purgada com nitrogênio por 15 minutos, a seguir, Pd(II)(PPh3)2Cl2 (71 mg, 0,1 mmol) foi adicionado e continuou-se a purga com nitrogênio por mais 15 minutos. Mais 1,0 N de solução de Na2CO3 (6,0 mL, 6,0 mmols) foi adicionado após purgar com nitrogênio por 15 minutos. A mistura resultante foi agitada a 85°C sob nitrogênio durante a noite. A mistura foi filtrada por Celite e lavada bem com MeOH. O filtrado foi concentrado em vácuo. O resíduo foi dividido entre EtOAc (200 mL) e solução de NaHCO3 saturada (2 x 50 mL), salmoura (50 mL). A camada orgânica foi seca (Na2SO4), então concentrada in vácuo. O resíduo foi purificado por sistema Biotage (CH2Cl2 25 M 100%; CH2Cl2 100% a 90% CH2Cl2:10% MeOH) coletando a fraca desejada rendendo 600 mg de 4-6 como uma graxa de cor marrom (91% rendimento).
[00473] 1H RMN (400 MHz, DMSO-D6) δ ppm 0,96-1,10 (m, 2H) 1,15 (t, J = 7,07 Hz, 2H) 1,74 (s, 3H) 1,79 (d, J = 6,57 Hz, 3H) 3,954,14 (m, 4H) 5,66 (s, 2H) 6,08 (d, J = 6,57 Hz, 1H) 6,88 (s, 1H) 7,43 (t, J = 8,72 Hz, 1H) 7,49-7,62 (m, 2H) 7,73 (s, 1H) 7,88 (s, 1H). LCMS calculado para C23H23Cl2FN4O3(M+H) 494, encontrado 494. HPLC 95% de pureza.
[00474] ácido 2-[(4-(6-amino-5-[1-(2,6-dicloro-3- fluorfenil)etóxi]piridin-3-il}-1H-pirazol-1-il) metil] ciclopropanocarboxilico 4-7: A uma solução de reação do composto 4-6 (377 mg, 0,76 mmol) em 5,0 mL de MeOH a temperatura ambiente sob nitrogênio adicionou-se uma outra solução de NaOH 2,0 N (2) (1,5 mL, 3,04 mmols). A mistura resultante foi agitada a 80°C por 3 horas. A mistura de reação foi concentrada em vácuo removendo a maioria do MeOH e acidulada com HCl 2M a pH 4.0. A mistura foi extraída com CH2Cl2 (2 x 200 mL). As camadas orgânicas foram lavadas com salmoura (50 mL) e secas (Na2SO4) concentradas em vácuo dando 324 mg de 4-7 como um sólido amarelo (92% rendimento).
[00475] 1H RMN (400 MHz, DMSO-D6) δ ppm 0,92-1,04 (m, 2H) 1,57-1,72 (m, 2H) 1,76-1,90 (m, 3H) 3,98-4,18 (m, 2H) 6,46 (s, 2H) 6,89-7,02 (m, 1H) 7,29-7,52 (m, 2H) 7,52-7,63 (m, 2H) 7,73 (d, J = 1,52 Hz, 1H) 7,94 (s, 1H) 12,19 (s, 1H). LCMS calculado para C21H19Cl2FN4O3 (M-H) 463, encontrado 463. HPLC 87% de pureza.
[00476] 2-[(4-(6-amino-5-[1-(2,6-dicloro-3-fluorfenil) etóxi]piridin-3- il}-1H-pirazol-1-il) metil] ciclopropano-carboxamida 4-8: (R = Me, R'= H): A uma solução de reação de 4-7 (1,0 eq). com iPr2EtN (2,0 eq) em 1,0 mL de DMF adicionou-se HATU (1,5 eq). Após agitação por 30 minutos adicionou-se alquilamina (1,1 eq). A mistura resultante foi agitada a temperatura ambiente durante a note. A mire foi dividida entre EtOAc (200 mL) e NaHCO3 sat. (2 x 50 mL) e salmoura (50 mL). A camada orgânica foi seca (Na2SO4) e concentrada em vácuo. A amostra era de base livre dividindo-se entre EtOAc (200 mL) e solução de NaHCO3 sat. (50 mL) e salmoura (50 mL). A camada orgânica foi seca (Na2SO4) e concentrada em vácuo. O resíduo foi tratado com 1,0 mL de H2O e liofilizada rendendo (4-8). Procedimento Geral 62
[00477] A uma solução de 5-bromo-3-[®-1-(2,6-dicloro-3-flúor- fenil)-etóxi]piridin-2-ilamina (12,83 g, 33,76 mmols) em DMF anidro (100 mL) adicionou-se dicarbonato de di-terc-butila (21,25 g, 97,35 mmols) e 4-dimetilaminopiridina (0,793g, 6,49 mmols). A reação foi agitada a temperatura ambiente por 18 horas sob nitrogênio. À mistura adicionou-se NaHCO3 (300 mL) e extraiu-se com EtOAc (3 x 250 mL). Os extratos combinados foram lavados com água (5 x 100 mL) NaHCO3 sat. e salmoura a seguir secou-se sobre Na2SO4. Após filtrar, evaporar e secar por alto vácuo, obteve-se 5-bromo-3-[(R)-1-(2,6- dicloro-3-flúor-fenil)-etóxi]-piridin-2-ilamina di-boc protegido como uma espuma esbranquiçada sólida (19,59 g, 100% rendimento).
[00478] 1H RMN (DMSO-d6, 400 MHz) δ 8,18 (d, 1H), 7,83 (d, 1H), 7,59 (dd, 1H), 7,48 (t, 1H), 6,25 (q, 1H), 1,75 (d, 3H), 1,39 (s, 9H), 1,19 (s, 9H).
[00479] A uma solução do 5-bromo-3-[(R)-1-(2,6-dicloro-3-flúor- fenil)-etóxi]-piridin-2-ilamina di-boc protegido (19,58 g 33,76 mmoL) em DMSO (68 mL) adicionou-se acetato de potássio (11,26 g 114,78 mmols) e bis(pinacolato)diboro (10,29 g, 40,51 mmols) A mistura foi desgaseificada e carregada com nitrogênio três X, então adicionou-se Pd(dppf)Cl2CH2Cl2 (1,38 g, 1,69 mmol). A mistura de reação foi desgaseificada e carregada com nitrogênio três vezes e agitou-se a 80°C no banho de óleo sob nitrogênio por 12 horas. A reação foi resfriada para temperatura ambiente, diluída com acetato de etila (100 mL) e filtrada por um chumaço de celite que foi lavado com acetato de etila. A solução de acetato de etila combinada (700 mL) foi lavada com água (5 x 100 mL), salmoura (100 mL) e seca sobre Na2SO4. Após filtração e concentração, o resíduo foi purificado numa coluna de gel de sílica eluindo com (EtOAc/hexanos) (0%-50%) dando 3-[(R)-1-(2,6- dicloro-3-flúor -fenil)-etóxi]-5-(4,4,5,5-tetrametil-]1,3,2] dioxaborolan-2- il)-piridin-2-ilamina di-boc protegido como uma espuma sólida (20,59 g 97% rendimento).
[00480] 1H RMN (DMSO-d6, 400 MHz) δ 8,20 (d, 1H), 7,70 (d, 1H), 7,63 (dd, 1H), 7,47 (t, 1H), 6,20 (q, 1H), 1,73 (d, 3H), 1,50-1,13 (m, 30H).
[00481] A uma solução do 3-[(R)-1-(2,6-dicloro-3-flúor-fenil)-etóxi]- 5-(4,4,5,5-tetrametil-]1,3,2]dioxaborolan- 2 -il) -piridin-2-ilamina di-boc protegido (20,34 g 32,42 mmol) em CH2Cl2 (80 mL) adicionou-se uma solução de HCl seco em dioxano (4N, 40,5 mL, 162 mmoL) A solução de reação foi filtrada a 40°C em banho de óleo sob nitrogênio por 12 horas. A mistura de reação foi resfriada para temperatura ambiente, diluída com EtOAc (400 mL), então lavada cuidadosamente porém rapidamente, com NaHCO3 sat. até a camada aquosa tornar-se básica (pH>8). A camada orgânica foi lavada com salmoura, e seca sobre Na2SO4. Após filtração, evaporação, e secagem a alto vácuo, 3-[(R)-1- (2,6-dicloro-3-flúor-fenil)-etóxi]-5-(4,4,5,5-tetrametil-[1,3,2] dioxaborolan-2- il)-piridin-2-ilamina foi obtido como uma espuma esbranquiçada sólida (13,48 g, 97% rendimento).
[00482] 1H RMN (DMSO-d6, 400 MHz) δ 8,01 (d, 1H), 7,27 (dd, 1H), 7,17 (d, 1H), 7,03 (t, 1H), 6,12 (q, 1H), 5,08 (bs, 2H), 1,81 (d, 3H), 1,30 (s, 6H), 1,28 (s, 6H).
[00483] A uma solução agitada de éster terc-butílico do ácido 3- [(R)-1-(2,6-dicloro-3-flúor-fenil)-etóxi]-5-(4,4,5,5-tetrametil-]1,3,2]dioxa- borolan-2-il)-piridin-2- ilamina (4,2711 g 10,0 mmols) e éster terc- butílico do ácido 4-(4-bromo-pirazol-1-il)-piperidino-1-carboxílico (ver procedi-mento 11) (3,9628 g 12,0 mmols) em DME (40 mL) foi adicionado para uma solução de Na2CO3 (3,1787 g, 30,0 mmols) em água (10 mL). A solução foi desgaseificada e carregada com nitrogênio três vezes. Para a solução adicionou-se Pd(PPh3)2Cl2 (351 mg, 0,50 mmol) A solução de reação foi desgaseificada e carregada com nitrogênio novamente três vezes. A solução de reação foi agitada a 87°C em banho de óleo por cerca de 16 horas ou até consumição do éster pinacol borano) resfriada para temperatura ambiente e diluída com EtOAc (200 mL). A mistura de reação foi filtrada por celite e lavada com EtOAc. A solução de EtOAc foi lavada com salmoura, seca sobre Na2SO4 e concentrada. O produto bruto foi purificado numa coluna de gel de sílica eluindo com EtOAc/hexanos (0% EtOAc a 100% EtOAc) rendendo éster terc-butílico do ácido 4-(4-[6-amino-5- [(R)-1-(2,6-dicloro-3-flúor-fenil)-etóxi]piridin-3-il]-pirazol-1 - il) - piperidino-1-carboxílico (3,4167 g 65% rendimento ~95% pureza) com uma Rf de 0,15 (50% EtOAc/hexanos) EM m/e 550 (M+1)+.
[00484] A uma solução de éster terc-butílico do ácido 4-(4-[6- amino-5-[(R)-1-(2,6-dicloro-3-flúor-fenil)-etóxi] piridin-3-il]-pirazol-1-il)- piperidino-1-carboxílico (566,7 mg, 1,03 mmol) em metanol (5 ml) ou diclorometano (30 mL adicionou-se HCl/dioxano 4N (15 mL). A solução foi agitada por cerca de uma hora ou até terminar a desproteção. Os solventes foram evaporados e o resíduo dissolvido em metanol e purificado numa HPLC preparativa C-18 de fase reversa eluindo com acetonitrila/água com ácido acético a 0,1% de 5% a 30% com um gradiente linear. Após liofilização acetato de 3-[(R)-1-(2,6-dicloro-3- flúor-fenil)-etóxi]-5-(1-piperidin-4-il-1H-pirazol-4-il}-piridin-2-ilamina foi obtido como um sólido branco (410 mg, 78% rendimento, 100% HPLC pureza, 96,4% ee).
[00485] 1H RMN (DMSO-d6, 400 MHz) δ 7,84 (s, 1H), 7,68 (d, 1H), 7,50 (dd, 1H), 7,46 (s, 1H), 7,37 (t, 1H), 6,83 (d, 1H), 6,02 (q, 1H), 5,57 (bs, 2H), 4,09 (m, 1H), 2,98 (m, 2H), 2,53 (m, 2H), 1,88 (m, 2H), 1,82 (s, 3H), 1,73 (d, 3H), 1,70 (m, 2H). EM m/e 450 (M+1)+. Procedimento Geral 63:
[00486] A uma suspensão de 3-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]- 5-(1-piperidin-4-il-1H-pirazol-4-il)-piridin-2 -ilamina como o cloridrato (procedimento 6) (150 mg, 0,288 mmol) em CH2Cl2 (2 mL) adicionou- se Net3 (0,121 mL, 0,863 mmol) e agitou-se por 30 minutos, a temperatura ambiente. A reação foi resfriada para 0°C. Éster clorocarbonilmetílico do ácido acético foi adicionado e agitado por uma hora a temperatura ambiente. A reação foi monitorada por LC-EM e após completa conversão no produto desejado adicionou-se água (2 ml). A reação foi extraída com EtOAc (4 x 10 mL). seca sobre Na2SO4 e concentrou-se dando rendimento quantitativo de éster 2-[4-(4-[6- amino-5-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]-piridin-3-il]-pirazol-1-il)- piperidin-1-il]-2-oxo-etilico do ácido acético (164 mg, quant.).
[00487] A uma solução do éster 2-[4-(4-[6-amino-5-[1-(2,6-dicloro- 3-flúor-fenil)-etóxi]-piridin-3-il]-pirazol-1-il)-piperidin-1-il]-2-oxo-etilico do ácido acético (164 mg, 0,298 mmol) em MeoH (4 mL), adicionou-se LiOH (7 mg, 0,298 mmol) dissolvido em 1 mL de água. A reação foi agitada por 30 minutos a temperatura ambiente, quando LC-EM demonstrou a completa conversão na 1-[4-(4-[6-amino-5-[1-(2,6- dicloro-3-flúor-fenil)-etóxi]-piridin-3-il]-pirazol-1-il)-piperidin-1-il]-2- hidróxi-etanona. O produto foi purificado numa C-18 preparação de fase reversa HPLC eluindo com acetonitrila/ água com ácido acético a 0,1% de 10% a 40%. Procedimento Geral 64:
[00488] Um frasco de 100 mL dom uma barra de agitação foi seco num forno e resfriado numa atmosfera de nitrogênio seco. O frasco foi equipado com uma tampa para seringa de borracha. O frasco foi imerso num banho de água gelada sob nitrogênio, e 1,6 mL (1,6 mmol) de solução de borano 1,0 M em THF foi introduzido. A seguir ácido 2- (4-[5-amino-6-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]-pirazin-2-il}-pirazol-1 - il)-2-metil-propiônico (procedimento 5) 0,1 g, 0,221 mmol) em THF anidro (1,0 mL) foi introduzido. A mistura de reação foi agitada a temperatura ambiente sob nitrogênio por 5 horas e HCl 6N (1,1 mL) foi adicionado lentamente a seguir H2O (1,1 mL) e MeOH (7,4 mL) foram introduzidos. A mistura de reação foi agitada continuamente durante a noite. A maioria dos solventes foram evaporados in vácuo, sendo usada uma solução NaOH 1N para ajustar o pH para 11. Adicionou-se água, e a solução foi extraída com EtOAc (3 x 30 ml). e seca sobre Na2SO4. Após filtração e concentração, o produto bruto foi purificado com uma HPLC preparação. de fase reversa, eluindo com acetonitrila/água contendo 0,1% de ácido acético de 10% a 60%. Após liofilização das frações puras, obteve-se acetato de 2-(4-[6-amino-5-[1- (2,6-dicloro-3-flúor-fenil)-etóxi]-piridin-3-il}-pirazol-1-il)-2-metil -propan- 1-ol como um sólido branco (21 mg, 22% rendimento). Procedimento Geral 65:
[00489] A uma solução agitada de éster terc-butílico do ácido 4- hidroxi-piperidino-1-carboxílico (7,94g, 39,45 mmols), em clom4 (100 mL), resfriada para 0°C adicionou-se lentamente Net3 (5,54 mL, 39,45 mmols) seguido por cloreto de metano sulfonila (3,06 mL, 39,45 mmols) e DMAP (48 mg, 0,39 mmol). A mistura foi agitada a temperatura ambiente durante a noite. À mistura adicionou-se água (30 ml). Extração com CH2Cl2 (3 x 30 mL) seguido por secagem Na2SO4 e remoção do solvente em vácuo rendeu éster terc-butílico do ácido 4-metanossulfonilóxi-piperidino-1-carboxílico como um sólido branco (11,00 g, >99% rendimento).
[00490] 1H RMN (CDCh, 400 MHz) δ 4,89 (m, 1H), 3,69 (m, 2H), 3,31 (m, 2H), 3,04 (s, 3H), 1,95 (m, 2H), 1,83 (m, 2H), 1,46 (s, 9H).
[00491] A uma solução agitada de 4-bromo-pirazol (10,44 g, 71,03 mmols) em DMF anidro (96 mL), resfriada para 0°C adicionou-se lentamente NaH (60% em óleo mineral) (3,13 g, 78,133 mmols). A solução foi agitada por uma hora a 0°C. Adicionou-se lentamente, éster terc-butílico do ácido 4-metanossulfinilóxi-1-carboxílico (19,82 g, 71,03 mmol) e a reação foi aquecida para 100°C durante a noite até o consumo do pirazol por RMN. A reação foi resfriada para temperatura ambiente adicionando-se água (20 mL) seguido por extração com EtOAc. Os extratos combinados foram lavados com NaCl aquoso saturado (4 x 20 mL) seco com Na2SO4 e concentrados rendendo éster terc-butílico do ácido 4-(4-bromo-pirazol-1-il)-piperidino-1- carboxílico como um óleo laranja. O óleo foi purificado usando cromatografia em gel de sílica eluindo com EtOAc/hexanos 10% a 25% EtOAc/hexanos para dar éster terc-butílico do ácido 4-(4-bromo- pirazol-1-il)piperidino-1-carboxílico como um sólido branco (10,55 g, 45% rendimento com um Rf = 0,4 (25% EtOAc/hexanos usando iodo como manchamento).
[00492] 1H RMN (CDCI3, 400 MHz) δ 7,46 (s, 1H), 7,43 (s, 1H), 4,23 (m, 3H), 2,88 (m, 2H), 2,10 (m, 2H), 1,88 (m, 2H), 1,47 (s, 9H).
[00493] Para uma soIução de éster terc-butíIico do ácido 4-(4- bromo-pirazoI-1-iI)piperidino-1-carboxíIico (500 mg, 1,515 mmoI) em CH2CI2 (3 mL) adicionou-se TFA (3 mL). A reação foi agitada a temperatura ambiente até ser indicado, por LCEM o término da reação. Os soIventes foram removidos in vácuo, e o resíduo foi dissoIvido em MeOH (15 mL). O pH da soIução foi ajustado para 9 com resina hidróxido rendendo 4-(4-bromo-pirazoI-1-iI)-piperidina.
[00494] A uma soIução de 4-(4-bromo-pirazoI-1-iI)-piperidina (375 mg, 1,63 mmol) em DMF (3,26 ml) adicionou-se Net3 (230 μL, 1,63 mmoI) e agitou-se por 5 minutos. Iodeto de metiIa (MeI), (1,63 mL, MeI 1M em DMF, recentemente feito) foi adicionado e a reação agitada durante a noite a temperatura ambiente. Adicionou-se água e a solução foi extraída com EtOAc (4 x 10 mL). A solução orgânica foi lavada com salmoura, seca com Na2SO4, concentrada e seca in vácuo rendendo 4-(4-bromo-pirazol-1-il)-metil-piperidina (251 mg, 63% rendimento). Procedimento Geral 66
[00495] Para uma solução de 3-[(R)-1-(2,6-dicloro-3-flúor-fenil)- etóxi]-5-(1H-pirazol-4-il)-pirazin-2-ilamina (295 mg, 0,80 mmol) em DMF anidro (4 mL) adicionou-se NaH (60% em óleo mineral, 30,7 mg, 0,80 mmol). A mistura foi agitada a temperatura ambiente sob nitrogênio por 0,5 horas e a seguir éster terc-butílico do ácido 4- metanossulfonilóxi-piperidino-1-carboxílico (223,5 mg, 0,80 mmol) foi introduzido. A mistura de reação foi aquecida para 90°C em banho de óleo por 0,5 h. sob nitrogênio e resfriada para temperatura ambiente. Adicionou-se água lentamente à mistura que foi extraída com EtOAc, lavada com salmoura e seca sobre Na2SO4. O produto bruto foi purificado numa coluna de gel de sílica dando éster terc-butílico do ácido 4-(4-[5-amino-6-[(R)-2,6-dicloro-3-flúor-fenil}-etóxi-pirazin - 2 - il) -pirazol-1-il]-piperidino-1-carboxílico como um sólido branco (265 mg, 59% rendimento).
[00496] A uma solução de éster terc-butílico do ácido 4-(4-[5- amino-6-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]pirazin-2 -il]-pirazol-1-il)- piperidino-1-carboxílico (265 mg, 0,48 mmol) em CH2Cl2, adicionou-se HCl/dioxano 4N (4 mL). A solução foi agitada a temperatura ambiente por uma hora. Após evaporação, o resíduo foi dissolvido em metanol e purificado numa HPLC preparativa C-18 de fase reversa eluindo com acetonitrila/água com ácido acético a 0,1% com um gradiente linear de 10% a 40%. Após liofilização, acetato de 3-[(R)-1-(2,6-dicloro-3-flúor- fenil)-etóxi]-5-(1-piperidin-4-il-1H-pirazol-4-il}-piridin-2-ilamina foi obtido como um sólido branco (125 mg, 51% rendimento. Procedimento Geral 67
[00497] Pentafluoreto de O-(7-azabenzotriazol-1-il)-N,N,N',N'- tetrametilurônio de fósforo (HATU) (66 mg, 0,17 mmoL) foi adicionado para uma solução de ácido 2-(4-{6-amino-5-[1-(2,6-dicloro-3-flúor- fenil)-etóxi]-5-(1-piridin-3-il)-pirazol-1-il)-propiônico (69 mg, 0,16 mmol), trietilamina (0,024 mL, 0,17 mmol) e 3-dimetilamino-propilamina (0,022 mL, 0,17 mmol) em 1,6 mL de DMF. Após agitar por 3 horas, a reação foi concentrada por evaporação rotativa. O resíduo foi purificado por cromatografia em gel de sílica usando gradiente de eluição de diclorometano, metanol, hidróxido de amônio produzindo ácido 2-(4- {6-amino-5-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]-piridin-3-il)-pirazol-1-il)-N- (3-dimetilamino-propil)-propionamida (41 mg, 50%). Procedimento Geral 68
[00498] Azodicarboxilato de dietila (0,48 mL, 3,1 mmols) foi adicionado para uma solução a 0°C de trifenilfosfina (0,80 g, 3,1 mmols) em THF (20 mL). Após agitação por 5 minutos, 4-bromo- pirazol (0,30 mg, 2,0 mmols) foi adicionado. Após mais 5 minutos de agitação, adicionou-se éster terc-butílico do ácido (2-hidroxietil-metil- carbâmico) (0,45 g, 2,6 mmols). A reação foi deixada aquecer para temperatura ambiente e agitar durante a noite. A reação foi resfriada para 0°C e filtrada. O filtrado foi concentrado por evaporação rotativa. O resíduo foi purificado por cromatografia em gel de sílica usando gradiente de eluição de diclorometano, acetato de etila rendendo éster terc-butílico do ácido [2-(4-bromo-pirazol-1-il)etil]-metil-carbâmico (541 mg,87%). Procedimento Geral 69
[00499] Hidreto de sódio (0,12 g, 4,9 mmols) foi adicionado para uma solução de 4-bromo-4H-pirazol (0,60 g, 4,1 mmols) em DMF (10 mL). Após agitação por 10 minutos, uma solução de éster terc-butílico do ácido 2-cloropropiônico em DMF (4 mL) foi adicionado. Após agitar por 4 horas, a reação foi dividida entre acetato de etila e água. As fases foram separadas e a fase aquosa foi extraída com acetato de etila. As fases orgânicas combinadas foram secas sobre MgSO4 e concentradas sob evaporação rotativa. O resíduo foi purificado por cromatografia em gel de sílica usando gradiente de eluição de acetato de etila e hexanos rendendo éster terc-butílico do ácido 2-(4-bromo- pirazol-1-il)-propiônico (733 mg, 77%). Procedimento Geral 70:
[00500] Uma solução de LiOH (34 mg, 1,4 mmol) em água (0,4 mL) foi adicionada para uma solução de éster metílico do ácido 2-(4- {6-amino-5-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]-piridin-3-il}-pirazol-1-il)- propiônico (70 mg, 0,15 mmol) numa mistura de THF (1,5 mL) e MeOH (0,4 mL). Após agitação durante a noite, a reação foi dividida entre diclorometano e salmoura meio-saturada. Uma pequena quantidade de etanol foi adicionado e o pH ajustado para 7 com HCl 1M. As fases foram separadas e a fase aquosa foi extraída com diclorometano. As fases orgânicas combinadas forma secas sobre Na2SO4, filtradas e concentradas por evaporação rotativa dando ácido 2-(4-{6-amino-5-[1- (2,6-dicloro-3-flúor-fenil)-etóxi]-piridin-3-il}-pirazol-1-il) - propiônico (69 mg, 100%). Procedimento Geral 71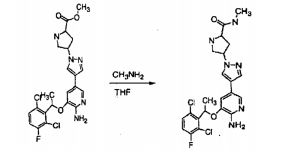
[00501] A uma solução agitada de éster metílico do ácido 4-(3-{6- amino-5-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]-piridin-3-il}-pirazol-1-il)- pirrolidino-2-carboxílico (105 mg, 0,21 mmol) em THF (5 mL) adicionou-se CH3NH2 2M em THF (1,06 mL, 2,12 mmols), sendo a mistura agitada e aquecida para 55°C por 18 horas. Verificou-se por LCEM que a reação estava terminada, removeu-se o THF sendo o resíduo purificado por HPLC preparativa, conferindo metilamida do ácido 4-(4-{6-amino-5-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]-piridin-3-il}- pirazol-1-il)-pirrolidino-2-carboxílico (30 mg,), rendimento 28,6%. Procedimento Geral 72:
[00502] 4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il) -1H-pirazol-1- carboxilato de terc-butila (21-1):
[00503] Dicarbonato de di-terc-butila (7,2 equivalente molar), 4- (dimetilamino)piridina (0,84 equivalente molar) foram adicionados para uma solução de 4-(4,4,5,5-tetrametil-2-(1H-pirazol-4-il)- [1,3,2]dioxaborolano (6 mmols) em 40 mL de DMF. A mistura de reação foi agitada a temperatura ambiente por 12 horas. Adicionou-se água para a mistura de reação arrefecendo a reação. EtOAc foi então adicionado para extrair a solução aquosa. Secou-se a camada de EtOAc com Na2SO4. O Na2SO4 foi filtrado e o filtrado foi evaporado dando um resíduo oleoso castanho-amarelo como composto 21-1 (1,32 g, 4,56 mmols; 76%).
[00504] 1H RMN (400 MHZ, clorofórmio-D) δ ppm 1,32 (s, 12H) 1,63 (s, 9H) 7,91 (s, 1H) 8,37 (s 1H). O resíduo foi usado na próxima etapa de reação sem mais purificação.
[00505] O composto 21-3 mostrado com o exemplo específico 3- [1-(2,6-dicloro-3-fluorfenil)etóxi]-5-(1H-pirazol- 4 - il) piridin-2-amina (21-3a):
[00506] Composto 21-1 (1,0 equivalente molar) foi adicionado para uma solução do composto 21-2a (composto 21-2 com substituintes R dando 2,6-dicloro-3-fluorfenil) (1,92 mmol) em 20 mL de DME. A mistura foi agitada a temperatura ambiente sob uma atmosfera de nitrogênio por 30 minutos, e a seguir adicionou-se diclorobis(trifenilfosfino) paládio(II) (0,05 equivalente molar). Carbonato de sódio (3 equivalente molar) em 4 mL de H2O foi adicionado para a mistura de reação e a solução resultante foi aquecida para 85°C por 12 h. Bases alternativas usadas foram CsF e Cs2CO3 em que 1 ou 2 equivalentes do éster borônico e a temperatura ambiente (CsF) ou 80°C) (todos). Adicionou-se água para a mistura de reação para esfriar a reação. EtOAc (150 mL x 2) foi a seguir adicionado para extrair a solução aquosa. Secar a camada de EtOAc sobre Na2SO4. O Na2SO4 foi filtrado e o filtrado evaporado dando um resíduo oleoso marrom escuro. O resíduo foi purificado por cromatografia em gel de sílica (eluindo com 0^10% MeOH em acetato de etila) dando o produto desejado, composto 21-3a (2,05 g 53,6% rendimento).
[00507] 1H RMN (400 MHz, clorofórmio-D) δ pm 1,60 (s, 1H) 1,84 (d, J = 6,57 Hz, 3H) 5,07 (s, 2H) 6,06 (q, J = 6,57 Hz, 1H) 6,89 (d, J = 1,77 Hz, 1H) 6,96-7,06 (m, 1H) 7,22-7,33 (m, 1H) 7,67 (s, 2H) 7,80 (d, J = 1,52 Hz, 1H).
[00508] Para produzir os compostos de fórmula 2104, podem ser usados o seguinte procedimento exemplar: Hidreto de sódio (1,2 equivalente molar ) é adicionado para uma solução do composto 21-3 (0,87 mmol) em 10 mL de DMF. A mistura é agitada a temperatura ambiente sob uma atmosfera de nitrogênio, durante 30 minutos e em seguida, o composto 21-6 (1 equivalente molar) é adicionado. A solução resultante é aquecida para 85-90°C por 12 horas. Adicionou- se água (20 mL) para a mistura de reação para resfriar a reação. EtOAc (50 mL x 2) é então adicionado para extrair a solução aquosa. Secar a camada de EtOAc sobre Na2SO4. O Na2SO4 é filtrado sendo o filtrado evaporado. O resíduo é purificado por cromatografia em gel de sílica (eluindo com EtOAc em hexanos) dando o produto desejado composto 21-4 (20-50% rendimento). Procedimento Geral 73: L = Br, Cl COOH, COCl, OMs, carbonato de etileno, aldeído
L = Br, Cl COOH, COCl, OMs, carbonato de etileno, aldeído
[00509] Os compostos de fórmula 22-3 podem ser preparados pelo seguinte procedimento exemplar: Composto 22-2 (1,2 equivalente molar) é adicionado para uma solução do composto 22-1 (0,24 mmol) e base (3,5 equivalente molar) e/ou reagente de acoplamento (1 equivalente molar) em 5 mL de DMF. A mistura é agitada sob uma atmosfera de nitrogênio por 12 horas. Adiciona-se água (20 mL) para a mistura de reação para resfriar a reação. EtOAc (50 mL x 2) é adicionado para extrair a solução aquosa. Secar a camada de EtOAc sobre Na2SO4. O Na2SO4 é filtrado e o filtrado evaporado. O resíduo é purificado por cromatografia em gel de sílica (eluindo com CH3OH, CH2CI2, EtOAc e hexanos) dando o produto desejado, composto 22-3.
[00510] O seguinte procedimento pode ser usado para preparer derivados piperidino-pirazol-2-aminopiridina
[00511] NaH (1,2 eq. 0,68 mmol) foi adicionado em partes a uma solução agitada de 4-iodopirazol (0,57 mmol) em DMF (2 L a 4°C. A mistura resultante foi agitada por uma hora a 4°C e o composto 23-4 (1,1 eq. 0,63 mmol) foi a seguir adicionado. A mistura resultante foi aquecida a 100°C por 12 horas. A reação foi arrefecida com água e extraída com EtOAc várias vezes. O resíduo foi purificado por cromatografia em gel de sílica (eluindo com EtOAc a 5% em pentano) dando o composto 23-1a) como um sólido branco 40 g, 66%).
[00512] Bis(pinacolato)diboro (1,4 eq. 134 g, 0,52 mol) e acetato de potássio (4 eq. 145 g, 1,48 mol) foram adicionado em seqüência para uma solução do composto 23-1a (140 g, 0,37 mol) em 1,5 L de DMSO. A mistura foi purgada com nitrogênio várias vezes e diclorobis(trifenilfosfino) paládio (II) (0,05 eq. 12,9 g, 0,018 mol) foi então adicionado. A mistura resultante foi aquecida a 80°C por duas horas. A mistura de reação foi resfriada para temperatura ambiente e filtrada através um leito de celite e lavada com EtOAc. O filtrado foi lavado com NaCl sat. (500 mL x 2) seco sobre Na2SO4, filtrado e concentrado. O resíduo foi purificado por cromatografia em gel de sílica (eluindo com 5% de EtOAc em hexanos) dando o composto 231b como um sólido branco (55 g, 40%).
[00513] O composto 23-2 (1,0 equivalente molar) foi adicionado para uma solução do composto 23-1b (13 equivalente molar) em 15 mL de DME. A mistura foi purgada com nitrogênio várias vezes e a seguir adicionou-se diclorobis(trifenilfosfino paládio (II) (0,05 equivalente molar) Carbonato de césio (3 equivalente molar) em 4 mL de água foi adicionado para a mistura de reação e a solução resultante foi aquecida a 85°C por 12 horas. Adicionou-se água (10 mL) à mistura de reação para arrefecer a reação. EtOAc (150 mL x 2) foi então adicionado para extrair a solução aquosa. Secar a camada de EtOAc sobre Na2SO4. O Na2SO4 foi filtrado sendo o filtrado evaporado para dar um resíduo oleoso marrom escuro. O resíduo foi purificado por cromatografia em gel de sílica (eluindo com 75 ^ 100% EtOAc em hexanos) dando o composto 23-3a (61% rendimento).
[00514] Cloridrato (19 eq. 12 mmol) foi adicionado para uma solução do composto 23-3a (0,63 mmol) em MeoH (4 mL). A mistura foi agitada a temperatura ambiente por 12 horas. O solvente foi evaporado adicionando-se água (10 mL). NaHcO3 (aq.) sat. foi adicionado para neutralizar a solução a pH 7. acetato de etila 100 mL x 2) foi adicionado para extrair a solução aquosa. As camadas orgânicas combinadas foram secas sobre Na2SO4, filtradas e evaporadas dando o composto 23-5a como um resíduo sólido (0,6 mmol, 95% rendimento).
[00515] Os compostos de fórmula 23-7 podem ser formados de acordo com o seguinte procedimento geral: O composto 23-8 (1,2 equivalente molar) é adicionado a uma solução do composto 23-5a (0,24 mmol) e base (3-5 equivalente molar) e/ou reagente de acoplamento (1 equivalente molar) em 5 mL de DMF. A mistura é agitada sob uma atmosfera de nitrogênio por 12 horas. Adicionou-se água (20 mL) para a mistura de reação para arrefecer a reação. EtOAc (50 mL x 2) é a seguir adicionado para extrair a solução aquosa. Secar a camada de EtOAc sobre Na2SO4. O Na2SO4 é filtrado e o filtrado evaporado dando um resíduo oleoso. O resíduo é purificado por cromatografia em gel de sílica (eluindo com CH3OH, CH2Cl2 EtOAc e hexanos ) dando o produto desejado, composto 23-7a. Procedimento Geral 75
[00516] Compostos 3-metóxi podem ser preparados dos correspondentes compostos 3-flúor pelo seguinte Procedimento Geral. A 4 mL de DMSO é adicionado 0,124 mL etanol seguido por 32 mg NaH. Após agitação por 30 minutos 250 mg de 24-1 é adicionado e a reação aquecida a 40°C. Após três horas a reação é resfriada e despejada em água para precipitar. Após neutralização para pH 6, o produto 24-2 é isolado. Procedimento Geral 76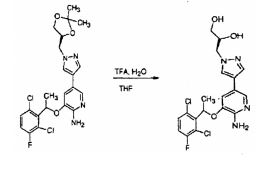
[00517] A uma solução agitada de 3-[1-(2,6-dicloro-3-flúor-fenil- etóxi]-5-[1-(2,2-dimetil-[1,3] dioxolan - 4 -ilmetil)-1H-pirazol-4-il]-piridin- 2-ilamina (150 mg, 0,31 mmol) em THF (3 mL) e H2O (2 mL) foi adicionado TFA (2 mL a 0°C sendo a mistura agitada e aquecida para temperatura ambiente, a seguir aquecida a 50°C por 5 horas. Verificou-se por LCEM que a reação chegou a seu término, removendo-se o THF,sendo o resíduo purificado por HPLC preparação para produzir 3-(4-(6-amino-5-[1-(2,6-dicloro-3-flúor-fenil)-etóxi]-piridin- 3-il]-pirazol-1-}-propan-1,2-diol (102 mg), rendimento: 74,2%. Procedimento Geral 77:
[00518] A uma solução agitada de 4-bromo-1H-pirazol em DMF adicionou-se hidreto de sódio a temperatura ambiente. A mistura foi agitada por 30 minutos, adicionou-se [1,3]dioxolan-2-ona sendo a mistura agitada e lentamente aquecida à temperatura ambiente. A reação foi monitorada por TLC. Após a reação estar pronta, adicionou- se EtOAc, lavou-se com NaHCO3 sat. água e salmoura, secou-se com Na2SO4, filtrou-se e concentrou-se. O resíduo foi purificado por sílica gel, eluentes EtOAc e DCM 10% dando 2-(4-bromo-pirazol-1-il)-etanol (0,22 g, rendimento 34%).
[00519] 1H RMN (400 MHz, clorofórmio-D) δ ppm 7,49 (s, 1H) 7,46 (s, 1H) 4,18-4,23 (m, 2H) 3,93-3,98 (m, 2H) 3,09 (s, 1H). Exemplo 1: 5-bromo-3-[(R)-1-{2,6-dicloro-3-flúor-fenil)-etóxi]-pirazin-2- ilamina
[00520] O composto titular foi preparado de acordo com o procedimento 2, a partir de (1S)-1-(2,6-dicloro-3-fluorfenil)etanol.
[00521] 1H RMN (400 MHz, DMSO-d6) δ 7,53 (s, 1H), 7,48 (m, 1H), 7,39 (t, 1H), 6,48 (s, 2H), 6,41 (q, 1H), 1,74 (d, 3H); LCMS: 381 [M+1]; c-Met Ki: 0,796 μM. Exemplo 2: ácido 4-[5-amino-6-[(R)-1-{2,6-dicloro-3-flúor-fenil)-etóxi]- pirazin-2-il-benzóico
[00522] O composto titular foi preparado de acordo com o procedimento 3.
[00523] 1H RMN (400 MHz, DMSO-d6) δ 8,16 (s, 1H), 7,84 (d, 2H), 7,77 (d, 2H), 7,53 (m, 1H), 7,37 (t, 1H), 6,64 (s, 2H), 6,53 (q, 1H), 1,78 (d, 3H); LCMS: 422 [M+1]; c-Met Ki: 0,154 μM. Exemplo 3: (4-[5-amino-6-[(R)-1-{2,6-dicloro-3-flúor-fenil)-etóxi]-pirazin- 2-il)-fenil)-piperazin-1 - il -metanona
[00524] O composto titular foi preparado de acordo com o procedimento 4.
[00525] 1H RMN (400 MHz, DMSO-d6) δ 8,11 (s, 1H), 7,73 ( 2H), 7,53 (m, 1H), 7,37 (t, 1H), 7,31 (d, 2H), 6,55 (m, 3H), 3,51 (br, 2H), 3,32 (br, 2H), 2,67 (br, 4H), 1,77 (d, 3H); LCMS: 490 [M+1]; c-Met Ki: 0,027 μM. Exemplo 4: éster terc-butílico do ácido 4-(4-[5-amino-6-[(R)-1-{2,6- dicloro-3-flúor-fenil)-etóxi]-pirazin-2-i-benzoil)-piperazino-1-carboxílico.
[00526] O composto titular foi preparado de acordo com o procedimento 16 seguido por 20.
[00527] 1H RMN (400 MHz, DMSO-d6) δ 8,12 (s, 1H), 7,72 (d, 2H), 7,50 (m, 1H), 7,33 (t, 3H), 6,55 (m, 3H), 3,51 (br, 2H), 3,39 (m, 3H), 3,32 (br, 3H), 1,77 (d, 3H), 1,40 (s, 9H); LCMS: 590 [M+1]; c-Met Ki: 0,335 μM. Exemplo 5: 3-[(R)-1-{2,6-dicloro-3-flúor-fenil)-etóxi]-5-[4-{piperazin-1- ilcarbonil)fenil]piridin-2-amina
[00528] O composto titular foi preparado de acordo com o procedimento 20 seguido pro 21 como uma mistura racêmica com o correspondente enantiômero S do Exemplo 119, seguido por separação pó cromatografia quiral. O composto titular também foi preparado como um composto enantiomericamente puro iniciando do material quiral de partida.
[00529] 1H RMN (400 MHz, DMSO-D6) δ ppm 1,83 (d, J = 6,57 Hz, 3H) 3,35 (s, 4H) 3,69 (s, 4H) 6,24 (q, J = 6,57 Hz, 1H) 6,91-7,08 (m, 2H) 7,10 (d, J = 126 Hz, 1H) 7,46 (t, J = 8,72 Hz, 1H) 7,50 (s, 4H) 7,58 (dd, J = 8,97, 4,93 Hz, 1H) 7,91 (d, J = 1,77 Hz, 1H) 9,35 (s, 2H); LCMS: 490 [M+1]; c-Met Ki: 0,01 μM. Exemplo 6: 4-{6-amino-5-[(1R)-1-{2,6-dicloro-3-fluorfenil)-etóxi]-piridin- 3-il}-N-[2-(dimetilamino)etil]-N-metilbenzamida
[00530] O composto titular foi preparado de acordo com o procedimento 20.
[00531] 1H RMN (400 MHZ, DMSO-D6) δ ppm 1,80 (d, J = 6,82 Hz, 3H) 1,97 (s, 3H) 2,19 (s, 3H) 2,30-2,42 (m, J = 1,77 Hz, 2H) 2,93 (s, 3H) 3,22-3,29 (m, 1H) 3,44-3,61 (m, 1H) 5,95 (s, 2H) 6,14 (q, J = 6,57 Hz, 1H) 6,98 (d, J = 1,01 Hz, 1H) 7,30-7,39 (m, 2H) 7,40-7,47 (m, 3H) 7,51-7,62 (m, 1H) 7,87 (d, J = 1,77 Hz, 1H); LCMS: 506 [M+1]; c- Met Ki: 0,01 μM. Exemplo 7: 4-{6-amino-5-[(1R)-1-{2,6-dicloro-3-fluorfenil)-etóxi]-piridin- 3-il}-fenil]metanol
[00532] O composto titular foi preparado de acordo com o procedimento 27.
[00533] 1H RMN (400 MHz, DMSO-D6) δ ppm 1,84 (d, J = 6,57 Hz, 3H) 4,49 (d, J = 5,81 Hz, 2H) 5,20 (t, J = 5,81 Hz, 1H) 6,25 (q, J = 6,57 Hz, 1H) 6,46-6,68 (m, 2H) 7,04 (d, J = 1,52 Hz, 1H) 7,34 (s, 4H) 7,46 (t, J = 8,72 Hz, 1H) 7,59 (dd, J = 8,97, 4,93 Hz, 1H) 7,76 (d, J = 1,52 Hz, 1H); LCMS: 408 [M+1]; c-Met Ki: 0,051 μM. Exemplo 8: 4-{6-amino-5-[(1R)-1-{2,6-dicloro-3-fluorfenil)-etóxi]-piridin- 3-il}-N-[3-(dimetilamino)propil]-N-metilbenzamida
[00534] O composto titular foi preparado de acordo com o procedimento 27.
[00535] 1H RMN (400 MHz, DMSO-D6) δ ppm 1,60-1,73 (m, 2H) 1,80 (d, J = 6,57 Hz, 3H) 1,94 (s, 3H) 2,13 (s, 3H) 2,20-2,29 (m, 2H) 2,92 (s, 3H) 3,36-3,50 (m, 2H) 5,96 (s, 2H) 6,14 (q, J = 6,57 Hz, 1H) 6,98 (s, 1H) 7,37 (s, 2H) 7,40-7,51 (m, 3H) 7,55 (dd, J = 8,84, 4,80 Hz, 1H) 7,86 (d, J = 1,77 Hz, 1H); LCMS: 520 [M+1]; c-Met Ki: 0,01 μM. Exemplo 9: 4-(4-(6-amino-5-[(1 R)-1-{2,6-dicloro-3-fluorfenil)-etóxi]- piridin-3-il]benzoil) piperazino - 1 -carboxilato de terc-butila
[00536] O composto titular foi preparado de acordo com o procedimento 20.
[00537] 1H RMN (400 MHz, clorofórmio-D) δ ppm 1,46 (s, 9H) 1,86 (d, J = 6,82 Hz, 3H) 3,30-3,89 (m, 8H) 4,90 (s, 2H) 6,11 (q, J = 6,57 Hz, 1H) 6,98 (d, J = 1,52 Hz, 1H) 7,01-7,10 (m, 1H) 7,30 (dd, J = 8,97, 4,93 Hz, 1H) 7,35-743 (m, 4H) 7,88 (d, J = 1,71 Hz, 1H); LCMS: 580 [M+1]; c-Met Ki: 0,03 μM. Exemplo 10: 3-[(R)-1-(2,6-dicloro-3-fluorfenil)-etóxi]-5-[1-(1-metil- piperidin-4-il)-1H-pirazol-4-il]piridin -2-ilamina
[00538] O composto titular foi preparado de acordo com o procedimento 62 usando 3-[(R)-1-(2,6-dicloro-3-fluorfenil)-etóxi]-5- (4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il]-piridin-2-ilamina e 4-(4- bromopirazol-1-il)-1-metil-piperidina (preparado de acordo com o procedimento 11).
[00539] 1H RMN (400 MHz, CDCh) δ 7,65 (s, 1H), 7,55 (s, 1H), 7,50 (s, 1H), 7,31 (m, 1H), 7,06 (m, 1H), 6,87 (s, 1H), 6,08 (m, 1H), 5,50 (bs, 2H), 4,18 (m, 1H), 3,11 (m, 2H), 2,40 (s, 3H), 2,30 (m, 2H), 2,20 (m, 4H), 2,07 (s, 3H), 1,86 (d, J = 8 Hz, 3H); LCMS: 464 [M+1]; c- Met Ki: 0,01 μM. Exemplo 11: 1-[4-(4-[6-amino-5-[(R)-1-{2,6-dicloro -3-fluorfenil)-etóxi]- piridin-3-il)-pirazol-1-il]-piperidin-1-il]-2-hidróxi-etanona
[00540] O composto titular foi preparado de acordo com o procedimento 63.
[00541] 1H RMN (400 MHz, CDCh) δ 7,72 (s, 1H), 7,57 (s, 1H), 7,47 (s, 1H), 7,31 (m, 1H), 7,06 (m, 1H), 6,86 (s, 1H), 6,08 (m, 1H), 5,00 (bs, 2H), 4,70 (m, 1H), 4,36 (m, 1H), 4,21 (s, 1H), 3,70 (m, 1H), 3,18 (m, 1H), 3,00 (m, 1H), 2,223 (m, 2H), 2,01 (m, 2H), 1,86 (d, J = 8 Hz, 3H); LCMS: 508 [M+1]; c-Met Ki: 0,004 μM.
[00542] 3-[(R)-1-(2,6-dicloro-3-flúor-fenil)-etóxi]-5-(1-piperidin-4-il)- 1H-pirazol-4-il)piridin-2-ilamina
[00543] O composto titular foi preparado de acordo com o procedimento 62 usando 3-[(R)-1-(2,6-dicloro-3-fluorfenil)-etóxi]-5- (4,4,5,5-tetrametil-[1,3,2]dioxaborolan- 2 - il] -piridin-2-ilamina e 4-(4- bromopirazol-1-il)-1-ciclopentil-piperidina (preparado de acordo com o Procedimento Geral 11 usando bromociclopentano como reagente de alquilação).
[00544] 1H RMN (400 MHz, CDCh) δ 7,73 (s, 1H), 7,55 (s, 1H), 7,48 (s, 1H), 7,31 (m, 1H), 7,07 (m, 1H), 6,88 (s, 1H), 6,08 (m, 1H), 4,64 (m, 1H), 2,04 (m 2H), 1,98 (m, 2H), 1,86 (d, J = 8 Hz, 3H), 1,73 (m, 2H); LCMS: 435 [M+1]; c-Met Ki: 0,02 μM. Exemplo 13 3-[(R)-1-(2,6-dicloro-3-flúor-fenil)-etóxi]-5-(1-piperidin-4-il- 1H-pirazol-4-il)piridin-2-ilamina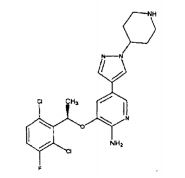
[00545] O composto titular foi preparado de acordo com o procedimento 62.
[00546] 1H RMN (400 MHz, CDCl3) δ 7,69 (s, 1H), 7,56 (s, 1H), 7,50 (s, 1H), 7,32 (m, 1H), 7,07 (m, 1H), 6,87 (m, 1H), 6,07 (m, 1H), 5,25 (bs, 2H), 4,30 (m, 1H), 3,41 (m, 2H), 2,96 (m, 2H), 2,26 (m, 2H), 2,12 (m, 2H), 1,86 (d, J = 8 Hz, 3H): LCMS: 450 [M+1]; c-Met Ki: 0,003 μM. Exemplo 14: 3-[(R)-1-(2,6-dicloro-3-flúor-fenil)-etóxi]-5-(1-piperidin-4-il- 1H-pirazol-4-il)pirazin-2-ilamina
[00547] O composto titular foi preparado de acordo com o procedimento 66.
[00548] 1H RMN (400 MHz, DMSO-d6) δ 7,86 (s, 1H), 7,76 (s, 1H), 7,63 (m, 2H), 7,54 (m, 1H), 7,37 (t, 1H), 6,46 (q, 1H), 6,15 (s, 1H) 4,10 (m, 1H), 3,01 (m, 2H), 1,95 (m, 2H), 1,85 (s, 2H), 1,75 (d, 3H), 1,67 (dd, 1H); LCMS: 451 [M+1]; c-Met Ki: 0,010 μM. Exemplo 15: 3-[(R)-1-(2,6-dicloro-3-flúor-fenil)-etóxi]-5-(1H-pirazol-4-il)- pirazin-2-ilamina
[00549] O composto titular foi preparado de acordo com o procedimento 3 usando 5-bromo-3-[(R)-1-(2,6-dicloro-3-flúor-fenil)- etóxi]-pirazin-2-ilamina e éster terc-butílico do ácido 4-(4,4,5,5- tetrametil-[1,3,2]dioxaborolan-2-il]-pirazol-1-carboxílico.
[00550] 1H RMN (400 MHz, DMSO-d6) δ 12,81 (s, 1H), 7,79 (s, 1H), 7,48 (m, 1H), 7,36 (t, 1H), 6,48 (q, 1H), 6,12 (s, 2H), 1,75 (d, 3H); LCMS: 368 [M+1]; c-Met Ki: 0,065 μM. Exemplo 16: 1-[4-(4-[5-amino-6-[(R)-1-(2,6-dicloro-3-flúor-fenil)-etóxi]- pirazin-2-il)-pirazol-1- il) -piperidin-1-il]-2-hidróxi-etanona
[00551] O composto titular foi preparado de acordo com os procedimentos 62 e 63 usando 5-bromo-3-[(R)-1-(2,6-dicloro-3-flúor- fenil)-etóxi]-pirazin-2-ilamina como o material de partida.
[00552] 1H RMN (400 MHZ, DMSO-dβ) δ 7,91 (s, 1H), 7,76 (s, 1H), 7,64 (s, 1H), 7,49 (m, 1H), 7,36 (t, 1H), 6,46 (q, 1H), 6,15 (s, 2H), 4,57 (br, 1H), 4,40 (m, 2H), 4,12 (br, 2H), 3,77 (m, 1H), 3,35 (m, 2H), 3,43 (m, 1H), 3,16 (m, 2H), 1,75 (d, 3H); LCMS: 509 [M+1]; c-Met Ki: 0,015 μM. Exemplo 17: 3-[(R)-1-(2,6-dicloro-3-flúor-fenil)-etóxi]-5-[1-(1-metil- piperidin-4-il)-1H-pirazon - 4 - il] -pirazin-2-ilamina
[00553] O composto titular foi preparado de acordo com o procedimento 62 usando 5-bromo-3-[(R)-1-(2,6-dicloro-3-flúor-fenil)- etóxi]-pirazin-2-ilamina e 4-(4-bromopirazol-1-il)-1-metil-piperidina (preparado de acordo com o procedimento 11).
[00554] 1H RMN (400 MHz, DMSO-d6) δ 7,88 (s, 1H), 7,76 (s, 1H), 7,64 (s, 1H), 7,49 (m, 1H), 7,36 (t, 1H), 6,46 (q, 1H), 6,15 (s, 2H), 4,02 (m, 1H), 2,84 (m, 2H), 2,19 (s, 3H), 2,00 (m, 4H), 1,85 (m, 3H), 1,75 (d, 3H); LCMS: 465 [M+1]; c-Met Ki: 0,03 μM. Exemplo 18: 1-[4-(4-[5-amino-6-[(R)-1-(2,6-dicloro -3-flúor-fenil)-etóxi]- pirazin-2-il)-pirazol-1-il)-piperidin -1-il]-2-dimetilamino-etanona
[00555] O composto titular foi preparado de acordo com o procedimento 63 usando 3-[(R)-1-(2,6-dicloro-3-flúor-fenil)-etóxi]-5-(1- piperidin-4-il-1H-pirazol-4-il)- pirazin - 1 -ilamina acoplado a ácido dimetilamino acético na presença de HOBt/EDC/trietilamina em DMF conforme descrito no procedimento 5 usando 5-bromo-3-[(R)-1-(2,6- dicloro-3-flúor-fenil)-etóxi]-pirazin-2-ilamina como material de partida.
[00556] 1H RMN (400 MHz, DMSO-d6) δ 7,90 (s, 1H), 7,76 (s, 1H), 7,65 (s, 1H), 7,49 (m, 1H), 7,36 (t, 1H), 6,47 (q, 1H), 6,15 (s, 2H), 4,39 (m, 1H), 4,16 (m, 1H), 3,16 (m, 2H), 3,02 (m, 1H), 2,75 (m, 1H), 2,19 (s, 6H), 2,01 (m, 2H), 1,88 (s, 1H), 1,75 (d, 3H); LCMS: 536 [M+1]; c- Met Ki: 0,015 μM. Exemplo 19: 3-[(R)-1-(2-cloro-3,6-diflúor-fenil)-etóxi]-5-(1-piperidin-4-il- 1H-pirazol-4-il)- piridin - 2 -ilamina
[00557] O composto titular foi preparado de acordo com o procedimento 62 usando 5-bromo-3-[(R)-1-(2-cloro-3,6-diflúor-fenil)- etóxi] -piridin-2-ilamina como material de partida (de acordo com os métodos para a síntese de 5-bromo-3-[(R)-1-(2,6-dicloro-3-flúor-fenil)- etóxi]-pirazin-2-ilamina a partir de (S)-1-(2-cloro-3,6-diflúor-fenil)-etanol obtido de SynChem. Inc).
[00558] 1H RMN (400 MHz, DMSO-dβ) δ 7,88 (s, 1H), 7,70 (s, 1H), 7,50 (s 1H), 7,38 (m, 1H), 7,25 (m, 1H), 6,99 (s, 1H), 5,88 (m, 1H), 5,48 (bs, 2H), 4,08 (m, 1H), 2,96 (m, 2H), 2,53 (m, 1H), 2,45 (m, 1H), 1,89 (m, 1H), 1,80 (m, 4H), 1,67 (m, 4H); LCMS: 434 [M+1]; c-Met Ki: 0,09 μM.
[00559] Considera-se que, será observada, em quaisquer séries dos compostos dados, uma faixa de atividades biológicas. Nestes aspectos presentemente preferidos, esta invenção refere-se a novos compostos capazes de modular, regular e/ou inibir atividade de proteína quinase. Os ensaios a seguir podem ser empregados para selecionar os compostos que demonstram o grau ótimo da atividade desejada.
[00560] O seguinte ensaio in vitro pode ser usado para determinar o nível de atividade e efeito dos diferentes compostos da presente invenção em um ou mais dos PKs. Ensaios similares podem ser realizados juntamente com as mesmas linhas para qualquer PK usando técnicas conhecidas. Uma referência literária é dada em (Technikova-Dobrova Z, Sardanelli AM, Papa S FEBS Lett 1991 Nov 4; 292: 69-72).
[00561] O procedimento geral é como a seguir: compostos e reagentes de ensaio quinase são introduzidos aos recipientes de teste. O ensaio é iniciado por adição da enzima quinase. Inibidores enzimáticos reduzem a atividade medida da enzima.
[00562] No ensaio espectrofotométrico continuamente acoplado, a produção dependente de tempo de ADP pela quinase é determinada por análise da velocidade de consumo de NADH medindo-se a redução na absorbância a 340 nm. ã medida que PK produz ADP ela é novamente convertida em ATP por reação com fosfoenol piruvato e piruvato quinase. Piruvato também é produzido nesta reação. O piruvato é a seguir convertido em lactato por reação com lactato desidrogenase, que converte, simultaneamente NADH em NAD. NADH possui uma absorbância mensurável a 340 nm, enquanto NAD não possui.
[00563] O protocolo atualmente preferido para realizar os experimentos espectrofotométricos continuamente acoplados para PKs específicos é dado abaixo. Contudo, a adaptação deste protocolo para determinar a atividade dos compostos contra outras RTKs, bem como para CTKs e STKs enquadra-se no escopo do conhecimento dos versados na técnica.
[00564] Este ensaio analisa a atividade tirosina quinase de HGFR no peptídeo de substrato Met-2, um peptídeo derivado do laço de ativação da HGFR.
[00565] 1. Enzima HGFR de Upstate (Met, ativa) Cat. n° 14-526.
[00566] 2. Peptídeo Met-2 (Laço de ativação a HGFR) Ac- ARDMYDKEYYSVHNK ( Peso molecular - 1960) Dissolver em até 200 mM HEPES pH 7,5 em solução de estoque 10 mM.
[00567] 3. PEP 1M (fosfoenol piruvato) em 200 mM de HEPES, pH 7,5.
[00568] 4. 100 mM de NADH (B-Nicotianamida Adenina Dinucleotídeo, Forma reduzida) em 200 mM de HEPES, pH 7,5
[00569] 5. MgCl2 4M (cloreto de magnésio) em ddH2O
[00570] 6. DTT 1M (Ditiotreitol) em 200 mM de HEPES, pH 7,5
[00571] 7. LDH 15 unidades/mL (desidrogenase láctica)
[00572] 8. PK 15 unidades/mL (piruvato quinase)
[00573] 9. NaCl 5M dissolvido em ddH2O
[00574] 10. Tween-20 (Grau de proteína) 10% solução
[00575] 11. tampão HEPES 1M (ácido (N-[2-hidroxietil] piperazino- N-[2-etanossulfônico Sal de sódio. Dissolver em ddH2O, ajustar o pH para 7,5, elevar o volume para 1L. Filtrar a 0,1 μm.
[00576] 12. Água Grau HPLC, Burdick e Jacson n° 365-4, 1 x 4 litros (ou equivalente)
[00577] 13. DMSO a 100% (Sigma)
[00578] 14. Costar N° 3880 - placas de meia-área de fundo planos transparentes pretos para determinação do Ki e % inibição
[00579] 15. Costar N° 3359 - placas de polipropileno de 96 poços, de fundo redondo para diluições seriais
[00580] 16. Costar N° 3635 - placas de fundo plano transparentes placa-UV para % inibição
[00581] 17. Prendedores w/ microcelulares Beckman DU-650
[00582] 18. Cuveta microcelular de 4 posições Beckman
[00583] Tampão de diluição Preparação. (DB) para Enzima (para preparação. de 30 mL)
[00584] 1. Concentração final de DB é DTT 2mM, NaCl2 25 mM, MgCl2 5 mM, Tween-20 0,01% e tampão HEPES 50 mM, pH 7,5.
[00585] 2. Compor 50 mM HEPES adicionando-se 1,5 mL de HEPES 1 M em 28,1 ml de ddH2O. Adicionar o resto dos reagentes. No frasco cônico de 50 mL, adicionar 60 μL de DTT 1M, 150 μL de NaCl2 5M, 150 μL de MgCh 1M e 30 μL de Tween-20 a 10% para dar o volume total de 30 mL.
[00586] 3. Turbilhonar por 5-10 segundos.
[00587] 4. Fazer alíquotas de DB a 1 mL/tubo e rotular os tubos como "DB HGFR"
[00588] 5. OBS: Pode ser preparado e armazenado de antemão.
[00589] 6. Congelar alíquotas não utilizadas em tubos de microcentrífuga a -20°C no congelador.
[00590] 1. Para placa de diluição do composto adicionar 4 μL de estoque 10 mM na coluna 1 da placa, e levar o volume a 100 μL com DMSO a 100%.
[00591] 2. Ajustar o método de diluição Precision 2000. Uma concentração final de 200 μM do composto em 50% de DMSO, 100 mM de HEPES (diluição serial 1:2)
[00592] 1. Concentração final no ensaio:
[00593] Reagente (coc. de estoque) (concentração final no Ensaio
[00594] a. PEP (1M) 1 mM
[00595] b. NADH (100mM) 300 μM
[00596] c. MgCl2 (4 M) 20 mM
[00597] d. DTT (1 M) 2 mM
[00598] e. ATP (500 mM) 300 μM
[00599] f. HEPES 200 Mm (pH 7,5) 100 mM
[00600] g. Piruvato quinase (PK) 15 unidades/mL
[00601] h. Desidrogenase láctica (LDH) 15 unidades/mL
[00602] i. Mep-2-peptídeo (10 mM) 0,500 mM
[00603] j. HGFR 50 n
[00604] 2. Para um tampão de reação de 10 mL adicionar 10 μL de PEP 1M, 33 μL de NADH 100 mM, 50 μL de MgCl2 4M, 20 μL de DTT 1M, 6 μL de ATP 500 mM, e 500 μL de Met-2-peptídeo 10 mM em 100 mM de tampão HEPES pH 7,5 e misturar c/turbilhonamento.
[00605] 3. Adicionar enzimas de acoplamento. LDH e PK na mistura de reação. Misturar por inversão suave.
[00606] 1. Ajustes espectrofotométricos:
[00607] i. comprimento de onda de absorbância (À): 340 nm
[00608] ii. Tempo de incubação: 10 minutos
[00609] iii. Tempo de teste: 10 minutos
[00610] iv. Temperatura: 37°C
[00611] 2. Adicionar 85 μL da mistura de reação CE para cada poço da placa de ensaio.
[00612] 3. Adicionar 5 μL do composto diluído a um poço da placa de ensaio.
[00613] 4. Adicionar 5 μL de 50% DMSO para controle negativo à última coluna da placa de ensaio.
[00614] 5. Misturar com pipeta de canal múltiplo ou agitador orbital.
[00615] 6. Pré-incubar por 10 minutos a 37°C.
[00616] 7. Adicionar 10 μL de HGFR a 500 nM para cada poço da placa de ensaio; a concentração de HGFR final é 50 nM num volume final total de 100 μL.
[00617] 8. Medir a atividade por 10 minutos a À = 340 nm e 37°C
[00618] Os seguintes ensaios in vitro podem ser usados para determinar o nível da atividade e efeito dos diferentes compostos da presente invenção, em um ou mais das PKs. Ensaios semelhantes podem ser pensados junto com as mesmas linhas para qualquer PK usando técnicas conhecidas.
[00619] Vários ensaios aqui descritos são realizados num formato ELISA (Ensaio Imunoabsorvente Ligado à Enzima) (Voller, et al 1980, "Enzyme-Linked Immunosorbent Assay" Manual of Clinical Immunology, 2a. ed. Rose and Friedman, Am. Soc. Of Microbiology, Washington, D.C.pág. 359-371) O procedimento geral é como a seguir: um composto é introduzido a células expressando a quinase de teste, seja naturalmente ou recombinantemente, durante um período selecionado de tempo após o que, caso a quinase teste seja um receptor, é adicionado um ligante conhecido por ativar o receptor. As células são lisadas e o lisado é transferido para os poços de uma placa ELISA previamente revestida com um anticorpo específico reconhecendo o substrato da reação de fosforilação enzimática. Componentes não-substrato do lisado celular são lavados e removidos, sendo a quantidade de fosforilação no substrato detectada com um anticorpo de reconhecimento específico para fosfotirosina comparado com as células de controle que não foram contactadas com um composto de teste.
[00620] Os protocolos atualmente preferidos para realização dos experimentos ELISA para PKs específicas são dados abaixo. Contudo, a adaptação desses protocolos na determinação da atividade dos compostos contra outras RTKs, bem como para CTKs e STKS, está dentro do escopo do conhecimento dos versados na técnica.
[00621] Outros ensaios aqui descritos medem a proporção de DNA produzido em resposta à ativação de uma quinase de teste, que é uma medida genérica de uma resposta proliferativa. O Procedimento Geral para este ensaio é como a seguir: Introduz-se um composto a células expressando a quinase teste, seja naturalmente ou recombinantemente, por um dado período de tempo, após o que, caso a quinase este seja um receptor é adicionado um ligante conhecido por ativar o receptor. Após incubação pelo menos durante a noite, um reagente rotulador de DNA conhecido como 5-bromodesoxiuridina (BrdU) ou timidina-H3 é adicionado. A quantidade de DNA rotulado é detectada com, ou um anticorpo anti-BrdU ou por medição da radioatividade e faz-se a comparação para células de controle não contactadas com um compostos de teste.
[00622] Este ensaio é empregado para medir os níveis de fosfotirosina num substrato ácido poli(glutâmico: tirosina 4:1) como um meio de identificar agonistas/antagonistas da transfosforilação de Met do substrato.
[00623] 1. Placas Corning de 96 poços, Corning Catalogo n° 25805-95
[00624] 2. Poli(Glu-Tyr), 4:1, Signa, catálogo n° P0275.
[00625] 3. PBS, Gibco, catálogo n° 450 1300EB
[00626] 4. 50 mM de HEPES
[00627] 5. Tampão de Bloqueio: Dissolver 25 g de Albumina de soro bovino, Sigma Cat. n° A-7888, em 500 mL de PBS, filtrar por u filtro de 4 μm.
[00628] 6. Proteína de fusão GST purificada contendo o domínio Met quinase, SUGEN, Inc.
[00629] 7. tampão TBST
[00630] 8. DMSO 10% aquoso (MilliQue H2O)
[00631] 9. Adenosina-5-'trifosfato (dH2O) aquoso 10 mM, Sigma Cat. n° A-5394.
[00632] 10. Tampão de diluição quinase 2X: para 100 mL, misturar 10 m de HEPES 1M a pH 7,5 com 0,4 mL BSA/PBS a 5%, 0,2 mL de ortovanadato de sódio 0,1M e 1 mL de cloreto de sódio 5M em 88,4 mL de dH2O.
[00633] 11. Mistura de Reação ATP 4X: para 10 mL, misturar 0,4 mL de cloreto de manganês 1M e 0,02 mL de APT 0,1 M em 9,56 mL de dH2O
[00634] 12. Mistura de controles negativos 4X: para 10 mL, misturar 0,4 mL de cloreto de manganês 1M em 9,6 mL de dH2O.
[00635] 13. Placas de polipropileno de fundo em V de 96 poços NUNC, Applied Scientific Cat. n° S72092
[00636] 14. 500 mM EDTA
[00637] 15. Tampão de Diluição de Anticorpo: para 100 mL, misturar 10 mL de BSA/PBS a 5%, 0,5 mL de Carnation® Instant Milk a 5% em PBS e 0,1 mL de ortovanadato de sódio 0,1M em 88,4 mL de TBST
[00638] 16. Anticorpo antofosfotirosina policlonal de coelho, SUGEN, Inc.
[00639] 17. Anticorpo conjugado a peroxidase de rábano de cavalo anti-coelho de cabra, Biosource, Inc.
[00640] 18. Solução ABTS: para1 L, misturar 19,21 g ácido cítrico, 35,49 g de Na2HPO4 e 500 mg de ABTS com suficiente dH2O compondo 1 L.
[00641] 19. ABTS/H2O2: misturar 15 mL de solução ABST com 2 μL de H2O2 por cinco minutos antes do uso.
[00642] 20. HCl 0,2 M
[00643] 1. Revestir placas ELISA com 1 μg de Poli(Glu-Tyr) em 100 μl de PBS, manter durante a noite a 4°C.
[00644] 2. Bloquear a placa com 150 μL de BSA/PBS a 5% por 60 minutos.
[00645] 3. Lavar a placa duas vezes com PBS seguir uma vez com tampão de Hepes 50 mM pH 7,4.
[00646] 4. Adicionar 50 μL da quinase diluída a todos os poços. (A quinase purificada é diluída com Tampão de Diluição de Quinase). A concentração final deve ser de 10 ng/poço.
[00647] 5. Adicionar 25 μL do composto de teste (em DMSO a 4%) ou apenas DMSO (4% em dH2O) para os controles.
[00648] 6. Incubar a mistura quinase/composto durante 15 minutos.
[00649] 7. Adicionar 15 μL de MnCl2 40 mM aos poços de controle negativo.
[00650] 8. Adicionar 25 μL de mistura ATP/ MnCl2 a todos os outros poços (exceto os controles negativos). Incubar por 5 minutos.
[00651] 9. Adicionar 25 μL de EDTA 500 nM para finalizar a reação.
[00652] 10. Lavar a placa 3X com TBST.
[00653] 11. Adicionar 100 μL de anti-Ptyr policlonal de coelho diluído 1:10.000 em Tampão de Diluição de Anticorpo para cada poço. Incubar, com agitação a temperatura ambiente por uma hora.
[00654] 12. Lavar a placa 3X com TBST.
[00655] 13. Diluir Anticorpo anti-coelho conjugado a HRP (Biosource) 1:6000 em Tampão de Diluição de Anticorpo. Adicionar 100 μ: por poço e incubar a temperatura ambiente, com agitação por uma hora.
[00656] 14. Lavar a placa 1X com PBS.
[00657] 15. Adicionar 100 μL de solução ABTS/H2O2 para cada poço.
[00658] 16. Caso necessário, interromper o desenvolvimento da reação com adição de 100 μL de HCl 0,2M por poço.
[00659] 17. Ler a placa em leitora ELIXA Dynatech MR7000 com o filtro de teste a 410 nM e filtro de referência a 630 nM.
[00660] Os ensaios a seguir utilizam células engenheiradas para expressar um receptor selecionado e a seguir avaliar o efeito de um composto de interesse na atividade da síntese de DNA induzida por ligante, por determinação da incorporação de BrdU ao DNA.
[00661] Os materiais, reagentes e procedimentos a seguir são genéricos para cada ensaio de incorporação de BrdU. Observam-se variações nos ensaios específicos.
[00662] 1. Ligante apropriado.
[00663] 2. Células engenheiradas adequadas.
[00664] 3. Reagente de Rotulação de BrdU: 10 mM em PBS, pH 7 (Roche Molecular Biochemicals, Indianápolis IN).
[00665] 4. FixDenat: solução de fixação (Roche Molecular Biochemicals Indianápolis, IN).
[00666] 5. Anti-BrdU-POD: anticorpo monoclonal de camundongo conjugado com peroxidase (Chemicon, Temecula, CA).
[00667] 6. Solução de Substrato de TMB: tetrametilbenzidina (TMB, pronto para uso, Roche Molecular Biochemicals, Indianápolis, IN).
[00668] 7. Solução de Lavagem PBS: PBS 1X, pH 7,4
[00669] 8. Albumina bovina (BSA, fração pó V (Sigma Chemical Co. USA).
[00670] 1. As células são cultivadas a 8000 células/poço em CS a 10%, 2 mM Gln em DMEM numa placa de 96 poços. AS células são incubadas durante a noite a 37°C em CO2 a 5%.
[00671] 2. Após 24 horas, as células são lavadas com PBS, e a seguir são destituídas de soro em meio isento de soro (0% DMEM CS com BSA a 0,1%) durante 24 horas.
[00672] 3. No dia 3, o ligante apropriado e o composto de teste são adicionados para as células simultaneamente. Os poços de controle negativo recebem DMEM isento de soro com BSA a 0,1% apenas; as células do controle positivo recebem o ligante, mas nenhum composto de teste. Os compostos de teste são preparados em DMEM isento de soro com ligante numa placa de 96 poços, e diluído serialmente para 7 concentrações de teste.
[00673] 4. Após 18 horas da ativação do ligante, é adicionado reagente de rotulação de BrdU diluído (1:100 em DMEM, 0,1% BSA) e as células são incubadas com BrdU (concentração final é de 10 μM) por 1,5 horas.
[00674] 5. Após incubação com reagente de rotulação, o meio é removido por decantação e inversão da placa com pancadinhas, numa torre de papel. Solução de FixDenat é adicionado (50 μL/poço) e as placas são incubadas a temperatura ambiente por 45 minutos num agitador de placa.
[00675] 6. A solução FixDenat é removida por decantação e batendo-se de leve na placa invertida numa torre de papel. Adiciona- se leite (5% de leite em pó em PBS, 200 μL/poço) como solução de bloqueio e a placa é incubada por 30 minutos a temperatura ambiente num agitador de placa.
[00676] 7. A solução de bloqueio é removida por decantação e os poços são lavados uma vez com PBS. Solução anti-BrdU-POD é adicionada (diluição 1:200 em PBS, BSA a 1%, 50 μL/poço) e a placa é incubada por 90 minutos a temperatura ambiente num agitador de placa.
[00677] 8. O conjugado de anticorpo é removido por decantação e enxágüe dos poços 5 X com PBS, e a placa é seca por inversão e pequenas pancadas numa torre de papel.
[00678] 9. Solução de substrato TMB é adicionado (100 μL/poços e incubado por 20 minutos a temperatura ambiente num agitador de placa até o desenvolvimento de cor ser suficiente para detecção fotométrica.
[00679] 10. A absorbância das amostras aos medidas a 410 nm (no modo "comprimento de onda duplo" com uma leitura de filtro a 490 nm, como um comprimento de onda de referência) numa leitora de placa ELISA Dynatech.
[00680] 1. HGF recombinante humano (Cat. n° 249-HG, R& D Systems, Inc. USA).
[00681] 2. células BxPC3 (ATCC CRL-1687) Materiais e Reagentes Restantes, como acima
[00682] 1. As células são cultivadas a 9000 células/poço em RPMI FBS a 10% numa placa de 96 poços. As células são incubadas durante a noite a 37°C em CO2 a 5%.
[00683] 2. Após 24 horas as células são lavada com PBS e em seguida são destituídas de soro em 100 μL de meio isento de soro (RPMI com BSA a 0,1%) por 24 horas
[00684] 3. No dia 3, ligante contendo 25 μL (preparado a 1 μg/mL em RPMI com 0,1% BSA; concentração final de HGF é 200 ng/mL) e os compostos de teste são adicionados para as células. Os poços de controle negativo recebem 25 μL de RPMI isento de soro com 0,1% de BSA apenas; as células do controle positivo recebem o ligante (HGF) sem nenhum composto de teste. Os compostos de teste são preparados a 5 vezes sua concentração final em RPMI isento de soro com ligante numa placa de 96 poços, e serialmente diluído para dar 7 concentrações de teste. Tipicamente, a maior concentração final do composto de teste é 100 μM e diluições 1:3 são usadas (ou seja, faixa de concentração do composto de teste final é 0,137-100 μM).
[00685] 4. Após 18 horas de ativação do ligante, 12,5 μL de reagente de rotulação BrdU diluído a (1:100 em RPMI 0,1% BSA) é adicionado para cada poço e as células são incubadas com BrdU (concentração final é de 10 μM) por 1 hora.s
[00686] 5. Igual ao Procedimento Geral.
[00687] 6. Igual ao Procedimento Geral
[00688] 7. A solução de bloqueio é removida por decantação e os poços são lavados uma vez com PBS. Solução anti-BrdU-POD (diluição 1:100 em PBS, 1% BSA) é adicionada (100 μL/poço) e a placa é incubada por 90 minutos a temperatura ambiente num agitador de placa.
[00689] 8. Igual ao Procedimento Geral.
[00690] 9. Igual ao Procedimento Geral
[00691] 10. Igual ao Procedimento Geral.
[00692] Células A549 (ATCC) foram empregadas neste teste. As células foram cultivadas no meio de crescimento (RPMI + FBS a 10%) em placas de 96 poços e cultivadas durante a noite a 37°C para ligação. AS células foram expostas ao meio de inanição (RPMI + 0,05% BSA) Diluições dos inibidores foram adicionados para as placas e incubadas a 37°C por 1 hora. As células foram então estimulada por adição de HGF 40 ng/mL por 15 minutos. As células foram lavada uma vez com Na3VO4 mM em HBSS e em seguida lisadas. Os lisados foram diluídos com Na3VO4 1 mM em HBSS e transferidos para uma placa revestida com anti-coelho de cabra de 96 poços (Pierce), que foi pré-revestida com anticorpo anti-HGFR (Zymed Laboratories). As placas foram incubadas durante a noite a 4°C e lavadas com Tween 20 a 1% em PBS por sete vezes. HRP-PY20 (Santa Cruz) foi diluído e adicionado para as placas durante 30 minutos de incubação. As placas foram então lavadas novamente e substrato de TMB peroxidase (Kirkergaard & Perry) foi adicionado e incubado por 10 minutos. A reação foi então interrompida adicionando-se H2SO4 0,09N AS pacas foram medidas a OD-450 nm usando um espectrofotômetro. Valores IC50 foram calculados por ajuste de curva usando uma análise de quatro parâmetros.
[00693] Os compostos da invenção foram medidos para atividade de inibição de HGFR; os dados demonstram-se em cada Exemplo. Dados Ki foram obtidos usando o Ensaio de Autofosforilação de HGFR Celular, ambos descritos supra.
[00694] Embora a invenção tenha sido ilustrada com referência a modalidades específicas e preferidas, os versados na técnica reconhecerão que, variações e modificações serão feitas, por meio de experimentação e prática rotineiras da invenção. Assim, a invenção pretende não estar limitada pela descrição precedente, mas ser determinada pelas reivindicações apensas e seus equivalentes.
[00695] Todas as referências aqui mencionadas, incluindo quaisquer documentos de prioridade, estão ora incorporadas por referência, em sua totalidade.
Claims (3)
1. Uso de um composto que apresenta a fórmula: ou um sal farmaceuticamente aceitável destes, caracterizado pelo fato de que é para preparação de um medicamento para o tratamento de crescimento celular anormal em um mamífero.
ou um sal farmaceuticamente aceitável destes, caracterizado pelo fato de que é para preparação de um medicamento para o tratamento de crescimento celular anormal em um mamífero.
2. Uso de acordo com a reivindicação 1, caracterizado pelo fato de que o tratamento de crescimento celular anormal é câncer.
3. Uso de acordo com a reivindicação 1, caracterizado pelo fato de que o composto é um composto enantioméricamente puro.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US60508604P | 2004-08-26 | 2004-08-26 | |
| US60/605,086 | 2004-08-26 | ||
| BRPI0513915-5A BRPI0513915A (pt) | 2004-08-26 | 2005-08-15 | compostos aminoeteroarila enantiomericamente puros como inibidores de proteìna quinase |
| PCT/IB2005/002837 WO2006021884A2 (en) | 2004-08-26 | 2005-08-15 | Enantiomerically pure aminoheteroaryl compounds as protein kinase inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| BR122020017756B1 true BR122020017756B1 (pt) | 2022-02-15 |
Family
ID=35967909
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BR122020017756-0A BR122020017756B1 (pt) | 2004-08-26 | 2005-08-15 | Uso de compostos de aminoeteroarila enantiomericamente puros na preparação de um medicamento para o tratamento de crescimento celular anormal em um mamífero |
| BRPI0513915-5A BRPI0513915A (pt) | 2004-08-26 | 2005-08-15 | compostos aminoeteroarila enantiomericamente puros como inibidores de proteìna quinase |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BRPI0513915-5A BRPI0513915A (pt) | 2004-08-26 | 2005-08-15 | compostos aminoeteroarila enantiomericamente puros como inibidores de proteìna quinase |
Country Status (48)
Families Citing this family (197)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2517256C (en) * | 2003-02-26 | 2013-04-30 | Sugen, Inc. | Aminoheteroaryl compounds as protein kinase inhibitors |
| MY150088A (en) | 2003-05-19 | 2013-11-29 | Irm Llc | Immunosuppressant compounds and compositions |
| ME01309B (me) * | 2004-08-26 | 2013-12-20 | Pfizer | Pirazolom supstituirani heteroarilni spojevi kao inhibitori proteinskih kinaza |
| BR122020017756B1 (pt) * | 2004-08-26 | 2022-02-15 | Pfizer Inc | Uso de compostos de aminoeteroarila enantiomericamente puros na preparação de um medicamento para o tratamento de crescimento celular anormal em um mamífero |
| KR101146852B1 (ko) * | 2005-12-05 | 2012-05-16 | 화이자 프로덕츠 인크. | C?met/hgfr 억제제의 다형체 |
| DE602006018354D1 (de) * | 2005-12-05 | 2010-12-30 | Pfizer Prod Inc | Verfahren zur behandlung von abnormalem zellwachstum |
| ES2336625T3 (es) * | 2006-03-22 | 2010-04-14 | Vertex Pharmaceuticals Incorporated | Inhibidores de la proteina quinasa c-met para el tratamiento de trastornos proliferativos. |
| EP2450437B1 (en) | 2006-04-14 | 2017-05-17 | Cell Signaling Technology, Inc. | Gene defects and mutant ALK kinase in human solid tumors |
| US8168383B2 (en) | 2006-04-14 | 2012-05-01 | Cell Signaling Technology, Inc. | Gene defects and mutant ALK kinase in human solid tumors |
| US7683060B2 (en) * | 2006-08-07 | 2010-03-23 | Incyte Corporation | Triazolotriazines as kinase inhibitors |
| GB0621607D0 (en) * | 2006-10-31 | 2006-12-06 | Chroma Therapeutics Ltd | Inhibitors of c-Met |
| ME02372B (me) | 2006-11-22 | 2016-06-20 | Incyte Holdings Corp | Imidazotriazini i imidazopiramidini kao inhibitori kinaze |
| US8455513B2 (en) | 2007-01-10 | 2013-06-04 | Aerie Pharmaceuticals, Inc. | 6-aminoisoquinoline compounds |
| WO2008088881A1 (en) * | 2007-01-19 | 2008-07-24 | Xcovery, Inc. | Kinase inhibitor compounds |
| CA2683559C (en) | 2007-04-13 | 2019-09-24 | Dana Farber Cancer Institute, Inc. | Methods for treating cancer resistant to erbb therapeutics |
| KR101586503B1 (ko) | 2007-09-13 | 2016-01-18 | 코덱시스, 인코포레이티드 | 아세토페논의 환원을 위한 케토리덕타제 폴리펩티드 |
| WO2009099982A1 (en) * | 2008-02-04 | 2009-08-13 | Osi Pharmaceuticals, Inc. | 2-aminopyridine kinase inhibitors |
| AR070317A1 (es) * | 2008-02-06 | 2010-03-31 | Osi Pharm Inc | Furo (3,2-c) piridina y tieno (3,2-c) piridinas |
| EP2269993B1 (en) | 2008-04-23 | 2013-02-27 | Kyowa Hakko Kirin Co., Ltd. | 2-aminoquinazoline derivative |
| WO2009143211A2 (en) | 2008-05-21 | 2009-11-26 | Incyte Corporation | Salts of 2-fluoro-n-methyl-4-[7-(quinolin-6-yl-methyl)- imidazo[1,2-b][1,2,4]triazin-2-yl]benzamide and processes related to preparing the same |
| EP2143441A1 (en) * | 2008-07-08 | 2010-01-13 | Pierre Fabre Medicament | Combination of a c-Met antagonist and an aminoheteroaryl compound for the treatment of cancer |
| US8450344B2 (en) | 2008-07-25 | 2013-05-28 | Aerie Pharmaceuticals, Inc. | Beta- and gamma-amino-isoquinoline amide compounds and substituted benzamide compounds |
| JP2012509342A (ja) * | 2008-11-20 | 2012-04-19 | オーエスアイ・フアーマスーテイカルズ・インコーポレーテツド | 置換ピロロ[2,3−b]−ピリジンおよび−ピラジン |
| CN102256942B (zh) | 2008-12-18 | 2013-07-24 | 诺瓦提斯公司 | 1-(4-{1-[(e)-4-环己基-3-三氟甲基-苄氧基亚氨基]-乙基}-2-乙基-苄基)-氮杂环丁烷-3-甲酸的多晶型物 |
| ES2663222T3 (es) | 2008-12-19 | 2018-04-11 | Vertex Pharmaceuticals Incorporated | Derivados de pirazina útiles como inhibidores de la quinasa ATR |
| EP3053913B1 (en) | 2009-05-01 | 2018-03-07 | Aerie Pharmaceuticals, Inc. | Dual mechanism inhibitors for the treatment of disease |
| EP3613729A1 (en) | 2009-06-10 | 2020-02-26 | Chugai Seiyaku Kabushiki Kaisha | Tetracyclic compounds |
| DE102009056886A1 (de) | 2009-12-03 | 2011-06-09 | Bayer Schering Pharma Aktiengesellschaft | cMet-Inhibitoren zur Behandlung der Endometriose |
| EA025304B1 (ru) | 2010-02-03 | 2016-12-30 | Инсайт Холдингс Корпорейшн | ИМИДАЗО[1,2-b][1,2,4]ТРИАЗИНЫ В КАЧЕСТВЕ c-Met ИНГИБИТОРОВ |
| EP2566858A2 (en) * | 2010-05-04 | 2013-03-13 | Pfizer Inc. | Heterocyclic derivatives as alk inhibitors |
| US9630956B2 (en) | 2010-05-12 | 2017-04-25 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| JP2013529200A (ja) * | 2010-05-12 | 2013-07-18 | バーテックス ファーマシューティカルズ インコーポレイテッド | Atrキナーゼ阻害剤として有用な化合物 |
| JP2013526540A (ja) | 2010-05-12 | 2013-06-24 | バーテックス ファーマシューティカルズ インコーポレイテッド | Atrキナーゼ阻害剤として有用な化合物 |
| WO2011143646A1 (en) | 2010-05-14 | 2011-11-17 | OSI Pharmaceuticals, LLC | Fused bicyclic kinase inhibitors |
| AR081039A1 (es) | 2010-05-14 | 2012-05-30 | Osi Pharmaceuticals Llc | Inhibidores biciclicos fusionados de quinasa |
| JP5960688B2 (ja) | 2010-05-17 | 2016-08-02 | インコゼン セラピューティクス プライベート リミテッド | プロテインキナーゼ調節物質としての新規3,5−二置換−3h−[1,2,3]トリアゾロ[4,5−b]ピリジン化合物 |
| US20130315895A1 (en) | 2010-07-01 | 2013-11-28 | Takeda Pharmaceutical Company Limited | COMBINATION OF A cMET INHIBITOR AND AN ANTIBODY TO HGF AND/OR cMET |
| CA2808210C (en) | 2010-08-20 | 2019-10-01 | Chugai Seiyaku Kabushiki Kaisha | Tetracyclic compounds having alk inhibitory activity and compositions thereof |
| WO2012042421A1 (en) | 2010-09-29 | 2012-04-05 | Pfizer Inc. | Method of treating abnormal cell growth |
| CN102464667B (zh) * | 2010-11-03 | 2014-06-04 | 中国科学院上海药物研究所 | 一类五元杂环并嘧啶类化合物及其制备方法和用途 |
| MX2013009551A (es) | 2011-02-24 | 2013-09-06 | Jiangsu Hanson Pharmaceutical Co Ltd | Compuestos que contienen fosforo como inhibidores de proteina cinasa. |
| EP2680843A4 (en) * | 2011-03-03 | 2015-05-06 | Concert Pharmaceuticals Inc | DERIVATIVES OF PYRAZOLSUBSTITUTED AMINO-HETEROARYL COMPOUNDS |
| US9145390B2 (en) | 2011-03-03 | 2015-09-29 | Concert Pharmaceuticals, Inc. | Derivatives of pyrazole-substituted amino-heteroaryl compounds |
| CN102718745A (zh) * | 2011-03-30 | 2012-10-10 | 中国科学院上海药物研究所 | 新型胺基吡啶类化合物、其制备方法、包含此类化合物的药物组合物及其用途 |
| EP2710003A1 (en) | 2011-05-16 | 2014-03-26 | OSI Pharmaceuticals, LLC | Fused bicyclic kinase inhibitors |
| CN102850328B (zh) * | 2011-07-01 | 2014-12-24 | 苏州东南药业股份有限公司 | 吡啶类化合物、其制备方法、包含该化合物的药物组合物及其用途 |
| AU2012291744A1 (en) | 2011-08-02 | 2014-02-20 | Pfizer Inc. | Crizotinib for use in the treatment of cancer |
| EP2758387A4 (en) * | 2011-09-21 | 2015-03-11 | Teligene Ltd | Pyridine compounds as kinase inhibitors |
| SG10201606774UA (en) | 2011-09-30 | 2016-10-28 | Vertex Pharma | Processes for making compounds useful as inhibitors of atr kinase |
| WO2013049859A1 (en) | 2011-09-30 | 2013-04-04 | Vertex Pharmaceuticals Incorporated | Treating pancreatic cancer and non-small cell lung cancer with atr inhibitors |
| CN103204844A (zh) * | 2012-01-17 | 2013-07-17 | 上海艾力斯医药科技有限公司 | 氨基杂芳基化合物及其制备方法与应用 |
| HK1210969A1 (en) | 2012-01-20 | 2016-05-13 | 奥克塞拉有限公司 | Substituted heterocyclic compounds for disease treatment |
| EP2620140A1 (en) | 2012-01-26 | 2013-07-31 | ratiopharm GmbH | Crizotinib containing compositions |
| HUE034118T2 (en) | 2012-03-06 | 2018-01-29 | Pfizer | Macrocyclic derivatives for the treatment of proliferative diseases |
| EP2831073B1 (en) | 2012-03-30 | 2020-12-09 | Rhizen Pharmaceuticals S.A. | Novel 3,5-disubstitued-3h-imidazo[4,5-b]pyridine and 3,5- disubstitued -3h-[1,2,3]triazolo[4,5-b]pyridine compounds as modulators of c-met protein kinases |
| CA2869309C (en) | 2012-04-05 | 2021-02-09 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of atr kinase and combination therapies thereof |
| CN103373986B (zh) * | 2012-04-22 | 2016-10-05 | 东南大学 | 克里唑替尼前药及其制备方法与用途 |
| CN103387535B (zh) * | 2012-05-10 | 2016-06-01 | 广东东阳光药业有限公司 | 取代的炔基吡啶化合物及其使用方法和用途 |
| CN103420987B (zh) * | 2012-05-15 | 2015-11-25 | 上海医药工业研究院 | 吡啶衍生物及其制备方法 |
| CN103420906B (zh) * | 2012-05-21 | 2015-09-09 | 南京圣和药业股份有限公司 | 新型酪氨酸蛋白激酶抑制剂 |
| WO2013181251A1 (en) | 2012-05-29 | 2013-12-05 | Ratiopharm Gmbh | Crizotinib hydrochloride salt in crystalline |
| WO2013192512A1 (en) * | 2012-06-22 | 2013-12-27 | Concert Pharmaceuticals, Inc. | Derivatives of pyrazole-substituted amino-heteroaryl compounds |
| CN103509008A (zh) * | 2012-06-22 | 2014-01-15 | 康瑟特制药公司 | 吡唑取代的氨基-杂芳基化合物的衍生物 |
| WO2014020467A2 (en) | 2012-07-30 | 2014-02-06 | Fresenius Kabi Oncology Ltd | Process for the preparation of pyrazole substituted aminoheteroaryl compounds |
| AU2013307383A1 (en) | 2012-08-27 | 2015-03-26 | Cemm - Forschungszentrum Fur Molekulare Medizin Gmbh | Aminoheteroaryl compounds as MTH1 inhibitors |
| CN104603620B (zh) | 2012-09-24 | 2018-02-23 | 文塔纳医疗系统公司 | 使用间变性淋巴瘤激酶(alk)作为标志物鉴定治疗响应性非小细胞肺癌的方法 |
| SI2902029T1 (sl) | 2012-09-25 | 2018-11-30 | Chugai Seiyaku Kabushiki Kaisha | Inhibitor ret |
| US8999632B2 (en) | 2012-10-04 | 2015-04-07 | Vertex Pharmaceuticals Incorporated | Method for measuring ATR inhibition mediated increases in DNA damage |
| WO2014081816A1 (en) * | 2012-11-21 | 2014-05-30 | Concert Pharmaceuticals, Inc. | Fluoro-derivatives of pyrazole-substituted amino-heteroaryl compounds |
| CN104016979B (zh) * | 2012-11-23 | 2017-05-03 | 广东东阳光药业有限公司 | 取代的环化合物及其使用方法和用途 |
| WO2014089324A1 (en) * | 2012-12-07 | 2014-06-12 | Calitor Sciences, Llc | Substituted cyclic compounds and methods of use |
| CA2896187A1 (en) | 2013-01-18 | 2014-07-24 | F. Hoffmann-La Roche Ag | 3-substituted pyrazoles and use as dlk inhibitors |
| WO2014115169A2 (en) * | 2013-01-24 | 2014-07-31 | Hetero Research Foundation | Crizotinib solid dispersion |
| AU2014211856C1 (en) * | 2013-02-02 | 2018-04-12 | Centaurus Biopharma Co., Ltd. | Substituted 2-aminopyridine protein kinase inhibitor |
| WO2014139391A1 (en) * | 2013-03-11 | 2014-09-18 | Teligene Ltd | Substituted pyridine compounds as kinases inhibitors |
| DK3811943T3 (da) | 2013-03-15 | 2023-04-03 | Aerie Pharmaceuticals Inc | Forbindelse til anvendelse til behandling af øjenlidelser |
| WO2014203177A1 (en) * | 2013-06-18 | 2014-12-24 | Shilpa Medicare Limited | Amorphous (r) -3- [1- (2, 6-dichloro-3-fluorophenyl) methoxy] -5- [1- (piperidin-4- yl) -1h-pyrazol-4-yl] pyridin-2-amine |
| CN104650049B (zh) * | 2013-08-28 | 2018-06-08 | 广东东阳光药业有限公司 | 取代的吡啶化合物及其使用方法和用途 |
| WO2015034729A1 (en) * | 2013-09-05 | 2015-03-12 | Calitor Sciences, Llc | Substituted pyridine compounds and methods of use |
| WO2015036898A2 (en) * | 2013-09-10 | 2015-03-19 | Shilpa Medicare Limited | Novel salts of crizotinib and their preparation |
| CN105745206B (zh) * | 2013-09-20 | 2019-08-13 | 生物马林药物股份有限公司 | 用于治疗疾病的葡萄糖神经酰胺合成酶抑制剂 |
| CN104513253A (zh) | 2013-10-01 | 2015-04-15 | 南京波尔泰药业科技有限公司 | 用于治疗增殖性疾病的大环化合物 |
| WO2015069922A2 (en) | 2013-11-06 | 2015-05-14 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Alk antibodies, conjugates, and chimeric antigen receptors, and their use |
| CN103755627B (zh) * | 2014-01-09 | 2016-02-17 | 定陶县友帮化工有限公司 | 2-氨基-3-羟基-5-氯吡啶的合成方法 |
| EA032374B1 (ru) * | 2014-01-29 | 2019-05-31 | ЮСиБи БАЙОФАРМА СПРЛ | Гетероарильные амиды в качестве ингибиторов агрегации белков |
| JP6247992B2 (ja) * | 2014-04-17 | 2017-12-13 | 株式会社ダイセル | ハロゲン化合物の製造方法 |
| WO2015163448A1 (ja) | 2014-04-25 | 2015-10-29 | 中外製薬株式会社 | 4環性化合物を高用量含有する製剤 |
| WO2015163447A1 (ja) | 2014-04-25 | 2015-10-29 | 中外製薬株式会社 | 4環性化合物の新規結晶 |
| WO2016015676A1 (zh) | 2014-07-31 | 2016-02-04 | 正大天晴药业集团股份有限公司 | 吡啶取代的2-氨基吡啶类蛋白激酶抑制剂 |
| TWI831347B (zh) | 2014-08-08 | 2024-02-01 | 日商中外製藥股份有限公司 | 包含4環性化合物的非晶質體之固體分散體及製劑 |
| CN105348265A (zh) * | 2014-08-19 | 2016-02-24 | 蔡苹 | 一种具有ALK和c-Met抑制活性的2,4-二取代杂环三氮唑类化合物的制备和应用 |
| CA2955676A1 (en) | 2014-08-25 | 2016-03-03 | Pfizer Inc. | Combination of a pd-1 antagonist and an alk inhibitor for treating cancer |
| CN105820113B (zh) * | 2015-01-07 | 2018-04-20 | 爱技特科技(北京)有限公司 | 一种克唑替尼手性中间体的制备方法 |
| CA2973857C (en) | 2015-01-16 | 2023-11-07 | Chugai Seiyaku Kabushiki Kaisha | Combination drug |
| CN104693184A (zh) * | 2015-03-17 | 2015-06-10 | 安润医药科技(苏州)有限公司 | 克唑替尼的合成方法 |
| CA3000386A1 (en) | 2015-09-30 | 2017-04-06 | Merck Patent Gmbh | Combination of a pd-1 axis binding antagonist and an alk inhibitor for treating alk-negative cancer |
| HK1258570A1 (zh) | 2015-09-30 | 2019-11-15 | Vertex Pharmaceuticals Inc. | 使用dna损伤剂及atr抑制剂的组合治疗癌症的方法 |
| US10550087B2 (en) * | 2015-11-17 | 2020-02-04 | Aerie Pharmaceuticals, Inc. | Process for the preparation of kinase inhibitors and intermediates thereof |
| US9643927B1 (en) | 2015-11-17 | 2017-05-09 | Aerie Pharmaceuticals, Inc. | Process for the preparation of kinase inhibitors and intermediates thereof |
| WO2017201502A1 (en) | 2016-05-20 | 2017-11-23 | Biohaven Pharmaceutical Holding Company Ltd. | Use of glutamate modulating agents with immunotherapies to treat cancer |
| CN106083708A (zh) * | 2016-06-30 | 2016-11-09 | 浙江大学 | 含2‑吡啶酮环侧链的2‑氨基吡啶衍生物及制备和应用 |
| KR102568079B1 (ko) | 2016-08-31 | 2023-08-17 | 에어리 파마슈티컬즈, 인코포레이티드 | 안과용 조성물 |
| WO2018119183A2 (en) | 2016-12-22 | 2018-06-28 | Amgen Inc. | Kras g12c inhibitors and methods of using the same |
| CN106831720B (zh) * | 2017-01-21 | 2019-04-16 | 河北科技大学 | 双-4-(1h-吡唑-1-基)哌啶-1-甲酸叔丁酯的合成方法及其应用 |
| KR20190135027A (ko) | 2017-03-31 | 2019-12-05 | 에어리 파마슈티컬즈, 인코포레이티드 | 아릴 시클로프로필-아미노-이소퀴놀리닐 아미드 화합물 |
| EA201992781A1 (ru) | 2017-05-22 | 2020-04-01 | Эмджен Инк. | Ингибиторы g12c kras и способы их применения |
| US10180422B1 (en) | 2017-08-22 | 2019-01-15 | Scripps Health | Methods of treating a neuroendocrine tumor |
| MA50077A (fr) | 2017-09-08 | 2020-07-15 | Amgen Inc | Inhibiteurs de kras g12c et leurs procédés d'utilisation |
| JP6635999B2 (ja) * | 2017-10-13 | 2020-01-29 | 株式会社ダイセル | カリウム塩の製造方法、及びカリウム塩 |
| CN108191833A (zh) * | 2018-01-17 | 2018-06-22 | 浙江树人学院 | 克唑替尼衍生物及其制备方法和应用 |
| WO2019206049A1 (en) * | 2018-04-25 | 2019-10-31 | Zhuhai Yufan Biotechnologies Co., Ltd | Hpk1 inhibitors, preparation method and application thereof |
| CN110396087A (zh) * | 2018-04-25 | 2019-11-01 | 珠海宇繁生物科技有限责任公司 | Hpk1激酶抑制剂、制备方法及其应用 |
| ES2995514T3 (en) | 2018-05-04 | 2025-02-10 | Amgen Inc | Kras g12c inhibitors and methods of using the same |
| WO2019213516A1 (en) | 2018-05-04 | 2019-11-07 | Amgen Inc. | Kras g12c inhibitors and methods of using the same |
| EP3790886B1 (en) | 2018-05-10 | 2024-06-26 | Amgen Inc. | Kras g12c inhibitors for the treatment of cancer |
| ES2938987T3 (es) | 2018-06-01 | 2023-04-18 | Amgen Inc | Inhibidores de KRAS G12c y métodos de uso de los mismos |
| US20190375749A1 (en) | 2018-06-11 | 2019-12-12 | Amgen Inc. | Kras g12c inhibitors and methods of using the same |
| ES3060664T3 (en) | 2018-06-12 | 2026-03-27 | Amgen Inc | Kras g12c inhibitors encompassing a piperazine ring and use thereof in the treatment of cancer |
| EP4316598B1 (en) | 2018-06-29 | 2025-06-18 | Chugai Seiyaku Kabushiki Kaisha | Pharmaceutical composition comprising a poorly soluble basic agent |
| BR112021001145A2 (pt) | 2018-09-04 | 2021-04-20 | Chugai Seiyaku Kabushiki Kaisha | método para a produção de composto tetracíclico |
| AU2019337703B2 (en) | 2018-09-14 | 2023-02-02 | Aerie Pharmaceuticals, Inc. | Aryl cyclopropyl-amino-isoquinolinyl amide compounds |
| JP7516029B2 (ja) | 2018-11-16 | 2024-07-16 | アムジエン・インコーポレーテツド | Kras g12c阻害剤化合物の重要な中間体の改良合成法 |
| US11053226B2 (en) | 2018-11-19 | 2021-07-06 | Amgen Inc. | KRAS G12C inhibitors and methods of using the same |
| JP7377679B2 (ja) | 2018-11-19 | 2023-11-10 | アムジエン・インコーポレーテツド | がん治療のためのkrasg12c阻害剤及び1種以上の薬学的に活性な追加の薬剤を含む併用療法 |
| TWI844602B (zh) | 2018-12-20 | 2024-06-11 | 美商安進公司 | Kif18a抑制劑 |
| JP7676308B2 (ja) | 2018-12-20 | 2025-05-14 | アムジエン・インコーポレーテツド | Kif18a阻害剤として有用なヘテロアリールアミド |
| EP3898616B1 (en) | 2018-12-20 | 2024-10-02 | Amgen Inc. | Heteroaryl amides useful as kif18a inhibitors |
| WO2020132651A1 (en) | 2018-12-20 | 2020-06-25 | Amgen Inc. | Kif18a inhibitors |
| JPWO2020130125A1 (ja) | 2018-12-21 | 2021-11-04 | 第一三共株式会社 | 抗体−薬物コンジュゲートとキナーゼ阻害剤の組み合わせ |
| CN113727758A (zh) | 2019-03-01 | 2021-11-30 | 锐新医药公司 | 双环杂环基化合物及其用途 |
| WO2020180768A1 (en) | 2019-03-01 | 2020-09-10 | Revolution Medicines, Inc. | Bicyclic heteroaryl compounds and uses thereof |
| EP3738593A1 (en) | 2019-05-14 | 2020-11-18 | Amgen, Inc | Dosing of kras inhibitor for treatment of cancers |
| NZ782284A (en) | 2019-05-21 | 2024-11-29 | Amgen Inc | Solid state forms |
| US12540129B2 (en) | 2019-08-02 | 2026-02-03 | Amgen Inc. | KIF18A inhibitors |
| AU2020326627B2 (en) | 2019-08-02 | 2026-01-29 | Amgen Inc. | KIF18A inhibitors |
| WO2021026099A1 (en) | 2019-08-02 | 2021-02-11 | Amgen Inc. | Kif18a inhibitors |
| EP4007756B1 (en) | 2019-08-02 | 2025-12-24 | Amgen Inc. | Kif18a inhibitors |
| EP4725489A2 (en) | 2019-08-02 | 2026-04-15 | OneHealthCompany, Inc. | Treatment of canine cancers |
| EP4031542B1 (en) | 2019-09-18 | 2025-10-15 | Merck Sharp & Dohme LLC | Small molecule inhibitors of kras g12c mutant |
| CN112552293A (zh) * | 2019-09-25 | 2021-03-26 | 珠海宇繁生物科技有限责任公司 | 一种protac小分子化合物及其应用 |
| EP4684786A3 (en) | 2019-10-24 | 2026-04-08 | Amgen Inc. | Pyridopyrimidine derivatives useful as kras g12c and kras g12d inhibitors in the treatment of cancer |
| CN114867726B (zh) | 2019-10-28 | 2023-11-28 | 默沙东有限责任公司 | Kras g12c突变体的小分子抑制剂 |
| WO2021085653A1 (en) | 2019-10-31 | 2021-05-06 | Taiho Pharmaceutical Co., Ltd. | 4-aminobut-2-enamide derivatives and salts thereof |
| MX2022005359A (es) | 2019-11-04 | 2022-06-02 | Revolution Medicines Inc | Inhibidores de ras. |
| WO2021091956A1 (en) | 2019-11-04 | 2021-05-14 | Revolution Medicines, Inc. | Ras inhibitors |
| CN120699039A (zh) | 2019-11-04 | 2025-09-26 | 锐新医药公司 | Ras抑制剂 |
| TW202128688A (zh) | 2019-11-08 | 2021-08-01 | 美商銳新醫藥公司 | 雙環雜芳基化合物及其用途 |
| WO2021097207A1 (en) | 2019-11-14 | 2021-05-20 | Amgen Inc. | Improved synthesis of kras g12c inhibitor compound |
| JP2023501528A (ja) | 2019-11-14 | 2023-01-18 | アムジエン・インコーポレーテツド | Kras g12c阻害剤化合物の改善された合成 |
| EP4065231A1 (en) | 2019-11-27 | 2022-10-05 | Revolution Medicines, Inc. | Covalent ras inhibitors and uses thereof |
| WO2021106231A1 (en) | 2019-11-29 | 2021-06-03 | Taiho Pharmaceutical Co., Ltd. | A compound having inhibitory activity against kras g12d mutation |
| CN114929279A (zh) | 2020-01-07 | 2022-08-19 | 锐新医药公司 | Shp2抑制剂给药和治疗癌症的方法 |
| WO2021196655A1 (zh) * | 2020-04-03 | 2021-10-07 | 中国药科大学 | 含苯并咪唑结构的化合物及其制备方法与用途 |
| WO2021215545A1 (en) | 2020-04-24 | 2021-10-28 | Taiho Pharmaceutical Co., Ltd. | Anticancer combination therapy with n-(1-acryloyl-azetidin-3-yl)-2-((1h-indazol-3-yl)amino)methyl)-1h-imidazole-5-carboxamide inhibitor of kras-g12c |
| WO2021215544A1 (en) | 2020-04-24 | 2021-10-28 | Taiho Pharmaceutical Co., Ltd. | Kras g12d protein inhibitors |
| IL299131A (en) | 2020-06-18 | 2023-02-01 | Revolution Medicines Inc | Methods for delaying, preventing and treating acquired resistance to RAS inhibitors |
| EP4183395A4 (en) | 2020-07-15 | 2024-07-24 | Taiho Pharmaceutical Co., Ltd. | PYRIMIDINE COMPOUND-CONTAINING COMBINATION FOR USE IN TUMOR TREATMENT |
| MX2023002248A (es) | 2020-09-03 | 2023-05-16 | Revolution Medicines Inc | Uso de inhibidores de sos1 para tratar neoplasias malignas con mutaciones de shp2. |
| MX2023003060A (es) | 2020-09-15 | 2023-04-05 | Revolution Medicines Inc | Derivados indolicos como inhibidores de ras en el tratamiento del cancer. |
| CN114437058A (zh) * | 2020-10-30 | 2022-05-06 | 珠海宇繁生物科技有限责任公司 | 氘代hpk1激酶抑制剂及其制备方法和应用 |
| CN116710431A (zh) * | 2020-11-19 | 2023-09-05 | 泰洛治疗公司 | 小分子化合物和组合物 |
| US20240051956A1 (en) | 2020-12-22 | 2024-02-15 | Qilu Regor Therapeutics Inc. | Sos1 inhibitors and uses thereof |
| CA3217393A1 (en) | 2021-05-05 | 2022-11-10 | Elena S. Koltun | Ras inhibitors |
| AU2022268962A1 (en) | 2021-05-05 | 2023-12-14 | Revolution Medicines, Inc. | Ras inhibitors for the treatment of cancer |
| WO2022235866A1 (en) | 2021-05-05 | 2022-11-10 | Revolution Medicines, Inc. | Covalent ras inhibitors and uses thereof |
| WO2022250170A1 (en) | 2021-05-28 | 2022-12-01 | Taiho Pharmaceutical Co., Ltd. | Small molecule inhibitors of kras mutated proteins |
| AR127308A1 (es) | 2021-10-08 | 2024-01-10 | Revolution Medicines Inc | Inhibidores ras |
| WO2023064058A1 (en) | 2021-10-12 | 2023-04-20 | Peloton Therapeutics Inc. | Tricyclic sultams and sulfamides as antitumor agents |
| TW202340214A (zh) | 2021-12-17 | 2023-10-16 | 美商健臻公司 | 做為shp2抑制劑之吡唑并吡𠯤化合物 |
| EP4227307A1 (en) | 2022-02-11 | 2023-08-16 | Genzyme Corporation | Pyrazolopyrazine compounds as shp2 inhibitors |
| WO2023172858A1 (en) | 2022-03-07 | 2023-09-14 | Amgen Inc. | A process for preparing 4-methyl-2-propan-2-yl-pyridine-3-carbonitrile |
| CN119136806A (zh) | 2022-03-08 | 2024-12-13 | 锐新医药公司 | 用于治疗免疫难治性肺癌的方法 |
| US20250195497A1 (en) | 2022-03-22 | 2025-06-19 | Kyoto University | Therapeutic or prophylactic medicine for fragile x syndrome |
| CN114767676B (zh) * | 2022-04-22 | 2024-04-19 | 珠海宇繁生物科技有限责任公司 | Hpk1激酶抑制剂在预防和/或治疗人的病原体感染中的应用 |
| WO2023240263A1 (en) | 2022-06-10 | 2023-12-14 | Revolution Medicines, Inc. | Macrocyclic ras inhibitors |
| AU2023358792A1 (en) | 2022-10-14 | 2025-04-17 | Black Diamond Therapeutics, Inc. | Methods of treating cancers using isoquinoline or 6-aza-quinoline derivatives |
| EP4687905A1 (en) | 2023-03-30 | 2026-02-11 | Revolution Medicines, Inc. | Compositions for inducing ras gtp hydrolysis and uses thereof |
| CN121263418A (zh) | 2023-04-07 | 2026-01-02 | 锐新医药公司 | 大环ras抑制剂 |
| KR20260005904A (ko) | 2023-04-07 | 2026-01-12 | 레볼루션 메디슨즈, 인크. | 매크로사이클릭 ras 억제제 |
| AU2024252105A1 (en) | 2023-04-14 | 2025-10-16 | Revolution Medicines, Inc. | Crystalline forms of ras inhibitors, compositions containing the same, and methods of use thereof |
| KR20250172857A (ko) | 2023-04-14 | 2025-12-09 | 레볼루션 메디슨즈, 인크. | Ras 억제제의 결정형 |
| AU2024265078A1 (en) | 2023-05-04 | 2025-12-11 | Revolution Medicines, Inc. | Combination therapy for a ras related disease or disorder |
| IL326136A (en) | 2023-08-07 | 2026-03-01 | Revolution Medicines Inc | RMC-6291 for use in the treatment of a disease or disorder associated with the RAS protein |
| KR20260042550A (ko) | 2023-08-24 | 2026-03-31 | 다이호야쿠힌고교 가부시키가이샤 | 세다주리딘 및 아자시티딘의 고정 용량 조합물 |
| US20250154171A1 (en) | 2023-10-12 | 2025-05-15 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2025085748A1 (en) | 2023-10-20 | 2025-04-24 | Merck Sharp & Dohme Llc | Small molecule inhibitors of kras proteins |
| TW202542151A (zh) | 2023-12-22 | 2025-11-01 | 美商銳格醫藥有限公司 | Sos1抑制劑及其用途 |
| WO2025240847A1 (en) | 2024-05-17 | 2025-11-20 | Revolution Medicines, Inc. | Ras inhibitors |
| US20250375445A1 (en) | 2024-06-07 | 2025-12-11 | Revolution Medicines, Inc. | Methods of treating a ras protein-related disease or disorder |
| WO2025265060A1 (en) | 2024-06-21 | 2025-12-26 | Revolution Medicines, Inc. | Therapeutic compositions and methods for managing treatment-related effects |
| WO2026006747A1 (en) | 2024-06-28 | 2026-01-02 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2026015801A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026015796A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026015825A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Use of ras inhibitor for treating pancreatic cancer |
| WO2026015790A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026050446A1 (en) | 2024-08-29 | 2026-03-05 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2026072904A2 (en) | 2024-09-26 | 2026-04-02 | Revolution Medicines, Inc. | Compositions and methods for treating lung cancer |
Family Cites Families (80)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB129611A (en) | 1919-04-15 | 1919-07-17 | John Norman Riddell Hannam | Improvements in Stropping or Sharpening Devices for Safety Razor Blades. |
| GB8827305D0 (en) | 1988-11-23 | 1988-12-29 | British Bio Technology | Compounds |
| US5376645A (en) | 1990-01-23 | 1994-12-27 | University Of Kansas | Derivatives of cyclodextrins exhibiting enhanced aqueous solubility and the use thereof |
| KR0166088B1 (ko) | 1990-01-23 | 1999-01-15 | . | 수용해도가 증가된 시클로덱스트린 유도체 및 이의 용도 |
| FI941572L (fi) * | 1991-10-07 | 1994-05-27 | Oncologix Inc | Anti-erbB-2-monoklonaalisten vasta-aineiden yhdistelmä ja käyttömenetelmä |
| AU675929B2 (en) * | 1992-02-06 | 1997-02-27 | Curis, Inc. | Biosynthetic binding protein for cancer marker |
| US6177401B1 (en) | 1992-11-13 | 2001-01-23 | Max-Planck-Gesellschaft Zur Forderung Der Wissenschaften | Use of organic compounds for the inhibition of Flk-1 mediated vasculogenesis and angiogenesis |
| US5455258A (en) | 1993-01-06 | 1995-10-03 | Ciba-Geigy Corporation | Arylsulfonamido-substituted hydroxamic acids |
| IL112248A0 (en) | 1994-01-25 | 1995-03-30 | Warner Lambert Co | Tricyclic heteroaromatic compounds and pharmaceutical compositions containing them |
| US5863949A (en) * | 1995-03-08 | 1999-01-26 | Pfizer Inc | Arylsulfonylamino hydroxamic acid derivatives |
| DK0821671T3 (da) * | 1995-04-20 | 2001-04-23 | Pfizer | Arylsulfonylhydroxamsyrederivater som MMP- og TNF-inhibitorer |
| US5747498A (en) * | 1996-05-28 | 1998-05-05 | Pfizer Inc. | Alkynyl and azido-substituted 4-anilinoquinazolines |
| US5880141A (en) * | 1995-06-07 | 1999-03-09 | Sugen, Inc. | Benzylidene-Z-indoline compounds for the treatment of disease |
| GB9518953D0 (en) | 1995-09-15 | 1995-11-15 | Pfizer Ltd | Pharmaceutical formulations |
| GB9520822D0 (en) | 1995-10-11 | 1995-12-13 | Wellcome Found | Therapeutically active compounds |
| GB9624482D0 (en) | 1995-12-18 | 1997-01-15 | Zeneca Phaema S A | Chemical compounds |
| DE69624081T2 (de) | 1995-12-20 | 2003-06-12 | Agouron Pharmaceuticals, Inc. | Matrix-metalloprotease Inhibitoren |
| US6291455B1 (en) | 1996-03-05 | 2001-09-18 | Zeneca Limited | 4-anilinoquinazoline derivatives |
| US6071935A (en) | 1996-06-27 | 2000-06-06 | Pfizer Inc. | Derivatives of 2-(2-oxo-ethylidene)-imidazolidin-4-one and their use as farnesyl protein transferase inhibitors |
| EP0818442A3 (en) | 1996-07-12 | 1998-12-30 | Pfizer Inc. | Cyclic sulphone derivatives as inhibitors of metalloproteinases and of the production of tumour necrosis factor |
| ES2191187T3 (es) | 1996-07-13 | 2003-09-01 | Glaxo Group Ltd | Compuestos heteroaromaticos biciclicos como inhibidores de la proteina tirosin-quinasa. |
| HRP970371A2 (en) | 1996-07-13 | 1998-08-31 | Kathryn Jane Smith | Heterocyclic compounds |
| ID19609A (id) | 1996-07-13 | 1998-07-23 | Glaxo Group Ltd | Senyawa-senyawa heterosiklik |
| DE19628569A1 (de) | 1996-07-16 | 1998-01-22 | Bayer Ag | Substituierte N-(5-Isothiazolyl)-thioamide |
| EP0923585B1 (en) | 1996-07-18 | 2002-05-08 | Pfizer Inc. | Phosphinate based inhibitors of matrix metalloproteases |
| WO1998007697A1 (en) | 1996-08-23 | 1998-02-26 | Pfizer Inc. | Arylsulfonylamino hydroxamic acid derivatives |
| ID18494A (id) | 1996-10-02 | 1998-04-16 | Novartis Ag | Turunan pirazola leburan dan proses pembuatannya |
| WO2000035298A1 (en) | 1996-11-27 | 2000-06-22 | Wm. Wrigley Jr. Company | Chewing gum containing medicament active agents |
| WO1998030566A1 (en) | 1997-01-06 | 1998-07-16 | Pfizer Inc. | Cyclic sulfone derivatives |
| EP0977733B1 (en) | 1997-02-03 | 2003-09-03 | Pfizer Products Inc. | Arylsulfonylamino hydroxamic acid derivatives |
| JP2000507975A (ja) | 1997-02-07 | 2000-06-27 | ファイザー・インク | N−ヒドロキシ−β−スルホニルプロピオンアミド誘導体類及びそれらのマトリックスメタロプロテイナーゼ阻害薬としての使用 |
| EA002546B1 (ru) | 1997-02-11 | 2002-06-27 | Пфайзер Инк. | Производные арилсульфонилгидроксамовой кислоты |
| EP0984930B1 (en) | 1997-05-07 | 2005-04-06 | Sugen, Inc. | 2-indolinone derivatives as modulators of protein kinase activity |
| CA2291709A1 (en) | 1997-05-30 | 1998-12-03 | Merck & Co., Inc. | Novel angiogenesis inhibitors |
| GB9711643D0 (en) | 1997-06-05 | 1997-07-30 | Janssen Pharmaceutica Nv | Glass thermoplastic systems |
| AU730248B2 (en) * | 1997-08-08 | 2001-03-01 | Pfizer Products Inc. | Aryloxyarylsulfonylamino hydroxamic acid derivatives |
| WO1999010349A1 (en) | 1997-08-22 | 1999-03-04 | Zeneca Limited | Oxindolylquinazoline derivatives as angiogenesis inhibitors |
| WO1999016755A1 (en) | 1997-09-26 | 1999-04-08 | Merck & Co., Inc. | Novel angiogenesis inhibitors |
| CA2309690A1 (en) | 1997-11-11 | 1999-05-20 | Pfizer Products Inc. | Thienopyrimidine and thienopyridine derivatives useful as anticancer agents |
| GB9725782D0 (en) | 1997-12-05 | 1998-02-04 | Pfizer Ltd | Therapeutic agents |
| JPH11236333A (ja) | 1997-12-30 | 1999-08-31 | Pfizer Prod Inc | 抗ガン剤として有用なイミダゾリン−4−オン誘導体 |
| GB9800575D0 (en) | 1998-01-12 | 1998-03-11 | Glaxo Group Ltd | Heterocyclic compounds |
| RS49779B (sr) | 1998-01-12 | 2008-06-05 | Glaxo Group Limited, | Biciklična heteroaromatična jedinjenja kao inhibitori protein tirozin kinaze |
| GB9801690D0 (en) | 1998-01-27 | 1998-03-25 | Pfizer Ltd | Therapeutic agents |
| PA8469501A1 (es) | 1998-04-10 | 2000-09-29 | Pfizer Prod Inc | Hidroxamidas del acido (4-arilsulfonilamino)-tetrahidropiran-4-carboxilico |
| PA8469401A1 (es) | 1998-04-10 | 2000-05-24 | Pfizer Prod Inc | Derivados biciclicos del acido hidroxamico |
| PT952148E (pt) | 1998-04-10 | 2004-09-30 | Pfizer Prod Inc | Derivados de acido ciclobutil-ariloxiarilsulfonilamino-hidroxamico |
| EA005032B1 (ru) | 1998-05-29 | 2004-10-28 | Сьюджен, Инк. | Пирролзамещенные 2-индолиноны (варианты), фармацевтическая композиция (варианты), способ модулирования каталитической активности протеинкиназы, способ лечения или профилактики нарушения в организме, связанного с протеинкиназой |
| UA60365C2 (uk) | 1998-06-04 | 2003-10-15 | Пфайзер Продактс Інк. | Похідні ізотіазолу, спосіб їх одержання, фармацевтична композиція та спосіб лікування гіперпроліферативного захворювання у ссавця |
| TR200101343T2 (tr) | 1998-08-27 | 2001-09-21 | Pfizer Products Inc. | Kansere karşı kullanılan maddeler olarak alkinil-ikame edilmiş kinolin-2-on türevleri |
| DE69923849T2 (de) | 1998-08-27 | 2006-01-12 | Pfizer Products Inc., Groton | Quinolin-2-on-derivate verwendbar als antikrebsmittel |
| DK1004578T3 (da) | 1998-11-05 | 2004-06-28 | Pfizer Prod Inc | 5-oxo-pyrrolidin-2-carboxylsyrehydroxamidderivater |
| EP1006113A1 (en) | 1998-12-02 | 2000-06-07 | Pfizer Products Inc. | Derivatives of 2-(2-oxo-ethylidene)-imidazolidin-4-one and their use to inhibit abnormal cell growth |
| US6649645B1 (en) | 1998-12-23 | 2003-11-18 | Pharmacia Corporation | Combination therapy of radiation and a COX-2 inhibitor for treatment of neoplasia |
| US6682736B1 (en) | 1998-12-23 | 2004-01-27 | Abgenix, Inc. | Human monoclonal antibodies to CTLA-4 |
| BR9916536A (pt) | 1998-12-23 | 2002-01-02 | Searle & Co | Método para o tratamento ou prevenção de um distúrbio de neoplasia em um mamìfero em necessidade deste tratamento ou prevenção, e, combinação |
| UA71945C2 (en) | 1999-01-27 | 2005-01-17 | Pfizer Prod Inc | Substituted bicyclic derivatives being used as anticancer agents |
| JP3270834B2 (ja) | 1999-01-27 | 2002-04-02 | ファイザー・プロダクツ・インク | 抗がん剤として有用なヘテロ芳香族二環式誘導体 |
| SK11002001A3 (sk) | 1999-02-11 | 2002-05-09 | Pfizer Products Inc. | Heteroarylom substituované chinolin-2-ónové deriváty použiteľné ako protinádorové prostriedky |
| US6586447B1 (en) | 1999-04-01 | 2003-07-01 | Pfizer Inc | 3,3-disubstituted-oxindole derivatives useful as anticancer agents |
| US6511993B1 (en) * | 1999-06-03 | 2003-01-28 | Kevin Neil Dack | Metalloprotease inhibitors |
| EP1081137A1 (en) | 1999-08-12 | 2001-03-07 | Pfizer Products Inc. | Selective inhibitors of aggrecanase in osteoarthritis treatment |
| UA75055C2 (uk) * | 1999-11-30 | 2006-03-15 | Пфайзер Продактс Інк. | Похідні бензоімідазолу, що використовуються як антипроліферативний засіб, фармацевтична композиція на їх основі |
| PT1106612E (pt) | 1999-11-30 | 2004-06-30 | Pfizer Prod Inc | Derivados de quinolina uteis para inibir a farnesil-proteina-transferase |
| HN2000000266A (es) | 2000-01-21 | 2001-05-21 | Pfizer Prod Inc | Compuesto anticanceroso y metodo de separacion de enantiomeros util para sintetizar dicho compuesto. |
| SI1255752T1 (sl) * | 2000-02-15 | 2007-12-31 | Pharmacia & Upjohn Co Llc | S pirolom substituirani zaviralci 2-indolinon protein kinaza |
| US6844357B2 (en) | 2000-05-01 | 2005-01-18 | Pfizer Inc. | Substituted quinolin-2-one derivatives useful as antiproliferative agents |
| SE0102438D0 (sv) | 2001-07-05 | 2001-07-05 | Astrazeneca Ab | New compounds |
| SE0102439D0 (sv) | 2001-07-05 | 2001-07-05 | Astrazeneca Ab | New compounds |
| US20060063782A1 (en) * | 2002-07-03 | 2006-03-23 | Murray Christopher W | 3-Hetero arylmethoxy ! pyridines and their analogues as p38 map kinase inhibitors |
| SE0203754D0 (sv) | 2002-12-17 | 2002-12-17 | Astrazeneca Ab | New compounds |
| CA2517256C (en) * | 2003-02-26 | 2013-04-30 | Sugen, Inc. | Aminoheteroaryl compounds as protein kinase inhibitors |
| UA80474C2 (en) * | 2003-02-26 | 2007-09-25 | Sugen Inc | Aminoheteroaryl compounds as inhibitors of protein kinase |
| WO2005002673A1 (en) | 2003-07-03 | 2005-01-13 | Astex Therapeutics Limited | Raf kinase inhibitors |
| BRPI0514687A (pt) * | 2004-08-26 | 2008-06-17 | Pfizer | compostos amino heteroarila como inibidores de proteìna tirosina cinase |
| WO2006021885A1 (en) * | 2004-08-26 | 2006-03-02 | Pfizer Inc. | Enantioselective biotransformation for preparation of protein tyrosine kinase inhibitor intermediates |
| ME01309B (me) * | 2004-08-26 | 2013-12-20 | Pfizer | Pirazolom supstituirani heteroarilni spojevi kao inhibitori proteinskih kinaza |
| BR122020017756B1 (pt) * | 2004-08-26 | 2022-02-15 | Pfizer Inc | Uso de compostos de aminoeteroarila enantiomericamente puros na preparação de um medicamento para o tratamento de crescimento celular anormal em um mamífero |
| KR101146852B1 (ko) * | 2005-12-05 | 2012-05-16 | 화이자 프로덕츠 인크. | C?met/hgfr 억제제의 다형체 |
| DE602006018354D1 (de) * | 2005-12-05 | 2010-12-30 | Pfizer Prod Inc | Verfahren zur behandlung von abnormalem zellwachstum |
-
2005
- 2005-08-15 BR BR122020017756-0A patent/BR122020017756B1/pt active IP Right Grant
- 2005-08-15 EA EA200700352A patent/EA013678B1/ru active Protection Beyond IP Right Term
- 2005-08-15 ME MEP-2010-255A patent/ME01788B/me unknown
- 2005-08-15 WO PCT/IB2005/002837 patent/WO2006021884A2/en not_active Ceased
- 2005-08-15 GE GEAP20059887A patent/GEP20104906B/en unknown
- 2005-08-15 PT PT05779735T patent/PT1786785E/pt unknown
- 2005-08-15 CN CN2005800288183A patent/CN101023064B/zh not_active Expired - Lifetime
- 2005-08-15 RS RSP-2010/0255A patent/RS51362B/sr unknown
- 2005-08-15 PL PL05779735T patent/PL1786785T3/pl unknown
- 2005-08-15 UA UAA200701928A patent/UA87153C2/ru unknown
- 2005-08-15 HR HR20100298T patent/HRP20100298T1/hr unknown
- 2005-08-15 MX MX2007002312A patent/MX2007002312A/es active IP Right Grant
- 2005-08-15 AT AT05779735T patent/ATE463486T1/de active
- 2005-08-15 EP EP05779735.9A patent/EP1786785B9/en not_active Expired - Lifetime
- 2005-08-15 AU AU2005276135A patent/AU2005276135B2/en active Active
- 2005-08-15 SI SI200531015T patent/SI1786785T1/sl unknown
- 2005-08-15 KR KR1020077004393A patent/KR100859891B1/ko not_active Expired - Lifetime
- 2005-08-15 JP JP2007529036A patent/JP4242911B2/ja not_active Expired - Lifetime
- 2005-08-15 BR BRPI0513915-5A patent/BRPI0513915A/pt not_active Application Discontinuation
- 2005-08-15 ES ES05779735T patent/ES2341351T3/es not_active Expired - Lifetime
- 2005-08-15 CA CA2578066A patent/CA2578066C/en not_active Expired - Lifetime
- 2005-08-15 NZ NZ552866A patent/NZ552866A/en not_active IP Right Cessation
- 2005-08-15 DK DK05779735.9T patent/DK1786785T3/da active
- 2005-08-15 DE DE602005020465T patent/DE602005020465D1/de not_active Expired - Lifetime
- 2005-08-15 AP AP2007003888A patent/AP2373A/xx active
- 2005-08-19 MY MYPI20053920A patent/MY145177A/en unknown
- 2005-08-23 AR ARP050103533A patent/AR050788A1/es active IP Right Grant
- 2005-08-23 UY UY29081A patent/UY29081A1/es active IP Right Grant
- 2005-08-24 HN HN2005000476A patent/HN2005000476A/es unknown
- 2005-08-24 PE PE2005000973A patent/PE20060501A1/es active IP Right Grant
- 2005-08-24 GT GT200500225A patent/GT200500225A/es unknown
- 2005-08-25 TW TW094129125A patent/TWI358406B/zh active
- 2005-08-25 PY PY200500525663A patent/PY0525663A/es unknown
- 2005-08-25 NL NL1029799A patent/NL1029799C2/nl not_active IP Right Cessation
- 2005-08-26 US US11/212,331 patent/US7858643B2/en active Active
- 2005-08-26 PA PA20058643301A patent/PA8643301A1/es unknown
-
2007
- 2007-01-04 ZA ZA200700127A patent/ZA200700127B/xx unknown
- 2007-01-16 CR CR8860A patent/CR8860A/es unknown
- 2007-02-15 IL IL181384A patent/IL181384A/en active Protection Beyond IP Right Term
- 2007-02-23 NI NI200700057A patent/NI200700057A/es unknown
- 2007-02-23 TN TNP2007000070A patent/TNSN07070A1/fr unknown
- 2007-02-26 EC EC2007007276A patent/ECSP077276A/es unknown
- 2007-02-26 MA MA29720A patent/MA28828B1/fr unknown
- 2007-03-09 NO NO20071290A patent/NO333231B1/no active Protection Beyond IP Right Term
-
2010
- 2010-05-14 CY CY20101100430T patent/CY1110044T1/el unknown
- 2010-09-01 US US12/874,073 patent/US20100324061A1/en not_active Abandoned
-
2012
- 2012-06-29 US US13/537,759 patent/US8785632B2/en not_active Expired - Lifetime
-
2013
- 2013-02-20 LU LU92155C patent/LU92155I2/fr unknown
- 2013-02-21 LT LTPA2013005C patent/LTC1786785I2/lt unknown
- 2013-03-06 FR FR13C0015C patent/FR13C0015I2/fr active Active
- 2013-03-19 BE BE2013C021C patent/BE2013C021I2/fr unknown
- 2013-04-15 NO NO2013008C patent/NO2013008I2/no not_active IP Right Cessation
- 2013-04-18 CY CY2013014C patent/CY2013014I1/el unknown
-
2014
- 2014-06-06 US US14/298,309 patent/US20140288086A1/en not_active Abandoned
-
2025
- 2025-10-10 NO NO2025046C patent/NO2025046I1/no unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1786785B1 (en) | Enantiomerically pure aminoheteroaryl compounds as protein kinase inhibitors | |
| EP1784396B1 (en) | Pyrazole-substituted aminoheteroaryl compounds as protein kinase inhibitors | |
| US20060178374A1 (en) | Aminoheteroaryl compounds as protein kinase inhibitors | |
| HK1105414B (en) | Enantiomerically pure aminoheteroaryl compounds as protein kinase inhibitors | |
| HK1105415B (en) | Pyrazole-substituted aminoheteroaryl compounds as protein kinase inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| B07A | Application suspended after technical examination (opinion) [chapter 7.1 patent gazette] | ||
| B07A | Application suspended after technical examination (opinion) [chapter 7.1 patent gazette] | ||
| B07A | Application suspended after technical examination (opinion) [chapter 7.1 patent gazette] | ||
| B09A | Decision: intention to grant [chapter 9.1 patent gazette] | ||
| B16A | Patent or certificate of addition of invention granted [chapter 16.1 patent gazette] |
Free format text: PRAZO DE VALIDADE: 20 (VINTE) ANOS CONTADOS A PARTIR DE 15/08/2005, OBSERVADAS AS CONDICOES LEGAIS. PATENTE CONCEDIDA CONFORME ADI 5.529/DF, QUE DETERMINA A ALTERACAO DO PRAZO DE CONCESSAO. |
|
| B25G | Requested change of headquarter approved |
Owner name: PFIZER INC. (US) |