[001] ESTE PEDIDO REIVINDICA OS BENEFÍCIOS DO PEDIDO PROVISÓRIO DOS ESTADOS UNIDOS Nº. 61/722.769, DEPOSITADO EM 5 DE NOVEMBRO DE 2012, PEDIDO PROVISÓRIO DOS ESTADOS UNIDOS Nº. 61/723.645, DEPOSITADO EM 7 DE NOVEMBRO DE 2012 E PEDIDO PROVISÓRIO DOS ESTADOS UNIDOS Nº. 61/829.409, DEPOSITADO EM 31 DE MAIO DE 2013, TODOS OS QUAIS SÃO AQUI INCORPORADOS POR REFERÊNCIA EM SUA TOTALIDADE.
CAMPO DA INVENÇÃO
[002] A presente invenção refere-se a novos compostos à base de spliceostin e/ou derivados de produtos naturais eficientes como cargas úteis em conjugados de anticorpo-droga (ADCs), e compostos ligantes de carga útil eficientes em conexão com ADCs. A presente invenção refere-se adicionalmente a composições incluindo as cargas úteis acima mencionadas, ligantes de carga útil e ADCs, e métodos para utilizar estas cargas úteis, ligantes de carga útil e ADCs, para tratar condições patológicas incluindo câncer.
FUNDAMENTO
[003] A conjugação de drogas a anticorpos, seja diretamente ou através de ligantes, envolve uma consideração de uma variedade de fatores, incluindo a identificação e localização do grupo químico para conjugação da droga, os mecanismos de liberação de droga, os elementos estruturais que proporcionam a liberação de droga e a modificação estrutural à droga livre liberada. Além disso, se a droga tiver que ser liberada após a internalização do anticorpo, o mecanismo de liberação de droga deve ser consoante com o tráfico intracelular do conjugado.
[004] Embora uma variedade de diferentes classes de droga tenha sido testada para entrega através de anticorpos, apenas poucas classes de drogas provaram eficácia como conjugados de anticorpo- droga, embora com perfil de toxicidade adequado.
[005] Os produtos naturais FR901463, FR901464, e FR901465 foram reportados como tendo atividades inibidoras potentes contra linhagens de células de câncer humanas e eficácias em vários modelos de tumor de xenoenxerto (Journal de Antibiotics (1996), 49(12), 12041211). O produto natural FR901464 e seu metil cetal designado, spli- ceostatin A, foram recentemente reportados por inibirem o spliceoso- me por interação com SF3b, que é um componente do subcomplexo essencial, U2 snRNA. (Nature Chemical Biology (2007), 3(9), 576583.; Nature (London, United Kingdom) (2010), 468(7324), 664-668.)
SUMÁRIO DA INVENÇÃO
[006] A presente invenção refere-se a compostos e composições farmacêuticas contendo-os, a sua preparação, e a usos para os compostos, primariamente, mas não exclusivamente, agentes anticâncer.
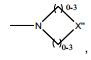
[007] De acordo com um aspecto, a presente invenção refere-se a um composto ou compostos de fórmula (I):
[008] (I)
[009] em que:
[010] uma linha pontilhada representa uma ligação opcional;
[011]cada X1 é independentemente selecionado do grupo que consiste em: -O-, -S- e -NR-;
[012] cada X2 é independentemente selecionado do grupo que consiste em: -O-, -S- e -NR-;
[013] R1 é selecionado do grupo que consiste em: -R, -OR, - OCOR13, -OCONR14R15, -OCON(R14)NR(R15), =O (ligação dupla a oxigênio) e -NR14R15;
[014] R2 e R3 são independentemente selecionados do grupo que consiste em: hidrogênio e C1-6alquil;
[015] R4 e R5 são independentemente selecionados do grupo que consiste em: hidrogênio, -OR, -NR14R15 e oxo;
[016] R6 e R7 são independentemente selecionados do grupo que consiste em: hidrogênio, halogênio, hidroxil e C1-6alquil opcionalmente substituído com 1-3 substituintes independentemente selecionados de hidroxil e halogênio,
[017] R6 e R7, juntos com o átomo de carbono ao qual estão ligados, formam um C2-5alquilideno opcionalmente substituído com 1-3 substituintes independentemente selecionados de R,
[018] R6 e R7 juntos são oxo, ou
[019] R6 e R7, juntos com o átomo de carbono ao qual estão ligados, formam uma porção heterocicloalquil de 3 a 5 membros compreendendo 1 ou 2 heteroátomos independentemente selecionados do grupo que consiste em oxigênio, nitrogênio e enxofre, em que a referida porção heterocicloalquil pode ser opcionalmente substituída com um a três substituintes independentemente selecionados de R;
[020] R8 é hidrogênio, C1-6alquil ou -OR;
[021] R9 é independentemente selecionado de hidrogênio, -C1-θalquil, -(C(R)2)m-C(O)OR, -(C(R)2)m-C(O)NR14R15, -(C(R)2)m-NR14R15, -(C(R)2)mrC(O)-SR, -(C(R)2)m-C(O)NR14N(R)R15, -(C(R)2)m-NR-C(O)- NR14R15, -(C(R)2)m-N(R)COR13 e -(C(R)2)m-NR14N(R)R15;
[022] R13 é selecionado do grupo que consiste em hidrogênio, C1- 6alquil, C3-8carbociclil, C3-8heterociclil, C1-6alquil-C6-14aril, C1-6alquil-C5- 14heteroaril, em que R13 é opcionalmente substituído com -NRR ou - SO2NRR;
[023] cada R14 e R15 é independentemente selecionado do grupo que consiste em: hidrogênio, hidroxil, -NRR, -NRNR2, -C3-10carbociclil, -C1-6alquileno-C3-10carbociclil, -C3-10heterociclil, -C1-6alquileno-C3- 10heterociclil, -(CH2CH2O)1-6CH2CH2C(O)OR, -(CH2CH2O)1-6CH2CH2NRR, -C1-6alquil, C6-14aril, -C1-6alquileno-C6-14aril e -C5- 14heteroaril;
[024] ou R14 e R15, juntos com o átomo ou átomos aos quais são ligados, formam um anel C3-10heterociclil,
[025] em que R14, R15, ou ambos, ou um anel formado com R14 e R15, são opcionalmente substituídos com -(C(R)2)m-R18, em que cada R18 é independentemente selecionado de (i) -NRR, (ii) - C(NRR)(C(O)OR), (iii) -S-R, (iv) aril ou heteroaril opcionalmente substituído com um ou mais de halogênio, -CF3, -(C(R)2)m-NRR ou - (C(R)2)m -SO2NRR, (v) -SO2R, (vi) -S-S-C1-6alquil-C(O)OR, (vii) - SO2NRR, (viii) -C(O)NRR, (ix) -C(O)OR, (x) -C4-6 cicloalquil opcionalmente substituído com -NRR, -SO2NRR ou -NR-C(O)(CH2)0-6NRR, (xi) -R, (xii) -OR, (xiii) -N(R)NRR, (xiv) -C(O)N(R)NRR, -(C(R)2)m-O- NRR e -S-S-C1-6alquil-NRR;
[026] cada R é independentemente selecionado do grupo que consiste em: hidrogênio e -C1-6alquil; e
[027] cada m é independentemente 0, 1, 2 ou 3;
[028] ou um sal farmaceuticamente aceitável do mesmo.
[029] De acordo com outro aspecto, a presente invenção refere- se a um composto ou compostos de fórmula (II):
[030] L-P
[031] (II)
[032] ou um sal farmaceuticamente aceitável do mesmo, em que:
[033] L é a porção ligante L1-L2-L3, em que L3 é ligado a P;
[034] P é um radical de fórmula (I):
[035] (I)
[036] em que:
[037] uma linha pontilhada representa uma ligação opcional;
[038] cada X1 é independentemente selecionado do grupo que consiste em: -O-, -S- e -NR-;
[039] cada X2 é independentemente selecionado do grupo que consiste em: -O-, -S- e -NR-;
[040] cada X’ é CR ou N;
[041] cada X’’ é CH-, CR-(C(R)2)m-NR-, CR-(C(R)2)m-O-; CR- (C(R)2)m-C(O)NR-, CR-(C(R)2)m-C(O)NR-NR-, CR-(C(R)2)m-SO2NR-, CR-(C(R)2)m-NR-NR-, CR-(C(R)2)m-NR-C(O)- ou N- se X’’ se liga a L2 ou um L3 adicional, ou de outra forma é O, S, CRR, CR-(C(R)2)m-NRR ou NRR;
[042] cada X’’’ é - (C(R)2)m-NR- ou CR-(C(R)2)m-O- se X’’’ se liga a L2, ou de outra forma é R;
[043] Y é -C(R)2-, -O-, -NR- ou -S-;
[044] R1 é selecionado do grupo que consiste em: -R, -OR, -OCOR13, -OCONR14R15, -OCON(R14)NR(R15), =O (ligação dupla a oxigênio) e -NR14R15;
[045] R2 e R3 são independentemente selecionados do grupo que consiste em: hidrogênio e C1-6alquil;
[046] R4 e R5 são independentemente selecionados do grupo que consiste em: hidrogênio, -OR, -NR14R15 e oxo;
[047] R6 e R7 são independentemente selecionados do grupo que consiste em: hidrogênio, halogênio, hidroxil e C1-6alquil opcionalmente substituído com 1-3 substituintes independentemente selecionados de hidroxil e halogênio,
[048] R6 e R7, juntos com o átomo de carbono ao qual estão liga-dos, formam um C2-5alquilideno opcionalmente substituído com 1-3 substituintes independentemente selecionados de R,
[049] R6 e R7 juntos são oxo, ou
[050] R6 e R7, juntos com o átomo de carbono ao qual estão ligados, formam uma porção heterocicloalquil de 3 a 5 membros compre- endendo 1 ou 2 heteroátomos independentemente selecionados do grupo que consiste em oxigênio, nitrogênio e enxofre, em que a referi- da porção heterocicloalquil pode ser opcionalmente substituída com um a três substituintes independentemente selecionados de R;
[051] R8 é hidrogênio, C1-6alquil ou -OR;
[052] R9 é -(C(R)2)m-C(O)- ou -(C(R)2)m- ;
[053] L1 é selecionado de: -halogênio, -NR2,
[054] L2 é L2A-L2B-L2C ou L2C-L2B-L2A, em que:
[055] L2A compreende um ou mais componentes selecionados de:
[056] -O-, -C(O)-, -C(O)NR-, -C(O)-C1-6alquil-, -C(O)NRC1-6alquil-,-C1-6alquil(OCH2CH2)1-6-, -C(O)-C1-6alquil-NRC(O)-, -C(O)-C1-6alquil(OCH2CH2)1-6-, -C1-6alquil(OCH2CH2)1-6-C(O)-, -C1-6alquil-S-S-C1-6alquil-NRC(O)CH2-, -C1-6alquil-(OCH2CH2)1-6-NRC(O)CH2-, -C(O)-C1- 6alquil-NRC(O)C1-6alquil-, -N=CR-fenil-O-C1-6alquil-, -N=CR-fenil-O-C1- 6alquil-C(O)-, -C(O)-C1-6alquil(OCH2CH2)1-6-NRC(O)-, -C(O)-C1-6alquil- fenil-(NR-C(O)-C1-6alquil)1-4-, -C(O)-C1-6alquil-(OCH2CH2)1-6-NRC(O)C1- 6alquil-, -C1-6alquil-, -S-, -C(O)-C1-6alquil-fenil-NR-, -O-C1-6alquil-S-, - C(O)-O-C1-6alquil-S- e (-CH2-CH2-O-)1-20, ou L2A é ausente;
[057] L2B é selecionado de AA0-aa, em que AA é um aminoácido natural ou não-natural e aa é 12; e
[058] L2C compreende um ou mais componentes selecionados de: -PABA- e -PABC-, ou L2C é ausente;
[059] L3 é selecionado de um ou mais de: -C1-6alquil-, -NR-C3-C8-heterociclil-NR-, -NR-C3-C8carbociclil-NR-, -NR-C1-6alquil-NR-, -NR-C1- 6alquil-, -S-, -NR-, -NR-NR- e -NR-C(O)-NR-, em que os dois grupos R opcionalmente se juntam para formar um anel de 4-10 membros, -NR- C1-6alquil-fenil-NR-, -NR-C1-6alquil-fenil-SO2-NR-, -SO2-, -NR-C1-6- alquil-fenil-C(O)-,
[060] ou L3 é ausente;
[061] R13 é selecionado do grupo que consiste em hidrogênio, C1- 6alquil, C3-8carbociclil, C3-8heterociclil, C1-6alquil-C6-14aril, C1-6alquil-C5- uheteroaril, em que R13 é opcionalmente substituído com -NRR ou - SO2NRR;
[062] cada R14 e R15 é independentemente selecionado do grupo que consiste em: hidrogênio, hidroxil, -NRR, -NRNR2, -C3-10carbociclil, -C1-6alquileno-C3-10carbociclil, -C3-10heterociclil, -C1-6alquileno-C3- 10heterociclil, -(CH2CH2O)1-6CH2CH2C(O)OR, -(CH2CH2O)1-6CH2CH2NRR, -C1-6alquil, C6-14aril, -C1-6alquileno-C6-14aril e -C5- 14heteroaril;
[063] ou R14 e R15, juntos com o átomo ou átomos aos quais são ligados, formam um anel C3-10heterociclil,
[064] em que R14, R15, ou ambos, ou um anel formado com R14 e R15, são opcionalmente substituídos com -(C(R)2)m-R18, em que cada R18 é independentemente selecionado de (i) -NRR, (ii) - C(NRR)(C(O)OR), (iii) -S-R, (iv) aril ou heteroaril opcionalmente substituído com um ou mais de halogênio, -CF3, -(C(R)2)m-NRR ou - (C(R)2)m -SO2NRR, (v) -SO2R, (vi) -S-S-C1-6alquil-C(O)OR, (vii) - SO2NRR, (viii) -C(O)NRR, (ix) -C(O)OR, (x) -C4-6 cicloalquil opcionalmente substituído com -NRR, -SO2NRR ou -NR-C(O)(CH2)0-6NRR, (xi) -R, (xii) -OR, (xiii) -N(R)NRR, (xiv) -C(O)N(R)NRR, (xv) - (C(R)2)m-O-NRR e (xiv) -S-S-C1-6alquil-NRR;
[065] cada R é independentemente selecionado do grupo que consiste em: hidrogênio e -C1-6alquil; e
[066] cada m é independentemente 0, 1, 2 ou 3.
[067] De acordo com outro aspecto, a presente invenção refere- se a um composto ou compostos de fórmula (II’):
[068] L-P’
[069] (II’)
[070] ou um sal farmaceuticamente aceitável do mesmo, em que:
[071] L é a porção ligante L1-L2-L3, em que L3 é ligado a P’;
[072] P’ é um radical de fórmula (I’):
[073] (I’)
[074] em que:
[075] uma linha pontilhada representa uma ligação opcional;
[076] cada X1 é independentemente selecionado do grupo que consiste em: -O-, -S- e -NR-;
[077] cada X2 é independentemente selecionado do grupo que consiste em: -O-, -S- e -NR-;
[078] cada X’ é CR ou N;
[079] cada X’’ é CH-, CR-(C(R)2)m-NR-, CR-(C(R)2)m-O-; CR- (C(R)2)m-C(O)NR-, CR-(C(R)2)m-C(O)NR-NR-, CR-(C(R)2)m-SO2NR-, CR-(C(R)2)m-NR-NR-, CR-(C(R)2)m-NR-C(O)- ou N- se X’’ se liga a L2 ou um L3 adicional, ou de outra forma é O, S, CRR, CR-(C(R)2)m-NRR ou NRR;
[080] cada X’’’ é - (C(R)2)m-NR- ou CR-(C(R)2)m-O- se X’’’ se liga a L2, ou de outra forma é R;
[081] Y é -C(R)2-, -O-, -NR- ou -S-;
[082] R1 é selecionado do grupo que consiste em: -(C(R)2)m-, -OR’’, -OCOR13’, -OC(O)NRR14’, -OCON(R)N(R)-, e -NR-
[083] R2 e R3 são independentemente selecionados do grupo que consiste em: hidrogênio e C1-6alquil;
[084] R4 e R5 são independentemente selecionados do grupo que consiste em: hidrogênio, -OR, -NR14R15 e oxo;
[085] R6 e R7 são independentemente selecionados do grupo que consiste em: hidrogênio, halogênio, hidroxil e C1-6alquil opcionalmente substituído com 1-3 substituintes independentemente selecionados de hidroxil e halogênio,
[086] R6 e R7, juntos com o átomo de carbono ao qual estão ligados, formam um C2-5alquilideno opcionalmente substituído com 1-3 substituintes independentemente selecionados de R,
[087] R6 e R7 juntos são oxo, ou
[088] R6 e R7, juntos com o átomo de carbono ao qual estão ligados, formam uma porção heterocicloalquil de 3 a 5 membros compre- endendo 1 ou 2 heteroátomos independentemente selecionados do grupo que consiste em oxigênio, nitrogênio e enxofre, em que a referida porção heterocicloalquil pode ser opcionalmente substituída com um a três substituintes independentemente selecionados de R;
[089] R8 é hidrogênio, C1-6alquil ou -OR;
[090] R9 é independentemente selecionado de hidrogênio, -C1-θalquil, -(C(R)2)m-C(O)OR, -(C(R)2)m-C(O)NR14R15, -(C(R)2)m-NR14R15, -(C(R)2)mrC(O)-SR, -(C(R)2)m-C(O)NR14N(R)R15, -(C(R)2)m-NR-C(O)- NR14R15, -(C(R)2)m-N(R)COR13 e -(C(R)2)m-NR14N(R)R15;
[091] L1 é selecionado de: -halogênio, -NR2,
[092] L2 é L2A-L2B-L2C ou L2C-L2B-L2A, em que:
[093] L2A compreende um ou mais componentes selecionados de:
[094] -O-, -C(O)-, -C(O)NR-, -C(O)-C1-6alquil-, -C(O)NRC1-6alquil-,-C1-6alquil(OCH2CH2)1-6-, -C(O)-C1-6alquil-NRC(O)-, -C(O)-C1-6alquil(OCH2CH2)1-6-, -C1-6alquil(OCH2CH2)1-6-C(O)-, -C1-6alquil-S-S-C1- 6alquil-NRC(O)CH2-, -C1-6alquil-(OCH2CH2)1-6-NRC(O)CH2-, -C(O)-C1- 6alquil-NRC(O)C1-6alquil-, -N=CR-fenil-O-C1-6alquil-, -N=CR-fenil-O-C1- 6alquil-C(O)-, -C(O)-C1-6alquil(OCH2CH2)1-6-NRC(O)-, -C(O)-C1-6alquil- fenil-(NR-C(O)-C1-6alquil)1-4-, -C(O)-C1-6alquil-(OCH2CH2)1-6-NRC(O)C1- 6alquil-, -C1-6alquil-, -S-, -C(O)-C1-6alquil-fenil-NR-, -O-C1-6alquil-S-, - C(O)-O-C1-6alquil-S- e (-CH2-CH2-O-)1-20, ou L2A é ausente;
[095] L2B é selecionado de AA0-aa, em que AA é um aminoácido natural ou não-natural e aa é 12; e
[096] L2C compreende um ou mais componentes selecionados de: -PABA- e -PABC-, ou L2C é ausente;
[097] L3 é selecionado de um ou mais de: -C1-6alquil-, -NR-C3-C8-heterociclil-NR-, -NR-C3-C8carbociclil-NR-, -NR-C1-6alquil-NR-, -NR-C1- 6alquil-, -S-, -NR-, -NR-NR- e -NR-C(O)-NR-, em que os dois grupos R opcionalmente se juntam para formar um anel de 4-10 membros, -NR- C1-6alquil-fenil-NR-, -NR-C1-6alquil-fenil-SO2-NR-, -SO2-, -NR-C1-6- alquil-fenil-C(O)-,
, ou L3 é ausente;
[098] R13’ é selecionado do grupo que consiste em uma ligação, -C1-6alquileno-, -C3-8carbociclil-, -C3-8heterociclil-, -C1-6alquil-C6-14aril-, - C1-6alquil-C5-14heteroaril-;
[099] cada R14 e R15 é independentemente selecionado do grupo que consiste em: hidrogênio, hidroxil, -NRR, -NRNR2, -C3-10carbociclil, -C1-6alquileno-C3-10carbociclil, -C3-10heterociclil, -C1-6alquileno-C3- 10heterociclil, -(CH2CH2O)1-6CH2CH2C(O)OR, -(CH2CH2O)1-6CH2CH2NRR, -C1-6alquil, C6-14aril, -C1-6alquileno-C6-14aril e -C5- 14heteroaril;
[0100] ou R14 e R15, juntos com o átomo ou átomos aos quais são ligados, formam um anel C3-10heterociclil,
[0101] em que R14, R15, ou ambos, ou um anel formado com R14 e R15, são opcionalmente substituídos com -(C(R)2)m-R18, em que cada R18 é independentemente selecionado de (i) -NRR, (ii) - C(NRR)(C(O)OR), (iii) -S-R, (iv) aril ou heteroaril opcionalmente substituído com um ou mais de halogênio, -CF3, -(C(R)2)m-NRR ou - (C(R)2)m -SO2NRR, (v) -SO2R, (vi) -S-S-C1-6alquil-C(O)OR, (vii) - SO2NRR, (viii) -C(O)NRR, (ix) -C(O)OR, (x) -C4-6 cicloalquil opcionalmente substituído com -NRR, -SO2NRR ou -NR-C(O)(CH2)0-6NRR, (xi) -R, (xii) -OR, (xiii) -N(R)NRR, (xiv) -C(O)N(R)NRR, (xv) - (C(R)2)m-O-NRR e (xiv) -S-S-C1-6alquil-NRR;
[0102] cada R14’ é independentemente selecionado do grupo que consiste em: uma ligação, -NR-, -C3-10carbociclil-, -C3-10heterociclil-, - (CH2CH2O)1-6CH2CH2C(O)OR’, -(CH2CH2O)1-6CH2CH2NR-, e -C1- 6alquileno-,
[0103] em que R14’ é opcionalmente substituído com -(C(R)2)m-R18, em que cada R18 é independentemente selecionado de (i) -NRR, (ii) - C(NRR)(C(O)OR), (iii) -S-R, (iv) aril ou heteroaril opcionalmente substituído com um ou mais de halogênio, -CF3, -NRR ou -SO2NRR, (v) - SO2R, (vi) -S-S-C1-6alquil-C(O)OR, (vii) -SO2NRR, (viii) -C(O)NRR, (ix) -C(O)OR, (x) -C4-6 cicloalquil opcionalmente substituído com -NRR, - SO2NRR ou -NR-C(O)(CH2)0-6NRR, (xi) -R, (xii) -OR, (xiii) -N(R)NRR, (xiv) -C(O)N(R)NRR, (xv) -(C(R)2)m-O-NRR e (xiv) -S-S-C1-6alquil- NRR;
[0104] cada R é independentemente selecionado do grupo que consiste em: hidrogênio e -C1-6alquil;
[0105] cada R' é independentemente selecionado de -H, C1-C8 al- quil, C1-C8 heteroalquil e aril;
[0106] cada R’’ é independentemente selecionado do grupo que consiste em: uma ligação e -C1-6alquileno-; e
[0107] cada m é independentemente 0, 1, 2 ou 3.
[0108] De acordo com ainda outro aspecto, a presente invenção refere-se a um composto ou compostos de fórmula (III):
[0109] (AB)-(L-P)b
[0110] (III)
[0111] ou um sal farmaceuticamente aceitável do mesmo, em que:
[0112] L é a porção ligante L1-L2-L3, em que L3 é ligado a P;
[0113] P é um radical de fórmula (I):
[0115] (I)
[0116] em que:
[0117] uma linha pontilhada representa uma ligação opcional;
[0118] AB é um anticorpo;
[0119] cada x1 é independentemente selecionado do grupo que consiste em: -O-, -S- e -NR-;
[0120] cada x2 é independentemente selecionado do grupo que consiste em: -O-, -S- e -NR-;
[0121] cada x’ é CR ou N;
[0122] cada x’’ é CH-, CR-(C(R)2)m-NR-, CR-(C(R)2)m-O-; CR- (C(R)2)m-C(O)NR-, CR-(C(R)2)m-C(O)NR-NR-, CR-(C(R)2)m-SO2NR-, CR-(C(R)2)m-NR-NR-, CR-(C(R)2)m-NR-C(O)- ou N- se x’’ se liga a L2 ou um L3 adicional, ou de outra forma é O, S, CRR, CR-(C(R)2)m-NRR ou NRR;
[0123] cada X’’’ é - (C(R)2)m-NR- ou CR-(C(R)2)m-O- se X’’’ se liga a L2, ou de outra forma é R;
[0124] Y é -C(R)2-, -O-, -NR- ou -S-;
[0125] R1 é selecionado do grupo que consiste em: -R, -OR, - OCOR13, -OCONR14R15, -OCON(R14)NR(R15), =O (ligação dupla a oxigênio) e -NR14R15;
[0126] R2 e R3 são independentemente selecionados do grupo que consiste em: hidrogênio e C1-6alquil;
[0127] R4 e R5 são independentemente selecionados do grupo que consiste em: hidrogênio, -OR, -NR14R15 e oxo;
[0128] R6 e R7 são independentemente selecionados do grupo que consiste em: hidrogênio, halogênio, hidroxil e C1-6alquil opcionalmente substituído com 1-3 substituintes independentemente selecionados de hidroxil e halogênio,
[0129] R6 e R7, juntos com o átomo de carbono ao qual estão ligados, formam um C2-5alquilideno opcionalmente substituído com 1-3 substituintes independentemente selecionados de R,
[0130] R6 e R7 juntos são oxo, ou
[0131] R6 e R7, juntos com o átomo de carbono ao qual estão ligados, formam uma porção heterocicloalquil de 3 a 5 membros compreendendo 1 ou 2 heteroátomos independentemente selecionados do grupo que consiste em oxigênio, nitrogênio e enxofre, em que a referida porção heterocicloalquil pode ser opcionalmente substituída com um a três substituintes independentemente selecionados de R;
[0132] R8 é hidrogênio, C1-6alquil ou -OR;
[0133] R9 é –(C(R)2)m-C(O)- ou –(C(R)2)m- ;
[0134] L1 é selecionado de: uma ligação a AB, -NR-(ligação a AB) e
[0135] L2 é L2A-L2B-L2C ou L2C-L2B-L2A, em que:
[0136] L2A compreende um ou mais componentes selecionados de:
[0137] -O-, -C(O)-, -C(O)NR-, -C(O)-C1-6alquil-, -C(O)NRC1-6alquil-,-C1-6alquil(OCH2CH2)1-6-, -C(O)-C1-6alquil-NRC(O)-, -C(O)-C1-6alquil(OCH2CH2)1-6-, -C1-6alquil(OCH2CH2)1-6-C(O)-, -C1-6alquil-S-S-C1- 6alquil-NRC(O)CH2-, -C1-6alquil-(OCH2CH2)1-6-NRC(O)CH2-, -C(O)-C1- 6alquil-NRC(O)C1-6alquil-, -N=CR-fenil-O-C1-6alquil-, -N=CR-fenil-O-C1- 6alquil-C(O)-, -C(O)-C1-6alquil(OCH2CH2)1-6-NRC(O)-, -C(O)-C1-6alquil- fenil-(NR-C(O)-C1-6alquil)1-4-, -C(O)-C1-6alquil-(OCH2CH2)1-6-NRC(O)C1- 6alquil-, -C1-6alquil-, -S-, -C(O)-C1-6alquil-fenil-NR-, -O-C1-6alquil-S-, - C(O)-O-C1-6alquil-S- e (-CH2-CH2-O-)1-20, ou L2A é ausente;
[0138] L2B é selecionado de AA0-aa, em que AA é um aminoácido natural ou não-natural e aa é 12; e
[0139] L2C compreende um ou mais componentes selecionados de: -PABA- e -PABC-, ou L2C é ausente;
[0140] L3 é selecionado de um ou mais de: -C1-6alquil-, -NR-C3-C8-heterociclil-NR-, -NR-C3-C8carbociclil-NR-, -NR-C1-6alquil-NR-, -NR-C1- 6alquil-, -S-, -NR-, -NR-NR- e -NR-C(O)-NR-, em que os dois grupos R opcionalmente se juntam para formar um anel de 4-10 membros, -NR- C1-6alquil-fenil-NR-, -NR-C1-6alquil-fenil-SO2-NR-, -SO2-, -NR-C1-6- alquil-fenil-C(O)-,
[0142] R13 é selecionado do grupo que consiste em hidrogênio, C1-6alquil, C3-8carbociclil, C3-8heterociclil, C1-6alquil-C6-14aril, C1-6alquil-C5- i4heteroaril, em que R13 é opcionalmente substituído com -NRR ou - SO2NRR;
[0143] cada R14 e R15 é independentemente selecionado do grupo que consiste em: hidrogênio, hidroxil, -NRR, -NRNR2, -C3-10carbociclil, -C1-6alquileno-C3-10carbociclil, -C3-10heterociclil, -C1-6alquileno-C3- 10heterociclil, -(CH2CH2O)1-6CH2CH2C(O)OR, -(CH2CH2O)1-6CH2CH2NRR, -C1-6alquil, C6-14aril, -C1-6alquileno-C6-14aril e -C5- 14heteroaril;
[0144] ou R14 e R15, juntos com o átomo ou átomos aos quais são ligados, formam um anel C3-10heterociclil,
[0145] em que R14, R15, ou ambos, ou um anel formado com R14 e R15, são opcionalmente substituídos com -(C(R)2)m-R18, em que cada R18 é independentemente selecionado de (i) -NRR, (ii) - C(NRR)(C(O)OR), (iii) -S-R, (iv) aril ou heteroaril opcionalmente substituído com um ou mais de halogênio, -CF3, -(C(R)2)m-NRR ou - (C(R)2)m -SO2NRR, (v) -SO2R, (vi) -S-S-C1-6alquil-C(O)OR, (vii) - SO2NRR, (viii) -C(O)NRR, (ix) -C(O)OR, (x) -C4-6 cicloalquil opcionalmente substituído com -NRR, -SO2NRR ou -NR-C(O)(CH2)0-6NRR, (xi) -R, (xii) -OR, (xiii) -N(R)NRR, (xiv) -C(O)N(R)NRR, (xv) - (C(R)2)m-O-NRR e (xiv) -S-S-C1-6alquil-NRR;
[0146] cada R é independentemente selecionado do grupo que consiste em: hidrogênio e -C1-6alquil; e
[0147] b é 1-20; e
[0148] cada m é independentemente 0, 1, 2 ou 3.
[0149] De acordo com ainda outro aspecto, a presente invenção refere-se a um composto ou compostos de fórmula (III’):
[0150] (AB)-(L-P’)b
[0151] (III’)
[0152] ou um sal farmaceuticamente aceitável do mesmo, em que:
[0153] L é a porção ligante L1-L2-L3, em que L3 é ligado a P’;
[0154] P’ é um radical de fórmula (I’):
[0155] (I’)
[0156] em que:
[0157] uma linha pontilhada representa uma ligação opcional;
[0158] AB é um anticorpo;
[0159] cada X1 é independentemente selecionado do grupo que consiste em: -O-, -S- e -NR-;
[0160] cada X2 é independentemente selecionado do grupo que consiste em: -O-, -S- e -NR-;
[0161] cada X’ é CR ou N;
[0162] cada X’’ é CH-, CR-(C(R)2)m-NR-, CR-(C(R)2)m-O-; CR- (C(R)2)m-C(O)NR-, CR-(C(R)2)m-C(O)NR-NR-, CR-(C(R)2)m-SO2NR-, CR-(C(R)2)m-NR-NR-, CR-(C(R)2)m-NR-C(O)- ou N- se X’’ se liga a L2 ou um L3 adicional, ou de outra forma é O, S, CRR, CR-(C(R)2)m-NRR ou NRR;
[0163] cada X’’’ é - (C(R)2)m-NR- ou CR-(C(R)2)m-O- se X’’’ se liga a L2, ou de outra forma é R;
[0164] Y é -C(R)2-, -O-, -NR- ou -S-;
[0165] R1 é selecionado do grupo que consiste em: -(C(R)2)m-C(O)-, -(C(R)2)m-, -OR’’, -OCOR13’, -OCONRR14’, -OCON(R14)N(R15)-, e -NR14-
[0166] R2 e R3 são independentemente selecionados do grupo que consiste em: hidrogênio e C1-6alquil;
[0167] R4 e R5 são independentemente selecionados do grupo que consiste em: hidrogênio, -OR, -NR14R15 e oxo;
[0168] R6 e R7 são independentemente selecionados do grupo que consiste em: hidrogênio, halogênio, hidroxil e C1-6alquil opcionalmente substituído com 1-3 substituintes independentemente selecionados de hidroxil e halogênio,
[0169] R6 e R7, juntos com o átomo de carbono ao qual estão ligados, formam um C2-5alquilideno opcionalmente substituído com 1-3 substituintes independentemente selecionados de R,
[0170] R6 e R7 juntos são oxo, ou
[0171] R6 e R7, juntos com o átomo de carbono ao qual estão liga dos, formam uma porção heterocicloalquil de 3 a 5 membros compreendendo 1 ou 2 heteroátomos independentemente selecionados do grupo que consiste em oxigênio, nitrogênio e enxofre, em que a referida porção heterocicloalquil pode ser opcionalmente substituída com um a três substituintes independentemente selecionados de R;
[0172] R8 é hidrogênio, C1-6alquil ou -OR;
[0173] R9 é independentemente selecionado de hidrogênio, -C1-θalquil, -(C(R)2)m-C(O)OR, -(C(R)2)m-C(O)NR14R15, -(C(R)2)m-NR14R15, -(C(R)2)mrC(O)-SR, -(C(R)2)m-C(O)NR14N(R)R15, -(C(R)2)m-NR-C(O)- NR14R15, -(C(R)2)m-N(R)COR13 e -(C(R)2)m-NR14N(R)R15;
[0174] L1 é selecionado de: uma ligação a AB, -NR-(ligação a AB) e
[0175] L2 é L2A-L2B-L2C ou L2C-L2B-L2A, em que:
[0176] L2A compreende um ou mais componentes selecionados de:
[0177] -O-, -C(O)-, -C(O)NR-, -C(O)-C1-6alquil-, -C(O)NRC1-6alquil-,-C1-6alquil(OCH2CH2)1-6-, -C(O)-C1-6alquil-NRC(O)-, -C(O)-C1-6alquil(OCH2CH2)1-6-, -C1-6alquil(OCH2CH2)1-6-C(O)-, -C1-6alquil-S-S-C1- 6alquil-NRC(O)CH2-, -C1-6alquil-(OCH2CH2)1-6-NRC(O)CH2-, -C(O)-C1- 6alquil-NRC(O)C1-6alquil-, -N=CR-fenil-O-C1-6alquil-, -N=CR-fenil-O-C1- 6alquil-C(O)-, -C(O)-C1-6alquil(OCH2CH2)1-6-NRC(O)-, -C(O)-C1-6alquil- fenil-(NR-C(O)-C1-6alquil)1-4-, -C(O)-C1-6alquil-(OCH2CH2)1-6-NRC(O)C1- 6alquil-, -C1-6alquil-, -S-, -C(O)-C1-6alquil-fenil-NR-, -O-C1-6alquil-S-, - C(O)-O-C1-6alquil-S- e (-CH2-CH2-O-)1-20, ou L2A é ausente;
[0178] L2B é selecionado de AA0-aa, em que AA é um aminoácido natural ou não-natural e aa é 12; e
[0179] L2C compreende um ou mais componentes selecionados de: -PABA- e -PABC-, ou L2C é ausente;
[0180] L3 é selecionado de um ou mais de: -C1-6alquil-, -NR-C3-C8-heterociclil-NR-, -NR-C3-C8carbociclil-NR-, -NR-C1-6alquil-NR-, -NR-C1- 6alquil-, -S-, -NR-, -NR-NR- e -NR-C(O)-NR-, em que os dois grupos R opcionalmente se juntam para formar um anel de 4-10 membros, -NR- C1-6alquil-fenil-NR-, -NR-C1-6alquil-fenil-SO2-NR-, -SO2-, -NR-C1-6- alquil-fenil-C(O)-,
[0182] R13’ é selecionado do grupo que consiste em uma ligação, -C1-6alquileno-, -C3-8carbociclil-, -C3-8heterociclil-, -C1-6alquil-C6-14aril-, - C1-6alquil-C5-14heteroaril-;
[0183] cada R14 e R15 é independentemente selecionado do grupo que consiste em: hidrogênio, hidroxil, -NRR, -NRNR2, -C3-10carbociclil, -C1-6alquileno-C3-10carbociclil, -C3-10heterociclil, -C1-6alquileno-C3-10heterociclil, -(CH2CH2O)1-6CH2CH2C(O)OR, -(CH2CH2O)1-6CH2CH2NRR, -C1-6alquil, C6-14aril, -C1-6alquileno-C6-14aril e -C5-14heteroaril;
[0184] ou R14 e R15, juntos com o átomo ou átomos aos quais são ligados formam um anel C3-10heterociclil,
[0185] em que R14, R15, ou ambos, ou um anel formado com R14 e R15, são opcionalmente substituídos com -(C(R)2)m-R18, em que cada R18 é independentemente selecionado de (i) -NRR, (ii) - C(NRR)(C(O)OR), (iii) -S-R, (iv) aril ou heteroaril opcionalmente substituído com um ou mais de halogênio, -CF3, -(C(R)2)m-NRR ou - (C(R)2)m -SO2NRR, (v) -SO2R, (vi) -S-S-C1-6alquil-C(O)OR, (vii) - SO2NRR, (viii) -C(O)NRR, (ix) -C(O)OR, (x) -C4-6 cicloalquil opcionalmente substituído com -NRR, -SO2NRR ou -NR-C(O)(CH2)0-6NRR, (xi) -R, (xii) -OR, (xiii) -N(R)NRR, (xiv) -C(O)N(R)NRR, (xv) - (C(R)2)m-O-NRR e (xiv) -S-S-C1-6alquil-NRR;
[0186] cada R14’ é independentemente selecionado do grupo que consiste em: uma ligação, -NR-, -C3-10carbociclil-, -C3-10heterociclil-, - (CH2CH2O)1-6CH2CH2C(O)OR’, -(CH2CH2O)1-6CH2CH2NR-, e -C1- 6alquileno-,
[0187] em que R14’ é opcionalmente substituído com -(C(R)2)m-R18, em que cada R18 é independentemente selecionado de (i) -NRR, (ii) - C(NRR)(C(O)OR), (iii) -S-R, (iv) aril ou heteroaril opcionalmente substituído com um ou mais de halogênio, -CF3, -NRR ou -SO2NRR, (v) - SO2R, (vi) -S-S-C1-6alquil-C(O)OR, (vii) -SO2NRR, (viii) -C(O)NRR, (ix) -C(O)OR, (x) -C4-6 cicloalquil opcionalmente substituído com -NRR, - SO2NRR ou -NR-C(O)(CH2)0-6NRR, (xi) -R, (xii) -OR, (xiii) -N(R)NRR, (xiv) -C(O)N(R)NRR, (xv) -(C(R)2)m-O-NRR e (xiv) -S-S-C1-6alquil- NRR;
[0188] cada R é independentemente selecionado do grupo que consiste em: hidrogênio e -C1-6alquil;
[0189] cada R' é independentemente selecionado de -H, C1-C8 al- quil, C1-C8 heteroalquil e aril;
[0190] cada R’’ é independentemente selecionado do grupo que consiste em: uma ligação e -C1-6alquileno-; e
[0191] b é 1-20; e
[0192] cada m é independentemente 0, 1, 2 ou 3.
[0193] Em outro aspecto, a presente invenção refere-se a um composto conjugado de anticorpo-droga de fórmulas III ou III’, em que o anticorpo AB é selecionado de: trastuzumab, mutantes de trastuzumab (por exemplo, os mutantes de trastuzumab divulgados aqui ou no pedido de patente internacional PCT/IB2012/056234), oregovomab, edrecolomab, cetuximab, um anticorpo monoclonal humanizado para o receptor vitronectina (αvβ3), alemtuzumab, um anticorpo anti-HLA-DR humanizado para o tratamento de linfoma de não-Hodgkin, 131I Lym- 1, um anticorpo anti-HLA-Dr10 de murino para o tratamento de linfoma de não-Hodgkin, um mAb anti-CD22 humanizado para o tratamento de Doença de Hodgkin ou linfoma de não-Hodgkin, labetuzumab, bevaci- zumab, ibritumomab tiuxetan, ofatumumab, panitumumab, rituximab, tositumomab, ipilimumab, gemtuzumab, anticorpo monoclonal humanizado para o receptor 5T4 de proteína oncofecal e M1/70 (anticorpo para receptor CD11b) e outros anticorpos.
[0194] Trastuzumab se refere a: (INN; nomes comerciais Herclon, Herceptin) se refere a um anticorpo monoclonal que interfere com o receptor HER2/neu.
[0195] Em outro aspecto, a presente invenção refere-se a um composto ou compostos de fórmulas II, II’, III ou III’, em que L compreende um ou mais di-radicais de aminoácido independentemente selecionados, preferencialmente um ou mais di-radicais de aminoácido independentemente selecionados, selecionados do grupo que consiste em valina, citrulina, fenilalanina, lisina, alanina e glicina.
[0196] De acordo com outro aspecto, a presente invenção refere- se a um composto ou compostos de fórmulas III ou III’, em que L é capaz de ser clivado a partir de P, ou um radical compreendendo P, por uma protease intracelular.
[0197] De acordo com um aspecto adicional, a presente invenção refere-se a um composto ou compostos de fórmulas III ou III’, em que o anticorpo é acoplado ao di-radical de aminoácido através de um resíduo de cisteína do anticorpo através de uma ligação de enxofre ou enxofre-enxofre, um resíduo de lisina do anticorpo através de uma ligação amida, ou um resíduo de glutamina através de uma ligação amida. Preferencialmente, o anticorpo é um anticorpo monoclonal, um anticorpo quimérico, um anticorpo humanizado, um anticorpo biespecí- fico ou um fragmento de anticorpo.
[0198] De acordo com ainda outro aspecto, a presente invenção refere-se a uma composição farmacêutica de um composto ou compostos de fórmulas I, I’, II, II’, III ou III’, e/ou um sal ou sais dos mesmos, compreendendo uma quantidade eficaz do composto(s) ou sal(is) e um excipiente, veículo ou diluente farmaceuticamente aceitável. Tais composições farmacêuticas podem adicionalmente incluir uma quantidade terapeuticamente eficaz de um agente quimioterapêutico selecionado do grupo que consiste em um inibidor de formação de tubulina, um inibidor de topoisomerase, e um aglutinante de DNA.
[0199] De acordo com outro aspecto, a presente invenção refere- se a um método para exterminar ou inibir a proliferação de células tu- morais ou células de câncer compreendendo tratar células tumorais ou células de câncer em um paciente com uma quantidade do composto de fórmulas I, I’, II, II’, III ou III’, e/ou um sal ou sais dos mesmos, a referida quantidade sendo eficaz para exterminar ou inibir a proliferação das células tumorais ou células de câncer.
[0200] Outro aspecto da invenção refere-se a um método de utilização de uma quantidade eficaz de qualquer um dos compostos acima mencionados e/ou qualquer um dos conjugados de anticorpo-droga para tratar câncer pela administração a um paciente em necessidade do mesmo de uma quantidade eficaz do referido composto e/ou conjugado.
BREVE DESCRIÇÂO DOS DESENHOS
[0201] A Fig. 1 é uma representação esquemática da relação filo- genética determinada com sequências rRNA 16S quase completas de FERM BP-3421 a outra Burkholderia spp.
[0202] A Fig. 2 é um grupo de gene biossintético para spliceosta- tins e etapas de hidroxilação de realce da via biossintética proposta catalizado pelo citocromo P450 Fr9R e dioxigenase Fr9P dependente de Fe(II)/α-cetoglutarato.
[0203] A Fig. 3 é um gráfico que apresenta dados de xenoenxerto para ADCs 3, 4 e 5.
[0204] A Fig. 4 é um gráfico que apresenta dados de xenoenxerto para ADCs 14 e 18.
DESCRIÇÃO DETALHADA
[0205] A presente invenção refere-se a produtos natural citotóxi- cos, incluindo análogos de spliceostatin citotóxicos, a conjugados de anticorpo-droga compreendendo os referidos produtos naturais citotó- xicos incluindo análogos de spliceostatin citotóxicos e a métodos para utilização dos mesmos para tratar câncer e outras condições patológicas. A invenção também se refere a métodos de utilização de tais compostos e/ou conjugados em vitro, in situ, e em vivo para a detecção, diagnose ou tratamento de células de mamíferos, ou condições patológicas associadas.
DEFINIÇÕES E ABREVIAÇÕES
[0206] A menos que definido de outra forma, os seguintes termos e frases como usados aqui destinam-se a ter os significados a seguir. Quando nomes comerciais são usados aqui, o nome comercial inclui a formulação do produto, a droga genérica, e o(s) ingrediente(s) farma- cêutico(s) ativo(s) do produto de nome comercial, a menos que de outra forma indicado pelo contexto.
[0207] O termo "anticorpo" (ou “Ab” ou “AB”) aqui é usado no sentido mais amplo e especificamente cobre anticorpos monoclonais intactos, anticorpos policlonais, anticorpos monoespecíficos, anticorpos multiespecíficos (por exemplo, anticorpos biespecíficos), e fragmentos de anticorpo que apresentam a atividade biológica desejada. Um anticorpo intacto possui primariamente duas regiões: uma região variável e uma região constante. A região variável se liga a e interage com um antígeno alvo. A região variável inclui uma região determinante de complementaridade (CDR) que reconhece e se liga a um sítio de ligação específico em um antígeno particular. A região constante pode ser reconhecida por e interagir com o sistema imune (ver, por exemplo, Janeway et al., 2001, Immuno. Biology, 5th Ed., Garland Publishing, New York). Um anticorpo pode ser de qualquer tipo ou classe (por exemplo, IgG, IgE, IgM, IgD, e IgA) ou subclasse (por exemplo, IgG1, IgG2, IgG3, IgG4, IgA1 e IgA2). O anticorpo pode ser derivado de qualquer espécie adequada. Em algumas modalidades, o anticorpo é de origem humana ou de murino. Um anticorpo pode ser, por exemplo, humano, humanizado ou quimérico.
[0208] Os termos "especificamente se liga" e "ligação específica" se referem a ligação de anticorpo a um antígeno predeterminado. Tipicamente, o anticorpo se liga com uma afinidade de pelo menos cerca de 1x107 M-1, e se liga ao antígeno predeterminado com uma afinidade que é pelo menos duas vezes superior do que sua afinidade para ligação a um antígeno não específico (por exemplo, BSA, caseína) outro que não o antígeno predeterminado ou um antígeno intimamente relacionado.
[0209] O termo "anticorpo monoclonal" como usado aqui se refere a um anticorpo obtido de uma população de anticorpos substancialmente homogêneos, isto é, os anticorpos individuais compreendendo a população são idênticos exceto por possíveis mutações de ocorrência natural que podem estar presentes em menores quantidades. Anticorpos monoclonais são altamente específicos, sendo direcionados contra um sítio antigênico específico. O "monoclonal" modificador indica o caráter do anticorpo como sendo obtido de uma população de anticorpos substancialmente homogênea, e não deve ser interpretado como requerendo produção do anticorpo por qualquer método particular.
[0210] O termo "anticorpos monoclonais" especificamente inclui anticorpos "quiméricos" em que uma porção da cadeia leve e/ou pesada é idêntica a ou homóloga com a sequência correspondente de anticorpos derivados de uma espécie particular ou pertencente a uma classe ou subclasse de anticorpo particular, embora o restante da ca- deia(s) seja idêntico a ou homólogo com a sequência correspondente de anticorpos derivados de ouyras espécies ou pertencentes a outra classe ou subclasse de anticorpo, bem como fragmentos de tais anti- corpos, contanto que eles exibam a atividade biológica desejada.
[0211] Um "anticorpo intacto" é um que compreende uma região variável de ligação a antígeno bem como um domínio constante de cadeia leve (CL) e domínios constantes de cadeia pesada, CH1, CH2, CH3 e CH4, conforme apropriado para a classe de anticorpo. Os domínios constantes podem ser domínios constantes de sequência nativa (por exemplo, domínios constantes de sequência nativa humanos) ou variantes de sequência de aminoácido dos mesmos.
[0212] Um anticorpo intacto pode ter uma ou mais "funções efeto- ras", que se referem àquelas atividades biológicas atribuíveis à região Fc (por exemplo, uma região Fc de sequência nativa ou região Fc variante de sequência de aminoácido) de um anticorpo. Exemplos de funções efetoras de anticorpo incluem citotoxicidade dependente de complemento, citotoxicidade mediada por célula dependente de anticorpo (ADCC) e fagocitose mediada por célula dependente de anticorpo.
[0213] Um "fragmento de anticorpo" compreende uma porção de um anticorpo intacto, preferencialmente compreendendo a região variável ou de ligação a antígeno dos mesmos. Exemplos de fragmentos de anticorpo incluem fragmentos Fab, Fab', F(ab')2, e Fv, diacorpos, triacorpos, tetracorpos, anticorpos lineares, moléculas de anticorpo de cadeia simples, scFv, scFv-Fc, fragmentos de anticorpo multiespecífi- cos formados a partir de fragmento(s) de anticorpo, um fragmento(s) produzido por uma biblioteca de expressão Fab, ou fragmentos de ligação a epítopo de qualquer dos acima que imunoespecificamente se ligam a um antígeno alvo (por exemplo, um antígeno de célula de câncer, um antígeno viral ou um antígeno microbiano).
[0214] O termo "variável" no contexto de um anticorpo se refere a determinadas porções dos domínios variáveis do anticorpo que diferem extensivamente na sequência e são usados na ligação e especifi- cidade de cada anticorpo particular para seu antígeno particular. Esta variabilidade é concentrada em três segmentos chamados "regiões hipervariáveis" nos domínios variáveis de cadeia leve e cadeia pesada. As porções mais altamente conservadas de domínios variáveis são chamadas as regiões de quadro (FRs). Os domínios variáveis de cadeias leves e pesadas nativas compreendem, cada, quatro FRs conectadas pelas três regiões hipervariáveis.
[0215] O termo "região hipervariável" quando usado aqui se refere aos resíduos de aminoácido de um anticorpo que são responsáveis por ligação a antígeno. A região hipervariável geralmente compreende resíduos de aminoácido de uma "região determinante de complementaridade" ou "CDR" (por exemplo, resíduos 24-34 (L1), 50-56 (L2) e 89-97 (L3) no domínio variável de cadeia leve e 31-35 (H1), 50-65 (H2) e 95-102 (L3) no domínio variável de cadeia pesada; Kabat et al. (Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, Md. (1991)) e/ou aqueles resíduos de um "ciclo hipervariável" (por exemplo, resíduos 26-32 (L1), 50-52 (L2) e 91-96 (L3) no domínio variável de cadeia leve e 26-32 (H1), 53-55 (142) e 96-101 (H3) no domínio variável de cadeia pesada; Chothia and Lesk, 1987, J. Mol. Biol. 196:901-917). Resíduos de FR são aqueles resíduos de domínio variável outros que não os resíduos de região hipervariável conforme aqui definido.
[0216] Um fragmento de anticorpo "Fv de cadeia simples" ou "scFv" compreende os domínios de um anticorpo V.sub.H e V.sub.L, em que estes domínios estão presentes em uma cadeia de polipeptí- deo simples. Tipicamente, o polipeptídeo Fv compreende ainda um ligante de polipeptídeo entre os domínios V.sub.H e V.sub.L que permite o scFv formar a estrutura desejada para ligação de antígeno. Para uma revisão de scFv, ver Pluckthun em The Pharmacology of Monoclonal Antibodies, vol. 113, Rosenburg and Moore eds., Springer- Verlag, New York, pp. 269-315 (1994).
[0217] O termo "diacorpo" se refere a pequenos fragmentos de anticorpo com dois sítios de ligação a antígeno, cujos fragments compreendem um domínio de cadeia variável (VH) conectado a um domínio leve variável (VL) na mesma cadeia de polipeptídeo. Pela utilização de um ligante que seja muito curto para permitir emparelhamento entre os dois domínios na mesma cadeia, os domínios são forced forçados a emparelhar com os domínios complementares de outra cadeia e criar dois sítios de ligação a antígeno. Diacorpos são descritos mais completamente em, por exemplo, EP 0 404 097; WO 93/11161; e Hollinger et al., 1993, Proc. Natl. Acad. Sci. USA 90:6444-6448.
[0218] Formas "humanizadas" de anticorpos não humanos (por exemplo, roedores) são anticorpos quiméricos que contêm derivados de sequência mínima de imunoglobulina não-humana. Para a maior parte, anticorpos humanizados são imunoglobulinas humanas (anticorpo receptor), em que resíduos da região hipervariável do receptor são substituídos por resíduos da região hipervariável de espécies não humanas (anticorpo doador), tais como camundongo, rato, coelho ou primata não humano tendo a desejada especificidade, afinidade e capacidade. Em alguns casos, resíduos da região de quadro (FR) da imunoglobulina humana são substituídos por resíduos não humanos correspondentes. Além disso, anticorpos humanizados podem com-preender resíduos que não sçao encontrados no anticorpo receptor ou no anticorpo doador. Estas modificações são feitas para adicionalmente refinar o desempenho do anticorpo. Em geral, o anticorpo humanizado irá compreendem substancialmente todos de pelo menos um, e tipicamente dois, domínios variáveis, em que todos ou substancialmente todos dos ciclos hipervariáveis correspondem àqueles de uma imunoglobulina não-humana e todas ou substancialmente todas das FRs são aquelas de uma sequência de imunoglobulina humana. O an- ticorpo humanizado opcionalmente irá também compreender pelo menos uma porção de uma região constante (Fc) de imunoglobulina, tipicamente aquela de uma imunoglobulina humana. Para detalhes adicionais, ver Jones et al., 1986, Nature 321:522-525; Riechmann et al., 1988, Nature 332:323-329; e Presta, 1992, Curr. Op. Struct. Biol. 2:593-596.
[0219] Como usado aqui, "isolado" significa separado de outros componentes de (a) uma fonte natural, tal como uma célula animal ou vegetal ou cultura celular, ou (b) uma mistura de reação química orgânica. Como usado aqui, "purificado" significa que, quando isolado o isolado contém pelo menos 95%, e, em outro aspecto, pelo menos 98%, de um composto (por exemplo, um conjugado) em peso do isolado.
[0220] Um anticorpo "isolado" é um que foi identificado e separado e/ou recuperado de um componente de seu ambiente natural. Componentes contaminnates de seu ambiente natural são materiais que não interferem com usos diagnósticos ou terapêuticos para o anticorpo, e podem incluir enzimas, hormônios, e outros solutos proteináceos ou não proteináceos. Em modalidades preferidas, o anticorpo será purificado (1) para mais do que 95% em peso de anticorpo conforme determinado pelo método Lowry, e mais preferencialmente mais do que 99% em peso, (2) até um grau suficiente para obter pelo menos 15 resíduos de sequência de aminoácido N-terminal ou interna por utilização de um sequenciado de copo giratório, ou (3) até homogeneidade por SDS-PAGE sob condições de redução ou não redução usando Coomassie blue ou, preferencialmente, corante prata. O anticorpo iso-lado inclui o anticorpo in situ dentro de células recombinantes desde que pelo menos um componente do ambiente natural do anticorpo não esteja presente. Ordinariamente, no entanto, o anticorpo isolado será preparado por pelo menos uma etapa de purificação.
[0221] Um anticorpo que "induz apoptose" é um que induz morte celular programada conforme determinado por ligação de anexina V, fragmentation de DNA, encolhimento celular, dilatação de retículo en- doplasmático, fragmentação celular, e/ou formação de vesículas de membrana (chamadas corpos apoptóticos). A célula é uma célula tumoral, por exemplo, uma célula de mama, ovário, estômago, endomé- trio, glândulas salivares, pulmão, rim, cólon, tiróide, pâcreas ou bexiga. Vários métodos estão disponíveis para avaliação dos eventos celulares associados a apoptose. Por exemplo, translocação de fosfatidil serina (PS) pode ser medida por ligação de anexina; fragmentação de DNA pode ser avaliada através de escalonamento de DNA; e condensação nuclear/de cromatina junto com fragmentação de DNA pode ser avaliada por qualquer aumento em células hipodiplóides.
[0222] O termo "quantidade terapeuticamente eficaz" se refere a uma quantidade de uma droga eficaz para tratar uma doença ou distúrbio em um mamífero. No caso de câncer, a quantidade terapeuti- camente eficaz da droga pode reduzir o número de células de câncer; reduzir o tamanho do tumor; inibir (isto é, reduzir até certo ponto e preferencialmente parar) a infiltração de células de câncer em órgãos periféricos; inibir (isto é, reduzir até certo ponto e preferencialmente parar) metástase de tumor; inibir, até certo ponto, crescimento do tumor; e/ou aliviar até certo ponto um ou mais dos sintomas associados com o câncer. Na medida em que a droga pode inibir o crescimento de e/ou exterminar células de câncer existentes, esta pode ser citostática e/ou citotóxica. Para terapia de câncer, a eficácia pode, por exemplo, ser medida pela avaliação do tempo de progressão da doença (TTP) e/ou determinação da taxa de resposta (RR).
[0223] O termo "quantidade substancial" se refere a uma maioria, isto é, mais do que 50% de uma população, de uma mistura ou uma amostra.
[0224] O termo "metabólito intracelular" se refere a um composto resultante de uma reação ou processo metabólico dentro de uma célula em um conjugado de anticorpo-droga (ADC). A reação ou processo metabólito pode ser um processo enzimático, tal como clivagem prote- olítica de um ligante de peptídeo do ADC. Metabólitos intracelulares incluem, mas não são limitados a, anticorpos e droga livre que sofreram clivagem intracelular após entrada, difusão, captação ou transporte em uma célula.
[0225] Os termos "clivado intracelularmente" e "clivagem intracelular" se referem a uma reação ou processo metabólico dentro de uma célula em um ADC ou semelhantes, por meio do que a ligação covalente, por exemplo, o ligante, entre a porção de droga e o anticorpo é quebrada, resultando na droga livre, ou outro metabólito do conjugado dissociado do anticorpo dentro da célula. As porções clivadas do ADC são, desta forma, metabólitos intracelulares.
[0226] O termo "biodisponibilidade" se refere à disponibilidade sistêmica (isto é, níveis de sangue/plasma) de uma dada quantidade de uma droga administrada a um paciente. Biodisponibilidade é um termo absoluto que indica medição de ambos o tempo (taxa) e quantidade total (extensão) de droga que alcança a circulação geral a partir de uma forma de dosagem administrada.
[0227] O termo "atividade citotóxica" se refere a um efeito de exterminação celular, citostático ou anti-proliferativo de um ADC ou um metabólito intracelular do referido ADC. A atividade citotóxica pode ser expressa como o valor de IC50, que é a concentration (molar ou de massa) por volume de unidade em que metade das células sobrevive.
[0228] Um "distúrbio" é qualquer condição que se beneficiaria do tratamento com uma droga ou conjugado de anticorpo-droga. Isto inclui doenças ou distúrbios agudos e crônicos incluindo aquelas condições patológicas que predispõem um mamífero ao distúrbio em ques- tão. Exemplos não limitantes de distúrbios a serem tratados aqui incluem cânceres malignos e benignos; leucemia e malignidades linfóides, distúrbios neuronais, gliais, astrocitais, hipotalâmicos e de outras glândulas, macrofágicos, epiteliais, estromais e blastocélicos; e distúrbios inflamatórios, angiogênicos e imunológicos.
[0229] Os termos "câncer" e "canceroso" se referem a ou descrevem o distúrbio ou condição fisiológica em mamíferos que tipicamente caracterizada por crescimento celular irregular. Um "tumor" compreende uma ou mais células cancerosas.
[0230] Exemplos de um "paciente" incluem, mas não são limitados a, um ser humano, rato, camundongo, porquinho-da-índia, macaco, porco, cabra, vaca, cavalo, cachorro, gato, pássaro e galinha. Em uma modalidade exemplar, o paciente é um ser humano.
[0231] Os termos "tratar" ou "tratamento," a menos que de outra forma indicado pelo contexto, se referem a tratamento terapêutico e medidas profiláticas para prevenir reincidência, em que o objeto deve inibir ou retardar (diminuir) um distúrbio ou alteração fisiológica indese- jada, tal como o desenvolvimento ou propagação de câncer. Para os propósitos desta invenção, resultados clínicos benéficos ou desejados incluem, mas não são limitados a, alívio de sintomas, diminuição da extensão da doença, estado de doença estabilização (isto é, não agravamento), retardo ou desaceleração da progressão da doença, melhoria ou paliação do estado da doença, e remissão (seja parcial ou total), seja detectável ou indetectável. "Tratamento" pode também significar prolongamento da sobrevivência quando comparado à expectativa de sobrevivência se não receber tratamento. Aqueles em necessidade de tratamento incluem aqueles que já apresentam a condição ou distúrbio bem como aqueles propícios a terem a condição ou distúrbio.
[0232] No contexto de câncer, o termo "tratar" inclui quaisquer ou todos de inibição do crescimento de células tumorais, células de cân- cer, ou de um tumor; inibição da replicação de células tumorais ou células de câncer, diminuição da carga global do tumor ou diminuição do número de células cancerosas, e melhora de um ou mais sintomas associados com a doença.
[0233] No contexto de uma doença autoimune, o termo "tratar" inclui quaisquer ou todos de inibição de replicação de células associada com um estado de doença autoimune incluindo, mas não limitado a, células que produzem um anticorpo autoimune, diminuição da carga de anticorpo autoimune e melhora de um ou mais sintomas de uma doença autoimune.
[0234] No contexto de uma doença infecciosa, o termo “tratar” inclui quaisquer ou todos de: inibição do crescimento, multiplicação ou replicação do patógeno que causa a doença infecciosa e melhora de um ou mais sintomas de uma doença infecciosa.
[0235] O termo "bula" é usado para se referir a instruções geralmente incluídas em embalagens comerciais de produtos terapêuticos, que contêm informações sobre a indicação(s), uso, dosagem, administração, contraindicações e/ou alertas a respeito da utilização de tais produtos terapêuticos.
[0236] Como usados aqui, os termos "célula", "linhagem celular" e "cultura celular" são usados intercambiavelmente e todas tais designações incluem progenitura. As palavras "transformantes" e "células transformadas" incluem a célula objeto primária e culturas ou progenitura derivadas dos mesmos sem levar em conta o número de transferências. Deve ser entendido que todas as progenituras podem não ser precisamente idênticas em teor de DNA, devido a mutações inadvertidas ou deliberadas. A progenitura mutante que tem a mesma função ou atividade biológica como pesquisado na célula originalmente transformada está incluída. Onde designações distintas são pretendidas, ficará claro a parti do contexto.
[0237] A menos que de outra forma indicado, o termo "alquil" sozinho ou como parte de outro termo se refere a um hidrocarboneto saturado, ramificado ou de cadeia reta tendo o número indicado de átomos de carbono (por exemplo, "C1-C8" alquil se referem a um grupo alquil tendo de 1 a 8 átomos de carbono). Quando o número de átomos de carbono não é indicado, o grupo alquil tem de 1 a 8 átomos de carbono, preferencialmente, de 1 a 6 átomos de carbono. C1-C8 alquils de cadeia reta representativos incluem, mas não são limitados a, metil, etil, n-propil, n-butil, n-pentil, n-hexil, n-heptil e n-octil; embora C1-C8 alquils ramificados incluem, mas não são limitados a, -isopropil, -sec- butil, -isobutil, -tent-butil, -isopentil, e -2-metilbutil; C2-C8 alquils insatu- rados incluem, mas não são limitados a, vinil, alil, 1-butenil, 2-butenil, isobutilenil, 1-pentenil, 2-pentenil, 3-metil-1-butenil, 2-metil-2-butenil, 2,3-dimetil-2-butenil, 1-hexil, 2-hexil, 3-hexil, acetilenil, propinil, 1- butinil, 2-butinil, 1-pentinil, 2-pentinil e 3-metil-1-butinil.
[0238] A menos que de outra forma indicado, "alquileno," sozinho ou como parte de outro termo, se refere a um radical hidrocarboneto cíclico ou de cadeia reta ou ramificada, saturado do número de átomos de carbono definido, tipicamente 1-18 átomos de carbono, e tendo dois centros radicais monovalentes derivados pela remoção de dois átomos de hidrogênio do mesmo ou de dois átomos de carbono diferentes de um alcano parente. Radicais alquileno típicos incluem, mas não são limitados a: metileno (-CH2-), 1,2-etileno -CH2CH2-), 1,3-propileno (-CH2CH2CH2-), 1,4-butileno (-CH2CH2CH2CH2-), e semelhantes. Um "C1-C10" alquileno de cadeia reta é um grupo hidrocarboneto saturado de cadeia reta da fórmula -(CH2)1-10-. Exemplos de um C1-C10 alquileno incluem metileno, etileno, propileno, butileno, pentileno, hexileno, hep- tileno, octileno, nonileno e decaleno. Em certas modalidades da invenção, alquilenos tem de 1 a 9, de 1 a 8, de 1 a 7, e de 1 a 6 carbonos.
[0239] A menos que de outra forma indicado, o termo "heteroal- quil," sozinho ou em combinação com outro termo, significa, a menos que de outra forma definido, um hidrocarboneto de cadeia ramificada ou reta estável, ou combinações dos mesmos, totalmente saturado ou contendo de 1 a 3 graus de insaturação, que consiste no número de átomos de carbono definido e de um a três heteroátomos selecionados do grupo que consiste em O, N, Si, S e/ou P, e, em que os átomos de nitrogênio e enxofre podem opcionalmente ser oxidados e o heteroá- tomo de nitrogênio pode opcionalmente ser quaternizado. O(s) hetero- átomo(s) O, N e S pode(m) ser colocado(s) em qualquer posição interior do grupo heteroalquil. O heteroátomo Si pode ser colocado em qualquer posição do grupo heteroalquil, incluindo a posição em que o grupo alquil é acoplado ao restante da molécula. Até dois heteroáto- mos podem ser consecutivos.
[0240] “Halo" ou "halogênio" se refere a flúor, cloro, bromo e iodo.
[0241] "Halo(C1-6-alquil)" se refere a grupos C1-6-alquil substituídoscom 1 a 3 ou 1 a 2 grupos halo, em que C1-6-alquil e halo são como aqui definidos. O termo inclui, por exemplo, CF3.
[0242] O termo “epóxi”, ou “grupo epóxi” ou “resíduo epóxi” como conhecido por aqueles versados na técnica se referem a um anel de três membros compreendendo átomos de carbono e um átomo de oxigênio ligados por ligações simples como a seguir:
[0243] Consequentemente, o termo “epóxido” se refere a um composto que compreende pelo menos um grupo epóxi como aqui anteriormente definido.
[0244] A menos que de outra forma indicado, o termo "heteroalqui- leno" sozinho ou como parte de outro substituinte significa um grupo divalente derivado de heteroalquil (como discutido acima). Para grupos heteroalquileno, heteroátomos podem também ocupar um ou ambos os terminais da cadeia.
[0245] A menos que de outra forma indicado, "aril," sozinho ou uma parte de outro termo, significa um radical hidrocarboneto aromático monovalente substituído ou insubstituído de 6 a 20 átomos de carbono, preferencialmente de 6 a 14 átomos de carbono, derivados pela remoção de um átomo de hidrogênio de um átomo de carbono simples de um sistema de anel aromático parente. Grupos aril típicos incluem, mas não são limitados a, radicals derivados de benzeno, benzeno substituído, naftaleno, antraceno, bifenil, e semelhantes. Um grupo aromático substituído (por exemplo, um grupo aril) pode ser substituído com um ou mais, preferencialmente, 1 a 5, dos seguintes grupos: C1-C8 alquil, -O-(C1-C8 alquil), -C(O)R', -OC(O)R', -C(O)OR', -C(O)NH2, -C(O)NHR', -C(O)N(R')2, -NHC(O)R', -S(O)2R', -S(O)R', -OH, halogênio, -N3, -NH2, -NH(R'), -N(R')2 e -CN;, em que cada R' é independentemente selecionado de -H, C1-C8 alquil, C1-C8 heteroalquil e aril, preferencialmente aril insubstituído. Em algumas modalidades, um grupo aromático substituído pode ainda incluir um ou mais de: -NHC(=NH)NH2, -NHCONH2, -S(=O)2R' e -SR'.
[0246] O termo "heteroaril", como usado aqui, se refere a um anel heterociclo aromático de 5 a 14 membros, tal como 5 a 6 membros, tendo pelo menos one heteroátomo selecionado de nitrogênio, oxigênio e enxofre, e contendo pelo menos 1 átomo de carbono. Heteroarils podem ser sistemas de anel monocíclicos, bicíclicos ou tricíclicos. Heteroarils representativos são triazolil, tetrazolil, oxadiazolil, piridil, furil, benzofuranil, tiofenil, benzotiofenil, quinolinil, pirrolil, indolil, oxazolil, benzoxazolil, imidazolil, benzimidazolil, tiazolil, benzotiazolil, isoxazolil, pirazolil, isotiazolil, piridazinil, pirimidinil, pirazinil, triazinil, cinolinil, fta- lazinil, quinazolinil, pirimidil, azepinil, oxepinil, e quinoxalinil. Heteroarils são opcionalmente substituídos. Substituintes típicos incluem, mas não são limitados a, -X, -R, -O-, -OR, -SR, -S-, -NR2, -NR3, =NR, -CX3, -CN, -OCN, -SCN, -N=C=O, -NCS, -NO, -NO2, =N2, -N3, -NRC(=O)R, -C(=O)NR2, -SO3-, -SO3H, -S(=O)2R, -OS(=O)2OR, -S(=O)2NR, -S(=O)R, -OP(=O)(OR)2, -P(=O)(OR)2, -PO32-, PO3H2, -AsO2H2, -C(=O)R, -C(=O)X, -C(=S)R, -CO2R, -CO2-, -C(=S)OR, -C(=O)SR, -C(=S)SR, -C(=O)NR2, -C(=S)NR2, -C(=NR)NR2, C1-C20 heteroalquil, C6-C20 aril, C3-C8 heterociclil, um grupo de proteção ou uma porção de pró-droga, em que cada X é independentemente um halogênio: -F, -Cl, -Br, ou -I; e cada R é independentemente -H ou C1-C6 alquil.
[0247] Os termos “arileno”, “heteroarileno” se referem a versões divalentes de “aril” e “heteroaril”, respectivamente, e outros termos incorporando “aril” e “heteroaril”.
[0248] "Hidroxi" se refere ao grupo -OH.
[0249] "Alquil substituído" significa an alquil em que um ou mais átomos de hidrogênio são, cada, independentemente substituídos com um substituinte. Substituintes típicos incluem, mas não são limitados a, -X, -R, -O-, -OR, -SR, -S-, -NR2, -NR3, =NR, -CX3, -CN, -OCN, -SCN, -N=C=O, -NCS, -NO, -NO2, =N2, -N3, -NRC(=O)R, -C(=O)NR2, -SO3-, -SO3H, -S(=O)2R, -OS(=O)2OR, -S(=O)2NR, -S(=O)R, -OP(=O)(OR)2, -P(=O)(OR)2, -PO32-, PO3H2, -AsO2H2, -C(=O)R, -C(=O)X, -C(=S)R, -CO2R, -CO2-, -C(=S)OR, -C(=O)SR, -C(=S)SR, -C(=O)NR2, -C(=S)NR2, -C(=NR)NR2, C1-C20 heteroalquil, C6-C20 aril, C3-C8 hetero- ciclil, um grupo de proteção ou uma porção de pró-droga, em que cada X é independentemente um halogênio: -F, -Cl, -Br, ou -I; e cada R é independentemente -H ou C1-C6 alquil.Um alquil substituído, substituído com um halogênio, é às vezes referido aqui como um haloalquil. Aril, alquileno, heteroalquileno e outros grupos contendo ou não contendo uma porção alquileno ou alquil como descrito aqui pode também ser similarmente substituído.
[0250] A menos que de outra forma indicado, “aralquil” sozinho ou parte de outro termo, significa um grupo alquil, como definido acima, substituído com um grupo aril, como definido acima.
[0251] A menos que de outra forma indicado, "C3-C8heterociclil" sozinho ou como parte de outro termo, se refere a um sistema de anel bicíclico ou monocíclico aromático ou não-aromático substituído ou insubstituído divalente ou monovalente tendo de 3 a 8 átomos de carbono (também referido como membros de anel) e um a quatro membros de anel heteroátomo independentemente selecionados de N, O, P ou S, e derivados por remoção de um átomo de hidrogênio de um átomo de anel de um sistema de anel parente. Similarmente, a menos que de outra forma indicado, "C3-C10heterociclil" sozinho ou como parte de outro termo, se refere a um sistema de anel bicíclico ou monocí- clico aromático ou não-aromático substituído ou insubstituído divalente ou monovalente tendo de 3 a 10 átomos de carbono (também referido como membros de anel) e um a quatro membros de anel heteroátomo independentemente selecionados de N, O, P ou S, e derivados por remoção de um átomo de hidrogênio de um átomo de anel de um sistema de anel parente. Um ou mais N, C ou S átomos no heterociclil podem ser oxidados. O anel que inclui o heteroátomo pode ser aromático ou não aromático. Grupos heterociclil com mais do que 10 carbonos, por exemplo, aneis ou sistemas de anel com 11, 12, 13, 14, 15, 16, 17, 18, 19 ou 20 carbonos, são também possíveis e estão englobados, junto com C3-C10heterociclils, quando o termo “heterociclil” é empregado sem referência a um número específico de carbonos. Similarmente, grupos heterociclil com menos do que 3 carbonos, por exemplo, aneis com 1 ou 2, são possíveis e estão englobados quando o termo “heterociclil” é empregado sem referência a um número específico de carbonos. O termo “heterocicloalquil” se refere a sistemas de anel ou aneis heterociclil não aromáticos em que todos os átomos de carbono são saturados (isto é, ligados a um hidrogênio ou outro substi- tuinte como indicado abaixo, com nenhuma ligação dupla ou tripla). Em certas modalidades, grupos heterocicloalquil tipicamente tem 3 a 5 membros e 1 a 2 heteroátomos. Em certas modalidades, heterociclo- alquil pode ser epóxi.
[0252] A menos que de outra forma indicado, o heterociclil é ligado ao seu grupo pendente em qualquer heteroátomo ou átomo de carbono que resulta em uma estrutura estável. Exemplos representativos de um C3-C8 heterociclil incluem, mas não são limitados a, tetrahidrofura- noil, oxetanil, piranil, pirrolidinil, piperidinil, benzofuranil, benzotiofeno, indolil, benzopirazolil, pirrolil, tiofenil (tiofeno), furanil, tiazolil, imidazolil, pirazolil, triazolil, quinolinil, pirimidinil, piridinil, piridonil, pirazinil, pirida- zinil, isotiazolil, isoxazolil e tetrazolil. Um C3-C8 heterociclil, ou um C3C10 heterociclil, pode ser substituído com até sete grupos incluindo, mas não limitados a, C1-C8 alquil, C1-C8 heteroalquil, -OR', aril, -C(O)R', -OC(O)R', -C(O)OR', -C(O)NH2, -C(O)NHR', -C(O)N(R')2, -NHC(O)R', -S(=O)2R', -S(O)R', halogênio, -N3, -NH2, -NH(R'), -N(R')2 e -CN;, em que cada R' é independentemente selecionado de -H, C1-C8 alquil, C1-C8 heteroalquil e aril. Em algumas modalidades, um hetero- ciclil substituído pode também incluir um ou mais de: -NHC(=NH)NH2, -NHCONH2, -S(=O)2R' e -SR'.
[0253] A menos que de outra forma indicado, “heteroaralquil” sozinho ou parte de outro termo, significa um grupo alquil, como definido acima, substituído com um grupo heterociclil aromático, como definido acima.
[0254] A menos que de outra forma indicado, “C3-C8 carbociclil” sozinho ou como parte de outro termo, é um anel carbocíclico bicíclico ou monocíclico não aromático saturado ou insaturado, substituído ou insubstituído, monovalente ou divalente de 3-, 4-, 5-, 6-, 7- ou 8- membros derivados pela remoção de um átomo de hidrogênio ou dois átomos de hidrogênio de um átomo de anel de um sistema de anel parente. Similarmente, a menos que de outra forma indicado, “C3-C10 carbo- ciclil” sozinho ou como parte de outro termo, é um anel carbocíclico bicíclico ou monocíclico não aromático saturado ou insaturado, substituído ou insubstituído, monovalente ou divalente de 3-, 4-, 5-, 6-, 7-, 8-, 9- ou 10 membros derivados pela remoção de um átomo de hidrogênio de um átomo de anel de um sistema de anel parente. C3-C8 carbociclil representativos incluem, mas não são limitados a, ciclopropil, ciclobutil, ciclopentil, ciclopentadienil, ciclohexil, ciclohexenil, 1,3-ciclohexadienil, 1,4-ciclohexadienil, cicloheptil, 1,3-cicloheptadienil, 1,3,5- cicloheptatrienil, ciclooctil, ciclooctadienil, biciclo(111)pentano, e bici- clo(222)octano. Um grupo C3-C8 carbociclil, ou um grupo C3-C10 carbo- ciclil, pode ser insubstituído ou substituído com até sete grupos incluindo, mas não limitado a, C1-C8 alquil, C1-C8 heteroalquil, -OR', aril, -C(O)R', -OC(O)R', -C(O)OR', -C(O)NH2, -C(O)NHR', -C(O)N(R')2, -NHC(O)R', -S(=O)2R', -S(=O)R', -OH, -halogênio, -N3, -NH2, -NH(R'), -N(R')2 e -CN;, em que cada R' é independentemente selecionado de -H, C1-C8 alquil, C1-C8 heteroalquil e aril. Grupos carbociclil com mais do que 10 carbonos, por exemplo, sistemas de anel com 11, 12, 13, 14, 15, 16, 17, 18, 19 ou 20 carbonos, são também possíveis e estão englobados, junto com C3-C10 carbociclils, quando o termo “carbociclil” é empregado sem referência a um número específico de carbonos. O termo “cicloalquil” se refere a aneis carbociclil ou sistemas de anel em que todos os átomos de carbono são saturados (isto é, ligados a um hidrogênio ou outro substituinte como indicado abaixo, com nenhuma ligação dupla ou tripla).
[0255] O termo "quiral" se refere a moléculas que possuem a propriedade de não sobreposição do parceiro de imagem de espelho, enquanto o termo "aquiral" se refere a moléculas que são passíveis de sobreposição em seu parceiro de imagem de espelho.
[0256] O termo "estereoisômeros" se refere a compostos que possuem constituição química idêntica, mas diferem com relação à dispo- sição dos átomos ou grupos no espaço.
[0257] "Diastereômero" se refere a um estereoisômero com dois ou mais centros de quiralidade e cujas moléculas não são imagens de espelho umas das outras. Diastereômeros apresentam diferentes propriedades físicas, por exemplo, pontos de fusão, pontos de ebulição, propriedades espectrais, e reatividades. Misturas de diastereômeros podem se separar sob procedimentos analíticos de alta resolução, tais como eletroforese e cromatografia.
[0258] Convenções e definições estereoquímicas usadas aqui geralmente seguem S. P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms, McGraw-Hill Book Company, New York (1984); e Eliel e Wilen, Stereochemistry Of Organic Compounds, John Wiley & Sons, Inc., New York (1994). Muitos compostos orgânicos existem em formas oticamente ativas, isto é, possuem a capacidade de girar o plano de luz polarizada plana. Na descrição de um composto oticamente ativo, os prefixos D e L, ou R e S são usados para denotar a configuração absoluta da molécula em torno de seu centro(s) quiral. Os prefixos d e l ou (+) e (-) são empregados para designar o sinal de rotação da luz polarizada plana pelo composto, com (-) ou 1 significando que o composto é levorrotatório. Um composto prefixado com (+) ou d é dex- trorrotatório. Para uma dada estrutura química, estes estereoisômeros são idênticos exceto que eles são imagens de espelho um do outro. Um estereoisômero específico pode também ser referido como um enantiômero, e uma mistura de tais isômeros é geralmente chamada uma mistura enantiomérica. Uma mistura de enantiômeros 50:50 é referida como uma mistura racêmica ou um racemato, que pode ocorrer onde não ocorreu estereosseleção ou estereoespecificidade em um processo ou reação química. Os termos "mistura racêmica" e "racemato" se referem a uma mistura equimolar de duas espécies enantioméri- cas, desprovida de atividade ótica.
[0259] Um "derivado" de aminoácido inclui um aminoácido tendo substituições ou modificações por ligação covalente de um aminoácido parente, tal como, por exemplo, por alquilação, glicosilação, acetila- ção, fosfoorilação, e semelhantes. Adicionalmente incluído dentro da definição de "derivado" é, por exemplo, um ou mais análogos de um aminoácido com ligações substituídas, bem como outras modificações conhecidas na técnica.
[0260] Um "aminoácido natural" se refere a arginina, glutamina, fenilalanina, tirosina, triptofano, lisina, glicina, alanina, histidina, serina, prolina, ácido glutâmico, ácido aspártico, treonina, cisteína, metionina, leucina, asparagina, isoleucina, e valina, a menos que de outra forma indicado pelo contexto.
[0261] A frase "sal farmaceuticamente aceitável", como usada aqui, se refere a sais inorgânicos ou orgânicos farmaceuticamente aceitáveis de um composto. O composto tipicamente contém pelo menos um grupo amino, e consequentemente sais de adição de ácido podem ser formados com este grupo amino. Sais exemplares incluem, mas não são limitados a, sulfato, citrato, acetato, oxalato, cloreto, brometo, iodeto, nitrato, bissulfato, fosfato, fosfato de ácido, isonicotinato, lactato, salicilato, citrato de ácido, tartarato, oleato, tanato, pantotena- to, bitartarato, ascorbato, succinato, maleato, gentisinato, fumarato, gluconato, glucuronato, sacarato, formiato, benzoato, glutamato, me- tanossulfonato, etanossulfonato, benzenossulfonato, p- toluenossulfonato, e sais de pamoato (isto é, 1,1'-metileno-bis-(2- hidroxi-3-naftoato)). Um sal farmaceuticamente aceitável pode envolver a inclusão de outra molécula, tal como um íon de acetato, um íon de succinate ou outro contra-íon. O contra-íon pode ser qualquer porção orgânica ou inorgânica que estabiliza a carga no composto parente. Além disso, um sal farmaceuticamente aceitável pode ter mais do que um átomo carregado em sua estrutura. Casos em que múltiplos átomos carregados são parte do sal farmaceuticamente aceitável podem ter vários contra-íons. Consequentemente, um sal farmaceutica- mente aceitável pode ter um ou mais átomos carregados e/ou um ou mais contra-íons.
[0262] Os termos “carregamento” ou “carregamento de droga” ou “carregamento de carga útil” representam ou se referem ao número médio de cargas úteis (“carga útil” e “cargas úteis” são usados inter- cambiavelmente aqui com “droga” e “drogas”) por anticorpo em uma molécula ADC. O carregamento de droga pode variar de 1 a 20 drugas por anticorpo. Isto é geralmente referido como o DAR, ou razão de droga a anticorpo. Composições dos ADCs descritos aqui tipicamente possuem DAR’s de 1-20, e em certas modalidades de 1-8, de 2-8, de 2-6, de 2-5 e de 2-4. Valores de DAR típicos são 2, 4, 6 e 8. O número médio de drogas por anticorpo, ou valor DAR, pode ser caracterizado por meios convencionais, tais como espectroscopia UV/visível, espec- trometria de massa, ensaio ELISA e HPLC. O valor DAR quantitativo pode também ser determinado. Em alguns casos, separação, purificação e caracterização de ADCs homogêneos tendo um valor DAR particular podem ser alcançadas por meios, tais como HPLC de fase reversa ou eletroforese. DAR pode ser limitado pelo número de sítio de li-gação no anticorpo. Por exemplo, onde a ligação é uma cisteína tiol, um anticorpo pode ter somente um ou vários grupos cisteína tiol, ou pode ter apenas um ou vários grupos tiol suficientemente reativos através dos quais uma unidade Ligante pode ser ligada. Em algumas modalidades, a cisteína tiol é um grupo tiol de um resíduo de cisteína que forma uma ligação dissulfito intercadeias. Em algumas modalidades, a cisteína tiol é um grupo tiol de um resíduo de cisteína que não forma uma ligação dissulfito intercadeias. Tipicamente, menos do que o máximo teórico de porções de droga é conjugado a um anticorpo durante uma reação de conjugação. Um anticorpo pode conter, por exemplo, muitos resíduos de lisina que não reagem com um ligante ou intermediário de ligante. Somente os grupos lisina mais reativos podem reagir com um reagent de ligante reativo.
[0263] Geralmente, anticorpos não contem muitos, se algum, grupos cisteína tiol livres e reativos que podem ser ligados a uma droga através de um ligante. A maior parte dos resíduos de cisteína tiol nos anticorpos existe como pontes dissulfeto e deve ser reduzida com um agente de redução, tal como ditiotreitol (DTT). O anticorpo pode ser submetido a condições de desnaturação para revelar grupos nucleofí- licos reativos, tais como lisina ou cisteína. O carregamento (razão dro- ga/anticorpo) de um ADC pode ser controlado de várias diferentes maneiras, incluindo: (i) limitar o excesso molar de droga-ligante relativo ao anticorpo, (ii) limitar o tempo de retenção de conjugação ou temperatura, e (iii) condições redutivas parciais ou limitantes para modificação de cisteína tiol. Onde mais do que um grupo nucleofílico reage com uma droga-ligante, então, o produto resultante é uma mistura de ADCs com uma distribuição de uma ou mais porções de droga por anticorpo. O número médio de drogas por anticorpo pode ser calculado a partir da mistura por, por exemplo, ensaio de anticorpo ELISA duplo, específico para anticorpo e específico para a droga. ADCs individuais podem ser identificados na mistura por espectroscopia de massa, e separados por HPLC, por exemplo, cromatografia de interação hidro- fóbica.
[0264] Abaixo está uma lista de abreviações e definições que não podem de outra forma ser definidas ou descritas neste pedido: DMSO (se refere a dimetil sulfóxido), HRMS (se refere a espectrometria de massa de alta resolução), DAD (se refere a detecção de matriz de diodo), TFA (se refere a ácido 2,2,2-trifluoroacético ou ácido trifluoroacéti- co), TFF (se refere a filtração de fluxo tangencial), EtOH (se refere a etanol), MW (se refere a peso molecular), HPLC (se refere a cromato- grafia líquida de alta performance), prep HPLC (se refere a cromato- grafia líquida de alta performance preparativa), etc. (se refere a e assim por diante), tritil (se refere a 1,1',1''-etano-1,1,1-triiltribenzeno), THF (se refere a tetrahidrofurano), NHS (se refere a 1-Hidroxi-2,5- pirrolidinadiona), Cbz (se refere a carboxibenzil), eq. (se refere a equivalente), n-BuLi (se refere a n-butillítio), OAc (se refere a acetato), MeOH (se refere a metanol), i-Pr (se refere a isopropil ou propan-2-il), NMM (se refere a 4-metilmorfolina), e “-“ (em uma tabela se refere a nenhum dado disponível no momento).
[0265] Como usado aqui, “H/C” se refere a trastuzumab (nome comercial HERCEPTIN®), que é um anticorpo monoclonal que interfere com o receptor HER2/neu, ligado através de um de seus resíduos de cisteína (a um ligante ou um composto da invenção).
[0266] Como usado aqui, “H/K” se refere a trastuzumab que é um anticorpo monoclonal que interfere com o receptor HER2/neu, ligado através de um de seus resíduos de lisina (a um ligante ou um composto da invenção).
[0267] Como usado ao longo deste pedido, a numeração do resíduo de aminoácido (por exemplo: Alanina na posição 114) é baseada no índice EU do método Kabat.
[0268] Como usado aqui, “H/TG1-(Q)” se refere a trastuzumab projetado que é um anticorpo monoclonal que interfere com o receptor HER2/neu, ligado através de um de seus resíduos de glutamina projetados ou naturais que está no rótulo do substrato de peptídeo transglutaminase (TG1) incorporado no anticorpo (a um ligante ou um composto da invenção).
[0269] Como usado aqui, “H-A114C/C114” se refere a trastuzumab projetado que é um anticorpo monoclonal que interfere com o receptor HER2/neu, ligado através de uma de suas cisteínas projetadas que foi substituída por alanina na posição 114 de cadeia pesada (a um ligante ou um composto da invenção).
[0270] Como usado aqui, “H-K392C+L443C/C392+C443” se refere a trastuzumab projetado que é um anticorpo monoclonal que interfere com o receptor HER2/neu, ligado através de um ou ambos de seus resíduos de cisteína projetados que foram substituídos por lisina na posição 392 da cadeia pesada e leucina na posição 443 da cadeia pesada (a um ligante ou um composto da invenção).
[0271] Como usado aqui, “H-E388C+N421C/C388+C421” se refere a trastuzumab projetado que é um anticorpo monoclonal que interfere com o receptor HER2/neu, ligado através de um ou ambos de seus resíduos de cisteína projetados que foram substituídos por ácido glu- tâmico na posição 388 e asparigina na posição 421 de cadeia pesada (a um ligante ou um composto da invenção).
[0272] Como usado aqui, “H-Q347C+K392C/C347+C392” se refere a trastuzumab projetado que é um anticorpo monoclonal que interfere com o receptor HER2/neu, ligado através de um ou ambos de seus resíduos de cisteína projetados que foram substituídos por glutamina na posição 347 e lisina na posição 392 de cadeia pesada (a um ligante ou um composto da invenção).
[0273] Como usado aqui, “H-L443C+kK183C/C443+kC183” se refere a trastuzumab projetado que é um anticorpo monoclonal que interfere com o receptor HER2/neu, ligado através de um ou ambos de sua cisteína projetada que foi substituída por leucina na posição 443 na cadeia pesada e lisina na posição 183 de cadeia leve (kappa) (a um ligante ou um composto da invenção).
[0274] Como usado aqui, “H-Q347C+L443C/C347+C443” se refere a trastuzumab projetado que é um anticorpo monoclonal que interfere com o receptor HER2/neu, ligado através de um ou ambos de sua cis- teína projetada que foi substituída por glutamina na posição 347 e leu- cina na posição 443 de cadeia pesada (a um ligante ou um composto da invenção).
[0275] Como usado aqui, “H- kK183C/kC183” se refere a trastuzumab projetado que é um anticorpo monoclonal que interfere com o receptor HER2/neu, ligado através de sua cisteína projetada que foi substituída em lisina na posição 183 de cadeia leve (kappa) (a um li- gante ou um composto da invenção).
[0276] Como usado aqui, “H-N421C/C421” se refere a trastuzumab projetado que é um anticorpo monoclonal que interfere com o receptor HER2/neu, ligado através de sua cisteína projetada que foi substituída em asparigina na posição 421 de cadeia pesada (a um li- gante ou um composto da invenção).
[0277] Geralmente, como usado aqui, “H- (AA1)###(AA2)/(AA2)###” (em que (AA1) e (AA2) são um primeiro e um segundo aminoácido) se refere a trastuzumab projetado que é um anticorpo monoclonal que interfere com o receptor HER2/neu, ligado através de seu (AA2) projetado que foi substituído em (AA1) na posição ### de cadeia pesada para composto da invenção, em que ### representa a posição do aminoácido(s) relevante. Notação similar referenciando “k” ou kappa” indicaria uma substituição na cadeia leve.
[0278] Similarmente, como usado aqui, “H- (AA1)###(AA2)+(AA3)####(AA4)/ (AA2)###+(AA4)####” (onde (AA1), (AA2), (AA3) e (AA4) são primeiro, segundo, terceiro e quarto aminoá- cidos) se refere a trastuzumab projetado que é um anticorpo monoclonal que interfere com o receptor HER2/neu, ligado através de seu (AA2) projetado que foi substituído em (AA1) na posição ### de cadeia pesada para composto da invenção, em que ### representa a posição do aminoácido(s) relevante, e também ligado através de seu (AA4) projetado que foi substituído em (AA3) na posição #### de cadeia pesada para composto da invenção, em que #### representa a posição do aminoácido(s) relevante. Notação similar referenciando “k” ou kap pa” indicaria uma substituição na cadeia leve.
[0279] Como usado aqui, “-PABC-” ou “PABC” se refere à estrutura:
[0280] ou variantes dos mesmos.
[0281] Como usado aqui, “-PABA-” ou “PABA” se refere à estrutura:
[0282] ou variantes dos mesmos.
COMPOSTOS E CONJUGADOS DE ANTICORPO-DROGA DOS MESMOS
[0283] De acordo com um aspecto, a presente invenção refere-se a um composto ou compostos de fórmula (I):
[0284] (I)
[0285] em que:
[0286] uma linha pontilhada representa uma ligação opcional;
[0287] cada x1 é independentemente selecionado do grupo que consiste em: -O-, -S- e -NR-;
[0288] cada x2 é independentemente selecionado do grupo que consiste em: -O-, -S- e -NR-;
[0289] R1 é selecionado do grupo que consiste em: -R, -OR, - OCOR13, -OCONR14R15, -OCON(R14)NR(R15), =O (ligação dupla a oxigênio) e -NR14R15;
[0290] R2 e R3 são independentemente selecionados do grupo que consiste em: hidrogênio e C1-6alquil;
[0291] R4 e R5 são independentemente selecionados do grupo que consiste em: hidrogênio, -OR, -NR14R15 e oxo;
[0292] R6 e R7 são independentemente selecionados do grupo que consiste em: hidrogênio, halogênio, hidroxil e C1-6alquil opcionalmente substituído com 1-3 substituintes independentemente selecionados de hidroxil e halogênio,
[0293] R6 e R7, juntos com o átomo de carbono ao qual estão ligados, formam um C2-5alquilideno opcionalmente substituído com 1-3 substituintes independentemente selecionados de R,
[0294] R6 e R7 juntos são oxo, ou
[0295] R6 e R7, juntos com o átomo de carbono ao qual estão ligados, formam uma porção heterocicloalquil de 3 a 5 membros compreendendo 1 ou 2 heteroátomos independentemente selecionados do grupo que consiste em oxigênio, nitrogênio e enxofre, em que a referida porção heterocicloalquil pode ser opcionalmente substituída com um a três substituintes independentemente selecionados de R;
[0296] R8 é hidrogênio, C1-6alquil ou -OR;
[0297] R9 é independentemente selecionado de hidrogênio, -C1- θalquil, -(C(R)2)m-C(O)OR, -(C(R)2)m-C(O)NR14R15, -(C(R)2)m-NR14R15, -(C(R)2)mrC(O)-SR, -(C(R)2)m-C(O)NR14N(R)R15, -(C(R)2)m-NR-C(O)- NR14R15, -(C(R)2)m-N(R)COR13 e -(C(R)2)m-NR14N(R)R15;
[0298] R13 é selecionado do grupo que consiste em hidrogênio, C1- 6alquil, C3-8carbociclil, C3-8heterociclil, C1-6alquil-C6-14aril, C1-6alquil-C5- 14heteroaril, em que R13 é opcionalmente substituído com -NRR ou - SO2NRR;
[0299] cada R14 e R15 é independentemente selecionado do grupo que consiste em: hidrogênio, hidroxil, -NRR, -NRNR2, -C3-10carbociclil, -C1-6alquileno-C3-10carbociclil, -C3-10heterociclil, -C1-6alquileno-C3- 10heterociclil, -(CH2CH2O)1-6CH2CH2C(O)OR, -(CH2CH2O)1-6CH2CH2NRR, -C1-6alquil, C6-14aril, -C1-6alquileno-C6-14aril e -C5- 14heteroaril;
[0300] ou R14 e R15, juntos com o átomo ou átomos aos quais são ligados, formam um anel C3-10heterociclil,
[0301] em que R14, R15, ou ambos, ou um anel formado com R14 e R15, são opcionalmente substituídos com -(C(R)2)m-R18, em que cada R18 é independentemente selecionado de (i) -NRR, (ii) - C(NRR)(C(O)OR), (iii) -S-R, (iv) aril ou heteroaril opcionalmente substituído com um ou mais de halogênio, -CF3, -(C(R)2)m-NRR ou - (C(R)2)m -SO2NRR, (v) -SO2R, (vi) -S-S-C1-6alquil-C(O)OR, (vii) - SO2NRR, (viii) -C(O)NRR, (ix) -C(O)OR, (x) -C4-6 cicloalquil opcionalmente substituído com -NRR, -SO2NRR ou -NR-C(O)(CH2)0-6NRR, (xi) -R, (xii) -OR, (xiii) -N(R)NRR, (xiv) -C(O)N(R)NRR, -(C(R)2)m-O- NRR e -S-S-C1-6alquil-NRR;
[0302] cada R é independentemente selecionado do grupo que consiste em: hidrogênio e -C1-6alquil; e
[0303] cada m é independentemente 0, 1, 2 ou 3;
[0304] ou um sal farmaceuticamente aceitável do mesmo.
[0305] De acordo com outro aspecto, a presente invenção refere- se a um composto ou compostos de fórmula (II):
[0306] L-P
[0307] (II)
[0308] ou um sal farmaceuticamente aceitável do mesmo, em que:
[0309] L é a porção ligante L1-L2-L3, em que L3 é ligado a P;
[0310] P é um radical de fórmula (I):
[0311] (I)
[0312] em que:
[0313] uma linha pontilhada representa uma ligação opcional;
[0314] cada X1 é independentemente selecionado do grupo que consiste em: -O-, -S- e -NR-;
[0315] cada X2 é independentemente selecionado do grupo que consiste em: -O-, -S- e -NR-;
[0316] cada X’ é CR ou N;
[0317] cada X’’ é CH-, CR-(C(R)2)m-NR-, CR-(C(R)2)m-O-; CR- (C(R)2)m-C(O)NR-, CR-(C(R)2)m-C(O)NR-NR-, CR-(C(R)2)m-SO2NR-, CR-(C(R)2)m-NR-NR-, CR-(C(R)2)m-NR-C(O)- ou N- se X’’ se liga a L2 ou um L3 adicional, ou de outra forma é O, S, CRR, CR-(C(R)2)m-NRR ou NRR;
[0318] cada X’’’ é - (C(R)2)m-NR- ou CR-(C(R)2)m-O- se X’’’ se liga a L2, ou de outra forma é R;
[0319] Y é -C(R)2-, -O-, -NR- ou -S-;
[0320] R1 é selecionado do grupo que consiste em: -R, -OR, - OCOR13, -OCONR14R15, -OCON(R14)NR(R15), =O (ligação dupla a oxigênio) e -NR14R15;
[0321] R2 e R3 são independentemente selecionados do grupo que consiste em: hidrogênio e C1-6alquil;
[0322] R4 e R5 são independentemente selecionados do grupo que consiste em: hidrogênio, -OR, -NR14R15 e oxo;
[0323] R6 e R7 são independentemente selecionados do grupo que consiste em: hidrogênio, halogênio, hidroxil e C1-6alquil opcionalmente substituído com 1-3 substituintes independentemente selecionados de hidroxil e halogênio,
[0324] R6 e R7, juntos com o átomo de carbono ao qual estão ligados, formam um C2-5alquilideno opcionalmente substituído com 1-3 substituintes independentemente selecionados de R,
[0325] R6 e R7 juntos são oxo, ou
[0326] R6 e R7, juntos com o átomo de carbono ao qual estão ligados, formam uma porção heterocicloalquil de 3 a 5 membros compreendendo 1 ou 2 heteroátomos independentemente selecionados do grupo que consiste em oxigênio, nitrogênio e enxofre, em que a referida porção heterocicloalquil pode ser opcionalmente substituída com um a três substituintes independentemente selecionados de R;
[0327] R8 é hidrogênio, C1-6alquil ou -OR;
[0328] R9 é -(C(R)2)m-C(O)- ou -(C(R)2)m- ;
[0329] L1 é selecionado de: -halogênio, -NR2,
,
[0330] L2 é L2A-L2B-L2C ou L2C-L2B-L2A, em que:
[0331] L2A compreende um ou mais componentes selecionados de:
[0332] -O-, -C(O)-, -C(O)NR-, -C(O)-C1-6alquil-, -C(O)NRC1-6alquil-,-C1-6alquil(OCH2CH2)1-6-, -C(O)-C1-6alquil-NRC(O)-, -C(O)-C1-6alquil(OCH2CH2)1-6-, -C1-6alquil(OCH2CH2)1-6-C(O)-, -C1-6alquil-S-S-C1- 6alquil-NRC(O)CH2-, -C1-6alquil-(OCH2CH2)1-6-NRC(O)CH2-, -C(O)-C1- 6alquil-NRC(O)C1-6alquil-, -N=CR-fenil-O-C1-6alquil-, -N=CR-fenil-O-C1- 6alquil-C(O)-, -C(O)-C1-6alquil(OCH2CH2)1-6-NRC(O)-, -C(O)-C1-6alquil- fenil-(NR-C(O)-C1-6alquil)1-4-, -C(O)-C1-6alquil-(OCH2CH2)1-6-NRC(O)C1- 6alquil-, -C1-6alquil-, -S-, -C(O)-C1-6alquil-fenil-NR-, -O-C1-6alquil-S-, - C(O)-O-C1-6alquil-S- e (-CH2-CH2-O-)1-20, ou L2A é ausente;
[0333] L2B é selecionado de AA0-aa, em que AA é um aminoácido natural ou não-natural e aa é 12; e
[0334] L2C compreende um ou mais componentes selecionados de: -PABA- e -PABC-, ou L2C é ausente;
[0335] L3 é selecionado de um ou mais de: -C1-6alquil-, -NR-C3-C8- heterociclil-NR-, -NR-C3-C8carbociclil-NR-, -NR-C1-6alquil-NR-, -NR-C1- 6alquil-, -S-, -NR-, -NR-NR- e -NR-C(O)-NR-, em que os dois grupos R opcionalmente se juntam para formar um anel de 4-10 membros, -NR- C1-6alquil-fenil-NR-, -NR-C1-6alquil-fenil-SO2-NR-, -SO2-, -NR-C1-6- alquil-fenil-C(O)-,
[0336] ou L3 é ausente;
[0337] R13 é selecionado do grupo que consiste em hidrogênio, C1-6alquil, C3-8carbociclil, C3-8heterociclil, C1-6alquil-C6-14aril, C1-6alquil-C5- uheteroaril, em que R13 é opcionalmente substituído com -NRR ou - SO2NRR;
[0338] cada R14 e R15 é independentemente selecionado do grupo que consiste em: hidrogênio, hidroxil, -NRR, -NRNR2, -C3-10carbociclil, -C1-6alquileno-C3-10carbociclil, -C3-10heterociclil, -C1-6alquileno-C3- 10heterociclil, -(CH2CH2O)1-6CH2CH2C(O)OR, -(CH2CH2O)1-6CH2CH2NRR, -C1-6alquil, C6-14aril, -C1-6alquileno-C6-14aril e -C5- 14heteroaril;
[0339] ou R14 e R15, juntos com o átomo ou átomos aos quais são ligados, formam um anel C3-10heterociclil,
[0340] em que R14, R15, ou ambos, ou um anel formado com R14 e R15, são opcionalmente substituídos com -(C(R)2)m-R18, em que cada R18 é independentemente selecionado de (i) -NRR, (ii) - C(NRR)(C(O)OR), (iii) -S-R, (iv) aril ou heteroaril opcionalmente substituído com um ou mais de halogênio, -CF3, -(C(R)2)m-NRR ou - (C(R)2)m -SO2NRR, (v) -SO2R, (vi) -S-S-C1-6alquil-C(O)OR, (vii) - SO2NRR, (viii) -C(O)NRR, (ix) -C(O)OR, (x) -C4-6 cicloalquil opcionalmente substituído com -NRR, -SO2NRR ou -NR-C(O)(CH2)0-6NRR, (xi) -R, (xii) -OR, (xiii) -N(R)NRR, (xiv) -C(O)N(R)NRR, (xv) - (C(R)2)m-O-NRR e (xiv) -S-S-C1-6alquil-NRR;
[0341] cada R é independentemente selecionado do grupo que consiste em: hidrogênio e -C1-6alquil; e
[0342] cada m é independentemente 0, 1, 2 ou 3.
[0343] De acordo com outro aspecto, a presente invenção refere- se a um composto ou compostos de fórmula (II’):
[0344] L-P’
[0345] (II’)
[0346] ou um sal farmaceuticamente aceitável do mesmo, em que:
[0347] L é a porção ligante L1-L2-L3, em que L3 é ligado a P’;
[0348] P’ é um radical de fórmula (I’):
[0349] (I’)
[0350] em que:
[0351] uma linha pontilhada representa uma ligação opcional;
[0352] cada X1 é independentemente selecionado do grupo que consiste em: -O-, -S- e -NR-;
[0353] cada X2 é independentemente selecionado do grupo que consiste em: -O-, -S- e -NR-;
[0354] cada X’ é CR ou N;
[0355] cada X’’ é CH-, CR-(C(R)2)m-NR-, CR-(C(R)2)m-O-; CR- (C(R)2)m-C(O)NR-, CR-(C(R)2)m-C(O)NR-NR-, CR-(C(R)2)m-SO2NR-, CR-(C(R)2)m-NR-NR-, CR-(C(R)2)m-NR-C(O)- ou N- se X’’ se liga a L2 ou um L3 adicional, ou de outra forma é O, S, CRR, CR-(C(R)2)m-NRR ou NRR;
[0356] cada X’’’ é - (C(R)2)m-NR- ou CR-(C(R)2)m-O- se X’’’ se liga a L2, ou de outra forma é R;
[0357] Y é -C(R)2-, -O-, -NR- ou -S-;
[0358] R1 é selecionado do grupo que consiste em: -(C(R)2)m-, -OR’’, -OCOR13’, -OC(O)NRR14’, -OCON(R)N(R)-, e -NR-
[0359] R2 e R3 são independentemente selecionados do grupo que consiste em: hidrogênio e C1-6alquil;
[0360] R4 e R5 são independentemente selecionados do grupo que consiste em: hidrogênio, -OR, -NR14R15 e oxo;
[0361] R6 e R7 são independentemente selecionados do grupo que consiste em: hidrogênio, halogênio, hidroxil e C1-6alquil opcionalmente substituído com 1-3 substituintes independentemente selecionados de hidroxil e halogênio,
[0362] R6 e R7, juntos com o átomo de carbono ao qual estão ligados, formam um C2-5alquilideno opcionalmente substituído com 1-3 substituintes independentemente selecionados de R,
[0363] R6 e R7 juntos são oxo, ou
[0364] R6 e R7, juntos com o átomo de carbono ao qual estão ligados, formam uma porção heterocicloalquil de 3 a 5 membros compreendendo 1 ou 2 heteroátomos independentemente selecionados do grupo que consiste em oxigênio, nitrogênio e enxofre, em que a referida porção heterocicloalquil pode ser opcionalmente substituída com um a três substituintes independentemente selecionados de R;
[0365] R8 é hidrogênio, C1-6alquil ou -OR;
[0366] R9 é independentemente selecionado de hidrogênio, -C1-θalquil, -(C(R)2)m-C(O)OR, -(C(R)2)m-C(O)NR14R15, -(C(R)2)m-NR14R15, -(C(R)2)mrC(O)-SR, -(C(R)2)m-C(O)NR14N(R)R15, -(C(R)2)m-NR-C(O)-NR14R15, -(C(R)2)m-N(R)COR13 e -(C(R)2)m-NR14N(R)R15;
[0367] L1 é selecionado de: -halogênio, -NR2,
[0368] L2 é L2A-L2B-L2C ou L2C-L2B-L2A, em que:
[0369] L2A compreende um ou mais componentes selecionados de:
[0370] -O-, -C(O)-, -C(O)NR-, -C(O)-C1-6alquil-, -C(O)NRC1-6alquil-,-C1-6alquil(OCH2CH2)1-6-, -C(O)-C1-6alquil-NRC(O)-, -C(O)-C1-6alquil(OCH2CH2)1-6-, -C1-6alquil(OCH2CH2)1-6-C(O)-, -C1-6alquil-S-S-C1- 6alquil-NRC(O)CH2-, -C1-6alquil-(OCH2CH2)1-6-NRC(O)CH2-, -C(O)-C1- 6alquil-NRC(O)C1-6alquil-, -N=CR-fenil-O-C1-6alquil-, -N=CR-fenil-O-C1- 6alquil-C(O)-, -C(O)-C1-6alquil(OCH2CH2)1-6-NRC(O)-, -C(O)-C1-6alquil- fenil-(NR-C(O)-C1-6alquil)1-4-, -C(O)-C1-6alquil-(OCH2CH2)1-6-NRC(O)C1- 6alquil-, -C1-6alquil-, -S-, -C(O)-C1-6alquil-fenil-NR-, -O-C1-6alquil-S-, - C(O)-O-C1-6alquil-S- e (-CH2-CH2-O-)1-20, ou L2A é ausente;
[0371] L2B é selecionado de AA0-aa, em que AA é um aminoácido natural ou não-natural e aa é 12; e
[0372] L2C compreende um ou mais componentes selecionados de: -PABA- e -PABC-, ou L2C é ausente;
[0373] L3 é selecionado de um ou mais de: -C1-6alquil-, -NR-C3-C8-heterociclil-NR-, -NR-C3-C8carbociclil-NR-, -NR-C1-6alquil-NR-, -NR-C1- 6alquil-, -S-, -NR-, -NR-NR- e -NR-C(O)-NR-, em que os dois grupos R opcionalmente se juntam para formar um anel de 4-10 membros, -NR- C1-6alquil-fenil-NR-, -NR-C1-6alquil-fenil-SO2-NR-, -SO2-, -NR-C1-6- alquil-fenil-C(O)-,
[0374]
e
, ou L3 é ausente;
[0375] R13’ é selecionado do grupo que consiste em uma ligação, - C1-6alquileno-, -C3-8carbociclil-, -C3-8heterociclil-, -C1-6alquil-C6-14aril-, - C1-6alquil-C5-14heteroaril-;
[0376] cada R14 e R15 é independentemente selecionado do grupo que consiste em: hidrogênio, hidroxil, -NRR, -NRNR2, -C3-10carbociclil, -C1-6alquileno-C3-10carbociclil, -C3-10heterociclil, -C1-6alquileno-C3- 10heterociclil, -(CH2CH2O)1-6CH2CH2C(O)OR, -(CH2CH2O)1-6CH2CH2NRR, -C1-6alquil, C6-14aril, -C1-6alquileno-C6-14aril e -C5- 14heteroaril;
[0377] ou R14 e R15, juntos com o átomo ou átomos aos quais são ligados, formam um anel C3-10heterociclil,
[0378] em que R14, R15, ou ambos, ou um anel formado com R14 e R15, são opcionalmente substituídos com -(C(R)2)m-R18, em que cada R18 é independentemente selecionado de (i) -NRR, (ii) - C(NRR)(C(O)OR), (iii) -S-R, (iv) aril ou heteroaril opcionalmente substituído com um ou mais de halogênio, -CF3, -(C(R)2)m-NRR ou - (C(R)2)m -SO2NRR, (v) -SO2R, (vi) -S-S-Ci-ealquil-C(O)OR, (vii) - SO2NRR, (viii) -C(O)NRR, (ix) -C(O)OR, (x) -C4-6 cicloalquil opcionalmente substituído com -NRR, -SO2NRR ou -NR-C(O)(CH2)0-6NRR, (xi) -R, (xii) -OR, (xiii) -N(R)NRR, (xiv) -C(O)N(R)NRR, (xv) - (C(R)2)m-O-NRR e (xiv) -S-S-Ci-6alquil-NRR;
[0379] cada Ri4’ é independentemente selecionado do grupo que consiste em: uma ligação, -NR-, -C3-i0carbociclil-, -C3-i0heterociclil-, - (CH2CH2O)i-6CH2CH2C(O)OR’, -(CH2CH2O)i-6CH2CH2NR-, e -Ci- 6alquileno-,
[0380] em que Ri4’ é opcionalmente substituído com -(C(R)2)m-Ri8, em que cada Ri8 é independentemente selecionado de (i) -NRR, (ii) - C(NRR)(C(O)OR), (iii) -S-R, (iv) aril ou heteroaril opcionalmente substituído com um ou mais de halogênio, -CF3, -NRR ou -SO2NRR, (v) - SO2R, (vi) -S-S-Ci-6alquil-C(O)OR, (vii) -SO2NRR, (viii) -C(O)NRR, (ix) -C(O)OR, (x) -C4-6 cicloalquil opcionalmente substituído com -NRR, - SO2NRR ou -NR-C(O)(CH2)0-6NRR, (xi) -R, (xii) -OR, (xiii) -N(R)NRR, (xiv) -C(O)N(R)NRR, (xv) -(C(R)2)m-O-NRR e (xiv) -S-S-Ci-6alquil- NRR;
[0381] cada R é independentemente selecionado do grupo que consiste em: hidrogênio e -Ci-6alquil;
[0382] cada R' é independentemente selecionado de -H, Ci-C8 al- quil, Ci-C8 heteroalquil e aril;
[0383] cada R’’ é independentemente selecionado do grupo que consiste em: uma ligação e -Ci-6alquileno-; e
[0384] cada m é independentemente 0, i, 2 ou 3.
[0385] De acordo com ainda outro aspecto, a presente invenção refere-se a um composto ou compostos de fórmula (III):
[0386] (AB)-(L-P)b
[0387] (III)
[0388] ou um sal farmaceuticamente aceitável do mesmo, em que:
[0389] L é a porção ligante L1-L2-L3, em que L3 é ligado a P;
[0390] P é um radical de fórmula (I):
[0391] (I)
[0392] em que:
[0393] uma linha pontilhada representa uma ligação opcional;
[0394] AB é um anticorpo;
[0395] cada x1 é independentemente selecionado do grupo que consiste em: -O-, -S- e -NR-;
[0396] cada x2 é independentemente selecionado do grupo que consiste em: -O-, -S- e -NR-;
[0397] cada x’ é CR ou N;
[0398] cada x’’ é CH-, CR-(C(R)2)m-NR-, CR-(C(R)2)m-O-; CR- (C(R)2)m-C(O)NR-, CR-(C(R)2)m-C(O)NR-NR-, CR-(C(R)2)m-SO2NR-, CR-(C(R)2)m-NR-NR-, CR-(C(R)2)m-NR-C(O)- ou N- se x’’ se liga a L2 ou um L3 adicional, ou de outra forma é O, S, CRR, CR-(C(R)2)m-NRR ou NRR;
[0399] cada X’’’ é - (C(R)2)m-NR- ou CR-(C(R)2)m-O- se X’’’ se liga a L2, ou de outra forma é R;
[0400] Y é -C(R)2-, -O-, -NR- ou -S-;
[0401] R1 é selecionado do grupo que consiste em: -R, -OR, - OCOR13, -OCONR14R15, -OCON(R14)NR(R15), =O (ligação dupla a oxigênio) e -NR14R15;
[0402] R2 e R3 são independentemente selecionados do grupo que consiste em: hidrogênio e C1-6alquil;
[0403] R4 e R5 são independentemente selecionados do grupo que consiste em: hidrogênio, -OR, -NR14R15 e oxo;
[0404] R6 e R7 são independentemente selecionados do grupo que consiste em: hidrogênio, halogênio, hidroxil e C1-6alquil opcionalmente substituído com 1-3 substituintes independentemente selecionados de hidroxil e halogênio,
[0405] R6 e R7, juntos com o átomo de carbono ao qual estão ligados, formam um C2-5alquilideno opcionalmente substituído com 1-3 substituintes independentemente selecionados de R,
[0406] R6 e R7 juntos são oxo, ou
[0407] R6 e R7, juntos com o átomo de carbono ao qual estão ligados, formam uma porção heterocicloalquil de 3 a 5 membros compreendendo 1 ou 2 heteroátomos independentemente selecionados do grupo que consiste em oxigênio, nitrogênio e enxofre, em que a referida porção heterocicloalquil pode ser opcionalmente substituída com um a três substituintes independentemente selecionados de R;
[0408] R8 é hidrogênio, C1-6alquil ou -OR;
[0409] R9 é –(C(R)2)m-C(O)- ou –(C(R)2)m- ;
[0410] L1 é selecionado de: uma ligação a AB, -NR-(ligação a AB) e
[0411] L2 é L2A-L2B-L2C ou L2C-L2B-L2A, em que:
[0412] L2A compreende um ou mais componentes selecionados de:
[0413] -O-, -C(O)-, -C(O)NR-, -C(O)-C1-6alquil-, -C(O)NRC1-6alquil-,-C1-6alquil(OCH2CH2)1-6-, -C(O)-C1-6alquil-NRC(O)-, -C(O)-C1-6alquil(OCH2CH2)1-6-, -C1-6alquil(OCH2CH2)1-6-C(O)-, -C1-6alquil-S-S-C1- 6alquil-NRC(O)CH2-, -C1-6alquil-(OCH2CH2)1-6-NRC(O)CH2-, -C(O)-C1- 6alquil-NRC(O)C1-6alquil-, -N=CR-fenil-O-C1-6alquil-, -N=CR-fenil-O-C1- 6alquil-C(O)-, -C(O)-C1-6alquil(OCH2CH2)1-6-NRC(O)-, -C(O)-C1-6alquil- fenil-(NR-C(O)-C1-6alquil)1-4-, -C(O)-C1-6alquil-(OCH2CH2)1-6-NRC(O)C1- 6alquil-, -C1-6alquil-, -S-, -C(O)-C1-6alquil-fenil-NR-, -O-C1-6alquil-S-, - C(O)-O-C1-6alquil-S- e (-CH2-CH2-O-)1-20, ou L2A é ausente;
[0414] L2B é selecionado de AA0-aa, em que AA é um aminoácido natural ou não-natural e aa é 12; e
[0415] L2C compreende um ou mais componentes selecionados de: -PABA- e -PABC-, ou L2C é ausente;
[0416] L3 é selecionado de um ou mais de: -C1-6alquil-, -NR-C3-C8-heterociclil-NR-, -NR-C3-C8carbociclil-NR-, -NR-C1-6alquil-NR-, -NR-C1- 6alquil-, -S-, -NR-, -NR-NR- e -NR-C(O)-NR-, em que os dois grupos R opcionalmente se juntam para formar um anel de 4-10 membros, -NR- C1-6alquil-fenil-NR-, -NR-C1-6alquil-fenil-SO2-NR-, -SO2-, -NR-C1-6- alquil-fenil-C(O)-,
, ou L3 é au sente;
[0417] R13 é selecionado do grupo que consiste em hidrogênio, C1- 6alquil, C3-8carbociclil, C3-8heterociclil, C1-6alquil-C6-14aril, C1-6alquil-C5- uheteroaril, em que R13 é opcionalmente substituído com -NRR ou - SO2NRR;
[0418] cada R14 e R15 é independentemente selecionado do grupo que consiste em: hidrogênio, hidroxil, -NRR, -NRNR2, -C3-10carbociclil, -C1-6alquileno-C3-10carbociclil, -C3-10heterociclil, -C1-6alquileno-C3- 10heterociclil, -(CH2CH2O)1-6CH2CH2C(O)OR, -(CH2CH2O)1-6CH2CH2NRR, -C1-6alquil, C6-14aril, -C1-6alquileno-C6-14aril e -C5- 14heteroaril;
[0419] ou R14 e R15, juntos com o átomo ou átomos aos quais são ligados, formam um anel C3-10heterociclil,
[0420] em que R14, R15, ou ambos, ou um anel formado com R14 e R15, são opcionalmente substituídos com -(C(R)2)m-R18, em que cada R18 é independentemente selecionado de (i) -NRR, (ii) - C(NRR)(C(O)OR), (iii) -S-R, (iv) aril ou heteroaril opcionalmente substituído com um ou mais de halogênio, -CF3, -(C(R)2)m-NRR ou - (C(R)2)m -SO2NRR, (v) -SO2R, (vi) -S-S-C1-6alquil-C(O)OR, (vii) - SO2NRR, (viii) -C(O)NRR, (ix) -C(O)OR, (x) -C4-6 cicloalquil opcionalmente substituído com -NRR, -SO2NRR ou -NR-C(O)(CH2)0-6NRR, (xi) -R, (xii) -OR, (xiii) -N(R)NRR, (xiv) -C(O)N(R)NRR, (xv) - (C(R)2)m-O-NRR e (xiv) -S-S-C1-6alquil-NRR;
[0421] cada R é independentemente selecionado do grupo que consiste em: hidrogênio e -C1-6alquil; e
[0422] b é 1-20; e
[0423] cada m é independentemente 0, 1, 2 ou 3.
[0424] De acordo com ainda outro aspecto, a presente invenção refere-se a um composto ou compostos de fórmula (III’):
[0425] (AB)-(L-P’)b
[0426] (III’)
[0427] ou um sal farmaceuticamente aceitável do mesmo, em que:
[0428] L é a porção ligante L1-L2-L3, em que L3 é ligado a P’;
[0429] P’ é um radical de fórmula (I’):
[0430] (I’)
[0431] em que:
[0432] uma linha pontilhada representa uma ligação opcional;
[0433] AB é um anticorpo;
[0434] cada x1 é independentemente selecionado do grupo que consiste em: -O-, -S- e -NR-;
[0435] cada x2 é independentemente selecionado do grupo que consiste em: -O-, -S- e -NR-;
[0436] cada x’ é CR ou N;
[0437] cada X’’ é CH-, CR-(C(R)2)m-NR-, CR-(C(R)2)m-O-; CR- (C(R)2)m-C(O)NR-, CR-(C(R)2)m-C(O)NR-NR-, CR-(C(R)2)m-SO2NR-, CR-(C(R)2)m-NR-NR-, CR-(C(R)2)m-NR-C(O)- ou N- se X’’ se liga a L2 ou um L3 adicional, ou de outra forma é O, S, CRR, CR-(C(R)2)m-NRR ou NRR;
[0438] cada X’’’ é - (C(R)2)m-NR- ou CR-(C(R)2)m-O- se X’’’ se liga a L2, ou de outra forma é R;
[0439] Y é -C(R)2-, -O-, -NR- ou -S-;
[0440] R1 é selecionado do grupo que consiste em: -(C(R)2)m- C(O)-, -(C(R)2)m-, -OR’’, -OCOR13’, -OCONRR14’, -OCON(R14)N(R15)-, e -NR14-
[0441] R2 e R3 são independentemente selecionados do grupo que consiste em: hidrogênio e C1-6alquil;
[0442] R4 e R5 são independentemente selecionados do grupo que consiste em: hidrogênio, -OR, -NR14R15 e oxo;
[0443] R6 e R7 são independentemente selecionados do grupo que consiste em: hidrogênio, halogênio, hidroxil e C1-6alquil opcionalmente substituído com 1-3 substituintes independentemente selecionados de hidroxil e halogênio,
[0444] R6 e R7, juntos com o átomo de carbono ao qual estão ligados, formam um C2-5alquilideno opcionalmente substituído com 1-3 substituintes independentemente selecionados de R,
[0445] R6 e R7 juntos são oxo, ou
[0446] R6 e R7, juntos com o átomo de carbono ao qual estão ligados, formam uma porção heterocicloalquil de 3 a 5 membros compreendendo 1 ou 2 heteroátomos independentemente selecionados do grupo que consiste em oxigênio, nitrogênio e enxofre, em que a referida porção heterocicloalquil pode ser opcionalmente substituída com um a três substituintes independentemente selecionados de R;
[0447] R8 é hidrogênio, C1-6alquil ou -OR;
[0448] R9 é independentemente selecionado de hidrogênio, -C1-θalquil, -(C(R)2)m-C(O)OR, -(C(R)2)m-C(O)NR14R15, -(C(R)2)m-NR14R15, -(C(R)2)mrC(O)-SR, -(C(R)2)m-C(O)NR14N(R)R15, -(C(R)2)m-NR-C(O)- NR14R15, -(C(R)2)m-N(R)COR13 e -(C(R)2)m-NR14N(R)R15;
[0449] L1 é selecionado de: uma ligação a AB, -NR-(ligação a AB) e
[0450] L2 é L2A-L2B-L2C ou L2C-L2B-L2A, em que:
[0451] L2A compreende um ou mais componentes selecionados de:
[0452] -O-, -C(O)-, -C(O)NR-, -C(O)-C1-6alquil-, -C(O)NRC1-6alquil-,-C1-6alquil(OCH2CH2)1-6-, -C(O)-C1-6alquil-NRC(O)-, -C(O)-C1-6alquil(OCH2CH2)1-6-, -C1-6alquil(OCH2CH2)1-6-C(O)-, -C1-6alquil-S-S-C1- 6alquil-NRC(O)CH2-, -C1-6alquil-(OCH2CH2)1-6-NRC(O)CH2-, -C(O)-C1- 6alquil-NRC(O)C1-6alquil-, -N=CR-fenil-O-C1-6alquil-, -N=CR-fenil-O-C1- 6alquil-C(O)-, -C(O)-C1-6alquil(OCH2CH2)1-6-NRC(O)-, -C(O)-C1-6alquil- fenil-(NR-C(O)-C1-6alquil)1-4-, -C(O)-C1-6alquil-(OCH2CH2)1-6-NRC(O)C1- 6alquil-, -C1-6alquil-, -S-, -C(O)-C1-6alquil-fenil-NR-, -O-C1-6alquil-S-, - C(O)-O-C1-6alquil-S- e (-CH2-CH2-O-)1-20, ou L2A é ausente;
[0453] L2B é selecionado de AA0-aa, em que AA é um aminoácido natural ou não-natural e aa é 12; e
[0454] L2C compreende um ou mais componentes selecionados de: -PABA- e -PABC-, ou L2C é ausente;
[0455] L3 é selecionado de um ou mais de: -C1-6alquil-, -NR-C3-C8-heterociclil-NR-, -NR-C3-C8carbociclil-NR-, -NR-C1-6alquil-NR-, -NR-C1- 6alquil-, -S-, -NR-, -NR-NR- e -NR-C(O)-NR-, em que os dois grupos R opcionalmente se juntam para formar um anel de 4-10 membros, -NR- C1-6alquil-fenil-NR-, -NR-C1-6alquil-fenil-SO2-NR-, -SO2-, -NR-C1-6- alquil-fenil-C(O)-,
, ou L3 é au sente;
[0456] R13’ é selecionado do grupo que consiste em uma ligação, -C1-6alquileno-, -C3-8carbociclil-, -C3-8heterociclil-, -C1-6alquil-C6-14aril-, - C1-6alquil-C5-14heteroaril-;
[0457] cada R14 e R15 é independentemente selecionado do grupo que consiste em: hidrogênio, hidroxil, -NRR, -NRNR2, -C3-10carbociclil, -C1-6alquileno-C3-10carbociclil, -C3-10heterociclil, -C1-6alquileno-C3-10heterociclil, -(CH2CH2O)1-6CH2CH2C(O)OR, -(CH2CH2O)1-6CH2CH2NRR, -C1-6alquil, C6-14aril, -C1-6alquileno-C6-14aril e -C5- 14heteroaril;
[0458] ou R14 e R15, juntos com o átomo ou átomos aos quais são ligados formam um anel C3-10heterociclil,
[0459] em que R14, R15, ou ambos, ou um anel formado com R14 e R15, são opcionalmente substituídos com -(C(R)2)m-R18, em que cada R18 é independentemente selecionado de (i) -NRR, (ii) - C(NRR)(C(O)OR), (iii) -S-R, (iv) aril ou heteroaril opcionalmente substituído com um ou mais de halogênio, -CF3, -(C(R)2)m-NRR ou -(C(R)2)m -SO2NRR, (v) -SO2R, (vi) -S-S-C1-6alquil-C(O)OR, (vii) - SO2NRR, (viii) -C(O)NRR, (ix) -C(O)OR, (x) -C4-6 cicloalquil opcionalmente substituído com -NRR, -SO2NRR ou -NR-C(O)(CH2)0-6NRR, (xi) -R, (xii) -OR, (xiii) -N(R)NRR, (xiv) -C(O)N(R)NRR, (xv) - (C(R)2)m-O-NRR e (xiv) -S-S-C1-6alquil-NRR;
[0460] cada R14’ é independentemente selecionado do grupo que consiste em: uma ligação, -NR-, -C3-10carbociclil-, -C3-10heterociclil-, - (CH2CH2O)1-6CH2CH2C(O)OR’, -(CH2CH2O)1-6CH2CH2NR-, e -C1- 6alquileno-,
[0461] em que R14’ é opcionalmente substituído com -(C(R)2)m-R18, em que cada R18 é independentemente selecionado de (i) -NRR, (ii) - C(NRR)(C(O)OR), (iii) -S-R, (iv) aril ou heteroaril opcionalmente substituído com um ou mais de halogênio, -CF3, -NRR ou -SO2NRR, (v) - SO2R, (vi) -S-S-C1-6alquil-C(O)OR, (vii) -SO2NRR, (viii) -C(O)NRR, (ix) -C(O)OR, (x) -C4-6 cicloalquil opcionalmente substituído com -NRR, - SO2NRR ou -NR-C(O)(CH2)0-6NRR, (xi) -R, (xii) -OR, (xiii) -N(R)NRR, (xiv) -C(O)N(R)NRR, (xv) -(C(R)2)m-O-NRR e (xiv) -S-S-C1-6alquil- NRR;
[0462] cada R é independentemente selecionado do grupo que consiste em: hidrogênio e -C1-6alquil;
[0463] cada R' é independentemente selecionado de -H, C1-C8 al- quil, C1-C8 heteroalquil e aril;
[0464] cada R’’ é independentemente selecionado do grupo que consiste em: uma ligação e -C1-6alquileno-; e
[0465] b é 1-20; e
[0466] cada m é independentemente 0, 1, 2 ou 3.
[0467] Deve ser notado que variáveis divalentes recitadas acima destinam-s e a descrever, quando apropriado, a posição de tais radicais em orientações múltiplas dentro da molécula. Desta forma, por exemplo, para citar apenas um único exemplo, a porção L2 “-PABC- Cit-Val-C(O)-C1-6alquil-“ entre L1 e L3 pode ser posicionada como L1- PABC-Cit-Val-C(O)-C1-6alquil-L3 ou como L1-C1-6alquil-C(O)-Val-Cit- PABC-L3. Similarmente, L2 é definido aqui como compreendendo L2A- L2B-L2C, cujo constructo pode igualmente ser posicionado em múltiplas orientações.
[0468] Desta forma, em certas modalidades, é provido um ADC da fórmula III ou III’ tendo a seguinte sequência de componentes:
[0469] AB-L1-L2-L3-P;
[0470] AB-L1-L2-L3-P’;
[0471] AB-L1-L2A-L2B-L2C-L3-P; ou
[0472] AB-L1-L2C-L2B-L2A-L3-P.
[0473] Determinados grupos e porções químicos descritos aqui são preferidos, dependendo das circunstâncias. Desta forma, em certas modalidades da invenção, incluindo com respeito às várias cargas úteis, ligante-cargas úteis e ADCs descritos e reivindicados aqui, um ou mais (ou todos, ou nenhum) do seguinte pode aplicar:
[0474] Em certas modalidades da invenção, X1 é preferencialmente -O-.
[0475] Em certas modalidades da invenção, X2 é preferencialmente -NR-.
[0476] Em certas modalidades da invenção, R1 é preferencialmente selecionado do grupo que consiste em:, -OR, -OCOR13, - OCONR14R15 e -NR14R15.
[0477] Em certas modalidades da invenção, R2 é preferencialmente C1-6alquil, e é, mais preferencialmente, metil.
[0478] Em certas modalidades da invenção, R3 é preferencialmente C1-6alquil, e é, mais preferencialmente, metil.
[0479] Em certas modalidades da invenção, R4 é preferencialmente hidrogênio ou -OR.
[0480] Em certas modalidades da invenção, R5 é preferencialmente hidrogênio ou -OR.
[0481] Em certas modalidades da invenção, é preferido que R6 e R7 sejam, cada um, independentemente selecionados do grupo que consiste em: hidroxil e C1-6alquil opcionalmente substituído com 1-3 substituintes independentemente selecionados de halogênio, ou R6 e R7, juntos com o átomo de carbono ao qual estão ligados, formam uma porção heterocicloalquil de 3 a 5 membros compreendendo 1 ou 2 he- teroátomos independentemente selecionados do grupo que consiste em oxigênio, nitrogênio e enxofre, em que a referida porção heteroci- cloalquil pode ser opcionalmente substituída com um a três substituin- tes independentemente selecionados de R.
[0482] Em certas modalidades da invenção, R8 é preferencialmente hidrogênio ou -OR.
[0483] Em certas modalidades da invenção, R9 é independentemente selecionado de,-(C(R)2)m-C(O)OR, -(C(R)2)m-C(O)NR14R15, - (C(R)2)m-NR14R15,-(C(R)2)m-C(O)NR14N(R)R15, -(C(R)2)m-NR-C(O)-NR14R15 e -(C(R)2)m-N(R)COR13.
[0484] Em certas modalidades da invenção, R13 é preferencialmente selecionado do grupo que consiste em hidrogênio, C1-6alquil.
[0485] Em certas modalidades da invenção, é preferido que cada R14 e R15 seja independentemente selecionado do grupo que consiste em: hidrogênio, -NRR, -NRNR2, -C3-10carbociclil, -C3-10heterociclil, -C1- 6alquil, C6-14aril, -C1-6alquileno-C6-14aril e -C5-14heteroaril; ou R14 e R15, juntos com o átomo ou átomos aos quais são ligados, formam um anel C3-10heterociclil: em que R14, R15, ou ambos, ou um anel formado com R14 e R15, são opcionalmente substituídos com -(C(R)2)m-R18, em que cada R18 é independentemente selecionado de (i) -NRR, (ii) - C(NRR)(C(O)OR), (iii) -S-R, (iv) aril ou heteroaril opcionalmente substituído com um ou mais de halogênio, -CF3, -(C(R)2)m-NRR ou - (C(R)2)m -SO2NRR, (v) -SO2R, (vi) -S-S-C1-6alquil-C(O)OR, (vii) - SO2NRR, (viii) -C(O)NRR, (ix) -C(O)OR, (x) -C4-6 cicloalquil opcionalmente substituído com -NRR, -SO2NRR ou -NR-C(O)(CH2)0-6NRR, (xi) -R, (xii) -OR, (xiii) -N(R)NRR, (xiv) -C(O)N(R)NRR, -(C(R)2)m-O- NRR e -S-S-C1-6alquil-NRR.
[0486] Em certas modalidades da invenção, é preferido que m seja 0. Em certas modalidades da invenção, é preferido que m seja 1. Em certas modalidades da invenção, é preferido que m seja 2. Em certas modalidades da invenção, é preferido que m seja 3.
[0487] Em certas modalidades da invenção, é preferido que X2 seja -NH-, X1 seja -O-, R1 seja -OCOR13, OH ou -OCONR14R15, R2 seja metil, R3 seja metil, R4 seja -OH, R5 seja hidrogênio, R8 seja hidrogênio, R6 e R7 juntos formem epóxido, R9 seja -(C(R)2)m-C(O)-, R13 seja C1-6 alquil (mais preferencialmente metil), R14 e R15, juntos com o áto- mo ou átomos aos quais são ligados, formam um anel C3-10heterociclil,
[0488] L1 seja
ou
, e L2A, L2B, L2C e L3 sejam todos ausentes. Alternativamente, R6 é -OH e R7 é C1-6alquil opcionalmente substituído com 1-3 substituintes independentemente selecionados de um halogênio, com cloro sendo mais preferível.
[0489] Em certas modalidades da invenção, é preferido que X2 seja -NH-, X1 seja -O-, R1 é -OCOR13, OH ou -OCONR14R15, R2 seja me- til, R3 seja metil, R4 seja -OH, R5 seja hidrogênio, R8 seja hidrogênio, R6 e R7 juntos formem epóxido, R9 seja -(C(R)2)m-C(O)-, R13 seja C1-6 alquil (mais preferencialmente metil), R14 e R15, juntos com o átomo ou átomos aos quais são ligados, formem um anel C3-10heterociclil, L1 seja um halogênio, L3 seja -NR-C1-6alquil-NR com R mais preferencialmente sendo hidrogênio e o grupo alquil mais preferencialmente sendo etil, L2A seja -C(O)-C1-6alquil- e L2B e L2C sejam ausentes.
[0490] Em certas modalidades da invenção, é preferido que X2 seja -NH-, X1 seja -O-, R1 seja -OCOR13, em que R13 é, mais preferencialmente, hidrogênio, R2 é metil, R3 é metil, R4 é -OH, R5 é hidrogênio, R8 é hidrogênio, R6 e R7 juntos formam epóxido, R9 é -(C(R)2)m-C(O)-, R13 é C1-6 alquil (mais preferencialmente metil), R14 e R15, juntos com o átomo ou átomos aos quais são ligados, formam um anel C3- 10heterociclil, L1 é um halogênio, L3 é -NR-C1-6alquil-NR com R mais preferencialmente sendo hidrogênio e o grupo alquil mais preferencialmente sendo etil, L2A é -C(O)-C1-6alquil- e L2B e L2C são ausentes.
[0491] Em certas modalidades da invenção, é preferido que R1 seja -OCOR13 ou -OR, em que R é, mais preferencialmente, hidrogênio, R2 é metil, R3 é metil, R4 é -OH, R5 é hidrogênio, R8 é hidrogênio, R6 e R7 formam um epóxido, R9 é -(C(R)2)m-C(O)-, L3 é -NR-NR-, em que cada R é, mais preferencialmente, hidrogênio ou metil ou os dois subs- tituintes de R juntos formam um anel de 6 membros, L1 é um halogê- nio, -NR2 ou
, L2C é PABC, L2B é -Cit-Val-, L2A é -C(O)-C1- 6alquil-NRC(O)C1-6alquil-.
[0492] Em certas modalidades da invenção, é preferido que R1 seja -OCOR13 ou -OR, em que R é, mais preferencialmente, hidrogênio, R2 é metil, R3 é metil, R4 é -OH, R5 é hidrogênio, R8 é hidrogênio, R6 e R7 formam um epóxido, R9 é -(C(R)2)m-C(O)-, L3 é -NR-NR-, em que cada R é, mais preferencialmente, hidrogênio ou metil ou os dois subs- tituintes R juntos formam um anel de 6 membros, L1 é um halogênio, - NR2 ou
, L2C é ausente; L2B é -Ala-Val- e L2A é -C(O)-C1- 6alquil-NRC(O)C1-6alquil- ou -C(O)C1-6alquil-.
[0493] Em certas modalidades da invenção, é preferido que L1 se- ja selecionado de: -halogênio, -NR2,
[0494] Em certas modalidades da invenção, é preferido que R1 seja -OCOR13’, R2 seja metil, R3 seja metil, R4 seja -OH, R5 seja hidrogê- nio, R8 seja hidrogênio, R9 seja -(C(R)2)m-C(O)NR14R15, em que R14 e R15 são mais preferencialmente hidrogênio, R13’ é uma ligação, L3 é
, em que m é 0, X' é N, X'' é -N- e X''' é ausente,L1 é um halogênio, L2C é PABC, L2B é -Cit-Val- e L2A é -C(O)-Ci-6alquil- NRC(O)C1-6alquil-.
[0495] Em certas modalidades da invenção, é preferido que X2 seja -NH-, X1 seja -O-, R1 seja -OCOR13, OH ou -OCONR14R15, R2 seja metil, R3 seja metil, R4 seja -OH, R5 seja hidrogênio, R8 seja hidrogênio, R6 e R7 juntos formem epóxido, R9 seja -(C(R)2)m-C(O)-, R13 seja C1-6 alquil (mais preferencialmente metil), R14 e R15, juntos com o átomo ou átomos aos quais são ligados, formem um anel C3-10heterociclil,
[0496] L1 seja selecionado de: uma ligação a AB, -NR-(ligação a AB) e
[0497] e L2A, L2B, L2C e L3 sejam todos ausentes. Alternativamente,R6 é -OH e R7 é C1-6alquil opcionalmente substituído com 1-3 substi- tuintes independentemente selecionados de um halogênio, com cloro sendo mais preferível.
[0498] Em certas modalidades da invenção, é preferido que X2 seja -NH-, X1 seja -O-, R1 seja -OCOR13, OH ou -OCONR14R15, R2 seja metil, R3 seja metil, R4 seja -OH, R5 seja hidrogênio, R8 seja hidrogênio, R6 e R7 juntos formem epóxido, R9 seja -(C(R)2)m-C(O)-, R13 seja C1-6 alquil (mais preferencialmente metil), R14 e R15, juntos com o átomo ou átomos aos quais são ligados, formem um anel C3-10heterociclil, L1 seja selecionado de: uma ligação a AB, -NR-(ligação a AB) e
L3 seja -NR-C1-6alquil-NR com R mais preferencialmente sendo hidrogênio e o grupo alquil mais preferencialmente sendo etil, L2A é -C(O)-C1-6alquil- e L2B e L2C sejam ausentes.
[0499] Em certas modalidades da invenção, é preferido que X2 seja -NH-, X1 seja -O-, R1 seja -OCOR13, em que R13 é, mais preferencialmente, hidrogênio, R2 é metil, R3 é metil, R4 é -OH, R5 é hidrogênio, R8 é hidrogênio, R6 e R7 juntos formam epóxido, R9 é -(C(R)2)m-C(O)-, R13 é C1-6 alquil (mais preferencialmente metil), R14 e R15, juntos com o átomo ou átomos aos quais são ligados, formam um anel C3- 10heterociclil, L1 é selecionado de: uma ligação a AB, -NR-(ligação a AB) e
[0500] , L3 é -NR-C1-6alquil-NR com R mais preferencialmente sendo hidrogênio e o grupo alquil mais preferencialmente sendo etil, L2A é -C(O)-C1-6alquil- e L2B e L2C são ausentes.
[0501] Em certas modalidades da invenção, é preferido que R1 seja -OCOR13 ou -OR, em que R é, mais preferencialmente, hidrogênio, R2 é metil, R3 é metil, R4 é -OH, R5 é hidrogênio, R8 é hidrogênio, R6 e R7 formam um epóxido, R9 é -(C(R)2)m-C(O)-, L3 é -NR-NR-, em que cada R é, mais preferencialmente, hidrogênio ou metil ou os dois subs- tituintes R juntos formam um anel de 6 membros, L1 é selecionado de: uma ligação a AB, -NR-(ligação a AB) e
, L2C é PABC, L2B é -Cit-Val-, L2A é -C(O)-C1-6alquil-NRC(O)C1-6alquil-.
[0502] Em certas modalidades da invenção, é preferido que R1 seja -OCOR13 ou -OR, em que R é, mais preferencialmente, hidrogênio, R2 é metil, R3 é metil, R4 é -OH, R5 é hidrogênio, R8 é hidrogênio, R6 e R7 formam um epóxido, R9 é -(C(R)2)m-C(O)-, L3 é -NR-NR-, em que cada R é, mais preferencialmente, hidrogênio ou metil ou os dois subs- tituintes R juntos formam um anel de 6 membros, L1 é selecionado de:uma ligação a AB, -NR-(ligação a AB) e
, L2C é au sente; L2B é -Ala-Val- e L2A é -C(O)-C1-6alquil-NRC(O)C1-6alquil-.
[0503] Em certas modalidades da invenção, é preferido que L1 seja selecionado de: uma ligação a AB, -NR-(ligação a AB) e
[0504] Em certas modalidades da invenção, é preferido que o anticorpo seja selecionado de trastuzumab, o mutante (C392 + L443) trastuzumab, e o mutante (C392 + C443) trastuzumab.
[0505] Em certas modalidades da invenção, é preferido que o anticorpo ligado através de um polipeptídeo contendo Fab ou contendo Fc projetado com um rótulo contendo glutamina doador de acil (por exemplo, Gln-contendo rótulos de peptídeo ou rótulos Q) ou uma glu- tamina endógena feita reativa (isto é, a capacidade de formar uma ligação covalente como um doador de acil na presença de uma amina e uma transglutaminase) por desenvolvimento de polipeptídeo (por exemplo, através de mutação, substituição, inserção, eliminação de aminoácido, ou qualquer combinação dos mesmos no polipeptídeo), na presença de transglutaminase.
[0506] Em certas modalidades, a presente invenção refere-se a qualquer um dos conjugados de anticorpo-droga acima mencionados e definições concomitantes, em que o conjugado de anticorpo-droga compreende entre 2, 3, 4, 5, 6, 7, 8, 9 ou 10 compostos da invenção.
[0507] Em certas modalidades, a presente invenção refere-se a qualquer um dos conjugados de anticorpo-droga acima mencionados e definições concomitantes, em que o conjugado de anticorpo-droga compreende 3 ou 4 compostos da invenção.
[0508] Os compostos tipicamente contendo carboxil- e/ou amino- da presente invenção apresentam vantagens únicas e distintas sobre compostos não contendo carboxil. Uma tal vantagem reside em melhor solubilidade de água. Outra vantagem é melhor estabilidade química em água e em fluidos biológicos, tais como soro, sangue, fluido cérebro-espinhal e em formulações de droga. Ainda outra vantagem é capacidade de prontamente preparar formas salínicas de compostos de carboxilato emparelhando-os com um ânion apropriado, tal como cloreto, acetato, e outro contraíon. Além disso, os compostos de carboxi- lato da presente invenção podem prontamente ser usados para preparar amida e derivados de éster com citotoxicidade potente e melhorada contra linhagens de células de câncer e cânceres. Compostos contendo carboxilato adicionalmente possuem uma vantagem na sua capaci-dade de se ligar a anticorpos, uma vez que o grupo de ácido carboxí- lico pode ser reagido com moléculas de ligante apropriadamente modificadas que suportam amina, álcool e outros grupos para obter ligantes de carga útil. Os compostos de ácido carboxílico podem também ser diretamente funcionalizados para obter derivados de ácido carboxílico ativados, que podem subsequentemente ser conjugados a anticorpos sem ligantes adicionais anexados. Por exemplo, os compostos contendo ácido carboxílico da invenção podem ser reagidos com N- hidroxissuccinimida para obter carboxil-NHS ésteres ativados. Os car- boxil-NHS ésteres e ligantes de carga útil produzidos, portanto, podem ser reagidos com anticorpos para produzir conjugados de anticorpo- droga.
[0509] A Unidade de Anticorpo (Ab ou AB)
[0510] Como referido acima, o termo "anticorpo" (ou “Ab” ou “AB”) aqui é usado no sentido mais amplo e especificamente cobre anticorpos monoclonais intactos, anticorpos policlonais, anticorpos monoes- pecíficos, anticorpos multiespecíficos (por exemplo, anticorpos biespe- cíficos), e fragmentos de anticorpo que apresentam a atividade biológica desejada. Além disso, embora certos aspectos da invenção descritos aqui se refiram a conjugados de anticorpo-droga, é ainda previsto que a porção de anticorpo do conjugado pode ser substituída com qualquer um que especificamente se liga ou reativamente associa ou se completa com um receptor, antígeno ou outra porção receptiva associada com uma dada população de célula alvo. Por exemplo, em vez de conter um anticorpo, conjugados da invenção podem conter uma molécula alvo que se liga a, se completa com, ou reage com um re-ceptor, antígeno ou outra porção receptiva de uma população de células que pretende ser terapeuticamente ou de outra forma biologicamente modificada. Exemplos de tais moléculas incluem proteínas de menor peso molecular, polipeptídeo ou peptídeos, lectinas, glicoprote- ínas, não-peptídeos, vitaminas, moléculas de transporte de nutrientes (tais como, mas não limitadas a, transferrina), ou qualquer outra molécula de ligação celular ou substâncias. Em determinados aspectos, o anticorpo ou outra tal molécula alvo atua para entregar uma droga à população de célula alvo particular com a qual o anticorpo ou outra molécula alvo interage.
[0511] Em outro aspecto, a presente invenção refere-se a um composto conjugado de anticorpo-droga de fórmulas III ou III’, em que o anticorpo AB é selecionado de: trastuzumab, mutantes de trastuzumab (por exemplo, os mutantes de trastuzumab divulgados aqui ou no pedido de patente internacional PCT/IB2012/056234), oregovomab, edrecolomab, cetuximab, um anticorpo monoclonal humanizado para o receptor vitronectina (αvβ3), alemtuzumab, anticorpos anti-HLA-DR incluindo um anticorpo anti-HLA-DR humanizado para o tratamento de linfoma de não-Hodgkin, 131I Lym-1, anticorpos anti-HLA-Dr10 incluindo um anticorpo anti-HLA-Dr10 de murino para o tratamento de lin- foma de não-Hodgkin, anticorpos anti-cd33, anticorpos anti-cd22 incluindo um mAb anti-CD22 humanizado para o tratamento de Doença de Hodgkin ou linfoma de não-Hodgkin, labetuzumab, bevacizumab, ibri- tumomab tiuxetan, ofatumumab, panitumumab, rituximab, tositu- momab, ipilimumab, e gemtuzumab.
[0512] Heteroátomos que podem estar presentes em uma unidade de anticorpo incluem enxofre (em uma modalidade, a partir de um grupo sulfidril de um anticorpo), oxigênio (em uma modalidade, a partir de um grupo carbonil, carboxil ou hidroxil de um anticorpo) e nitrogênio (em uma modalidade, a partir de grupo amino primeiro ou secundário de um anticorpo). Estes heteroátomos podem estar presentes no anticorpo no estado natural do anticorpo, por exemplo, um anticorpo de ocorrência natural, ou pode ser introduzido no anticorpo através de modificação química.
[0513] Em uma modalidade, uma unidade de anticorpo possui um grupo sulfidril e a unidade de anticorpo se liga através do átomo de enxofre do grupo sulfidril
[0514] Em outra modalidade, o anticorpo possui resíduos de lisina que podem reagir com ésteres ativados (tais ésteres incluem, mas não são limitados a, N-hidroxissuccinimda, pentafluorofenil, e p-nitrofenil ésteres) e, desta forma, formam uma ligação amida que consiste no átomo de nitrogênio da unidade de anticorpo e um carbonil.
[0515] Em outro aspecto, a unidade de anticorpo tem um ou mais resíduos de lisina que podem ser quimicamente modificados para introduzir um ou mais grupos sulfidril. Os reagentes que podem ser usados para modificar lisinas incluem, mas não são limitados a, N- succinimidil S-acetiltioacetato (SATA) e cloridrato de 2-Iminotiolano (Reagente de Traut).
[0516] Em outra modalidade, a unidade de anticorpo pode ter um ou mais grupos de carboidrato que podem ser quimicamente modificados para um ou mais grupos sulfidril.
[0517] Em ainda outra modalidade, a unidade de anticorpo pode ter um ou mais grupos de carboidrato que podem ser oxidados para prover um grupo aldeído (ver, por exemplo, Laguzza, et al., 1989, J. Med. Chem. 32(3):548-55). O aldeído correspondente pode formar uma ligação com um sítio reativo, tal como, por exemplo, hidrazina e hidroxilamina. Outros protocolos para a modificação de proteínas para a ligação ou associação de drogas são descritos em Coligan et al., Current Protocols em Protein Science, vol. 2, John Wiley & Sons (2002) (aqui incorporado por referência).
[0518] Quando os conjugados compreendem proteína não imunor- reativa, polipeptídeo, ou unidades de peptídeo em vez de um anticorpo, proteína não imunorreativa útil, polipeptídeo, ou unidades de pep- tídeo incluem, mas não são limitados a, transferrina, fatores de crescimento epidérmico ("EGF"), bombesina, gastrina, peptídeo de liberação de gastrina, fator de crescimento derivado de plaquetas, IL-2, IL-6, fatores de crescimento de transformação ("TOP"), tais como TGF-α e TGF-β, fator de crescimento de vacínia ("VGF"), insulina e fatores de crescimento semelhantes a insulina I e II, somatostatina, lectinas e apoproteína de lipoproteína de baixa densidade.
[0519] Anticorpos policlonais úteis são populações heterogêneas de moléculas de anticorpo derivadas do soro de animais imunizados. Anticorpos monoclonais úteis são populações homogêneas de anticorpos para um determinante antigênico particular (por exemplo, um antí- geno de célula de câncer, um antígeno viral, um antígeno microbiano, uma proteína, um peptídeo, um carboidrato, um produto químico, ácido nucleico, ou fragmentos dos mesmos). Um anticorpo monoclonal (mAb) para um antígeno de interesse pode ser preparado pela utilização de qualquer técnica conhecida na arte que provê a produção de moléculas de anticorpo moléculas por linhagens celulares contínuas em cultura.
[0520] Anticorpos monoclonais úteis incluem, mas não são limitados a, anticorpos monoclonais humanos, anticorpos monoclonais humanizados, fragmentos de anticorpo, ou anticorpos monoclonais quiméricos. Anticorpos monoclonais humanos podem ser produzidos por uma variedade de técnicas conhecidas na arte (por exemplo, Teng et al., 1983, Proc. Natl. Acad. Sci. USA. 80:7308-7312; Kozbor et al., 1983, Immunology Today 4:72-79; e Olsson et al., 1982, Meth. Enzymol. 92:3-16).
[0521] O anticorpo pode também ser um anticorpo biespecífico. Métodos para produção de anticorpos biespecíficos são conhecidos na arte e são discutidos abaixo.
[0522] O anticorpo pode ser um fragmento funcionalmente ativo, derivado ou análogo de um anticorpo que immunoespecificamente se liga a células alvo (por exemplo, antígenos de célula de câncer, antí- genos virais, ou antígenos microbiais) ou outros anticorpos que se ligam a células tumorais ou matriz. Neste sentido, "funcionalmente ativo" significa que o fragmento, derivado ou análogo é capaz de induzir anticorpos anti-anti-idiotipo, que reconhecem o mesmo antígeno que o anticorpo do qual o fragmento, derivado ou análogo é derivado, reconhecidos. Especificamente, em uma modalidade exemplar, a antigeni- cidade do idiotipo da molécula de imunoglobulina pode ser melhorada por eliminação de sequências de quadro e CDR que são C-terminais à sequência de CDR que especificamente reconhece o antígeno. Para determinar quais sequências de CDR se ligam ao antígeno, peptídeos sintéticos contendo as sequências de CDR podem ser usados em ensaios de ligação com o antígeno por qualquer método de ensaio de ligação conhecido na técnica (por exemplo, o ensaio de núcleo BIA) (para localização das sequências de CDR, ver, por exemplo, Kabat et al., 1991, Sequences of Proteins of Immunological Interest, Fifth Edition, National Institute of Health, Bethesda, Md.; Kabat E et al., 1980, J. Immunology 125(3):961-969).
[0523] Outros anticorpos úteis incluem fragmentos de anticorpos, tais como, mas não limitados a, fragmentos F(ab')2, fragmentos Fab, Fvs, anticorpos de cadeia simples, diacorpos, triacorpos, tetracorpos, scFv, scFv-FV, ou qualquer outra molécula com a mesma especificidade que o anticorpo.
[0524] Adicionalmente, anticorpos recombinantes, tais como anticorpos monoclonais humanizados e quiméricos, compreendendo tanto porções humanas e não-humanas, que podem ser feitos usando técnicas de DNA recombinante padrão, são anticorpos úteis. Um anticorpo quimérico é uma molécula em que diferentes porções são derivadas de diferentes espécies animais, tais como, por exemplo, aquelas tendo uma região variável derivada de regiões constantes de imunoglobulina humana e monoclonal de murino. (Ver, por exemplo, Patente dos Estados Unidos N°. 4.816.567; e Patente dos Estados Unidos N°. 4.816.397, que são aqui incorporadas por referência em sua totalidade.) Anticorpos humanizados são moléculas de anticorpo de espécies não-humanas tendo uma ou mais regiões determinantes de comple-mentaridade (CDRs) das espécies não-humanas e uma região de quadro de uma molécula de imunoglobulina humana. (Ver, por exemplo, Patente dos Estados Unidos N°. 5.585.089, que é aqui incorpora- da por referência em sua totalidade.) Tais anticorpos humanizados monoclonais e quiméricos podem ser produzidos por técnicas de DNA recombinante conhecidas na arte, por exemplo, usando métodos descritos na Publicação Internacional N°. WO 87/02671; Publicação de Patente Européia N°. 0 184 187; Publicação de Patente Européia N°. 0 171 496; Publicação de Patente Européia N°. 0 173 494; Publicação Internacional N°. WO 86/01533; Patente dos Estados Unidos N°. 4.816.567; Publicação de Patente Européia N°. 012 023; Berter et al., 1988, Science 240:1041-1043; Liu et al., 1987, Proc. Natl. Acad. Sci. USA 84:3439-3443; Liu et al., 1987, J. Immunol. 139:3521-3526; Sun et al., 1987, Proc. Natl. Acad. Sci. USA 84:214-218; Nishimura et al., 1987, Cancer. Res. 47:999-1005; Wood et al., 1985, Nature 314:446449; e Shaw et al., 1988, J. Natl. Cancer Inst. 80:1553-1559; Morrison, 1985, Science 229:1202-1207; Oi et al., 1986, BioTechniques 4:214; Patente dos Estados Unidos N°. 5.225.539; Jones et al., 1986, Nature 321:552-525; Verhoeyan et al., 1988, Science 239:1534; e Beidler et al., 1988, J. Immunol. 141:4053-4060; cada um dos quais é aqui incorporado por referência em sua totalidade.
[0525] Anticorpos completamente humanos são particularmente desejáveis e podem ser produzidos usando camundongos transgêni- cos que são incapazes de expressar genes de cadeias leve e pesada de imunoglobulina endógena, mas que podem expressar genes de cadeia leve e pesada humana. Os camundongos transgênicos são imunizados na forma normal com um antígeno selecionado, por exemplo, todos ou uma porção de um polipeptídeo da invenção. Anticorpos mo- noclonais direcionados contra o antígeno podem ser obtidos usando tecnologia de hibridoma convencional. Os transgenes de imunoglobu- lina humana abrigados pelos camundongos transgênicos se reorganizam durante diferenciação de célula B, e subsequentemente subme-tem-se a comutação de classe e mutação somática. Desta forma, usando tal técnica, é possível produzir anticorpos IgG, IgA, IgM e IgE terapeuticamente úteis. Para uma visão geral desta tecnologia para produção de anticorpos humanos, ver Lonberg e Huszar, 1995, Int. Rev. Immunol. 13:65-93. Para uma discussão detalhada desta tecnologia para produção de anticorpos humanos e anticorpos monoclonais humanos e protocolos para produção de tais anticorpos, ver, por exemplo, Patentes dos Estados Unidos N°s. 5.625,126; 5.633.425; 5.569.825; 5.661.016; 5.545.806; cada uma das quais é aqui incorporada por referência em sua totalidade. Outros anticorpos humanos podem ser obtidos comercialmente de, por exemplo, Abgenix, Inc. (agora Amgen, Freemont, Calif.) e Medarex (Princeton, N.J.).
[0526] Anticorpos completamente humanos que reconhecem um epítopo selecionado podem ser gerados usando uma técnica referida como "seleção guiada." Nesta abordagem, um anticorpo monoclonal não humano selecionado, por exemplo, um anticorpo de camundongo, é usado para guiar a seleção de um anticorpo completamente humano reconhecendo o mesmo epítopo. (Ver, por exemplo, Jespers et al., 1994, Biotechnology 12:899-903). Anticorpos humanos podem também ser produzidos usando várias técnicas conhecidas na arte, incluindo bibliotecas de exibição de fagos (ver, por exemplo, Hoogenboom e Winter, 1991, J. Mol. Biol. 227:381; Marks et al., 1991, J. Mol. Biol. 222:581; Quan e Carter, 2002, The rise of monoclonal antibodies as therapeutics, em Anti-IgE and Allergic Disease, Jardieu e Fick, eds., Marcel Dekker, New York, N.Y., Chapter 20, pp. 427-469).
[0527] Em outras modalidades, o anticorpo é uma proteína de fusão de um anticorpo, ou um fragmento funcionalmente ativo dos mesmos, por exemplo, em que o anticorpo é fundido através de uma ligação covalente (por exemplo, uma ligação de peptídeo), seja no N- terminal ou C-terminal para uma sequência de aminoácido de outra proteína (ou porção dos mesmos, preferencialmente pelo menos por- ção de 10, 20 ou 50 aminoácidos da proteína) que não é de um anticorpo. Preferencialmente, o anticorpo ou fragmento dos mesmos é co- valentemente ligado à outra proteína no N-terminal do domínio constante.
[0528] Anticorpos incluem análogos e derivados que são tanto modificados, isto é, por uma ligação covalente de qualquer tipo de molécula desde que tal ligação covalente permita o anticorpo reter sua immunoespecificidade de ligação a antígeno. Por exemplo, mas não como forma de limitação, derivados e análogos dos anticorpos incluem aqueles que foram adicionalmente modificados, por exemplo, por gli- cosilação, acetilação, peguilação, fosforilação, amidação, derivação por grupos de bloqueio/proteção conhecidos, clivagem proteolítica, ligação a uma unidade de anticorpo celular ou outra proteína etc. Qualquer de uma variedade de modificações químicas pode ser realizada por técnicas conhecidas incluindo, mas não limitadas a, clivagem química específica, acetilação, formilação, síntese metabólica na presença de tunicamicina etc., adicionalmente, o análogo ou derivado pode conter um ou mais aminoácidos não naturais.
[0529] Anticorpos podem ter modificações (por exemplo, substituições, exclusões ou adições) em resíduos de aminoácido que interagem com receptores Fc. Em particular, anticorpos podem ter modificações em resíduos de aminoácido identificados conforme envolvidos na interação entre o domínio anti-Fc e o receptor FcRn (ver, por exemplo, Publicação Internacional N°. WO 97/34631, que é aqui incorporada por referência em sua totalidade).
[0530] Anticorpos imunoespecíficos para um antígeno de célula de câncer podem ser obtidos comercialmente ou produzidos por qualquer método conhecido de um versado na técnica, tal como, por exemplo, síntese química ou técnicas de expressão recombinante. A sequência de nucleotídeo que codifica anticorpos imunoespecíficos para um antí- geno de célula de câncer pode ser obtida, por exemplo, do banco de dados GenBank ou baco de dados similar, publicações de literatura, ou por sequenciamento ou clonagem de rotina.
[0531] Em uma modalidade específica, anticorpos conhecidos para o tratamento de câncer podem ser usados. Anticorpos imunoespecí- ficos para um antígeno de célula de câncer podem ser obtidos comercialmente ou produzidos por qualquer método conhecido de um versado na técnica, tal como, por exemplo, técnicas de expressão recombi- nante. A sequência de nucleotídeo que codifica anticorpos imunoespe- cíficos para um antígeno de célula de câncer pode ser obtida, por exemplo, do banco de dados GenBank ou um banco de dados similar, as publicações da literatura, ou por sequenciamento e clonagem de rotina. Exemplos de anticorpos disponíveis para o tratamento de câncer incluem, mas não são limitados a, OVAREX que é um anticorpo de murino para o tratamento de câncer de ovário; PANOREX (Glaxo Wellcome, NC) que é um anticorpo IgG2a de murino para o tratamento de câncer colorretal; Cetuximab ERBITUX (Imclone Systems Inc., NY) que é um anticorpo quimérico IgG anti-EGFR para o tratamento de cânceres positivos para fator de crescimento epidérmico, tais como câncer de pescoço e cabeça; Vitaxin (MedImmune, Inc., MD) que é um anticorpo humanizado para o tratamento de sarcoma; CAMPATH I/H (Leukosite, MA) que é um anticorpo IgG1 humanizado para o tratamento de leucemia linfocítica crônica (CLL); SMART ID10 (Protein Design Labs, Inc., CA) que é um anticorpo anti-HLA-DR humanizado para o tratamento de linfoma de não-Hodgkin; ONCOLYM (Techniclone, Inc., CA) que é um anticorpo anti-HLA-Dr10 de murino radiorrotulado para o tratamento de linfoma de não-Hodgkin; ALLOMUNE (BioTransplant, CA) que é um mAb anti-CD2 humanizado para o tratamento de Doença de Hodgkin ou linfoma de não-Hodgkin; e CEACIDE (Immunomedics, NJ) que é um anticorpo anti-CEA humanizado para o tratamento de câncer colorretal.
[0532] Em tentativas para descobrir alvos celulares eficazes para diagnose e terapia de câncer, pesquisadores buscam identificar transmembrana ou, de outra forma, polipeptídeos associados a tumor que são especificamente expressos na superfície de um ou mais ti- po(s) particular de célula de câncer se comparado a uma ou mais célu- la(s) não cancerosa normal. Geralmente, tais polipeptídeos associados a tumor são mais abundantemente expressos na superfície das células de câncer se comparado a na superfície das células não cancerosas. A identificação de tais polipeptídeos de antígeno de superfície celular associada a tumor deu origem à capacidade de especificamente ter como alvo células de câncer para destruição através de terapias à base de anticorpo.
[0533] A Unidade Ligante (L)
[0534] Um ligante (às vezes referido como “[ligante]” aqui) é um composto bifuncional que pode ser usado para ligar uma droga e um anticorpo para formar um conjugado de anticorpo-droga (ADC). Tais conjugados são úteis, por exemplo, na formação de imunoconjugados direcionados contra antígenos associados a tumor. Tais conjugados permitem a entrega seletiva de drogas citotóxicas a células tumorais.
[0535] Em um ADC, o ligante serve para ligar a carga útil ao anticorpo.
[0536] Em um aspecto, uma segunda seção da unidade ligante é introduzida a qual possui um segundo sítio reativo, por exemplo, um grupo eletrofílico que é reativo a um grupo nucleofílico presente em uma unidade de anticorpo (por exemplo, um anticorpo). Grupos nucle- ofílicos úteis em um anticorpo incluem, mas não são limitados a, grupos amino, sulfidril e hidroxil. O heteroátomo do grupo nucleofílico de um anticorpo é reativo a um grupo eletrofílico em uma unidade ligante e forma uma ligação covalente para uma unidade ligante. Grupos ele- trofílicos úteis incluem, mas não são limitados a, grupos maleimida e haloacetamida. O grupo eletrofílico provê um sítio conveniente para ligação de anticorpo.
[0537] Em outra modalidade, uma unidade ligante tem um sítio reativo que tem um grupo nucleofílico que é reativo a um grupo eletrofí- lico presente em um anticorpo. Grupos eletrofílicos úteis em um anticorpo incluem, mas não são limitados a, grupos aldeído e cetona car- bonil. O heteroátomo de um grupo nucleofílico de uma unidade ligante pode reagir com um grupo eletrofílico em um anticorpo e formam uma ligação covalente ao anticorpo. Grupos nucleofílicos úteis em uma unidade ligante incluem, mas não são limitados a, hidrazida, oxima, amino, hidrazina, tiossemicarbazona, carboxilato de hidrazina, e arilhidra- zida. O grupo eletrofílico em um anticorpo provê um sítio conveniente para ligação a uma unidade ligante.
[0538] Grupos amino funcionais são também sítios reativos úteis para uma unidade ligante porque eles podem reagir com ácido carbo- xílico, ou ésteres ativados de um composto to form uma ligação amida. Tipicamente, os compostos à base de peptídeo da invenção podem ser preparados pela formação de uma ligação de peptídeo entre dois ou mais fragmentos de aminoácidos e/ou peptídeo. Tais ligações de peptídeo podem ser preparadas, por exemplo, de acordo com o método de sítese de fase líquida (ver, por exemplo, Schroder and Lubke, "The Peptides", volume 1, pp 76-136, 1965, Academic Press) que é bem conhecido no campo de química de peptídeo.
[0539] No contexto da invenção, particularmente, mas não limitado a, componentes de ligante, tais como L1, L2 (incluindo L2A, L2B e L2C) e L3, a linguagem “selecionado de um ou mais de” ou “um ou mais de” indica que vários componentes, que podem ser os mesmos ou diferentes, são ou podem ser dispostos sequencialmente. Desta forma, por exemplo, L3 pode ser -Ci-6alquil-, -NR- ou os outros componentes in- dividualmente listados, mas também -Ci-6alquil-NR-, ou qualquer outra combinação de 2 ou mais componentes listados.
[0540] Síntese de Compostos e Conjugados de Anticorpo-droga dos mesmos
[0541] Os compostos e conjugados da invenção podem ser produzidos usando os procedimentos sintéticos descritos abaixo na Exemplificação. Conforme descrito em mais detalhes abaixo, os compostos e conjugados da invenção podem ser preparados usando uma seção de uma unidade ligante tendo um sítio reativo para ligação ao composto. Em um aspecto, uma segunda seção da unidade ligante é introduzida a qual tem um segundo sítio reativo, por exemplo, um grupo eletrofílico que é reativo a um grupo nucleofílico presente em uma unidade de anticorpo (por exemplo, um anticorpo). Grupos nucleofílicos úteis em um anticorpo incluem, mas não são limitados a, grupos amino, sulfidril e hidroxil. O heteroátomo do grupo nucleofílico de um anticorpo é reativo a um grupo eletrofílico em uma unidade ligante e forma uma ligação covalente para uma unidade ligante. Grupos eletrofílicos úteis incluem, mas não são limitados a, grupos maleimida e haloacetamida. O grupo eletrofílico provê um sítio conveniente para ligação de anticorpo.
[0542] Em outra modalidade, uma unidade ligante tem um sítio reativo que tem um grupo nucleofílico que é reativo a um grupo eletrofí- lico presente em um anticorpo. Grupos eletrofílicos úteis em um anticorpo incluem, mas não são limitados a, grupos aldeído e cetona car- bonil. O heteroátomo de um grupo nucleofílico de uma unidade ligante pode reagir com um grupo eletrofílico em um anticorpo e forma uma ligação covalente ao anticorpo. Grupos nucleofílicos úteis em uma unidade ligante incluem, mas não são limitados a, hidrazida, oxima, amino, hidrazina, tiossemicarbazona, carboxilato de hidrazina, e arilhidra- zida. O grupo eletrofílico em um anticorpo provê um sítio conveniente para ligação a uma unidade ligante.
[0543] Grupos amino funcionais são também sítios reativos úteis para uma unidade ligante porque eles podem reagir com ácido carbo- xílico, ou ésteres ativados de um composto para formar uma ligação amida. Tipicamente, os compostos à base de peptídeo da invenção podem ser preparados pela formação de uma ligação de peptídeo entre dois ou mais fragmentos de aminoácidos e/ou peptídeo. Tais ligações de peptídeo podem ser preparadas, por exemplo, de acordo com o método de sítese de fase líquida (ver, por exemplo, Schroder and Lubke, "The Peptides", volume 1, pp 76-136, 1965, Academic Press) que é bem conhecido no campo de química de peptídeo.
[0544] Conforme descrito em mais detalhes abaixo, os conjugados podem ser preparados usando uma seção do ligante tendo um sítio reativo para ligação a um composto da invenção e introduzindo outra seção da unidade ligante tendo um sítio reativo para um anticorpo. Em um aspecto, uma unidade ligante tem um sítio reativo que tem um grupo eletrofílico que é reativo com um grupo nucleofílico presente em uma unidade de anticorpo, tal como um anticorpo. O grupo eletrofílico provê um sítio conveniente para ligação de anticorpo. Grupos nucleofí- licos úteis em um anticorpo incluem, mas não são limitados a, grupos amino, sulfidril e hidroxil. O heteroátomo do grupo nucleofílico de um anticorpo é reativo a um grupo eletrofílico em uma unidade ligante e forma uma ligação covalente para uma unidade ligante. Grupos eletro- fílicos úteis incluem, mas não são limitados a, grupos maleimida e ha- loacetamida.
[0545] Em outra modalidade, uma unidade ligante tem um sítio reativo que tem um grupo nucleofílico que é reativo com um grupo ele- trofílico presente em uma unidade de anticorpo. O grupo eletrofílico em um anticorpo provê um sítio conveniente para ligação a uma unidade ligante. Grupos eletrofílicos úteis em um anticorpo incluem, mas não são limitados a, grupos aldeído e cetona carbonil. O heteroátomo de um grupo nucleofílico de uma unidade ligante pode reagir com um grupo eletrofílico em um anticorpo e forma uma ligação covalente ao anticorpo. Grupos nucleofílicos úteis em uma unidade ligante incluem, mas não são limitados a, hidrazida, oxima, amino, hidrazina, tiossemi- carbazona, carboxilato de hidrazina, e arilhidrazida.
[0546] Conjugação com Transglutaminase
[0547] Em certas modalidades, um composto da invenção pode ser covalentemente reticulado a um polipeptídeo contendo Fab ou contendo Fc projetado com um rótulo contendo glutamina doador de acil (por exemplo, rótulos de peptídeo contendo Gln ou rótulos Q) ou uma glutamina endógena feita reativa (isto é, a capacidade de formar uma ligação covalente como um doador de acil na presença de uma amina e uma transglutaminase) por desenvolvimento de polipeptídeo (por exemplo, através de mutação, substituição, inserção, eliminação de aminoácido, ou qualquer combinação dos mesmos no polipeptídeo), na presença de transglutaminase, desde que o composto da invenção compreenda um agente doador de amina (por exemplo, molécula pequena compreendendo ou ligada a uma amina reativa), formando assim uma população estável e homogênea de um conjugado de poli- peptídeo contendo Fc projetado com o agente doador de amina sendo sítio especificamente conjugado ao polipeptídeo contendo Fc ou contendo Fab através do rótulo contendo glutamina doador de acil ou a glutamina endógena exposta/acessível/reativa. Por exemplo, compostos da invenção podem ser conjugados como descrito no Pedido de Patente Internacional N°. de Série PCT/IB2011/054899, cujo inteiro conteúdo é aqui incorporado por referência. Em certas modalidades, para facilitar a conjugação do composto da invenção a um polipeptídeo contendo Fab ou contendo Fc projetado com um rótulo contendo glu- tamina doador de acil ou uma glutamina endógena feita reativa por desenvolvimento de polipeptídeo na presença de transglutaminase, Z é NH2.
[0548] Conjugação à Região Constante de Domínio Kappa de Ca- deia Leve Humana
[0549] Em certas modalidades, um composto da invenção pode ser covalentemente ligado à cadeia lateral de K188 da região constante de domínio kappa de cadeia leve humana (CLK) (cadeia totalmente leve numerada de acordo com Kabat). Por exemplo, compostos da invenção podem ser conjugados conforme descrito no Pedido de Patente dos Estados Unidos N°. de Série 13/180,204, cujo inteiro conteúdo é aqui incorporado por referência. Em certas modalidades, para facilitar a conjugação a K188 CLκ, Z é
; R7 é independen temente selecionado para cada ocorrência do grupo que consiste em F, Cl, I, Br, NO2, CN e CF3; e h é 1, 2, 3, 4 ou 5.
[0550] Em certas modalidades, a invenção provê uma composição compreendendo um composto da invenção covalentemente conjugado a um anticorpo (ou porção de ligação a antígeno dos mesmos), em que pelo menos cerca de 50%, ou pelo menos cerca de 60%, ou pelo menos cerca de 70%, ou pelo menos cerca de 80%, ou pelo menos cerca de 90% do composto da invenção na composição é conjugado ao anticorpo ou porção de ligação a antígeno do mesmo em K188 CLk.
[0551] Em certas modalidades, os compostos da invenção podem ser conjugados ao sítio de combinação de um anticorpo catalítico, tal como anticorpos aldolase, ou porção de ligação a antígeno dos mesmos. Anticorpos aldolase contem porções de sítio combinantes que, quando livres (por exemplo, por conjugação), catalisam uma reação de adição de aldol entre um doador de cetona alifática e um aceptor de aldeído. O teor da Publicação de Pedido de Patente dos Estados Unidos N°. US 2006/205670 é aqui incorporado por referência, em particular, páginas 78-118 que descrevem ligantes, e parágrafos [0153]- [0233] que descrevem anticorpos, fragmentos úteis, variantes e modificações dos mesmos, h38C2, sítios de combinação e regiões determinantes de complementaridade (CDRs), e tecnologia de anticorpo relacionada. O termo “sítio de combinação” inclui as CDRs e os resíduos de quadro adjacentes que estão envolvidos em ligação de antí- geno.
[0552] Composições e Métodos de Administração
[0553] Em outras modalidades, outro aspecto da invenção refere- se a composições farmacêuticas incluindo uma quantidade eficaz de um composto da invenção e/ou conjugado de anticorpo-droga dos mesmos e um veículo ou transportador farmaceuticamente aceitável. Em certas modalidades, as composições são adequadas para administração humana ou veterinária.
[0554] As composições farmacêuticas presentes podem ser em qualquer forma que permita que a composição seja administrada a um paciente. Por exemplo, a composição pode ser na forma de um sólido ou líquido. Rotas típicas de administração incluem, sem limitação, parenteral, ocular e intra-tumor. Administração parenteral inclui injeções subcutâneas, injeção intravenosa, intramuscular ou intrasternal ou técnicas de infusão. Em um aspecto, as composições são administradas parenteralmente. Em uma modalidade específica, as composições são administradas intravenosamente.
[0555] Composições farmacêuticas podem ser formuladas de forma a permitir que um composto da invenção e/ou conjugado de anti- corpo-droga dos mesmos esteja biodisponível quando da administração da composição a um paciente. Composições podem ter a forma de uma ou mais unidades de dosagem, em que, por exemplo, um comprimido pode ser uma unidade de dosagem única, e um recipiente de um composto da invenção e/ou conjugado de anticorpo-droga dos mesmos em forma líquida pode portar uma pluralidade de unidades de dosagem.
[0556] Materiais usados na preparação das composições farmacêuticas podem ser não tóxicos nas quantidades usadas. Será evidente para aqueles ordinariamente versados na técnica que a dosagem ótima do ingrediente(s) ativo na composição farmacêutica dependerá de uma variedade de fatores. Fatores relevantes incluem, sem limitação, o tipo de animal (por exemplo, ser humano), a forma particular do composto da invenção e/ou conjugado de anticorpo-droga dos mesmos, a forma de administração, e a composição empregada.
[0557] O veículo ou transportador farmaceuticamente aceitável pode ser sólido ou particulado, de forma que as composições sejam, por exemplo, em forma de comprimido ou pó. O transportador(s) pode ser líquido. Além disso, o transportador(s) pode ser particulado.
[0558] A composição pode ser na forma de um líquido, por exemplo, uma solução, emulsão ou suspensão. Em uma composição para administração por injeção, um ou mais de um tensoativo, conservante, agente umidificante, agente dispersante, agente de suspensão, tampão, estabilizador e agente isotônico pode também ser incluído.
[0559] As composições líquidas, sejam elas soluções, suspensões ou outra forma semelhante, podem também incluir um ou mais do seguinte: diluentes estéreis, tais como água para injeção, solução salina, preferencialmente, solução salina fisiológica, solução de Ringer, cloreto de sódio isotônico, óleos fixos, tais como mono ou diglicerídeos sintéticos que podem servir como o solvente ou meio de suspensão, poli- etileno glicois, glicerina, ciclodextrina, propileno glicol ou outros solventes; agentes antibacterianos, tais como álcool benzílico ou metil parabeno; antioxidantes, tais como ácido ascórbico ou bissulfito de sódio; agentes quelantes, tais como ácido etilenodiaminatetraacético; tampões, tais como acetatos, citratos, fosfatos ou aminoácidos e agentes para ajuste de tonicidade, tais como cloreto de sódio ou dextrose. Uma composição parenteral pode ser inclusa em ampola, seringa descartável ou um frasco de doses múltiplas feito de vidro, plástico ou outro material. Solução salina fisiológica é um adjuvante exemplar. Uma composição injetável é preferencialmente estéril.
[0560] A quantidade de um composto da invenção e/ou conjugado de anticorpo-droga do mesmo que é eficaz no tratamento de uma condição ou distúrbio particular dependerá da natureza do distúrbio ou condição, e pode ser determined por técnicas clínicas padrão. Além disso, ensaios em vitro ou em vivo podem opcionalmente ser empregados para ajudar a identificar faixas de dosagem ótimas. A dose precisa a ser empregada nas composições também dependerá da rota de administração, e a gravidade da doença ou distúrbio, e deve ser decidida de acordo com o julgamento do médico e cada circunstância do paciente.
[0561] As composições compreendem uma quantidade eficaz de um composto da invenção e/ou conjugado de anticorpo-droga do mesmo tal que uma dosagem adequada será obtida. Tipicamente, esta quantidade é pelo menos cerca de 0,01% de um composto da invenção e/ou conjugado de anticorpo-droga do mesmo em peso da composição. Em uma modalidade exemplar, composições farmacêuticas são preparadas de modo que uma unidade de dosagem parenteral contém de cerca de 0,01% a cerca de 2% em peso da quantidade de um composto da invenção e/ou conjugado de anticorpo-droga do mesmo.
[0562] Para administração intravenosa, a composição pode compreender de cerca de 0,01 a cerca de 100 mg de um composto da invenção e/ou conjugado de anticorpo-droga do mesmo por kg de peso do corpo do paciente. Em um aspecto, a composição pode incluir de cerca de 1 a cerca de 100 mg de um composto da invenção e/ou conjugado de anticorpo-droga do mesmo por kg de peso do corpo do pa- ciente. Em outro aspecto, a quantidade administrada será na faixa de cerca de 0,1 a cerca de 25 mg/kg de peso corporal de um composto da invenção e/ou conjugado de anticorpo-droga do mesmo.
[0563] Geralmente, a dosagem de um composto da invenção e/ou conjugado de anticorpo-droga do mesmo administrada a um paciente é tipicamente cerca de 0,01 mg/kg a cerca de 20 mg/kg de peso do corpo do paciente. Em um aspecto, a dosagem administrada a um paciente é entre cerca de 0,01 mg/kg a cerca de 10 mg/kg de peso do corpo do paciente. Em outro aspecto, a dosagem administrada a um paciente é entre cerca de 0,1 mg/kg e cerca de 10 mg/kg de peso do corpo do paciente. Em outro aspecto, a dosagem administrada a um paciente é entre cerca de 0,1 mg/kg e cerca de 5 mg/kg de peso do corpo do paciente. Em outro aspecto a dosagem administrada é entre cerca de 0,1 mg/kg a cerca de 3 mg/kg de peso do corpo do paciente. Em outro aspecto, a dosagem administrada é entre cerca de 1 mg/kg a cerca de 3 mg/kg de peso do corpo do paciente.
[0564] Um composto da invenção e/ou conjugado de anticorpo- droga do mesmo pode ser administrado por qualquer rota conveniente, por exemplo, por infusão ou injeção de bólus. A administração pode ser sistêmica ou local. Vários sistemas de entrega são conhecidos, por exemplo, encapsulação em lipossomas, micropartículas, microcápsu- las, cápsulas etc., e podem ser usados para administrar um composto da invenção e/ou conjugado de anticorpo-droga do mesmo. Em certas modalidades, mais do que um composto da invenção e/ou conjugado de anticorpo-droga do mesmo é administrado a um paciente.
[0565] Em modalidades específicas, pode ser desejável administrar um ou mais compostos da invenção e/ou conjugados de anticorpo- droga do mesmo localmente à área em necessidade de tratamento. Isto pode ser alcançado, por exemplo, e não como forma de limitação, por infusão local durante cirurgia; aplicação tópica, por exemplo, em conjunção com um curativo após cirurgia; por injeção; por meio de um cateter; ou por meio de um implante, o implante sendo de um material poroso, não poroso ou gelatinoso, incluindo membranas, tais como membranas sialásticas, ou fibras. Em uma modalidade, a administração pode ser por injeção direta no sítio (ou local anterior) de um tecido pré-neoplásico ou neoplásico, de câncer ou tumor. Em outra modalidade, a administração pode ser por injeção direta no sítio (ou local anterior) de uma manifestação de uma doença autoimune.
[0566] Em ainda outra modalidade, o composto da invenção e/ou conjugado de anticorpo-droga do mesmo pode ser entregue em um sistema de liberação controlada, tal como, mas não limitado a, uma bomba ou vários materiais poliméricos podem ser usados. Em ainda outra modalidade, um sistema de liberação controlada pode ser colocado na proximidade do alvo do composto da invenção e/ou conjugado de anticorpo-droga do mesmo, por exemplo, o fígado, requerendo assim somente uma fração da dose sistêmica (ver, por exemplo, Good- son, em Medical Applications of Controlled Release, supra, vol. 2, pp. 115-138 (1984)). Outros sistemas de liberação controlada discutidos na revisão por Langer (Science 249:1527-1533 (1990)) podem ser usados.
[0567] O termo "transportador" se refere a um diluente, adjuvante ou excipiente, com o qual um composto ou conjugado de anticorpo- droga do mesmo é administrado. Tais transportadores farmacêuticos podem ser líquidos, tais como água e óleos, incluindo aqueles de origem sintética, vegetal, animal ou de petróleo. Os transportadores podem ser soluções salinas, e semelhantes. Além disso, agentes auxiliares, estabilizantes e outros agentes podem ser usados. Em uma modalidade, quando administrado a um paciente, o composto ou conjugado e transportadores farmaceuticamente aceitáveis são estéreis. Água é transportador exemplar quando o composto ou conjugado são administrados intravenosamente. Soluções salinas e dextrose aquosa e soluções de glicerol podem também ser empregadas como transportadores líquidos, particularmente para soluções injetáveis. As presentes composições, se desejado, podem também conter quantidades menores de agentes de umidificação ou emulsificantes, ou agentes de tamponamento de pH.
[0568] As presentes composições podem tomar a forma de soluções, pastilhas, pós, formulações de liberação controlada, ou qualquer outra forma adequada para uso. Outros exemplos de transportadores farmacêuticos adequados são descritos em "Remington's Pharmaceutical Sciences" por E. W. Martin.
[0569] Em uma modalidade, o composto da invenção e/ou conjugado de anticorpo-droga do mesmo é formulado de acordo com procedimentos de rotina como uma composição farmacêutica adaptada para administração intravenosa a animais, particularmente seres humanos. Tipicamente, os transportadores ou veículos para administração intravenosa são soluções tampão aquosas isotônicas estéreis. Quando necessário, as composições podem também incluir um agente solubilizante. Composições para administração intravenosa podem opcionalmente compreender um anestésico local, tal como lignocaína para aliviar a dor no sítio da injeção. Geralmente, os ingredientes são fornecidos separadamente ou misturados juntos em forma de dosagem unitária, por exemplo, como um pó liofilizado seco ou concentrado livre de água em um recipiente hermeticamente vedado, tal como uma ampola ou sachê indicando a quantidade de agente ativo. Quando um composto da invenção e/ou conjugado de anticorpo-droga do mesmo deve ser administrado por infusão, pode ser dispensado, por exemplo, com uma garrafa de infusão contendo solução salina ou água de grau farmacêutica estéril. Quando o composto da invenção e/ou conjugado de anticorpo-droga do mesmo é administrado por injeção, uma ampola de água estéril para injeção ou solução salina pode ser provida de modo que os ingredientes possam ser misturados antes da administração.
[0570] A composição pode incluir vários materiais que modificam a forma física de uma unidade de dosagem líquida ou sólida. Por exemplo, a composição pode incluir materiais que formam um revestimento em torno dos ingredientes ativos. Os materiais que formam o revestimento são tipicamente inertes, e podem ser selecionados de, por exemplo, açúcar, goma-laca, e outros agentes de revestimento entéricos. Alternativamente, os ingredientes ativos podem ser encapsulados em uma cápsula de gelatina.
[0571] Seja na forma líquida ou sólida, as presentes composições podem incluir um agente farmacológico usado no tratamento de câncer.
[0572] Usos Terapêuticos de Compostos e Conjugados de Anti- corpo-droga dos mesmos
[0573] Outro aspecto da invenção refere-se a um método de utilização dos compostos da invenção e conjugados de anticorpo-droga dos mesmos para tratamento de câncer.
[0574] Os compostos da invenção e/ou conjugados de anticorpo- droga dos mesmos são úteis para inibição da multiplicação de uma célula tumoral ou célula de câncer, causando apoptose em uma célula tumoral ou de câncer, ou para tratamento de câncer em um paciente. Os compostos da invenção e/ou conjugados de anticorpo-droga dos mesmos podem ser usados consequentemente em uma variedade de configurações para o tratamento de cânceres animais. Os referidos conjugados podem ser usados para entregar um composto da invenção a uma célula tumoral ou célula de câncer. Sem estar limitado pela teoria, em uma modalidade, o anticorpo do conjugado se liga a ou se associa com um antígeno associado a célula tumoral ou célula de cân- cer, e o conjugado pode ser retomado (internalizado) dentro de uma célula tumoral ou célula de câncer através de endocitose mediada por receptor ou outro mecanismo de internalização. O antígeno pode ser ligado a uma célula tumoral ou célula de câncer ou pode ser uma proteína de matriz extracelular associada com a célula tumoral ou célula de câncer. Em certas modalidades, uma vez dentro da célula, uma ou mais sequências de peptídeo específicas são enzimaticamente ou hi- droliticamente clivadas por uma ou mais proteases associadas a célula tumoral ou célula de câncer, resultando em liberação de um composto da invenção do conjugado. O composto liberado da invenção é, então, livre para migrar dentro da célula e induz atividades citotóxicas ou ci- tostáticas. O conjugado também pode ser clivado por uma protease intracelular para liberar um composto da invenção. Em uma modalidade alternativa, o composto da invenção é clivado a partir do conjugado para fora da célula tumoral ou célula de câncer, e o composto da invenção subsequentemente penetra a célula.
[0575] Em certas modalidades, os conjugados proveem alvo de droga de câncer ou tumor específica de conjugação, reduzindo assim a toxicidade geral dos compostos da invenção.
[0576] Em outra modalidade, a unidade de anticorpo se liga à célula tumoral ou célula de câncer.
[0577] Em outra modalidade, a unidade de anticorpo se liga a um antígeno de célula tumoral ou célula de câncer que está na superfície da célula tumoral ou célula de câncer.
[0578] Em outra modalidade, a unidade de anticorpo se liga a um antígeno de célula tumoral ou célula de câncer que é uma proteína de matriz extracelular associada com a célula tumoral ou célula de câncer.
[0579] A especificidade da unidade de anticorpo para uma célula tumoral ou célula de câncer particular pode ser importante para deter- minar aqueles tumores ou cânceres que são mais eficazmente tratados.
[0580] Tipos particulares de câncer que podem ser tratados com um composto da invenção e/ou conjugado de anticorpo-droga do mesmo, incluem, mas não são limitados a, carcinomas da bexiga, mama, colo do útero, cólon, endométrio, rim, pulmão, esôfago, ovário, próstata, pâncreas, pele, estômago e testículos; e cânceres de origem sanguínea, incluindo, mas não limitados a, leucemias e linfomas.
[0581] Terapia Multi-modalidade para Câncer. Cânceres, incluindo, mas não limitados a, a tumor, metástase, ou outra doença ou distúrbio caracterizado por crescimento celular descontrolado, pode ser tratado ou inibido pela administração de um composto da invenção e/ou conjugado de anticorpo-droga do mesmo.
[0582] Em outras modalidades, métodos para tratamento de câncer são providos, incluindo a administração a um paciente em necessidade dos mesmos de uma quantidade eficaz de um composto da invenção e/ou conjugado de anticorpo-droga do mesmo e um agente quimioterapêutico. Em uma modalidade, o agente quimioterapêutico é aquele com o qual tratamento do câncer não foi constatado ser refratário. Em outra modalidade, o agente quimioterapêutico é aquele com o qual o tratamento de câncer foi constatado ser refratário. Um composto da invenção e/ou conjugado de anticorpo-droga do mesmo pode ser administrado a um paciente que também foi submetido a cirurgia como tratamento para o câncer.
[0583] Em algumas modalidades, o paciente também recebe um tratamento adicional, tal como terapia de radiação. Em uma modalidade específica, o composto da invenção e/ou conjugado de anticorpo- droga do mesmo é administrado concomitantemente com o agente quimioterapêutico ou com terapia de radiação. Em outra modalidade específica, o agente quimioterapêutico ou terapia de radiação é admi- nistrado antes ou após a administração de um composto da invenção e/ou conjugado de anticorpo-droga do mesmo.
[0584] Um agente quimioterapêutico pode ser administrado ao longo de uma série de sessões. Qualquer um ou uma combinação dos agentes quimioterapêuticos, tais como um padrão de agente de cuidado quimioterapêutico(s), pode ser administrado.
[0585] Adicionalmente, métodos de tratamento de câncer com um composto da invenção e/ou conjugado de anticorpo-droga do mesmo são providos como uma alternativa à quimioterapia ou terapia de radiação, em que a quimioterapia ou a terapia de radiação provou ou pode revelar-se demasiadamente tóxica, por exemplo, resulta em efeitos colaterais insuportáveis ou inaceitáveis, para o indivíduo a ser tratado. O paciente sendo tratado pode, opcionalmente, ser tratado com outro tratamento de câncer, tal como cirurgia, terapia de radiação ou quimioterapia, dependendo de que tratamento é considerado aceitável ou suportável.
[0586] Os compostos da invenção e/ou conjugados de anticorpo- droga dos mesmos podem também ser usados em uma forma em vitro ou ex vivo, tal como para o tratamento de determinados cânceres, incluindo, mas não limitados a, leucemias e linfomas, tal tratamento envolvendo transplantes de células estaminais autólogas. Isto pode envolver um processo multi-etapas, em que as células estaminais hema- topoiéticas autólogas de animal são colhidas e purgadas de todas as células de câncer, o restante da população de células da medula óssea do animal é, então, erradicado através da administração de uma alta dose de um composto da invenção e/ou conjugado de anticorpo- droga do mesmo com ou sem terapia de radiação de alta dose acompanhante, e o enxerto de células estaminais é infundido de volta no animal. Tratamento suporte é, então, provido enquanto a função da medula óssea é restaurada e o paciente se recupera.
[0587] A invenção é ainda descrita nos seguintes exemplos, os quais não se destinam a limitar o escopo da invenção.
[0588] Espécies Liberadas
[0589] Modalidades adicionais da invenção incluem as espécies químicas liberadas, dentro ou nas imediações da célula de câncer ou célula de tumor pelo que acredita-se ser clivagem hidrolítica e/ou en- zimática por uma ou mais proteases associadas a célula de câncer ou célula de tumor. Tais compostos incluem as espécies descritas aqui, e também incluem compostos, tais como aqueles descritos na estrutura:
[0590] Um composto ou compostos de fórmula (II):
[0591] L-P
[0592] (II)
[0593] ou um sal farmaceuticamente aceitável do mesmo, em que:
[0594] L é a porção ligante L1-L2-L3, em que L3 é ligado a P;
[0595] P é um radical de fórmula (I):
[0596] (I)
[0597] em que:
[0598] uma linha pontilhada representa uma ligação opcional;
[0599] cada X1 é independentemente selecionado do grupo que consiste em: -O-, -S- e -NR-;
[0600] cada X2 é independentemente selecionado do grupo que consiste em: -O-, -S- e -NR-;
[0601] cada X’ é CR ou N;
[0602] cada X’’ é CH-, CR-(C(R)2)m-NR-, CR-(C(R)2)m-O-; CR- (C(R)2)m-C(O)NR-, CR-(C(R)2)m-C(O)NR-NR-, CR-(C(R)2)m-SO2NR-, CR-(C(R)2)m-NR-NR-, CR-(C(R)2)m-NR-C(O)- ou N- se X’’ se liga a L2 ou um L3 adicional, ou de outra forma é O, S, CRR, CR-(C(R)2)m-NRR ou NRR;
[0603] cada X’’’ é - (C(R)2)m-NR- ou CR-(C(R)2)m-O- se X’’’ se liga a L2, ou de outra forma é R;
[0604] Y é -C(R)2-, -O-, -NR- ou -S-;
[0605] R1 é selecionado do grupo que consiste em: -R, -OR, - OCOR13, -OCONR14R15, -OCON(R14)NR(R15), =O (ligação dupla a oxigênio) e -NR14R15;
[0606] R2 e R3 são independentemente selecionados do grupo que consiste em: hidrogênio e C1-6alquil;
[0607] R4 e R5 são independentemente selecionados do grupo que consiste em: hidrogênio, -OR, -NR14R15 e oxo;
[0608] R6 e R7 são independentemente selecionados do grupo que consiste em: hidrogênio, halogênio, hidroxil e C1-6alquil opcionalmente substituído com 1-3 substituintes independentemente selecionados de hidroxil e halogênio,
[0609] R6 e R7, juntos com o átomo de carbono ao qual estão ligados, formam um C2-5alquilideno opcionalmente substituído com 1-3 substituintes independentemente selecionados de R,
[0610] R6 e R7 juntos são oxo, ou
[0611] R6 e R7, juntos com o átomo de carbono ao qual estão ligados, formam uma porção heterocicloalquil de 3 a 5 membros compreendendo 1 ou 2 heteroátomos independentemente selecionados do grupo que consiste em oxigênio, nitrogênio e enxofre, em que a referida porção heterocicloalquil pode ser opcionalmente substituída com um a três substituintes independentemente selecionados de R;
[0612] R8 é hidrogênio, C1-6alquil ou -OR;
[0613] R9 é -(C(R)2)m-C(O)- ou -(C(R)2)m- ;
[0614] L1 é selecionado de: -ácido, -NR-ácido e
[0615] L2 é L2A-L2B-L2C ou L2C-L2B-L2A, em que:
[0616] L2A compreende um ou mais componentes selecionados de:
[0617] -O-, -C(O)-, -C(O)NR-, -C(O)-C1-6alquil-, -C(O)NRC1-6alquil-,-C1-6alquil(OCH2CH2)1-6-, -C(O)-C1-6alquil-NRC(O)-, -C(O)-C1- 6alquil(OCH2CH2)1-6-, -C1-6alquil(OCH2CH2)1-6-C(O)-, -C1-6alquil-S-S-C1- 6alquil-NRC(O)CH2-, -C1-6alquil-(OCH2CH2)1-6-NRC(O)CH2-, -C(O)-C1- 6alquil-NRC(O)C1-6alquil-, -N=CR-fenil-O-C1-6alquil-, -N=CR-fenil-O-C1- 6alquil-C(O)-, -C(O)-C1-6alquil(OCH2CH2)1-6-NRC(O)-, -C(O)-C1-6alquil- fenil-(NR-C(O)-C1-6alquil)1-4-, -C(O)-C1-6alquil-(OCH2CH2)1-6-NRC(O)C1- 6alquil-, -C1-6alquil-, -S-, -C(O)-C1-6alquil-fenil-NR-, -O-C1-6alquil-S-, - C(O)-O-C1-6alquil-S- e (-CH2-CH2-O-)1-20, ou L2A é ausente;
[0618] L2B é selecionado de AA0-aa, em que AA é um aminoácido natural ou não-natural e aa é 12; e
[0619] L2C compreende um ou mais componentes selecionados de: -PABA- e -PABC-, ou L2C é ausente;
[0620] L3 é selecionado de um ou mais de: -C1-6alquil-, -NR-C3-C8- heterociclil-NR-, -NR-C3-C8carbociclil-NR-, -NR-C1-6alquil-NR-, -NR-C1- 6alquil-, -S-, -NR-, -NR-NR- e -NR-C(O)-NR-, em que os dois grupos R opcionalmente se juntam para formar um anel de 4-10 membros, -NR- C1-6alquil-fenil-NR-, -NR-C1-6alquil-fenil-SO2-NR-, -SO2-, -NR-C1-6- alquil-fenil-C(O)-,
, ou L3 é ausente;
[0621] R13 é selecionado do grupo que consiste em hidrogênio, C1-6alquil, C3-8carbociclil, C3-8heterociclil, C1-6alquil-C6-14aril, C1-6alquil-C5- uheteroaril, em que R13 é opcionalmente substituído com -NRR ou - SO2NRR;
[0622] cada R14 e R15 é independentemente selecionado do grupo que consiste em: hidrogênio, hidroxil, -NRR, -NRNR2, -C3-10carbociclil, -C1-6alquileno-C3-10carbociclil, -C3-10heterociclil, -C1-6alquileno-C3- 10heterociclil, -(CH2CH2O)1-6CH2CH2C(O)OR, -(CH2CH2O)1-6CH2CH2NRR, -C1-6alquil, C6-14aril, -C1-6alquileno-C6-14aril e -C5- 14heteroaril;
[0623] ou R14 e R15, juntos com o átomo ou átomos aos quais são ligados, formam um anel C3-10heterociclil,
[0624] em que R14, R15, ou ambos, ou um anel formado com R14 e R15, são opcionalmente substituídos com -(C(R)2)m-R18, em que cada R18 é independentemente selecionado de (i) -NRR, (ii) - C(NRR)(C(O)OR), (iii) -S-R, (iv) aril ou heteroaril opcionalmente substituído com um ou mais de halogênio, -CF3, -(C(R)2)m-NRR ou - (C(R)2)m -SO2NRR, (v) -SO2R, (vi) -S-S-C1-6alquil-C(O)OR, (vii) - SO2NRR, (viii) -C(O)NRR, (ix) -C(O)OR, (x) -C4-6 cicloalquil opcionalmente substituído com -NRR, -SO2NRR ou -NR-C(O)(CH2)0-6NRR, (xi) -R, (xii) -OR, (xiii) -N(R)NRR, (xiv) -C(O)N(R)NRR, (xv) - (C(R)2)m-O-NRR e (xiv) -S-S-C1-6alquil-NRR;
[0625] Ácido é um resíduo de aminoácido selecionado de - SCH2CH(COOH)(NH2), -NH(CH2)4CH(COOH)(NH2) e -C(O)(CH2)2CH(COOH)(NH2);
[0626] cada R é independentemente selecionado do grupo que consiste em: hidrogênio e -C1-6alquil; e
[0627] cada m é independentemente 0, 1, 2 ou 3.
[0628] adicionalmente, um composto ou compostos de fórmula (II’):
[0629] L-P’
[0630] (II’)
[0631] ou um sal farmaceuticamente aceitável do mesmo, em que:
[0632] L é a porção ligante L1-L2-L3, em que L3 é ligado a P’;
[0633] P’ é um radical de fórmula (I’):
[0634] (I’)
[0635] em que:
[0636] uma linha pontilhada representa uma ligação opcional;
[0637] cada X1 é independentemente selecionado do grupo que consiste em: -O-, -S- e -NR-;
[0638] cada X2 é independentemente selecionado do grupo que consiste em: -O-, -S- e -NR-;
[0639] cada X’ é CR ou N;
[0640] cada X’’ é CH-, CR-(C(R)2)m-NR-, CR-(C(R)2)m-O-; CR- (C(R)2)m-C(O)NR-, CR-(C(R)2)m-C(O)NR-NR-, CR-(C(R)2)m-SO2NR-, CR-(C(R)2)m-NR-NR-, CR-(C(R)2)m-NR-C(O)- ou N- se X’’ se liga a L2 ou um L3 adicional, ou de outra forma é O, S, CRR, CR-(C(R)2)m-NRR ou NRR;
[0641] cada X’’’ é - (C(R)2)m-NR- ou CR-(C(R)2)m-O- se X’’’ se liga a L2, ou de outra forma é R;
[0642] Y é -C(R)2-, -O-, -NR- ou -S-;
[0643] R1 é selecionado do grupo que consiste em: -(C(R)2)m-, -OR’’, -OCOR13’, -OC(O)NRR14’, -OCON(R)N(R)-, e -NR-
[0644] R2 e R3 são independentemente selecionados do grupo que consiste em: hidrogênio e C1-6alquil;
[0645] R4 e R5 são independentemente selecionados do grupo que consiste em: hidrogênio, -OR, -NR14R15 e oxo;
[0646] R6 e R7 são independentemente selecionados do grupo que consiste em: hidrogênio, halogênio, hidroxil e C1-6alquil opcionalmente substituído com 1-3 substituintes independentemente selecionados de hidroxil e halogênio,
[0647] R6 e R7, juntos com o átomo de carbono ao qual estão ligados, formam um C2-5alquilideno opcionalmente substituído com 1-3 substituintes independentemente selecionados de R,
[0648] R6 e R7 juntos são oxo, ou
[0649] R6 e R7, juntos com o átomo de carbono ao qual estão ligados, formam uma porção heterocicloalquil de 3 a 5 membros compreendendo 1 ou 2 heteroátomos independentemente selecionados do grupo que consiste em oxigênio, nitrogênio e enxofre, em que a referida porção heterocicloalquil pode ser opcionalmente substituída com um a três substituintes independentemente selecionados de R;
[0650] R8 é hidrogênio, C1-6alquil ou -OR;
[0651] R9 é independentemente selecionado de hidrogênio, -C1- θalquil, -(C(R)2)m-C(O)OR, -(C(R)2)m-C(O)NR14R15, -(C(R)2)m-NR14R15, -(C(R)2)mrC(O)-SR, -(C(R)2)m-C(O)NR14N(R)R15, -(C(R)2)m-NR-C(O)- NR14R15, -(C(R)2)m-N(R)COR13 e -(C(R)2)m-NR14N(R)R15; O Vy -acid
[0652] L1 é selecionado de: -ácido, -NR-ácido e
[0653] L2 é L2A-L2B-L2C ou L2C-L2B-L2A, em que:
[0654] L2A compreende um ou mais componentes selecionados de:
[0655] -O-, -C(O)-, -C(O)NR-, -C(O)-C1-6alquil-, -C(O)NRC1-6alquil-,-C1-6alquil(OCH2CH2)1-6-, -C(O)-C1-6alquil-NRC(O)-, -C(O)-C1-6alquil(OCH2CH2)1-6-, -C1-6alquil(OCH2CH2)1-6-C(O)-, -C1-6alquil-S-S-C1- 6alquil-NRC(O)CH2-, -C1-6alquil-(OCH2CH2)1-6-NRC(O)CH2-, -C(O)-C1- 6alquil-NRC(O)C1-6alquil-, -N=CR-fenil-O-C1-6alquil-, -N=CR-fenil-O-C1- 6alquil-C(O)-, -C(O)-C1-6alquil(OCH2CH2)1-6-NRC(O)-, -C(O)-C1-6alquil- fenil-(NR-C(O)-C1-6alquil)1-4-, -C(O)-C1-6alquil-(OCH2CH2)1-6-NRC(O)C1- 6alquil-, -C1-6alquil-, -S-, -C(O)-C1-6alquil-fenil-NR-, -O-C1-6alquil-S-, - C(O)-O-C1-6alquil-S- e (-CH2-CH2-O-)1-20, ou L2A é ausente;
[0656] L2B é selecionado de AA0-aa, em que AA é um aminoácido natural ou não-natural e aa é 12; e
[0657] L2C compreende um ou mais componentes selecionados de: -PABA- e -PABC-, ou L2C é ausente;
[0658] L3 é selecionado de um ou mais de: -C1-6alquil-, -NR-C3-C8- heterociclil-NR-, -NR-C3-C8carbociclil-NR-, -NR-C1-6alquil-NR-, -NR-C1- 6alquil-, -S-, -NR-, -NR-NR- e -NR-C(O)-NR-, em que os dois grupos R opcionalmente se juntam para formar um anel de 4-10 membros, -NR- C1-6alquil-fenil-NR-, -NR-C1-6alquil-fenil-SO2-NR-, -SO2-, -NR-C1-6- alquil-fenil-C(O)-,
, ou L3 é ausente;
[0659] R13’ é selecionado do grupo que consiste em uma ligação, -C1-6alquileno-, -C3-8carbociclil-, -C3-8heterociclil-, -C1-6alquil-C6-14aril-, - C1-6alquil-C5-14heteroaril-;
[0660] cada R14 e R15 é independentemente selecionado do grupo que consiste em: hidrogênio, hidroxil, -NRR, -NRNR2, -C3-10carbociclil, -C1-6alquileno-C3-10carbociclil, -C3-10heterociclil, -C1-6alquileno-C3- 10heterociclil, -(CH2CH2O)1-6CH2CH2C(O)OR, -(CH2CH2O)1-6CH2CH2NRR, -C1-6alquil, C6-14aril, -C1-6alquileno-C6-14aril e -C5- 14heteroaril;
[0661] ou R14 e R15, juntos com o átomo ou átomos aos quais são ligados, formam um anel C3-10heterociclil,
[0662] em que R14, R15, ou ambos, ou um anel formado com R14 e R15, são opcionalmente substituídos com -(C(R)2)m-R18, em que cada R18 é independentemente selecionado de (i) -NRR, (ii) - C(NRR)(C(O)OR), (iii) -S-R, (iv) aril ou heteroaril opcionalmente subs tituído com um ou mais de halogênio, -CF3, -(C(R)2)m-NRR ou - (C(R)2)m -SO2NRR, (v) -SO2R, (vi) -S-S-Ci-ealquil-C(O)OR, (vii) - SO2NRR, (viii) -C(O)NRR, (ix) -C(O)OR, (x) -C4-6 cicloalquil opcionalmente substituído com -NRR, -SO2NRR ou -NR-C(O)(CH2)0-6NRR, (xi) -R, (xii) -OR, (xiii) -N(R)NRR, (xiv) -C(O)N(R)NRR, (xv) - (C(R)2)m-O-NRR e (xiv) -S-S-Ci-6alquil-NRR;
[0663] cada Ri4’ é independentemente selecionado do grupo que consiste em: uma ligação, -NR-, -C3-i0carbociclil-, -C3-i0heterociclil-, - (CH2CH2O)i-6CH2CH2C(O)OR’, -(CH2CH2O)i-6CH2CH2NR-, e -Ci- 6alquileno-,
[0664] em que Ri4’ é opcionalmente substituído com -(C(R)2)m-Ri8, em que cada Ri8 é independentemente selecionado de (i) -NRR, (ii) - C(NRR)(C(O)OR), (iii) -S-R, (iv) aril ou heteroaril opcionalmente substituído com um ou mais de halogênio, -CF3, -NRR ou -SO2NRR, (v) - SO2R, (vi) -S-S-Ci-6alquil-C(O)OR, (vii) -SO2NRR, (viii) -C(O)NRR, (ix) -C(O)OR, (x) -C4-6 cicloalquil opcionalmente substituído com -NRR, - SO2NRR ou -NR-C(O)(CH2)0-6NRR, (xi) -R, (xii) -OR, (xiii) -N(R)NRR, (xiv) -C(O)N(R)NRR, (xv) -(C(R)2)m-O-NRR e (xiv) -S-S-Ci-6alquil- NRR;
[0665] Ácido é um resíduo de aminoácido selecionado de - SCH2CH(COOH)(NH2), -NH(CH2)4CH(COOH)(NH2) e -C(O)(CH2)2CH(COOH)(NH2);
[0666] cada R é independentemente selecionado do grupo que consiste em: hidrogênio e -Ci-6alquil;
[0667] cada R' é independentemente selecionado de -H, Ci-C8 al- quil, Ci-C8 heteroalquil e aril;
[0668] cada R’’ é independentemente selecionado do grupo que consiste em: uma ligação e -Ci-6alquileno-; e
[0669] cada m é independentemente 0, i, 2 ou 3.
EXEMPLOS
[0670] Produção de Produto Natural
[0671] Os procedimentos a seguir definem a produção de “produtos naturais” úteis como cargas úteis na presente invenção. O termo “produto natural” denota que o produto é produzido através de um processo de fermentação, mas não sugere que estes produtos são conhecidos ou podem ser encontrados na natureza. Produtos naturais são notas abaixo com o prefixo “NP”.
Exemplo 1
[0672] Fermentação, Extração e Isolamento de: ácido [(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2.5]oct-5-il]acético (#NP1); ácido [(3S,5S,7S)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-1,6-dioxaspiro[2.5]oct-5-il]acético (#NP2); ácido [(2S,5S,6R)-6-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-5-hidroxi-4-metilidenotetrahidro-2H-piran-2- il]acético (#NP3); ácido [(2S,6S)-6-{(1E,3E)-5-[(2S,3S,5R,6R)-5- {[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-4-metilidenotetrahidro-2H-piran-2- il]acético (#NP4)
[0673] Etapal: Fermentação usando Pseudomonas sp. N°.2663,(Cepa FERM BP-3421):
[0674] Pseudomonas sp. N°.2663, (Cepa FERM BP-3421), foi adquirida do International Patent Organism Depositary (IPOD) no National Institute of Advanced Industrial Science and Technology (AIST Tsukuba, Central 6, 1-1-1, Higashi, Tsukuba, Ibaraki 305-8566, Japan). Subsequent estudos taxonômicos realizados por bioquímica (BBL Crystal Kit) e análise de sequência rRNA 16S revelaram que FERM BP-3421 foi um Burkholderia sp.
[0675] Isolados de colônia única foram cultivados por diluição e plaqueamento de uma cultura congelada de FERM BP-3421 de tipo selvagem em placas de ágar nutriente. Vários frascos Erlenmeyer de 250 mL contendo 50 mL de meio de semeadura (1% polipeptona, 0,5% extrato de levedura, 0,5% NaCl) foram inoculados com cultura cultivada de ágar e incubados a 30°C com agitação em 220 rpm por 18-20 horas. A cultura de semeadura foi inoculada em 500 mL de meio de produção (1% amido solúvel, 1% glicerina, 0,5% glicose, 1% HySoy Soypeptona, 0,5% licor de milho, 0,2% sulfato de amônio, 0,006% sulfato de magnésio.6H2O, 0,2% CaCO3, pH 7,0) por frasco Fernbach de 2,8 L sem defletores a 2,5% (v/v). A fermentação foi incubada a 25°C com agitação em 200 rpm por 72 horas.
[0676] Etapa2. Extração de caldo de fermentação: Ao final da fermentação da etapa 1 do exemplo 1, 50 g/L de resina DIAION HP-20 úmida foram adicionados ao sobrenadante da fermentação de produção e a mistura foi agitada a 100 rpm por 30 minutos. O HP-20 foi coletado por centrifugação e, em seguida, extraído com etil acetato em temperatura ambiente. Em mais detalhes, uma fermentação de 13 L de FERM BP-3421 foi realizada a 25oC por 72 horas de acordo com a etapa1 do Exemplo 1. Todo o caldo foi centrifugado a 3800 rpm por 30 minutos. As células foram descartadas e o sobrenadante foi misturado com resina HP20 úmida pré-lavada (260 g peso seco). A suspensão resultante foi agitada em um agitador em temperatura ambiente por 1 hora. A resina HP20 ligada a composto foi extraída duas vezes com etil acetato (1 L por vez) e a solução de etil acetato foi filtrada sobre Celite seguido por evaporação sob pressão reduzida para gerar um extrato bruto levemente colorido (2,4 g).
[0677] Etapa3: Isolamento e Caracterização de ácido [(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2.5]oct-5-il]acético (#NP1),; ácido [(3S,5S,7S)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-1,6-dioxaspiro[2.5]oct-5-il]acético (#NP2): O extrato bruto da etapa2 do exemplo 1 foi dissolvido em uma mistura de 1:1 sulfóxido de acetonitrila/dimetil (14 mL total). A solução viscosa foi filtrada e, em seguida, purificada por HPLC preparativa: (Coluna: Waters C18 DELTA PAK (WAT011801), 300 X 50 mm, 15 μm, 100 A; Fase Móvel A: 0,02% de ácido acético (v/v) em 1:1 acetonitrila/H2O; Fase Móvel B: 0,02% de ácido acético (v/v) em 3:1 acetonitrila/H2O e Fase Móvel C: 0,02% de ácido acético (v/v) em acetonitrila. Gradiente : 100% A por 5 min, 0% A a 100% B por 18 min e 100%, B a 100% C por 2 min, e 100% C por 2 min. Taxa de fluxo: 50 mL/min.). As frações com tempos de retenção de 13,5 e 18,0 min foram coletadas e liofili- zadas para gerar #NP1 (172,5 mg) e #NP2 (227,2 mg), respectivamente, como pós brancos. As frações com tempos de retenção de 14,8 min e 20,5 min foram também coletadas e liofilizadas para render dois pós cinzentos semi-purificados I e II.
[0678] #NP1.; HPLC (Protocolo N): tempo de retenção = 9,36 minutos (pureza 92,5%); HRESIMS (Protocolo O) m/z 536,2837 [M+H]+; 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 12,20 (br s, D2O permutável), 7,80 (d, J = 7,9, 1H, D2O permutável), 6,35 (dq, J = 6,0, 6,0, 1H), 6,32 (br d, J = 15,6, 1H), 6,10 (d, J = 11,2, 1H), 5,85 (dd, J = 11,8, 7,4, 1H), 5,58 (dd, J = 15,5, 5,8, 1H), 5,49 (br dd, J = 7,0, 7,0, 1H), 4,24 (m, 2H), 3,63 (m, 2H), 3,49 (ddd, J = 5,5, 5,5, 2,5, 1H), 3,24 (d, J = 6,0, 1H), 2,74 (d, J = 3,5, 1H), 2,57 (d, J = 3,5, 1H), 2,55 (dd, J = 16,4, 8,5, 1H), 2,46 (m, 1H), 2,28 (m, 1H), 2,18 (m, 1H), 1,97(s, 3H), 1,82 (m, 1H), 1,79 (m, 2H), 1,68 (s, 3H), 1,65 (m, 1H), 1,56 (m), 1,23 (d, J = 6,4, 3H), 1,06 (d, J = 6,5, 3H), 0,93 (d, J = 7,0, 3H),13C RMN (100 MHz, DMSO-d6) δ172,15, 169,66, 164,55, 142,76, 136,22, 133,88, 128,84, 123,73, 122,83, 79,90, 76,93, 74,88, 70,35, 68,10, 67,93, 57,36, 49,63, 46,39, 39,09, 35,21, 33,91, 31,71, 28,67, 21,02, 19,96, 17,79, 14,22, 12,41.
[0679] #NP2. HPLC (Protocolo N): tempo de retenção = 10,93 minutos (pureza 90,4%); HRESIMS (Protocolo O) m/z 520,2895 [M+H]+; 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 12,13 (br s, 1H, D2O permutável), 7,80 (d, J = 7,9, 1H, D2O permutável), 6,35 (dq, J = 6,0, 6,0, 1H), 6,27 (br d, J = 15,8, 1H), 6,10 (d, J = 11,2, 1H), 5,85 (dd, J = 11,8, 7,3, 1H), 5,57 (dd, J = 15,6, 5,8, 1H), 5,50 (br dd, J = 7,0, 7,0, 1H), 4,50 (ddd, J = 5,5, 5,5, 5,5, 1H), 4,29 (m, 1H), 3,63 (m, 2H), 3,48 (m, 1H), 2,61 (s, 2H), 2,58 (dd, J = 16,0, 8,5, 1H), 2,49 (m, 1H), 2,28 (m, 1H), 2,18 (m, 1H), 1,96 (s, 3H), 1,79 (m, 2H), 1,76 (m, 1H), 1,68 (s, 3H), 1,65 (m, 1H), 1,63 (m, 1H), 1,40 (dd, J = 11,5, 7,2, 1H), 1,24 (d, J = 6,4, 3H), 1,06 (d, J = 6,4, 3H), 0,94 (d, J = 7,0, 3H), 13C RMN (100 MHz, DMSO-d6), 172,88, 169,64, 164,56, 142,75, 135,37, 133,74, 128,99, 126,57, 122,84, 79,97, 74,90, 70,36, 68,16, 68,11, 54,71, 52,25, 46,40, 39,08, 37,22, 36,46, 35,23, 31,72, 28,71, 21,01, 19,95, 17,78, 14,23, 12,40.
[0680] Etapa 4: Isolamento de ácido [(2S,5S,6R)-6-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-5-hidroxi-4- metilidenotetrahidro-2H-piran-2-il]acético (#NP3).
[0681] O pó semi-purificado I isolado na etapa2 do exemplo 1 foi adicionalmente purificado por HPLC de fase reversa (Coluna: YMC- Pack-ODS-A, 250 X 30 mm, S-10 μm, 12 nm.: Fase Móvel A: 0,02% de ácido acético em água; Fase Móvel B: 0,02% de ácido acético em acetonitrila: Sistema de gradiente: 30% a 100% B por 23 min e mantido 100% B por 1 min. Taxa de fluxo: 20 mL/min) para gerar #NP3 (7,6 mg,) como um pó branco.
[0682] #NP3: HPLC (Protocolo N): tempo de retenção = 10,9 minutos (pureza 94,2%); HRESIMS (Protocolo O) m/z 520,2910 [M+H]+; 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 7,76 (d, J = 7,9, 1H, D2O permutável), 6,35 (dq, J = 6,4, 6,4, 1H), 6,22 (br d, J = 15,8, 1H), 6,10 (d, J = 11,0, 1H), 5,85 (dd, J = 11,8, 7,4, 1H), 5,57 (dd, J = 15,5, 5,8, 1H), 5,48 (br dd, J = 7,0, 7,0, 1H), 5,04 (br s, 1H), 4,80 (br s, 1H), 4,18 (m, 1H), 3,88 (dd, J = 5,8, 5,8, 1H), 3,63 (m, 2H), 3,49 (ddd, J = 6,0, 6,0, 2,5, 1H), 2,37 (m, 2H), 2,33 (m, 1H), 2,27 (m, 1H), 2,23 (m, 1H), 2,17 (m, 1H), 1,97 (s, 3H), 1,79 (m, 2H), 1,68 (s, 3H), 1,65 (m, 1H), 1,24 (d, J = 6,4, 3H), 1,06 (d, J = 6,5, 3H), 0,94 (d, J = 7,0, 3H). 13C RMN (100 MHz, DMSO-d6) δ 172,35, 169,57, 164,50, 144,64, 142,64, 136,08, 133,77, 128,74, 125,24, 122,81, 108,88, 79,95, 76,97, 74,84, 72,37, 69,47, 68,02, 46,34, 38,09, 36,97, 35,17, 31,67, 28,70, 20,95, 19,91, 17,72, 14,20, 12,34.
[0683] Etapa 5: Isolamento de ácido [(2S,6S)-6-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-4- metilidenotetrahidro-2H-piran-2-il]acético (#NP4).
[0684] O pó semi-purificado II isolado na etapa2 do exemplo 1 foi adicionalmente purificado por HPLC de fase reversa (Coluna: YMC- Pack-ODS-A, 250 X 30 mm, S-10 μm, 12 nm.: Fase Móvel A: 0,02% de ácido acético em água; Fase Móvel B: 0,02% de ácido acético em acetonitrila: Sistema de gradiente: 30% a 100% B por 23 min e manti- do 100% B por 1 min. Taxa de fluxo: 20 mL/min) para gerar #NP4 (12,2 mg) como um pó branco.
[0685] #NP4: HPLC (Protocolo N): tempo de retenção = 12,7 minutos (pureza 96,5%); HRESIMS (Protocolo O) m/z 504,2959 [M+H]+. 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 7,75 (d, J = 7,9, 1H, D2O permutável), 6,37 (dq, J = 7,5, 6,4, 1H), 6,23 (br d, J = 16,0, 1H), 6,10 (d, J = 11,8, 1H), 5,85 (dd, J = 11,8, 7,5, 1H), 5,53 (dd, J = 16,0, 5,6, 1H), 5,51 (dd, J = 6,5, 6,5, 1H), 4,80 (br s, 1H), 4,76 (br s, 1H), 4,32 (ddd, J = 5,6, 5,5, 5,5, 1H), 4,13 (m, 1H), 3,63 (m, 2H), 3,49 (ddd, J = 6,0, 6,0, 2,5, 1H), 2,38 (m, 2H), 2,36 (dd, J = 11,5, 5,0, 1H), 2,32 (m, 1H), 2,29 (m, 1H), 2,18 (br dd, J = 11,9, 6,5, 1H), 2,13 (dd, J = 11,5, 5,9, 1H), 2,00 (dd, J = 10,5, 7,0, 1H), 1,97 (s, 3H), 1,79 (m, 2H), 1,67 (s, 3H), 1,65 (m, 1H), 1,24 (d, J = 6,4, 3H), 1,05 (d, J = 6,3, 3H), 0,94 (d, J = 7,0, 3H). 13C RMN (100 MHz, DMSO-d6)D δ172,11, 169,61, 164,54, 142,69, 141,40, 135,72, 133,71, 129,01, 126,48, 122,83, 110,53, 79,94, 74,86, 71,99, 68,76, 68,06, 46,37, 38,97, 38,92, 38,70, 35,19, 31,70, 28,71, 20,97, 19,92, 17,74, 14,21, 12,36.
Exemplo 2
[0686] Fermentação, Extração e Isolamento de análogos de produto natural: (5R)-5-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-1,5-anidro-1-(carboximatil)-3-C-(clorometil)-2- desoxipentitol (#NP5); (6R)-6-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-1-desoxi-4-C-(hidroximatil)hex-2-ulopiranose (#NP6),; ácido (4-[(acetiloxi)metil]-6-{(1E,3E)-5-[(2S,3S,5R,6R)-5- {[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-4-hidroxitetrahidro-2H-piran-2- il)acético (#NP7); ácido [6-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-4-(clorometil)-4-hidroxitetrahidro-2H-piran-2- il]acético (#NP8); 4-C-[(acetiloxi)metil]-6-{(1E,3E)-5-[(2S,3S,5R,6R)-5- {[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-1-desoxihex-2-ulopiranose (#NP9); 6-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien- 1-il}-4-C-[({[6-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent- 2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3- dien-1-il}-4-(clorometil)-4-hidroxitetrahidro-2H-piran-2-il]acetil}oxi)metil]- 1-desoxihex-2-ulopiranose (#NP10); (2S,3Z)-5-{[(2R,3R,5S,6S)-2,5- dimetil-6-{(2E,4E)-3-metil-5-[(2S)-4-metil-6-oxo-3,6-dihidro-2H-piran-2- il]penta-2,4-dien-1-il}tetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#NP11).
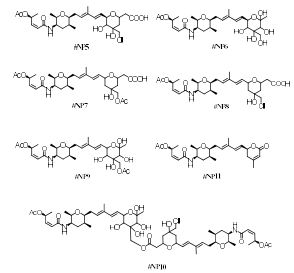
[0687] Etapa 1: Extrato sólido bruto (3,7 g) preparado como na etapa2, Exemplo 1 foi dissolvido em metanol e fracionado em uma coluna Sephadex LH20 usando metanol, com eluentes coletados em intervalos de 15 min usando um coletor de fração automático por um período de 15,0 horas (Total de 65 frações coletadas). Fração-19 deste foi adicionalmente purificada por HPLC de fase reversa (Coluna: YMC- Pack-ODS-A, 250 x 30 mm, S-10 um, 12 nm; Fase Móvel A: 0,2% de acetato de amônio (P/v); Fase Móvel B: 0,02% de ácido acético em acetonitrila; Gradiente: 30% B a 60% B por 20 minutos, a 100% B por 5 minutos e mantido a 100% B por 4 minutos e 100% B a 30% B por 2 minutos; Taxa de fluxo: 20 mL/min.) para render treze frações: Fração A (4,3-6,4 min), B (10,8-11,9 min), C (12,5-13,5 min), D (13,5-14,6 min), E (15,0-16,1 min), F (16,5-17,8 min), G (19,0-19,8 min), H (19,821,0 min), I (21,8-23,0 min), J (23,3-25,4 min), J1 (25,4-26,2 min), K (27,9-28,5 min), L (28,7-29,5 min).
[0688] Etapa2: Isolamento de (5R)-5-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,5-anidro-1-(carboximatil)-3-C- (clorometil)-2-desoxipentitol (#NP5): (6R)-6-{(1E,3E)-5-[(2S,3S,5R,6R)- 5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-1-desoxi-4-C-(hidroximatil)hex-2- ulopiranose (#NP6); ácido (4-[(acetiloxi)metil]-6-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-4- hidroxitetrahidro-2H-piran-2-il)acético (#NP7)
[0689] Fração D da etapa 1 do exemplo 2 foi adicionalmente purificada por HPLC de fase reversa (Coluna, Coluna: C18-Phenomenex; Luna 10μM; 250 x 10 mm.; Fase Móvel A: 0,2% de acetato de amônio em água (P/v); Fase Móvel B: 0,02% de ácido acético em acetonitrila; Gradiente: 25% B a 35% B por 5 minutos, a 45% B por 17 minutos, a 70% B por 2 minutos: Taxa de fluxo: 2,5 mL/minuto) e frações eluindo em 13, 14 e 15 minutos foram coletadas e liofilizadas.
[0690] Fração eluindo em 13 minutos rendeu #NP5: Rendimento: 1,0 mg: HRESIMS (Protocolo O) m/z 572,2614 (M+H)+, m/z 594,2438 (M+Na)+; 1H RMN (500 MHz, DMSO-d6, mult, J em Hz) δ 7,80 (d, J = 8,0, 1H), 6,36 (m, 1H), 6,23 (d, J = 15,8, 1H), 6,11 (dd, J = 1,3, 11,7, 1H), 5,86 (dd, J = 7,5, 11,6, 1H), 5,63 (dd, J = 5,6, 15,8, 4H), 5,47 (m, 1H), 4,21 (m, 1H), 4,10 (dd, J = 5,6, 8,3, 1H), 3,64 (m, 2H), 3,62 (d, J = 10,6, 1H), 3,49 (m, 1H), 3,42 (d, J = 10,6, 1H), 3,16 (d, J = 8,3, 1H), 2,68 (m, 1H), 2,57 (m, 1H), 2,30 (m, 1H), 2,19 (m, 1H), 1,98 (s, 3H), 1,83 (m, 2H), 1,80 (m, 2H), 1,69 (s, 3H), 1,65 (m, 1H), 1,24 (d, J = 6,5, 3H), 1,06 (d, J = 6,3, 3H), 0,95 (d, J = 7,3, 2H). 13C RMN (126 MHz, DMSO-d6) δ173,3, 170,1, 165,0, 143,1, 135,6, 134,4, 128,5, 126,8, 123,2, 80,2, 75,1, 71,7, 71,3, 70,0, 68,6, 68,2, 50,4, 46,6, 39,9, 35,2, 35,0, 31,9, 29,0, 21,0, 20,1, 17,9, 14,4, 12,5.
[0691] Etapa 3: Isolamento de 6R)-6-{(1E,3E)-5-[(2S,3S,5R,6R)-5- {[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-1-desoxi-4-C-(hidroximatil)hex-2- ulopiranose (#NP6): Fração coletada no tempo de retenção 14,0 minutos acima da etapa 2 do exemplo 2 foi adicionalmente purificada usando HPLC de fase reversa (Coluna: Chromolith: RP 18e, 100-10 mm.: Fase Móvel A: 0,2% de acetato de amônio em água (P/v); Fase Móvel B: 0,02% de ácido acético em acetonitrila; Gradiente: 25% B a 33% B por 20 minutos; taxa de fluxo: 2,5 mL/min) para gerar #NP6: Rendimento 4,0 mg, HRESIMS (Protocolo O) m/z 542.2948 (M+H)+; 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 7,77 (d, J = 8,0, 1H), 6,32 (m, 1H), 6,17 (d, J = 15,8, 1H), 6,05 (dd, J = 1,3, 11,6, 1H), 5,82 (dd, J = 7,5, 11,6, 1H), 5,57 (dd, J = 5,9, 15,8, 1H), 5,42 (m, 1H), 4,09 (m, 1H), 3,60 (m, 2H), 3,46 (m, 1H), 3,38 (d, J = 10,1, 1H), 3,28 (d, J = 10,1, 1H), 3,21 (br s, 1H), 3,17 (br s, 1H), 2,25 (m, 1H), 2,16 (m, 1H), 1,94 (s, 1H), 1,76 (m, 2H), 1,66 (s, 3H), 1,62 (m, 1H), 1,21 (d, J = 6,3, 3H), 1,17 (d, J = 8,5, 3H), 1,02 (d, J = 6,3, 3H), 0,91 (d, J = 7,3, 3H). 13C RMN (126 MHz, DMSO-d6) δ 174,1, 165,2, 143,5, 135,9, 134,6, 128,9, 127,4, 123,6, 98,8, 80,0, 77,3, 75,5, 69,7, 69,4, 69,2, 68,8, 59,6, 47,0, 35,8, 32,6, 29,5, 26,5, 21,7, 20,7, 18,5, 14,9, 13,1.
[0692] Etapa 4: Isolamento de ácido (4-[(acetiloxi)metil]-6-{(1E,3E)- 5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-4-hidroxitetrahidro-2H-piran-2-il)acético (#NP7): Fração coletada no tempo de retenção 15,0 minutos acima da etapa 2 do exemplo 2 foi adicionalmente purificada usando HPLC de fase reversa (YMC-Pack-ODS- A; 250 X 10 mm, S-5 um, 12 nm. Fase Móvel A: 0,2% de acetato de amônio em água (P/v); Fase Móvel B: 0,02% de ácido acético em ace- tonitrila; Gradiente: 30% B a 50% B por 20 minutos, a 95% B por 5 minutos: Taxa de fluxo: 2,5 mL/min) para render #NP7: HRESIMS (Protocolo O) m/z 580,3112 (M+H)+, m/z 602,2928 (M+Na)+; 1H RMN (500 MHz, DMSO-d6, mult, J em Hz) δ 7,80 (d, J = 8,0, 1H), 6,36 (m, 1H), 6,19 (d, J = 15,8, 1H), 6,11 (d, J = 11,6, 1H), 5,87 (dd, J = 7,5, 11,6, 1H), 5,49 (m, 1H), 5,48 (m, 1H), 4,41 (dd, J = 7,2, 12,8, 1H), 4,28 (d, J = 5,8, 1H), 3,79 (m, 2H), 3,65 (m, 1H), 3,64 (m, 1H), 3,49 (m, 1H), 2,60 (m, 1H), 2,56 (m, 1H), 2,29 (m, 1H), 2,19 (m, 1H), 2,02 (s, 3H), 1,97 (s, 3H), 1,80 (m, 2H), 1,67 (br s, 3H), 1,68-1,65 (br m, 2H), 1,51 (m, 1H), 1,45 (m, 1H), 1,40 (m, 1H), 1,24 (d, J = 6,5, 3H), 1,06 (d, J = 6,3, 3H), 0,94 (d, J = 7,3, 3H). 13C RMN (126 MHz, DMSO-d6) δ 173,4, 170,8, 170,1, 165,0, 143,2, 134,4, 134,2, 129,0, 128,9, 123,3, 80,3, 75,1, 68,3, 68,2, 68,0, 66,3, 71,1, 46,5, 39,4, 38,6, 35,4, 35,3, 31,8, 28,9, 21,1, 20,8, 20,0, 17,9, 14,4, 12,5.
[0693] Etapa 5: Isolamento ácido [6-{(1E,3E)-5-[(2S,3S,5R,6R)-5- {[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-4-(clorometil)-4-hidroxitetrahidro- 2H-piran-2-il]acético (#NP8), 4-C-[(acetiloxi)metil]-6-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1-desoxihex- 2-ulopiranose (#NP9), e 6-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-4-C-[({[6-{(1E,3E)-5-[(2S,3S,5R,6R)-5- {[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-4-(clorometil)-4-hidroxitetrahidro- 2H-piran-2-il]acetil}oxi)metil]-1-desoxihex-2-ulopiranose (#NP10):
[0694] Fração F da etapa 1 do exemplo 2 foi adicionalmente purificada por HPLC de fase reversa (C18-Phenomenex; Luna 250 x 10 mm. 10uM;; Fase Móvel A: 0,2% de acetato de amônio em água (P/v); Fase Móvel B: 0,02% de ácido acético em acetonitrila; Gradiente: 40% B a 45% B por 20 min, a 95% B por 5 min; taxa de fluxo: 2,5 mL/min: As frações eluindo em 8, 13 e 28 minutos foram coletadas e liofilizadas para dar:
[0695] #NP8 (fração eluindo em 8,0 min): Rendimento: 1,0 mg; HRESIMS (Protocolo O) m/z 556,2671 (M+H)+, m/z 578,2489 (M+Na)+; 1H RMN (500 MHz, DMSO-d6, mult, J em Hz) δ 7,80 (d, J = 8,0, 1H), 6,37 (m, 1H), 6,21 (d, J = 15,9, 1H), 6,11 (dd, J = 1,3, 11,6, 1H), 5,87 (dd, J = 7,5, 11,6, 1H), 5,50 (m, 1H), 5,48 (m, 1H), 4,39 (m, 1H), 4,30 (m, 1H), 3,65 (m, 2H), 3,50 (m, 1H), 3,48 (br s, 2H), 2,91 (dd, J = 8,9, 15,0, 1H), 2,60 (dd, J = 6,0, 15,0, 1H), 2,30 (m, 1H), 2,19 (m, 1H), 1,98 (s, 3H), 180 (m, 2H), 1,73 (m, 1H), 1,69 (br s, 3H), 1,65 (m, 1H), 1,58 (m, 1H), 1,55 (m, 1H), 1,43 (br dd, J = 13,1, 10,4, 1H), 1,25 (d, J = 6,5, 3H), 1,07 (d, J = 6,3, 3H), 0,95 (d, J = 7,3, 3H). 13C RMN (126 MHz, DMSO-d6) δ 173,4, 170,1, 165,0, 143,2, 134,5, 134,2, 128,9 (x2), 123,2, 80,2, 75,2, 68,7, 68,5, 68,2, 66,5, 54,6, 46,5, 39,9, 38,7, 35,6, 35,4, 31,9, 28,9, 21,1, 20,1, 17,9, 14,4, 12,5.
[0696] #NP9 (fração eluindo em 13 minutos): Rendimento: 1,0 mg: HRESIMS (Protocolo O) m/z 584,3066 (M+H)+, m/z 606,2887 (M+Na)+; 1H RMN (500 MHz, DMSO-d6, mult, J em Hz) δ 7,80 (d, J = 7,9, 1H), 6,36 (m, 1H), 6,25 (d, J = 15,7, 1H), 6,11 (dd, J = 1,0, 11,6, 1H), 5,87 (dd, J = 7,5, 11,6, 1H), 5,63 (dd, J = 6,1, 15,8, 1H), 5,48 (m, 1H), 4,19 (dd, J = 6,1, 9,6, 1H), 3,97 (d, J = 10,0, 1H), 3,88 (d, J = 10,0, 1H), 3,65 (m, 2H), 3,50 (m, 1H), 3,17 (br s, 1H), 3,15 (br s, 1H), 2,30 (m, 1H), 2,19 (m, 1H), 2,00 (s, 3H), 1,98 (s, 3H), 1,80 (m, 2H), 1,70 (s, 3H), 1,65 (m, 1H), 1,26 (br d, J = 1,6, 3H), 1,24 (br s, 3H), 1,06 (d, J = 6,2, 3H), 0,96 (d, J = 7,3, 3H). 13C RMN (126 MHz, DMSO-d6) δ 170,4, 170,1, 165,0, 143,1, 136,1, 134,4, 128,9, 126,4, 123,2, 98,5, 80,4, 75,4, 75,2, 69,2 (x2), 69,0, 68,2, 60,7, 46,5, 35,4, 31,9, 28,9, 25,9, 21,1 (x2), 20,0, 17,9, 14,4, 12,6.
[0697] #NP10 (fração eluindo em 23 minutos): Rendimento: 1,0 mg, HRESIMS (Protocolo O) m/z 562,2582 (M+2Na)2+, m/z 1101,5263 (M+Na)+; 1H RMN (500 MHz, DMSO-d6, mult, J em Hz) δ 7,79 (d, J = 7,9, 2H), 6,37 (m, 2H), 6,27 (d, J = 16,2, 1H), 6,21 (d, J = 15,6, 1H), 6,12 (d, J = 11,6, 1H), 6,11 (d, J = 15,3, 1H), 5,88 (dd, J = 7,5, 11,6, 2H), 5,64 (dd, J = 6,2, 15,8, 1H), 5,54-5,48 (m, 3H), 4,40-4,31 (m, 2H), 4,20 (dd, J = 6,2, 9,4, 1H), 4,04 (m, 1H), 3,90 (d, J = 10,0 Hz, 1H), 3,69-3,62 (m, 4H), 3,54-3,47 (m, 4H), 3,21 - 3,13 (m, 2H), 3,06 (dd, J = 8,7, 15,6, 1H), 2,73 (dd, J = 5,3, 15,7, 1H), 2,31 (m, 2H), 2,20 (m, 2H), 1,99 (s, 6H), 1,84-1,79 (m, 5H), 1,72 (s, 3H), 1,69 (s, 3H), 1,68-1,64 (m, 2H), 1,62 (m, 1H), 1,55 (d, J = 13,8 Hz, 1H), 1,44 (m, 1H), 1,27 (brs, 3H), 1,26 (brs, 6H), 1,08 (m, 6H), 0,96 (m, 6H). 13C RMN (126 MHz, DMSO-d6) δ 171,3, 170,0 (x2), 165,0 (x2), 143,2 (x2), 136,5, 134,6, 134,7, 134,4, 128,9 (x3), 126,6, 123,3 (x2), 98,4, 80,5 (x2), 75,4, 75,5 (x2), 69,5 (x2), 69,2, 68,6, 68,5 (x3), 66,5, 60,7, 54,8, 46,9 (x2), 40,0, 38,5, 35,9, 35,7 (x2), 32,3 (x2), 29,3, 29,2, 26,2, 21,5 (x2), 20,4 (x2), 18,2 (x2), 14,7 (x2), 12,9 (x2).
[0698] Etapa 6: Isolamento de (2S,3Z)-5-{[(2R,3R,5S,6S)-2,5- dimetil-6-{(2E,4E)-3-metil-5-[(2S)-4-metil-6-oxo-3,6-dihidro-2H-piran-2- il]penta-2,4-dien-1-il}tetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#NP11).
[0699] Fração K da etapa 1 do exemplo 2 foi adicionalmente purificada por HPLC de fase reversa (C18-Phenomenex; Luna 250 x 10 mm. 10uM; Phenomenex; Luna 250 x 10 mm. 10uM;; Fase Móvel A: 0,2% de acetato de amônio em água (P/v); Fase Móvel B: 0,02% de ácido acético em acetonitrila; Gradiente: 40 a 95% B por 15 min. taxa de fluxo: 2,5 mL/min) para gerar #NP11 Rendimento: 2,0 mg. HRESIMS (Protocolo O) m/z 460,2694 (M+H)+, m/z 482,2514 (M+Na)+; 1H RMN (500 MHz, DMSO-d6) δ ppm 1H RMN (500 MHz, DMSO-d6, mult, J em Hz) δ 7,82 (d, J = 6,9 Hz, 1H), 6,37 (m, 1H), 6,38 (m, 1H), 6,13 (dd, J = 1,2, 11,6, 1H), 5,88 (dd, J = 7,5, 11,6, 1H), 5,78 (s, 1H), 5,68 (dd, J = 6,7, 15,8, 1H), 5,62 (t, J = 7,0, 1H), 4,97 (m, 1H), 3,67 (m, 1H), 3,66 (m, 1H), 3,52 (m, 1H), 2,47 (m, 1H), 2,44 (m, 1H), 2,33 (m, 1H), 2,23 (m, 1H), 2,00 (s, 3H), 1,97 (s, 3H), 1,82 (m, 2H), 1,74 (s, 3H), 1,67 (m, 1H), 1,26 (d, J = 6,5, 3H),1,08 (d, J = 6,3, 3H), 0,97 (d, J = 7,1, 3H). 13C RMN (126 MHz, DMSO-d6) δ 170,1, 165,0, 164,4, 159,0, 143,1, 137,6, 133,9, 131,2, 124,4, 123,2, 115,6, 80,2, 75,2, 77,2, 68,0, 46,5, 35,3, 34,5, 32,1, 28,9, 22,3, 21,0, 20,1, 17,9, 14,3, 12,4.
Exemplo 3
[0700] Caracterização Filogenética Molecular de FERM BP-3421
[0701] Etapa 1: DNA genômico foi isolado a partir de uma cultura pura de FERM BP-3421 e o quase completo gene rRNA 16S foi amplificado por PCR usando iniciadores 8FPL (5’AGAGTTTGATCCTGGCTCAG3‘) (SEQ. ID N°.1) e 1492RPL (5’GGTTACCTTGTTACGACTT3‘) (SEQ. ID N°.2). Produtos PCR foram purificados com o kit DNA Clean e Concentrator™-25 (Zymo Re-search) e diretamente sequenciados para prover cobertura de filamento duplo com os seguintes iniciadores rRNA 16S: 8FPL, pC FWD (5’CTACGGGAGGCAGCAGTGGG3‘) (SEQ. ID N°.3), pC REV (5’CCCACTGCTGCCTCCCGTAG3‘) (SEQ. ID N°4), pD FWD (5’CAGCAGCCGCGGTAATAC3‘) (SEQ. ID N°.5), pD REV (5’GTATTACCGCGGCTGCTG3‘) (SEQ. ID N°.6), pF FWD (5’CATGGCTGTCGTCAGCTCGT3‘) (SEQ. ID N°.7), pF REV (5’ACGAGCTGACGACAGCCATG3‘) (SEQ. ID N°.8) e 1492RPL. A sequência de rRNA 16S de filamento totalmente duplo (SEQ ID N°.: 1) foi pesquisada em um banco de dados nacional (National Center for Biotechnology Information) para determinar a filiação taxonômica de FERM BP-3421 como um Burkmantidoeria sp. As sequências de rRNA 16S das cepas tipo Burkmantidoeria spp. mais proximamente relacionadas e a sequência de Burkmantidoeria sp. NRRL B50319 (cepa A396)(US20110207604A1 Asolkar et al., 2011), que compartilha 100% de identidade com FERM BP-3421, foram extraídas do GenBank. Um alinhamento de sequência múltipla foi realizado usando ClustalX (versão 1.81) e a posição filogenética de FERM BP-3421 em relação a outro Burkmantidoeria spp. foi determinada com métodos de triagem padrão, tais como TREECON (versão 1.3b).
[0702] FERM BP-3421 (SEQ. ID N°.9) AGAGTTTGATCCTGGCTCAGATTGAACGCTGGCGGCATGCCTTAC ACATGCAAGTCGAACGGCAGCACGGGTGCTTGCACCTGGTGGCG AGTGGCGAACGGGTGAGTAATACATCGGAACATGTCCTGTAGTGG GGGATAGCCCGGCGAAAGCCGGATTAATACCGCATACGATCTAC GGATGAAAGCGGGGGATCTTCGGACCTCGCGCTATAGGGTTGGC CGATGGCTGATTAGCTAGTTGGTGGGGTAAAGGCCTACCAAGGC GACGATCAGTAGCTGGTCTGAGAGGACGATCAGCCACACTGGGA CTGAGACACGGCCCAGACTCCTACGGGAGGCAGCAGTGGGGAAT TTTGGACAATGGGGGAAACCCTGATCCAGCAATGCCGCGTGTGT GAAGAAGGCCTTCGGGTTGTAAAGCACTTTTGTCCGGAAAGAAAT CCTTTGGGCTAATACCCCGGGGGGATGACGGTACCGGAAGAATA AGCACCGGCTAACTACGTGCCAGCAGCCGCGGTAATACGTAGGG TGCGAGCGTTAATCGGAATTACTGGGCGTAAAGCGTGCGCAGGC GGTTTGTTAAGACAGATGTGAAATCCCCGGGCTTAACCTGGGAAC TGCATTTGTGACTGGCAAGCTAGAGTATGGCAGAGGGGGGTAGA ATTCCACGTGTAGCAGTGAAATGCGTAGAGATGTGGAGGAATACCGATGGCGAAGGCAGCCCCCTGGGCCAATACTGACGCTCATGCAC GAAAGCGTGGGGAGCAAACAGGATTAGATACCCTGGTAGTCCAC GCCCTAAACGATGTCAACTAGTTGTTGGGGATTCATTTCCTTAGTA ACGTAGCTAACGCGTGAAGTTGACCGCCTGGGGAGTACGGTCGC AAGATTAAAACTCAAAGGAATTGACGGGGACCCGCACAAGCGGTG GATGATGTGGATTAATTCGATGCAACGCGAAAAACCTTACCTACC CTTGACATGGTCGGAATCCTGAAGAGATTCGGGAGTGCTCGAAAG AGAACCGATACACAGGTGCTGCATGGCTGTCGTCAGCTCGTGTC GTGAGATGTTGGGTTAAGTCCCGCAACGAGCGCAACCCTTGTCCT TAGTTGCTACGCAAGAGCACTCTAAGGAGACTGCCGGTGACAAAC CGGAGGAAGGTGGGGATGACGTCAAGTCCTCATGGCCCTTATGG GTAGGGCTTCACACGTCATACAATGGTCGGAACAGAGGGTTGCCA ACCCGCGAGGGGGAGCTAATCCCAGAAAACCGATCGTAGTCCGG ATTGCACTCTGCAACTCGAGTGCATGAAGCTGGAATCGCTAGTAA TCGCGGATCAGCATGCCGCGGTGAATACGTTCCCGGGTCTTGTA CACACCGCCCGTCACACCATGGGAGTGGGTTTTACCAGAAGTGG CTAGTCTAACCGCAAGGAGGACGGTCACCACGGTAGGATTCATGA CTGGGGTGAAGTCGTAACAAGGTAACC
[0703] A Figura 1 ilustra a relação filogenética determinada com sequências rRNA 16S quase completas de FERM BP-3421 para outro Burkmantidoeria spp. A árvore filogenética de ligação próxima foi enraizada com Burkmantidoeria pickettii e mostra valores de inicialização (com base em 100 repetições e superior a 50%) em seus respectivos nós. A barra de escala representa 0,02 substituição por nucleotídeo.
Exemplo 4
[0704] Fermentação, Extração e Isolamento de: ácido [(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2.5]oct-5-il]acético (#NP1); e ácido [(3S,5S,7S)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)- 4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-1,6-dioxaspiro[2.5]oct-5-il]acético (#NP2) com cepa projetada #1 de FERM BP-3421
[0705] Etapa1: Mineração de genoma para agrupamento de genes biossintéticos de spliceostatin:
[0706] O genoma de FERM BP-3421 foi sequenciado usando tecnologias de última geração (454 e Illumina). O agrupamento de genes biossintéticos para spliceostatins (Figura 2) foi inferido a partir da sequência de DNA por mineração de genoma (para uma revisão, ver Challis GL 2008 J Med Chem 51: 2618-2628) que nos levou a identificar uma rota híbrida de trans-aciltransferase (AT) policetídeo sintase (PKS)/ peptídeo sintetase não ribossômico (NRPS) (para uma revisão, ver J Piel 2010 Nat Prod Rep 27:996-1047 e referências ali). Mutantes knockout de gene PKS e NRPS não apresentaram nenhuma produção de spliceostatin detectável, confirmando o envolvimento destes genes na biossíntese de spliceostatin. Nossas descobertas estão em concordância com aquelas repostadas por Zhang F et al. (2011 J Am Chem Soc 133: 2452-62) e a terminologia de gene introduzida neste paper JACS é usada a partir de agora.
[0707] A Figura 2 é um grupo de gene biossintético para spliceos- tatins e rota biossintética proposta destacando etapas de hidroxilação catalisadas pelo citocromo P450 Fr9R e dioxigenase Fr9P dependente de Fe(II)/α-cetoglutarato. As setas no topo representam sequências de codificação de DNA de genes PKS-NRPS; genes auxiliares não são mostrados.
[0708] Etapa2: Geração da cepa de mutante knockout de dioxige-nase (fr9P) de FERM BP-3421 (Cepa #1)
[0709] Dois fragmentos de DNA longos de ~700-bp a montante e a jusante do ponto de substituição do gene foram amplificados por PCR (Pfu Ultra™ Polymerase, Promega) usando DNA genômico FERM BP- 3421 como template e pares de iniciador P1_diox (TGG CGA ACA GAT CGA GTT TG) (SEQ. ID N°.10) e P2_diox (CTT GCG GAG AAC TGT GAA TGC GCA ATA GAA GCG CTG TCA TGG AAT G) (SEQ. ID N°.11), e P3_diox (CCG AAA AGT GCC ACC TGA CGT CTA AGA TAA CTC GTG GAT ATT CGG CAA G) (SEQ. ID N°12) e P4_diox (AGA ATC CCG CGA TCC CAA C) (SEQ. ID N°.13); bases sublinhadas representam regiões de homologia para o marcador de resistência tetraciclina (tet). O marcador tet foi amplificado por PCR usando pEX18Tc (Schweizer HP 1998 Gene 212:77-86) como template e par de iniciador Ptet_f (TTG CGC ATT CAC AGT TCT C) (SEQ. ID N°.14) e Ptet_r (TCT rótulo ACG TCA GGT GGC AC) (SEQ. ID N°.15). Os três fragmentos foram montados por SOE-PCR (usando Pfu Ultra™ Polimerase, Promega) e ligados no sítio SmaI de pEX100T (Schweizer HP & Hoang TT 1995 Gene 158:15-22) para gerar plasmídeo pAE- PF12. pAE-PF12 foi transferido em FERM BP-3421 por conjugação de E. coli S17.1.Tetraciclina (25 μg/mL) foi usada para seleção de mutan- tes; sacarose 5% para contra-seleção da estrutura do vetor; e genta- micina (10 μg/mL) para remover E. coli após conjugação. Mutantes foram confirmados por colônia PCR (RED Taq®, Sigma) em três reações separadas usando pares de iniciador P1_diox/P4_diox, P1_diox/Ptet_r, e TP1_pEX100T (GGA CGA ATC GAA CTC AGG AAC TTG) (SEQ. ID N°.16) / TP2_pEX100T (CGA AGA GCG ATT GAG GAA AAG G) (SEQ. ID N°.17), gerando cepa #1.
[0710] Etapa3: Fermentação usando cepa projetada #1: Cepa projetada #1 foi cultivada em meio de semeadura (1% polipeptona, 0,5% extrato de levedura, 0,5% NaCl) contendo tetraciclina (25 mg/L) a 30°C e 220 rpm por ~24 horas. Uma segunda cultura de semeadura foi gerada por inoculação de meio de semeadura fresco contendo tetracicli- na (25 mg/L) com a primeira cultura de semeadura a 10% (v/v) e incubada a 30°C com agitação em 220 rpm por ~24 horas. 850 mL da cul- tura de semeadura foram usados para inocular 29 L de meio de produção (4% glicerina, 2% peptona de soja HySoy, 0,2% sulfato de amônio, 0,01% sulfato de magnésio.6H2O, 0,2% CaCO3) contidos em um Bior- reator de 30L (BIOSTAT® C plus, Sartorius BBI Systems). A fermentação foi realizada a 25°C por 5 dias. A agitação inicial foi ajustada a 344 rpm; fluxo de ar inicial a 1.3 slpm; DO foi controlado a 3% com agitação elevada.
[0711] Etapa4. Extração de caldo de fermentação: Ao final da fermentação da etapa 3 do exemplo 4, 1,5 kg de resina DIAION HP-20 úmida foi adicionado a todo o caldo e a mistura foi agitada durante a noite. A HP-20 foi coletada por filtração através de uma rede metálica de cunha de aço inoxidável de 50μm-150μm. A resina HP-20 ligada a composto foi extraída quatro vezes com etil acetato (3 L por vez, com agitação por 45 min). A resina foi, então, lavada (uma vez com 2 L metanol e 3 vezes com água DI abundante) e reutilizada para recaptura de composto ainda remanescente no filtrado aquoso, seguindo o mesmo procedimento descrito acima. O solvente dos extratos de etil acetato combinados foi removido por evaporação sob pressão reduzida para gerar um pó amarelo claro (137 g).
[0712] Etapa 5: Isolamento de ácido [(3R,5S,7R,8R)-7-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi-1,6- dioxaspiro[2.5]oct-5-il]acético (#NP1); e ácido [(3S,5S,7S)-7-{(1E,3E)- 5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6- dioxaspiro[2.5]oct-5-il]acético (#NP2): 1,7 g de extrato da etapa 4 do exemplo 4 foi dissolvido em um solvente misto de 2:1 DMF/ACN (22 mL total), filtrado, e, em seguida, purificado por HPLC de fase reversa (Waters ODS-A 50 x 300 mm, 15 um, 120 A, Fase Móvel A: 0,02% AcOH em água, Fase Móvel B: 0,02% AcOH em sistema de solvente de acetonitrila, Gradiente: 50% B por 2 min, a 75% B por 18 min; 100% B por 2 min. Taxa de fluxo: 50 mL/min; 5 injeções repetidas). As frações com tempos de retenção de 13,5 e 18,0 min foram coletadas e liofilizadas para gerar #NP1 (191 mg) e #NP2 (466 mg) respectivamente como pós brancos.
[0713] #NP1.; HPLC (Protocolo N): tempo de retenção = 9,38 mi nutos (pureza 98,5%)
[0714] #NP2.; HPLC (Protocolo N): tempo de retenção = 10,97 mi nutos (pureza 96,5%)
Exemplo 5
[0715] Fermentação, Extração e Isolamento de: ácido [(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2.5]oct-5-il]acético (#NP1) com cepa projetada #2 de Ferm FERM BP-3421
[0716] Etapa1: Geração de cepa projetada #2: Primeiramente, o marcador tet no vetor mini-CTX1 (Hoang TT et al. 2000 Plasmid 43:5972) foi substituído por neo (resistência a neomicina e canamicina) a partir de pCR2,1 (Invitrogen) por recombinação mediada por À-Red (Datsenko KA & Wanner BL. 2000 Proc Natl Acad Sci U S A 97:66405). Iniciadores usados foram P1_neo_pCR2,1 (GTT GGT TTG CGC ATT CAC AGT TCT CCG CAA GAA TTG ATT GCA AGG GCT AAA GGA AG) (SEQ. ID N°.18) e P2_neo_tet_CTX1_pCR2,1 (TCT TCC GCT TCC TCG CTC ACT GAC TCG CTG CGC TCG GTC ACG GAA ATG TTG AAT ACT CAT ACT C) (SEQ. ID N°.19); sequência sublinhadas representam regiões de homologia para recombinação mediada por À-Red. O vetor obtido foi nomeado pAE-PF24.
[0717] Um sistema indutível por PBAD/araC arabinose foi amplificado por PCR (Phusion® Hot Start polimerase, Finnzymes) usando pKD46 como template e par de iniciador P1_BADp_f (GCT CTA GAC ATC GAT TTA TGA CAA CTT GAC, XbaI sítio sublinhado) (SEQ. ID N°.20) e P2_BADp_r (CCC AAA ACG GGT ATG G) (SEQ. ID N°.21). O gene (incluindo o RBS putativo mas nenhum promotor) codificando para o gene do citocromo P450 (fr9R) contido no agrupamento de genes biossintéticos de spliceostatin foi amplificado por PCR (Phusion® Hot Start polimerase, Finnzymes) usando DNA genômico de FERM BP-3421 e par de iniciador P3_P450_BAD_f (CTA CTG TTT CTC CAT ACC CGT TTT GGG TTG GTT TTT GAA ATT GC, extensão para SOE-PCR sublinhado) (SEQ. ID N°22) e P4_P450_r (ATG GTG AAG CTT AAG TCG ACA ACC GGC ATT CC, HindlII sítio sublinhado) (SEQ. ID N°.23). Os dois fragmentos assim obtidos foram montados por SOE-PCR (Phusion® Hot Start polimerase, Finnzymes) e subse-quentemente ligados nos sítios SpeI e HindIII de pAE-PF24, gerando pAE-PF29. pAE-PF29 foi transferido na cepa projetada #1 por conjugação de E. coli S17.1. Canamicina (500 μg/mL) foi usada para seleção de mutantes; e gentamicina (10 μg/mL) para remover E. coli após conjugação. Mutantes foram confirmados por duas reações PCR de colônia (RED Taq®, Sigma) usando conjuntos de iniciador TP1_CTX1_marcador (GCA TTC ACA GTT CTC CGC AAG) (SEQ. ID N°.24) e TP2_CTX1_marcador (CTC GCT CAC TGA CTC GCT G) (SEQ. ID N°.25), e T3_mini-CTX1_f (GCA ATT AAC CCT CAC TAA AGG) (SEQ. ID N°.26) e MCS_mini-CTX1_r (CTA rótulo GGC GAA TTG GGT AC) (SEQ. ID N°.27), gerando cepa projetada #2.
[0718] Etapa2: Fermentação usando cepa projetada #2: Cepa projetada #2 foi cultivada em meio de semeadura (1% polipeptona, 0,5% extrato de levedura, 0,5% NaCl) contendo tetraciclina (25 mg/L) a 30°C e 220 rpm por ~24 horas. Uma segunda cultura de semeadura foi gerada por inoculação de meio de semeadura fresco contendo tetracicli- na (25 mg/L) com a primeira cultura de semeadura a 10% (v/v) e incubada a 30°C com agitação em 220 rpm por ~24 horas. A cultura de semeadura foi usada para inocular 550 mL de meio de produção (4% glicerina, 2% peptona de soja HySoy, 1,5% L-arabinose, 0,2% sulfato de amônio, 0,01% sulfato de magnésio.6H2O, 0,2% CaCO3) por frasco Fernbach de 2,8 L sem defletores a 2,5% (v/v). A fermentação foi incubada a 25°C com agitação em 200 rpm por 4 dias.
[0719] Etapa3. Extração de caldo de fermentação: Ao final da fermentação da etapa 2 do exemplo 5, 100 g/L de resina DIAION HP-20 úmida foram adicionados a ~6 L de fermentação de produção e a mistura foi agitada por 3 horas. A HP-20 foi coletada por filtração através de uma rede metálica de cunha de aço inoxidável de 50μm-150μm. A resina HP-20 ligada a composto foi extraída três vezes com etil acetato (2 L por vez). Em mais detalhes, cada extração foi realizada por transferência da resina para um frasco de ensaio, adicionando 2 L de etil acetato, agitação por 1 hora e filtragem através de uma rede metálica de cunha de aço inoxidável de 50μm-150μm. Solvente dos extratos de etil acetato combinados foi removido por evaporação sob pressão reduzida para gerar um extrato bruto amarelo claro (17,25 g).
[0720] Etapa 4: Isolamento de ácido [(3R,5S,7R,8R)-7-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi-1,6- dioxaspiro[2.5]oct-5-il]acético (#NP1), 0,12 g de extrato da etapa 3 do exemplo 5 foi dissolvido em um solvente misto de 2:1 DMF/ACN (22 mL total), filtrado, e, em seguida, purificado por HPLC de fase reversa (YMC ODS-A 30 x 250 mm, 10 um, 120 A, Fase Móvel A: 0,02% AcOH em água, Fase Móvel B: 0,02% AcOH em acetonitrila. Gradiente: 30% B por 2 min, a 100% B por 18 min; 100% B por 2 min. Taxa de fluxo: 20 mL/min). A fração com tempo de retenção de 15,0 min foi coletada e liofilizada para gerar #NP1 (73,6 mg) como um pó branco.
[0721] #NP1.; HPLC (Protocolo N): tempo de retenção = 9,36 mi nutos (pureza 92,5%)
[0722] Exemplo 6
[0723] Fermentação, Extração e Isolamento de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,5S,7S)-7-hidroxi-7-metil-1,6- dioxaspiro[2.5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#NP12); e ácido [(3S,5S,7S)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent- 2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3- dien-1-il}-1,6-dioxaspiro[2.5]oct-5-il]acético (#NP2) com cepa projetada #3 de FERM BP-3421
[0724] Etapa1: Geração de cepa projetada #3: Dois fragmentos de DNA longos de ~700-bp a montante e a jusante do ponto de substituição de gene foram amplificados por PCR (Pfu Ultra™ Polimerase, Promega) usando DNA genômico FERM BP-3421 como template e pares de iniciador P1_P450 (GCA TCC AAT CAC TTG AAC AGG) (SEQ. ID N°.28) e P2_P450 (CTT GCG GAG AAC TGT GAA TGC GCA AGC CAT CAT TCT CGA CAT TTC C) (SEQ. ID N°.29), e P3_P450 (CCG AAA AGT GCC ACC TGA CGT CTA AGA AGA TTG TGA CGG TAC TGA AGC) (SEQ. ID N°.30) e P4_P450 (AGA GAA CGA TCG CTC CAC AG) (SEQ. ID N°.31); bases sublinhadas representam regiões de homologia ao marcador de resistência à tetraciclina (tet). O marcador tet foi amplificado por PCR usando pEX18Tc (Schweizer HP 1998 Gene 212:77-86) como template e par de iniciador Ptet_f (TTG CGC ATT CAC AGT TCT C) (SEQ. ID N° 32) e Ptet_r (TCT rótulo ACG TCA GGT GGC AC) (SEQ. ID N°.33). Os três fragmentos foram montados por SOE-PCR (usando Pfu Ultra™ Polimera- se, Promega) e ligados no sítio SmaI de pEX100T (Schweizer HP & Hoang TT 1995 Gene 158:15-22) para gerar o plasmídeo pAE- PF11. pAE-PF11 foi transferido na FERM BP-3421 por conjugação a partir de E. coli S17.1.Tetraciclina (25 μg/mL) foi usada para seleção de mutantes; sacarose 5% para contra-seleção da estrutura do vetor; e gentamicina (10 μg/mL) para remover E. coli após conjugação. Mutantes foram confirmados em duas reações de PCR de colônia (RED Taq®, Sigma) usando pares de iniciador P1_P450/Ptet_r e TP1_pEX100T (GGA CGA ATC GAA CTC AGG AAC TTG) (SEQ. ID N°.34) / TP2_pEX100T (CGA AGA GCG ATT GAG GAA AAG G) (SEQ. ID N°.35), gerando cepa #3.
[0725] Etapa2: Fermentação usando cepa projetada #3: Cepa projetada #3 foi cultivada em meio de semeadura (1% polipeptona, 0,5% extrato de levedura, 0,5% NaCl) contendo tetraciclina (25 mg/L) a 30°C e 220 rpm por ~24 horas. Uma segunda cultura de semeadura foi gerada por inoculação de meio de semeadura fresco contendo tetracicli- na (25 mg/L) com a primeira cultura de semeadura a 10% (v/v) e incubada a 30°C com agitação em 220 rpm por ~24 horas. A cultura de semeadura foi usada para inocular 400 mL de meio de produção (4% glicerina, 2% peptona de soja HySoy, 0,2% sulfato de amônio, 0,01% sulfato de magnésio.6H2O, 0,2% CaCO3) a 2,5% (v/v) contida em um frasco Fernbach de 2,8 L sem defletores. A fermentação foi incubada a 25°C com agitação em 200 rpm por 5 dias.
[0726] Etapa3. Extração de caldo de fermentação: A cultura de produção da etapa 2 do exemplo 6 foi centrifugada por 30 min a 4,200 rpm para remover células. 50 g de resina DIAION HP-20 úmida foram adicionados ao sobrenadante (12,5% p/v) e a mistura foi agitada a 200 rpm por 1 h. A HP-20 ligada a composto foi coletada por centrifugação e, em seguida, extraída duas vezes com etil acetato (250 mL para cada extração). Após secagem, os extratos combinados com MgSO4 (que foram, então, removidos por filtração com papel Whatman), o sol- vente foi removido por evaporação sob pressão reduzida para gerar um extrato bruto levemente colorido.
[0727] Etapa4: Isolamento de ácido [(3S,5S,7S)-7-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6- dioxaspiro[2.5]oct-5-il]acético (#NP2) e (2S,3Z)-5-{[(2R,3R,5S,6S)-6- {(2E,4E)-5-[(3R,5S,7S)-7-hidroxi-7-metil-1,6-dioxaspiro[2.5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5- oxopent-3-en-2-il acetato (#NP12): Metade do extrato bruto da etapa3 do exemplo 6 foi purificada HPLC de fase normal preparativa: (Coluna: Princeton SFC 2-etilpiridina, 250 x 21.2 mm, 5 μm; Fase Móvel A: heptano; Fase Móvel B: etanol (desnaturado). Gradiente: 5% B por 1,5 min, a 100% B por 8,5 min, 100% B por 2 min, a 5% B por 0,5 min e 5% B por 2,5 min. Taxa de fluxo: 27 mL/min). As frações com tempos de retenção de 6,58 min e 8,18 min foram coletadas e liofilizadas para gerar #NP12 (163 mg, 89% puro como um pó amarelado bem claro), e #NP2 (205 mg, 89% puro por UV), respectivamente.
[0728] #NP12: HPLC (Protocolo P): tempo de retenção = 12,65 min (pureza 89%); LC/MS: m/z 474,2 [M+H+-H2O]+ e 514,2 [M+Na+]+
[0729] #NP2: HPLC (Protocolo P): tempo de retenção = 12,46 min (pureza 89%); LC/MS: m/z 520,2 [M+H+]+
[0730] Etapa 5: Isolamento de (2S,3Z)-5-{[(2R,3R,5S,6S)-6- {(2E,4E)-5-[(3R,5S,7S)-7-hidroxi-7-metil-1,6-dioxaspiro[2.5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5- oxopent-3-en-2-il acetato (#NP12): metade da fração de 6,58 min da etapa 4 do exemplo 6 foi purificada por HPLC de fase reversa: (Coluna: Phenomenex Luna C18, 150 x 21.2 mm, 5 μm; Fase Móvel A: água; Fase Móvel B: acetonitrila. Gradiente: 20% B por 1,5 min, a 70% B por 8,5 min, a 100% B por 2 min, a 20% B por 0,5 min. Taxa de fluxo: 27 mL/min). A fração com tempo de retenção de 8,25 min foi cole- tada e liofilizada para gerar #NP12 (28 mg) como um pó branco. #NP12.; HPLC (Protocolo N): tempo de retenção = 12,6 min (pureza 98,5%); HRESIMS (Protocolo O) m/z 492,296 [M+H]+; 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 7,78 (d, J = 8,0 Hz, 1H), 6,35 (m, 1H), 6,21 (d, J = 15,8, 1H), 6,11 (dd, J = 0,9, 11,7, 1H), 5,85 (dd, J = 11,6, 7,5 Hz, 1H), 5,53 (m, 1H), 5,49 (m, 1H), 5,41 (d, J = 1,6 Hz, OH), 4,64 (m, 1H), 3,65 (m, 1H), 3,64 (m, 1H), 3,49 (m, 1H), 2,45 (m, 2H), 2,30 (m, 1H), 2,20 (m, 1H), 1,98 (s, 3H), 1,96 (m, 1H), 1,81 (m, 3H), 1,69 (s, 3H), 1,65 (m, 1H), 1,31 (s, 3H), 1,25 (m, 1H), 1,25 (d, J = 6,3 Hz, 3H), 1,14 (m, 1H), 1,07 (d, J = 6,5 Hz, 3H), 0,95 (d, J = 7,3 Hz, 3H). 13C RMN (100 MHz, DMSO-d6) δ 169,6, 164,6, 142,1, 134,1, 133,7, 128,2, 127,1, 122,6, 95,3, 79,5, 74,3, 67,6, 66,7, 54,5, 48,4, 46,1, 41,4, 37,5, 35,0, 31,1, 29,6, 28,5, 20,8, 19,5, 17,8, 13,9, 12,2.
Exemplo 7
[0731] Fermentação, Extração e Produção de Estabelecimento de: ácido [(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2.5]oct-5-il]acético (#NP1); ácido [(3S,5S,7S)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-1,6-dioxaspiro[2.5]oct-5-il]acético (#NP2) usando Burkmantidoeria sp. MSMB 43
[0732] Etapa 1: Fermentação usando Burkmantidoeria sp. MSMB 43: Burkmantidoeria sp. (nome proposto “Burkmantidoeria hump- tydooensis”) MSMB 43 foi adquirida do Centers for Disease Control and Prevention (CDC) e Menzies School of Health Research. MSMB 43 foi cultivada em placas de ágar nutriente de uma criopreservação e incubada a 30°C por 48 horas. A cultura cultivada de ágar foi inoculada em um tubo de cultura de 25 x 150 mm contendo 10 mL de meio de semeadura (1% polipeptona, 0,5% extrato de levedura, 0,5% NaCl). A cultura de semeadura foi incubada a 30°C com agitação em 220 rpm por 18-20 horas. A cultura de semeadura foi inoculada em 50 mL de meio de produção (1% amido solúvel, 1% glicerina, 0,5% glicose, 1% peptona de soja HySoy, 0,5% licor de milho, 0,2% sulfato de amônio, 0,006% sulfato de magnésio.6H2O, 0,2% CaCO3, pH 7,0) por frasco Erlenmeyer de 250 mL a 2,5% (volume/volume). As fermentações foram incubadas a 25°C com agitação em 200 rpm por 72 horas.
[0733] Etapa 2: Análise LC-MS de fermentações.
[0734] Fermentações foram centrifugadas para peletizar as células, e os sobrenadantes filtrados através de membranas de fluoreto de polivinilideno de 0,22 μm. Uma porção de cada sobrenadante foi misturada com dimetil sulfóxido (10:1) e analisada por LC-MS usando um instrumento Acuity UPLC (Waters): Coluna: XBridge C18, 4.6X150 mm, 3.5 uM Fase Móvel A: 0,1% ácido fórmico em água (v/v); Fase Móvel B: 0,1% ácido fórmico em acetonitrila (v/v); Gradiente 5% a 100% B por 12,0 minutos; 100% B por 3,0 minutos (Inj. Volume: 5.0 uL. No dia cinco das fermentações, MSMB43 produziu NP1 e NP2 a uma titulação de 150 mg/L de e 50 mg/L respectivamente como evidente pelo tempo de retenção e dados de espec. de massa.
[0735] NP1:: m/z: 535,9 (M+H)+, Tempo de retenção: 13,31 min.
[0736] NP2: m/z: 519,9 (M+H)+Tempo de retenção: 14,58 min
[0737] Procedimentos Experimentais Sintéticos
[0738] Experimentos foram geralmente realizados sob atmosfera inerte (nitrogênio ou argônio), particularmente, em casos em que reagentes sensíveis a umidade ou oxigênio ou intermediários foram empregados. Reagentes e solventes comerciais foram geralmente usados sem purificação adicional, incluindo solventes anidros quando apropriado (geralmente produtos Sure-SealTM de Aldrich Chemical Company, Milwaukee, Wisconsin). Dados de espectrometria de massa são reportados tanto de espectrometria de massa por cromatografia líquida (LCMS) ou ionização químia por pressão atmosférica (APCI). Permutas químicas para dados ressonância magnética nuclear (NMR) são expressos em partes por milhão (ppm, δ) referenciados a picos residuais dos solventes deuterados empregados.
[0739] Para procedimentos referenciais de síntese em outros Exemplos ou Métodos, um Protocolo de reação (comprimento de reação e temperatura) pode variar. Em geral, as reações foram seguidas por cromatografia de camada fina, LCMS ou HPLC, e submetidas a trabalho quando apropriado. Purificações podem variar entre experimentos: em geral, solventes e as razões de solvente usados para elu- entes/gradientes foram escolhidos para prover tempos de retenção apropriados. A menos que de outra forma especificado, frações de HPLC de fase reversa foram concentradas através de liofiliza- ção/liofilização. Compostos finais e intermediários foram armazenados a (0°C) ou temperatura ambiente em frascos fechados ou frascos sob nitrogênio.
[0740] Nomes de compostos foram gerados com software ACD Labs.
[0741] Condições de HPLC Usadas para Análise
[0742] Protocolo AA e AB: Coluna: Phenomenex Luna C18 (2), 150 x 3.0 mm, 5μm; Fase Móvel A: 0,02% ácido trifluoroacético em água (v/v); Fase Móvel B: 0,02% ácido trifluoroacético em acetonitrila (v/v); Gradiente: 5% a 100% B (por 10 minutos)A ou (por 20 minutos)B; Taxa de fluxo: 0,75 mL/minuto. Temperatura: não controlada; Detecção: DAD 215, 254 nm; Volume de injeção 10 μL; Instrumento: HP 1100.
[0743] Protocolo B: Coluna: Waters Sunfire C18, 50x 4,6 mm, 5 μm; Fase Móvel A: 0,05% ácido fórmico em água (v/v); Fase Móvel B: 0,05% ácido fórmico em acetonitrila (v/v); Gradiente: 5% a 95% B por 4 minutos, Mantido a 95% B por 1 minuto. Taxa de fluxo: 2,0 mL/min. Temperatura: temperatura ambiente; Detecção: DAD 215 nm; Volume de injeção 4 μL; Instrumento: Espectrômetro de Massa Waters LC e ZQ.
[0744] Protocolo C: Coluna: Waters Acquity UPLC HSS T3, C18, 2,1 x 50 mm, 1,7 μm; Fase Móvel A: 0,1% ácido fórmico em água (v/v); Fase Móvel B: 0,1% ácido fórmico em acetonitrila (v/v); Gradiente: 5% B por 0,1 minuto, 5% a 95% B por 2,5 minutos, 95% B por 0,35 minuto; Taxa de fluxo: 1,25 mL/minuto. Temperatura: 60°C; Detecção: 200- 450nm; MS (+) faixa 100-2000 daltons; Volume de injeção: 5 μL; Instrumento: Waters Acquity.
[0745] Protocolo D: Coluna: Waters Acquity UPLC HSS T3, C18, 2,1 x 50 mm, 1,7μm; Fase Móvel A: 0,1% ácido fórmico em água (v/v); Fase Móvel B: 0,1% ácido fórmico em acetonitrila (v/v); Gradiente: 5% B por 0,1 minuto, 5% a 95% B por 1,5 minuto, 95% B por 0,35 minuto; Taxa de fluxo: 1,25 mL/minuto. Temperatura: 60°C; Detecção: 200- 450nm; MS (+) faixa 100-2000 daltons; Volume de injeção: 5 μL; Instrumento: Waters Acquity.
[0746] Protocolo E: Coluna: Phenomenex Luna C18 (2), 150 x 3,0 mm, 5 μm; Fase Móvel A: 0,02% ácido trifluoroacético em água (v/v); Fase Móvel B: 0,02% ácido trifluoroacético em acetonitrila (v/v); Gradiente: 0% a 100% B por 23,5 minutos; Taxa de fluxo: 1,5 mL/minuto. Temperatura: não controlada; Detecção: DAD 210 nm; Volume de injeção: 10 μL; Instrumento: HPLC Agilent 1100.
[0747] Protocolo F: Coluna: Phenomenex Luna C18 (2), 150 x 3,0 mm, 5 μm; Fase Móvel A: 0,1% ácido fórmico em água (v/v); Fase Móvel B: 0,1% ácido fórmico em acetonitrila (v/v); Gradiente: 5% B por 1,5 minuto, 5% a 100% B por 8,5 minutos, em seguida, 100% B por 1 minuto; Taxa de fluxo: 0,75 mL/minuto. Temperatura: 45°C; Detecção: DAD 215 nm, 254 nm; MS (+) faixa 150-2000 daltons; Volume de injeção: 10 μL; Instrumento: LCMS Agilent 1200.
[0748] Protocolo G: Coluna: Atlantis dC18, 50 x 4,6 mm, 5 μm; Fa- se Móvel A: 0,05% ácido trifluoroacético em água (v/v); Fase Móvel B: 0,05% ácido trifluoroacético em acetonitrila (v/v); Gradiente: 5% a 95% B por 4,0 minutos, linear; em seguida, mantido a 95% B por 1 minuto. Taxa de fluxo: 2 mL/minuto. Temperatura: temperatura ambiente; Detecção: DAD 215 nm; MS (+) faixa 160 -1000 daltons; Volume de injeção 4 uL; Instrumento: PDA Waters 996.
[0749] Protocolo H: Coluna: Phenomenex Luna C18 (2), 150 x 3,0 mm, 5 μm; Fase Móvel A: água ; Fase Móvel B: acetonitrila; Gradiente: 5% B por 1,5 minuto, 5% a 100% B por 8,5 minutos, em seguida, 100% B por 1 minuto; Taxa de fluxo: 0,75 mL/minuto. Temperatura: 25°C; Detecção: DAD 215 nm, 254 nm; MS (+) faixa 150-2000 daltons; Volume de injeção: 10 μL; Instrumento: LCMS Agilent 1200.
[0750] Protocolo I: Coluna: Xtimate C18, 2,1 x 30 mm, 3 μm; Fase Móvel A: 0,1% ácido trifluoroacético em água (v/v); Fase Móvel B: 0,1% ácido trifluoroacético em acetonitrila (v/v); Gradiente 0% a 60% B por 0,9 minuto, 60% B por 0,6 minuto; 100% B por 0,5 minuto; Taxa de fluxo: 1,2mL/minuto. Detecção: DAD 220 nM; Temperatura: 25°C; Volume de injeção: 1 μL; Instrumento: Agilent.
[0751] Protocolo J: Coluna: Xtimate C18, 2,1 x 30 mm, 3 μm; Fase Móvel A: 0,1% ácido trifluoroacético em água (v/v); Fase Móvel B: 0,1% ácido trifluoroacético em acetonitrila (v/v); Gradiente: 10% a 80% B por 0,9 minuto, 80% B por 0,6 minuto; 100% B por 0,5 minuto; Taxa de fluxo: 1,2 mL/minuto. Detecção: DAD 220 nM; Temperatura: 25°C ; Volume de injeção: 1 μL; Instrumento: Agilent.
[0752] Protocolo K: Coluna: Phenomenex Luna PFP, 100 x 3 mm, 5 μm; Fase Móvel A: 0,05% ácido fórmico em água (v/v); Fase Móvel B: 0,05% ácido fórmico em acetonitrila (v/v); Gradiente: 5% a 95% B por 9 minutos, Mantido a 95% B por 1 minuto. Taxa de fluxo: 1,0 mL/ min. Temperatura: temperatura ambiente; Detecção: DAD 215 nm; Volume de injeção: 4 μL; Instrumento: Espectrômetro de Massa Waters LC e ZQ.
[0753] Protocolo L: Coluna: Phenomenex Gemini-NX, C18, 4,6 mm x 50 mm, 110A, 3μm, Fase Móvel A: 0,1% ácido fórmico em água (v/v); Fase Móvel B: 0,1% ácido fórmico em acetonitrila (v/v); Gradiente 5%; a 100% B por 0.0 - 4,10 min; mantido a 100% B de 4,10 - 4,50min; Taxa de fluxo: 1,25 mL/minuto. Temperatura: 60°C; Detecção: 200-450nm; MS (+) faixa 100-2000 daltons; Volume de injeção: 5 μL; Instrumento: Waters Acquity.
[0754] Protocolo M: Coluna: Phenomenex Gemini-NX, 4,6 mm x 50 mm, C18, 3 μm, 110A; Fase Móvel A : 0,1% ácido fórmico em água (v/v); Fase Móvel B: 0,1% ácido fórmico em acetonitrila (v/v); Gradiente: 5% a 100% B por 4,10 minutos, mantido a 100% B por 0,4 minuto, em seguida, 100% a 5% B por 0,5 minuto; Taxa de fluxo: 1,5 mL/minuto. Temperatura: 60°C; Detecção: HP1100 DAD (1315A), digitalização 200-450nm; intervalo de 1 nm; MS ESI(+/-),digitalização 1001200 m/z, tempo de digitalização 0,5 segundo, Centróide; Volume de injeção: 5 μL; Instrumento: HPLC Pump, DAD Detector, Coluna Oven de Agilent Technologies, Wilmington, DE; Auto-amostrador e detector MS de águas Corporation, Milford, MA; Detector ELS de Varian medical devices, Palo Alto, CA.
[0755] Protocolo N: Coluna: YMC ODS-A, 4,6 x 150 mm, 5 μm; Fase Móvel A: 0,01% ácido trifluoroacético em água (v/v); Fase Móvel B: 0,01% ácido trifluoroacético em acetonitrila (v/v); Gradiente: 10% a 100% B por 15 minutos; Taxa de fluxo: 1,0 mL/minuto. Temperatura: não controlada; Detecção: DAD 230 nm; Volume de injeção: 5 μL; Instrumento: Agilent 1100 HPLC.
[0756] Protocolo O: Espectro de Massa com Ionização por Pulverização de Alta Resolução (HRESIMS) foi obtido usando um espectrô- metro de massa Bruker (Billerica, MA) APEXII FTICR equipado com um magneto supercondutor ativamente protegido Tesla 9.4 (Magnex Scientific Ltd., UK), uma fonte externa Bruker APOLLO ESI, e um laser Synrad 50W CO2 CW. A amostra foi injetada por fluxo no espectrôme- tro de massa com solvente transportador que consiste em 1:1 (v:v) água:acetonitrila (0,25% ácido fórmico) a uma taxa de fluxo de 50 μL/min. Software Bruker Xmassa foi usado para aquisição de dados e análise. O espectro de massa foi externamente calibrado usando HP tuning mix.
[0757] Protocolo P: Coluna: YMC ODS-A, 4,6 x 150 mm, 5 μm; Fase Móvel A: 0,01% ácido trifluoroacético em água (v/v); Fase Móvel B: 0,01% ácido trifluoroacético em acetonitrila (v/v); Gradiente: 10% a 100% B por 19 minutos; Taxa de fluxo: 1,0 mL/minuto. Temperatura: não controlada; Detecção: DAD 230 nm; Volume de injeção: 5 μL; Instrumento: Agilent 1100 HPLC.
[0758] Protocolo Q: Coluna: Coluna: Agilent Poroshell 300SB-C8, 75 x 2,1 mm, 2,6 μm; Fase Móvel A: 0,1% ácido fórmico em água (v/v); Fase Móvel B: 0,1% ácido fórmico em acetonitrila (v/v); Gradiente: 20% B a 45% B por 4 minutos; Taxa de fluxo: 1,0 mL/minuto. Temperatura: 60°C; Detecção: 220 nm; MS (+) faixa 400-2000Da; Volume de injeção: 10 μL; Instrumento: Agilent 1100 LC, MS Waters Micromas- sZQ. Desconvolução foi realizada usando MaxEnt1.
[0759] Condições de HPLC Usadas para Purificação
[0760] Método A: Coluna: Phenomenex Gemini, C18, 30 x 100 mm, 5 μm; Fase Móvel A: 0,02% de ácido acético em água (v/v); Fase Móvel B: 0,02% de ácido acético em acetonitrila (v/v); Gradiente: variável, Gradiente crescente de B em A por 15-20 minutos; Taxa de fluxo: 20 mL/minuto. Temperatura: não controlada; Detecção: DAD 215 nm, 254 nm; Volume de injeção: variável; Instrumento: Gilson.
[0761] Método B*: Coluna: YMC ODS- A, 30 x 250 mm, 10 μm; Fase Móvel A: 0,02% de ácido acético em água (v/v); Fase Móvel B: 0,02% de ácido acético em acetonitrila (v/v); Gradiente: variável, Gra- diente crescente de B em A por 15-20 minutos; Taxa de fluxo: 20 mL/minuto. Temperatura: não controlada; Detecção: DAD 230 nm; Volume de injeção: variável, 0,5-2 mL; Instrumento: HPLC preparativa Varian ProStar Model 330.
[0762] Método C*: Coluna: Phenomenex Luna C18(2), 150 x 21,2 mm, 5 μm; Fase Móvel A: Água; Fase Móvel B: acetonitrila; Gradiente: variável, Gradiente crescente de B em A por 10 minutos; Taxa de fluxo: 27 mL/ minuto. Temperatura: temperatura ambiente; Detecção: DAD 210-360 nm; MS (+) faixa 150-2000 daltons; Instrumento: LCMS Waters Fração Lynx.
[0763] Método D*: Coluna: Waters Sunfire, C18, 19x100 mm, 5 μm; Fase Móvel A: 0,05% ácido fórmico em água (v/v); Fase Móvel B: 0,05% ácido fórmico em acetonitrila (v/v); Gradiente: variável, Gradiente crescente de B em A por 10-20 minutos; Taxa de fluxo: 25 mL/minuto. Detecção: DAD 215 nm MS (+) faixa 160-1000 daltons; Instrumento: Waters FraçãoLynx.
[0764] Método E: Coluna: Waters Sunfire,C18, 19x100 mm, 5 μm; Fase Móvel A: 0,05% ácido trifluoroacético em água (v/v); Fase Móvel B: 0,05% ácido trifluoroacético em acetonitrila (v/v); Gradiente: 10 a 50% B por 8,5 minutos, 50 a 100% B por 0,5 minutos, mantido a 100% B por 1 minuto. Taxa de fluxo: 25 mL/minuto. Detecção: DAD 215 nm MS (+) faixa 160-1000 daltons; Instrumento: Waters FraçãoLynx.
[0765] Método F*: Coluna : Waters C18 DELTA PAK (WAT011801), 300 X 50 mm, 15 μm; Fase Móvel A: 0,02% de ácido acético em água (v/v); Fase Móvel B: 0,02% de ácido acético em ace- tonitrila (v/v); Gradiente: variável, Gradiente crescente de B em A por 15-20 minutos; Taxa de fluxo: 50 mL/minuto. Temperatura: não controlada; Detecção: DAD 230 nm; Volume de injeção: variável, 0,5-5 mL; Instrumento: HPLC preparativa Varian ProStar Model 330.
[0766] Método G*: Coluna: YMC ODS-A, 50 x 300 mm, 12 μm, 120 A. Fase Móvel A: 0,02% de ácido acético em água (v/v); Fase Móvel B: 0,02% de ácido acético em acetonitrila (v/v); Gradiente: 40% B por 3 minutos, 40-100% B por 20 minutos e 100% B por 3 minutos. Taxa de fluxo; 20 mL/minuto. Temperatura: não controlada; Detecção: DAD 230 nm; Volume de injeção: variável, 0,5-5 mL; Instrumento: HPLC preparativa Varian ProStar Model 330.
[0767] Método H: Coluna: Cromolith RP-18e 100-10 mm. Fase Móvel A: 0,02% de ácido acético em água (v/v); Fase Móvel B: 0,02% de ácido acético em acetonitrila (v/v); Gradiente: 20-55% B por 30 minutos, 55-100% B por 4 min, 100-20% B por 2 min e 20% B por 2 minutos. Taxa de fluxo; 2,5 mL/minuto. Temperatura: não controlada; Detecção: DAD 230 nm; Volume de injeção: variável, 0,025-0,1 mL; Instrumento: HPLC analítica Agilent 1100.
[0768] Método I: Coluna: C18 semiprep YMC-Pack ODS-A 250x10 mm (S-5 μm, 12 nm). Fase Móvel A: 0,02% de ácido acético em água (v/v); Fase Móvel B: 0,02% de ácido acético em acetonitrila (v/v); Gradiente: 18-25% B por 22 minutos, 25-95% B por 1 min, 95% B por 4 min, 95-18% B por 1 min e 18% B por 6 minutos. Taxa de fluxo: 2,5 mL/minuto. Temperatura: não controlada. Detecção: DAD 230 nm. Volume de injeção: variável, 0,025-0,1 mL. Instrumento: HPLC analítica Agilent 1200.
[0769] Método J: Coluna: C18 semiprep YMC-Pack ODS-A 250x10 mm (S-5 μm, 12 nm). Fase Móvel A: 0,02% de ácido acético em água (v/v); Fase Móvel B: 0,02% de ácido acético em acetonitrila (v/v); Gradiente: 20-30% B por 30 minutos, 30-95% B por 1 min, 95% B por 4 min, 95-20% B por 2 min e 20% B por 6 minutos. Taxa de fluxo: 2,5 mL/minuto. Temperatura: não controlada. Detecção: DAD 230 nm. Volume de injeção: variável, 0,025-0,1 mL. Instrumento: HPLC analítica Agilent 1200.
[0770] Método K: Coluna: Cromolith RP-18e 100-10 mm. Fase Móvel A: água (v/v); Fase Móvel B: acetonitrila (v/v); Gradiente: 30- 65% B por 20 minutos, 65-95% B por 1 min, 95-30% B por 2 min. Taxa de fluxo; 2,5 mL/minuto. Temperatura: não controlada; Detecção: DAD 230 nm; Volume de injeção: variável, 0,025-0,1 mL; Instrumento: HPLC analítica Agilent 1200.
[0771] Em alguns casos, algumas alterações mínimas a condições de HPLC de purificação foram feitas, tais como, mas não limitadas a, uma alteração no gradiente, comprimento de gradiente e taxa de fluxo que é indicada pelo símbolo *.
[0772] Procedimentos Gerais
[0773] Procedimento Geral A: Preparação de N hidroxissuccinimida (NHS) éster ativado.
[0774] A uma solução de 0,i M do ácido em tetrahidrofurano a 0°C, foi adicionado N,N'-diciclohexilcarbodiimida (DCC) (2,2 eq.) seguido por N-hidroxi succinimida (2,2 eq.) e a reação foi deixada aquecer para temperatura ambiente e agitada. O progresso da reação foi monitorado por LC-MS (ou HPLC ou TLC); a reação estava geralmente completa em i-72 horas. Os solventes foram removidos sob pressão reduzida e o resíduo foi purificado por cromatografia de fase reversa para gerar o N-hidroxissuccinimida éster desejado.
[0775] Procedimento Geral B: Preparação de amidas a partir de NHS ésteres.
[0776] A um (0,1M) do N-hidroxissuccinimida éster (1 eq.) seja em tetrahidrofurano, N,N-dimetilformamida, ou N,N-Dimetilacetamida a 0°C, foi adicionada a amina (1 a 10 eq.). O progresso da reação foi monitorado por LC-MS (ou HPLC ou TLC); a reação estava geralmente completa em 1-72 horas. A mistura de reação foi concentrada in vacuo e o resíduo foi purificado por cromatografia de fase reversa para dar o produto de amida desejado.
[0777] Procedimento Geral C: Preparação de pentafluorofenil (PFP) ester
[0778] A uma solução de 0,05M do ácido em tetrahidrofurano a 0°C, foi adicionado DCC (1 eq.) seguido por uma solução de pentaflu- rofenol (2 a 4 eq.) dissolvida em tetrahidrofurano (0.3 M). A reação foi aquecida para temperatura ambiente e agitada. O progresso da reação foi monitorado por LC-MS (ou HPLC ou TLC); a reação estava geralmente completa em 1-48 horas. A reação foi concentrada sob pressão reduzida e o resíduo foi purificado por cromatografia de fase reversa para prover o pentaflurofenil (PFP) éster desejado.
[0779] Procedimento Geral D: Protocolo de Biblioteca para Preparação de Amidas a partir de NHS ester. A amina (1 eq.) foi dissolvida em tetrahidrofurano (1 mL, 0,04 M) e N,N-diisopropiletilamina (5 eq.) foi adicionado seguido por metanol (0,2 mL). Toda a solução foi, então, adicionada em gotas a uma solução resfriada (0°C) do N-hidroxi succi- nimida éster (1 eq.) dissolvido em tetrahidrofurano (1 mL, 0,04 M). A reação foi agitada a (0°C) por 30 minutos e, em seguida, deixada aquecer para temperatura ambiente e agitada até 72 horas. O progresso da reação foi monitorado por LC-MS (ou HPLC ou TLC); a rea- ção estava geralmente completa em 1-72 horas. Solventes foram removidos in vacuo e o resíduo foi purificado por cromatografia de fase reversa e as frações que pertenciam ao produto desejado foram combinadas e liofilizadas para dar as amidas alvo.
[0780] Procedimento Geral E: Preparação de amidas através de formação in situ de NHS ésteres.
[0781] A uma solução (0,08 M) do ácido (1 eq.) (0°C ou temperatura ambiente) em tetrahidrofurano ou N,N-dimetilformamida, ou N,N- Dimetilacetamida, foi adicionado DCC (2,2 eq.) seguido por N- hidroxissuccinimida (2,2 eq.) e a reação foi ou agitada a 0°C ou deixada aquecer para temperatura ambiente e agitada até que a análise por LC/MS indicasse que a maior parte do ácido de partida ácido foi consumida. A mistura de reação foi novamente resfriada para 0°C e a amina (1 a 20 eq.) foi adicionada, aquecida para temperatura ambiente e agitada. O progresso da reação foi monitorado por LC-MS (ou HPLC ou TLC); a reação estava geralmente completa em 1-72 horas. A mistura de reação foi concentrada in vacuo e o resíduo foi purificado por cromatografia de fase reversa para dar a amida desejada.
[0782] Procedimento Geral F: Preparação de amidas a partir de NHS ésteres. Uma mistura de amina (ou sal de amina-ácido) (1,0 eq.) e N,N’-Diisopropiletilamina (5eq) em metanol (0,2 mL) foi agitada por 15 minutos e a solução resultante transferida para uma solução do NHS éster (1,0 eq.) em tetrahidrofurano (1,0 mL). A reação foi agitada à temperatura ambiente com adição de mais amina (1-3 eq.) até que a análise por LC/MS indicasse que a maioria do material de partida de NHS-éster foi consumida. A mistura de reação foi concentrada in vacuo e o resíduo foi purificado por cromatografia de fase reversa para dar o produto de amida desejado.
[0783] Exemplo A1
[0784] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,4R,5R,7S)-7-{2-[(2,5-dioxopirrolidin-1-il)oxi]-2-oxoetil}-4-hidroxi- 1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5- dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato #B1.
[0785] Etapa 1. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-{2-[(2,5-dioxopirrolidin-1-il)oxi]-2-oxoetil}-4-hidroxi- 1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato #B1. A uma solução resfriada (0 °C) de #NP1 (103 mg, 0,192 mmol, 1 eq.) em tetrahidrofurano (2 mL, 0,096 M) foi adicionado DCC (87,1 mg, 0,422 mmol, 2,2 eq.) e a reação foi agitada por 15 minutos. N- hidroxissuccinimida (48,6 mg, 0,422 mmol, 2,2 eq.) foi adicionada e a reação foi agitada a (0°C) por 15 minutos, aquecida para temperatura ambiente e agitada por 72 horas. A reação foi concentrada in vacuo, e o resíduo foi purificado por cromatografia de fase reversa C18 em pressão do meio (Gradiente: 5% a 90% de água em acetonitrila com 0,02% de ácido acético em cada fase). As frações que pertenciam ao produto desejado foram liofilizadas para dar #B1 como um sólido. Rendimento: 66,6 mg, 0,103 mmol, 54%. HPLC (Protocolo AA) tempo de retenção = 8,170 minutos (pureza 91%). LCMS (Protocolo D): m/z 633,3 [M+H]+, tempo de retenção = 0,81 minuto. 1H RMN (400 MHz, DMSO-d6) δ ppm 7,78 (d, J=8,02 Hz, 1 H) 6,37 - 6,30 (m, 2 H) 6,09 (m, 1 H) 5,85 (dd, J=11,54, 7,43 Hz, 1 H) 5,59 (dd, J=16,04, 5,28 Hz, 1 H) 5,50 (t, J=7,04 Hz, 1 H) 5,07 (d, J=6,06 Hz, 1 H, D2O permutável) 4,34 - 4,25 (m, 2 H) 3,63 (d, J=5,48 Hz, 2 H) 3,48 (td, J=7,09, 2,64 Hz, 1 H) 3,27 (d, J=5,28 Hz, 1 H) 2,97 (d, J=6,85 Hz, 2 H) 2,82 - 2,77 (m, 4 H) 2,59 (d, J=5,09 Hz, 1 H) 2,33 - 2,11 (m, 2 H) 1,96 (s, 3 H) 1,92 (d, J=8,22 Hz, 1 H) 1,82 - 1,77 (m, 2 H) 1,68 (s, 3 H) 1,66-1,6 (br, s, 1 H) 1,59 - 1,55 (m, 1 H) 1,23 (d, J=6,46 Hz, 3 H) 1,05 (d, J=6,26 Hz, 3 H) 0,93 (d, J=7,24 Hz, 3 H).
[0786] Exemplo A2
[0787] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3S,5S,7S)-7-{2-[(2,5-dioxopirrolidin-1-il)oxi]-2-oxoetil}-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B2).
[0788] Etapa 1. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3S,5S,7S)-7-{2-[(2,5-dioxopirrolidin-1-il)oxi]-2-oxoetil}-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B2). A uma solução resfriada (0 °C) de #NP2 (430 mg, 0,828 mmol, 1 eq.) em tetrahidrofu- rano (6 mL, 0,13 M), foi adicionado DCC (376 mg, 1,82 mmol, 2,2 eq.) seguido por N-hidroxissuccinimida (210 mg, 1,82 mmol, 2,2 eq.). A reação foi deixada aquecer para temperatura ambiente. Após 18 horas, filtrou-se sólido branco e filtrado concentrado para um resíduo amarelo. O resíduo foi purificado por cromatografia de fase reversa (Método A) para gerar #B2 como um sólido branco. Rendimento: 204 mg, 0,331 mmol, 40%. HPLC (Protocolo AA): tempo de retenção = 9,463 minutos (pureza 77%). LCMS (Protocolo D): m/z 617,3 [M+H]+ tempo de retenção = 0,91 minuto. 1H RMN (400 MHz, DMSO-d6)D δ ppm 7,72 (d, J=7,81 Hz, 1 H) 6,32 - 6,21 (m, 2 H) 6,04 (m, 1 H) 5,85 (dd, J=11,48, 7,21 Hz, 1 H) 5,57 (dd, J=15,80, 4,90 Hz, 1 H) 5,47 (t, J=7,04 Hz, 1 H) 4,53 - 4,47 (m, 1 H) 4,32 - 4,25 (m, 1 H) 3,62-3,55 (m, 2 H) 3,45 - 3,41 (m, 1 H) 2,95 (d, J=6,60 Hz, 2 H) 2,74 (s, 3 H) 2,59 (dd, J= 16,00, 4,68 Hz, 2 H) 2,30 - 2,07 (m, 2 H) 1,91 (s, 3 H) 1,77 -1,65 (m, 4 H) 1,63 (br s, 4 H) 1,61- 1,57 (m, 1 H) 1,48 (dd, J= 13,27, 7,02 Hz, 1 H) 1,19 (d, J=6,24 Hz, 3 H) 1,050 (d, J=6,24 Hz, 3 H) 0,89 (d, J=7,41 Hz, 3 H).
[0789] Exemplo A3
[0790] Síntese de pentafluorofenil [(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]acetato (#B3)
[0791] Etapa 1. Síntese de pentafluorofenil [(3R,5S,7R,8R)-7- {(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi- 1,6-dioxaspiro[2,5]oct-5-il]acetato (#B3). A uma solução de #NP1 (25 mg, 0,047 mmol, 1 eq.) em tetrahidrofurano (0,7 mL, 0,06 M), foi adicionado DCC (9,7 mg, 0,047 mmol, 1 eq.) seguido por uma solução de pentafluorofenol (17,3 mg, 0,094 mmol, 2 eq.) em tetrahidrofurano (0,3 mL, 0,3 M). A reação foi agitada à temperatura ambiente por 18 horas, filtrada e o bolo de filtro enxaguado com acetonitrila. Os filtrados combinados foram concentrados in vacuo e o material bruto foi purificado por cromatografia de fase reversa (Método A) para render #B3 como um sólido branco. Rendimento: 21,6 mg, 0,030 mmol, 65%. HPLC (Protocolo AB): tempo de retenção = 15,617 minutos (pureza 87%). LCMS (Protocolo D): m/z 702,2 [M+H]+ tempo de retenção = 1,0 minutos, 1H RMN ((400 MHz, DMSO-d6) δ: 7,79 (d, J=7,8 Hz, 1H), 6,38 (t, J=6,2 Hz, 1H), 6,32 (d, J=16,4 Hz, 1H), 6,12 (dd, J=11,7, 1,2 Hz, 1H), 5,88 (dd, J=11,5, 7,6 Hz, 1H), 5,64 (dd, J=16,0, 5,1 Hz, 1H), 5,45 (t, J=7,0 Hz, 1H), 5,10 (d, J=6,2 Hz, 1H), 4,43 (dd, J=7,0, 3,9 Hz, 1H), 4,32 (t, J=4,7 Hz, 1H), 3,70-3,60 (m, 1H), 3,51-3,43 (m, 1H), 3,12 (d, J=6,6 Hz, 1H), 2,82 (d, J=5,1 Hz, 1H), 2,65 (d, J=5,1 Hz, 1H), 2,36- 2,15 (m, 2 H), 2,02-1,91 (m, 3 H), 1,81 (br, s, 1H), 1,71 (s, 3 H), 1,681,59 (m, 4 H), 1,27 (d, J=6,2 Hz, 3 H), 1,06 (d, J=6,2 Hz, 3 H), 0,95 (d, J=7,4 Hz, 3 H).
[0792] Exemplo A4
[0793] Preparação de (2Z,4S)-N-[(2R,3R,5S,6S)-6-{(2E,4E)-5- [(3R,4R,5R,7S)-7-{2-[(2,5-dioxopirrolidin-1-il)oxi]-2-oxoetil}-4-hidroxi- 1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5- dimetiltetrahidro-2H-piran-3-il]-4-hidroxipent-2-enamida (#B5).
[0794] Etapa 1. Síntese de ácido [(3R,5S,7R,8R)-8-hidroxi-7- {(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-hidroxipent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6-dioxaspiro[2,5]oct-5-il]acético (#B4). A uma solução de #NP1 (10,2 mg, 0,019 mmol, 1 eq.) dissolvida em uma mistura 1:1 de tetrahidrofura- no/água (1,5 mL, 0,012 M), foi adicionado hidróxido de lítio (6 mg, 0,25 mmol, 13 eq.). A reação foi agitada à temperatura ambiente por 1 ^ hora e os solventes foram removidos in vacuo. O resíduo bruto foi purificado por cromatografia de fase reversa (Método A) para gerar #B4 como um sólido. Rendimento: 2 mg, 0,004 mmol, 20%. HPLC (Protocolo A): tempo de retenção = 7,850 minutos (pureza 93%). LCMS (Protocolo D); m/z 494,1[M+H]+ tempo de retenção = 0,68 minutos, 1H RMN (400 MHz, CDCl3) δ 6,46 (d, J = 8,6 Hz, 1 H), 6,28 (d, J = 16 Hz, 1 H), 6,09 (dd, J = 11,9 e 5,3 Hz, 1 H), 5,73, (dd, J = 12,1 e 1,6 Hz, 1 H), 5,57 (dd, J = 15,6 e 5,9 Hz, 1 H), 5,39-5,32 (m, 1 H), 4,75-4,66 (m, 1 H), 4,50-4,41 (m, 1 H), 4,19-4,13 (m, 1H), 3,89-3,82 (m, 1 H), 3,68-3,59 (m, 1 H), 3,52-3,43 (m, 2 H), 2,99 (dd, J = 15,2 e 9,4 Hz, 1 H), 2,94 (d, J = 4,3 Hz, 1 H), 2,59-2,49 (m, 2 H), 2,36-2,25 (m, 1 H), 2,20-2,09 (m, 2 H), 1,94-1,79 (m, 2 H), 1,76-1,68 (m, 1 H), 1,66 (s, 3 H), 1,63 (d, J = 3,9 Hz, 1 H), 1,27 (d, J = 6,6 Hz, 3 H), 1,08 (d, J = 6,2 Hz, 3 H), 0,93 (d, J = 7,4 Hz, 3 H).
[0795] Etapa 2. Síntese de (2Z,4S)-N-[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-{2-[(2,5-dioxopirrolidin-1-il)oxi]-2-oxoetil}-4-hidroxi- 1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]-4-hidroxipent-2-enamida (#B5) foi preparada de acordo com o Procedimento Geral para a síntese de #B1 no Exemplo A1 exceto que #B4 foi usado em vez de #NP1. A reação bruta foi concentrada in vacuo e foi, então, purificada por cromatografia de fase reversa (Método A) para gerar #B5 como um sólido. Rendimento: 16,2 mg, 0,027 mmol, 64%. LCMS (Protocolo D): m/z 591,3[M+H]+ tempo de retenção = 0,71 minutos. 1H RMN (500 MHz, DMSO-d6, mult, J em Hz) δ 7,78 (d, J = 8,0 Hz, 1 H), 6,35 (d, J = 15,6 Hz, 1 H), 5,97 (dd, J = 11,9 e 1,2 Hz, 1 H), 5,87 (dd, J = 11,7 e 7,1 Hz, 1 H), 5,61 (dd, J = 15,6 e 5,1 Hz, 1 H), 5,55-5,49 (m, 1 H), 5,22-5,14 (m, 1 H), 5,11 (d, J = 4,7 Hz, 1 H), 5,08 (d, J = 6,0 Hz, 1 H), 4,36-4,25 (m, 2 H), 3,693,60 (m, 2 H), 3,53-3,45 (m, 1 H), 3,31-3,27 (m, 1 H), 2,99 (d, J = 6,7 Hz, 2 H), 2,84-2,76 (m, 4 H), 2,61 (d, J = 5,0 Hz, 1 H), 2,34-2,26 (m, 1 H), 2,24-2,15 (m, 1 H), 1,95 (dd, J = 13,0 e 8,2 Hz, 1 H), 1,87-1,73 (m, 2 H), 1,72-1,62 (m, 4 H), 1,59 (dd, J = 13,0 e 3,6 Hz, 1 H), 1,11 (d, J = 6,4 Hz, 3 H), 1,06 (d, J = 6,4 Hz, 3 H), 0,95 (d, J = 7,3 Hz, 3 H).
[0796] Exemplo A5
[0797] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,4R,5R,7S)-7-(2-hidrazinil-2-oxoetil)-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B6).
[0798] Etapa 1. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-(2-hidrazinil-2-oxoetil)-4-hidroxi-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B6). Hidrazina (0,615 mL de uma solução 1M em tetrahidrofurano 0,615 mmol, 5 eq.) foi adicionada a uma solução de #B1 (78 mg, 0,12 mmol, 1 eq.) dissolvida em diclorometano (3 mL, 0,04M) e agitada à temperatura ambiente por 1 hora e, em seguida, adicional de hidrazina (0,615 mL de uma solução 1M em tetrahidrofurano 0,615 mmol, 5 eq.) foi adicionado. Após 1 hora, a reação foi diluída com água e extraída com diclorome- tano (3 X), as camadas orgânicas foram combinadas, secas sobre sul-fato de sódio, filtradas e concentradas in vacuo. O resíduo bruto foi purificado por cromatografia de fase reversa (Método A) para gerar #B6 como um sólido. Rendimento: 43 mg, 58%. HPLC (Protocolo A) tempo de retenção = 6,870 minutos (pureza =72%). LCMS (Protocolo C): m/z 550,4 [M+H]+, tempo de retenção = 1,15 minutos, 1H RMN (400 MHz, DMSO-d6) δ 8,99 (s, 1 H), 7,78 (d, J = 8,2 Hz, 1 H), 6,42-6,25 (m, 2 H), 6,11 (dd, J = 11,5 e 1,4 Hz, 1 H), 5,86 (dd, J = 11,7 e 7,4 Hz, 1 H), 5,60 (dd, J = 16 e 5,9 Hz, 1 H), 5,55-5,49 (m, 1 H), 5,02 (d, J = 5,5 Hz, 1 H), 4,32-4,07 (m, 4 H), 3,70-3,60 (m, 2 H), 3,54-3,45 (m, 1 H), 3,22 (app t, J = 4,9 Hz, 1 H), 2,74 (d, J = 5,1 Hz, 1 H), 2,58 (d, J = 5,1 Hz, 1 H), 2,44 (dd, J = 14,2 e 8,4 Hz, 1 H), 2,35-2,25 (m, 1 H), 2,242,14 (m, 2 H), 1,97 (s, 3 H), 1,91-1,77 (m, 3 H), 1,70-1,60 (m, 4 H), 1,46 (dd, J = 12,9 e 3,5 Hz, 1 H), 1,25 (d, J = 6,2 Hz, 3 H), 1,07 (d, J = 6,6 Hz, 3 H), 0,95 (d, J = 7,4 Hz, 3 H).
[0799] Exemplo A6
[0800] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5- [(3S,5S,7S)-7-(2-hidrazinil-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5- oxopent-3-en-2-il acetato (#B7).
[0801] Etapa 1. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3S,5S,7S)-7-(2-hidrazinil-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5- oxopent-3-en-2-il acetato (#B7). A uma solução de #NP2 (140 mg, 0,269 mmol, 1 eq.) em tetrahidrofurano (4 mL, 0,07 M), foi adicionado DCC (122 mg, 0,592 mmol, 2,2 eq.), seguido por N-Hidróxi succinimida (68,1 mg, 0,592 mmol, 2,2 eq). Após 18 horas, hidrazina (0,576 mL de uma solução 1M em tetrahidrofurano, 0,576 mmol, 2,1 eq.) foi adicionada. Após 30 minutos, adicional de hidrazina (1 mL de uma solução 1M em tetrahidrofurano, 1 mmol, 3.7 eq.) foi adicionado. Após 10 minutos, a reação foi concentrada in vacuo e o material desejado bruto foi purificado por cromatografia de fase reversa (Método C*) para gerar #B7 como um sólido. Rendimento: 78 mg, 0,145 mmol, 54%. HPLC (Protocolo F): m/z 534.4 [M+H]+, tempo de retenção = 8,143 minutos (pureza 100%).
[0802] Exemplo A7
[0803] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,4R,5R,7S)-4-hidroxi-7-{2-[(2-hidroxietil)amino]-2-oxoetil}-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B8) e (2S,3Z)-5- {[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,4R,5R,7S)-7-(2-amino-2-oxoetil)-4- hidroxi-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5- dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B9) e (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,4R,5R,7S)-4-hidroxi-7- {2-oxo-2-[(4-sulfamoilbenzil)amino]etil}-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5- oxopent-3-en-2-il acetato (#B10) e (2S,3Z)-5-{[(2R,3R,5S,6S)-6- {(2E,4E)-5-[(3R,4R,5R,7S)-7-{2-[(4-aminobenzil)amino]-2-oxoetil}-4- hidroxi-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5- dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B11) e (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5-{(3R,4R,5R,7S)-4- hidroxi-7-[2-oxo-2-(piperazin-1-il)etil]-1,6-dioxaspiro[2,5]oct-5-il}-3- metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro-2H-piran-3-il}amino)-5- oxopent-3-en-2-il acetato, sal de acetato (#B12) e (2S,3Z)-5- ({(2R,3R,5S,6S)-6-[(2E,4E)-5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-(4- metilpiperazin-1-il)-2-oxoetil]-1,6-dioxaspiro[2,5]oct-5-il}-3-metilpenta- 2,4-dien-1-il]-2,5-dimetiltetrahidro-2H-piran-3-il}amino)-5-oxopent-3-en- 2-il acetato, sal de acetato (#B13) e (2S,3Z)-5-({(2R,3R,5S,6S)-6- [(2E,4E)-5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-(hidroxiamino)-2-oxoetil]-1,6- dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato(#B14).

[0804] Etapa 1a. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-4-hidroxi-7-{2-[(2-hidroxietil)amino]-2-oxoetil}-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B8). De acordo com o Procedimento Geral D, a partir de 2-aminoetanol (2,0 mg, 0,033 mmol, 1,03 eq.), tetrahidrofurano (1 mL), N,N-diisopropiletilamina (0,028 mL, 0,160 mmol, 5 eq.), metanol (0,2 mL), e #B1 (20 mg, 0,032 mmol, 1 eq.), foi sintetizado o material desejado bruto, que foi purificado por cromatografia de fase reversa (Método D*) para gerar #B8 como um sólido. Rendimento: 17,6 mg, 0,031 mmol, 97%. HPLC (Protocolo B): m/z 579,6 [M+H]+, tempo de retenção = 2,00 minutos (pureza 100%).
[0805] Etapa 1b. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-(2-amino-2-oxoetil)-4-hidroxi-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B9). A uma solução de #B1 (15 mg, 0,024 mmol, 1 eq.) dissolvida em tetrahidrofurano (1 mL, 0,024 M), foi adicionado amônia (0,069 mL de uma solução 7M em metanol, 0,480 mmol, 20 eq.) após agitação por 3 % horas, os solventes foram removidos in vacuo, e o material desejado bruto foi purificado por cromatografia de fase reversa (Método C*) para dar #B9 como um sólido. Rendimento: 6 mg, 0,012 mmol, 50%. HPLC (Protocolo F) m/z 535,3 [M+H]+, tempo de retenção = 7,796 minutos (pureza 100%). 1H RMN (400 MHz, DMSO-d6) δ 7,78 (d, J = 8,2 Hz, 1 H), 7,30 (s, 1 H), 6,77 (s, 1 H), 6,40-6,28 (m, 2 H), 6,10 (d, J = 11,7 Hz, 1 H), 5,87 (dd, J = 11,5 e 7,6 Hz, 1 H), 5,61 (dd, J = 15,8 e 5,7 Hz, 1 H), 5,54-5,47 (m, 1 H), 4,99 (d, J = 5,9 Hz, 1 H), 4,29-4,20 (m, 2 H), 3,693,61 (m, 2 H), 3,53-3,46 (m, 1 H), 3,23 (app t, J = 5,1 Hz, 1 H), 2,74 (d, J = 5,1 Hz, 1 H), 2,57 (d, J = 5,1 Hz, 1 H), 2,48-2,44 (m, 1 H), 2,362,25 (m, 1 H), 2,25-2,16 (m, 2 H), 1,97 (s, 3 H), 1,87-1,77 (m, 2 H), 1,69 (s, 3 H), 1,68-1,60 (m, 2 H), 1,49 (dd, J = 13,1 e 3,7 Hz, 1 H), 1,25 (d, J = 6,6 Hz, 3 H), 1,07 (d, J = 6,2 Hz, 3 H), 0,95 (d, J = 7,4 Hz, 3 H).
[0806] Etapa 1c. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-4-hidroxi-7-{2-oxo-2-[(4-sulfamoilbenzil)amino]etil}- 1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5- dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B10). De acordo com o Procedimento Geral D, a partir de 4- (aminometil)benzenossulfonamida, sal de cloridrato (9,2 mg, 0,041 mmol, 1 eq.) tetrahidrofurano (1 mL), N,N-diisopropiletilamina (0,035 mL, 0,200 mmol, 5 eq.), metanol (0,2 mL), e #B1 (25 mg, 0,040 mmol, 1 eq.), foi sintetizado o material desejado bruto, que foi purificado por cromatografia de fase reversa (Método D*) para gerar #B10 como um sólido. Rendimento: 15,5 mg, 0,022 mmol, 55%. HPLC (Protocolo B): m/z 704,4 [M+H]+, tempo de retenção = 2,36 minutos (pureza 100%). 1H RMN (400 MHz, METANOL-d4) δ: 8,58 (br, s,, 1H), 7,80 (d, J=8,2 Hz, 1H), 7,63 (d, J=8,6 Hz, 2 H), 7,45 (d, J=8,2 Hz, 2H), 6,42-6,32 (m, 2 H), 6,02-5,90 (m, 2 H), 5,67 (dd, J=15,8, 6,0 Hz, 1H), 5,53 (t, J=7,0 Hz, 1H), 4,62-4,54 (m, 1H), 4,46 (d, J=4,3 Hz, 1H), 4,42-4,32 (m, 2 H), 3,78-3,65 (m, 2 H), 3,58 (t, J=5,8 Hz, 1H), 3,43 (d, J=5,8 Hz, 1H), 2,922,81 (m, 2 H), 2,66 (d, J=5,1 Hz, 1H), 2,40 (m, 2 H), 2,24 (m, 1H), 2,022,02 (m, 3 H), 1,98 (s, 1H), 1,98-1,67 (m, 2 H), 1,90-1,85 (m, 1 H), 1,83-1,80 (m, 1 H), 1,77 (s, 2 H), 1,41-1,32 (m, 4 H), 1,13-0,98 (m, 3 H).
[0807] Etapa 1d. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-{2-[(4-aminobenzil)amino]-2-oxoetil}-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B11). De acordo com o Procedimento Geral D, a partir de 4-(aminometil)anilina (3,9 mg, 0,032 mmol, 1 eq.) tetrahidrofurano (1 mL), N,N-diisopropiletilamina (0,011 mL, 0,064 mmol, 2 eq.), metanol (0,2 mL), e #B1 (20 mg, 0,032 mmol, 1 eq.), foi sintetizado o material desejado bruto, que foi purificado por cromatografia de fase reversa (Método A) para gerar #B11 como um sólido. Rendimento: 17,6 mg, 0,027 mmol, 86%. HPLC (Protocolo AA): tempo de retenção = 6,748 minutos (pureza 91%). 1H RMN (400 MHz, DMSO-d6) δ ppm 8,14 (t, J=5,77 Hz, 1 H) 7,78 (d, J=8,02 Hz, 1 H) 6,90 - 6,84 (m, 2 H) 6,48 - 6,43 (m, 2 H) 6,39 - 6,30 (m, 1 H) 6,27 (s, 1 H) 6,09 (m,1 H) 5,88 - 5,80 (m, 1 H) 5,59 (dd, J=15,85, 5,48 Hz, 1 H) 5,51 (t, J=6,94 Hz, 1 H) 5,01 (d, J=5,28 Hz, 1 H) 4,89 (s, 2 H) 4,30 - 4,22 (m, 1 H) 4,12 - 3,99 (m, 1 H) 3,63 (d, J=5,87 Hz, 2 H) 3,49 (td, J=7,04, 2,54 Hz, 1 H) 3,22 (t, J=4,40 Hz, 1 H) 2,73 (d, J=5,09 Hz, 1 H) 2,59 - 2,50 (m, 2 H) 2,35 - 2,13 (m, 3 H) 1,96 (s, 3 H) 1,88 - 1,75 (m, 3 H) 1,69 (s, 3 H) 1,64 (td, J=4,89, 2,54 Hz, 1 H) 1,44 (dd, J=12,81, 3,62 Hz, 1 H) 1,23 (d, J=6,46 Hz, 3 H) 1,04 (d, J=6,46 Hz, 3 H) 0,93 (d, J=7,43 Hz, 3 H).
[0808] Etapa 1e. (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-oxo-2-(piperazin-1-il)etil]-1,6- dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato, sal de acetato (#B12). A uma solução de #B1 (15,5 mg, 0,024 mmol, 1 eq.) dissolvida em te- trahidrofurano (0,24 mL, 0,048 M), foi adicionada piperazina (2,5 mg, 0,029 mmol, 1,2 eq.). Após agitação por 30 minutos, a reação foi diluída com água, extraída com diclorometano, as fases orgânicas combinadas foram secas sobre sulfato de sódio, filtradas e os solventes foram removidos in vacuo. O material desejado bruto foi purificado por cromatografia de fase reversa (Método A) para dar #B12 como um sólido branco. Rendimento: 8,2 mg, 0,012 mmol, 52%. HPLC (Protocolo AA) tempo de retenção = 6,795 minutos (pureza 80%). LCMS (Protocolo C): m/z 604,3 [M+H]+, tempo de retenção = 1,01 minutos. 1H RMN (400 MHz, DMSO-d6) δ 7,79 (d, J = 8,2 Hz, 1 H), 6,41-6,28 (m, 2 H), 6,11 (d, J = 10,5 Hz, 1 H), 5,87 (dd, J = 11,5 e 7,6 Hz, 1 H), 5,60 (dd, J = 16 e 5,1 Hz, 1 H), 5,55-5,48 (m, 1 H), 4,97 (d, J = 5,9 Hz, 1 H), 4,314,20 (m, 2 H), 3,70-3,60 (m, 2 H), 3,55-3,35 (m, 6 H), 3,27-3,22 (m, 1 H), 2,75 (d, J = 5,1 Hz, 1 H), 2,69-2,54 (m, 5 H), 2,36-2,13 (m, 4 H), 1,98 (s, 3 H), 1,88-1,76 (m, 3 H), 1,72-1,61 (m, 4 H), 1,58-1,51 (m, 1 H), 1,25 (d, J = 6,2 Hz, 3 H), 1,07 (d, J = 6,2 Hz, 3 H), 0,95 (d, J = 7,4 Hz, 3 H).
[0809] Etapa 1f. (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-(4-metilpiperazin-1-il)-2-oxoetil]-1,6- dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato, sal de acetato (#B13). A uma solução de #B1 (18,8 mg, 0,03 mmol, 1 eq.) dissolvida em te- trahidrofurano (0,500 mL, 0,06 M), foi adicionada 1-metilpiperazina (3,6 mg, 0,036 mmol, 1.2 eq.) após agitação por 30 minutos, a reação foi diluída com água, extraída com diclorometano, as fases orgânicas combinadas foram secas sobre sulfato de sódio, filtradas e os solventes foram removidos in vacuo. O material desejado bruto foi purificado por cromatografia de fase reversa (Método A) para dar #B13 como um sólido branco. Rendimento: 11,6 mg, 0,017 mmol, 57%. HPLC (Proto-colo AA) tempo de retenção = 6,422 minutos (pureza 94%). LCMS (Protocolo C): m/z 618,4 [M+H]+, tempo de retenção = 0,97 minutos. 1H RMN (400 MHz, DMSO-d6) δ 7,79 (d, J = 7,8 Hz, 1 H), 6,42-6,27 (m, 2 H), 6,11 (d, J = 10,5 Hz, 1 H), 5,87 (dd, J = 11,3 e 7,4 Hz, 1 H), 5,60 (dd, J = 15,8 e 5,3 Hz, 1 H), 5,55-5,48 (m, 1 H), 4,97 (d, J = 6,2 Hz, 1 H), 4,30-4,21 (m, 2 H), 3,70-3,61 (m, 2 H), 3,56-3,33 (m, 5 H), 3,25 (app t, J = 5,5 Hz, 1 H), 2,79-2,65 (m, 2 H), 2,60-2,53 (m, 2 H), 2,352,12 (m, 9 H), 1,98 (s, 3 H), 1,88-1,78 (m, 3 H), 1,72-1,61 (m, 4 H), 1,56 (dd, J = 12,9 e 3,5 Hz, 1 H), 1,25 (d, J = 6,2 Hz, 3 H), 1,07 (d, J = 6,2 Hz, 3 H), 0,95 (d, J = 7,4 Hz, 3 H).
[0810] Etapa 1g. (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-(hidroxiamino)-2-oxoetil]-1,6- dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B14). A uma solução de #B1 (30,7 mg, 0,049 mmol, 1 eq.) dissolvida em tetrahidrofura- no (0,450 mL) e N,N-dimetilformamida (0,175 mL), foi adicionada N,N- diisopropiletilamina (32 mg, 0,245 mmol, 5 eq.) e cloridrato de hidroxi- lamina (10,6 mg, 0,152 mmol, 3 eq.) após agitação por 30 minutos, a reação foi diluída com água, extraída com etil acetato (3 X), as fases orgânicas combinadas foram lavadas novamente com água, secas sobre sulfato de sódio, filtradas e concentradas in vacuo. O material de- sejado bruto foi purificado por cromatografia de fase reversa (Método A) para dar #B14 como um sólido branco. Rendimento: 11,8 mg, 0,021 mmol, 43%. HPLC (Protocolo AA) tempo de retenção = 7,189 minutos (pureza 96%). LCMS (Protocolo C): m/z 551,2 [M+H]+, tempo de retenção = 1,18 minutos. 1H RMN (400 MHz, DMSO-d6) δ 10,42 (s, 1 H), 8,74 (s, 1 H) 7,80 (d, J = 7,8 Hz, 1 H), 6,43-6,29 (m, 2 H), 6,16-6,10 (m, 1 H), 5,88 (dd, J = 11,7 e 7,4 Hz, 1 H), 5,61 (dd, J = 16 e 5,5 Hz, 1 H), 5,57-5,51 (m, 1 H), 5,04 (d, J = 5,5 Hz, 1 H), 4,32-4,23 (m, 2 H), 3,723,62 (m, 2 H), 3,57-3,48 (m, 1 H), 3,27-3,21 (m, 1 H), 2,76 (d, J = 5,1 Hz, 1 H), 2,60 (d, J = 5,1 Hz, 1 H), 2,44-2,27 (m, 2 H), 2,26-2,11 (m, 2 H), 2,00 (s, 3 H), 1,92-1,80 (m, 3 H), 1,74-1,63 (m, 4 H), 1,49 (dd, J = 12,7 e 3,3 Hz, 1 H), 1,27 (d, J = 6,6 Hz, 3 H), 1,09 (d, J = 6,2 Hz, 3 H), 0,97 (d, J = 7,4 Hz, 3 H).
[0811] Exemplo A8
[0812] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3S,5S,7S)-7-(2-amino-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5- oxopent-3-en-2-il acetato (#B15).
[0813] Etapa 1. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3S,5S,7S)-7-(2-amino-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5- oxopent-3-en-2-il acetato (#B15). A uma solução de #B2 (108 mg, 0,175 mmol, 1 eq.) dissolvida em tetrahidrofurano (3 mL, 0,06 M), foi adicionada amônia (0,500 mL de uma solução 7M em metanol, 3,5 mmoles, 20 eq.) após agitação por 1 hora, os solventes foram removidos in vacuo, e o material desejado bruto foi purificado por cromato- grafia de fase reversa C18 em pressão do meio (Gradiente: 0% a 90% de água em acetonitrila com 0,02% de ácido acético em cada fase) para dar #B15 como um sólido. Rendimento: 23,9 mg, 0,045 mmol, 26%. HPLC (Protocolo AA) tempo de retenção = 8,231 minutos (pureza 89%). LCMS (Protocolo C): m/z 519,3 [M+H]+, tempo de retenção = 1,41 minuto. 1H RMN (400 MHz, DMSO-d6) δ 7,80 (d, J = 7,8 Hz, 1 H), 7,33 (s, 1 H), 6,78 (s, 1 H), 6,42-6,33 (m, 1 H), 6,28 (d, J = 16 Hz, 1 H), 6,16-6,10 (m, 1 H), 5,88 (dd, J = 11,7 e 7,4 Hz, 1 H), 5,61 (dd, J = 15,8 e 5,7 Hz, 1 H), 5,56-5,50 (m, 1 H), 4,60-4,51 (m, 1 H), 4,38-4,27 (m, 1 H), 3,72-3,62 (m, 2 H), 3,56-3,48 (m, 1 H), 2,71-2,54 (m, 4 H), 2,382,27 (m, 1 H), 2,26-2,16 (m, 2 H), 2,00 (s, 3 H), 1,89-1,74 (m, 3 H), 1,71 (s, 3 H), 1,69-1,61 (m, 2 H), 1,39 (dd, J = 13,5 e 6,4 Hz, 1 H), 1,27 (d, J = 6,2 Hz, 3 H), 1,09 (d, J = 6,6 Hz, 3 H), 0,97 (d, J = 7,4 Hz, 3 H).
[0814] Exemplo A9
[0815] Preparação de ácido [(3S,5S,7S)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-hidroxipent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6-dioxaspiro[2,5]oct-5-il]acético (#B16) e (2Z,4S)-N-[(2R,3R,5S,6S)-6- {(2E,4E)-5-[(3S,5S,7S)-7-{2-[(2,5-dioxopirrolidin-1-il)oxi]-2-oxoetil}-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]-4-hidroxipent-2-enamida (#B17).
[0816] Etapa 1. Síntese de ácido [(3S,5S,7S)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-hidroxipent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6- dioxaspiro[2,5]oct-5-il]acético (#B16). A uma solução de #NP2 (25 mg, 0,048 mmol, 1 eq.) dissolvida em uma mistura 1:1 de tetrahidrofura- no/água (3 mL, 0,016 M), foi adicionado hidróxido de lítio (15 mg, 0,63 mmol, 13 eq.). A reação foi agitada à temperatura ambiente por 1 hora e os solventes foram removidos in vacuo. O resíduo bruto foi purificado por cromatografia de fase reversa (Método A) para gerar #B16 como um sólido. Rendimento: 17 mg, 0,035 mmol, 74%. LCMS (Protocolo D); m/z 478,1 [M+H]+, tempo de retenção = 0,75 minutos. 1H RMN (400 MHz, CDCl3-d) δ 6,27 (d, J=8,98 Hz, 1 H) 6,09 (d, J=15,61 Hz, 1 H) 6,00 (dd, J=12,10, 5,46 Hz, 1 H) 5,62 (dd, J=12,10, 1,17 Hz, 1 H) 5,40 (dd, J=15,61, 5,85 Hz, 2H) 5,25 (t, J=6,63 Hz, 1 H) 4,61 (t, J=6,63 Hz, 1 H) 4,48 - 4,32 (m, 2 H) 3,79- 3,73 (m, 1 H) 3,57 - 3,48 (m, 2 H) 3,42 - 3,33 (m, 2 H) 2,85 (dd, J=15,22, 8,98 Hz, 2 H) 2,52 - 2,44 (m, 2 H) 2,42 (d, J=5,07 Hz, 1 H) 2,25 - 2,00 (m, 1 H) 1,94 - 1,87 (m, 2 H) 1,80 - 1,74 (m, 2 H) 1,68 - 1,59 (m, 2 H) 1,55 (s, 3 H) 1,46 (dd, J=13,46, 3,71 Hz, 1 H) 1,27 (dd, J=13,66, 4,29 Hz, 1 H) 1,17 (d, J=6,63 Hz, 3 H) 0,98 (d, J=6,24 Hz, 3H) 0,83 (d, J=7,02 Hz, 3 H).
[0817] Etapa 2. Síntese de (2Z,4S)-N-[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3S,5S,7S)-7-{2-[(2,5-dioxopirrolidin-1-il)oxi]-2-oxoetil}-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]-4-hidroxipent-2-enamida (#B17) foi preparada de acordo com o Procedimento Geral para a síntese de #B1 no Exemplo A1 exceto que #B16 foi usado em vez de #NP1. A reação bruta foi concentrada in vacuo e foi, então, purificada por cromatografia de fase reversa (Método A) para gerar #B17 como um sólido. Rendimento: 28 mg, 0,043 mmol, 43% LCMS (Protocolo D); m/z 575,1 [M+H]+, tempo de retenção = 0,82 minutos. 1H RMN (400 MHz, DMSO-d6) δ 7,76 (d, J = 7,8 Hz, 1 H), 6,30 (d, J = 16 Hz, 1 H), 5,97 (d, J = 12,1 Hz, 1 H), 5,86 (dd, J = 11,7 e 7,0 Hz, 1 H), 5,63 (dd, J = 16 e 5,1 Hz, 1 H), 5,56-5,48 (m, 1H), 5,22-5,06 (m, 2 H), 4,60-4,53 (m, 1 H), 4,39-4,30 (m, 1 H), 3,70-3,60 (m, 2H), 3,54-3,45 (m, 1 H), 3,03-2,98 (m, 2 H), 2,80 (s, 4 H), 2,70-2,60 (m, 2 H), 2,59 (s, 1 H), 2,37-2,13 (m, 3 H), 1,87-1,60 (m, 7H), 1,54 (dd, J = 13,3 e 7 Hz, 1 H), 1,11 (d, J = 6,2 Hz, 3 H), 1,06 (d, J = 6,2 Hz, 3 H), 0,95 (d, J = 7,4 Hz, 3 H).
[0818] Exemplo A10
[0819] Preparação de ácido 4-{4-[(1E)-1-(2-{[(3S,5S,7S)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6-dioxaspiro[2,5]oct-5-il]acetil}hidrazinilideno)etil]fenoxi}butanóico (#B18) e (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3S,5S,7S)-7-(2-{(2E)-2-[1- (4-{4-[(2,5-dioxopirrolidin-1-il)oxi]-4-oxobutoxi}fenil)etilideno]hidrazinil}- 2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5- dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B19).
[0820] Etapa 1. Síntese de ácido 4-{4-[(1E)-1-(2-{[(3S,5S,7S)-7- {(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6- dioxaspiro[2,5]oct-5-il]acetil}hidrazinilideno)etil]fenoxi}butanóico (#B18). A uma solução de #B7 (35 mg, 0,066 mmol, 1 eq.) em etanol (1 mL, 0,06 M), foi adicionado ácido 4-(4-acetilfenoxi)butanóico (73,3 mg, 0,330 mmol, 5 eq.) seguido por ácido acético glacial (0,250 mL) e a reação foi aquecida para 37°C. Após 3 % horas, a reação foi filtrada e purificada por cromatografia de fase reversa (Método A) para gerar #B18 como um sólido branco. Rendimento: 25,5 mg, 0,034 mmol, 52%. LCMS (Protocolo D); m/z 737,38 [M+H]+, tempo de retenção = 0,88 minutos.
[0821] Etapa 2. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3S,5S,7S)-7-(2-{(2E)-2-[1-(4-{4-[(2,5-dioxopirrolidin-1-il)oxi]-4- oxobutoxi}fenil)etilideno]hidrazinil}-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5- il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}- 5-oxopent-3-en-2-il acetato (#B19). A uma solução de #B18 (25 mg, 0,034 mmol, 1 eq.) dissolvida em tetrahidrofurano (0,7 mL, 0,049 M), foi adicionado DCC (15,5 mg, 0,075 mmol, 2,2 eq.) seguido por N- Hidroxi succinimida (8,60 mg, 0,075 mmol, 2,2 eq.). A reação foi agitada por 4 horas. Solventes foram removidos in vacuo e o resíduo foi purificado por cromatografia de fase reversa (Método A) para gerar #B19 como um sólido branco. Rendimento: 13 mg, 0,015 mmol, 46%. LCMS (Protocolo D); m/z 835,8 [M+H]+, tempo de retenção = 0,92 minuto.
[0822] Exemplo A11
[0823] Preparação de ácido 4-{4-[(1E)-1-(2-{[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi- 1,6-dioxaspiro[2,5]oct-5-il]acetil}hidrazinilideno)etil]fenoxi}butanóico (#B20) e (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,4R,5R,7S)-7-(2- {(2E)-2-[1-(4-{4-[(2,5-dioxopirrolidin-1-il)oxi]-4- oxobutoxi}fenil)etilideno]hidrazinil}-2-oxoetil)-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B21).
[0824] Etapa 1. Síntese de ácido 4-{4-[(1E)-1-(2-{[(3R,5S,7R,8R)- 7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien- 1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5-il]acetil}hidrazinilideno)etil]fenoxi}butanóico (#B20). A uma solução de #B6 (18,1 mg, 0,033 mmol, 1 eq.) em etanol (0,500 mL, 0,06 M), foi adicionado ácido 4-(4-acetilfenoxi)butanóico (36,7 mg, 0,165 mmol, 5 eq.) seguido por ácido acético glacial (0,125 mL) e a reação foi aquecida para 37°C. Após 1 hora, a reação foi filtrada e purificada por cro- matografia de fase reversa (Método A) para gerar #B20 como um sólido branco. Rendimento: 24,9 mg, 0,028 mmol, 85%. LCMS (Protocolo C); m/z 754,5 [M+H]+, tempo de retenção = 1,47 minuto.
[0825] Etapa 2. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-(2-{(2E)-2-[1-(4-{4-[(2,5-dioxopirrolidin-1-il)oxi]-4- oxobutoxi}fenil)etilideno]hidrazinil}-2-oxoetil)-4-hidroxi-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B21). A uma solução de #B20 (21,3 mg, 0,028 mmol, 1 eq.) dissolvida em tetrahidrofu- rano (0,550 mL, 0,05 M), foi adicionado DCC (13,5 mg, 0,062 mmol, 2,2 eq.) seguido por N-Hidroxi succinimida (7,3 mg, 0,062 mmol, 2,2 eq.). A reação foi agitada por 5 horas e adicional de DCC (5 mg, 0,022 mmol, 0.8 eq.) seguido por N-Hidroxi succinimida (5 mg, 0,042 mmol, 1,5 eq.). Após 18 horas, solventes foram removidos in vacuo e o resíduo foi purificado por cromatografia de fase reversa (Método A) para gerar #B21 como um sólido branco. Rendimento: 11 mg, 0,013 mmol, 47%. HPLC (Protocolo H): m/z 851,3 [M+H]+, tempo de retenção = 9,074 minutos (pureza 88%). LCMS (Protocolo C); m/z 851,5 [M+H]+, tempo de retenção = 1,58 minuto. 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 10,42-10,25 (m, 1 H), 7,83-7,66 (m, 3 H), 7,02-6,92 (m, 2 H), 6,43-6,22 (m, 2 H), 6,1-6,05 (m, 1 H), 5,93-5,81 (m, 1 H), 5,68-5,37 (m, 2 H), 5,08-4,90 (m, 1 H), 4,52-4,25 (m, 3 H), 4,13-4,04 (m, 2 H), 3,71-3,55 (m, 2 H), 3,52-3,40 (m, 1 H), 2,94-2,55 (m, 9 H), 2,35-2,03 (m, 7 H), 1,98 (s, 3 H), 1,95-1,85 (m, 1 H), 1,84-1,73 (m, 2 H), 1,721,54 (m, 5 H), 1,30-1,20 (m, 3 H), 1,12-1,00 (m, 3 H), 0,98-0,87 (m, 3 H).
[0826] Preparação de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5- {(3S,5S,7S)-7-[2-(hidroxiamino)-2-oxoetil]-1,6-dioxaspiro[2,5]oct-5-il}-3- metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro-2H-piran-3-il}amino)-5- oxopent-3-en-2-il acetato (#B22).
[0827] Etapa 1. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3S,5S,7S)-7-[2-(hidroxiamino)-2-oxoetil]-1,6-dioxaspiro[2,5]oct-5-il}- 3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro-2H-piran-3-il}amino)-5- oxopent-3-en-2-il acetato (#B22). A uma solução de #B2 (100,8 mg, 0,175 mmol, 1 eq.) dissolvida em tetrahidrofurano (1,8 mL) e N,N- dimetilformamida (0,600 mL), foi adicionada N,N-diisopropiletilamina (114 mg, 0,875mmol, 5 eq.) e cloridrato de hidroxilamina (10,6 mg, 0,152 mmol, 3 eq.) após agitação por 1 hora, a reação foi diluída com água, extraída com etil acetato (3x), as fases orgânicas combinadas foram lavadas novamente com água, secas sobre sulfato de sódio, filtradas e concentradas in vacuo. O material desejado bruto foi purificado por cromatografia de fase reversa C18 em pressão do meio (Gradiente: 10% a 100% de água em acetonitrila com 0,02% de ácido acético em cada fase) para dar #B22 como um sólido branco. Rendimento: 68 mg, 0,127 mmol, 73%. LCMS (Protocolo C): m/z 535,4 [M+H]+, tempo de retenção = 1,36 minuto. 1H RMN (400 MHz, DMSO-d6) δ 10,40 (s, 1H), 8,73 (d, J = 2 Hz, 1 H), 7,79 (d, J = 8,2 Hz, 1 H), 6,416,32 (m, 1 H), 6,26 (d, J = 15,6 Hz, 1 H), 6,12 (d, J = 11,3 Hz, 1 H), 5,87 (dd, J = 11,5 e 7,6 Hz, 1 H), 5,59 (dd, J = 16 e 5,5 Hz, 1 H), 5,555,49 (m, 1 H), 4,56-4,49 (m, 1 H), 4,36-4,27 (m, 1 H), 3,70-3,61 (m, 2 H), 3,54-3,47 (m, 1 H), 2,65-2,60 (m, 2 H), 2,48-2,41 (m, 1 H), 2,362,17 (m, 2 H), 2,16-2,09 (m, 1 H), 1,98 (s, 3 H), 1,85-1,72 (m, 3 H), 1,72-1,61 (m, 6 H), 1,43-1,35 (m, 1 H), 1,25 (d, J = 6,6 Hz, 3 H), 1,07 (d, J = 6,2 Hz, 3 H), 0,95 (d, J = 7,4 Hz, 3 H).
[0828] Exemplo A12
[0829] Preparação de N-[3-(2-{2-[(bromoacetil)amino]etoxi}etoxi)propanoil]-D-valil-N-(4-{[({4- [({[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5- il]acetil}amino)metil]fenil}carbamoil)oxi]metil}fenil)-N~5~-carbamoil-D- ornitinamida (#B27).
[0830] Etapa 1. Síntese de N-[(9H-fluoren-9-ilmetoxi)carbonil]-L- valil-N-[4-({[(4-{[(terc-butoxicarbonil)amino]metil}fenil)carbamoil]oxi}metil)fenil]-N~5~- carbamoil-L-ornitinamida (#B23). A uma solução de terc-butil (4- aminobenzil)carbamato (75,4 mg, 0,339 mmol, 1,3 eq.) em N,N- dimetilformamida (2 mL, 0,16 M), foi adicionada 2,6-dimetilpiridina (140 mg, 1,3 mmol, 5 eq.), N,N-diisopropiletilamina (169 mg, 1,3 mmol, 5 eq.) e 3H-[1,2,3]triazolo[4,5-b]piridin-3-ol (HOAt, 71,1 mg, 0,522 mmol, 2 eq.), e foi agitada por 5 minutos. Toda a mistura de reação foi adici- onada a uma solução de N-[(9H-fluoren-9-ilmetoxi)carbonil]-L-valil- N~5~-carbamoil-N-[4-({[(4-nitrofenoxi)carbonil]oxi}metil)fenil]-L- ornitinamida (200 mg, 0,261 mmol, 1 eq.) em N,N-dimetilformamida (2 mL, 0,13 M) e a reação foi aquecida para 50°C por 5 horas e concentrada in vacuo. O material desejado bruto foi purificado por cromato- grafia de fase reversa C18 em pressão do meio (Gradiente: 10% a 100% de água em acetonitrila com 0,02% ácido trifluoroacético em cada fase) para dar #B23 como um sólido. Rendimento: 40 mg, 0,047 mmol, 18%.
[0831] Etapa 2. Síntese de N-[(9H-fluoren-9-ilmetoxi)carbonil]-L- valil-N-{4-[({[4-(aminometil)fenil]carbamoil}oxi)metil]fenil}-N~5~- carbamoil-L-ornitinamida, sal de ácido ditrifluoroacético. (#B24). A uma suspensão de #B23 (40 mg, 0,047 mmol, 1 eq.) em diclorometano (2 mL, 0,023 M), foi adicionada uma solução 1:1 de diclorometano/ácido trifluoroacético (2 mL). Após 45 minutos, a reação foi concentrada in vacuo para uma goma laranja #B24 que foi usada sem purificação adicional. Rendimento: 46 mg (rendimento quantitativo assumido). LCMS (Protocolo D): m/z 750,4 [M+H]+, tempo de retenção = 0,73 minuto.
[0832] Etapa 3. Síntese de N-[(9H-fluoren-9-ilmetoxi)carbonil]-D- valil-N-(4-{[({4-[({[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5- {[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5- il]acetil}amino)metil]fenil}carbamoil)oxi]metil}fenil)-N~5~-carbamoil-D- ornitinamida (#B25). A uma solução de #B24 (46 mg, 0,047 mmol, 1 eq.) em tetrahidrofurano (1 mL, 0,047 M), foi adicionada N,N- diisopropiletilamina (12,8 mg, 0,017 mmol, 2,1 eq.). A mistura inteira foi, então, adicionada em gotas a uma solução de #B1 (29,7 mg, 0,047 mmol, 1 eq.) em tetrahidrofurano (1 mL). Após 18 horas, metanol (0,4 mL) foi adicionado. Após 48 horas, a reação foi concentrada in vacuo e o produto bruto foi purificado por cromatografia de fase reversa (Méto- do A) para gerar #B25 como um sólido branco. Rendimento: 12,2 mg, 0,009 mmol, 20%. HPLC (Protocolo AA): tempo de retenção = 9,140 (pureza = 89%). LCMS (Protocolo D): m/z 1268,7 [M+H]+, tempo de retenção = 0,92 minuto.
[0833] Etapa 4. Síntese de D-valil-N-(4-{[({4-[({[(3R,5S,7R,8R)-7- {(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi- 1,6-dioxaspiro[2,5]oct-5-il]acetil}amino)metil]fenil}carbamoil)oxi]metil}fenil)-N~5~-carbamoil-D- ornitinamida (#B26). A uma solução de #B25 (12 mg, 0,009 mmol, 1 eq.) em N,N-dimetilformamida (0,3 mL, 0,3 M), foi adicionada piperidi- na (0,2 mL de uma solução estoque 0,050 mL em 1 mL de N,N- dimetilformamida). Após 30 minutos, a reação foi concentrada in vacuo e purificada por cromatografia de fase reversa (Método A) e as frações que pertenciam ao produto desejado foram liofilizadas para gerar #B26 como um sólido. Rendimento: 6,6 mg, 0,006 mmol, 70%. HPLC (Protocolo AA): tempo de retenção = 6,957 (pureza = 89%). LCMS (Protocolo D): m/z 1045,8 [M+H]+, tempo de retenção = 0,69 minuto.
[0834] Etapa 5. Síntese de N-[3-(2-{2-[(bromoacetil)amino]etoxi}etoxi)propanoil]-D-valil-N-(4-{[({4- [({[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5-il]acetil}amino)metil]fenil}carbamoil)oxi]metil}fenil)-N~5~-carbamoil-D- ornitinamida (#B27). A uma solução de #B26 (6 mg, 0,006 mmol, 1 eq.) em tetrahidrofurano (0,6 mL, 0,01 M), foi adicionada N,N- diisopropiletilamina (0,25 mL de uma solução estoque [preparada por dissolução de 0,01 mL N,N-diisopropiletilamina em 1 mL tetrahidrofu- rano], 0,012 mmol, 2 eq.) e foi agitada à temperatura ambiente por 5 minutos. A mistura inteira foi adicionada em gotas a uma solução res- friada (0 °C) de 2-bromo-N-[2-(2-{3-[(2,5-dioxopirrolidin-1-il)oxi]-3- oxopropoxi}etoxi)etil]acetamida (2,4 mg, 0,006 mmol, 1 eq.). A reação foi agitada a (0°C) por 5 minutos e, em seguida, deixada aquecer para temperatura ambiente. Após 16 horas, a reação foi concentrada in vacuo e purificada por cromatografia de fase reversa (Método A) para gerar #B27 como um sólido branco. Rendimento: 1,4 mg, 0,001 mmol, 20%. LCMS (Protocolo D): m/z 1348,7 [M+Na]+, tempo de retenção = 0,77 minuto.
[0835] Exemplo A13
[0836] Preparação de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5-{(3R,4R,5R,7S)-7-[2-({4-[(3-{2-[(N-{6-[(bromoace-til)amino]hexanoil}glicil)amino]fenil}propanoil)sulfamoil]benzil}amino)-2- oxoetil]-4-hidroxi-1,6-dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]- 2,5-dimetiltetrahidro-2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B37)
[0837] Etapa 1. Síntese de metil (2E)-3-(2-{[N-(terc- butoxicarbonil)glicil]amino}fenil)prop-2-enoato (#B28). A uma solução de N-(terc-butoxicarbonil)glicina (13,4 g, 77,1 mmoles, 1 eq.), cloridrato de 1-[3-(dimetilamino)propil]-3-etilcarbodiimida (22,1 g, 115,7 mmol, 1,5 eq.), 1-Hidroxibenzotriazol (11,4 g, 84,8 mmoles, 1,1 eq.), 4- (dimetilamino)piridina (0,9 g, 7,4 mmoles, 0,10 eq.) em N,N- dimetilformamida (350 mL), foi adicionado metil (2E)-3-(2- aminofenil)prop-2-enoato (15 g, 84,7 mmoles, 1,1 eq.) à temperatura ambiente. A reação foi aquecida para 50°C, agitada por 18 horas. A reação foi diluída com água (400 mL), lavada com ácido cítrico (200 mL), extraída com etil acetato (300 mL X 3). A camada orgânica foi lavada com salmoura (150 mL), seca sobre sulfato de sódio, filtrada e o filtrado concentrado in vacuo. O produto bruto foi purificado por cro- matografia de sílica gel eluída com éter de petróleo:etil acetato de 8:1 a 1:1 para gerar o composto #B28 (20 g, 71%) como um sólido. 1H RMN (400 MHz, CDCl3): δ 8,29 (br, 1H), 7,84 (d, 2H), 7,55(t, 1H), 7,40 (t, 1H), 7,22 (t, 1H), 6,41(d, 2H), 5,37 (br, 1H), 4,00 (d, 2H), 3,79 (s, 3H), 1,46 (s, 9H).
[0838] Etapa 2. Síntese de metil 3-(2-{[N-(terc- butoxicarbonil)glicil]amino}fenil)propanoato (#B29). Uma suspensão de composto #B28 (20 g, 59.8 mmoles, 1 eq.) e Pd/C (2,0 g) em etil acetato (350 mL) e metanol (300 mL) foi desgaseificada sob vácuo e purgada com H2 várias vezes. A mistura de reação foi agitada à temperatura ambiente sob H2 (30 Psi) por 9 horas. A mistura de reação foi filtrada, e o bolo de filtro foi lavado com etil acetato (100 mL). O filtrado foi evaporado até secagem para gerar o composto #B29 como um óleo que foi usado na etapa seguinte sem purificação adicional: Rendimento (20,75 g, presumir quantitativa). 1H RMN (400 MHz, CDCl3): δ 9,13 (br, 1H), 7,75 (d, 1H), 7,22(d, 1H), 7,17 (m, 2H), 5,34 (br, 1H), 4,04 (d, 2H), 3,64 (s, 3H), 2,95 (t, 2H), 2,72(t, 2H),1,46 (s, 9H).
[0839] Etapa 3. Síntese de ácido 3-(2-{[N- (tercbutoxicarbonil)glicil]amino}-fenil)propanóico (#B30). A uma solução de #B29 (20,75 g, 59,8 mmoles, 1 eq.) em tetrahidrofurano (200 mL), foi adicionada uma solução de hidróxido de sódio (12,24 g, 0,306 mmol) em água (155 mL). A mistura de reação foi agitada à temperatura ambiente por 3 horas. O pH da mistura de reação foi ajustado para pH 5~6 por ácido cítrico, extraído com etil acetato (400 mL X 2). A camada orgânica foi lavada com salmoura (150 mL X 2), seca sobre sul- fato de sódio, concentrada até secagem para gerar #B30 como sólido que foi usado sem purificação adicional na etapa seguinte. Rendimento: 21,5 g (presumir quantitativa). 1H RMN (400 MHz, CD3OD) δ 7,29 (m, 1H), 7,23 (m, 3H), 3,90 (s, 2H), 2,91 (m, 2H), 2,66 (m, 2H), 1,49 (m, 9H), LCMS (Protocolo I): m/z 345 [M+Na]+, tempo de retenção = 1,034 minutos.
[0840] Etapa 4. Síntese de ácido 3-[2-(glicilamino)fenil]propanóico (#B31). A uma solução de #B30 (10 g, 31,1 mmol, 1 eq.) em etil acetato (100 mL), foi adicionado HCl/dioxano (70 mL). A reação foi agitada à temperatura ambiente por 3 horas, e, em seguida, concentrada até secagem. O resíduo foi recristalizado a partir de terc-butil metil éter (50 mL) para gerar #B31 como sólido (5,9 g, 26,5 mmol, 85,5% em três etapas). 1H RMN (400 MHz, DMSO-d6): δ 10,06 (br, 1H), 8,31 (br, 3H), 7,41(d, 1H), 7,28 (m, 3H), 3,84 (s, 2H), 2,88(m, 2H), 2,52 (m, 2H).
[0841] Etapa 5. Síntese de ácido 3-(2-{[N-(6-{[(9H-fluoren-9- ilmetoxi)carbonil]amino}hexanoil)glicil]amino}fenil)propanóico (#B32). A uma solução de #B31 (1,11 g, 4,98 mmoles, 1 eq.) e bicarbonato de sódio (528 mg, 7,47 mmoles, 1,5 eq.) em tetrahidrofurano (25 mL) e água (10 mL), foi adicionada em gotas uma solução de 9H-fluoren-9- ilmetil {6-[(2,5-dioxopirrolidin-1-il)oxi]-6-oxohexil}carbamato (2,7 g, 5,98 mmol, 1,2 eq.) em tetrahidrofurano (45 mL) e 1,2-Dimetoxietano (10 mL). A mistura de reação foi agitada à temperatura ambiente por 16 horas. O pH da mistura de reação foi ajustado para pH 5~6 por ácido cítrico, extraído com diclorometano (100 mL X 2). A camada orgânica foi lavada com salmoura (100 mL), seca sobre sulfato de sódio, concentrada até secagem. O produto bruto foi purificado por cromatografia de sílica gel eluída com diclorometano: metanol a partir de 100:1 a 10:1 para gerar #B32. (1,1 g, 1,97 mmol, 39,7%) como sólido. 1H RMN (400 MHz, DMSO-d6), δ 9,31(s, 1H), 8,18 (s, 1H), 7,89 (d, 2H), 7,70 (d, 2H), 7,40 (m, 6H), 7,13 (m, 5H), 4,30 (m, 2H), 4,21 (m, 1H), 3,90 (m, 2H), 2,98 (m, 2H), 2,80 (m, 2H), 2,77 (m, 1H), 2,48 (m, 1H), 2,20 (m, 2H), 1,54(m, 2H), 1,42 (m, 2H), 1,25 (m, 2H). LCMS (Protocolo I): m/z 580,1 [M+Na]+, tempo de retenção = 1,315 minuto.
[0842] Etapa 6. Síntese de 9H-fluoren-9-ilmetil {6-[(2-{[2-(3-{[(4- {[(terc-butoxicarbonil)amino]metil}fenil)sulfonil]amino}-3-oxopropil)fenil]amino}-2-oxoetil)amino]-6-oxohexil}carbamato (#B33). A mistura de #B32 (900 mg, 1,62 mmol, 1 eq.) e terc-butil (4- sulfamoilbenzil)carbamato (787 mg, 2,59 mmoles, 1,6 eq.), 4- (dimetilamino)piridina (198 mg, 1,62 mmol, 1 eq.), cloridrato de 1-[3- (dimetilamino)propil]-3-etilcarbodiimida (369 mg, 1,2 mmol, 0,7 eq.) em diclorometano (30 mL) foi agitada à temperatura ambiente por 3,5 horas. O pH da mistura de reação foi ajustado para pH 5~6 por ácido cítrico, extraído com diclorometano (30 mL X 2). A camada orgânica foi lavada com salmoura (100 mL), seca sobre sulfato de sódio, concentrada até secagem. O produto bruto foi purificado por cromatografia de sílica gel eluída com diclorometano:metanol a partir de 100:1 a 20:1 para gerar #B33 como um sólido (800 mg, 0,972 mmol, 60,0%). 1H RMN (400 MHz, DMSO-d6), δ 12,09 (br, 1H), 9,30 (s, 1H), 8,19 (m,1H), 7,89 (m, 4H), 7,70 (m, 2H), 7,46 (m, 1H), 7,44 (m, 4H), 7,42 (m,3H), 7,35 (m, 1H), 7,16 (m, 1H), 7,08 (m, 2H), 4,23 (m, 2H), 3,88 (m,3H), 3,87 (m, 2H), 2,70 (m, 2H), 2,68 (m, 2H), 2,50 (m 2H), 2,17 (m,2H), 1,53 (m, 2H), 1,51 (s, 9H), 1,25 (m, 4H), LCMS (Protocolo J): m/z 726,1 [M-Boc]+, tempo de retenção = 1,211 minutos.
[0843] Etapa 7. Síntese de 9H-fluoren-9-ilmetil (6-{[2-({2-[3-({[4- (aminometil)fenil]sulfonil}amino)-3-oxopropil]fenil}amino)-2- oxoetil]amino}-6-oxohexil)carbamato (#B34). A uma suspensão de (#B33) (52,6 mg, 0,063 mmol, 1 eq.), em diclorometano (3 mL, 0,02 M), foi adicionado ácido trifluoroacético (0,6 mL) e foi agitada por 2 horas e, em seguida, concentrada in vacuo. O resíduo foi azeotropado com acetonitrila (3X) para gerar #B34 (45,7 mg, 0,063 mmol, presumir quantitativa) que foi usado como é na etapa seguinte sem purificação adicional. LCMS (Protocolo D): m/z 726,3 [M+H]+, tempo de retenção = 0,73 minuto.
[0844] Etapa 8. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-{2-[(4-{[3-(2-{[N-(6-{[(9H-fluoren-9- ilmeto-xi)carbonil]amino}hexanoil)glicil]amino}fenil)propanoil]sulfamoil}benzil)a mino]-2-oxoetil}-4-hidroxi-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4- dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B35). A uma solução de #B34 (45,7 mg, 0,063 mmol, 1 eq.) em tetrahidrofurano (0,5 mL, 0,12 M), foi adicionada N,N- diisopropiletilamina (24,4 mg, 0,189 mmol, 3 eq.). Toda a mistura de reação foi adicionada a uma solução resfriada (0 °C) de #B1 (40 mg, 0,063 mmol, 1 eq.) em tetrahidrofurano (0,5 mL) e a reação foi deixada aquecer para temperatura ambiente. Após uma hora, a reação foi concentrada in vacuo para gerar #B35 (55 mg, 0,053 mmol, 70%) que foi usado como é na etapa seguinte sem purificação adicional. LCMS (Protocolo D): m/z 1243,6 [M+H]+, tempo de retenção = 0,95 minuto.
[0845] Etapa 9. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-{2-[(4-{[3-(2-{[N-(6-aminohexanoil)glicil]amino}fenil)propanoil]sulfamoil}benzil)amino]-2- oxoetil}-4-hidroxi-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}- 2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato, sal de formiato (#B36). A uma solução de #B35 (55 mg, 0,053 mmol, 1 eq.) em N,N-dimetilformamida, foi adicionada piperidina (0,2 mL de uma solução estoque [preparada por dissolução de 0,05 mL em 1 mL N,N-dimetilformamida], 0,106 mmol, 2 eq.). Após 30 minutos, a reação foi concentrada in vacuo e purificada por cromatografia de fase reversa (Método A) para gerar #B36 como um sólido branco. Rendimento: 30 mg, 0,027 mmol, 52%. HPLC (Protocolo AA): tempo de retenção = 7,143 minutos (pureza = 92%). LCMS (Protocolo D): m/z 1021,4 [M+H]+ tempo de retenção = 0,67 minutos. 1H RMN (DMSO-d6) δ: 10,23-10,21 (b,s,, 1 H D2O permutável) 8,36-8,31 (m, 1H), 8,25-8,20 (m, 1H), 7,76-7,68 (m, 1H), 7,53-7,42 (m, 3H), 7,14-7,02 (m, 3H), 6,996,93 (m, 1H), 6,35-6,21 (m, 2H), 6,08-6,01 (m, 1H), 5,82-5,76 (m, 1H), 5,61-5,52 (m, 1H), 5,50-5,42 (m, 1H), 4,28-4,16 (m, 4H), 3,82 (d, J=5,9 Hz, 2H), 3,57 (d, J=6,2 Hz, 2H), 3,49-3,42 (m, 1H), 3,21-3,18 (m, 1H), 2,74-2,66 (m, 3H), 2,62-2,49 (m, 3H), 2,31-2,11 (m, 6H), 1,91 (s, 3H), 1,84-1,69 (m, 3H), 1,64 (s, 3H), 1,60-1,41 (m, 6H), 1,32-1,22 (m,, 2H), 1,18 (d, J=6,6 Hz, 3H), 0,99 (d, J=6,21 Hz, 3H) -0,88 (d, J=6,21 Hz, 3H).
[0846] Etapa 10. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3R,4R,5R,7S)-7-[2-({4-[(3-{2-[(N-{6-[(bromoace-til)amino]hexanoil}glicil)amino]fenil}propanoil)sulfamoil]benzil}amino)-2- oxoetil]-4-hidroxi-1,6-dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]- 2,5-dimetiltetrahidro-2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B37). A uma solução de #B36 (20 mg, 0,018 mmol, 1 eq.) em tetra- hidrofurano, foi adicionada N,N-diisopropiletilamina (9,8 mg, 0,076 mmol, 4,2 eq.) e metanol (0,1 mL). A mistura inteira foi adicionada a uma solução resfriada (0 °C) de 1-[(bromoacetil)oxi]pirrolidina-2,5- diona (4,2 mg, 0,018 mmol, 1 eq) e foi agitada a 0°C por 5 minutos e, em seguida, deixada aquecer para temperatura ambiente. Após 18 horas, a reação foi concentrada in vacuo e purificada por cromatografia de fase reversa (Método A) para gerar #B37 como um sólido branco. Rendimento: 3,8 mg, 0,003 mmol, 18%. HPLC (Protocolo AA): tempo de retenção = 7,554 e 7,77 minutos (pureza 91%). LCMS (Protocolo D): m/z 1141,4 [M+H]+ tempo de retenção = 0,8 minutos. 1H RMN (400 MHz, METANOL-d4) □ : 7,66 (d, J=8,2 Hz, 2H), 7,56-7,49 (m, 1H), 7,33-7,26 (m, 2 H), 7,03 (m, 3 H), 6,32-6,23 (m, 1H), 5,94-5,81 (m, 2H), 5,58 (dd, J=16,0, 8,0 Hz, 1 H), 5,47-5,39 (m, 1H), 4,46 (s, 2H), 4,43- 4,34 (m, 1H), 4,3 (m, 2 H), 3,88 (s, 2 H), 3,68 (s, 2H), 3,67-3,62 (m, 2 H), 3,61-3,55 (m, 1H), 3,52-3,44 (m, 1H), 3,36-3,32 (m, 1 H), 3,11 (t,J=16 Hz, 1 H), 2,8 (d, J=8,0, 1H), 2,78-2,72 (m, 2H), 2,68 (t, J=8,0, 2 H), 2,57 (d, J=4,0 Hz, 1 H), 2,45-2,39 (m, 2 H), 2,36 (dd, J=16,0, 8,0 Hz, 1 H), 2,30 (d, J=8,0 Hz, 1 H), 2,25 (t, J=8,0 Hz, 2 H), 2,20-2,09 (m, 1 H), 1,9 (s, 1H), 1,85-1,78 (m, 2 H), 1,75-1,71, (m, 1H), 1,68 (s, 3 H),1,64-1,54 (m, 3 H), 1,50-1,43 (m, 2 H), 1,27-1,35 (m, 2 H), 1,25 (d,J=8,0 Hz, 3 H), 1,00 (d, J=8,0 Hz, 3 H), 1,05-0,87 (d, J=8,0 Hz, 3 H).
[0847] Exemplo A14
[0848] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3S,5S,7S)-7-(2-amino-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5- oxopent-3-en-2-il piperidina-1-carboxilato (#B40).
[0849] Etapa 1. Síntese de (2Z,4S)-N-[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3S,5S,7S)-7-(2-amino-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]-4-hidroxipent-2-enamida (#B39). A uma solução de #B15 (38 mg, 0,075 mmol, 1 eq.) em 4:1 tetrahidrofurano:água (2,2 mL), foi adicionado hidróxido de lítio (15,6 mg, 0,652 mmol, 8,7 eq.) e a mistura agitada à temperatura ambiente por 12 horas. A reação foi diluída com etil acetato e lavada com água. A camada aquosa foi extraída com etil acetato (3X) e as camadas orgânicas combinadas foram secas sobre sulfato de sódio anidro e concentradas in vacuo. Purificação por cromatografia de fase reversa (Método A) forneceu # B39 como um sólido. Rendimento: 16,7 mg, 0,035 mol, 47%. 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 7,76 (d, J = 8,2 Hz, 1 H), 7,31 (s, 1 H), 6,77 (s, 1 H), 6,27 (d, J = 16,0 Hz, 1 H), 5,97 (d, J = 11 Hz, 1 H), 5,86 (dd, J = 11,7 e 7,0 Hz, 1 H), 5,59 (dd, J = 16,0 e 5,5 Hz, 1 H), 5,54-5,47 (m, 1 H), 5,22-5,12 (m, 1 H), 5,10 (d, J = 4,7 Hz, 1 H), 4,58-4,48 (m, 1 H), 4,35-4,25 (m, 1 H), 3,69-3,59 (m, 2 H), 3,54-3,46 (m, 1 H), 2,64-2,52 (m, 3 H), 2,372,14 (m, 3 H), 1,87-1,73 (m, 3 H), 1,72-1,60 (m, 6 H), 1,37 (dd, J = 13,3 e 6,2 Hz, 1 H), 1,11 (d, J = 6,2 Hz, 3 H), 1,06 (d, J = 6,2 Hz, 3 H), 0,95 (d, J = 7,4 Hz, 3 H). HPLC (Protocolo AA) tempo de retenção = 7,15 minutos (pureza = 100%). LCMS (Protocolo C): m/z 499,3 [M+Na]+, tempo de retenção = 1,18 minuto.
[0850] Etapa 2. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3S,5S,7S)-7-(2-amino-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5- oxopent-3-en-2-il piperidina-1-carboxilato #B40: A uma solução de #B39 (106 mg, 0,222 mmol, 1 eq.) em diclorometano (3 mL), foi adicionada trietilamina (79 mg, 0,777 mmol, 3,5 eq), 4-N,N’-dimetilamino Piridina (18,9 mg, 0,155 mmol, 0,7 eq.) e bis(4-nitrofenil) carbonato (207 mg, 0,666 mmol, 3 eq.) e a reação agitada à temperatura ambiente por 2 horas. A 1/5 desta mistura, foi adicionada piperidina (18,9 mg, 0,222 mmol, 1 eq.) e a mistura agitada à temperatura ambiente por 3,5 horas, concentrada in vacuo e o resíduo purificado por cromatografia de fase reversa (Método D*) para prover #B40. Rendimento 2,7 mg, 0,021 mmol, 9,5%. HPLC (Protocolo B) m/z 588,4 [M+H]+, tempo de retenção = 2,82 minutos (pureza= 100%).
[0851] Exemplo A15
[0852] Preparação de N-[3-(2-{2- [(bromoacetil)amino]etoxi}etoxi)propanoil]-L-valil-N-[4-({[(2-{[(3S,5S,7S)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent- 2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3- dien-1-il}-1,6-dioxaspiro[2,5]oct-5-il]acetil}hidrazinil)carbonil]oxi}metil)fenil]-N~5~-carbamoil-L- ornitinamida(#B43).
[0853] Etapa 1. Síntese de N-[(9H-fluoren-9-ilmetoxi)carbonil]-L- valil-N-[4-({[(2-{[(3S,5S,7S)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-1,6-dioxaspiro[2,5]oct-5- il]acetil}hidrazinil)carbonil]oxi}metil)fenil]-N~5~-carbamoil-L-ornitinamida (#B41). A uma solução de #B7 de (56 mg, 0,1 mmol, 1 eq.) em N,N-dimetilformamida (2,6 mL), foi adicionado N-[(9H-fluoren- 9-ilmetoxi)carbonil]-L-valil-N~5~-carbamoil-N-[4-({[(4-nitrofenoxi)carbonil]oxi}metil)fenil]-L-ornitinamida (FMocValCitPABC- PNP, WO04010957, 121 mg, 0,15 mmol, 1,5 eq.) N,N’-diisopropiletilamina (56 mg, 0,4 mmol, 4,0 eq.), 2,6-Dimetilpiridina (45 mg, 0,4 mmol, 4,0 eq.,) e 3H-[1,2,3]triazolo[4,5-b]piridin-3-ol (14,3 mg, 0,105 mmol, 1,05 eq.,). Após agitação a 50°C por 1,5 hora, a mistura de reação foi concentrada in vacuo e o material bruto foi purificado por cromatografia de fase reversa (Método A) para prover #B41 como um sólido. Rendimento: 72 mg, 0,06 mmol, 59%. LCMS (Protocolo D): m/z 1183,5 [M+Na]+, tempo de retenção = 0,95 minuto.
[0854] Etapa 2. Síntese de L-valil-N-[4-({[(2-{[(3S,5S,7S)-7- {(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6- dioxaspiro[2,5]oct-5-il]acetil}hidrazinil)carbonil]oxi}metil)fenil]-N~5~- carbamoil-L-ornitinamida (#B42). A uma solução de #B41 (50 mg, 0,043 mmol, 1 eq.) em N,N-dimetilformamida (0,7 mL), foi adicionada piperidina (66 mg, 0,78 mmol, 20 eq.) e a mistura agitada por 20 minutos. A mistura de reação foi concentrada in vacuo e o material bruto foi purificado por cromatografia de fase reversa (Método A) para prover #B42. Rendimento: 31 mg, 0,033 mmol, 76%. LCMS (Protocolo D): m/z 939,3 [M+H]+, tempo de retenção = 0,66 minutos.
[0855] Etapa 3. Síntese de N-[3-(2-{2-[(bromoacetil)amino]etoxi}etoxi)propanoil]-L-valil-N-[4-({[(2- {[(3S,5S,7S)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent- 2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3- dien-1-il}-1,6-dioxaspiro[2,5]oct-5-il]acetil}hidrazinil)carbonil]oxi}metil)fenil]-N~5~-carbamoil-L- ornitinamida (#B43). A uma solução de #B42 (10 mg, 0,011 mmol, 1 eq.) em N,N-dimetilformamida (0,2 mL), foi adicionado 2-bromo-N-[2- (2-{3-[(2,5-dioxopirrolidin-1-il)oxi]-3-oxopropoxi}etoxi)etil]acetamida (4,3 mg, 0,011 mmol, 1 eq.) e a mistura agitada à temperatura ambiente por 30 minutos. A reação foi diluída com dimetilsulfóxido (0,2 mL) e purificada por cromatografia de fase reversa (Método A) para prover #B43 como um sólido. Rendimento: 8,8 mg, 0,007 mmol, 66%. HPLC (Protocolo AA) tempo de retenção = 7,69 minutos (pureza= 71%). LCMS (Protocolo A): m/z 1220,4 [M+H]+, tempo de retenção = 0,77 minuto.
[0856] Exemplo A16
[0857] Preparação de N-(6-aminohexanoil)-L-valil-N-[4-({[(2- {[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5- il]acetil}hidrazinil)carbonil]oxi}metil)fenil]-N~5~-carbamoil-L- ornitinamida (#B47).
[0858] Etapa1. Síntese de N-(6-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}hexanoil)-L-valil-N~5~-carbamoil-N-[4- (hidroximatil)fenil]-L-ornitinamida (#B44). N,N-diisopropiletilamina (3,8 mL, 22,12 mmoles, 1,9 eq.) foi adicionada a uma solução de ácido 6- {[(9H-fluoren-9-ilmetoxi)carbonil]amino}hexanóico (4,2 g, 11,80 mmol, 1 eq.) e N,N,N’,N'-tetrametil-O-(7-azabenzotriazol-1-il)urônio hexafluo- rofosfato (HATU, 5,6 g, 14,75 mmoles, 1,25 eq.) em N,N- dimetilformamida (50 mL, 0,24 M) à temperatura ambiente e agitada por dez minutos. Em seguida, L-valil-N~5~-carbamoil-N-[4- (hidroximatil)fenil]-L-ornitinamida (de WO04010957, 5,6 g, 14,75 mmoles, 1,25 eq.) foi adicionado à mistura. Após 15 horas, a mistura de reação foi precipitada adicionando diclorometano e filtrada para obter #B44 como um sólido esbranquiçado. Rendimento: 6,9 g, 9,6 mmoles, 82%. LCMS 715,6 (M+H)+.
[0859] Etapa 2. Síntese de 4-[(N~5~-carbamoil-N~2~-{(3S)-3-[(6- {[(9H-fluoren-9-ilmetoxi)carbonil]amino}hexanoil)amino]-4-metilpent-1- en-2-il}-L-ornitil)amino]benzil 4-nitrofenil carbonato (#B45), uma solu- ção de #B44 (500 mg, 0,7 mmol, 1 eq.) e 4-nitrofenil carbonato (638 mg, 2,1 mmoles, 3 eq.) em N,N-dimetilformamida (3 mL, 0,2 M) foi tratada com N,N-diisopropiletilamina (365 μL, 2,1 mmoles, 3 eq.). A reação foi agitada à temperatura ambiente durante a noite. A mistura de reação foi concentrada in vacuo, absorvida em SiO2 e purificada por cromatografia de sílica gel (Gradiente: 0 a 25% metanol em diclorome- tano) para dar #B45 como um sólido. Rendimento: 402 mg, 0,476 mmol, 68%. LCMS (Protocolo L): m/z 880,7 [M+H]+ tempo de retenção 3,39.
[0860] Etapa 3. Síntese de N-(6-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}hexanoil)-L-valil-N-[4-({[(2-{[(3R,5S,7R,8R)-7- {(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi- 1,6-dioxaspiro[2,5]oct-5-il]acetil}hidrazinil)carbonil]oxi}metil)fenil]-N~5~- carbamoil-L-ornitinamida (#B46). O composto do título foi preparado em rendimento de 10% de 71 mg (0,13 mmol) de #B6 e 171 mg (0,194 mmol) de #B45 usando o procedimento descrito para a preparação do composto #B41. LCMS (Protocolo D): m/z 1290,5 [M+H]+, tempo de retenção = 0,91 minuto.
[0861] Etapa 4. Síntese de N-(6-aminohexanoil)-L-valil-N-[4-({[(2- {[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5- il]acetil}hidrazinil)carbonil]oxi}metil)fenil]-N~5~-carbamoil-L- ornitinamida (#B47). A uma solução de #B46 (19 mg, 0,015 mmol, 1 eq.) em N,N-dimetilformamida (0,35 mL), foi adicionada piperidina (25 mg, 0,3 mmol, 20 eq.) e a mistura foi agitada à temperatura ambiente por 30 minutos. A reação foi diluída com dimetilsulfóxido (0,7 mL) e purificada por cromatografia de fase reversa (Método A) para prover #B47 como um sólido. Rendimento: 3 mg, 0,0028 mmol, 18%. HPLC (Protocolo AA) tempo de retenção = 6,65, 6,69 minutos (pureza = 91%). LCMS (Protocolo D): m/z 1069,9 [M+H]+, tempo de retenção = 0,61 minuto.
[0862] Exemplo A17 Preparação de N-(6-aminohexanoil)-L-valil-N-[4-({[(2-{[(3S,5S,7S)-7- {(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6- dioxaspiro[2,5]oct-5-il]acetil}hidrazinil)carbonil]oxi}metil)fenil]-N~5~- carbamoil-L-ornitinamida (#B48).
[0863] Etapa 1. Composto #B9 (113 mg, 0,217 mmol) foi convertido para #B7 bruto como no Procedimento Geral E. LCMS (Protocolo D): m/z 534,1 [M+H]+, tempo de retenção = 0,77 min. O material bruto foi usado na etapa seguinte sem purificação adicional.
[0864] Etapa 2. Síntese de N-(6-aminohexanoil)-L-valil-N-[4-({[(2- {[(3S,5S,7S)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent- 2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3- dien-1-il}-1,6-dioxaspiro[2,5]oct-5-il]acetil}hidrazinil)carbonil]oxi}metil)fenil]-N~5~-carbamoil-L-ornitinamida (#B48). A uma solução de #B7 bruto (60 mg, 0,11 mmol, 1 eq.) em N,N-dimetilformamida (2,8 mL), foi adicionada 2,6- dimetilpiridina (48 mg, 0,448 mmol, 4 eq.), N,N’-Diisopropiletilamina (57,9 mg, 0,448 mmol, 4 eq.) e HOAt (15,2 mg, 0,112 mmol, 1 eq.). A reação foi aquecida para 50°C e agitada por 1,5 hora. A reação foi resfriada para temperatura ambiente e piperidina (191 mg, 2,24 mmoles, 20 eq.) foi adicionada lentamente e agitada por 2,0 horas. A reação foi concentrada in vacuo e purificada por cromatografia de fase reversa (Método C*) para render #B48 como um sólido. Rendimento 6,4 mg, 0,005 mmol, 5,2%. HPLC (Protocolo F): m/z 1052,6 [M+H] +, tempo de retenção = 7,114 minutos (pureza 100%).
[0865] Exemplo A18
[0866] Preparação de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5- {(3R,4R,5R,7S)-4-hidroxi-7-[2-oxo-2-(3H-[1,2,3]triazolo[4,5-b]piridin-3- iloxi)etil]-1,6-dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5- dimetiltetrahidro-2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetate
[0867] Etapa 1. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-oxo-2-(3H-[1,2,3]triazolo[4,5-b]piridin- 3-iloxi)etil]-1,6-dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5- dimetiltetrahidro-2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B49). Uma mistura de #NP1 (1,5 g, ~50% de pureza, 2,8 mmoles, 1,0 eq.), Hexafluorofosfato de O-(7-azabenzotriazol-1-il)-N,N,N‘,N‘- tetrametilurônio (HATU, 1,07 g, 4,5 mmoles, 1,6 eq.), e N,N’- Diisopropiletilamina (base de Hunig, 1,0 mL) em N,N-dimetilformamida (7,0 mL) foi agitada à temperatura ambiente por 40 minutos. A mistura de reação foi filtrada e, em seguida, purificada usando cromatografia de fase reversa (Método F*) para gerar #B49 como um pó branco. Rendimento: 730,7 mg, 80% de rendimento. HPLC (Protocolo N): tempo de retenção = 11,15 minutos (pureza 92%). HRESIMS (Protocolo O) m/z 654,313 [M+H]+. 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 8,84 (dd, J = 4,4, 1,2, 1H), 8,74 (dd, J = 8,3, 1,2, 1H), 7,77 (d, J = 8,2, 1H, D2O permutável), 7,66 (dd, J = 8,3, 4,4, 1H), 6,45 (br d, J = 15,8, 1H), 6,36 (ddq, J = 1,5, 6,5, 6,5, 1H), 6,09 (dd, J = 1,3, 11,7, 1H), 5,86 (dd, J = 11,7, 7,4, 1H), 5,62 (dd, J = 16,0, 5,0, 1H), 5,38 (br dd, J = 7,4, 7,4, 1H), 5,18 (d, J = 6,0, D2O permutável), 4,48 (m, 1H), 4,37 (dd, J = 4,0, 4,0, 1H), 3,61 (m, 1H), 3,54 (dq, 2,1, 6,5, 1H), 3,37 (ddd, J = 6,9, 6,9, 3,1, 1H), 3,34 (m, 2H), 3,26 (dd, J = 16,0, 9,7, 1H), 2,21 (m, 1H), 2,14 (m, 1H), 2,10 (dd, J = 12,7, 8,9, 1H), 1,99 (s, 3H), 1,74 (m, 2H), 1,68 (s, 3H), 1,59 (dd, J = 13,0, 3,3, 1H), 1,50 (m, 1H), 1,25 (d, J = 6,4, 3H), 0,95 (d, J = 6,5, 3H), 0,86 (d, J = 7,0, 3H).
[0868] Exemplo A19
[0869] Preparação de (2S,3Z)-5-({(2R,3R,5S,6S)-2,5-dimetil-6-[(2E,4E)-3-metil-5-{(3S,5S,7S)-7-[2-oxo-2-(3H-[1,2,3]triazolo[4,5- b]piridin-3-iloxi)etil]-1,6-dioxaspiro[2,5]oct-5-il}penta-2,4-dien-1- il]tetrahidro-2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B50).
[0870] Etapa 1. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-2,5-dimetil- 6-[(2E,4E)-3-metil-5-{(3S,5S,7S)-7-[2-oxo-2-(3H-[1,2,3]triazolo[4,5- b]piridin-3-iloxi)etil]-1,6-dioxaspiro[2,5]oct-5-il}penta-2,4-dien-1-il]tetrahidro-2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B50). Uma mistura de #NP2 (284 mg, 92% pura, 0,47 mmol, 1,0 eq), hexa- fluorofosfato de O-(7-azabenzotriazol-1-il)-N,N,N‘,N‘-tetrametilurônio (HATU, 220 mg, 0,58 mmol, 1,2 eq) e N,N’-Diisopropiletilamina (base de Hunig, 0,1 mL) em N,N-dimetilformamida (2 mL) foi agitada à temperatura ambiente por 60 minutos. A mistura de reação foi filtrada e, em seguida, purificada usando cromatografia de fase reversa (Método F*) para gerar #B50 como um pó branco. Rendimento: 307,0 mg, 88% de rendimento HPLC (Protocolo N): tempo de retenção = 13,12 minu- tos (pureza 94%). HRESIMS (Protocolo O) m/z 638,320 [M+H]+. 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 8,83 (dd, J = 4,4, 1,2, 1H), 8,73 (dd, J = 8,5, 1,2, 1H), 7,76 (d, J = 8,1, 1H, D2O permutável), 7,66 (dd, J = 8,5, 4,4, 1H), 6,39 (br d, J = 15,8, 1H), 6,36 (ddq, J = 1,5, 6,5, 6,5, 1H), 6,09 (dd, J = 1,5, 11,7, 1H), 5,87 (dd, J = 11,7, 7,5, 1H), 5,66 (dd, J = 16,1, 4,9, 1H), 5,44 (br dd, J = 7,1, 7,1, 1H), 4,71 (ddd, J = 4,4, 4,4, 4,4, 1H), 4,51 (m, 1H), 3,63 (m, 1H), 3,57 (dq, J = 2,3, 6,6, 1H), 3,41 (ddd, J = 6,9, 6,9, 3,1, 1H), 3,36 (dd, J = 16,0, 4,1, 1H), 3,33 (dd, J = 16,1, 10,0, 1H), 2,74 (d, J = 4,8, 1H), 2,70 (d, J = 4,8, 1H), 2,24 (m, 1H), 2,15 (m, 1H), 1,98 (s, 3H), 1,85 (m, 1H), 1,83 (m, 1H), 1,70 (dd, J = 13,0, 7,5, 1H), 1,66 (dd, J = 13,5, 7,9, 1H), 1,52 (m, 1H), 1,25 (d, J = 6,8, 3H), 1,00 (d, J = 6,5, 3H), 0,89 (d, J = 7,5, 3H).
[0871] Exemplo A20
[0872] Preparação de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-({2-[(iodoacetil)amino]etil}amino)-2- oxoetil]-1,6-dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5- dimetiltetrahidro-2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B52).
[0873] Etapa 1. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-{2-[(2-aminoetil)amino]-2-oxoetil}-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B51). A uma solu- ção de #B49 (50,5 mg, 92% de pureza, 0,08 mmol, 1,0 eq.) e 9H- fluoren-9-ilmetil (2-aminoetil)carbamato (32,1 mg, 0,11 mmol, 1,4 eq.) em N,N-dimetilformamida (1,0 mL), foi adicionado N,N’- Diisopropiletilamina (base de Hunig, 20 μL) com agitação. A mistura de reação foi, então, agitada à temperatura ambiente por 10 minutos. A uma solução de reação, foi lentamente adicionada piperidina (30 μL, 0,35 mmol, 4,4 eq.) e a solução foi agitada à temperatura ambiente por 1 hora. A mistura de reação foi, então, purificada usando cromatografia de fase reversa (Método B*) para gerar #B51 como um pó branco. Rendimento: 38,8 mg, 86% de rendimento HPLC (Protocolo N): tempo de retenção = 6,61 minutos (pureza 97%). LCMS (Protocolo M): m/z 578,8 [M+H]+.
[0874] Etapa 2. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-({2-[(iodoacetil)amino]etil}amino)-2- oxoetil]-1,6-dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro-2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B52). Uma solução de ácido iodoacético (40,3 mg, 0,22 mmol, 7,3 eq.) e N,N’-diciclohhexilcarbodiimida (DCC, 61,6 mg, 0,3 mmol, 10,0 eq.) em N,N-dimetilformamida (2,5 mL) foi agitada à temperatura ambiente por 10 minutos e a solução amarelo claro foi adicionada a #B51 (21,1 mg, 85,0% de pureza, 0,031 mmol, 1,0 eq.) em N,N- dimetilformamida (0,2 mL). A solução resultante foi agitada à temperatura ambiente por 20 minutos. A mistura de reação foi, então, purificada usando cromatografia de fase reversa (Método B*) para gerar #B52 como um pó branco. Rendimento: 10,3 mg, 37% de rendimento HPLC (Protocolo N): tempo de retenção = 8,96 minutos (pureza 37%). LCMS (Protocolo M): m/z 746,3 [M+H]+. 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 8,22 (m, 1H, D2O permutável), 7,91 (m, 1H, D2O permutável), 7,80 (d, J = 7,8, 1H, D2O permutável), 6,36 (dq, J = 6,0, 6,0, 1H), 6,29 (br d, J = 16,0, 1H), 6,11 (d, J = 11,7, 1H), 5,86 (dd, J = 11,7, 7,8, 1H), 5,60 (dd, J = 16,0, 5,5, 1H), 5,51 (br dd, J = 6,6, 6,6, 1H), 5,02 (d, J = 5,0, D2O permutável, 1H), 4,26 (m, 2H), 3,65 (m, 2H), 3,60 (s, 2H), 3,51 (br dd, J = 6,2, 6,2, 1H), 3,24 (m, 1H), 3,08 (br s, 4H), 2,76 (d, J = 5,1, 1H), 2,60 (d, J = 5,1, 1H), 2,51 (m, 1H), 2,47 (m, 1H), 2,28 (m, 1H), 2,18 (m, 1H), 1,98 (s, 3H), 1,86 (m, 1H), 1,80 (m, 2H), 1,70 (s, 3H), 1,65 (m, 1H), 1,49 (dd, J = 12,5, 2,7, 1H), 1,25 (d, J = 6,6, 3H), 1,07 (d, J = 6,5, 3H), 0,95 (d, J = 7,0, 3H).
[0875] Exemplo A21
[0876] Preparação de (2Z,4S)-4-hidroxi-N-{(2R,3R,5S,6S)-6-[(2E,4E)-5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-({2-[(iodoacetil)amino]etil}amino)-2-oxoetil]-1,6-dioxaspiro[2,5]oct-5-il}-3- metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro-2H-piran-3-il}pent-2- enamida (#B54).
[0877] Etapa 1. Síntese de ácido [(3R,5S,7R,8R)-8-hidroxi-7- {(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-hidroxipent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6-dioxaspiro[2,5]oct-5-il]acético (#B4). Uma mistura de #NP1 (192 mg, ~50% de pureza, 0,18 mmol, 1,0 eq.), carbonato de potássio (300 mg, 2,4 mmoles, 13,5 eq.), e metanol (5 mL) foi agitada à temperatura ambiente por 2 horas. A mistura de reação foi neutralizada com ácido acético, filtrada, e, em seguida, purificada usando cromatografia de fase reversa (Método G) para gerar #B4 como um pó branco. Rendimento: 50,2 mg. HPLC (Protocolo N): tempo de retenção = 7,39 minu- tos (pureza 96%). LCMS (Protocolo M): m/z 494,3 [M+H]+.
[0878] Etapa 2. Síntese de (2Z,4S)-N-[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-{2-[(2-aminoetil)amino]-2-oxoetil}-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]-4-hidroxipent-2-enamida (#B53). Uma solução de #B4 (23,4 mg, 0,047 mmol, 1,0 eq.), hexafluorofosfato de O-(7- azabenzotriazol-1-il)-N,N,N‘,N‘-tetrametilurônio (HATU, 34,0 mg, 0,09 mmol, 2,0 eq.), e N,N’-Diisopropiletilamina (20,0 μL) em N,N- dimetilformamida (1,0 mL) foi agitada à temperatura ambiente por 30 minutos. A esta solução, foi adicionado etano-1,2-diamina (80 μl, 1,3 mmol, ~30 eq.) e a solução resultante foi agitada por 1 hora. A mistura de reação foi filtrada e, em seguida, purificada usando cromatografia de fase reversa (Método B*) para gerar #B53 como um pó branco. Rendimento: 31 mg. HPLC (Protocolo N): tempo de retenção = 5,58 minutos (pureza 50%). LCMS (Protocolo M): m/z 536,4 [M+H]+
[0879] Etapa 3. Síntese de (2Z,4S)-4-hidroxi-N-{(2R,3R,5S,6S)-6- [(2E,4E)-5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-({2- [(iodoacetil)amino]etil}amino)-2-oxoetil]-1,6-dioxaspiro[2,5]oct-5-il}-3- metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro-2H-piran-3-il}pent-2- enamida (#B54). Uma solução de ácido iodoacético (35 mg, 0,18 mmol, 6 eq.) e N,N’-diciclohhexilcarbodiimida (DCC, 49,8 mg, 0,24 mmol, 8 eq.) em N,N-dimetilformamida (2 mL) foi agitada à temperatura ambiente por 10 minutos e, em seguida, adicionada a #B53 (31,0 mg, ~50% de pureza, ~0,03 mmol, 1,0 eq.) em N,N-dimetilformamida (0,2 mL). A solução resultante foi agitada à temperatura ambiente por 0,5 hora. A mistura de reação foi purificada usando cromatografia de fase reversa (Método B*) para gerar #B54 bruto como um pó branco (23,0 mg). Este material foi repurificado com um sistema de Gradiente diferente, que gerou #B54 como um pó branco. Rendimento: 15,9 mg, 27% de rendimento, em três etapas. HPLC (Protocolo N): tempo de retenção = 7,10 minutos (pureza 92%). LCMS (Protocolo M) m/z 704,2 [M+H]+. 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 8,22 (m, 1H, D2O permutável), 7,91 (m, 1H, D2O permutável), 7,77 (d, J = 7,8, 1H, D2O permutável), 6,28 (br d, J = 16,0, 1H), 5,98 (d, J = 11,7, 1H), 5,86 (dd, J = 11,7, 7,0, 1H), 5,60 (dd, J = 16,0, 5,5, 1H), 5,51 (br dd, J = 6,6, 6,6, 1H), 5,16 (dq, J = 6,2, 6,2, 1H), 5,11 (d, J = 3,9, 1H, D2O permutável), 5,03 (d, J = 4,5, D2O permutável, 1H), 4,26 (m, 2H), 3,65 (m, 2H), 3,60 (s, 2H), 3,51 (br dd, J = 6,5, 6,5, 1H), 3,25 (m, 1H), 3,08 (br s, 4H), 2,75 (d, J = 4,7, 1H), 2,60 (d, J = 4,7, 1H), 2,51 (m, 1H), 2,47 (m, 1H), 2,27 (m, 1H), 2,22 (m, 1H), 1,86 (m, 1H), 1,80 (m, 2H), 1,70 (s, 3H), 1,65 (m, 1H), 1,49 (dd, J = 12,5, 2,3, 1H), 1,11 (d, J = 6,6, 3H), 1,07 (d, J = 6,0, 3H), 0,95 (d, J = 7,4, 3H).
[0880] Exemplo A22
[0881] Preparação de metil [(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]acetato (#B55).
[0882] Etapa 1. Síntese de metil [(3R,5S,7R,8R)-7-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]acetato (#B55). Uma mistura de #NP1 (9,9 mg, 92% de pureza, 0,018 mmol, 1,0 eq.), carbonato de potássio (40 mg, 0,33 mmol, 18 eq.), e iodometano (30 μL, 0,31 mmol, 17 eq.) em N,N- dimetilformamida (500 μL) foi agitada à temperatura ambiente por 2 horas. A mistura de reação foi neutralizada com ácido acético, filtrada, e, em seguida, purificada usando cromatografia de fase reversa (Mé- todo B*) para gerar #B55 como um pó branco. Rendimento: 7,8 mg, 77% de rendimento. HPLC (Protocolo N): tempo de retenção = 10,7 minutos (pureza 94%). LCMS (Protocolo M) m/z 550,5 [M+H]+. 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 7,80 (d, J = 8,1, 1H, D2O permutável), 6,36 (ddq, J = 1,2, 6,8, 6,8, 1H), 6,28 (br d, J = 15,9, 1H), 6,11 (dd, J = 1,2, 11,7, 1H), 5,87 (dd, J = 11,7, 7,7, 1H), 5,58 (dd, J = 16,1, 5,0, 1H), 5,52 (br dd, J = 7,4, 7,4, 1H), 5,02 (d, J = 5,8, D2O permutável), 4,29 (m, 1H), 4,27 (dd, J = 5,3, 5,3, 1H), 3,65 (m, 2H), 3,60 (s, 3H), 3,51 (ddd, J = 7,0, 7,0, 2,5, 1H), 3,25 (dd, J = 5,8, 5,3, 1H), 2,76 (d, J = 5,0, 1H), 2,65 (dd, 15,4, 8,7, 1H), 2,58 (d, J = 5,0, 1H), 2,57 (dd, J = 15,4, 5,0), 2,30 (m, 1H), 2,21 (m, 1H), 1,98 (s, 3H), 1,86 (dd, J = 13,2, 7,6, 1H), 1,69 (s, 3H), 1,66 (m, 1H), 1,53 (dd, 13,2, 3,9, 1H), 1,25 (d, J = 6,1, 3H), 1,07 (d, J = 6,4, 3H), 0,95 (d, J = 7,4, 3H).
Exemplo A23
[0883] Preparação de ácido [(3R,5S,7R,8R)-7-{(1E,3E)-5- [(2S,3S,5R,6R)-3,6-dimetil-5-({(2Z,4S)-4-[(piperidin-1-ilcarbonil)oxi]pent-2-enoil}amino)tetrahidro-2H-piran-2-il]-3-metilpenta- 1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5-il]acético (#B60) e (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5-{(3R,4R,5R,7S)-4-hidroxi-7-[2- ({2-[(iodoacetil)amino]etil}amino)-2-oxoetil]-1,6-dioxaspiro[2,5]oct-5-il}- 3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro-2H-piran-3-il}amino)-5- oxopent-3-en-2-il piperidina-1-carboxilato (#B62).
[0884] Etapa 1. Síntese de metil [(3R,5S,7R,8R)-7-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-(1-etoxietoxi)-1,6-dioxaspiro[2,5] oct-5-il]acetato (#B56). A uma mistura de #NP1 (195 mg, ~50% de pureza, 0,18 mmol, 1,0 eq.) e carbonato de potássio (200 mg, 1,6 mmol, 9 eq.) em N,N-dimetilformamida (4.0 mL), foi adicionado iodometano (500 μL, 18 eq.). A solução resultante foi agitada por 120 minutos. A mistura de reação foi filtrada e o filtrado dividido entre água e etil acetato (10 mL cada fase). A camada orgânica foi seca sobre sulfato de magnésio anidro e evaporada sob pressão reduzida para gerar #B55 como uma película (191,2 mg, ~50% de pureza). Em seguida, #B55 bruto (191,0 mg, 0,18 mmol, 1,0 eq.) foi misturado com p-toluenossulfonato de piridínio (PPTS, 56,1 mg, 0,22 mmol, 1.2 eq.), e etil vinil éter (2,5 ml, 43 mmoles) em diclo- rometano anidro (2,0 mL) foi agitado à temperatura ambiente por 1 ho-ra. A mistura de reação foi parcialmente evaporada sob pressão reduzida e, em seguida, dividida entre solução aquosa de bicarbonato de sódio/etil acetato (10 mL) (saturados, 10 mL). A camada orgânica foi seca sobre sulfato de magnésio anidro e, em seguida, evaporada sob pressão reduzida para gerar #B56 (187,1 mg) HPLC (Protocolo N): tempo de retenção = 13,2 minutos (pureza 50%). LCMS (Protocolo M): m/z 549,5 [M+H-CHCH3OCH2CH3]+ que foi usado como é na reação seguinte.
[0885] Etapa 2. Síntese de metil [(3R,5S,7R,8R)-8-(1-etoxietoxi)-7- {(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-hidroxipent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6- dioxaspiro[2,5]oct- 5-il]acetato (#B57). Uma suspensão de #B56 (187,1 mg, ~50% de pureza, 0,15 mmol, 1,0 eq.), carbonato de potássio (120 mg, 0,98 mmol, 6,5 eq.) em metanol (4 mL) foi agitada à temperatura ambiente por 1 hora. A mistura de reação foi, então, filtrada e evaporada até secagem sob pressão reduzida para gerar #B57 (171,5 mg) que foi usado como é na reação seguinte. HPLC (Protocolo N): tempo de retenção = 9,9 minutos (pureza 50%). LCMS (Protocolo M): m/z 507,5 [M+H-CHCH3OCH2CH3]+.
[0886] Etapa 3. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-4-(1-etoxietoxi)-7-(2-metoxi-2-oxoetil)-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il piperidina-1-carboxilato (#B58). Uma solução de #B57 (171 mg, 50% de pureza, 0,15 mmol, 1,0 eq.), bis(p-nitrofenil)carbonato (320,1 mg, 1,0 mmol, 7 eq.), 4- (dimetilamino)piridina (9,8 mg, 0,08 mmol, 0,5 eq.), e N,N’- diisopropiletilamina (base de Hunig, 150 μL) em diclorometano (4,0 mL) foi agitada à temperatura ambiente por 6 horas. A uma solução de reação, foi lentamente adicionada piperidina (500 μL, 5,8 mmoles, 38 eq.) e a solução amarela resultante foi agitada à temperatura ambiente por 15 minutos. Água gelada (20 mL) foi adicionada e o solvente orgânico foi removido por evaporação sob pressão reduzida. O precipitado assim formado foi coletado por filtração e, em seguida, seco sob vácuo para gerar #B58 (347,0 mg), que foi usado como é na reação seguinte HPLC (Protocolo N): tempo de retenção = 13,20 minutos (pureza 25%). LCMS (Protocolo M): m/z 691,7 [M+H]+.
[0887] Etapa 4. Síntese de ácido [(3R,5S,7R,8R)-7-{(1E,3E)-5- [(2S,3S,5R,6R)-3,6-dimetil-5-({(2Z,4S)-4-[(piperidin-1-ilcarbonil)oxi]pent-2-enoil}amino)tetrahidro-2H-piran-2-il]-3-metilpenta- 1,3-dien-1-il}-8-(1-etoxietoxi)-1,6-dioxaspiro[2,5]oct-5-il]acético (#B59). A uma solução de #B58 (347 mg, ~25% de pureza, 0,13 mmol, 1,0 eq.) em acetonitrila (10 mL), foi adicionado 1M hidróxido de lítio (1 mL) e água (1 mL). A solução turva resultante foi agitada à temperatura ambiente por 1 hora e se tornou gradualmente clara. Adicional de 1M hidróxido de lítio (1,0 mL) foi adicionado e a solução foi adicionalmente agitada por 2 horas. A mistura de reação foi dividida entre n-butanol (30 mL) e água (30 mL). A camada superior foi lavada com H2O (20 mL), e, em seguida, evaporada até secagem sob pressão reduzida para gerar #B59 (280,2 mg) que foi usado como é na reação seguinte HPLC (Protocolo N): tempo de retenção = 7,43 minutos (pureza 30%). LCMS (Protocolo M): m/z 677,4 [M+H]+.
[0888] Etapa 5. Síntese de ácido [(3R,5S,7R,8R)-7-{(1E,3E)-5- [(2S,3S,5R,6R)-3,6-dimetil-5-({(2Z,4S)-4-[(piperidin-1-ilcarbonil)oxi]pent-2-enoil}amino)tetrahidro-2H-piran-2-il]-3-metilpenta- 1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5-il]acético (#B60). Uma solução de #B59 (280,2 mg, ~30% de pureza, 0,12 mmol, 1,0 eq.) e p- toluenossulfonato de piridínio (PPTS, 250,4 mg, 1 mmol, 8.0 eq.) em metanol (5 mL) foi agitada à temperatura ambiente por 4 horas e, em seguida, deixada em repouso a 4°C por 18 horas. A mistura de reação foi, então, purificada usando cromatografia de fase reversa (Método B*) para gerar #B60 como um pó branco. Rendimento: 78,4 mg, (40% de rendimento, sobre etapas 1-5) HPLC (Protocolo N): tempo de re- tenção = 10,84 minutos (pureza 96,7%). LCMS (Protocolo M): m/z 605,4 [M+H]+.
[0889] Etapa 6. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-{2-[(2-aminoetil)amino]-2-oxoetil}-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il piperidina-1-carboxilato (#B61). Uma solução de #B60 (38,2 mg, 0,06 mmol, 1,0 eq.), hexafluo- rofosfato de O-(7-azabenzotriazol-1-il)-N,N,N‘,N‘-tetrametilurônio (HATU, 40,1 mg, 0,1 mmol, 1,7 eq.), e N,N’-Diisopropiletilamina (base de Hunig, 20,0 μL) em N,N-dimetilformamida (1,0 mL) foi agitada à temperatura ambiente por 20 minutos. A esta solução, foi adicionada 1,2-etilenodiamina (120 μL, 2 mmoles, 33 eq.) e a solução resultante foi agitada por 20 minutos. A mistura de reação foi filtrada e, em seguida, purificada usando cromatografia de fase reversa (Método B*) para gerar #B61 como um pó branco. Rendimento: 14,2 mg, xx%) HPLC (Protocolo N): tempo de retenção = 7,86 minutos (pureza 70%). LCMS (Protocolo M): m/z 647,8 [M+H]+.
[0890] Etapa 7. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-({2-[(iodoacetil)amino]etil}amino)-2- oxoetil]-1,6-dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5- dimetiltetrahidro-2H-piran-3-il}amino)-5-oxopent-3-en-2-il piperidina-1- carboxilato (#B62). Uma solução de ácido iodoacético (20,3 mg, 0,1 mmol, 7 eq.) e N,N’-diciclohhexilcarbodiimida (DCC, 31,6 mg, 0,15 mmol, 10 eq.) em N,N-dimetilformamida (2 mL) foi agitada à temperatura ambiente por 10 minutos e, em seguida, adicionada a um frasco contendo #B61 (14,0 mg, ~70% puro, 0,015 mmol, 1,0 eq.) em N,N- dimetilformamida (0,2 mL). A solução resultante foi agitada à temperatura ambiente por 30 minutos. A mistura de reação foi purificada usando cromatografia de fase reversa (Método B*) para gerar #B62 como um pó branco. Rendimento: 3,7 mg, (11%, durante as etapas 6-7) HPLC (Protocolo N): tempo de retenção = 10,48 minutos (pureza 94%). LCMS (Protocolo M): m/z 815,4 [M+H]+, 837,4 [M+Na]+. 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) □ □ 8,22 (m, 1H, D2O permutável), 7,91 (m, 1H, D2O permutável), 7,78 (d, J = 7,8, 1H, D2O permutável), 6,29 (br d, J = 16,1, 1H), 6,22 (dq, J = 7,0, 7,0, 1H), 6,09 (d, J = 11,7, 1H), 5,89 (dd, J = 11,3, 7,4, 1H), 5,61 (dd, J = 16,0, 5,1, 1H), 5,51 (br dd, J = 6,2, 6,2, 1H), 5,03 (d, J = 5,5, D2O permutável), 4,26 (m, 2H), 3,65 (m, 2H), 3,60 (s, 2H), 3,51 (br dd, J = 6,2, 6,2, 1H), 3,30 (m, 4H), 3,24 (dd, J = 5,0, 5,0, 1H), 3,12-3,08 (m, 4H), 3,00 (d, 1H), 2,80(s, 4H), 2,75 (d, J = 5,1, 1H), 2,60 (d, J = 5,1, 1H), 2,49 (m, 2H), 2,28(m, 1H), 2,21 (m, 1H), 1,89 (dd, J = 13,2, 8,6, 1H), 1,80 (m, 2H), 1,70(s, 3H), 1,64 (m, 1H), 1,50 (dq, J = 13,2, 3,1, 1H), 1,52 (m, 2H), 1,42(m, 4H), 1,25 (d, J = 6,2, 3H), 1,07 (d, J = 6,0, 3H), 0,95 (d, J = 7,4, 3H).
[0891] Exemplo A24
[0892] Preparação de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-oxo-2-(propilamino)etil]-1,6-dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B63).
[0893] Etapa 1. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-oxo-2-(propilamino)etil]-1,6-dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B63). A uma solução de #B49 (43,2 mg, 92,1% de pureza, 0,065 mmol, 1,0 eq.) em N,N-dimetilformamida (1,0 mL), foi adicionada propilamina pura (30 μL, 0,5 mmol, 7,0 eq.). A solução resultante foi agitada por 10 minutos. A mistura de reação foi dividida entre H2O e etil acetato (10 mL cada). A camada orgânica foi seca sobre sulfato de magnésio anidro e evaporada sob pressão reduzida para gerar #B63 Rendimento: 40,4 mg, 100%. HPLC (Protocolo N): tempo de retenção = 9,73 minutos (pureza 89%). HRESIMS (Protocolo O) m/z 577,3478 [M+H]+. 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 7,83 (t, J = 6,0, 1H, D2O permutável), 7,81 (d, J = 8,1, 1H, D2O permutável), 6,36 (ddq, J = 1,4, 6,5, 6,5, 1H), 6,28 (br d, J = 15,9, 1H), 6,11 (dd, J = 1,4, 11,7, 1H), 5,87 (dd, J = 11,7, 7,5, 1H), 5,59 (dd, J = 15,9, 5,4, 1H), 5,51 (br dd, J = 7,1, 7,1, 1H), 5,02 (d, J = 5,4, D2O permutável), 4,26 (dd, J = 5,0, 5,0, 1H), 4,24 (m, 1H), 3,65 (m, 1H), 3,64 (m, 1H), 3,49 (ddd, J = 7,0, 7,0, 2,6, 1H), 3,24 (dd, J = 5,0, 5,0, 1H), 3,01 (m, 1H), 2,96 (m, 1H), 2,75 (d, J = 5,2, 1H), 2,58 (d, J = 5,2, 1H), 2,52 (m, 1H), 2,29 (ddd, J = 15,5, 7,1, 7,1, 1H), 2,21 (m, 1H), 2,20 (dd, J = 14,0, 4,8, 1H), 1,98 (s, 3H), 1,83 (dd, J = 13,4, 5,0, 1H), 1,80 (m, 2H), 1,69 (s, 3H), 1,65 (m, 1H), 1,48 (dd, 12,7, 3,9, 1H), 1,38 (dq, J = 7,5, 7,5, 2H), 1,25 (d, J = 6,6, 3H), 1,07 (d, J = 6,8, 3H), 0,95 (d, J = 7,5, 3H), 0,82 (t, J = 7,5, 3H).
[0894] Exemplo A25
[0895] Preparação de (2S,3Z)-5-({(2R,3R,5S,6S)-2,5-dimetil-6-[(2E,4E)-3-metil-5-{(3S,5S,7S)-7-[2-oxo-2-(propilamino)etil]-1,6- dioxaspiro[2,5]oct-5-il}penta-2,4-dien-1-il]tetrahidro-2H-piran-3- il}amino)-5-oxopent-3-en-2-il acetato (#B64).
[0896] Etapa 1. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-2,5-dimetil- 6-[(2E,4E)-3-metil-5-{(3S,5S,7S)-7-[2-oxo-2-(propilamino)etil]-1,6- dioxaspiro[2,5]oct-5-il}penta-2,4-dien-1-il]tetrahidro-2H-piran-3- il}amino)-5-oxopent-3-en-2-il acetato (#B64). Uma solução de #NP2 (28,7 mg, 91% de pureza, 0,055 mmol, 1,0 eq.), Hexafluorofosfato de O-(7-azabenzotriazol-1-il)-N,N,N‘,N‘-tetrametilurônio (HATU, 24,8 mg, 0,065 mmol, 1,2 eq.), e N,N’-Diisopropiletilamina (base de Hunig, 30 μL) em N,N-dimetilformamida (0,5 mL) foi agitada à temperatura ambiente por 30 minutos. A esta solução, foi adicionada propilamina pura (30 μL, 0,5 mmol, 7,0 eq.) e a mistura de reação resultante foi agitada à temperatura ambiente por 1 hora. A mistura de reação foi filtrada e, em seguida, purificada usando cromatografia de fase reversa (Método B*) para gerar #B64 como um pó branco. Rendimento: 34,1 mg, 86%. HPLC (Protocolo N): tempo de retenção = 13,11 minutos (pureza 100%). LCMS (Protocolo M): m/z 561,6 [M+H]+. 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 7,83 (t, J = 6,0, 1H, D2O permutável), 7,81 (d, J = 8,0, 1H, D2O permutável), 6,36 (ddq, J = 1,6, 7,4, 6,6, 1H), 6,24 (br d, J = 16,0, 1H), 6,10 (dd, J = 1,6, 12,0, 1H), 5,87 (dd, J = 11,8, 7,6, 1H), 5,58 (dd, J = 15,9, 5,4, 1H), 5,50 (br dd, J = 7,3, 7,3, 1H), 4,55 (ddd, J = 5,3, 5,3, 5,3, 1H), 4,29 (dddd, J = 9,5, 5,3, 5,3, 5,3, 1H), 3,65 (m, 1H), 3,64 (m, 1H), 3,48 (ddd, J = 7,1, 7,1, 2,6, 1H), 3,01 (m, 1H), 2,96 (m, 1H), 2,63 (d, J = 5,0, 1H), 2,61 (d, J = 5,0, 1H), 2,59 (dd, J = 14,2, 8,8, 1H), 2,31 (ddd, J = 16,1, 7,5, 7,0, 1H), 2,21 (dd, J = 14,1, 5,0, 1H), 2,19 (m, 1H), 1,96 (s, 3H), 1,81 (m, 1H), 1,79 (m, 1H), 1,77 (dd, J = 13,0, 4,0, 1H), 1,69 (s, 3H), 1,66 (m, 2H), 1,37 (dq, J = 7,5, 7,5, 2H), 1,36 (m, 1H), 1,25 (d, J = 6,6, 3H), 1,07 (d, J = 6,8, 3H), 0,95 (d, J = 7,5, 3H), 0,82 (t, J = 7,5, 3H).
[0897] Exemplo A26
[0898] Preparação de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-oxo-2-(propilamino)etil]-1,6-dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il piperidina-1-carboxilato (#B66).
[0899] Etapa 1. Síntese de (3R,4R,5R,7S)-5-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-hidroxipent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-7-[2-oxo-2- (propilamino)etil]-1,6-dioxaspiro[2,5] oct-4-il acetato (#B64a). A uma solução de #B63 (44,0 mg, ~90% de pureza, 0,076 mmol, 1,0 eq.) em piridina (0,5 mL), foi adicionado anidrido acético (150 μL, 1,6 mmol, 21,0 eq.)). A mistura de reação foi, então, agitada à temperatura ambiente por 3 horas. A mistura de reação foi transferida para água gelada (10 mL), agitada por 20 minutos, e, em seguida, dividida entre etil acetato (30 mL) e água (30 mL). A camada orgânica foi lavada com água (3 x 20 mL) e evaporada até secagem. O resíduo foi dissolvido em di- metil sulfóxido (150 μL) e a solução foi lentamente adicionada a solução tampão 1M Tris (pH 7,0) que continha uma esterase produzida por Bucillus stearothermorphillus (Sigma 69509, 0,5 mg/mL, 15 mL total). A reação foi agitada por uma hora e, em seguida, dividida entre etil acetato (2 x 20 mL) e água (20 mL). As camadas orgânicas combinadas foram lavadas com água (2 x 20 mL) e, em seguida, evaporadas sob pressão reduzida para gerar #B64a como um pó esbranquiçado Rendimento: 44,9 mg, (presumir quantitativa) HPLC (Protocolo N): tempo de retenção = 9,51 minutos (pureza 88%). LCMS (Protocolo M): m/z 577,6 [M+H]+.
[0900] Etapa 2. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3R,4R,5R,7S)-4-(acetiloxi)-7-[2-oxo-2-(propilamino)etil]-1,6- dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino) -5-oxopent-3-en-2-il piperidina-1-carboxilato (#B65). Uma solução de #B64a (14,7 mg, 0,025 mmol, 1,0 eq.), bis(p- nitrofenil)carbonato (38,4 mg, 0,13 mmol, 5 eq.), p-dimetilaminopiridina (1,6 mg, 0,013 mmol, 0,5 eq.), e N,N’-Diisopropiletilamina (base de Hunig, 30 μL) em diclorometano (1 mL) foi agitada à temperatura ambiente por 16 horas. A esta solução de reação, foi lentamente adicionada piperidina (60 μL, 0,7 mmol, 28 eq.) e a solução foi agitada à temperatura ambiente por 15 minutos. Água gelada (10 mL) foi adicionada e o solvente orgânico foi removido por evaporação sob pressão reduzida. O precipitado foi coletado por filtração, lavado com água, e, em seguida, evaporado sob pressão reduzida para gerar #B65. Rendimento 26,2 mg, (presumir quantitativa) HPLC (Protocolo N): tempo de retenção = 13,1 minutos (pureza 45%). LCMS (Protocolo M): m/z 688,5 [M+H]+.
[0901] Etapa 3. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-oxo-2-(propilamino)etil]-1,6- dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il piperidina-1-carboxilato (#B66). Uma solução de #B65 (26,1 mg, ~45% de pureza, 0,017 mmol, 1,0 eq.), carbonato de potássio (51 mg, 0,41 mmol, 24 eq.) em metanol (1,5 mL) foi agitada à temperatura ambiente por 1 hora. A mistura de reação foi filtrada e o filtrado dividido entre etil acetato e água (10 mL cada fase). A camada orgânica foi seca sobre sulfato de magnésio anidro e evaporada sob pressão reduzida até secagem, que foi, então, purificada usando cromatografia de fase reversa (Método B*) para gerar #B66 como um pó branco. Rendimento: 7,3 mg, (44% nas etapas 1-3). HPLC (Protocolo N): tempo de retenção = 11,44 minutos (pureza 96,7%). LCMS (Protocolo M): m/z 646,4 [M+H]+. 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 7,83 (t, J = 5,6, 1H, D2O permutável), 7,79 (d, J = 8,0, 1H, D2O permutável), 6,28 (br d, J = 16,1, 1H), 6,22 (ddq, J = 1,4, 6,8, 6,8, 1H), 6,08 (dd, J = 1,4, 11,8, 1H), 5,88 (dd, J = 11,7, 7,4, 1H), 5,59 (dd, J = 15,7, 5,3, 1H), 5,51 (br dd, J = 7,0, 7,0, 1H), 5,02 (d, J = 5,3, D2O permutável), 4,27 (dd, J = 5,0, 5,0, 1H), 4,23 (m, 1H), 3,65 (m, 1H), 3,64 (m, 1H), 3,49 (ddd, J = 7,0, 7,0, 2,6, 1H), 3,30 (m, 4H), 3,24 (dd, J = 5,0, 5,0, 1H), 3,01 (m, 1H), 2,96 (m, 1H), 2,75 (d, J = 5,2, 1H), 2,58 (d, J = 5,2, 1H), 2,51 (m, 1H), 2,29 (m, 1H), 2,22 (m, 1H), 2,20 (dd, J = 14,0, 4,8, 1H), 1,82 (dd, J = 13,3, 8,2, 1H), 1,79 (m, 2H), 1,69 (s, 3H), 1,65 (m, 1H), 1,51 (m, 2H), 1,49 (dd, 13,3, 5,0, 1H), 1,42 (m, 4H), 1,38 (dq, J = 7,5, 7,5, 2H), 1,25 (d, J = 6,5, 3H), 1,07 (d, J = 6,4, 3H), 0,95 (d, J = 7,4, 3H), 0,82 (t, J = 7,5, 3H).
[0902] Exemplo A27
[0903] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,4R,5R,7S)-7-{2-[(2,5-dioxopirrolidin-1-il)oxi]-2-oxoetil}-4-hidroxi- 1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-ilpiperidina-1- carboxilato (#B67) e 2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5- {(3R,4R,5R,7S)-4-hidroxi-7-[2-oxo-2-(propilamino)etil]-1,6- dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il piperidina-1-carboxilato (#B67).
[0904] Etapa 1. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-{2-[(2,5-dioxopirrolidin-1-il)oxi]-2-oxoetil}-4-hidroxi- 1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-ilpiperidina-1- carboxilato (#B67). Uma mistura de #B60 (Exemplo A23, 23,0 mg, 96,7% de pureza, 0,038 mmol, 1,0 eq.) e N,N’-diciclohexilcarbodiimida (DCC, 13,1 mg, 0,06 mmol, 1,7 eq.) em N,N-dimetilformamida (0,8 mL) foi agitada à temperatura ambiente por 20 minutos. A esta solução, foi adicionado N-hidroxil succinimida (34,5 mg, 0,3 mmol, 7.7 eq.) em N,N-dimetilformamida (0,2 mL). A solução resultante foi agitada à temperatura ambiente por 16 horas. A mistura de reação foi filtrada e, em seguida, purificada usando cromatografia de fase reversa (Método B*) para gerar #B67 como um pó branco. Rendimento: 9,4 mg, 39%. HPLC (Protocolo N): tempo de retenção = 11,04 minutos (pureza 100%). 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) □ □ 7,78 (d, J = 8,0, 1H, D2O permutável), 6,35 (br d, J = 16,1, 1H), 6,22 (dq, J = 7,0, 7,0, 1H), 6,08 (d, J = 11,7, 1H), 5,89 (dd, J = 11,7, 7,4, 1H), 5,60 (dd, J = 16,0, 5,1, 1H), 5,52 (br dd, J = 7,0, 7,0, 1H), 5,08 (d, J = 6,2, D2O permutável), 4,29 (m, 2H), 3,65 (m, 2H), 3,49 (br dd, J = 7,0, 7,0, 1H), 3,29 (m, 5H), 3,00 (d, J = 6,6, 2H), 2,80 (s, 4H), 2,79 (d, J = 5,2, 1H), 2,60 (d, J = 5,2, 1H), 2,28 (m, 1H), 2,21 (m, 1H), 1,95 (dd, J = 13,2, 8,6, 1H), 1,81 (m, 2H), 1,69 (s, 3H), 1,64 (m, 1H), 1,58 (dq, J = 13,2, 3,1, 1H), 1,52 (m, 2H), 1,43 (m, 4H), 1,25 (d, J = 6,2, 3H), 1,07 (d, J = 6,2, 3H), 0,95 (d, J = 7,0, 3H).
[0905] Exemplo A28
[0906] Preparação de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-oxo-2-(propilamino)etil]-1,6- dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il 2-metilpropanoato (#B71).
[0907] Etapa 1. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3R,4R,5R,7S)-4-(1-etoxietoxi)-7-[2-oxo-2-(propilamino)etil]-1,6- dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B68). Uma solução de #B63 (Exemplo A24, 35,0 mg, 92% de pureza, 0,06 mmol, 1,0 eq.), p-toluenossulfonato de piridínio (PPTS, 7,1 mg, 0,028 mmol, 0,4 eq.), e etil vinil éter (0,5 mL, 8,6 mmoles, grande quantidade em excesso) em diclorometano anidro (1,0 mL) foi agitada à temperatura ambiente por 2 horas. A mistura de reação foi dividida entre diclorometano (10 mL)/água(10 mL). A camada orgânica foi seca sobre sulfato de magnésio anidro e, em seguida, evaporada sob pressão reduzida para gerar #B68. Rendimento: 36,1 mg, HPLC (Protocolo N): tempo de retenção = 12,33 minutos (pureza 90%). LCMS (Protocolo M): m/z 577,6 [M+H-CHCH3OCH2CH3]+.
[0908] Etapa 2. Síntese de (2Z,4S)-N-{(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3R,4R,5R,7S)-4-(1-etoxietoxi)-7-[2-oxo-2-(propilamino)etil]-1,6- dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}-4-hidroxipent-2-enamida (#B69). Uma suspensão de #B68 (36,1 mg, 0,05 mmol, 1,0 eq.) e carbonato de potássio (50 mg, 0,4 mmol, 8 eq.) em metanol (1,0 mL) foi agitada à temperatura ambiente por 1 hora. A mistura de reação foi, então, filtrada e evaporada até secagem sob pressão reduzida para gerar #B69. Rendimento: 33,4 mg. HPLC (Protocolo N): tempo de retenção = 10,458 e 10,459 minutos (pureza 87,6%). LCMS (Protocolo M): m/z 535,5 [M+H- CHCH3OCH2CH3]+.
[0909] Etapa 3. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3R,4R,5R,7S)-4-(1-etoxietoxi)-7-[2-oxo-2-(propilamino)etil]-1,6- dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il 2-metilpropanoato (#B70). Uma solução de #B69 (33,0 mg, 0,049 mmol, 1,0 eq.) e anidrido isobu- tírico (100 μL, 0,75 mmol, 15 eq.) em piridina (500 μL) foi agitada a 35°C por 24 horas. A mistura de reação foi, então, evaporada sob pressão reduzida para gerar #B70. Rendimento: 37,3 mg, HPLC (Protocolo N): tempo de retenção = 14,06 (pureza 88,3%). LCMS (Protocolo M): m/z 605,6 [M+H-CHCH3OCH2CH3]+.
[0910] Etapa 4. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-oxo-2-(propilamino)etil]-1,6-dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il 2-metilpropanoato (#B71). Uma solução de #B70 (17,8 mg, 0,023 mmol, 1,0 eq.), p- toluenossulfonato de piridínio (60 mg, 0,24 mmol, 10 eq.) em metanol anidro (2,0 mL) foi agitada à temperatura ambiente por 60 minutos. A mistura de reação foi, então, purificada usando cromatografia de fase reversa (Método B*) para gerar #B71 como um pó branco. Rendimento: 8 mg, (50% nas etapas 1-4). HPLC (Protocolo N): tempo de retenção = 11,50 minutos (pureza 100%). HRESIMS (Protocolo O) m/z 605,3798 (M+H)+. 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 7,83 (t, J = 6,0, 1H, D2O permutável), 7,81 (d, J = 8,1, 1H, D2O permutável), 6,35 (ddq, J = 1,3, 6,5, 6,5, 1H), 6,28 (br d, J = 15,9, 1H), 6,12 (dd, J = 1,3, 11,7, 1H), 5,86 (dd, J = 11,7, 7,5, 1H), 5,59 (dd, J = 15,9, 5,4, 1H), 5,51 (br dd, J = 7,1, 7,1, 1H), 5,02 (d, J = 5,4, D2O permutável), 4,26 (dd, J = 5,0, 5,0, 1H), 4,24 (m, 1H), 3,65 (m, 1H), 3,64 (m, 1H), 3,49 (ddd, J = 7,0, 7,0, 2,6, 1H), 3,24 (dd, J = 5,0, 5,0, 1H), 3,01 (m, 1H),2,96 (m, 1H), 2,75 (d, J = 5,2, 1H), 2,58 (d, J = 5,2, 1H), 2,52 (m, 1H),2,48 (sept, J = 6,5, 1H), 2,29 (ddd, J = 15,5, 7,1, 7,1, 1H), 2,21 (m, 1H),2,20 (dd, J = 14,0, 4,8, 1H), 1,83 (dd, J = 13,4, 5,0, 1H), 1,80 (m, 2H),1,69 (s, 3H), 1,65 (m, 1H), 1,48 (dd, 12,7, 3,9, 1H), 1,38 (dq, J = 7,5, 7,5, 2H), 1,25 (d, J = 6,6, 3H), 1,07 (d, J = 6,8, 9H), 0,95 (d, J = 7,5, 3H), 0,82 (t, J = 7,5, 3H).
[0911] Exemplo A29
[0912] Preparação de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-oxo-2-(piperidin-1-il)etil]-1,6- dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B72).
[0913] Etapa 1. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-oxo-2-(piperidin-1-il)etil]-1,6-dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B72). Uma mistura de #NP1 (163,0 mg, 92% de pureza, 0,27 mmol, 1,0 eq.), hidrato de 1- hidroxibenzotriazol (HOBT, 160,0 mg, 1 mmol, 4 eq.), e 1-etil-3-(3- dimetilaminopropil) carbodiimida-HCl (EDC, 195,0 mg, 1 mol, 4 eq.) em N,N-dimetilformamida (4,0 mL) foi agitada a 0oC por 30 min. A esta solução, foram subsequentemente adicionadas trietilamina (50 μL) e pi- peridina (180 μL, 2,1 mmoles, 7,5 eq.) a 0°C. A mistura de reação resultante foi agitada à temperatura ambiente por 3 horas e a 0°C por 16 horas. A mistura de reação foi, então, dividida entre etil acetato (2 x 20 mL) e água (20 mL). A camada orgânica combinada foi lavada com água (2 x 10 mL), seca sobre sulfato de magnésio anidro, e, em seguida, evaporada sob pressão reduzida para gerar #B72 bruto como um vidro esbranquiçado (223,5 mg, 79,5% de pureza). Uma porção deste material (33,1 mg) foi purificada usando cromatografia de fase reversa (Método B*) para gerar #B72 como um pó branco. Rendimento: 27,4 mg, 100% HPLC (Protocolo N): tempo de retenção = 10,58 minutos (pureza 98%). LCMS (Protocolo M): m/z 603,7 [M+H]+. 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 7,80 (d, J = 7,8, 1H, D2O permutável), 6,36 (dq, J = 6,0, 6,0, 1H), 6,31 (br d, J = 16,0, 1H), 6,11 (d, J = 11,7, 1H), 5,87 (dd, J = 11,7, 7,8, 1H), 5,60 (dd, J = 16,0, 5,5, 1H), 5,52 (br dd, J = 7,0, 7,0, 1H), 4,98 (d, J = 5,8, 1H, D2O permutável), 4,25 (m, 2H), 3,65 (m, 2H), 3,49 (br dd, J = 6,2, 6,2, 1H), 3,40 (m, 4H), 3,25 (dd, 5,8, 5,1, 1H), 3,08 (br s, 4H), 2,76 (d, J = 5,1, 1H), 2,68 (dd, J = 15,2, 7,0, 1H), 2,58 (d, J = 5,1, 1H), 2,50 (m, 1H), 2,29 (m, 1H), 2,20 (m, 1H), 1,98 (s, 3H), 1,86 (m, 1H), 1,80 (m, 2H), 1,70 (s, 3H), 1,65 (m, 1H), 1,56 (m, 3H), 1,48 (m, 2H), 1,40 (m, 2H), 1,25 (d, J = 6,2, 3H), 1,07 (d, J = 6,2, 3H), 0,95 (d, J = 7,0, 3H).
[0914] Exemplo A30
[0915] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,4R,5R,7S)-7-{2-[(trans-3-aminociclobutil)amino]-2-oxoetil}-4- hidroxi-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5- dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B73).
[0916] Etapa 1. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,4R,5R,7S)-7-{2-[(trans-3-aminociclobutil)amino]-2-oxoetil}-4- hidroxi-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5- dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B73). Uma mistura de #NP1 (50,2 mg, 94% pura, 0,09 mmol, 1,0 eq), hidrato de 1-hidroxibenzotriazol (HOBT, 65,5 mg, 0,43 mmol, 4,7 eq.), 1-etil-3-(3-dimetilaminopropil) carbodiimida-HCl (EDC, 70 mg, 0,37 mmol, 4 eq.) em N,N-dimetilformamida (3,0 mL) foi agitada a 0°C por 30 minutos. A esta solução, foram subsequentemente adicionados trans-1,3-diaminociclobutano (112 mg, 1,3 mmol, 14 eq.) em N,N- dimetilformamida (1,0 mL) e trietilamina (200 μL) a 0°C. A mistura de reação resultante foi agitada à temperatura ambiente por 15 minutos. A mistura de reação foi neutralizada com ácido acético, filtrada, e, em seguida, purificada usando cromatografia de fase reversa (Método B*) para gerar #B73 como um vidro incolor. Rendimento: 64,3 mg, 100%. HPLC (Protocolo N): tempo de retenção = 6,66 minutos (pureza 85,2%). LCMS (Protocolo M): m/z 604,6 [M+H]+. 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 8,26 (d, J = 7,0, 1H, D2O permutável), 7,80 (d, J = 7,9, 1H, D2O permutável), 6,36 (dq, J = 6,0, 6,0, 1H), 6,28 (br d, J = 16,0, 1H), 6,11 (d, J = 11,3, 1H), 5,88 (dd, J = 11,7, 7,8, 1H), 5,60 (dd, J = 16,0, 5,8, 1H), 5,49 (br dd, J = 6,6, 6,6, 1H), 5,04 (m, 1H, D2O permutável), 4,37 (m, 1H), 4,27-4,21 (m, 2H), 3,65 (m, 3H), 3,50 (br dd, J = 5,5, 5,5, 1H), 3,26 (d, J = 4,3, 1H), 2,76 (d, J = 4,7, 1H), 2,58 (d, J = 4,7, 1H), 2,48 (m, 1H), 2,29 (m, 1H), 2,22-2,11 (m, 6H), 1,98(s, 3H), 1,82 (m, 1H), 1,80 (m, 2H), 1,70 (s, 3H), 1,65 (m, 1H), 1,49 (dd, J = 12,5, 2,7, 1H), 1,25 (d, J = 6,2, 3H), 1,07 (d, J = 6,0, 3H), 0,95 (d, J = 7,4, 3H).
[0917] Exemplo A31
[0918] Preparação de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-({trans-3- [(iodoacetil)amino]ciclobutil}amino)-2-oxoetil]-1,6-dioxaspiro[2,5]oct-5- il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro-2H-piran-3-il}amino)- 5-oxopent-3-en-2-il acetato (#B74).
[0919] Etapa 1. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-({trans-3- [(iodoacetil)amino]ciclobutil}amino)-2-oxoetil]-1,6-dioxaspiro[2,5]oct-5- il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro-2H-piran-3-il}amino)- 5-oxopent-3-en-2-il acetato (#B74). Uma solução de ácido iodoacético (38,6 mg, 0,21 mmol, 5.9 eq.) e N,N’-diciclohhexilcarbodiimida (DCC, 64,2 mg, 0,31 mmol, 9 eq.) em N,N-dimetilformamida anidra (2,0 mL) foi agitada à temperatura ambiente por 10 minutos. A solução amarelo claro resultante foi lentamente adicionada a #B73 (Exemplo A30, 27,1 mg, 0,035 mmol, 1,0 eq) em N,N-dimetilformamida (0,5 mL) e, em seguida, agitada à temperatura ambiente por 15 minutos. O produto foi purificado usando cromatografia de fase reversa (Método B*) para gerar #B74 como um pó branco. Rendimento: 14,2 mg, 41%. HPLC (Protocolo N): tempo de retenção = 9,61 minutos (pureza 100%). LCMS (Protocolo M); m/z 772,4 [M+H]+. 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 8,60 (d, J = 7,0, D2O permutável), 8,28 (d, J = 7,0, 1H, D2O permutável), 7,80 (d, J = 7,9, 1H, D2O permutável), 6,36 (dq, J = 6,0, 6,0, 1H), 6,29 (br d, J = 16,0, 1H), 6,11 (d, J = 11,7, 1H), 5,87 (dd, J = 11,7, 7,8, 1H), 5,60 (dd, J = 16,0, 5,8, 1H), 5,49 (br dd, J = 7,0, 7,0, 1H), 5,01 (d, J = 5,4, 1H, D2O permutável), 4,27-4,23 (m, 3H), 4,19 (m, 1H), 3,65 (m, 2H), 3,59 (s, 2H), 3,49 (br dd, J = 6,0, 6,0, 1H), 3,26 (dd, J = 5,1, 5,1, 1H), 2,76 (d, J = 5,1, 1H), 2,58 (d, J = 5,1, 1H), 2,53 (m, 1H), 2,32 (m, 1H), 2,22-2,11 (m, 6H), 1,98(s, 3H), 1,82 (m, 1H), 1,80 (m, 2H), 1,69 (s, 3H), 1,65 (m, 1H), 1,52 (dd, J = 14,8, 2,7, 1H), 1,25 (d, J = 6,2, 3H), 1,07 (d, J = 6,0, 3H), 0,95 (d, J = 7,0, 3H).
[0920] Exemplo A32
[0921] Preparação de N-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1- il)hexanoil]-L-valil-N-{4-[({[trans-3-({[(3R,5S,7R,8R)-7-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]acetil}amino)ciclobutil]carbamoil}oxi)metil]fenil}- N5-carbamoil-L-ornitinamida (#B75).
[0922] Etapa 1. Síntese de N-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1- il)hexanoil]-L-valil-N-{4-[({[trans-3-({[(3R,5S,7R,8R)-7-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]acetil}amino)ciclobutil]carbamoil}oxi)metil]fenil}- N5-carbamoil-L-ornitinamida (#B75). A uma solução de N-[6-(2,5- dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]-L-valil-N~5~-carbamoil-N-{4- [({[(4-nitrobenzil)oxi]carbonil}oxi)metil]fenil}-L-ornitinamida (MalCValCi- tPABA-PNP, Eur. Pat. Appl. (1994), EP624377, 23,1 mg, 0,03 mmol, 1,3 eq.) e #B73 (15,5 mg, 85% de pureza, 0,022 mmol, 1,0 eq.) em N,N-dimetilformamida anidra (0,6 mL), foi adicionada N,N- diisopropiletilamina (base de Hunig, 30 μL). A mistura de reação foi agitada à temperatura ambiente por 1 hora. A mistura de reação foi purificada usando cromatografia de fase reversa (Método B*) para gerar #B75 como um pó branco. Rendimento: 12,6 mg, 28%. HPLC (Pro- tocolo N): tempo de retenção = 9,3 minutos (pureza 91%). LCMS (Protocolo M): m/z 1202,5 [M+H]+.
[0923] Exemplo A33
[0924] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,4R,5R,7S)-7-{2-[(trans-4-aminociclohexil)amino]-2-oxoetil}-4- hidroxi-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5- dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B76).
[0925] Etapa 1. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-{2-[(trans-4-aminociclohexil)amino]-2-oxoetil}-4- hidroxi-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B76). Uma solução de #NP1 (40,2 mg, 92% de pureza, 0,07 mmol, 1,0 eq.), hidrato de 1-hidroxibenzotriazol (HOBT, 62,5 mg, 0,4 mmol, 5,8 eq.), 1-etil-3-(3-dimetilaminopropil) carbodiimida-HCl (EDC, 72,2 mg, 0,38 mmol, 5 eq.) em N,N-dimetilformamida (3,0 mL) foi agitada a 0°C por 30 minutos. A esta solução, foram subsequentemente adicionados trans-1,4-diaminociclohexano (290,0 mg, 2,5 mmoles, 30 eq.) em N,N-dimetilfórmamida (1,5 mL) e trietilamina (50 μL) a 0°C. A mistura de reação foi, então, agitada à temperatura ambiente por 0,5 hora e 0°C por 16 horas. A mistura de reação foi neutralizada com ácido acético, filtrada, e, em seguida, purificada usando cromatografia de fase reversa (Método B*) para gerar #B76 como um pó branco. Rendimento: 48,2 mg, 100%. HPLC (Protocolo N): tempo de retenção = 7,16 minutos (pureza 87,5%). LCMS (Protocolo M): m/z 632,2 [M+H]+. 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 7,81 (d, J = 7,8, 1H, D2O permutável), 7,72 (d, J = 7,8, 1H, D2O permutável), 6,36 (dq, J = 6,0, 6,0, 1H), 6,28 (br d, J = 15,6, 1H), 6,11 (d, J = 11,7, 1H), 5,87 (dd, J = 11,7, 7,8, 1H), 5,59 (dd, J = 16,0, 5,5, 1H), 5,49 (br dd, J = 6,6, 6,6, 1H), 5,02 (m, 1H, D2O permutável), 4,26 (m, 1H), 4,22 (m, H), 3,65 (m, 2H), 3,49 (br dd, J = 6,2, 6,2, 1H), 3,45-3,32 (m, 2H), 3,26 (d, J = 3,9, 1H), 2,75 (d, J = 5,1, 1H), 2,58 (d, J = 5,1, 1H), 2,46 (m, 1H), 2,29 (m, 1H), 2,22 (m, 1H), 2,16 (dd, J = 14,0, 4,7, 1H), 1,98 (s, 3H), 1,83 (m, 1H), 1,81 (m, 2H), 1,79-1,72 (m, 4H), 1,70 (s, 3H), 1,65 (m, 1H), 1,46 (dd, J = 12,5, 3,0, 1H), 1,25 (d, J = 6,2, 3H), 1,16-1,10 (m, 4H), 1,07 (d, J = 6,0, 3H), 0,95 (d, J = 7,0, 3H).
[0926] Exemplo A34
[0927] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,4R,5R,7S)-7-{2-[(5-aminopentil)amino]-2-oxoetil}-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B77).
[0928] Etapa 1. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-{2-[(5-aminopentil)amino]-2-oxoetil}-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B77). Uma solução de #NP1 (30,5 mg, 92% pura, 0,056 mmol, 1,0 eq.), hidrato de 1- hidroxibenzotriazol (HOBT, 38,0 mg, 0,24 mmol, 4.4 eq.), 1-etil-3-(3- dimetilaminopropil) carbodiimida-HCl (EDC, 54,0 mg, 0,28 mmol, 5 eq.) em N,N-dimetilformamida (3,0 mL) foi agitada a 0°C por 30 minutos. A esta solução, foram subsequentemente adicionadas trietilamina (50 μL) e 1,5-pantanediamina (50 μL, 0,5 mmol, 9 eq.) a 0°C. A mistura de reação foi agitada à temperatura ambiente por 1 hora. A mistura de reação foi neutralizada com ácido acético, filtrada, e, em seguida, purificada usando cromatografia de fase reversa (Método B*). O pico com tempo de retenção de 22,0 minutos foi coletado, neutralizado com NH4OH, e liofilizado para gerar #B77 como um pó branco. Rendimento 23,1 mg, 68% de rendimento. HPLC (Protocolo N): tempo de retenção = 7,67 minutos (pureza 91%). LCMS (Protocolo M): m/z 620,6 [M+H]+.
[0929] Exemplo A35
[0930] Preparação de N-{[(3S,5S,7S)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6-dioxaspiro[2,5]oct-5-il]acetil}- 2-metilalanina (#B79).
[0931] Etapa 1. Síntese de N-{[(3S,5S,7S)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6- dioxaspiro[2,5]oct-5-il]acetil}-2-metilalanina (#B78). Uma mistura de #NP2 (118,3 mg, 94,0% de pureza, 0,2 mmol, 1,0 eq.), Hexafluorofos- fato de O-(7-azabenzotriazol-1-il)-N,N,N‘,N‘-tetrametilurônio (HATU, 31,7 mg, 0,083 mmol, 0,4 eq.), e N,N-diisopropiletilamina (base de Hu- nig, 10 μL) em N,N-dimetilformamida (2,0 mL) foi agitada à temperatura ambiente por 30 minutos. A esta solução, foram subsequentemente adicionadas trietilamina (100 μL) e 2-metilalanina (32,5 mg, 0,3 mmol, 1,3 eq.) em 1:1 piridina/dimetil sulfóxido (1,0 mL). A suspensão resultante foi agitada à temperatura ambiente por 2,0 horas. A mistura de reação foi purificada usando cromatografia de fase reversa (Método B*) para gerar #B78 como um pó branco. Rendimento: 54,3 mg, 45% de rendimento. HPLC (Protocolo N): tempo de retenção = 11,29 minutos (pureza 94,1%). LCMS (Protocolo M); m/z 605,6 [M+H]+.
[0932] Etapa 2. Síntese de N-{[(3S,5S,7S)-7-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6- dioxaspiro[2,5]oct-5-il]acetil}-2-metilalanina (#B79). Uma solução de #B78 (6,6 mg, 0,011 mmol, 1,0 eq.), Hexafluorofosfato de O-(7- azabenzotriazol-1-il)-N,N,N‘,N‘-tetrametilurônio (HATU, 6,0 mg, 0,016 mmol, 1,5 eq.), e N,N-diisopropiletilamina (base de Hunig, 3,0 μL) em N,N-dimetilformamida (200 μL) foi agitada à temperatura ambiente por 30 minutos. A esta solução, foi adicionada propilamina (3,6 μL, 0,06 mmol, 5 eq.) e a solução resultante foi agitada por 1 hora. A mistura de reação foi purificada usando cromatografia de fase reversa (Método B*) para gerar #B79 como um pó branco. Rendimento: 4,9 mg, 70% de rendimento. HPLC (Protocolo N): tempo de retenção = 12,31 minutos (pureza 100%). LCMS (Protocolo M): m/z 646,7 [M+H]+; 668,7 [M+Na]+.
[0933] Exemplo A36
[0934] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,4R,5R,7S)-4-hidroxi-7-(2-{[2-metil-1-oxo-1-(propilamino)propan-2- il]amino}-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1- il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B81).
[0935] Etapa 1. Síntese de N-{[(3R,5S,7R,8R)-7-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]acetil}-2-metilalanina (#B80). Uma mistura de #NP1 (122,4 mg, 92,0% de pureza, 0,22 mmol, 1,0 eq.), Hexafluoro- fosfato de O-(7-azabenzotriazol-1-il)-N,N,N‘,N‘-tetrametilurônio (HATU, 33,2 mg, 0,087 mmol, 0,4 eq.), e N,N-diisopropiletilamina (base de Hu- nig, 10 μL) em N,N-dimetilformamida (2,0 mL) foi agitada à temperatura ambiente por 30 minutos. A esta solução, foram subsequentemente adicionadas trietilamina (100 μL) e 2-metilalanina (36,4 mg, 0,35 mmol, 1,2 eq.) em 1:1 piridina/dimetil sulfóxido (1,0 mL). A suspensão resultante foi agitada à temperatura ambiente por 2,0 horas. A mistura de reação foi purificada usando cromatografia de fase reversa (Método B*) para gerar #B80 como um pó branco. Rendimento: 52,7 mg, 42% de rendimento. HPLC (Protocolo N): tempo de retenção = 9,28 minutos (pureza 90%). LCMS (Protocolo M): m/z 621,6 [M+H]+.
[0936] Etapa 2. Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6- {(2E,4E)-5-[(3R,4R,5R,7S)-4-hidroxi-7-(2-{[2-metil-1-oxo-1-(propilamino)propan-2-il]amino}-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5- oxopent-3-en-2-il acetato (#B81). Uma solução de #B80 (6,0 mg, 90% de pureza, 0,01 mmol, 1,0 eq.), Hexafluorofosfato de O-(7- azabenzotriazol-1-il)-N,N,N‘,N‘-tetrametilurônio (HATU, 5,1 mg, 0,013 mmol, 1,3 eq.), e N,N-diisopropiletilamina (base de Hunig, 3,0 μL) em N,N-dimetilformamida (200 μL) foi agitada à temperatura ambiente por 30 minutos. A esta solução, foi adicionada propilamina (3,6 μL, 0,06 mmol, 6 eq.) e a solução resultante foi agitada por 1 hora. A mistura de reação foi purificada usando cromatografia de fase reversa (Método B*) para gerar #B81 como um pó branco. Rendimento: 4,7 mg, 87% de rendimento. HPLC (Protocolo N): tempo de retenção = 10,95 minutos (pureza 99%). LCMS (Protocolo M): m/z 662,7 [M+H]+.
[0937] Exemplo A37
[0938] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,4R,5R,7S)-7-{2-[(4-{[({4-[(N-{6-[(bromoace-til)amino]hexanoil}glicil)amino]benzil}oxi)carbonil]amino}benzil)amino]- 2-oxoetil}-4-hidroxi-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1- il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B123)
[0939] Etapa 1: Síntese de 9H-fluoren-9-ilmetil {2-[(4-{[(terc- butoxicarbonil)amino]metil}fenil)amino]-2-oxoetil}carbamato (#B118): A uma solução de Fmoc-Glicina (16 g, 54 mmoles, 1,0 eq.) em DMF seco (160 mL) a 0oC, foi adicionado N,N-diisopropiletilamina (14 g, 108 mmoles, 2.0 eq) e Tetrafluoroborato N,N,N‘,N‘-Tetrametil-O- (benzotriazol-1-il)urônio (16 g, 54 mmoles, 1,0 eq). A mistura foi agitada a 0oC por 30 min e uma solução de terc-butil [4- (glicilamino)benzil]carbamato (12 g, 54 mmoles, 1,0 eq.) em DMF seco (50 mL) foi adicionada. A mistura foi agitada à temperatura ambiente durante a noite, vertida em água gelada (400 mL) e extraída com EtO- Ac (400 mLx2). A camada orgânica foi lavada com salmoura (200 mLx2), seca sobre Na2SO4 e concentrada em vácuo. O resíduo foi recristalizado a partir de EtOAc (200 mL) e éter de petróleo (400 mL) para gerar #B118 (18 g, 66,6%) como um sólido branco. 1H RMN (400Hz, DMSO-d6): δ 9,93 (s, 1H), 7,91 (d, 2H), 7,75 (d, 2H), 7,61 (m, 1 H), 7,52 (d, 2H), 7,43 (m, 2H), 7,36 (m, 3H), 7,18 (d, 3 H), 4,32 (d, 2H), 4,26 (m, 1 H), 4,07 (d, 2H), 3,80 (d, 2H), 1,39 (s, 9H).
[0940] Etapa 2: Síntese de terc-butil [4-(glicilamino)benzil]carbamato (#B119): A uma solução de #B118 (7,0 g, 14,0 mmoles, 1,0 eq) em DMF (70 mL), foi adicionada piperidina (4,7 mL, 47,5 mmoles, 3,4 eq.) à temperatura ambiente. A mistura foi agitada à temperatura ambiente por 30 min, evaporada in vacuo. O resíduo foi lavado com éter de petróleo (100 mLx2) e recristalizado a partir de EtOAc (50 mL) e éter de petróleo (200 mL) para dar #B119 (3,3 g, 84,6%) como um sólido branco. 1H RMN (400Hz, CDCl3): δ 9,37 (s, 1H), 7,57 (d, 2H), 7,25 (d, 2H), 4,80 (br, 1H), 4,27 (d, 2H), 3,47 (s, 2H), 1,45 (s, 9H).
[0941] Etapa 3: Síntese de 9H-fluoren-9-ilmetil [6-({2-[(4-{[(terc- butoxicarbonil)amino]metil}fenil)amino]-2-oxoetil}amino)-6-oxohexil]carbamato (#B120): A uma solução de ácido 6-{[(9H-fluoren- 9-ilmetoxi)carbonil]amino}hexanóico (2,66 g, 7,53 mmoles, 1,0 eq.) em DCM seco (50 mL) a 0oC, foi adicionada N,N-diisopropiletilamina (1,93 g, 15,1 mmoles, 2.0 eq) e Hexafluorofosfato de O-(7-azabenzotriazol- 1-il)-N,N,N‘,N‘-tetrametilurônio (HATU, 2,86 g, 7,53 mmoles, 1,0 eq.). A mistura foi agitada a 0oC por 30 min e #B119 (2,1 g, 7,53 mmoles, 1,0 eq.) foi adicionado em uma porção. A mistura foi agitada à temperatura ambiente durante a noite. A mistura foi filtrada e o sólido foi lavado com DCM e seco in vacuo para gerar #B120 (4 g, 86,4%) como umsólido branco. 1H RMN (400Hz, DMSO-d6): δ 9,91 (s, 1H), 8,11 (br,1H), 7,90 (d, 2H), 7,69 (d, 2H), 7,51 (d, 2H), 7,41 (m, 2H), 7,33 (m,3H), 7,17 (d, 2H), 4,28 (m, 3H), 4,06 (d, 2H), 3,86 (br, 2H), 2,97 (m,2H), 2,15 (m, 2H), 1,51 (m, 2H), 1,38 (m, 12H); LCMS (Protocolo I): m/z 637,1 (M+Na]+, tempo de retenção = 1,18 minutos.
[0942] Etapa 4: Síntese de 9H-fluoren-9-ilmetil {6-[(2-{[4- (aminometil)fenil]amino}-2-oxoetil)amino]-6-oxohexil}carbamato sal de trifluoroacetato (#B121): A uma suspensão de #B120 (1 g, 1,63 mmol, 1,0 eq) em DCM seco (20 mL) a 0 °C, foi adicionado ácido trifluoroacé- tico (6 mL, amplo excesso). A mistura foi agitada à temperatura ambiente por 2 horas e concentrada in vacuo. O resíduo foi suspenso em água (30 mL) e liofilizado para gerar #B121 (1,2 g, 100%) como um sólido ligeiramente amarelo. 1H RMN (400Hz, DMSO-d6): δ10,08 (s, 1H), 8,15 (br, 4H), 7,90 (d, 2H), 7,69 (d, 2H), 7,63 (d, 2H), 7,41 (m, 8H), 4,30 (m, 3H), 3,98 (m, 3H), 3,87 (d, 2H), 2,97 (m, 2H), 2,17 (m, 2H), 1,51 (m, 2H), 1,40 (m, 2H), 1,26 (m, 2H): LCMS (Protocolo I): m/z 537,1 (M+Na]+, tempo de retenção = 1,10 minutos.
[0943] Etapa 5: Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-{2-[(4-{[N-(6-{[(9H-fluoren-9- ilmetoxi)carbonil]amino}hexanoil)glicil]amino}benzil)amino]-2-oxoetil}-4- hidroxi-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5- dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B122): A uma solução de #B121 (32,7 mg, 0,044 mmol, 1 eq.) em tetrahidrofurano (1,0 mL) e metanol (0,1 mL), foi adicionada N,N- diisopropiletilamina (26,0 mg, 0,2 mmol, 4,5 eq.). Toda a mistura de reação foi adicionada a uma solução resfriada (0 °C) de #B1 (28 mg, 0,044 mmol, 1 eq.) em tetrahidrofurano (1,0 mL) e a reação foi deixada aquecer para temperatura ambiente. Após uma hora, a reação foi concentrada in vacuo e o resíduo foi purificado por cromatografia de fase reversa (Método A) para gerar #B122 (12,4 mg, 0,011 mmol, 27%): LCMS (Protocolo D): m/z 1032,6 [M+H]+, tempo de retenção = 0,92 minuto.
[0944] Etapa 6: Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-{2-[(4-{[({4-[(N-{6- [(bromoace- til)amino]hexanoil}glicil)amino]benzil}oxi)carbonil]amino}benzil)amino]- 2-oxoetil}-4-hidroxi-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1- il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B123). A uma solução de #B122 (12,4 mg, 0,012 mmol, 1 eq.) em dimetilformamida (0,7 mL, 0,01 M), foi adicionada piperidina (11 uL de uma solução estoque [preparada por dissolução de 100 uL piperidina em 1 mL DMF], 0,013 mmol, 1,1 eq.). A reação foi agitada por 16 horas e uma solução de N-hidroxissuccinimida éster de ácido bromoacé- tico (2,8 mg, 0,012 mmol, 1 eq.) em tetrahidrofurano (0,5 mL), em seguida, adicionada em gotas. A reação foi agitada por 16 horas, concentrada in vacuo e o resíduo foi purificado por cromatografia de fase reversa (Método A) para gerar #B123 como um sólido. Rendimento: 2,5 mg, 0,027 mmol, 22%. LCMS (Protocolo D): m/z 932,2 [M+H]+ tempo de retenção = 0,76 minuto.
[0945] Exemplo A38
[0946] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,4R,5R,7S)-7-(2-carbamimidamido-2-oxoetil)-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B124) e (2S,3Z)-5- ({(2R,3R,5S,6S)-6-[(2E,4E)-5-{(3R,4R,5R,7S)-7-[2-(N'- {[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5-il]acetil}carbamimidamido)-2-oxoetil]-4-hidroxi-1,6-dioxaspiro[2,5]oct-5- il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro-2H-piran-3-il}amino)- 5-oxopent-3-en-2-il acetato (#B125)
[0947] Etapa 1: Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-(2-carbamimidamido-2-oxoetil)-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B124) e (2S,3Z)-5- ({(2R,3R,5S,6S)-6-[(2E,4E)-5-{(3R,4R,5R,7S)-7-[2-(N'-{[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5-il]acetil}carbamimidamido)-2-oxoetil]-4-hidroxi-1,6-dioxaspiro[2,5]oct-5- il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro-2H-piran-3-il}amino)- 5-oxopent-3-en-2-il acetato (#B125): a uma mistura de #NP1 (135 mg, ~60% de pureza, ~0,15 mmol) e hexafluorofosfato de O-(7- azabenzotriazol-1-il)-N,N,N‘,N‘-tetrametilurônio (HATU, 72 mg, 0,19 mmol, 1,2 eq.) em N,N-dimetilformamida (DMF, 1,0 mL) a 0oC, foi adicionada N,N’- diisopropiletilamina (30 uL, #eq). Após agitação em temperatura ambiente por 10 minutos, a mistura foi transferida para uma solução de cloridrato de guanidina (400 mg, 4,1 mmoles, 28 eq.) e N,N’-diisopropiletilamina (100 uL) em 1:1 metilsulfóxido/água (3,0 mL). A solução resultante foi agitada por 20 minutos e purificada usando cromatografia de fase reversa (Método B*) para gerar (#B124) e (#B125) como pós brancos.
[0948] #B124: Rendimento: 55,6 mg, 38% de rendimento HPLC (Protocolo N): tempo de retenção = 8,01 minutos (pureza 87%). LCMS (Protocolo M): m/z 577,44 [M+H]+. 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 7,80 (d, J = 7,9, 1H, D2O permutável), 6,36 (dq, J = 6,0, 6,0, 1H), 6,27 (br d, J = 16,0, 1H), 6,11 (d, J = 11,3, 1H), 5,87 (dd, J = 11,3, 7,4, 1H), 5,60 (dd, J = 15,6, 5,5, 1H), 5,51 (br dd, J = 7,4, 7,4, 1H), 4,93 (d, J = 5,8, 1H, D2O permutável), 4,29 (m, 1H), 4,22 (m, 1H), 3,65 (m, 2H), 3,50 (br dd, J = 6,0, 6,0, 1H), 3,22 (dd, J = 4,7, 4,7, 1H), 2,73 (d, J = 5,1, 1H), 2,56 (d, J = 5,1, 1H), 2,46 (m, 1H), 2,32 (m, 2H), 2,20 (m, 1H), 1,98(s, 3H), 1,82 (m, 1H), 1,80 (m, 2H), 1,69 (s, 3H), 1,65 (m, 1H), 1,52 (dd, J = 13,2, 3,5, 1H), 1,25 (d, J = 6,2, 3H), 1,07 (d, J = 6,0, 3H), 0,95 (d, J = 7,4, 3H).
[0949] #B125: Rendimento: 49,0 mg, 36% de rendimento HPLC (Protocolo N): tempo de retenção = 10,14 minutos (pureza 90%). LCMS (Protocolo M): m/z 1094,76 [M+H]+. 1H RMN (400 MHz, DMSO- d6, mult, J em Hz) δ 7,78 (d, J = 7,9, 2H, D2O permutável), 6,36 (dq, J = 6,0, 6,0, 2H), 6,27 (br d, J = 16,0, 2H), 6,11 (d, J = 11,7, 2H), 5,87 (dd, J = 11,7, 7,8, 2H), 5,60 (dd, J = 16,0, 5,0, 2H), 5,49 (br dd, J = 6,7, 6,7, 2H), 5,01 (br s, 2H, D2O permutável), 4,32 (m, 2H), 4,25 (m, 2H), 3,65 (m, 4H), 3,49 (br dd, J = 6,6, 6,6, 2H), 3,28 (d, J = 4,3, 2H), 2,76 (d, J = 4,7, 2H), 2,59 (d, J = 4,7, 2H), 2,54 (m, 2H), 2,30 (m, 2H), 2,28 (m, 2H), 2,21 (m, 2H), 1,98(s, 6H), 1,83 (m, 2H), 1,80 (m, 4H), 1,69 (s, 6H), 1,65 (m, 2H), 1,52 (br d, J = 12,8, 2H), 1,25 (d, J = 6,2, 6H), 1,06 (d, J = 6,2, 6H), 0,94 (d, J = 7,0, 6H).
[0950] Compostos diméricos similares àqueles divulgados aqui estão também incluídos no escopo da presente invenção, por exem- plo, compostos diméricos tendo substituições como descrito neste pedido.
[0951] Exemplo A39
[0952] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,4R,5R,7S)-7-{2-[N'-(bromoacetil)carbamimidamido]-2-oxoetil}-4- hidroxi-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5- dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B126)
[0953] Etapa 1: Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-{2-[N'-(bromoacetil)carbamimidamido]-2-oxoetil}-4- hidroxi-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B126) L. A uma solução de #B124 (24,0 mg, 0,042 mmol, 1,0 eq.) e N-hidroxissuccinimida éster de ácido bromoacético (24,1 mg, 0,092 mmol, 2.0 eq.) em N,N-dimetilformamida (1 mL), foi adicionada N,N’- diisopropiletilamina (10 uL). A solução resultante foi agitada à temperatura ambiente por 30 minutos e purificada usando cromatografia de fase reversa (Método B*) para gerar #B126 como um pó branco. Rendimento: 5,7 mg, 20%. HPLC (Protocolo N): tempo de retenção = 10,01 minutos (pureza 99%). LCMS (Protocolo M): m/z 697,35, 699,35 (1:1) [M+H]+. 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 9,57(br s, 1H, D2O permutável), 9,41(br s, 1H, D2O permutável), 9,24(br s, 1H, D2O permutável), 9,08 (br s, 1H, D2O permutável), 7,79 (d, J = 7,5, 1H, D2O permutável), 6,36 (dq, J = 6,0, 6,0, 1H), 6,26 (br d, J = 16,0, 1H), 6,11 (d, J = 11,7, 1H), 5,87 (dd, J = 11,3, 7,4, 1H), 5,60 (m, 1H), 5,49 (m, 1H), 4,35-4,28 (m, 2H), 3,65 (m, 2H), 3,50 (br dd, J = 6,0, 6,0, 1H), 3,41 (s, 2H), 3,26 (d, J = 4,7, 1H), 2,78 (d, J = 4,3, 1H), 2,61 (d, J = 4,3, 1H), 2,55 (m, 1H), 2,32 (m, 2H), 2,20 (m, 1H), 1,98(s, 3H), 1,85 (m, 1H), 1,80 (m, 2H), 1,69 (s, 3H), 1,65 (m, 1H), 1,53 (dd, J = 13,2, 3,5, 1H), 1,25 (d, J = 6,2, 3H), 1,07 (d, J = 6,2, 3H), 0,95 (d, J = 6,6, 3H).
[0954] Exemplo A40
[0955] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,4R,5R,7S)-4-hidroxi-7-{2-[{2-[(iodoacetil)(metil)amino]etil}(metil)amino]-2-oxoetil}-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B128)
[0956] Etapa 1. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-{2-[(metil(2-(metilamino)etil)amino)]-2-oxoetil}-4- hidroxi-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il aceta- to.(#B127): A uma solução de #B49 (54,5 mg, 0,084 mmol, 1,0 eq.) em N,N-dimetilformamida (2,0 mL), foi adicionada N,N'-dimetil-1,2- etilenodiamina (120 uL, 1,1 mmol, 12 eq.). A mistura de reação foi agitada à temperatura ambiente por 5 minutos e o produto foi purificado usando cromatografia de fase reversa (Método B*) para gerar. (#B127) Rendimento: 29,1 mg, 57%. HPLC (Protocolo N): tempo de retenção = 6,92 minutos (pureza 76%). LCMS (Protocolo M): m/z 606,3 [M+H]+.
[0957] Etapa 2. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-4-hidroxi-7-{2-[{2- [(iodoacetil)(metil)amino]etil}(metil)amino]-2-oxoetil}-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B128). Uma solução de ácido iodoacético (43,1 mg, 0,23 mmol, 4,8 eq.) e N,N'- diciclohexilcarbodiimida (DCC, 64,10 mg, 0,3 mmol, 6.3 eq.) em N,N- dimetilformamida (2,0 mL) foi agitada à temperatura ambiente por 10 minutos e, em seguida, transferida para uma solução de (#B127) (29,1 mg, 76,0% pura, 0,048 mmol, 1 eq.) em N,N-dimetilformamida (0,2 mL). A mistura de reação foi agitada por 20 minutos e purificada por cromatografia de fase reversa para gerar (#B128) como um pó branco. Rendimento: 12,2 mg, 54%. HPLC análise (Protocolo N): tempo de retenção = 9,57 minutos (pureza 95,2%). LCMS (Protocolo M): m/z 774,2 [M+H]+. 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 7,80 (d, J = 7,4, 1H, D2O permutável), 6,36 (dq, J = 6,2, 6,2, 1H), 6,33 (br d, J = 15,5, 1H), 6,11 (d, J = 11,3, 1H), 5,86 (dd, J = 11,7, 7,8, 1H), 5,60 (dd, J = 16,0, 4,7, 1H), 5,52 (br dd, J = 6,6, 6,6, 1H), 4,98 (m, 1H, D2O per-mutável), 4,26 (m, 2H), 3,65 (m, 2H), 3,51 (br dd, J = 6,2, 6,2, 1H), 3,46-3,35 (m, 6H), 3,24 (m, 1H), 3,02 (s, 1,5H), 2,97 (s, 1,5H), 2,95 (s, 1,5H), 2,82 (s, 1,5H), 2,76 (m, 1H), 2,65 (m, 1H), 2,59 (m, 1H), 2,55 (m, 1H), 2,30 (m, 1H), 2,22 (m, 1H), 1,98 (s, 3H), 1,86 (m, 1H), 1,80 (m, 2H), 1,70 (s, 3H), 1,65 (m, 1H), 1,49 (dd, J = 12,5, 2,7, 1H), 1,25 (d, J = 6,2, 3H), 1,07 (d, J = 6,2, 3H), 0,95 (d, J = 7,0, 3H).
[0958] Exemplo A41
[0959] Preparação de (2Z)-N-[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3S,5S,7S)-7-(2-amino-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]-4-oxopent- 2-enamida (#B129).
[0960] Etapa 1. Síntese de (2Z)-N-[(2R,3R,5S,6S)-6-{(2E,4E)-5- [(3S,5S,7S)-7-(2-amino-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]-4-oxopent- 2-enamida (#B129): A uma solução de #B39 (60 mg, 0,13 mmol, 1 eq.) em diclorometano (2 mL) a 0°C, foi adicionado Dess-Martin periodina- no (119 mg, 0,27 mmol, 2 eq.), e o banho de gelo foi removido. Após 35 min, bicarbonato de sódio saturado e diclorometano foram adicionados, e a camada aquosa foi extraída com diclorometano. Os extratos orgânicos combinados foram secos sobre sulfato de sódio e filtrados, e os solventes foram removidos in vacuo. O material bruto foi purificado por cromatografia de fase reversa (Método A) para dar #B129 como um sólido branco. Rendimento: 19,04 mg, 0,04 mmol, 32%. LCMS (Protocolo D): m/z 475,3 [M+H]+, tempo de retenção = 0,70 mi-nuto. 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ d, J = 8,0 Hz, 1 H), 7,32 (s, 1 H), 6,77 (s, 1 H), 6,32 (s, 2 H), 6,27 (d, J = 16,0 Hz, 1 H), 5,60 (dd, J = 16,0 e 5,5 Hz, 1 H), 5,55-5,47 (m, 1 H), 4,58-4,50 (m, 1 H), 4,35-4,26 (m, 1 H), 3,69-3,59 (m, 2 H), 3,54-3,48 (m, 1 H), 2,652,51 (m, 3 H), 2,36-2,15 (m, 6 H), 1,88-1,73 (m, 3 H), 1,72-1,60 (m, 6 H), 1,37 (dd, J = 13,3 e 6,2 Hz, 1 H), 1,08 (d, J = 6,6 Hz, 3 H), 0,96 (d, J = 7,4 Hz, 3 H).
[0961] Exemplo A42
[0962] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,4R,5R,7S)-7-(2-amino-2-oxoetil)-4-hidroxi-1,6-dioxaspiro[2,5]oct- 5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3- il]amino}-5-oxopent-3-en-2-il metil[2- (metilsulfanil)etil]carbamato(#B130).
[0963] Etapa 1. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-(2-amino-2-oxoetil)-4-{[terc-butil(dimetil)silil]oxi}- 1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B131).: A uma solução de #B9 (119 mg, 0,22 mmol, 1 eq.) em diclo- rometano (3 mL) a 0°C, foi adicionado 2,6-lutidina (104 μL, 0,89 mmol, 4 eq.) seguido por terc-butil-dimetilsilil-trifluorometanossulfonato (160 μL, 067 mmol, 3 eq.) em gotas. Após 70 min, a reação foi diluída com bicarbonato de sódio saturado e diclorometano, extraída, filtrada sobre um tubo separador de solvente e os solventes foram removidos in vacuo. O material desejado bruto foi purificado por cromatografia líquida de pressão média de fase reversa eluída com 0,02% de ácido acético em água (v/v) e 0,02% de ácido acético em acetonitrila (v/v) (5% a 100%) para gerar #B131 como um sólido branco. Rendimento: 34 mg, 0,05 mmol, 23%. LCMS (Protocolo C): m/z 671,3 [M+Na]+, tempo de retenção = 2,08 minutos.
[0964] Etapa 2. Síntese de (2Z,4S)-N-[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-(2-amino-2-oxoetil)-4-{[terc-butil(dimetil)silil]oxi}- 1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5- dimetiltetrahidro-2H-piran-3-il]-4-hidroxipent-2-enamida (#B132). A uma solução de #B131 (32 mg, 0,049 mmol, 1 eq.) em 4:1 tetrahidrofu- rano:água (1 mL), foi adicionado hidróxido de lítio (11,7 mg, 0,49 mmol, 10 eq.), e a mistura agitada à temperatura ambiente por 21 horas. A reação foi concentrada in vacuo, e o resíduo foi retomado em etil acetato e água. A camada aquosa foi extraída com etil acetato (3X) e as camadas orgânicas combinadas foram secas sobre sulfato de sódio anidro e concentradas in vacuo. Purificação por cromatografia líquida de pressão média de fase reversa eluída com 0,02% de ácido acético em água (v/v) e 0,02% de ácido acético em acetonitrila (v/v) (10% a 100%) forneceu #B132 como um sólido branco. Rendimento: 11,3 mg, 0,019 mol, 38%. LCMS (Protocolo D): m/z 629,3 [M+Na]+, tempo de retenção = 0,98 minuto.
[0965] Etapa 3. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-(2-amino-2-oxoetil)-4-{[terc-butil(dimetil)silil]oxi}- 1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il metil[2- (metilsulfanil)etil]carbamato (#B133).: A uma solução de #B132 (63,2 mg, 0,104 mmol, 1 eq.) em diclorometano (1,8 mL), foi adicionada trie- tilamina (73 μL, 0,520 mmol, 5 eq), 4-N,N-dimetilamino piridina (8,9 mg, 0,073 mmol, 0,7 eq.) e bis-(4-nitrofenil)-carbonato (106 mg, 0,343 mmol, 3,3 eq.) e a reação agitada à temperatura ambiente por 2,5 horas. A 1/3 desta mistura, foi adicionado cloridrato de N-metil-2- (metilsulfanil)etanamina (24,6 mg, 0,174 mmol, 1,67 eq.), e a mistura foi agitada à temperatura ambiente por 1 hora. A reação foi diluída com água, extraída com diclorometano, filtrada sobre um tubo separador de solvente, diluída com dimetil sulfóxido (1 mL), e concentrada in vacuo. O resíduo purificado por cromatografia de fase reversa (Método A) para prover #B133. Rendimento: 15,2 mg, 0,021 mmol, 20%. LCMS (Protocolo C): m/z 760,76 [M+Na]+, tempo de retenção = 2,19 minutos.
[0966] Etapa 4. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-(2-amino-2-oxoetil)-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-ilmetil[2-(metilsulfanil)etil]carbamato (#B130).: A uma solução de #B133 (15,2 mg, 0,021 mmol, 1 eq.) em tetrahidrofurano (0,4 mL) resfriado para 0°C, foi adicionado fluoreto de tetrabutilamônio (1 M em tetrahidrofura- no 53 μL, 0,053 mmol, 2,5 eq.), e a reação foi aquecida para temperatura ambiente após 10 min. Após 1,5 hora, a reação foi concentrada in vacuo, e o resíduo purificado por cromatografia de fase reversa (Método A) para prover #B130. Rendimento: 8,5 mg, 0,014 mmol, 65%. LCMS (Protocolo D): m/z 646,3 [M+Na]+, tempo de retenção = 0,79 minuto. 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 7,77 (d, J = 8,2 Hz, 1 H), 7,31 (s, 1 H), 6,77 (s, 1 H), 6,32 (d, J = 16,0 Hz, 1 H), 6,266,18 (m, 1 H), 6,09 (d, J = 11,5 Hz, 1 H), 5,89 (dd, J = 11,5 e 7,0 Hz, 1 H), 5,60 (dd, J = 16,0 e 5,5 Hz, 1 H), 5,54-5,47 (m, 1 H), 5,00 (d, J = 5,5 Hz, 1 H), 4,29-4,21 (m, 2 H), 3,69-3,61 (m, 2 H), 3,54-3,47 (m, 1 H), 3,42-3,33 (m, 2 H), 3,26-3,21 (m, 1 H), 2,89-2,78 (m, 3 H), 2,74 (d, J = 5,1 Hz, 1 H), 2,64-2,56 (m, 3 H), 2,36-2,17 (m, 4 H), 2,07 (s, 3 H), 1,90-1,78 (m, 3 H), 1,72-1,61 (m, 4 H), 1,54-1,46 (m, 1 H), 1,26 (d, J = 6,6 Hz, 3 H), 1,07 (d, J = 6,2 Hz, 3 H), 0,96 (d, J = 7,0 Hz, 3 H).
[0967] Exemplo A43
[0968] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,4R,5R,7S)-7-(2-amino-2-oxoetil)-4-hidroxi-1,6-dioxaspiro[2,5]oct- 5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3- il]amino}-5-oxopent-3-en-2-il piperidina-1-carboxilato (#B134).
[0969] Etapa 1. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-(2-amino-2-oxoetil)-4-{[terc-butil(dimetil)silil]oxi}- 1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5- dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il piperidina-1- carboxilato (#B135).: Usando o procedimento descrito na etapa 3 do exemplo A42, o composto do título foi preparado em 18% de rendimento a partir de 63,2 mg (0,104 mmol, 1,0 eq) de #B132, trietilamina (73 μL, 0,520 mmol, 5 eq), 4-N,N-dimetilamino piridina (8,9 mg, 0,073 mmol, 0,7 eq.) e bis-(4-nitrofenil)-carbonato (106 mg, 0,343 mmol, 3,3 eq.) e piperidina (14,8 mg, 0,174 mmol, 1,7 eq.) usando o procedimento descrito para a preparação de composto #B133. LCMS (Protocolo D): m/z 740,5 [M+Na]+, tempo de retenção = 1,13 minuto.
[0970] Etapa 2. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-(2-amino-2-oxoetil)-4-hidroxi-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il piperidina-1-carboxilato (#B134).: O composto do título foi preparado em 76% de rendimento a partir de 13,5 mg (0,019 mmol) de #B135 e 12,8 mg (53 μL de 1 M em tetrahidrofurano 0,053 mmol, 2,5 eq.) de fluoreto de tetrabutilamônio usando o procedimento descrito para o composto #B130. LCMS (Protocolo D): m/z 626,60 [M+Na]+, tempo de retenção = 0.81 minuto. 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 7,77 (d, J = 7,80 Hz, 1 H), 7,31 (s, 1 H), 6,77 (s, 1 H), 6,32 (d, J = 15,6 Hz, 1 H), 6,26-6,17 (m, 1 H), 6,09 (d, J = 11,7 Hz, 1 H), 5,89 (dd, J = 11,7 e 7,4 Hz, 1 H), 5,60 (dd, J = 16,0 e 5,5 Hz, 1 H), 5,54-5,47 (m, 1 H), 5,00 (d, J = 5,5 Hz, 1 H), 4,29-4,20 (m, 2 H), 3,69-3,61 (m, 2 H), 3,54-3,47 (m, 1 H), 3,273,21 (m, 1 H), 2,75 (d, J = 5,1 Hz, 1 H), 2,58 (d, J = 5,1 Hz, 1 H), 2,362,16 (m, 4 H), 1,88-1,78 (m, 3 H), 1,73-1,61 (m, 4 H), 1,57-1,38 (m, 7 H), 1,25 (d, J = 6,2 Hz, 3 H), 1,07 (d, J = 6,2 Hz, 3 H), 0,96 (d, J = 7,4 Hz, 3 H).
[0971] Exemplo A44
[0972] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5- [(3S,5S,7S)-7-(2-amino-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5- oxopent-3-en-2-il piperazina-1-carboxilato, sal de acetato (#B136
[0973] Etapa 1. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3S,5S,7S)-7-(2-amino-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5- oxopent-3-en-2-il piperazina-1-carboxilato, sal de acetato (#B136).: O composto do título foi preparado em 27% de rendimento a partir de 37 mg (0,078 mmol) de trietilamina (39,7 mg, 0,39 mmol, 5 eq), 4-N,N- dimetilamino piridina (6,7 mg, 0,055 mmol, 0,7 eq.), bis-(4-nitrofenil)- carbonato (84,7 mg, 0,273 mmol, 3,5 eq.) e piperazina (16,8 mg, 0,195 mmol, 2,5 eq.) usando o procedimento descrito para a preparação de composto #B133. HPLC (Protocolo AA): tempo de retenção = 6,318 minutos (pureza 95%). LCMS (Protocolo C): m/z 589,4 [M+H]+ tempo de retenção = 0,97 minuto. 1H RMN (400 MHz, DMSO-d6) δ 7,77 (d, J = 8,20 Hz, 1 H), 7,31 (s, 1 H), 6,77 (s, 1 H), 6,31-6,17 (m, 2 H), 6,09 (d, J = 11,7 Hz, 1 H), 5,89 (dd, J = 11,7 e 7,4 Hz, 1 H), 5,60 (dd, J = 15,6 e 5,5 Hz, 1 H), 5,55-5,45 (m, 1 H), 4,57-4,50 (m, 1 H), 4,35-4,25 (m, 1 H), 3,69-3,60 (m, 2 H), 3,54-3,46 (m, 2 H), 2,65-2,53 (m, 4 H) 2,36-2,14 (m, 4 H), 1,88 (s, 3 H), 1,85-1,73 (m, 3 H), 1,72-1,61 (m, 5 H), 1,411,33 (m, 1 H), 1,25 (d, J = 6,2 Hz, 3 H), 1,07 (d, J = 6,2 Hz, 3 H), 0,95 (d, J = 7,0 Hz, 3 H).
[0974] Exemplo A45
[0975] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3S,5S,7S)-7-(2-amino-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5- oxopent-3-en-2-il -4-metilpiperazina-1-carboxilato (#B137).
[0976] Etapa 1. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3S,5S,7S)-7-(2-amino-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5- oxopent-3-en-2-il-4-metilpiperazina-1-carboxilato, sal de acetato (#B137).: O composto do título foi preparado em 27% de rendimento a partir de 37 mg (0,078 mmol) de trietilamina (39,7 mg, 0,39 mmol, 5 eq), 4-N,N-dimetilamino piridina (6,7 mg, 0,055 mmol, 0,7 eq.), bis-(4- nitrofenil)-carbonato (84,7 mg, 0,273 mmol, 3,5 eq.) e 1-Me-piperazina (19,5 mg, 0,195 mmol, 2,5 eq.) usando o procedimento descrito para a preparação do composto #B133. LCMS (Protocolo C): m/z 603,4 [M+H]+ tempo de retenção = 1,29 minutos. 1H RMN (400 MHz, DMSO- d6) δ 7,77 (d, J = 7,8 Hz, 1 H), 7,32 (s, 1 H), 6,77 (s, 1 H), 6,32-6,19 (m, 2 H), 6,10 (d, J = 11,7 Hz, 1 H), 5,89 (dd, J = 11,7 e 7,4 Hz, 1 H), 5,60 (dd, J = 16,0 e 5,9 Hz, 1 H), 5,55-5,47 (m, 1 H), 4,58-4,50 (m, 1 H), 4,35-4,26 (m, 1 H), 3,71-3,61 (m, 2 H), 3,55-3,47 (m, 2 H), 2,652,53 (m, 3 H), 2,36-2,14 (m, 8 H), 1,89 (s, 3 H), 1,85-1,74 (m, 3 H), 1,72-1,61 (m, 5 H), 1,42-1,33 (m, 1 H), 1,26 (d, J = 6,2 Hz, 3 H), 1,07 (d, J = 6,2 Hz, 3 H), 0,96 (d, J = 7,4 Hz, 3 H).
[0977] Exemplo A46
[0978] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,5S,7S)-7-{[(1H-imidazol-1-ilcarbonil)amino]metil}-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B138). e (2Z,4S)-N- [(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,5S,7S)-7-(aminometil)-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]-4-hidroxipent-2-enamida, sal de acetato (#B139) e (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,5S,7S)-7-(aminometil)-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato, sal de acetato (#B140).
[0979] Etapa 1a. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,5S,7S)-7-{[(1H-imidazol-1-ilcarbonil)amino]metil}-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B138).: A uma solução de #B22 (15,9 mg, 0,030 mmol, 1 eq.) em acetonitrila (0,6 mL) à temperatura ambiente, foi adicionado carbonil diimidazol (7,4 mg, 0,045 mmol, 1,5 eq.), e a reação foi deixada agitar por 10 min. A reação foi, então, aquecida para 60°C por 5,5 horas e resfriada para temperatura ambiente. Água e diclorometano foram adicionados, e a camada aquosa foi extraída. Os extratos orgânicos combinados foram secos sobre sulfato de sódio e filtrados, e os solventes foram removidos in vacuo. O material desejado bruto foi purificado por cromatogra- fia de fase reversa (Método A) para dar #B138 como um sólido branco. Rendimento: 3,4 mg, 0,0059 mmol, 20%. LCMS (Protocolo C): m/z 585,4 [M+H]+, tempo de retenção = 1,40 minuto. HPLC (Protocolo AA) tempo de retenção = 7,426 minutos (pureza 83%).
[0980] Etapa 1b. Síntese de (2Z,4S)-N-[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,5S,7S)-7-(aminometil)-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta- 2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]-4-hidroxipent-2- enamida, sal de acetato (#B139).: A uma solução de #B22 (35,2 mg, 0,066 mmol, 1 eq.) em acetonitrila (2,2 mL) à temperatura ambiente, foi adicionado carbonil diimidazol (16,2 mg, 0,099 mmol, 1,5 eq.), e a reação foi deixada agitar por 30 min. A reação foi, então, aquecida para 60°C por 5 horas. A reação foi resfriada para temperatura ambiente, adicionada a uma solução de acetonitrila (33 mL), água (17 mL), e 1 N NaOH (17 mL) e deixada agitar à temperatura ambiente por 15 min. A reação foi diluída com água e a acetonitrila removida in vacuo. A solução aquosa foi extraída com diclorometano, neutralizada com ácido acético (0,5 mL) e concentrada in vacuo. O resíduo foi retomado em acetonitrila, seco sobre sulfato de sódio, filtrado e concentrado in vacuo. O material desejado bruto foi purificado por cromatografia de fase reversa (Método A) para dar #B139 como um sólido branco. Rendimento: 7,7 mg, 0,015 mmol, 23%. LCMS (Protocolo C): m/z 449,3 [M+H]+, tempo de retenção = 0,93 minutos. 1H RMN (400 MHz, DMSO- d6, mult, J em Hz) δ 7,76 (d, J = 8,2 Hz, 1 H), 6,26 (d, J = 15,6 Hz, 1 H), 5,97 (d, J = 11,7 Hz, 1 H), 5,86 (dd, J = 11,7 e 7,0 Hz, 1 H), 5,67 (dd, J = 15,6 e 5,8 Hz, 1 H), 5,56-5,49 (m, 1 H), 5,21-5,13 (m, 1 H), 4,54-4,46 (m, 1 H), 3,79-3,71 (m, 1 H), 3,70-3,60 (m, 2 H), 3,54-3,46 (m, 1 H), 2,80 (dd, J = 12,9 e 7,4 Hz, 1 H), 2,62 (s, 2 H), 2,58-2,53 (m, 1 H), 2,37-2,14 (m, 2 H), 1,89-1,56 (m, 11 H), 1,44 (dd, J = 13,3 e 7,4 Hz, 1 H), 1,11 (d, J = 6,6 Hz, 3 H), 1,07 (d, J = 6,2 Hz, 3 H), 0,96 (d, J = 7,4 Hz, 3 H).
[0981] Etapa 1c. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,5S,7S)-7-(aminometil)-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta- 2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en- 2-il acetato, sal de acetato (#B140).: A uma solução de #B22 (50,8 mg, 0,095 mmol, 1 eq.) em acetonitrila (3,1 mL) à temperatura ambiente, foi adicionado carbonil diimidazol (23,4 mg, 0,143 mmol, 1,5 eq.), e a reação foi deixada agitar por 30 min. A reação foi, então, aquecida para 65°C por 4 horas. A reação foi resfriada para temperatura ambiente, adicionada a uma solução de acetonitrila (83 mL), água (6 mL), e 1 N NaOH (6 mL) e deixada agitar à temperatura ambiente por 35 min. A reação foi neutralizada com ácido acético (0,35 mL) e concentrada in vacuo. O resíduo foi retomado em acetonitrila, seco sobre sulfato de sódio, filtrado e concentrado in vacuo. O material desejado bruto foi purificado por cromatografia de fase reversa (Método A) para dar #B140 Rendimento: 15 mg, 0,030 mmol, 32%. LCMS (Protocolo C): m/z 491,3 [M+H]+, tempo de retenção = 1,13 minuto. HPLC (Protocolo AA) tempo de retenção = 6,969 minutos (pureza 87%). 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 7,78 (d, J = 7,8 Hz, 1 H), 6,42-6,30 (m, 1 H), 6,26 (d, J = 16,0 Hz, 1 H), 6,11 (d, J = 11,5 Hz, 1 H), 5,87 (dd, J = 11,5 e 7,4 Hz, 1 H), 5,67 (dd, J = 16,0 e 5,8 Hz, 1 H), 5,565,49 (m, 1 H), 4,54-4,46 (m, 1 H), 3,81-3,73 (m, 1 H), 3,69-3,61 (m, 2 H), 3,54-3,46 (m, 1 H), 2,80 (dd, J = 12,9 e 7,4 Hz, 1 H), 2,63 (s, 2 H), 2,58-2,53 (m, 1 H), 2,37-2,14 (m, 2 H), 1,98 (s, 3 H), 1,84-1,56 (m, 8 H), 1,44 (dd, J = 13,3 e 7,4 Hz, 1 H), 1,25 (d, J = 6,2 Hz, 3 H), 1,07 (d, J = 6,6 Hz, 3 H), 0,96 (d, J = 7,0 Hz, 3 H).
[0982] Exemplo A47
[0983] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,4R,5R,7S)-4-hidroxi-7-{2-[(trans-4-hidroxiciclohexil)amino]-2- oxoetil}-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5- dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B141). e (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,4R,5R,7S)-4- hidroxi-7-{2-[(cis-3-hidroxiciclobutil)amino]-2-oxoetil}-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B142). e (2S,3Z)-5- ({(2R,3R,5S,6S)-6-[(2E,4E)-5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-(2- metilhidrazinil)-2-oxoetil]-1,6-dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4- dien-1-il]-2,5-dimetiltetrahidro-2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B143). e (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5- {(3R,4R,5R,7S)-4-hidroxi-7-[2-(1-metilhidrazinil)-2-oxoetil]-1,6- dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B144). e (2S,3Z)-5- ({(2R,3R,5S,6S)-6-[(2E,4E)-5-{(3R,4R,5R,7S)-7-[2-(1,2- dimetilhidrazinil)-2-oxoetil]-4-hidroxi-1,6-dioxaspiro[2,5]oct-5-il}-3- metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro-2H-piran-3-il}amino)-5- oxopent-3-en-2-il acetato (#B145).

[0984] Etapa 1a. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-4-hidroxi-7-{2-[(trans-4-hidroxiciclohexil)amino]-2- oxoetil}-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5- dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato(#B141).: A uma solução de #B1 (19,7 mg, 0,031 mmol, 1 eq.) dissol- vida em tetrahidrofurano (0,5 mL), foi adicionado trans-4- aminociclohexanol (5,7 mg, 0,049 mmol, 1,6 eq.) após agitação por 1,5 hora, a reação foi diluída com água, extraída com diclorometano, e as fases orgânicas combinadas foram secas sobre sulfato de sódio e filtradas. Os solventes foram removidos in vacuo. O material desejado bruto foi purificado por cromatografia de fase reversa (Método A) para dar #B141 como um sólido branco. Rendimento: 11,8 mg, 0,019 mmol, 60%. HPLC (Protocolo AA) tempo de retenção = 7,408 minutos (pureza 94%). LCMS (Protocolo D): m/z 633,3 [M+H]+, tempo de retenção = 0,73 minutos. 1H RMN (400 MHz, DMSO-d6) δ 7,79 (d, J = 8,2 Hz, 1 H), 7,68 (d, J = 7,8 Hz, 1 H), 6,42-6,32 (m, 1 H), 6,28 (d, J = 16,0 Hz, 1 H), 6,11 (d, J = 11,5 Hz, 1 H), 5,87 (dd, J = 11,5 e 7,4 Hz, 1 H), 5,59 (dd, J = 16,0 e 5,5 Hz, 1 H), 5,54-5,45 (m, 1 H), 5,00 (d, J = 5,1 Hz, 1 H), 4,49 (d, J = 4,3 Hz, 1 H), 4,30-4,15 (m, 2 H), 3,70-3,60 (m, 2 H), 3,543,39 (m, 2 H), 3,26-3,20 (m, 1 H), 2,74 (d, J = 5,1 Hz, 1 H), 2,58 (d, J = 5,1 Hz, 1 H), 2,37-2,10 (m, 3 H), 1,98 (s, 3 H), 1,89-1,59 (m, 10 H), 1,51-1,41 (m, 1 H), 1,25 (d, J = 6,6 Hz, 3 H), 1,20-1,10 (m, 4 H), 1,07 (d, J = 6,2 Hz, 3 H), 0,95 (d, J = 7,4 Hz, 3 H),
[0985] Etapa 1b. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-4-hidroxi-7-{2-[(cis-3-hidroxiciclobutil)amino]-2- oxoetil}-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B142). A uma solução de #B1 (16,2 mg, 0,026 mmol, 1 eq.) em te- trahidrofurano/N,N-dimetilformamida (5:1, 0,6 mL) à temperatura ambiente, foi adicionada N,N-diisopropiletilamina (13,7 μL, 0,078 mmol, 3 eq.) e cloridrato de trans-3-aminociclobutanol (4,8 mg, 0,039 mmol, 1,5 eq.) (12:48 pm), e a reação foi agitada por 2 horas. Adicional de N,N- dimetilformamida (100 uL), N,N-diisopropiletilamina (13 uL, 0,078 mmol, 3 eq.) e cloridrato de trans-3-aminociclobutanol (3 mg, 0,024 mmol, 0.9 eq.) foram adicionados, e a reação foi agitada por um adici- onal de 30 min. A reação foi diluída com dimetil sulfóxido e concentrada in vacuo para remover o tetrahidrofurano. O material desejado bruto foi purificado por cromatografia de fase reversa (Método A) para dar #B142 como um sólido branco. Rendimento: 9,5 mg, 0,016 mmol, 61%. HPLC (Protocolo AA) tempo de retenção = 7,057 minutos (pureza 91%). LCMS (Protocolo D): m/z 627,1 [M+Na]+, tempo de retenção = 0,72 minuto. 1H RMN (400 MHz, DMSO-d6) δ 8,03 (d, J = 7,8 Hz, 1 H), 7,79 (d, J = 8,2 Hz, 1 H), 6,42-6,32 (m, 1 H), 6,28 (d, J = 16,0 Hz, 1 H), 6,11 (d, J = 11,7 Hz, 1 H), 5,87 (dd, J = 11,7 e 7,4 Hz, 1 H), 5,59 (dd, J = 16,0 e 5,5 Hz, 1 H), 5,55-5,48 (m, 1 H), 5,07-4,97 (m, 2 H), 4,30-4,17 (m, 2 H), 3,81-3,71 (m, 1 H), 3,70-3,59 (m, 3 H), 3,54-3,46 (m, 1 H), 3,27-3,20 (m, 1 H), 2,75 (d, J = 4,9 Hz, 1 H), 2,58 (d, J = 4,9 Hz, 1 H), 2,48-2,39 (m, 2 H), 2,36-2,13 (m, 3 H), 1,98 (s, 3 H), 1,89-1,61 (m, 9 H), 1,49-1,41 (m, 1 H), 1,25 (d, J = 6,6 Hz, 3 H), 1,07 (d, J = 6,2 Hz, 3 H), 0,95 (d, J = 7,4 Hz, 3 H).
[0986] Etapa 1c. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-(2-metilhidrazinil)-2-oxoetil]-1,6- dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B143) e (2S,3Z)-5- ({(2R,3R,5S,6S)-6-[(2E,4E)-5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-(1- metilhidrazinil)-2-oxoetil]-1,6-dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4- dien-1-il]-2,5-dimetiltetrahidro-2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B144).: A uma solução de #B1 (32,4 mg, 0,051 mmol, 1 eq.) em tetrahidrofurano/N,N-dimetilformamida (2:1, 0,75 mL) à temperatura ambiente, foi adicionada N,N-diisopropiletilamina (71,8 μL, 0,408 mmol, 8 eq.) e sulfato de N-metilhidrazina (22,1 mg, 0,15 mmol, 3 eq.), e a reação foi agitada por 20 min. Adicional de N,N-dimetilformamida (250 uL) foi adicionado. Após um adicional de 30 min, mais N,N- diisopropiletilamina (35 uL, 0,20 mmol, 4 eq.) e sulfato de N- metilhidrazina foi adicionado (15 mg, 0,10 mmol, 2 eq), e a reação foi agitada por 45 min. A reação foi diluída com água e etil acetato, e a camada aquosa foi extraída. As camadas orgânicas combinadas foram lavadas com salmoura, secas sobre sulfato de sódio, filtradas e concentradas in vacuo. O material desejado bruto foi purificado por croma- tografia de fase reversa (Método A) para dar #B143 e #B144 como sólidos brancos.
[0987] #B143 Rendimento: 1,9 mg, 0,0034 mmol, 7%. LCMS (Protocolo D): m/z 564,2 [M+H]+, tempo de retenção = 0,73 minuto. 1H RMN (500 MHz, DMSO-d6) δ 9,31 (d, J = 5,9 Hz, 1 H), 7,81 (d, J = 8,1 Hz, 1 H), 6,41-6,32 (m, 1 H), 6,29 (d, J = 16,0 Hz, 1 H), 6,11 (d, J = 11,6 Hz, 1 H), 5,87 (dd, J = 11,6 e 7,6 Hz, 1 H), 5,59 (dd, J = 16,0 e 5,6 Hz, 1 H), 5,54-5,48 (m, 1 H), 5,04 (d, J = 5,4 Hz, 1 H), 4,80-4,72 (m, 1 H), 4,29-4,20 (m, 2 H), 3,69-3,61 (m, 2 H), 3,53-3,46 (m, 1 H), 3,273,21 (m, 1 H), 2,75 (d, J = 5,1 Hz, 1 H), 2,58 (d, J = 5,1 Hz, 1 H), 2,452,13 (m, 7 H), 1,98 (s, 3 H), 1,89-1,74 (m, 3 H), 1,72-1,58 (m, 4 H), 1,53-1,45 (m, 1 H), 1,25 (d, J = 6,6 Hz, 3 H), 1,07 (d, J = 6,4 Hz, 3 H), 0,95 (d, J = 7,3 Hz, 3 H).
[0988] #B144 Rendimento: 2,3 mg, 0,0040 mmol, 8%. LCMS (Protocolo D): m/z 564,2 [M+H]+, tempo de retenção = 0,76 minuto. 1H RMN (500 MHz, DMSO-d6) δ 7,81 (d, J = 7,8 Hz, 1 H), 6,41-6,27 (m, 2 H), 6,11 (d, J = 11,5 Hz, 1 H), 5,87 (dd, J = 11,5 e 7,3 Hz, 1 H), 5,60 (dd, J = 16,1 e 5,9 Hz, 1 H), 5,56-5,48 (m, 1 H), 4,94 (d, J = 6,4 Hz, 1 H), 4,67 (s, 1 H), 4,32-4,20 (m, 2 H), 3,69-3,62 (m, 2 H), 3,53-3,47 (m, 1 H), 3,26-3,21 (m, 1 H), 3,06 (dd, J = 154 e 7,3 Hz, 1 H), 2,98 (s, 2 H), 2,75 (d, J = 4,9 Hz, 1 H), 2,62-2,53 (m, 2 H), 2,35-2,13 (m, 3 H), 1,98 (s, 3 H), 1,89-1,74 (m, 3 H), 1,72-1,58 (m, 5 H), 1,25 (d, J = 6,4 Hz, 3 H), 1,07 (d, J = 6,4 Hz, 3 H), 0,95 (d, J = 7,3 Hz, 3 H).
[0989] Etapa 1d. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3R,4R,5R,7S)-7-[2-(1,2-dimetilhidrazinil)-2-oxoetil]-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B145).: A uma solução de #B1 (29,7 mg, 0,047 mmol, 1 eq.) em tetrahidrofurano/N,N- dimetilformamida (1:1, 1 mL) à temperatura ambiente, foi adicionada N,N-diisopropiletilamina (116 μL, 0,658 mmol, 14 eq.) seguido por di- cloridrato de N,N’-dimetilhidrazina (31,3 mg, 0,235 mmol, 5 eq.), e a reação foi agitada por 30 min. A reação foi diluída com água e etil acetato, e a camada aquosa extraída. As camadas orgânicas combinadas foram lavadas com salmoura, secas sobre sulfato de sódio, filtradas e concentradas in vacuo. O material desejado bruto foi purificado por cromatografia de fase reversa (Método A) para dar #B145 como um sólido branco. Rendimento: 2,2 mg, 0,0038 mmol, 8%. LCMS (Protocolo D): m/z 578,2 [M+H]+, tempo de retenção = 0,82 minuto. 1H RMN (500 MHz, DMSO-d6) δ 7,81 (d, J = 8,1 Hz, 1 H), 6,41-6,27 (m, 2 H), 6,11 (d, J = 11,7 Hz, 1 H), 5,87 (dd, J = 11,7 e 7,6 Hz, 1 H), 5,60 (dd, J = 15,9 e 5,4 Hz, 1 H), 5,56-5,48 (m, 1 H), 4,94 (d, J = 6,6 Hz, 1 H), 4,79 (q, J = 5,7 Hz, 1 H), 4,32-4,19 (m, 2 H), 3,68-3,61 (m, 2 H), 3,533,47 (m, 1 H), 3,26-3,21 (m, 1 H), 3,01 (d, J = 15,2 e 7,3 Hz, 1 H), 2,93 (s, 3 H), 2,76-2,73 (m, 1 H), 2,60-2,53 (m, 2 H), 2,44 (d, J = 5,7 Hz, 3 H), 2,35-2,15 (m, 3 H), 1,98 (s, 3 H), 1,87-1,75 (m, 3 H), 1,72-1,56 (m, 5 H), 1,25 (d, J = 6,4 Hz, 3 H), 1,07 (d, J = 6,4 Hz, 3 H), 0,95 (d, J = 7,3 Hz, 3 H).
[0990] Exemplo A48
[0991] Preparação de N-{6-[(bromoacetil)amino]hexanoil}-D-valil-N-[4-({[(2-{[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5- il]acetil}hidrazinil)carbonil]oxi}metil)fenil]-N~5~-carbamoil-L- ornitinamida (#B146).
[0992] Etapa 1. Síntese de N-{6-[(bromoacetil)amino]hexanoil}-D- valil-N-[4-({[(2-{[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5- {[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5- il]acetil}hidrazinil)carbonil]oxi}metil)fenil]-N~5~-carbamoil-L- ornitinamida (#B146). A uma solução de #B47 (10,4 mg, 0,009 mmol, 1 eq.) em N,N-dimetilformamida (0,3 mL) à temperatura ambiente, foi adicionada N,N-diisopropiletilamina (6,3 μL, 0,036 mmol, 4 eq.) seguido por 1-[(bromoacetil)oxi]pirrolidina-2,5-diona (4,2 mg, 0,017 mmol, 1,9 eq.) e foi agitada por 15 minutos e, em seguida, purificada por cromatografia de fase reversa (Método A) para gerar #B146 como um sólido branco. Rendimento: 4,6 mg, 0,004 mmol, 43%. HPLC (Protocolo AA): tempo de retenção = 7,597 minutos (pureza 87%). LCMS (Protocolo C): m/z 1188,6 [M+H]+ tempo de retenção = 1,37 minutos. 1H RMN (400 MHz, DMSO-d6) δ 9,99 (s, 1 H), 9,68 (s, 1 H), 9,12 (s, 1 H), 8,26-8,18 (m, 1 H), 8,11-8,04 (m, 1 H), 7,84-7,74 (m, 2 H), 7,64-7,54 (m, 2 H), 7,34-7,21 (m, 2 H), 6,41-6,27 (m, 2 H), 6,11 (d, J = 11,7 Hz, 1 H), 6,00-5,93 (m, 1 H), 5,87 (dd, J = 11,7 e 7,4 Hz, 1 H), 5,65-5,47 (m, 2 H), 5,40 (s, 2 H), 5,04 (d, J = 5,5 Hz, 1 H), 5,02-4,96 (m, 2 H), 4,434,34 (m, 1 H), 4,30-4,16 (m, 3 H), 3,81 (s, 2 H), 3,69-3,60 (m, 2 H), 3,54-3,45 (m, 1 H), 3,26-3,20 (m, 1 H), 3,09-2,88 (m, 4 H), 2,77-2,73 (m, 1 H), 2,62-2,55 (m, 1 H), 2,36-2,07 (m, 5 H), 1,98 (s, 3 H), 1,84- 1,76 (m, 1 H), 1,73-1,61 (m, 5 H), 1,54-1,33 (m, 5 H), 1,29-1,21 (m, 5 H), 1,07 (d, J = 6,2 Hz, 3 H), 0,95 (d, J = 7,4 Hz, 3 H), 0,84 (dd, J = 11,3 e 6,6 Hz, 6 H),
[0993] Exemplo A49
[0994] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,4R,5R,7S)-7-(aminometil)-4-hidroxi-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5- oxopent-3-en-2-il acetato, sal de acetato (#B147).
[0995] Etapa 1. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-(aminometil)-4-hidroxi-1,6-dioxaspiro[2,5]oct-5-il]- 3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5- oxopent-3-en-2-il acetato (#B147).: A uma solução de #B14 (40 mg, 0,073 mmol, 1 eq.) em acetonitrila (2,3 mL), foi adicionado CDI (29,8 mg, 0,182 mmol, 2,5 eq.) à temperatura ambiente, e a reação foi agitada por 40 min. A reação foi, então, aquecida para 65°C por 4 horas. Após resfriamento para temperatura ambiente, a mistura de reação foi adicionada a uma solução de acetonitrila (63 mL), água (4,5 mL), e 1 N NaOH (4,5 mL) e agitada por 30 min. Ácido acético (270 uL) foi adicionado, e a mistura concentrada in vacuo. O resíduo obtido foi retomado em acetonitrila, seco sobre sulfato de sódio, filtrado e concentrado in vacuo. O material desejado bruto foi purificado por cromatografia de fase reversa (Método A) para dar #B147 como um sólido branco. Rendimento: 5,1 mg, 0,0088 mmol, 12%. HPLC (Protocolo AA) tempo de retenção = 6,778 minutos (pureza 89%). LCMS (Protocolo C): m/z 507,2 [M+H]+, tempo de retenção = 0,98 minuto. 1H RMN (400 MHz, DMSO-d6) δ 7,77 (d, J = 7,4 Hz, 1 H), 6,41-6,24 (m, 2 H), 6,11 (d, J = 11,7 Hz, 1 H), 5,87 (dd, J = 11,7 e 7,4 Hz, 1 H), 5,65 (dd, J = 16,0 e 6,2 Hz, 1 H), 5,56-5,48 (m, 1 H), 5,00-4,91 (m, 1 H), 4,27-4,20 (m, 1 H), 3,78-3,60 (m, 3 H), 3,55-3,46 (m, 1 H), 3,22-3,18 (m, 1 H), 2,80-2,71 (m, 2 H), 2,62-2,53 (m, 2 H), 2,33-2,15 (m, 3 H), 1,98 (s, 3 H), 1,891,77 (m, 5 H), 1,73-1,61 (m, 4 H), 1,48-1,41 (m, 1 H), 1,25 (d, J = 6,2 Hz, 3 H), 1,07 (d, J = 6,2 Hz, 3 H), 0,95 (d, J = 7,0 Hz, 3 H).
[0996] Exemplo A50
[0997] Preparação de metil [(3S,5S,7S)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-hidroxipent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6- dioxaspiro[2,5]oct-5-il]acetato (#B148).
[0998] Etapa 1. Síntese de metil [(3S,5S,7S)-7-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6- dioxaspiro[2,5]oct-5-il]acetato (#B148).: A uma solução de #NP2 (204 mg, 0,393 mmol, 1 eq.) em N,N-dimetilformamida (4,5 mL) à temperatura ambiente, foram adicionados carbonato de potássio (272 mg, 1,96 mmol, 5 eq.) e iodometano (740 μL, 11,8 mmol, 30 eq.), e a reação foi agitada por 1,5 hora. A reação foi filtrada, lavada com água (3x), seca sobre sulfato de sódio, filtrada e concentrada in vacuo. LCMS (Protocolo D): m/z 534,42 [M+H]+, tempo de retenção = 0,89 min. O material bruto foi usado na etapa seguinte sem purificação adicional.
[0999] Etapa 2. Síntese de metil [(3S,5S,7S)-7-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-hidroxipent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6-dioxaspiro[2,5]oct-5-il]acetato (#B148). A uma solução do material bruto da etapa 1, do exemplo A#50 em metanol (3,5 mL) à temperatura ambiente, foi adicionado carbonato de potássio (136 mg, 0,056 mmol, 2,5 eq.), e a reação foi agitada por 2 horas. A reação foi filtrada com metanol, diluída com dimetil sulfóxido (2 mL), e concentrada in vacuo. Purificação por cromatografia líquida de pressão média de fase reversa eluída com 0,02% de ácido acético em água (v/v) e 0,02% de ácido acético em acetonitrila (v/v) (10% a 90%) forneceu #B148 como um sólido branco. Rendimento: 91,6 mg, 0,18 mmol, 48%. LCMS (Protocolo D): m/z 492,47 [M+H]+, tempo de retenção = 0,80 minuto. 1H RMN (400 MHz, DMSO-d6) δ 7,76 (d, J = 7,8 Hz, 1 H), 6,25 (d, J = 16,0 Hz, 1 H), 5,97 (d, J = 11,9 Hz, 1 H), 5,87 (d, J = 11,9 e 7,0 Hz, 1 H), 5,655,48 (m, 2 H), 5,22-5,13 (m, 1 H), 5,10 (d, J = 4,7 Hz, 1 H), 4,56-4,48 (m, 1 H), 4,36-4,25 (m, 1 H), 3,69-3,61 (m, 2 H), 3,60 (s, 3 H), 3,553,46 (m, 1 H), 2,74-2,56 (m, 4 H), 2,38-2,13 (m, 2 H), 1,90-1,60 (m, 9 H), 1,44 (dd, J = 13,2 e 7,0 Hz, 1 H), 1,11 (d, J = 6,2 Hz, 3 H), 1,06 (d, J = 6,2 Hz, 3 H), 0,96 (d, J = 7,0 Hz, 3H).
[01000] Exemplo A51
[01001] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5- [(3S,5S,7S)-7-(2-metoxi-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5- oxopent-3-en-2-il 4-(metilsulfanil)butanoato (#B149).
[01002] Etapa 1. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3S,5S,7S)-7-(2-metoxi-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5- oxopent-3-en-2-il 4-(metilsulfanil)butanoato (#B149). A uma solução de #B148 (12 mg, 0,024 mmol, 1 eq.) em diclorometano (0,5 mL), foi adicionado ácido 4-(metiltio)butanóico (32,2 mg, 0,24 mmol, 10 eq.), 4- N,N-dimetilamino piridina (2,9 mg, 0,023 mmol, 1 eq.), e DIC (41,3 μL, 0,264 mmol, 11 eq.), e a reação foi deixada agitar por 1 hora. A reação foi diluída com dimetilsulfóxido (0,8 mL) e concentrada in vacuo. O material desejado bruto foi purificado por cromatografia de fase reversa (Método A) para dar #B149 como uma goma. Rendimento: 9.7 mg, 0,016 mmol, 66%. LCMS (Protocolo D): m/z 608,2 [M+H]+, tempo de retenção = 1,05 minuto. 1H RMN (400 MHz, DMSO-d6) δ 7,79 (d, J = 7,8 Hz, 1 H), 6,44-6,32 (m, 1 H), 6,25 (d, J = 16,0 Hz, 1 H), 6,12 (d, J = 11,7 Hz, 1 H), 5,87 (dd, J = 11,7 e 7,4 Hz, 1 H), 5,62-5,50 (m, 2 H), 4,57-4,48 (m, 1 H), 4,36-4,25 (m, 1 H), 3,70-3,62 (m, 2 H), 3,60 (s, 3 H), 3,55-3,47 (m, 1 H), 2,74-2,56 (m, 4 H), 2,48-2,43 (m, 2 H), 2,412,15 (m, 4 H), 2,02 (s, 3 H), 1,88-1,60 (m, 11 H), 1,44 (dd, J = 12,9 e 6,6 Hz, 1 H), 1,26 (d, J = 6,6 Hz, 3 H), 1,07 (d, J = 6,2 Hz, 3 H), 0,96 (d, J = 7,4 Hz, 3H).
[01003] Exemplo A52
[01004] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5- [(3R,4R,5R,7S)-7-{[(bromoacetil)amino]metil}-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B150).
[01005] Etapa 1. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-{[(bromoacetil)amino]metil}-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B150).: A uma solução de #B147 (7 mg, 0,01 mmol, 1 eq.) em N,N-dimetilformamida (0,4 mL) à temperatura ambiente, foram adicionados N,N-diisopropiletilamina (8,5 μL, 0,048 mmol, 4 eq.) e 1- [(bromoacetil)oxi]pirrolidina-2,5-diona (4,2 mg, 0,018 mmol, 1,5 eq.), e a reação foi agitada por 10 min. O material desejado bruto foi purificado por cromatografia de fase reversa (Método A) para dar #B150 como um sólido branco. Rendimento: 2,9 mg, 0,005 mmol, 40%. LCMS (Pro tocolo D): m/z 649,2 [M+Na]+, tempo de retenção = 0,81 minuto. 1H RMN (400 MHz, DMSO-d6) δ 8,42-8,32 (m, 1 H), δ 7,79 (d, J = 7,8 Hz, 1 H), 6,44-6,32 (m, 1 H), 6,28 (d, J = 16,0 Hz, 1 H), 6,11 (d, J = 11,3Hz, 1 H), 5,87 (dd, J = 11,3 e 7,4 Hz, 1 H), 5,61 (dd, J = 16,0 e 5,5 Hz, 1 H), 5,56-5,49 (m, 1 H), 5,02 (d, J = 5,9 Hz, 1 H), 4,33-4,26 (m, 1 H), 3,95-3,83 (m, 3 H), 3,72-3,59 (m, 2 H), 3,55-3,45 (m, 1 H), 3,40-3,32 (m, 1 H), 3,28-3,14 (m, 2 H), 2,77 (d, J = 5,1 Hz, 1 H), 2,61 (d, J = 5,1 Hz, 1 H), 2,31-2,12 (m, 2 H), 1,98 (s, 3 H), 1,88-1,75 (m, 3 H), 1,73- 1,61 (m, 4 H), 1,51-1,41 (m, 1 H), 1,25 (d, J = 6,2 Hz, 3 H), 1,07 (d, J =6,2 Hz, 3 H), 0,95 (d, J = 7,0 Hz, 3 H).
[01006] Exemplo A53
[01007] Preparação de N-{6-[(bromoacetil)amino]hexanoil}-L-valil-N-(4-{[({[(3R,5S,7S)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-1,6-dioxaspiro[2,5]oct-5- il]metil}carbamoil)oxi]metil}fenil)-N~5~-carbamoil-L-ornitinamida (#B151).
[01008] Etapa 1. Síntese de N-(6-{[(9H-fluoren-9- ilmetoxi)carbonil]amino}hexanoil)-L-valil-N-(4-{[({[(3R,5S,7S)-7- {(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6- dioxaspiro[2,5]oct-5-il]metil}carbamoil)oxi]metil}fenil)-N~5~-carbamoil- L-ornitinamida (#B152). A uma solução de #B140 (10,7 mg, 0,022 mmol, 1 eq.) em N,N-dimetilformamida à temperatura ambiente, foram adicionados 2,6-lutidina (10,2 μL, 0,088 mmol, 4 eq.), N,N- diisopropiletilamina (15,5 μL, 0,088 mmol, 4 eq.), 4-N,N-dimetilamino piridina (2,7 mg, 0,022 mmol, 1 eq.), e #B45 (22,9 mg, 0,026 mmol, 1,2 eq.), e a reação foi agitada por 40 min. O material desejado bruto foi purificado por cromatografia de fase reversa (Método A) para dar #B152 como um sólido branco. Rendimento: 14,9 mg, 0,012 mmol, 55%. LCMS (Protocolo C): m/z 1231,6 [M+H]+, tempo de retenção = 197 minuto.
[01009] Etapa 2. Síntese de N-(6-aminohexanoil)-L-valil-N-(4- {[({[(3R,5S,7S)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-1,6-dioxaspiro[2,5]oct-5-il]metil}carbamoil)oxi]metil}fenil)-N~5~-carbamoil-L-ornitinamida, sal de acetato (#B153).: O composto do título foi preparado em 86% de rendimento a partir de 14,9 mg (0,012 mmol, 1,0 eq) de #B152 e 20,4 mg (0,24 mmol, 20,0 eq) de Piperidina usando o procedimento descrito para a preparação do composto #B47. LCMS (Protocolo C): m/z 1009.83 [M+H]+, tempo de retenção = 1,35 minuto.
[01010] Etapa 3. Síntese de N-{6-[(bromoacetil)amino]hexanoil}-L- valil-N-(4-{[({[(3R,5S,7S)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-1,6-dioxaspiro[2,5]oct-5- il]metil}carbamoil)oxi]metil}fenil)-N~5~-carbamoil-L-ornitinamida (#B151). O composto do título foi preparado em 70% de rendimento a partir de 11 mg (0,01 mmol, 1,0 eq), de #B153 e 3,5 mg (0,015 mmol, 1,5 eq), 1-[(bromoacetil)oxi]pirrolidina-2,5-diona e 5,2 mg (0,04 mmol, 4,0 eq) de N,N-diisopropiletilamina usando o procedimento descrito para a preparação do composto #B150. HPLC (Protocolo AA) tempo de retenção = 8,413 minutos (pureza 87%). LCMS (Protocolo C): m/z 1151,5 [M+Na]+, tempo de retenção = 1,61 minuto. 1H RMN (400 MHz, DMSO-d6) δ 9,96 (s, 1 H), 8,25-8,18 (m 1 H), 8,06 (d, J = 7,4 Hz, 1 H), 7,83-7,74 (m, 2 H), 7,62-7,54 (m, 2 H), 7,31-7,20 (m, 3 H), 6,41-6,31 (m, 1 H), 6,25 (d, J = 15,8 Hz, 1 H), 6,11 (d, J = 11,7 Hz, 1 H), 6,005,92 (m, 1 H), 5,87 (dd, J = 11,7 e 7,4 Hz, 1 H), 5,61 (dd, J = 15,8 e 5,9 Hz, 1 H), 5,55-5,47 (m, 1 H), 5,39 (s, 2 H), 5,00-4,88 (m, 2 H), 4,594,49 (m, 1 H), 4,43-4,34 (m, 1 H), 4,24-4,15 (m, 2 H), 3,99-3,88 (m, 1 H), 3,81 (s, 2 H), 3,69-3,58 (m, 2 H), 3,53-3,36 (m, 2 H), 3,10-2,88 (m, 5 H), 2,63 (s, 2 H), 2,31-2,08 (m, 4 H), 2,02-1,92 (m, 4 H), 1,83-1,56 (m, 10 H), 1,55-1,29 (m, 7 H), 1,28-1,20 (m, 5 H), 1,05 (d, J = 6,6 Hz, 3 H), 0,94 (d, J = 7,4 Hz, 3 H), 0,85 (dd, J = 11,3 e 6,6 Hz, 6 H).
[01011] Exemplo A54
[01012] Preparação de N-{6-[(bromoacetil)amino]hexanoil}-L-valil-N- (4-{[({[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5- il]metil}carbamoil)oxi]metil}fenil)-N~5~-carbamoil-L-ornitinamida (#B154).
[01013] Etapa 1. Síntese de N-(6-{[(9H-fluoren-9- ilmetoxi)carbonil]amino}hexanoil)-L-valil-N-(4-{[({[(3R,5S,7R,8R)-7- {(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi- 1,6-dioxaspiro[2,5]oct-5-il]metil}carbamoil)oxi]metil}fenil)-N~5~-carbamoil-L-ornitinamida (#B155). O composto do título foi preparado em 41% de rendimento a partir de 13 mg (0,023 mmol) de #B147, 9,9 mg, (0,092 mmol, 4 eq.) de 2,6-lutidina, 12.0 mg (0,092 mmol, 4 eq.), de N,N-diisopropiletilamina, 2,8 mg, (0,023 mmol, 1 eq.) de 4-N,N- dimetilamino piridina e 24,6 mg (0,028 mmol, 4eq) de #B45 (22.9 mg, 0,026 mmol, 1,2 eq.) usando o procedimento descrito para a preparação de #B152. LCMS (Protocolo D): m/z 1247,93 [M+H]+, tempo de retenção = 0,91 minuto.
[01014] Etapa 2. Síntese de N-(6-aminohexanoil)-L-valil-N-(4- {[({[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5-il]metil}carbamoil)oxi]metil}fenil)-N~5~-carbamoil-L-ornitinamida, sal de acetato (#B156). O composto do título foi preparado em 66% de rendimento a partir de 11,9 mg (0,01 mmol, 1,0 eq) de ##B155 e 17,0 mg (0,2 mmol, 20,0 eq) de piperidina usando o procedimento descrito para a preparação de #B153. HPLC (Protocolo AA) tempo de retenção = 7,001 minutos (pureza 82%). LCMS (Protocolo D): m/z 1025,4 [M+H]+, tempo de retenção = 0,69 minuto.
[01015] Etapa 3. Síntese de N-{6-[(bromoacetil)amino]hexanoil}-L- valil-N-(4-{[({[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)- 4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5- il]metil}carbamoil)oxi]metil}fenil)-N~5~-carbamoil-L-ornitinamida(#B154). O composto do título foi preparado em 46% de rendimento a partir de 6,1 mg (0,006 mmol, 1,0 eq.) de #B156, 5,0 mg (0,015 mmol, 7,0 eq) de N,N-diisopropiletilamina e 3,5 mg, 0,015 mmol, 1,5 eq.) de 1-[(bromoacetil)oxi]pirrolidina-2,5-diona usando o procedimento descrito para a preparação do composto #B150. HPLC (Protocolo AA) tempo de retenção = 7,669 minutos (pureza 84%). LCMS (Protocolo D): m/z 1151,5 [M+Na]+, tempo de retenção = 0,79 minuto. 1H RMN (400 MHz, DMSO-d6) δ 9,99 (s, 1 H), 8,27-8,19 (m 1 H), 8,16-8,06 (m, 1 H), 7,89-7,73 (m, 2 H), 7,62-7,54 (m, 2 H), 7,31-7,20 (m, 3 H), 6,416,31 (m, 1 H), 6,27 (d, J = 16,4 Hz, 1 H), 6,11 (d, J = 10,9 Hz, 1 H), 6,05-5,94 (m, 1 H), 5,86 (dd, J = 10,9 e 7,0 Hz, 1 H), 5,67-5,56 (m, 1 H), 5,55-5,47 (m, 1 H), 5,40 (s, 2 H), 5,02-4,88 (m, 3 H), 4,44-4,33 (m, 2 H), 4,30-4,23 (m, 1 H), 4,22-4,15 (m, 2 H), 3,96-3,84 (m, 1 H), 3,81 (s, 2 H), 3,69-3,58 (m, 2 H), 3,53-3,43 (m, 2 H), 3,10-2,89 (m, 5 H), 2,79-2,71 (m, 1 H), 2,61-2,56 (m, 1 H), 2,31-2,10 (m, 4 H), 2,04-1,91 (m, 4 H), 1,84-1,32 (m, 15 H), 1,30-1,18 (m, 4 H), 1,06 (d, J = 6,2 Hz, 3 H), 0,94 (d, J = 7,4 Hz, 3 H), 0,84 (dd, J = 10,9 e 6,6 Hz, 6 H).
[01016] Exemplo A55
[01017] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5- [(3R,4R,5R,7S)-7-{2-[(2E)-2-{1-[4-({5-[(bromoacetil)amino]pentil}oxi)fenil]etilideno}hidrazinil]-2-oxoetil}-4- hidroxi-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5- dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetate (#B157).
[01018] Etapa 1. Síntese de 9H-fluoren-9-ilmetil [5-(4- acetilfenoxi)pentil]carbamato (#B158).: A uma solução de (9H-fluoren- 9-il)metil 5-hidroxipentilcarbamato (5 g, 15,4 mmoles, 1 eq.), 1-(4- hidroxifenil)etanona (2,1 g, 15,4 mmoles, 1 eq.), e trifenilfosfina (4,53 g, 16,9 mmoles, 1,1 eq.) em tolueno (50 mL), foi adicionado DIAD (3,43 g, 16,9 mmol, 1,1 eq.) em gotas a 0-10°C. A solução foi agitada à temperatura ambiente por 1 hora, diluída com etil acetato, e lavada com cloreto de amônio aquoso saturado e salmoura. As camadas orgânicas foram secas sobre sulfato de sódio, filtradas e concentradas in vacuo. O resíduo foi purificado por cromatografia de sílica gel eluída com éter de petróleo:etil acetato a partir de 10:1 a 7:1 e adicionalmente purificado por cromatografia de fase reversa para gerar #B158 (3,6 g, 53%) como um sólido branco. 1H RMN (400 MHz, CDCl3): δ 7,93 (d, 2 H), 7,76 (d, 2 H), 7,59 (d, 2 H), 7,42 (m, 2 H), 7,33 (m, 2 H), 6,92 (d, 2 H), 4,79 (m, 1 H), 4,43 (m, 2 H), 4,23 (m, 1 H), 4,04 (m, 2 H), 3,25 (m, 2 H), 2,55 (s, 3 H), 1,84 (m, 2 H), 1,58-1,52 (m, 4 H).
[01019] Etapa 2. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-{2-[(2E)-2-(1-{4-[(5-{[(9H-fluoren-9-lmetoxi)carbonil]amino}pentil)oxi]fenil}etilideno)hidrazinil]-2-oxoetil}-4- hidroxi-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5- dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B159). O composto do título foi preparado em 33% de rendimento a partir de 30,8 mg (0,056 mmol, 1,0 eq.) de #B6 e 124,0 mg (0,28 mmol, 5,0 eq) de #B158 usando o procedimento descrito para a preparação do composto #B20. LCMS (Protocolo D): m/z 975,4 [M+H]+, tempo de retenção = 1,05 minuto.
[01020] Etapa 3. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-{2-[(2E)-2-(1-{4-[(5-aminopentil)oxi]fenil}etilideno)hidrazinil]-2-oxoetil}-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato, sal de acetato (#B160). O composto do título foi preparado em 64% de rendimento a partir de 17,8 mg (0,018 mmol, 1,0 eq)) de #B159 e 30,7 mg (0,36 mmol, 20,0 eq.) de piperidina usando o procedimento descrito para a preparação de #B47. LCMS (Protocolo D): m/z 753.62 [M+H]+, tempo de retenção = 0,66 minuto.
[01021] Etapa 4. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-{2-[(2E)-2-{1-[4-({5-[(bromoacetil)amino]pentil}oxi)fenil]etilideno}hidrazinil]-2-oxoetil}-4- hidroxi-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5- dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B157).: O composto do título foi preparado em 46% de rendimento a partir de 6,2 mg (0,008 mmol, 1,0 eq) de #B160 e 2,8 mg (0,012 mmol, 1,5 eq) de 1-[(bromoacetil)oxi]pirrolidina-2,5-diona e 4,2 mg (0,032 mmol, 4,0 eq) de N,N-diisopropiletilamina usando o procedimento descrito para a preparação do composto #B150.. HPLC (Protocolo AA) tempo de retenção = 8,668 minutos (pureza 53%). LCMS (Protocolo D): m/z 873,3 [M+H]+, tempo de retenção = 0,88 minuto. 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 10,39-10,25 (m, 1 H), 8,30-8,20 (m, 1 H), 7,82-7,66 (m, 3 H), 6,99-6,90 (m, 2 H), 6,42-6,22 (m, 2 H), 6,166,06 (m, 1 H), 5,92-5,81 (m, 1 H), 5,68-5,34 (m, 3 H), 5,09-4,92 (m, 1 H), 4,51-4,25 (m, 3 H), 4,03-3,94 (m, 2 H), 3,82 (s, 2 H), 3,70-3,55 (m, 2 H), 3,50-3,40 (m, 1 H), 3,15-3,05 (m, 2 H), 2,90-2,71 (m, 2 H), 2,642,56 (m, 2 H), 2,30-2,10 (m, 5 H), 1,98 (s, 3 H), 1,94-1,84 (m, 1 H), 1,83-1,55 (m, 8 H), 1,53-1,33 (m, 4 H), 1,29-1,20 (m, 3 H), 1,12-1,00 (m, 3 H), 0,98-0,88 (m, 3 H).
[01022] Exemplo A56
[01023] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5- [(3R,4R,5R,7S)-7-({[N-(bromoacetil)-beta-alanil]amino}metil)-4-hidroxi- 1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B161)
[01024] Etapa 1. Síntese de 2,5-dioxopirrolidin-1-il N-[(9H-fluoren-9- ilmetoxi)carbonil]-beta-alaninato (#B162). A uma solução de N-[(9H- fluoren-9-ilmetoxi)carbonil]-β-alanina (297 mg, 0,95 mmol, 1 eq.) em tetrahidrofurano (3,5 mL) à temperatura ambiente, foi adicionado N- hidroxissuccinimida (112 mg, 0,954 mmol, 1 eq.) e N,N'- Diciclohexilcarbodiimida (228 mg, 1,05 mmol, 1,1 eq.), e a reação foi deixada agitar por 4 horas. A reação foi filtrada e lavada com etil acetato e concentrada in vacuo. O material desejado bruto foi purificado por cromatografia líquida de pressão média de fase reversa eluída com 0,02% de ácido acético em água (v/v) e 0,02% de ácido acético em acetonitrila (v/v) (10% a 95%) para dar #B162 como um sólido branco. Rendimento: 320 mg, 0,78 mmol, 82%. LCMS (Protocolo D): m/z 431,0 [M+Na]+, tempo de retenção = 0,91 minuto.
[01025] Etapa 2. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3R,4R,5R,7S)-7-[({N-[(9H-fluoren-9-ilmetoxi)carbonil]-beta- alanil}amino)metil]-4-hidroxi-1,6-dioxaspiro[2,5]oct-5-il}-3-metilpenta- 2,4-dien-1-il]-2,5-dimetiltetrahidro-2H-piran-3-il}amino)-5-oxopent-3-en- 2-il acetato (#B163). A uma solução de #B147 (15,1 mg, 0,027 mmol, 1 eq.) em N,N-dimetilformamida (0,5 mL), foram adicionados N,N- diisopropiletilamina (14,3 μL, 0,081 mmol, 3 eq.) e #B162 (22,1 mg, 0,054 mmol, 2 eq.), e a reação foi deixada agitar por 30 min. A reação foi purificada por cromatografia de fase reversa (Método A) para dar uma mistura do #B163 desejado e #B162 não reagido LCMS (Protocolo D): m/z 800,4 [M+H]+, tempo de retenção = 0.97 minuto. Este material foi usado na etapa seguinte sem purificação adicional.
[01026] Etapa 3. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3R,4R,5R,7S)-7-[(beta-alanilamino)metil]-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato, sal de acetato (#B164). O composto do título foi preparado em 63% de rendimento a partir de 15,7 mg (0,02 mmol, 1,0 eq) de #B163 e 34,1 mg (0,4 mmol, 20,0 eq) de piperidina usando o procedimento descrito para a preparação do composto #B47. LCMS (Protocolo D): m/z 578,41 [M+H]+, tempo de retenção = 0,62 minuto.
[01027] Etapa 4. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-({[N-(bromoacetil)-beta-alanil]amino}metil)-4- hidroxi-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5- dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B161). O composto do título foi preparado em 43% de rendimento a partir de 7,3 mg (0,013 mmol, 1,0 eq) de #B164 e 4,5 mg (0,019 mmol, 1,5 eq) de 1-[(bromoacetil)oxi]pirrolidina-2,5-diona e 6,8 mg (0,052 mmol, 4,0 eq) de N,N-diisopropiletilamina usando o procedimento descrito para a preparação do composto #B150. HPLC (Protocolo AA) tempo de retenção = 6,564 minutos (pureza 72%). LCMS (Protocolo D): m/z 698,1 [M+H]+, tempo de retenção = 0,79 minuto. 1H RMN (400 MHz, DMSO-d6) δ 8,31-8,23 (m, 1 H), 8,02-7,93 (m, 1 H), δ 7,78 (d, J = 7,8 Hz, 1 H), 6,41-6,32 (m, 1 H), 6,28 (d, J = 15,8 Hz, 1 H), 6,11 (d, J = 11,7 Hz, 1 H), 5,87 (dd, J = 11,7 e 7,4 Hz, 1 H), 5,61 (dd, J = 15,8 e 5,5 Hz, 1 H), 5,56-5,45 (m, 1 H), 5,00 (d, J = 6,2 Hz, 1 H), 4,31-4,24 (m, 1 H), 3,93-3,79 (m, 3 H), 3,72-3,59 (m, 2 H), 3,55-3,45 (m, 1 H), 3,273,08 (m, 3 H), 3,28-3,14 (m, 2 H), 2,77 (d, J = 5,1 Hz, 1 H), 2,61 (d, J = 5,1 Hz, 1 H), 2,36-2,14 (m, 4 H), 1,98 (s, 3 H), 1,88-1,75 (m, 3 H), 1,731,61 (m, 4 H), 1,51-1,41 (m, 1 H), 1,25 (d, J = 6,6 Hz, 3 H), 1,07 (d, J = 6,2 Hz, 3 H), 0,95 (d, J = 7,0 Hz, 3 H).
[01028] Exemplo A57
[01029] Preparação de N-{6-[(bromoacetil)amino]hexanoil}-L-valil-N- {4-[({[4-({[(2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3S,5S,7S)-7-(2- amino-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}- 2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2- il]oxi}carbonil)piperazin-1-il]carbonil}oxi)metil]fenil}-N~5~-carbamoil-L- ornitinamida (#B165).
[01030] Etapa 1. Síntese de N-(6-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}hexanoil)-L-valil-N-{4-[({[4-(terc- butoxicarbonil)piperazin-1-il]carbonil}oxi)metil]fenil}-N~5~-carbamoil-L- ornitinamida (#B166). A uma solução de terc-butil piperazina-1- carboxilato (32 mg, 0,17 mmol, 1 eq.) em N,N-dimetilformamida (0,9 mL), foi adicionada N,N-diisopropiletilamina (90,8 μL, 0,52 mmol, 3 eq.) e 4-N,N-dimetilamino piridina (4,2 mg, 0,034 mmol, 0,2 eq.) seguido por #B45 (151 mg, 0,17 mmol, 1 eq.), e a reação foi agitada por 30 min. A reação foi diluída com DMSO (2,5 mL) e purificada por croma- tografia líquida de pressão de meio de fase reversa eluída com 0,02% de ácido acético em água (v/v) e 0,02% de ácido acético em acetonitri- la (v/v) (10% a 95%) para dar #B166 como um sólido branco. Rendimento: 114 mg, 0,12 mmol, 71%. LCMS (Protocolo C): m/z 927,5 [M+H]+, tempo de retenção = 1,89 minuto.
[01031] Etapa 2. Síntese de N-(6-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}hexanoil)-L-valil-N~5~-carbamoil-N-(4- {[(piperazin-1-ilcarbonil)oxi]metil}fenil)-L-ornitinamida (#B167). Em dois recipientes separados, a uma suspensão de #B166 (106 mg total, 0,11 mmol, 1 eq.) em acetonitrila (6 mL) à temperatura ambiente, foi adicionado TFA (800 μL), e as reações foram agitadas por 1,5-2 horas. As reações foram concentradas in vacuo, rediluídas com acetonitrila, e concentradas (3x) in vacuo. O material desejado bruto foi purificado por cromatografia líquida de pressão média de fase reversa eluída com 0,02% de ácido acético em água (v/v) e 0,02% de ácido acético em acetonitrila (v/v) (10% a 100%) para dar #B167 como um sólido branco. Rendimento: 62 mg, 0,066 mmol, 57%. LCMS (Protocolo D): m/z 827,4 [M+H]+, tempo de retenção = 0,72 minuto.
[01032] Etapa 3. Síntese de N-(6-{[(9H-fluoren-9- ilmetoxi)carbonil]amino}hexanoil)-L-valil-N-{4-[({[4-({[(2S,3Z)-5- {[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3S,5S,7S)-7-(2-amino-2-oxoetil)-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il]oxi}carbonil)piperazin-1- il]carbonil}oxi)metil]fenil}-N~5~-carbamoil-L-ornitinamida (#B168). A uma solução de #B39 (13,5 mg, 0,028 mmol, 1 eq.) em diclorometano (0,4 mL) à temperatura ambiente, foram adicionados 4-N,N- dimetilamino piridina (3,4 mg, 0,028 mmol, 1 eq.), N,N-diisopropiletilamina (24,7 μL, 0,14 mmol, 5 eq.) e bis(4- nitrofenil)carbonato (10,6 mg, 0,034 mmol, 1,2 eq.), e a reação foi deixada agitar por 6 horas. Uma solução de #B167 (34,5 mg, 0,037 mmol, 1,3 eq.) e N,N-diisopropiletilamina (12 uL, 0,07 mmol, 2,5 eq.) em N,N- dimetilformamida (500 uL) foi adicionada, e a reação foi deixada agitar por 1 hora. A reação foi diluída com DMSO (500 ul), e o diclorometano foi removido in vacuo. O material desejado bruto foi purificado por cromatografia de fase reversa (Método A) para dar #B168 como um sólido branco. Rendimento: 13 mg, 0,01 mmol, 35%. LCMS (Protocolo C): m/z 1329,6 [M+H]+, tempo de retenção = 1,81 minuto.
[01033] Etapa 4. Síntese de N-(6-aminohexanoil)-L-valil-N-{4-[({[4- ({[(2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3S,5S,7S)-7-(2-amino-2- oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5- dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2- il]oxi}carbonil)piperazin-1-il]carbonil}oxi)metil]fenil}-N~5~-carbamoil-L- ornitinamida, sal de acetato (#B169). O composto do título foi prepara- do em 80% de rendimento a partir de 13 mg (0,01 mmol, 1,0 eq.) de #B168 e 17,0 mg (0,2 mmol, 20,0 eq) de piperidina usando o procedimento descrito para a preparação do composto #B47. LCMS (Protocolo D): m/z 1107,5 [M+H]+, tempo de retenção = 0,69 minuto.
[01034] Etapa 5. Síntese de N-{6-[(bromoacetil)amino]hexanoil}-L- valil-N-{4-[({[4-({[(2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3S,5S,7S)- 7-(2-amino-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien- 1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2- il]oxi}carbonil)piperazin-1-il]carbonil}oxi)metil]fenil}-N~5~-carbamoil-L- ornitinamida (#B165). O composto do título foi preparado em 69% de rendimento a partir de 9,4 mg (0,008 mmol, 1,0 eq) de #B169 e 2,8 mg (0,012 mmol, 1,5 eq) de 1-[(bromoacetil)oxi]pirrolidina-2,5-diona e 4,2 mg (0,032 mmol, 4,0 eq) de N,N-diisopropiletilamina usando o procedimento descrito para a preparação do composto #B150. HPLC (Protocolo AA) tempo de retenção = 7,741 minutos (pureza 91%). LCMS (Protocolo C): m/z 1229,4 [M+H]+, tempo de retenção = 1,48 minuto. 1H RMN (400 MHz, DMSO-d6) δ 9,99 (s, 1 H), 8,27-8,19 (m 1 H), 8,138,04 (m, 1 H), 7,85-7,73 (m, 2 H), 7,64-7,55 (m, 2 H), 7,35-7,26 (m, 3 H), 6,77 (s, 1 H), 6,32-6,21 (m, 2 H), 6,10 (d, J = 12,1 Hz, 1 H), 6,015,85 (m, 2 H), 5,59 (dd, J = 16,0 e 5,5 Hz, 1 H), 5,55-5,47 (m, 1 H), 5,40 (s, 2 H), 5,02 (s, 2 H), 4,58-4,49 (m, 1 H), 4,45-4,25 (m, 2 H), 4,24-4,14 (m, 2 H), 3,81 (s, 2 H), 3,69-3,60 (m, 2 H), 3,53-3,45 (m, 2 H), 3,43-3,33 (m, 6 H), 3,10-2,89 (m, 4 H), 2,64-2,53 (m, 2 H), 2,382,09 (m, 5 H), 2,03-1,92 (m, 1 H), 1,87-1,56 (m, 11 H), 1,55-1,31 (m, 7 H), 1,30-1,19 (m, 5 H), 1,07 (d, J = 6,2 Hz, 3 H), 0,95 (d, J = 7,0 Hz, 3 H), 0,84 (dd, J = 10,9 e 6,6 Hz, 6 H).
[01035] Exemplo A58
[01036] Preparação de N-{6-[(bromoacetil)amino]hexanoil}-L-valil-N- [4-({[(4-{[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5- il]acetil}piperazin-1-il)carbonil]oxi}metil)fenil]-N~5~-carbamoil-L- ornitinamida (#B170).
[01037] Etapa 1. Síntese de N-(6-{[(9H-fluoren-9- ilmetoxi)carbonil]amino}hexanoil)-L-valil-N-[4-({[(4-{[(3R,5S,7R,8R)-7- {(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi- 1,6-dioxaspiro[2,5]oct-5-il]acetil}piperazin-1-il)carbonil]oxi}metil)fenil]- N~5~-carbamoil-L-ornitinamida (#B171).: A uma solução de #B1 (15 mg, 0,024 mmol, 1 eq.) em N,N-dimetilformamida (0,1 mL) à temperatura ambiente, foi adicionada uma solução de #B167 (28,2 mg, 0,03 mol, 1,25 eq.) e N,N-diisopropiletilamina (16,8 μL, 0,096 mmol, 4 eq.) em N,N-dimetilformamida (0,6 mL), e a reação foi agitada por 1,5 hora. A reação foi diluída com dimetil sulfóxido e purificada por cromatogra- fia de fase reversa (Método A) para dar #B171 como um sólido branco. Rendimento: 17,8 mg, 0,013 mmol, 55%. LCMS (Protocolo D): m/z 1345,8 [M+H]+, tempo de retenção = 0,92 minuto.
[01038] Etapa 2. Síntese de N-(6-aminohexanoil)-L-valil-N-[4-({[(4- {[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5- il]acetil}piperazin-1-il)carbonil]oxi}metil)fenil]-N~5~-carbamoil-L- ornitinamida (#B172).: O composto do título foi preparado em 79% de rendimento a partir de 17,8 mg (0,013 mmol) de #B171 e 22,1 mg (0,26 mmol, 20,0 eq) de piperidina usando o procedimento descrito para a preparação do composto #B47. LCMS (Protocolo C): m/z 1122,6 [M+H]+, tempo de retenção = 1,23 minuto.
[01039] Etapa 3. Síntese de N-{6-[(bromoacetil)amino]hexanoil}-L- valil-N-[4-({[(4-{[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5- {[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5- il]acetil}piperazin-1-il)carbonil]oxi}metil)fenil]-N~5~-carbamoil-L- ornitinamida (#B170). O composto do título foi preparado em 57% de rendimento a partir de 12,1 mg (0,01 mmol) de #B172 e 3,5 mg (0,015 mmol, 1,5 eq) de 1-[(bromoacetil)oxi]pirrolidina-2,5-diona e 5,2 mg (0,04 mmol, 4,0 eq) de N,N-diisopropiletilamina usando o procedimento descrito para a preparação do composto #B150. HPLC (Protocolo AA) tempo de retenção = 7,925 minutos (pureza 81%). LCMS (Protocolo C): m/z 1244,4 [M+H]+, tempo de retenção = 1,45 minuto. 1H RMN (400 MHz, DMSO-d6) δ 9,99 (s, 1 H), 8,27-8,19 (m 1 H), 8,13-8,04 (m, 1 H), 7,85-7,73 (m, 2 H), 7,64-7,55 (m, 2 H), 7,35-7,26 (m, 2 H), 6,446,24 (m, 2 H), 6,10 (d, J = 11,7 Hz, 1 H), 6,03-5,93 (m, 1 H), 5,87 (dd, J = 11,7 e 7,4 Hz, 1 H), 5,61 (dd, J = 15,6 e 5,5 Hz, 1 H), 5,55-5,47 (m, 1 H), 5,40 (s, 2 H), 5,02 (s, 2 H), 4,98 (d, J = 5,9 Hz, 1 H), 4,43-4,33 (m, 2 H), 4,30-4,16 (m, 3 H), 3,81 (s, 2 H), 3,69-3,60 (m, 2 H), 3,55-3,34 (m, 6 H), 3,26-3,22 (m, 1 H), 3,09-2,89 (m, 4 H), 2,75 (d, J = 5,1 Hz, 1 H), 2,63-2,55 (m, 2 H), 2,31-2,09 (m, 4 H), 1,98 (s, 3 H), 1,92-1,78 (m, 3 H), 1,73-1,31 (m, 12 H), 1,30-1,19 (m, 5 H), 1,07 (d, J = 6,6 Hz, 3 H), 0,95 (d, J = 7,4 Hz, 3 H), 0,84 (dd, J = 10,9 e 6,6 Hz, 6 H).
[01040] Exemplo A59
[01041] Preparação de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5- {(3R,5S,7S)-7-[(butanoilamino)metil]-1,6-dioxaspiro[2,5]oct-5-il}-3- metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro-2H-piran-3-il}amino)-5- oxopent-3-en-2-il acetato (#B173).
[01042] Etapa 1. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3R,5S,7S)-7-[(butanoilamino)metil]-1,6-dioxaspiro[2,5]oct-5-il}-3- metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro-2H-piran-3-il}amino)-5- oxopent-3-en-2-il acetato (#B173). A uma solução de #B140 (6,7 mg, 0,014 mmol, 1 eq.) em diclorometano (0,4 mL) a 0°C, foram adicionados 4-N,N-dimetilamino piridina (0,4 mg, 0,003 mmol, 0,2 eq.), N,N- diisopropiletilamina (12,3 μL, 0,07 mmol, 5 eq.) e ácido butírico (6,8 μL, 0,074 mmol, 5,3 eq.), e a reação foi deixada agitar por 2 horas à temperatura ambiente. À reação, foi adicionado DCC (8 mg, 0,042 mmol, 3 eq.), e a reação foi agitada por 45 min. A reação foi diluída com etil acetato e bicarbonato de sódio saturado, extraída, lavada com salmoura, seca sobre sulfato de sódio, filtrada, e concentrada in vacuo. O material desejado bruto foi purificado por cromatografia de fase reversa (Método A) para dar #B173 como um sólido branco. Rendimento: 3,4 mg, 0,006 mmol, 43%. HPLC (Protocolo AA): tempo de retenção = 8,927 minutos (pureza 87%). LCMS (Protocolo D): m/z 583,2 [M+Na]+, tempo de retenção = 0,88 minuto. 1H RMN (400 MHz, DMSO-d6) δ 7,84-7,75 (m, 2 H), 6,42-6,32 (m, 1 H), 6,23 (d, J = 16,0 Hz, 1 H), 6,11 (d, J = 11,5 Hz, 1 H), 5,87 (dd, J = 11,5 e 7,4 Hz, 1 H), 5,65-5,45 (m, 3 H), 4,60-4,51 (m, 1 H), 3,96-3,85 (m, 1 H), 3,70-3,60 (m, 2 H), 3,543,46 (m, 1 H), 3,12-3,01 (m, 1 H), 2,63 (s, 2 H), 2,32-2,12 (m, 2 H), 2,09-2,01 (m, 2 H), 1,98 (s, 3 H), 1,75-1,56 (m, 7 H), 1,55-1,45 (m, 2 H), 1,41-1,33 (m, 1 H), 1,25 (d, J = 6,6 Hz, 3 H), 1,07 (d, J = 6,6 Hz, 3 H), 0,95 (d, J = 7,4 Hz, 3 H), 0,88-0,80 (m, 3 H).
[01043] Exemplo A60
[01044] Preparação de ácido [(3R,7S)-7-{(1E,3E)-5-[(2S,3S,5R,6R)- 5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-5-hidroxi-1,6-dioxaspiro[2,5]oct-5- il] acético (#B174) por biocatálise com Fr9P recombinante
[01045] Etapa 1. Produção de enzima Fr9P recombinante em e purificação a partir de Escherichia coli. O gene Fr9P otimizado por códon (como descrito na etapa 1 do exemplo 4) foi sintetizado e ligado nos sítios NcoI-HindIII de pGS-21a (GenScript) para gerar pAE-PF16. Proteína Fr9P rotulada por His6-GST recombinante foi produzida em e purificada a partir de E. coli BL21(DE3) após transformação com plasmí- deo pAE-PF16. Dois frascos Fernbach de 2,8 L contendo 0,5 L de meio (Caldo Terrific com 100 mg/L ampicilina) foram, cada um, inoculados com 20 mL de uma cultura LB de uma noite toda e incubados a 200 rpm, 25°C. Quando o OD600 alcançou ~0.9, as células foram induzidas com 0,2 mM IPTG e a incubação foi resumida a 25°C e 200 rpm. Após 18-20 h, células foram coletadas por centrifugação e congeladas a -80°C. Um pélete de célula foi ressuspenso em ~50 mL de tampão de lise gelado [10 mM tampão de fosfato pH 7,4; 500 mM NaCl; 20 mM imidazol; 10% glicerol; lisozima 1 mg/mL; 0,5% (v/v) Tween 20; 20 mM β-mercaptoetanol] e incubadas em gelo por 30 min. Após sonicação em gelo, o lisado de célula foi centrifugado a 14,000 rpm e 4°C por 45 min. O sobrenadante foi transferido para um novo tubo e centrifugado novamente a 14,000 rpm e 4°C por 30 min. 5 mL de pasta fluida de resina Ni-NTA (Qiagen) foram adicionados à fração de sobrenadante (lisado claro) contida em uma pequena proveta em gelo e delicadamente agitada por 1 hora. A suspensão foi transferida para um tubo Falcon e centrifugada a 3,000 rpm e 4°C por 10 min. O sobrenadante foi descartado e a resina lavada três vezes, cada um com 30 mL tam pão de lavagem gelado [10 mM tampão de fosfato pH 7,4; 500 mM NaCI; 40 mM imidazol; 10% glicerol; 20 mM β-ME] seguido por centrifugação a 3,000 rpm e 4°C por 10 min. A resina foi transferida para uma coluna descartável e lavada mais três vezes, cada uma com 2,5 mL de tampão de lavagem. A enzima foi eluída com 3x 2,5 mL de tampão de eluição [10 mM de tampão de fosfato pH 7,4; 500 mM NaCl; 250 mM imidazol; 10% glicerol; 20 mM β-ME]. O tampão foi trocado para 50 mM MOPS pH 7,5 usando uma coluna PD-10 e a solução concentrada usando uma coluna Vivaspin com redução de peso molecular de 30 kDa. O tampão de armazenamento continha 50 mM MOPS pH 7,5, 2 mM DTT e 10% glicerol (para armazenamento a - 80°C) ou 50% glicerol (para armazenamento a -20°C). O rendimento da enzima purificada foi ~25 mg por litro de cultura.
[01046] Etapa 2. Síntese de ácido [(3R,7S)-7-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-5-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]ácido (#B174) usando Fr9P recombinante. A uma solução aquosa de #NP2 (1 mg, 0,4 mM, 1 eq.) em 50 mM tampão MOPS pH 7,5, foram adicionados α-cetoglutarato (0,8 mM concentração final, 2 eq.), ascorbato de sódio (0,08 mM, 0,2 eq.), NH4Fe(II)SO4 (0,04 mM, 0,1 eq.) e Fr9P recombinante da etapa 1 do exemplo #A60 (1,2 μM, 0,003 eq.). Após incubação à temperatura ambiente por 2 horas, a reação foi acidificada para pH ~4-5 com ácido acético e extraída três vezes com volume igual de etilacetato. O solvente foi evaporado sob pressão reduzida, o resíduo ressuspenso em 0,25 mL acetonitrila, filtrado e purificado por cromatografia de fase reversa (Método H). A fração com tempo de retenção de 18,5 min foi coletada e neutralizada com hidróxido de amônio antes de ser concentrada sob pressão reduzida. O concentrado aquoso foi acidificado para pH ~4 com ácido acético e extraído duas vezes com volume igual de etilacetato. O solvente foi removido sob pressão reduzida para gerar #B174 como um sólido branco. Rendimento: 0,2 mg. HPLC (Protocolo P): tempo de retenção = 10,39 minutos. HRESIMS m/z 536,286 [M+H]+; 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 12,24 (brs, OH), 8,00 (d, J = 8,0 Hz, 1H), 6,37 (m, 1H), 6,23 (d, J = 15,9, 1H), 6,12 (dd, J = 0,7, 11,5, 1H), 5,88 (dd, J = 11,6, 7,5 Hz, 1H), 5,54 (m, 1H),5,50 (m, 1H), 4,67 (m, 1H), 3,66 (m, 2H), 3,51 (m, 1H), 2,60 (m, 1H),2,53 (m, 1H), 2,33 (m, 1H), 2,31 (m, 1H), 2,20 (m, 1H), 2,00 (s, 3H),1,84 (m, 1H), 1,81 (m, 2H), 1,70 (s, 3H), 1,67 (m, 1H), 1,39 (m, 1H),1,26 (d, J = 6,6 Hz, 3H), 1,16 (m, 1H), 1,08 (d, J = 6,4 Hz, 3H), 0,96 (d, J = 7,2 Hz, 3H), 13C RMN (100 MHz, DMSO-d6) δ 171,7, 170,5, 165,0, 142,7, 134,4, 133,3, 128,5, 127,3, 122,6, 95,3, 79,9, 74,8, 67,9, 67,2, 46,7, 46,1, 38,9, 37,7, 35,0, 31,5, 28,7, 20,8, 19,7, 17,6, 14,2, 12,2.
[01047] Exemplo A61
[01048] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5- [(3S,5S,7S)-7-(2-amino-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5- oxopent-3-en-2-il metil[2-(piridin-2-ildissulfanil)etil]carbamato (#B175).
[01049] Etapa 1. Síntese de N-metil-2-(piridin-2-ildissulfanil)etanamina, sal de trifluoroacetato (#B176). A uma solução de terc-butil metil[2-(piridin-2-ildissulfanil)etil]carbamato (Angew. Chem. Int. Ed. 2007, 46, 6469) (90 mg, 0,3 mmol, 1 eq.) em diclorome- tano (1 mL) à temperatura ambiente, foi adicionado TFA (1 mL), e a reação foi agitada por 1 hora. A reação foi concentrada in vacuo e azeotropada com acetonitrila (3X) para dar #B176 como um óleo. LCMS (Protocolo D): m/z 201,1 [M+H]+, tempo de retenção = 0,43 min. O material bruto foi usado na etapa seguinte sem purificação adicional.
[01050] Etapa 2. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3S,5S,7S)-7-(2-amino-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5- oxopent-3-en-2-ilmetil[2-(piridin-2-ildissulfanil)etil]carbamato (#B175): A uma solução do #B39 (19,5 mg, 0,041 mmol, 1 eq.) em diclorometano (0,5 mL) à temperatura ambiente, foi adicionado 4-N,N-dimetilamino piridina (5 mg, 0,041 mmol, 1 eq.), N,N-diisopropiletilamina (21,7 μL, 0,123 mmol, 3 eq.) e bis(4-nitrofenil)carbonato (18,9 mg, 0,62 mmol, 1,5 eq.), e a reação foi deixada agitar por 2,5 horas. Adicional de bis(4- nitrofenil)carbonato (3,1 mg, 0,008 mmol, 0,2 eq.) foi adicionado, e a reação agitada por mais 1,5 hora. Uma solução de #B176 (44,1 mg, 0,103 mmol, 2,5 eq.) e N,N-diisopropiletilamina (54 μL, 0,31 mmol, 7,5 eq.) em diclorometano (0,4 mL) foi adicionada, e a reação foi agitada por 1 hora. Adicional de solução de #B176 (17 mg, 0,04 mmol, 1 eq.) e N,N-diisopropiletilamina (44 μL, 0,25 mmol, 6 eq.) em diclorometano (0,2 mL) foi adicionado, e a reação foi agitada por outros 15 minutos. A mistura de reação foi diluída com DMSO (1 mL), e o diclorometano removido in vacuo. O material desejado bruto foi purificado por croma- tografia líquida de pressão média de fase reversa eluída com 0,02% de ácido acético em água (v/v) e 0,02% de ácido acético em acetonitri- la (v/v) (10% a 100%) para gerar #B175 como um sólido branco. Rendimento: 7,6 mg, 0,011 mmol, 26%. LCMS (Protocolo D): m/z 703,6 [M+H]+, tempo de retenção = 0,91 minutos, 1H RMN (400 MHz, DMSO- de, mult, J em Hz) □□8,49-8,43 (m, 1 H), 7,87-7373 (m, 2 H), 7,32 (s, 1 H), 7,28-7,21 (m, 1 H), 6,78 (s, 1 H), 6,32 (d, J = 16,0 Hz, 1 H), 6,26 (d, J = 16,4 Hz, 1 H), 6,23-6,13 (m, 1 H), 6,11-5,98 (m, 1 H), 5,93-5,84 (m, 1 H), 5,76-5,67 (m, 1 H), 5,59 (dd, J = 15,9 e 5,6 Hz, 1 H)
[01051] Exemplo A# 62
[01052] Preparação de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5-{(3R,4R,5R,7S)-7-[2-(2,2-dimetilhidrazinil)-2-oxoetil]-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B177).
[01053] Etapa 1. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3R,4R,5R,7S)-7-[2-(2,2-dimetilhidrazinil)-2-oxoetil]-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B177). A uma solução de #B1 (26,8 mg, 0,042 mmol, 1 eq.) dissolvida em tetrahidrofura- no (1 mL) à temperatura ambiente, foi adicionado N,N-dimetil hidrazina (16 μ, 0,21 mmol, 5 eq.) após agitação por 40 min, mais N,N-dimetil hidrazina (6,4 μL, 0,084 mmol, 2 eq.) foi adicionado, e a reação foi agitada por 5 min. A reação foi diluída com água, extraída com etil acetato, e as fases orgânicas combinadas foram secas sobre sulfato de sódio e filtradas. Os solventes foram removidos in vacuo. O material desejado bruto foi purificado por cromatografia de fase reversa (Método A) para dar #B177 como um sólido branco. Rendimento: 14,5 mg, 0,025 mmol, 60%. LCMS (Protocolo D): m/z 578,5 [M+H]+, tempo de retenção = 0,72 minutos. 1H RMN (500 MHz, DMSO-d6) δ 8,85 (s, 0,6 H), 8,33 (s, 0,4 H), 7,93-7,84 (m, 1 H), 6,40-6,27 (m, 2 H), 6,10 (d, J = 11,6 Hz, 1 H), 5,87 (dd, J = 11,6 e 7,6 Hz, 1 H), 5,63-5,54 (m, 1 H), 5,53-5,46 (m, 1 H), 5,09 (d, J = 5,1 Hz, 0,6 H), 5,01 (d, J = 6,6 Hz, 0,4 H), 4,35-4,16 (m, 2 H), 3,68-3,60 (m, 2 H), 3,53-3,46 (m, 1 H), 3,273,21 (m, 1 H), 2,99-2,90 (m, 0,6 H), 2,76 (d, J = 5,4 Hz, 1 H), 2,60-2,53 (m, 1 H), 2,47-2,38 (m, 6,4 H), 2,34-2,26 (m, 1 H), 2,25-2,15 (m, 1 H), 2,06 (dd, J = 14,2 e 4,9 Hz, 1 H), 1,98 (s, 3 H), 1,87-1,72 (m, 3 H), 1,71-1,58 (m, 4 H), 1,51-1,43 (m, 1 H), 1,24 (d, J = 6,4 Hz, 3 H), 1,07 (d, J = 6,4 Hz, 3 H), 0,94 (d, J = 7,3 Hz, 3 H).
[01054] Exemplo A#63
[01055] Preparação de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5- oxopent-3-en-2-il acetato (#B178).
[01056] Etapa 1. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-({trans-3-[(1H-imidazol-1- ilcarbonil)amino]ciclobutil}amino)-2-oxoetil]-1,6-dioxaspiro[2,5]oct-5-il}- 3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro-2H-piran-3-il}amino)-5- oxopent-3-en-2-il acetato (#B178). A uma solução de #B73 (15,3 mg, 0,025 mmol, 1 eq.) em diclorometano (0,5 mL) à temperatura ambiente, foi adicionado N,N-diisopropiletilamina (8,8 μL, 0,05 mmol, 2 eq.) e carbonildimidazol (4,9 mg, 0,03 mmol, 1,2 eq.), e a reação foi agitada por 25 min. A reação foi diluída com diclorometano, lavada com água, seca sobre sulfato de sódio, filtrada, diluída com DMSO (0,8 mL) e concentrada para remover o diclorometano. O material desejado bruto foi purificado por cromatografia de fase reversa (Método A) para dar #B178 como um sólido branco. Rendimento: 3,2 mg, 0,0045 mmol, 18%. LCMS (Protocolo D): m/z 698,6 [M+H]+, tempo de retenção = 0,68 minutos. 1H RMN (500 MHz, DMSO-d6) δ 8,67 (d, J = 6,9 Hz, 1 H), 8,37 (d, J = 7,1 Hz, 1 H), 8,24 (s, 1 H), 7,78 (d, J = 8,1 Hz, 1 H), 7,707,67 (m, 1 H), 7,02 (s, 1 H), 6,40-6,27 (m, 2 H), 6,10 (dd, 11,6 e 1,5 Hz, 1 H), 5,87 (dd, J = 11,6 e 7,3 Hz, 1 H), 5,60 (dd, J = 15,7 e 5,6 Hz, 1 H), 5,52-5,45 (m, 1 H), 5,01 (d, J = 5,4 Hz, 1 H), 4,43-4,34 (m, 1 H), 4,33-4,21 (m, 2 H), 3,67-3,54 (m, 2 H), 3,48-3,43 (m, 1 H), 3,30-3,26 (m, 1 H), 2,78 (d, J = 5,1 Hz, 1 H), 2,60-2,54 (m, 2 H), 2,39-2,13 (m, 7 H), 1,98 (s, 3 H), 1,83-1,74 (m, 3 H), 1,69 (s, 3 H), 1,65-1,53 (m, 3 H), 1,25 (d, J = 6,4 Hz, 3 H), 1,04 (d, J = 6,4 Hz, 3 H), 0,93 (d, J = 7,3 Hz, 3 H).
[01057] Exemplo A#64
[01058] Preparação de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-oxo-2-(tetrahidropiridazin-1(2H)-il)etil]-1,6-dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5- dimetiltetrahidro-2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetate (#B179).
[01059] Etapa 1. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3R,4R,5R,7S)-4-hidroxi-7-[2-oxo-2-(tetrahidropiridazin-1(2H)-il)etil]- 1,6-dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro-2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetate (#B179). A uma solução de #B1 (18,1 mg, 0,029 mmol, 1 eq.) em N,N- dimetilformamida (0,6 mL) à temperatura ambiente, foi adicionada N,N- diisopropiletilamina (51,1 μL, 0,29 mmol, 10 eq.) e Dicloridrato de he- xahidropiridazina (18,5 mg, 0,12 mmol, 4 eq.), e a reação foi agitada por 30 min. A reação foi purificada por cromatografia de fase reversa (Método A) para dar #B179 como um sólido branco. Rendimento: 10,7 mg, 0,018 mmol, 61%. LCMS (Protocolo D): m/z 604,6 [M+H]+, tempo de retenção = 0,80 minuto. 1H RMN (400 MHz, DMSO-d6) δ 7,79 (d, J = 8,2 Hz, 1 H), 6,42-6,27 (m, 2 H), 6,11 (d, J = 11,7 Hz, 1 H), 5,87 (dd, J = 11,7 e 7,4 Hz, 1 H), 5,61 (dd, J = 16,0 e 5,9 Hz, 1 H), 5,55-5,47 (m, 1 H), 4,92 (d, J = 6,2 Hz, 1 H), 4,76 (app t, J = 7,0 Hz, 1 H), 4,34-4,18 (m, 2 H), 3,70-3,60 (m, 2 H), 3,54-3,40 (m, 3 H), 3,27-3,21 (m, 1 H),3,03 (dd, J = 15,2 e 7,4 Hz, 1 H), 2,81-2,70 (m, 3 H), 2,55 (d, J = 5,5Hz, 1 H), 2,33-2,15 (m, 2 H), 1,98 (s, 3 H), 1,85-1,74 (m, 3 H), 1,71-1,56 (m, 5 H), 1,55-1,48 (m, 4 H), 1,25 (d, J = 6,2 Hz, 3 H), 1,07 (d, J =6,2 Hz, 3 H), 0,95 (d, J = 7,4 Hz, 3 H).
[01060] Exemplo A#65
[01061] Preparação de N-{6-[(bromoacetil)amino]hexanoil}-L-valil-N-{4-[({[({[(3S,5S,7S)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-1,6-dioxaspiro[2,5]oct-5-il]acetil}amino)metil]carbamoil}oxi)metil]fenil}-N-5-carbamoil-L- ornitinamida (#B180).
[01062] Etapa 1. Síntese de N-(6-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}hexanoil)-L-valil-N5-carbamoil-N-[4-(9,9-dimetil- 3,7-dioxo-2,8-dioxa-4,6-diazadec-1-il)fenil]-L-ornitinamida (#B181). Uma solução de cloridrato de terc-butil aminometilcarbamato (J.Org.Chem., 1980, 45, 1703, 32,9 mg, 0,18 mmol, 1 eq.) e N,N- diisopropiletilamina (47 μL, 0,27 mmol, 3 eq.) em N,N-dimetilformamida (1 mL) foi adicionado em gotas a uma solução de #B45 (161,5 mg, 0,18 mmol, 1 eq.) em N,N-dimetilformamida (2 mL) a 0°C. 4-N,N- dimetilamino piridina (2 mg, 0,016 mmol, 0,1 eq.) foi adicionada, e a solução resultante foi agitada à temperatura ambiente por uma hora. A mistura de reação foi diluída com terc-butil metil éter e filtrada. O bolo de filtro foi purificado por HPLC preparativa para gerar #B181 como um sólido branco. Rendimento: 20 mg, 0,00023 mmol, 13%. 1H RMN (400 MHz, MeOD-d4) δ 7,82 (d, 2 H), 7,67 (d, 2H), 7,58 (d, 2 H), 7,42(m, 6 H), 5,04 (br, 3 H), 4,63 (s, 4 H), 4,52 (m, 5 H), 4,36 (m, 2 H), 3,20(m, 4 H), 2,32 (m, 2 H), 2,10 (m, 1H), 1,90 (m, 1 H), 1,77 (m, 1 H), 1,65(m, 4 H), 1,44 (m, 7 H), 1,35 (m, 3 H),0,98 (m, 6 H).
[01063] Etapa 2. Síntese de N-(6-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}hexanoil)-L-valil-N-[4-({[(aminometil)carbamoil]oxi}metil)fenil]-N5-carbamoil-L-ornitinamida sal de trifluoroacetato (#B182). A #B181 (20 mg, 0,00023 mmol), foi adicionado ácido trifluoroacético pré-refrigerado (1,3 mL) a 0°C, e a reação foi deixada agitar por 10 min. A reação foi concentrada, retomada em acetonitrila e reconcentrada três vezes para dar #B182 como uma goma que foi usada na etapa seguinte sem purificação adicional: Rendimento: 25 mg, 0,028 mmol, 100%. LCMS (Protocolo D): m/z 787,6 [M+H]+, tempo de retenção = 0,75 minuto.
[01064] Etapa 3. Síntese de N-(6-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}hexanoil)-L-valil-N-{4-[({[({[(3S,5S,7S)-7- {(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6- dioxaspiro[2,5]oct-5-il]acetil}amino)metil]carbamoil}oxi)metil]fenil}-N5- carbamoil-L-ornitinamida (#B183). A uma solução de #NP2 (14,5 mg, 0,028 mmol, 1 eq.) em N,N-dimetilformamida (0,4 mL) à temperatura ambiente, foram adicionados N,N-diisopropiletilamina (19,7 μL, 0,11 mmol, 4 eq.) e hexafluorofosfato de O-(7-azabenzotriazol-1-il)- N^N^N-tetrametilurônio (12 mg, 0,031 mmol, 1,1 eq.). #B182 (25,2 mg, 0,028 mmol, 1 eq.) em DMF (0,6 mL) foi adicionado, e a reação foi deixada agitar por 45 min. A reação foi purificada por cromatografia de fase reversa (Método A) para dar #B183 como um sólido branco. Rendimento: 9,8 mg, 0,0076 mmol, 27%. LCMS (Protocolo D): m/z 1288,94 [M+H]+, tempo de retenção = 0,94 minuto.
[01065] Etapa 4. Síntese de N-(6-aminohexanoil)-L-valil-N-{4- [({[({[(3S,5S,7S)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-1,6-dioxaspiro[2,5]oct-5- il]acetil}amino)metil]carbamoil}oxi)metil]fenil}-N5-carbamoil-L- ornitinamida sal de acetato (#B184). O composto do título foi preparado em 75% de rendimento a partir de 11,9 mg (0,009 mmol, 1,0 eq) de #B183 e 15,3 mg (0,18 mmol, 20,0 eq) de piperidina usando o procedimento descrito para a preparação do composto #B47. LCMS (Protocolo D): m/z 1066,8 [M+H]+, tempo de retenção = 0,73 minuto.i. Etapa 5. Síntese de N-{6-[(bromoacetil)amino]hexanoil}-L-valil-N-{4-[({[({[(3S,5S,7S)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-1,6-dioxaspiro[2,5]oct-5- il]acetil}amino)metil]carbamoil}oxi)metil]fenil}-N-5-carbamoil-L- ornitinamida (#B180). O composto do título foi preparado em 61% de rendimento a partir de 7,6 mg (0,01 mmol) de #B184 e 2,6 mg (0,011 mmol, 1,5 eq.) de 1-[(bromoacetil)oxi]pirrolidina-2,5-diona e 3,7 mg (0,028 mmol, 4,0 eq) de N,N-diisopropiletilamina usando o procedi- mento descrito para a preparação do composto #B150. LCMS (Protocolo D): m/z 1188,8 [M+H]+, tempo de retenção = 0,81 minuto. 1H RMN (500 MHz, DMSO-d6) δ 9,98 (s, 1 H), 8,45-8,37 (m, 1 H), 8,27-8,20 (m, 1 H), 8,14-8,05 (m, 1 H), 7,85-7,73 (m, 2 H), 7,61-7,55 (m, 2 H), 7,307,24 (m, 2 H), 6,40-6,32 (m, 1 H), 6,25 (d, J = 16,1 Hz, 1 H), 6,10 (dd, J = 11,5 e 1,2 Hz, 1 H), 6,01-5,93 (m, 1 H), 5,87 (dd, J = 11,7 e 7,6 Hz, 1 H), 5,60 (dd, J = 15,6 e 5,4 Hz, 1 H), 5,56-5,49 (m, 1 H), 5,41 (s, 2 H), 5,02 (s, 2 H), 4,56-4,50 (m, 1 H), 4,42-4,23 (m, 3 H), 4,22-4,16 (m, 1 H), 3,81 (s, 2 H), 3,69-3,60 (m, 3 H), 3,52-3,45 (m, 2 H), 3,09-2,89 (m, 5 H), 2,66-2,53 (m, 3 H), 2,34-2,09 (m, 6 H), 2,01-1,92 (m, 4 H), 1,851,55 (m, 10 H), 1,54-1,31 (m, 7 H), 1,27-1,20 (m, 4 H), 1,06 (d, J = 6,4 Hz, 3 H), 0,94 (d, J = 7,3 Hz, 3 H), 0,86 (d, J = 6,9 Hz, 3 H), 0,83 (d, J = 6,9 Hz, 3 H).
[01066] Exemplo A#66
[01067] Preparação de N-{6-[(bromoacetil)amino]hexanoil}-L-valil-N-[4-({[(2-{[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5-il]acetil}-2- metilhidrazinil)carbonil]oxi}metil)fenil]-N5-carbamoil-L-ornitinamida (#B185).
[01068] Etapa 1. Síntese de N-(6-{[(9H-fluoren-9- ilmetoxi)carbonil]amino}hexanoil)-L-valil-N-{4-[({[2-(terc-butoxicarbonil)- 2-metilhidrazinil]carbonil}oxi)metil]fenil}-N5-carbamoil-L-ornitinamida (#B186). A uma solução terc-butil 1-metilhidrazinacarboxilato (34,8 mg, 0,24 mmol, 1,3 eq.) em N,N-dimetilformamida (1 mL) à temperatura ambiente, foi adicionada N,N-diisopropiletilamina (64,4 μL, 0,37 mmol, 2 eq.) e 4-N,N-dimetilamino piridina (11,1 mg, 0,091 mmol, 0,5 eq.) se- guido por #B45 (161 mg, 0,18 mmol, 1 eq.), e a reação foi deixada agitar. Após 4 h, mais terc-butil 1-metilhidrazinacarboxilato (14 mg, 0,096 mmol, 0,5 eq.) em N,N-dimetilformamida (0,2 mL) foi adicionado, e a reação foi agitada por 1,5 h. A reação foi diluída com DMSO (1 mL) e purificada por cromatografia líquida de pressão de meio de fase reversa eluída com 0,02% de ácido acético em água (v/v) e 0,02% de ácido acético em acetonitrila (v/v) (10% a 100%) para dar #B186 como um sólido branco. Rendimento: 26,1 mg, 0,029 mmol, 16%. LCMS (Protocolo D): m/z 887,6 [M+H]+, tempo de retenção = 0,92 minuto.
[01069] Etapa 2. Síntese de N-(6-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}hexanoil)-L-valil-N5-carbamoil-N-[4-({[(2- metilhidrazinil)carbonil]oxi}metil)fenil]-L-ornitinamida sal de trifluoroace- tato (#B187). A #B186 (16,5 mg, 0,019 mmol, 1 eq.), foi adicionado ácido trifluoroacético (1 mL) à temperatura ambiente, e a reação foi deixada agitar por 20 min. A reação foi concentrada, retomado em acetonitrila e reconcentrada três vezes para dar #B187 como uma goma que foi usada na etapa seguinte sem purificação adicional. LCMS (Protocolo D): m/z 809,6 [M+Na]+, tempo de retenção = 0,80 minuto.
[01070] Etapa 3. Síntese de N-(6-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}hexanoil)-L-valil-N-[4-({[(2-{[(3R,5S,7R,8R)-7- {(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi- 1,6-dioxaspiro[2,5]oct-5-il]acetil}-2-metilhidrazinil)carbonil]oxi}metil)fenil]-N5-carbamoil-L-ornitinamida (#B188). A uma solução de #NP1 (8,7 mg, 0,016 mmol, 1 eq.) em N,N- dimetilformamida (0,2 mL) à temperatura ambiente, foi adicionada N,N- diisopropiletilamina (11,3 μL, 0,064 mmol, 4 eq.) e hexafluorofosfato de O-(7-azabenzotriazol-1-il)-N,N,N‘,N‘-tetrametilurônio (7,4 mg, 0,019 mmol, 1,2 eq.) seguido por uma solução de #B187 (17,1 mg, 0,019 mmol, 1,2 eq.) e N,N-diisopropiletilamina (5,7 μL, 0,032 mmol, 2 eq.) em N,N-dimetilformamida (0,4 mL), e a reação foi deixada agitar por 30 min. A reação foi purificada por cromatografia de fase reversa (Método A) para dar #B188 como um sólido branco. Rendimento: 8,3 mg, 0,0064 mmol, 40%. LCMS (Protocolo D): m/z 1304,9 [M+H]+, tempo de retenção = 0,93 minuto.
[01071] Etapa 4. Síntese de N-(6-aminohexanoil)-L-valil-N-[4-({[(2- {[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5-il]acetil}-2- metilhidrazinil)carbonil]oxi}metil)fenil]-N5-carbamoil-L-ornitinamida sal de acetato (#B189). O composto do título foi preparado em 80% de rendimento a partir de 8,3 mg (0,006 mmol, 1,0 eq.) de #B188 e 10,2 mg (0,12 mmol, 20,0 eq) de piperidina usando o procedimento descrito para a preparação do composto #B47. LCMS (Protocolo D): m/z 1082,81 [M+H]+, tempo de retenção = 0,66 minuto.
[01072] Etapa 5. Síntese de N-{6-[(bromoacetil)amino]hexanoil}-L- valil-N-[4-({[(2-{[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5- {[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5- il]acetil}-2-metilhidrazinil)carbonil]oxi}metil)fenil]-N5-carbamoil-L-ornitinamida (#B185). O composto do título foi preparado em 63% de rendimento a partir de 5,5 mg (0,005 mmol, 1 eq.) de #B189, 1,7 mg (0,011 mmol, 1,5 eq) de 1-[(bromoacetil)oxi]pirrolidina-2,5-diona e 2,6 mg (0,02 mmol, 4,0 eq) de N,N-diisopropiletilamina usando o procedimento descrito para a preparação do composto #B150. LCMS (Protocolo D): m/z 1204,86 [M+H]+, tempo de retenção = 0,77 minuto. 1H RMN (500 MHz, DMSO-d6) δ 10,01 (s, 1 H), 9,94-9,81 (m, 1 H), 8,288,20 (m, 1 H), 8,13-8,05 (m, 1 H), 7,86-7,76 (m, 2 H), 7,66-7,55 (m, 2 H), 7,37-7,25 (m, 2 H), 6,59-6,46 (m, 1 H), 6,40-6,29 (m, 2 H), 6,10 (dd, J = 11,6 e 1,5 Hz, 1 H), 6,01-5,95 (m, 1 H), 5,87 (dd, J = 11,6 e 7,6 Hz, 1 H), 5,65-5,57 (m, 1 H), 5,56-5,50 (m, 1 H), 5,41 (m, 2 H), 5,12-4,96 (m, 4 H), 4,42-4,33 (m, 1 H), 4,32-4,24 (m, 1 H), 4,23-4,16 (m, 2 H), 3,81 (s, 2 H), 3,69-3,60 (m, 2 H), 3,52-3,47 (m, 1 H), 3,07-2,89 (m, 5 H), 2,77-2,73 (m, 1 H), 2,60-2,54 (m, 1 H), 2,34-2,08 (m, 4 H), 2,011,92 (m, 4 H), 1,86-1,31 (m, 14 H), 1,27-1,20 (m, 4 H), 1,06 (d, J = 6,1 Hz, 3 H), 0,94 (d, J = 7,3 Hz, 3 H), 0,86 (d, J = 6,9 Hz, 3 H), 0,83 (d, J = 6,6 Hz, 3 H).
[01073] Exemplo A#67
[01074] Preparação de N-{7-[(2,5-dioxopirrolidin-1-il)oxi]-7-oxoheptanoil}-L-valil-N-[4-({[(2-{[(3R,5S,7R,8R)-7-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]acetil}hidrazinil)carbonil]oxi}metil)fenil]-N5- carbamoil-L-ornitinamida (#B190).
[01075] Etapa 1. Síntese de N-[(9H-fluoren-9-ilmetoxi)carbonil]-L- valil-N-[4-({[(2-{[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5- {[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5- il]acetil}hidrazinil)carbonil]oxi}metil)fenil]-N5-carbamoil-L-ornitinamida (#B191). A uma solução de #B6 (19,4 mg, 0,035 mmol, 1 eq.) em N,N- dimetilformamida (0,5 mL) à temperatura ambiente, foram adicionados N,N-diisopropiletilamina (24,7 μL, 0,14 mmol, 4 eq.), 2,6-lutidina (16,3 μL, 0,14 mmol, 4 eq.), 4-N,N-dimetilamino piridina (4,3 mg, 0,035 mmol, 1 eq.) e N-[(9H-fluoren-9-ilmetoxi)carbonil]-L-valil-N5-carbamoil- N-[4-({[(4-nitrofenoxi)carbonil]oxi}metil)fenil]-L-ornitinamida (40,6 mg, 0,053 mmol, 1,5 eq.), e a reação foi agitada por 2,5 h. Mais N-[(9H- fluoren-9-ilmetoxi)carbonil]-L-valil-N5-carbamoil-N-[4-({[(4-nitrofenoxi)carbonil]oxi}metil)fenil]-L-ornitinamida (13,5 mg, 0,018 mmol, 0,5 eq.) foi adicionado, e a reação foi agitada por mais 1 h. A reação foi purificada por cromatografia de fase reversa (Método A) para dar #B191 como um sólido branco. Rendimento: 9,4 mg, 0,0081 mmol, 23%. LCMS (Protocolo D): m/z 1177,8 [M+H]+, tempo de retenção = 0,90 minuto.
[01076] Etapa 2. Síntese de L-valil-N-[4-({[(2-{[(3R,5S,7R,8R)-7- {(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi- 1,6-dioxaspiro[2,5]oct-5-il]acetil}hidrazinil)carbonil]oxi}metil)fenil]-N5- carbamoil-L-ornitinamida sal de acetato (#B192). O composto do título foi preparado em 56% de rendimento a partir de 9,4 mg (0,008 mmol, 1,0 eq) de #B191 e 13,6 mg (0,16 mmol, 20,0 eq) de piperidina usando o procedimento descrito para a preparação do composto #B47. LCMS (Protocolo D): m/z 955,8 [M+H]+, tempo de retenção = 0,65 minuto.
[01077] Etapa 3. Síntese de N-{7-[(2,5-dioxopirrolidin-1-il)oxi]-7- oxoheptanoil}-L-valil-N-[4-({[(2-{[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]acetil}hidrazinil)carbonil]oxi}metil)fenil]-N5- carbamoil-L-ornitinamida (#B190). A uma solução de #B192 (4,5 mg, 0,004 mmol, 1 eq.) em N,N-dimetilformamida (0,3 mL) à temperatura ambiente, foi adicionada N,N-diisopropiletilamina (3,5 μL, 0,02 mmol, 5 eq.) seguido por 1,1'-[(1,7-dioxoheptano-1,7-diil)bis(oxi)]dipirrolidina- 2,5-diona (preparado como em J.Am.Chem.Soc. 2006, 128, 2802, 8,9 mg, 0,025 mmol) 6,2 eq.), e a reação foi deixada agitar por 35 min. A reação foi purificada por cromatografia de fase reversa (Método A) para dar #B190 como um sólido branco. Rendimento: 1,65 mg, 0,0014 mmol, 34%. LCMS (Protocolo D): m/z 1194,80 [M+H]+, tempo de retenção = 0,75 minutos. 1H RMN (500 MHz, CD3CN) δ 9,06 (s, 1 H), 8,17 (s, 1 H), 7,71-7,63 (m, 2 H), 7,35-7,25 (m, 2 H), 7,19 (d, J = 7,6 Hz, 1 H), 6,73 (d, J = 6,6 Hz, 1 H), 6,47 (d, J = 8,8 Hz, 1 H), 6,41-6,30 (m, 2 H), 5,96-5,85 (m, 2 H), 5,67-5,50 (m, 2 H), 5,33-5,24 (m, 1 H), 5,08-4,99 (m, 2 H), 4,74 (s, 1 H), 4,57-4,48 (m, 1 H), 4,39-4,25 (m, 2 H), 4,15-4,08 (m, 1 H), 3,82-3,75 (m, 1 H), 3,67-3,59 (m, 1 H), 3,553,47 (m, 1 H), 3,35-3,20 (m, 2 H), 3,12-2,99 (m, 2 H), 2,82-2,73 (m, 5 H), 2,66-2,52 (m, 6 H), 2,46-2,38 (m, 2 H), 2,36-2,20 (m, 4 H), 1,98 (s, 3 H), 1,77-1,57 (m, 11 H), 1,53-1,36 (m, 6 H), 1,30 (d, J = 6,6 Hz, 3 H), 1,06 (d, J = 6,6 Hz, 3 H), 1,00-0,91 (m, 9 H).
[01078] Exemplo A#68
[01079] Preparação de N-{6-[(bromoacetil)amino]hexanoil}-L-valil-N-[2-(3-{[(2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3S,5S,7S)-7-(2- metoxi-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}- 2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il]oxi}-3- oxopropil)fenil]-L-alaninamida (#B193).
[01080] Etapa 1. Síntese de ácido 6-[(bromoacetil)amino]hexanóico (#B194). Ácido 6-aminohexanóico (14,2 g, 0,11 mol, 1 eq.)) foi adicionado a KOH (6,2 g, 0,11 mol, 1 eq.) em água (30 mL) a 0°C. Brometo de bromoacetil (26,1 g, 0,13 mol, 1,2 eq.) foi adicionado em gotas enquanto solução de carbonato de potássio (2,8 N) foi adicionada em gotas para ajustar o pH > 7,8. Após a adição, a solução foi agitada a 0°C por uma hora. A mistura de reação foi acidificada por 0,5 M HCl para ajustar o pH para 1 e extraída com etil acetato. A fase orgânica foi seca sobre sulfato de sódio e concentrada in vacuo. O resíduo foi purificado por cromatografia de coluna de sílica eluída com diclorome- tano:metanol 50:1 para gerar #B194 (10,2 g, 38%) como um sólido branco.
[01081] Etapa 2. Síntese de pentafluorofenil 6- [(bromoacetil)amino]hexanoato (#B195). A uma solução de #B194 (8 g, 31,7 mmoles, 1 eq.) em diclorometano (400 mL), foi adicionado pen- tafluorofenil trifluoroacetato (13,3 g, 45,7 mmoles, 1,45 eq.) e piridina (10 g, 127 mmoles, 4 eq.) a 0°C. A mistura de reação foi agitada a 0°C por 10 minutos. A mistura de reação foi lavada com 0,5 M HCl e concentrada in vacuo. O resíduo foi purificado por cromatografia flash elu- ída com etil acetato (49,2% em PE) para gerar #B195 (9,5 g, 71,7%) como um sólido branco. 1H RMN (400 MHz, CDCI3) □ 6.53 (br, 1 H), 3,89 (s, 2 H), 3,35 (m, 2 H), 2,70 (m, 2 H), 1,83 (m, 2 H), 1,64 (m, 2 H), 1,48 (m, 2 H).
[01082] Etapa 3. Síntese de metil (2E)-3-(2-{[N-(terc-butoxicarbonil)- L-alanil]amino}fenil)prop-2-enoato (#B196). Uma mistura de metil (2E)- 3-(2-aminofenil)prop-2-enoato (14 g, 79,1 mmoles, 1 eq.), N-(terc- butoxicarbonil)-L-alanina (22,4 g, 119 mmoles, 1,5 eq.), 1- hidroxibenzotriazol (16,1 g, 119 mmoles, 1,5 eq.), cloridrato de 1-[3- (dimetilamino)propil]-3-etilcarbodiimida (22,8 g, 119 mmoles, 1,5 eq.), e 4-N,N-dimetilamino piridina (1,93 g, 15,8 mmoles, 0,2 eq.) em N,N- dimetilformamida (600 mL) foi agitada a 50°C por 3 dias. A mistura de reação foi diluída com etil acetato (1500 mL) e água (500 mL). A camada orgânica foi separada e lavada com água (300 mL x 2), seca sobre sulfato de sódio e concentrada até secagem. O resíduo foi purificado por cromatografia de coluna de sílica eluída com éter de petró- leo:etil acetato a partir 20:1 a 5:1 para gerar #B196 bruto (21 g, 76,4%) como um óleo amarelo que foi usado sem purificação adicional.
[01083] Etapa 4. Síntese de metil 3-(2-{[N-(terc-butoxicarbonil)-L- alanil]amino}fenil)propanoato (#B197). A uma solução de #B196 bruto (21 g, 60,3 mmoles, 1 eq.) em metanol (1 L), foi adicionado Pd/C (4 g) a 20°C, e a mistura de reação foi agitada à temperatura ambiente sob hidrogênio (35 psi) por 12 h. A mistura de reação foi filtrada e concentrada até secagem para gerar #B197 bruto (19 g, 90,5%) como um óleo amarelo que foi usado sem purificação adicional.
[01084] Etapa 5. Síntese de ácido 3-(2-{[N-(terc-butoxicarbonil)-L- alanil]amino}fenil)propanóico (#B198). A uma solução de #B197 bruto (19 g, 54,2 mmoles, 1 eq.) em tetrahidrofurano (150 mL), foi adicionado hidróxido de sódio (110 mL, 2 M) a 0°C, e a reação foi agitada a 50°C por 3 horas. O tetrahidrofurano foi removido in vacuo, e a solução resultante foi ajustada para pH = 3-4 por 1 M HCl e extraída com etil acetato (100 mL x 3). O extrato foi lavado com salmoura (20 mL x 1), seco sobre sulfato de sódio e concentrado até secagem para gerar #B198 bruto (16 g, 88,9%) como óleo marrom.
[01085] Etapa 3. Síntese de ácido 3-[2-(L-alanilamino)fenil]propanóico sal de trifluoroacetato (#B199). A uma solução de #B198 (16 g, 47,5 mmoles, 1 eq.) em diclorometano (150 mL), foi adicionado TFA (100 mL) a 0°C, e a reação foi agitada a 25°C por 12 h. A mistura de reação foi concentrada até secagem, e o resíduo foi usado diretamente na etapa seguinte sem purificação adicional.
[01086] Etapa 4. Síntese de ácido 3-[2-({N-[(9H-fluoren-9- ilmetoxi)carbonil]-L-alanil}amino)fenil]propanóico (#B200). A uma solução de #B199 (5 g, 21,1 mmoles, 1 eq.) em acetona (50 mL) e água (100 mL), foi adicionado bicarbonato de sódio (5,30 g, 63,4 mmoles, 3 eq.) a 0°C. Em seguida, cloridrato de 9H-fluoren-9-ilmetilcarbono (4,94 g, 19,1 mmoles, 0.9 eq.) em acetona (50 mL) foi adicionado em gotas a 0°C. A reação foi ajustada para pH = 3-4 com 1 M HCl, e a fase aquosa foi extraída com etil acetato. As camadas orgânicas combinadas foram secas sobre sulfato de sódio, concentradas in vacuo, e o resíduo foi purificado por cromatografia de coluna de sílica eluída com metanol:diclorometano (1,5%~2%) para gerar um produto bruto, que foi adicionalmente purificado por HPLC preparativa para gerar um sólido branco, que foi adicionalmente purificado por separação por SFC para gerar #B200 (560 mg, 5,8%) como um sólido branco. 1H RMN (400Hz, DMSO-d6): □ 9,65 (s, 1H), 7,92 (d, 2H), 7,76 (m, 3H), 7,437,14 (m, 8H), 4,32 (m, 4H), 2,80 (m, 2H), 2,50 (m, 2H), 1,37 (m, 3H).
[01087] Etapa 5. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3S,5S,7S)-7-(2-metoxi-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5- oxopent-3-en-2-il 3-[2-({N-[(9H-fluoren-9-ilmetoxi)carbonil]-L-alanil}amino)fenil]propanoato (#B201). A uma solução de #B148 (21,2 mg, 0,043 mmol, 1 eq.) em diclorometano (0,5 mL) à temperatura ambiente, foram adicionados 4-N,N-dimetilamino piridina (3,5 mg, 0,029 mmol, 0,67 eq.), uma solução de #B200 (39,4 mg, 0,086 mmol, 2 eq.) em N,N-dimetilformamida (0,3 mL), e N,N'-diciclohexilcarbodiimida (DCC) (23,2 mg, 0,107 mmol, 2,5 eq.), e a reação foi deixada agitar por 2,5 h. Mais DCC (23 mg, 0,107 mmol, 2,5 eq.) foi adicionado, e a reação foi deixada agitar por um adicional de 2 horas. A reação foi diluída com DMSO (0,7 mL), e purificada por cromatografia de fase reversa (Método A) para dar #B201 como um sólido branco. Rendimento: 8,6 mg, 0,009 mmol, 21%. LCMS (Protocolo D): m/z 954,57 [M+Na]+, tempo de retenção = 1,10 minuto.
[01088] Etapa 6. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3S,5S,7S)-7-(2-metoxi-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5- oxopent-3-en-2-il 3-[2-(L-alanilamino)fenil]propanoato sal de acetato (#B202). O composto do título foi preparado em 70% de rendimento a partir de 15,1 mg (0,016 mmol, 1,0 eq.) de #B201 e 27,2 mg (0,32 mmol, 20,0 eq.) de piperidina usando o procedimento descrito para a preparação do composto #B47. LCMS (Protocolo D): m/z 955,8 [M+H]+, tempo de retenção = 0,65 minuto.
[01089] Etapa 7. Síntese de N-[(9H-fluoren-9-ilmetoxi)carbonil]-L- valil-N-[2-(3-{[(2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3S,5S,7S)-7-(2- metoxi-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}- 2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il]oxi}-3- oxopropil)fenil]-L-alaninamida (#B203). A uma solução de #B202 (9 mg, 0,01 mmol, 1 eq.) em N,N-dimetilformamida (0,4 mL) à temperatura ambiente, foi adicionada N,N-diisopropiletilamina (8,5 μL, 0,048 mmol, 4 eq.) seguido por 2,5-dioxopirrolidin-1-il-N-[(9H-fluoren-9- ilmetoxi)carbonil]-L-valinato (10,5 mg, 0,024 mmol, 2 eq.), e a reação foi deixada agitar por 20 min. A reação foi purificada por cromatografia de fase reversa (Método A) para dar #B203 como um sólido branco. Rendimento: 7,4 mg, 0,007 mmol, 60%. LCMS (Protocolo D): m/z 1031,9 [M+H]+, tempo de retenção = 1,11 minuto.
[01090] Etapa 8. Síntese de L-valil-N-[2-(3-{[(2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3S,5S,7S)-7-(2-metoxi-2-oxoetil)-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il]oxi}-3-oxopropil)fenil]-L- alaninamida sal de acetato (#B204). O composto do título foi preparado em 87% de rendimento a partir de 7,4 mg (0,007 mmol, 1,0 eq.) de #B203 e 11,9 mg (0,14 mmol, 20,0 eq.) de piperidina usando o procedimento descrito para a preparação do composto #B47. LCMS (Protocolo D): m/z 809,9 [M+H]+, tempo de retenção = 0,81 minuto.
[01091] Etapa 9. Síntese de N-{6-[(bromoacetil)amino]hexanoil}-L- valil-N-[2-(3-{[(2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3S,5S,7S)-7-(2- metoxi-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}- 2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il]oxi}-3- oxopropil)fenil]-L-alaninamida (#B193). A uma solução de #B204 (5,3 mg, 0,006 mmol, 1 eq.) em N,N-dimetilformamida (0,4 mL) à temperatura ambiente, foi adicionada N,N-diisopropiletilamina (6,3 μL, 0,036 mmol, 6 eq.) seguido por #B195 (2,9 mg, 0,007 mmol, 1,2 eq.), e a reação foi deixada agitar por 10 min. A reação foi purificada por croma- tografia de fase reversa (Método A) para dar #B193 como um sólido branco. Rendimento: 4,1 mg, 0,004 mmol, 65%. LCMS (Protocolo D): m/z 1044,9 [M+H]+, tempo de retenção = 0,95 minuto. 1H RMN (500 MHz, DMSO-d6) δ 9,42 (s, 1 H), 8,26-8,19 (m, 1 H), 8,14 (d, J = 6,9 Hz, 1 H), 7,85-7,77 (m, 2 H), 7,30-7,10 (m, 4 H), 6,42-6,33 (m, 1 H), 6,25 (d, J = 15,9 Hz, 1 H), 6,10 (dd, J = 11,7 e 1,2 Hz, 1 H), 5,83 (dd, J = 11,5 e 7,3 Hz, 1 H), 5,58 (dd, J = 15,9 e 5,1 Hz, 1 H), 5,56-5,50 (m, 1 H), 4,55-4,42 (m, 2 H), 4,34-4,26 (m, 1 H), 4,20 (dd, J = 8,8 e 6,9 Hz, 1H), 3,81 (s, 2 H), 3,68-3,62 (m, 2 H), 3,60 (s, 3 H), 3,54-3,46 (m, 1 H),3,07-2,99 (m, 2 H), 2,87-2,56 (m, 7 H), 2,35-2,08 (m, 5 H), 2,02-1,92 (m, 2 H), 1,88-1,61 (m, 8 H), 1,53-1,35 (m, 5 H), 1,33 (d, J = 7,1 Hz, 3H), 1,28-1,19 (m, 4 H), 1,06 (d, J = 6,4 Hz, 3 H), 0,95 (d, J = 7,3 Hz, 3H), 0,85 (d, J = 6,6 Hz, 3 H), 0,82 (d, J = 6,6 Hz, 3 H).
[01092] Exemplo A#69
[01093] Preparação de N-{6-[(bromoacetil)amino]hexanoil}-L-valil-N-{4-[({[2-{[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5- il]acetil}tetrahidropiridazin-1(2H)-il]carbonil}oxi)metil]fenil}-N5- carbamoil-L-ornitinamida (#B205).
[01094] Etapa 1. Síntese de N-(6-{[(9H-fluoren-9- ilmetoxi)carbonil]amino}hexanoil)-L-valil-N5-carbamoil-N-(4- {[(tetrahidropiridazin-1(2H)-ilcarbonil)oxi]metil}fenil)-L-ornitinamida (#B206). A uma solução de dicloridrato de hexahidropiridazina (11,1 mg, 0,07 mmol, 1 eq.) em N,N-dimetilformamida (0,4 mL) à temperatura ambiente, foi adicionada N,N-diisopropiletilamina (49,3 μL, 0,28 mmol, 4 eq.) e 4-N,N-dimetilamino piridina (4,3 mg, 0,035 mmol, 0,5 eq.) seguido por #B45 (61,6 mg, 0,07 mmol, 1 eq.), e a reação foi deixada agitar por 30 min. A reação foi purificada por cromatografia líquida de pressão média de fase reversa eluída com 0,02% de ácido acético em água (v/v) e 0,02% de ácido acético em acetonitrila (v/v) (5% a 95%) para dar #B206 como um sólido branco. Rendimento: 19,8 mg, 0,024 mmol, 34%. LCMS (Protocolo D): m/z 827,63 [M+H]+, tempo de retenção = 0,84 minuto.
[01095] Etapa 2. Síntese de N-(6-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}hexanoil)-L-valil-N-{4-[({[2-{[(3R,5S,7R,8R)-7- {(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi- 1,6-dioxaspiro[2,5]oct-5-il]acetil}tetrahidropiridazin-1(2H)- il]carbonil}oxi)metil]fenil}-N5-carbamoil-L-ornitinamida (#B207). A uma solução de #NP1 (15,5 mg, 0,029 mmol, 2 eq.) em N,N- dimetilformamida (0,15 mL) à temperatura ambiente, foi adicionada N,N-diisopropiletilamina (19,7 μL, 0,11 mmol, 8 eq.) e hexafluorofosfa- to de O-^-azabenzotriazoM-iO-N^N^N-tetrametHurônio (11,3 mg, 0,029 mmol, 2,1 eq.) seguido por uma solução de #B206 (11,4 mg, 0,014 mmol, 1 eq.) em N,N-dimetilformamida (0,6 mL), e a reação foi deixada agitar por 22 horas. A reação foi purificada por cromatografia de fase reversa (Método A) para dar #B207 como um sólido branco. Rendimento: 4,2 mg, 0,003 mmol, 22%. LCMS (Protocolo D): m/z 1345,2 [M+H]+, tempo de retenção = 0,97 minuto.
[01096] Etapa 3. Síntese de N-(6-aminohexanoil)-L-valil-N-{4-[({[2- {[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5- il]acetil}tetrahidropiridazin-1(2H)-il]carbonil}oxi)metil]fenil}-N5-carbamoil-L-ornitinamida sal de acetato (#B208). O composto do título foi preparado em 67% de rendimento a partir de 9,8 mg (0,007 mmol, 1,0 eq.) de #B207 e 11,9 mg (0,14 mmol, 20,0 eq.) de piperidina usando o procedimento descrito para a preparação do composto #B47. LCMS (Protocolo D): m/z 1122,95 [M+H]+, tempo de retenção = 0,74 minuto.
[01097] Etapa 4. Síntese de N-{6-[(bromoacetil)amino]hexanoil}-L- valil-N-{4-[({[2-{[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5- {[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5- il]acetil}tetrahidropiridazin-1(2H)-il]carbonil}oxi)metil]fenil}-N5-carbamoil-L-ornitinamida (#B205). O composto do título foi preparado em 52% de rendimento a partir de 5,6 mg (0,005 mmol, 1 eq.) de #B208, 1,7 mg (0,007 mmol, 1,5 eq) de 1-[(bromoacetil)oxi]pirrolidina- 2,5-diona e 2,6 mg (0,02 mmol, 4.0 eq) de N,N-diisopropiletilamina usando o procedimento descrito para a preparação do composto #B150. LCMS (Protocolo D): m/z 1244,9 [M+H]+, tempo de retenção = 0,83 minuto. 1H RMN (500 MHz, DMSO-d6) δ 10,04 (br s, 1 H), 8,24 (br s, 1 H), 8,13 (br s, 1 H), 7,87-7,77 (m, 2 H), 7,65-7,55 (m, 2 H), 7,367,24 (m, 2 H), 6,88-6,77 (m, 1 H), 6,41-6,26 (m, 2 H), 6,10 (d, J = 11,5 Hz, 1 H), 6,04-5,95 (m, 1 H), 5,86 (dd, J = 11,5 e 7,3 Hz, 1 H), 5,665,48 (m, 2 H), 5,42 (br s, 1 H), 5,18-5,06 (m, 1 H), 5,05-4,94 (m, 1 H), 4,39-4,15 (m, 5 H), 4,11-3,98 (m, 1 H), 3,81 (s, 2 H), 3,68-3,60 (m, 2 H), 3,53-3,45 (m, 1 H), 3,28-3,20 (m, 2 H), 3,08-2,89 (m, 4 H), 2,852,72 (m, 2 H), 2,34-2,08 (m, 5 H), 2,02-1,92 (m, 4 H), 1,86-1,31 (m, 18 H), 1,28-1,20 (m, 4 H), 1,09-1,03 (m, 3 H), 0,94 (d, J = 7,3 Hz, 3 H), 0,85 (d, J = 6,6 Hz, 3 H), 0,82 (d, J = 6,6 Hz, 3 H).
[01098] Exemplo A#70
[01099] Preparação de (2Z,4S)-N-[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,4R,5R,7S)-7-(2-hidrazinil-2-oxoetil)-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]-4-hidroxipent-2-enamida (#B209).
[01100] Etapa 1. Síntese de (2Z,4S)-N-[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-(2-hidrazinil-2-oxoetil)-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]-4-hidroxipent-2-enamida (#B209). A uma solução de #B4 (13,1 mg, 0,027 mmol, 1 eq.) em tetrahidrofurano (0,4 mL) a 0°C, foi adicionado DCC (11,7 mg, 0,054 mmol, 2 eq.), e a reação foi agitada por 10 min. N-hidroxissuccinimida (6,3 mg, 0,054 mmol, 2 eq.) foi adicionada, e a reação foi deixada agitar por 5 horas à temperatura ambiente. A reação foi diluída com acetonitrila, filtrada e concentrada. O resíduo foi retomado em diclorometano (0,5 mL), e uma solução de hidrazina (1 M em THF, 270 μL, 0,27 mmol, 10 eq.) foi adicionada. A reação foi agitada por 10 min, diluída com dimetil sulfóxido, concentrada para remover o diclorometano, e filtrada. O resíduo bruto foi purificado por cromatografia de fase reversa (Método A) para gerar #B209 como um sólido. Rendimento: 8,1 mg, 59%. LCMS (Protocolo D): m/z 508,6 [M+H]+, tempo de retenção = 0,59 minuto. 1H RMN (400 MHz, DMSO-d6) δ 9,00 (s, 1 H), 7,76 (d, J = 7,4 Hz, 1 H), 6,29 (d, J = 15,8 Hz, 1 H) 5,98 (d, J = 11,3 Hz, 1 H), 5,86 (dd, J = 11,3 e 7,4 Hz, 1 H), 5,60 (dd, J = 15,8 e 5,5 Hz, 1 H), 5,56-5,48 (m, 1 H), 5,23-5,07 (m, 2 H), 5,06-4,98 (m, 1 H), 4,32-4,09 (m, 3 H), 3,70-3,59 (m, 2 H), 3,553,45 (m, 1 H), 3,25-3,19 (m, 1 H), 2,74 (d, J = 5,1 Hz, 1 H), 2,58 (d, J = 5,1 Hz, 1 H), 2,44 (dd, J = 14,4 e 8,6 Hz, 1 H), 2,36-2,14 (m, 3 H), 1,93-1,58 (m, 8 H), 1,50-1,42 (m, 1 H), 1,11 (d, J = 6,2 Hz, 3 H), 1,07 (d, J = 6,2 Hz, 3 H), 0,96 (d, J = 7,0 Hz, 3 H).
[01101] Exemplo A#71
[01102] Preparação de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5- {(3R,4R,5R,7S)-7-[(6S,9S)-19-bromo-6-metil-2,5,8,11,18-pentaoxo-9- (propan-2-il)-3,4,7,10,17-pentaazanonadec-1-il]-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B210).
[01103] Etapa 1. Síntese de 9H-fluoren-9-ilmetil-{6-[(2,5- dioxopirrolidin-1-il)oxi]-6-oxohexil}carbamato (#B211). A uma solução de ácido 6-((((9H-fluoren-9-il)metoxi)carbonil)amino)hexanóico (6 g, 16,9 mmoles, 1 eq.) em tetrahidrofurano (250 mL), foi adicionada N- hidroxissuccinimida (2,13 g, 18,5 mmoles, 1,1 eq.) e DCC (3,5 g, 18,59 mmoles, 1,1 eq.) a 0°C, e a reação foi agitada a 20°C durante a noite. A mistura de reação foi resfriada para -20°C, filtrada e concentrada até secagem. O resíduo foi agitado em MTBE (300 mL) por 20 min e novamente filtrado. O bolo de filtro foi seco in vacuo para gerar #B211 (5,6 g, 73%) como um sólido branco.
[01104] Etapa 2. Síntese de N-(6-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}hexanoil)-L-valina (#B212). A uma solução de L-valina (1,5 g, 12,8 mmoles, 1 eq.) em água (60 mL) e tetrahidrofura- no (30 mL) a 0°C, foi adicionado NaHCO3 (1,37 g, 16,3 mmoles, 1,3 eq.). Em seguida, uma solução de #B211 (5,67 g, 12,6 mmoles, 0.98 eq.) em dimetoxietano (80 mL) e tetrahidrofurano (80 mL) foi adicionada em gotas a 0-10°C, e a reação foi agitada a 20°C por 18 horas. O pH da mistura de reação foi ajustado para 4 por adição de ácido cítrico, e a mistura de reação foi concentrada. Etil acetato (450 mL) e metanol (50 mL) foram adicionados, e a mistura foi agitada por 10 min. A camada orgânica foi separada, seca sobre sulfato de sódio e concentrada até secagem. O resíduo foi purificado por cromatografia de coluna flash eluída com diclorometano:metanol a partir de 100:1 a 8:1 para gerar #B212 (2,6 g, 45%) como um sólido branco.
[01105] Etapa 3. Síntese de 2,5-dioxopirrolidin-1-il-N-(6-{[(9H- fluoren-9-ilmetoxi)carbonil]amino}hexanoil)-L-valinato (#B213). A uma solução de #B212 (2 g, 4,42 mmoles, 1 eq.) em tetrahidrofurano (60 mL) a 0°C, foram adicionados N-hidroxissuccinimida (0,53 g, 4,65 mmoles, 1.05 eq.) e DCC (0,88 g, 4,65 mmoles, 1,05 eq.), e a reação foi agitada a 20°C durante a noite. A mistura de reação foi resfriada para -20°C, filtrada e concentrada até secagem. O resíduo foi agitado em MTBE (300 mL) por 20 min e filtrado. O bolo de filtro foi seco in vacuo para gerar #B213 (1,9 g, 79%) como um sólido branco.
[01106] Etapa 4. Síntese de N-(6-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}hexanoil)-L-valil-L-alanina (#B214). A uma so-lução de L-alanina (0,32 g, 3.6 mmoles, 1,04 eq.) em água (15 mL) e tetrahidrofurano (10 mL) a 0°C, foi adicionado NaHCO3 (0,44 g, 5,19 mmoles, 1,5 eq.). Em seguida, uma solução de #B213 (1,9 g, 3,46 mmoles, 1 eq.) em dimetoxietano (30 mL) foi adicionada em gotas a 010°C, e a reação foi agitada a 20°C por 18 horas. O pH da mistura de reação foi ajustado para 4 por adição de ácido cítrico, e a mistura de reação foi concentrada. Diclorometano (400 mL) e metanol (50 mL) foram adicionados, e a mistura foi agitada por 10 min. A camada orgânica foi separada, seca sobre sulfato de sódio e concentrada até secagem. O resíduo foi purificado por cromatografia de coluna flash eluída com diclorometano:metanol a partir de 100:1 a 8:1 para gerar um resíduo que foi recristalizado com metanol/tetrahidrofurano (3:1) três vezes para dar #B214 (490 mg, 27%) como um sólido branco. 1H RMN (400 MHz, DMSO): □ 12,48 (b, 1 H), 8,21 (b, 1 H), 7,91 (d, 2 H), 7,77 (d, 1 H), 7,68 (m, 2 H), 7,41 (m, 2 H), 7,33 (m, 2 H), 7,31 (m, 1 H), 4,29 (m, 2 H), 4,18 (m, 3 H), 2,94 (m, 2 H), 2,16 (m, 2 H), 1,93 (m, 1 H), 1,47 (m, 2 H), 1,37 (m, 2 H), 1,25 (m, 3 H), 1,21 (m, 2 H), 0,86 (m, 6 H).
[01107] Etapa 5. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3R,4R,5R,7S)-7-[(12S,15S)-1-(9H-fluoren-9-il)-15-metil- 3,10,13,16,19-pentaoxo-12-(propan-2-il)-2-oxa-4,11,14,17,18- pentaazaicosan-20-il]-4-hidroxi-1,6-dioxaspiro[2,5]oct-5-il}-3- metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro-2H-piran-3-il}amino)-5- oxopent-3-en-2-il acetato (#B215). A uma solução de #B214 (11,5 mg, 0,022 mmol, 1,2 eq.) em N,N-dimetilformamida (0,2 mL) à temperatura ambiente, foram adicionados N,N-diisopropiletilamina (12,7 μL, 0,072 mmol, 4 eq.) e hexafluorofosfato de O-(7-azabenzotriazol-1-il)- N,N,N‘,N‘-tetrametilurônio (8,5 mg, 0,022 mmol, 1,2 eq.) seguido por uma solução de #B6 (10 mg, 0,018 mmol, 1 eq.) em N,N- dimetilformamida (0,5 mL), e a reação foi deixada agitar por 35 min. A reação foi purificada por cromatografia de fase reversa (Método A) para dar #B215 como um sólido branco. Rendimento: 14,6 mg, 0,014 mmol, 77%. LCMS (Protocolo D): m/z 1056.0 [M+H]+, tempo de retenção = 0,94 minuto.
[01108] Etapa 6. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-(2-{2-[(2S)-2-({(2S)-2-[(6-aminohexanoil)amino]-3- metilbutanoil}amino)propanoil]hidrazinil}-2-oxoetil)-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato, sal de acetato (#B216). O composto do título foi preparado em 85% de rendimento a partir de 20,8 mg (0,02 mmol, 1,0 eq.) de #B215 e 34,1 mg (0,4 mmol, 20,0 eq.) de piperidina usando o procedimento descrito para a preparação do composto #B47. LCMS (Protocolo D): m/z 833,9 [M+H]+, tempo de retenção = 0,65 minuto.
[01109] Etapa 7. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3R,4R,5R,7S)-7-[(6S,9S)-19-bromo-6-metil-2,5,8,11,18-pentaoxo- 9-(propan-2-il)-3,4,7,10,17-pentaazanonadec-1-il]-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B210). O composto do título foi preparado em 57% de rendimento a partir de 15,2 mg (0,017 mmol, 1 eq.) de #B216, 6,1 mg (0,026 mmol, 1,5 eq) de 1- [(bromoacetil)oxi]pirrolidina-2,5-diona e 8,9 mg (0,068 mmol, 4,0 eq) de N,N-diisopropiletilamina usando o procedimento descrito para a preparação do composto #B150. LCMS (Protocolo D): m/z 975,68 [M+Na]+, tempo de retenção = 0,76 minuto. 1H RMN (500 MHz, DMSO-d6) δ 9,93-9,80 (m, 2 H), 8,26-8,19 (m, 1 H), 8,14 (d, J = 7,8 Hz, 1 H), 7,98 (d, J = 7,6 Hz, 1 H), 7,85-7,73 (m, 2 H), 6,56 (br s, 1 H), 6,41-6,26 (m, 2 H), 6,11 (d, J = 11,5 Hz, 1 H), 5,86 (dd, J = 11,7 e 7,6 Hz, 1 H), 5,61 (dd, J = 15,9 e 5,6 Hz, 1 H), 5,56-5,48 (m, 1 H), 5,10-5,03 (m, 1 H), 4,39-4,13 (m, 4 H), 3,81 (s, 2 H), 3,69-3,60 (m, 2 H), 3,54-3,45 (m, 1 H), 3,25-3,19 (m, 1 H), 3,09-3,00 (m, 2 H), 2,74 (d, J = 5,0 Hz, 1 H), 2,58 (d, J = 5,0, 1 H), 2,35-2,25 (m, 2 H), 2,24-2,05 (m, 3 H), 1,98 (s, 3 H), 1,96-1,75 (m, 4 H), 1,73-1,60 (m, 4 H), 1,55-1,33 (m, 5 H), 1,291,18 (m, 7 H), 1,07 (d, J = 6,4 Hz, 3 H), 0,95 (d, J = 7,3 Hz, 3 H), 0,870,77 (m, 6 H).
[01110] Exemplo A#72
[01111] Preparação de (2R)-2-(piridin-2-ildissulfanil)propil-2-{[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5- il]acetil}hidrazinacarboxilato (#B217).
[01112] Etapa 1. Síntese de metil (2R)-2-(acetilsulfanil)propanoato (#B218). A uma solução de tioacetato de potássio (3.9 g, 34,4 mmoles, 1.2 eq.) em N,N-dimetilformamida (60 mL), foi adicionada uma solução de S-metil-2-cloropropanoato (3,5 g, 28,7 mmoles, 1 eq.) em N,N- dimetilformamida (10 mL) à temperatura ambiente, e a mistura foi agitada à temperatura ambiente durante a noite. A mistura foi vertida em água (150 mL) e extraída com éter de petróleo (100 mL) três vezes. Os extratos foram lavados com salmoura, secos sobre sulfato de sódio e concentrados in vacuo para gerar #B218 (4,4 g, 94,8%) como um óleo ligeiramente amarelo.
[01113] Etapa 2. Síntese de (2R)-2-sulfanilpropan-1-ol (#B219). A uma suspensão de LAH (3,4 g, 89,5 mmoles, 5 eq.) em tetrahidrofura- no (116 mL), foi adicionada uma solução de #B218 (2,9 g, 17,9 mmoles, 1 eq.) em tetrahidrofurano (29 mL) a 0°C, e a mistura foi agitada a temperatura ambiente por 1 h. A reação foi resfriada bruscamente com 2 N HCl (50 mL) cuidadosamente. A mistura foi extraída com dicloro- metano (100 mL) cinco vezes, e os extratos foram secos sobre sulfato de sódio. A solução foi concentrada in vacuo a cerca de 150 mL, e a solução foi usada na etapa seguinte diretamente sem purificação adi- cional.
[01114] Etapa 3. Síntese de (2R)-2-(piridin-2-ildissulfanil)propan-1- ol (#B220). A uma solução de aldritiol-2 (5,9 g, 26,8 mmoles, 1,5 eq.) e ácido acético (1,07 g, 17,9 mmoles, 1 eq.) em etanol (120 mL) a 0°C, foi adicionada uma solução de #B219 em THF (150 mL, ~17,9 mmoles, 1 eq.), e a mistura foi agitada à temperatura ambiente durante a noite. A mistura foi concentrada in vacuo, e o resíduo foi purificado por cromatografia de sílica gel eluída com éter de petróleo:etil acetato (10:1 a 4:1) para gerar um óleo amarelo que foi repurificado por SFC para gerar #B220 (860 mg, 24%) como um óleo ligeiramente amarelo. 1H RMN (400Hz, CDCI3): □ 8,50 (m, 1 H), 7,59 (m, 1 H), 7,40 (d, 1 H), 7,16 (m, 1 H), 5,98 (m, 1 H), 3,70 (m, 1 H), 3,41 (m, 1 H), 3,12 (m, 1 H), 1,31 (d, 3 H).
[01115] Etapa 4. Síntese de 4-nitrofenil-(2R)-2-(piridin-2- ildissulfanil)propil carbonato (#B221). A uma solução de #B220 (111 mg, 0,554 mmol, 1 eq.) em diclorometano (0,9 mL) à temperatura ambiente, foi adicionada piridina (99,4 μL, 1,22 mmol, 2,2 eq.) seguido por uma solução de 4-nitrofenilcloroformiato (140 mg, 0,665 mmol, 1,2 eq.) em diclorometano (0,9 mL) em gotas, e a reação foi agitada durante a noite. A reação foi diluída com diclorometano e água, extraída duas vezes e lavada com salmoura, e os extratos orgânicos combinados foram secos sobre sulfato de sódio, filtrados e concentrados. O resíduo foi purificado por cromatografia de sílica gel em eluição com diclo- rometano para dar #B221 como uma goma. Rendimento: 45 mg, 0,123 mmol, 22%. LCMS (Protocolo D): m/z 367,2 [M+H]+, tempo de retenção = 0,99 minuto.
[01116] Etapa 5. Síntese de (2R)-2-(piridin-2-ildissulfanil)propil-2- {[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5- il]acetil}hidrazinacarboxilato (#B217). A uma solução de #B6 (9,8 mg, 0,018 mmol, 1 eq.) em N,N-dimetilformamida (0,1 mL) à temperatura ambiente, foram adicionados N,N-diisopropiletilamina (12,7 μL, 0,072 mmol, 4 eq.), 2,6-lutidina (8,4 μL, 0,072 mmol, 4 eq.), 4-N,N- dimetilamino piridina (2,2 mg, 0,018 mmol, 1 eq.), foi adicionada uma solução de #B221 (10 mg, 0,027 mmol, 1,5 eq.) em N,N- dimetilformamida (0,3 mL), e a reação foi deixada agitar por 5,5 h. A reação foi purificada por cromatografia de fase reversa (Método A) para dar #B217 como um sólido branco. Rendimento: 5,9 mg, 0,0076 mmol, 42%. LCMS (Protocolo D): m/z 777,51 [M+H]+, tempo de reten-ção = 0,84 minuto. 1H RMN (500 MHz, CD3CN) δ 8,45-8,38 (m, 1 H), 8,15 (br s, 1 H), 7,84-7,73 (m, 2 H), 7,35 (br s, 1 H), 7,16 (ddd, J = 7,3, 4,9, e 1,2 Hz, 1 H), 6,48-6,28 (m, 3 H), 5,97-5,84 (m, 2 H), 5,63 (dd, J = 15,7 e 5,9 Hz, 1 H), 5,59-5,52 (m, 1 H), 4,40-4,26 (m, 2 H), 4,20-4,04 (m, 2 H), 3,83-3,75 (m, 1 H), 3,69-3,61 (m, 1 H), 3,56-3,49 (m, 1 H), 3,32 (d, J = 4,7 Hz, 1 H), 3,24 (br s, 1 H), 2,79 (d, J = 4,9 Hz, 1 H), 2,65-2,53 (m, 2 H), 2,47-2,38 (m, 1 H), 2,36-2,19 (m, 4 H), 1,97 (s, 3 H), 1,77-1,67 (m, 4 H), 1,66-1,58 (m, 1 H), 1,35-1,26 (m, 6 H), 1,07 (d, J = 6,4 Hz, 3 H), 0,98 (d, J = 7,3 Hz, 3 H).
[01117] Exemplo A#73
[01118] Preparação de N2-acetil-L-lisil-L-valil-N5-carbamoil-N-[4-({[(2-{[(3R,5S,7R,8R)-8-hidroxi-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5- {[(2Z,4S)-4-hidroxipent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2- il]-3-metilpenta-1,3-dien-1-il}-1,6-dioxaspiro[2,5]oct-5-il]acetil}hidrazinil)carbonil]oxi}metil)fenil]-L-ornitinamida, sal de acetato (#B222).
[01119] Etapa 1. Síntese de N2-acetil-N6-(terc-butoxicarbonil)-L- lisina (#B223). A uma mistura de N6-(terc-butoxicarbonil)-L-lisina (22,5 g, 91,5 mmoles, 1 eq.) e K2CO3 (63,1 g, 0,457 mol, 5 eq.) em tetrahi- drofurano/água (200 mL/200 mL) a 0°C, foi adicionado acetil cloreto (8,62 g, 0,109 mol, 1,2 eq.), e a mistura foi agitada à temperatura ambiente por 4 horas. A mistura foi concentrada in vacuo para remover o tetrahidrofurano, e a camada aquosa foi ajustada para pH = 1 com 2 M HCl e extraída com EtOAc (100 mL) três vezes. O extrato foi lavado com salmoura (100 mL), seco sobre sulfato de sódio e concentrado in vacuo para gerar #B223 (23,1 g, 87,7%) como um óleo amarelo.
[01120] Etapa 2. Síntese de N2-acetil-L-lisina sal de cloridrato (#B224). A uma solução de #B223 (23,1 g, 0,080 mmol, 1 eq.) em etil acetato (400 mL) a 0°C, foi adicionado HCl (g) em etil acetato (250 mL) sob nitrogênio. A mistura foi agitada à temperatura ambiente por 4 horas e filtrada. O sólido foi lavado com etil acetato e seco in vacuo para gerar #B224 (18,5 g, >100%) como um sólido branco que foi usado sem purificação adicional.
[01121] Etapa 3. Síntese de N2-acetil-N6-[(9H-fluoren-9- ilmetoxi)carbonil]-L-lisina (#B225). A uma mistura de #B224 (8 g, 35,6 mmoles, 1 eq.) e NaHCO3 (5,99 g, 71,3 mmoles, 2 eq.) em aceto- na/água (80 mL/80 mL) a 0°C, foi adicionada uma solução de Fmoc-Cl (9,41 g, 36,3 mmoles, 1.02 eq.) em acetona (80 mL), e a mistura foi agitada à temperatura ambiente por 2 horas. A mistura foi ajustada para pH = 3-4 com 2 N HCl e extraída com etil acetato (100 mL) três vezes. Os extratos foram lavados com salmoura (100 mL), secos sobre sulfato de sódio e concentrados in vacuo para dar o produto bruto (7 g) como um óleo amarelo. Ao produto bruto, foram adicionados dicloro- metano e terc-butilmetil éter (100 mL), e a suspensão foi agitada por 30 min e, em seguida, filtrada. O bolo de filtro foi seco in vacuo para gerar #B225 (3,25 g, 22,2%) como um sólido branco.
[01122] Etapa 4. Síntese de N2-acetil-N6-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N5-carbamoil-N-[4-(hidroximatil)fenil]-L- ornitinamida (#B226). A uma mistura de #B225 (1,04 g, 2,54 mmoles, 1 eq.) em N,N-dimetilformamida (20 mL) a 0°C, foram adicionados N- metilmorfolina (769 mg, 7,61 mmoles, 3 eq.), 1-etil-3-(3-dimetilaminopropil) carbodiimida-HCl (632 mg, 3,30 mmoles, 1,3 eq.), hidrato de 1-hidroxibenzotriazol (445 mg, 3,30 mmoles, 1,3 eq.) e L- valil-N5-carbamoil-N-[4-(hidroximetil)fenil]-L-ornitinamida (de WO04010957, 1,01 g, 2,66 mmoles, 1,05 eq.) sob nitrogênio, e a mistura foi agitada à temperatura ambiente por 2 horas. A mistura foi vertida em terc-butilmetil éter (300 mL) e filtrada. O sólido foi lavado com diclorometano (50 mL) e água (50 mL) e seco in vacuo para gerar #B226 (1,87 g, 95,6%) como um sólido branco.
[01123] Etapa 5. Síntese de N2-acetil-N6-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N5-carbamoil-N-[4-({[(4-nitrofenoxi)carbonil]oxi}metil)fenil]-L-ornitinamida (#B227). A uma mistura de #B226 (1,87 g, 2,43 mmoles, 1 eq.) e bis-(4-nitrofenil)carbonato (2,21 g, 7,28 mmoles, 3 eq.) em N,N-dimetilformamida (30 mL) a 0°C, foi adicionada N,N-diisopropiletilamina (313 mg, 2,43 mmoles, 1 eq.) sob nitrogênio, e a mistura foi agitada à temperatura ambiente durante a noite. A mistura foi vertida em terc-butilmetiléter (50 mL) e filtrada. O sólido (1.95 g) foi purificado por HPLC preparativa para dar #B227 (580 mg, 25,7%) como um sólido branco. 1H RMN (400Hz, DMSO-d6): □ 10,1 (s, 1 H), 8,29 (d, 2 H), 8,00 (d, 1 H), 7,86 (d, 1 H), 7,65 (d, 2 H), 7,64 (d, 1 H), 7,61 (m, 4 H), 7,40 (m, 2 H), 7,38 (m, 4 H), 7,30 (m, 3 H), 6,01 (br, 1 H), 5,21 (s, 2 H), 4,35 (br, 1 H), 4,27-4,15 (m, 5 H), 2,96 (m, 4 H), 1,98 (m, 1 H), 1,82 (s, 3 H), 1,65 (br, 3 H), 1,43-1,24 (m, 7 H), 0,83 (m, 6 H).
[01124] Etapa 6. Síntese de N2-acetil-N6-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N5-carbamoil-N-[4-({[(2-{[(3R,5S,7R,8R)- 8-hidroxi-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-hidroxipent-2- enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien- 1-il}-1,6-dioxaspiro[2,5]oct-5-il]acetil}hidrazinil)carbonil]oxi}metil)fenil]- L-ornitinamida (#B228). A uma solução de #B209 (8,1 mg, 0,016 mmol, 1 eq.) em N,N-dimetilformamida (0,4 mL) à temperatura ambiente, foram adicionados 2,6-lutidina (7,5 μL, 0,064 mmol, 4 eq.), N,N- diisopropiletilamina (11,3 μL, 0,064 mmol, 4 eq.) e 4-N,N-dimetilamino piridina (2 mg, 0,016 mmol, 1 eq.) seguido por #B227 (17,8 mg, 0,019 mmol, 1,2 eq.), e a reação foi agitada por 5 horas. A reação foi purificada por cromatografia de fase reversa (Método A) para dar #B228 como um sólido branco. Rendimento: 5,5 mg, 0,004 mmol, 26%. LCMS (Protocolo D): m/z 1306,1 [M+H]+, tempo de retenção = 0,81 minuto.
[01125] Etapa 7. Síntese de N2-acetil-L-lisil-L-valil-N5-carbamoil-N- [4-({[(2-{[(3R,5S,7R,8R)-8-hidroxi-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5- {[(2Z,4S)-4-hidroxipent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2- il]-3-metilpenta-1,3-dien-1-il}-1,6-dioxaspiro[2,5]oct-5-il]acetil}hidrazinil)carbonil]oxi}metil)fenil]-L-ornitinamida, sal de acetato (#B222). O composto do título foi preparado em 79% de rendimento a partir de 9,5 mg (0,007 mmol, 1,0 eq.) de #B228 e 11,9 mg (0,14 mmol, 20,0 eq.) de piperidina usando o procedimento descrito para a preparação do composto #B47. LCMS (Protocolo D): m/z 1084,1 [M+H]+, tempo de retenção = 0,58 minuto. 1H RMN (500 MHz, DMSO- d6) δ 10,10 (s, 1 H), 8,22-8,12 (m, 1 H), 8,03 (d, J = 7,8 Hz, 1 H), 7,877,74 (m, 2 H), 7,64-7,53 (m, 2 H), 7,34-7,18 (m, 2 H), 6,31 (d, J = 15,9 Hz, 1 H), 6,09-6,01 (m, 1 H), 5,98 (d, J = 11,8 Hz, 1 H), 5,86 (dd, J = 11,8 e 7,1 Hz, 1 H), 5,66-5,56 (m, 1 H), 5,55-5,49 (m, 1 H), 5,44 (br s, 1 H), 5,23-4,91 (m, 3 H), 4,43-4,33 (m, 1 H), 4,30-4,21 (m, 2 H), 4,20-4,12 (m, 1 H), 3,69-3,59 (m, 1 H), 3,53-3,45 (m, 1 H), 3,07-2,88 (m, 2 H), 2,76-2,71 (m, 1 H), 2,61-2,56 (m, 1 H), 2,35-2,14 (m, 4 H), 2,041,53 (m, 18 H), 1,52-1,18 (m, 10 H), 1,11 (d, J = 6,4 Hz, 3 H), 1,06 (d, J = 6,4 Hz, 3 H), 0,95 (d, J = 7,3 Hz, 3 H), 0,85 (d, J = 6,9 Hz, 3 H), 0,82 (d, J = 6,9 Hz, 3 H).
[01126] Exemplo A#74
[01127] Preparação de metil [(3R,5S,7R,8R)-8-hidroxi-7-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-metoxipent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6- dioxaspiro[2,5]oct-5-il]acetato (#B229).
[01128] Etapa 1. Síntese de metil [(3R,5S,7R,8R)-7-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-{[terc- butil(dimetil)silil]oxi}-1,6-dioxaspiro[2,5]oct-5-il]acetato (#B230). A uma solução do #B55 (66,8 mg, 0,122 mmol, 1 eq.) em diclorometano a 0°C, foram adicionados 2,6-lutidina (71,1 μL, 0,61 mmol, 5 eq.) seguido por terc-butil(cloro)dimetilsilano (86,3 μL, 0,366 mmol, 3 eq.), e a reação foi deixada aquecer para temperatura ambiente. Após 1 hora, a reação foi resfriada para 0°C, resfriada bruscamente com NaHCO3 aquoso, extraída com diclorometano três vezes, seca sobre sulfato de sódio, filtrada e concentrada. O resíduo foi purificado por cromatografia líquida de pressão média de fase reversa eluída com 0,02% de ácido acético em água (v/v) e 0,02% de ácido acético em acetonitrila (v/v) (10% a 100%) para dar #B230 como uma goma. Rendimento: 68 mg, 0,001 mmol, 84%. LCMS (Protocolo D): m/z 686,58 [M+Na]+, tempo de retenção = 1,16 minuto.
[01129] Etapa 2. Síntese de metil [(3R,5S,7R,8R)-8-{[terc-butil(dimetil)silil]oxi}-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- hidroxipent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-1,6-dioxaspiro[2,5]oct-5-il]acetato (#B231). A uma solução #B230 (68 mg, 0,1 mmol, 1 eq.) em metanol (1 mL) à temperatura ambiente, foi adicionado K2CO3 (35,2 mg, 0,255 mmol, 2,5 eq.), e a reação foi deixada agitar por 1 hora. A reação foi filtrada e lavada com etil acetato. A camada orgânica foi lavada com água e salmoura, seca sobre sulfato de sódio e concentrada. O resíduo foi purificado por cromatografia líquida de pressão média de fase reversa eluída com 0,02% de ácido acético em água (v/v) e 0,02% de ácido acético em acetonitrila (v/v) (10% a 100%) para dar #B231 como um sólido branco. Rendimento: 33,2 mg, 0,053 mmol, 52%. LCMS (Protocolo D): m/z 622,55 [M+H]+, tempo de retenção = 1,09 minuto.
[01130] Etapa 3. Síntese de metil [(3R,5S,7R,8R)-8-{[terc- butil(dimetil)silil]oxi}-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- metoxipent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-1,6-dioxaspiro[2,5]oct-5-il]acetato (#B232). A uma solução de #B231 (24,7 mg, 0,04 mmol, 1 eq.) em N,N- dimetilformamida (0,5 mL) à temperatura ambiente, foi adicionado MeI (37,5 μL, 0,6 mmol, 15 eq.) e Ag2O (55,6 mg, 0,24 mmol, 6 eq.), e a reação foi deixada agitar por 23 horas no escuro. Mais MeI (38 μL, 0,6 mmol, 15 eq.) e Ag2O (55 mg, 0,24 mmol, 6 eq.) foi adicionado, e a reação foi agitada por um adicional de 25 horas. A reação foi filtrada sobre celite e purificada por cromatografia de fase reversa (Método A) para dar #B232 como um sólido branco. Rendimento: 9,4 mg, 0,015 mmol, 37%. LCMS (Protocolo D): m/z 636,7 [M+H]+, tempo de retenção = 1,19 minuto.
[01131] Etapa 4. Síntese de metil [(3R,5S,7R,8R)-8-hidroxi-7- {(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-metoxipent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6- dioxaspiro[2,5]oct-5-il]acetato (#B229). A uma solução de #B232 (12,6 mg, 0,02 mmol, 1 eq.) em tetrahidrofurano (0,4 mL) a 0°C, foi adicionado fluoreto de tetrabutilamônio (1 M em tetrahidrofurano 20,7 μL, 0,02 mmol, 1 eq.), e a reação foi deixada aquecer para temperatura ambiente e agitar por 1 hora. Mais fluoreto de tetrabutilamônio (1 M em tetrahidrofurano 10,3 uL, 0,01 mmol, 0,5 eq) foi adicionado, e a reação foi agitada por 45 minutos. A reação foi concentrada, retomada em DMSO, e purificada por cromatografia de fase reversa (Método A) para dar #B229 como um sólido branco. Rendimento: 4,9 mg, 0,01 mmol, 47%. LCMS (Protocolo D): m/z 522,50 [M+H]+, tempo de retenção = 0,79 minuto. 1H RMN (500 MHz, DMSO-d6) δ 7,75 (d, J = 8,0 Hz, 1 H), 6,28 (d, J = 15,8 Hz, 1 H), 6,16 (d, J = 11,7 Hz, 1 H), 5,75 (dd, J = 11,7 e 8,1 Hz, 1 H), 5,58 (dd, J = 15,8 e 5,1 Hz, 1 H), 5,55-5,47 (m, 1 H), 5,10-4,99 (m, 2 H), 4,31-4,21 (m, 2 H), 3,69-3,62 (m, 2 H), 3,60 (s, 3H), 3,54-3,47 (m, 1 H), 3,28-3,22 (m, 1 H), 3,14 (s, 3 H), 2,76 (d, J =5,1 Hz, 1 H), 2,69-2,55 (m, 3 H), 2,35-2,14 (m, 2 H), 1,90-1,75 (m, 3H), 1,73-1,60 (m, 4 H), 1,57-1,48 (m, 1 H), 1,12 (d, J = 6,4 Hz, 3 H),1,07 (d, J = 6,4 Hz, 3 H), 0,95 (d, J = 7,3 Hz, 3 H).
[01132] Exemplo A#75
[01133] Preparação de N2-acetil-L-lisil-L-valil-N-[4-({[(2-{[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5- il]acetil}hidrazinil)carbonil]oxi}metil)fenil]-N5-carbamoil-L-ornitinamida, sal de acetato (#B233).
[01134] Etapa 1. Síntese de N2-acetil-N6-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N-[4-({[(2-{[(3R,5S,7R,8R)-7-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]acetil}hidrazinil)carbonil]oxi}metil)fenil]-N5- carbamoil-L-ornitinamida (#B234). A uma solução de #B6 (20,5 mg, 0,037 mmol, 1 eq.) em N,N-dimetilformamida (0,8 mL) à temperatura ambiente, foram adicionados 2,6-lutidina (17,3 μL, 0,148 mmol, 4 eq.), N,N-diisopropiletilamina (26 μL, 0,148 mmol, 4 eq.) e 4-N,N- dimetilamino piridina (4,5 mg, 0,037 mmol, 1 eq.) seguido por #B227 (45 mg, 0,048 mmol, 1,3 eq.), e a reação foi agitada por 4 horas. A reação foi purificada por cromatografia de fase reversa (Método A) para dar #B234 como um sólido branco. Rendimento: 18,5 mg, 0,014 mmol, 37%. LCMS (Protocolo D): m/z 1348,1 [M+H]+, tempo de retenção = 0,88 minuto.
[01135] Etapa 2. Síntese de N2-acetil-L-lisil-L-valil-N-[4-({[(2-{[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5- il]acetil}hidrazinil)carbonil]oxi}metil)fenil]-N5-carbamoil-L-ornitinamida, sal de acetato (#B233). A uma solução de #B234 (18,5 mg, 0,014 mmol, 1 eq.) em N,N-dimetilformamida (0,7 mL) à temperatura ambiente, foi adicionada piperidina (27,6 μL, 0,28 mmol, 20 eq.), e a reação foi agitada por 20 minutos. A reação foi purificada por cromatografia de fase reversa (Método A) para dar um sólido branco que foi adicionalmente purificado por cromatografia de fase reversa (Método C, coluna Phenomenex Luna PFP(2)) para dar #B233 como um sólido branco. Rendimento: 8 mg, 0,07 mmol, 50%. LCMS (Protocolo D): m/z 1125,91 [M+H]+, tempo de retenção = 0,63 minuto. 1H RMN (500 MHz, DMSO- d6) δ 10,11 (s, 1 H), 8,43 (s, 1 H), 8,19-8,11 (m, 1 H), 8,05 (d, J = 8,1 Hz, 1 H), 7,86-7,76 (m, 2 H), 7,64-7,53 (m, 2 H), 7,34-7,18 (m, 2 H), 6,42-6,27 (m, 2 H), 6,16-6,04 (m, 2 H), 5,86 (dd, J = 11,5 e 7,3 Hz, 1 H), 5,66-5,38 (m, 3 H), 5,12-4,89 (m, 3 H), 4,43-4,33 (m, 1 H), 4,324,22 (m, 2 H), 4,20-4,14 (m, 1 H), 3,68-3,59 (m, 1 H), 3,54-3,45 (m, 1 H), 3,07-2,86 (m, 2 H), 2,79-2,72 (m, 1 H), 2,71-2,65 (m, 1 H), 2,612,55 (m, 1 H), 2,34-2,14 (m, 4 H), 2,04-1,94 (m, 4 H), 1,92-1,75 (m, 7 H), 1,74-1,54 (m, 8 H), 1,53-1,19 (m, 12 H), 1,06 (d, J = 6,4 Hz, 3 H), 0,94 (d, J = 7,1 Hz, 3 H), 0,86 (d, J = 6,8 Hz, 3 H), 0,82 (d, J = 6,8 Hz, 3 H).
[01136] Exemplo A#76
[01137] Preparação de metil [(3R,5S,7R,8R)-8-hidroxi-7-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-hidroxipent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6- dioxaspiro[2,5]oct-5-il]acetato (#B235).
[01138] Etapa 1. Síntese de metil [(3R,5S,7R,8R)-8-hidroxi-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-hidroxipent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6- dioxaspiro[2,5]oct-5-il]acetato (#B235). A uma solução de #B55 (60 mg, 0,11 mmol, 1 eq.) em metanol (1 mL) à temperatura ambiente, foi adicionado K2CO3 (37,7 mg, 0,273 mmol, 2,5 eq.), e a reação foi deixada agitar por 1 hora. A reação foi filtrada e lavada com etil acetato. A camada orgânica foi lavada com água e salmoura, seca sobre sulfato de sódio e concentrada. O resíduo foi purificado por cromatografia de fase reversa (Método A) para dar #B235 como um sólido branco. Rendimento: 31,2 mg, 0,06 mmol, 56%. LCMS (Protocolo D): m/z 530,43 [M+Na]+, tempo de retenção = 0,72 minuto. 1H RMN (500 MHz, DMSO- d6) δ 7,78 (d, J = 7,6 Hz, 1 H), 6,28 (d, J = 16,0 Hz, 1 H), 5,98 (d, J = 11,8 Hz, 1 H), 5,87 (dd, J = 11,8 e 7,6 Hz, 1 H), 5,58 (dd, J = 16,0 e 5,2 Hz, 1 H), 5,55-5,49 (m, 1 H), 5,23-5,14 (m, 1 H), 5,10 (d, J = 4,7 Hz, 1 H), 5,02 (d, J = 6,1 Hz, 1 H), 4,31-4,22 (m, 2 H), 3,69-3,62 (m, 2 H), 3,60 (s, 3 H), 3,54-3,47 (m, 1 H), 3,28-3,22 (m, 1 H), 2,76 (d, J = 5,1 Hz, 1 H), 2,69-2,55 (m, 3 H), 2,35-2,15 (m, 2 H), 1,90-1,73 (m, 3 H), 1,73-1,61 (m, 4 H), 1,57-1,49 (m, 1 H), 1,11 (d, J = 6,5 Hz, 3 H), 1,06 (d, J = 6,2 Hz, 3 H), 0,96 (d, J = 7,5 Hz, 3 H).
[01139] Exemplo A#77
[01140] Preparação de (2R)-2-(piridin-2-ildissulfanil)propil-[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5-il]acetato (#B236).
[01141] Etapa 1. Síntese de (2R)-2-(piridin-2-ildissulfanil)propil- [(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5-il]acetato (#B236). A uma solução de #NP1 (10,4 mg, 0,019 mmol, 1 eq.) e #B220 (11,5 mg, 0,057 mmol, 3 eq.) em diclorometano (0,3 mL) à temperatura ambiente, foi adicionado 4-N,N-dimetilamino piridina (2,3 mg, 0,019 mmol, 1 eq.) e N,N-di-/so-propilcarbodiimida (8,9 μL, 0,057 mmol, 3 eq.), e a reação foi deixada agitar por 75 minutos. A reação foi concentrada, retomada em DMSO, e purificada por cromatografia de fase reversa (Método A) para dar #B236 como um sólido branco. Rendimento: 7,6 mg, 0,011 mmol, 55%. LCMS (Protocolo D): m/z 719,58 [M+H]+, tempo de retenção = 0,94 minuto. 1H RMN (500 MHz, DMSO- d6) δ 8,46-8,40 (m, 1 H), 7,86-7,74 (m, 3 H), 7,27-7,20 (m, 1 H), 6,41-6,32 (m, 1 H), 6,27 (d, J = 16,1 Hz, 1 H), 6,10 (dd, J = 11,7 e 1,5 Hz, 1 H), 5,87 (dd, J = 11,7 e 7,6 Hz, 1 H), 5,61 (dd, J = 16,1 e 5,9 Hz, 1 H), 5,52-5,45 (m, 1 H), 5,02 (d, J = 6,1 Hz, 1 H), 4,31-4,20 (m, 2 H), 4,184,06 (m, 2 H), 3,68-3,58 (m, 2 H), 3,52-3,44 (m, 1 H), 3,28-3,23 (m, 1 H), 2,76 (d, J = 4,9 Hz, 1 H), 2,70 (dd, J = 15,2 e 9,3 Hz, 1 H), 2,622,53 (m, 2 H), 2,34-2,14 (m, 2 H), 1,98 (s, 3 H), 1,86-1,72 (m, 4 H), 1,70-1,59 (m, 4 H), 1,29-1,21 (m, 6 H), 1,06 (d, J = 6,4 Hz, 3 H), 0,94 (d, J = 7,3 Hz, 3 H).
[01142] Exemplo A#78
[01143] Preparação de N-{6-[(bromoacetil)amino]hexanoil}-L-valil-N-{4-[({[({[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5- il]acetil}amino)metil]carbamoil}oxi)metil]fenil}-N5-carbamoil-L- ornitinamida (#B237).
[01144] Etapa 1. Síntese de N-(6-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}hexanoil)-L-valil-N-{4-[({[({[(3R,5S,7R,8R)-7- {(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi- 1,6-dioxaspiro[2,5]oct-5-il]acetil}amino)metil]carbamoil}oxi)metil]fenil}- N5-carbamoil-L-ornitinamida (#B238). A uma solução de #NP1 (20,4 mg, 0,038 mmol, 1 eq.) em N,N-dimetilformamida (0,4 mL) à temperatura ambiente, foram adicionados N,N-diisopropiletilamina (40,2 μL, 0,228 mmol, 6 eq.) e hexafluorofosfato de O-(7-azabenzotriazol-1-il)- N,N,N‘,N‘-tetrametilurônio (19 mg, 0,049 mmol, 1,3 eq.) seguido por uma solução de #B182 (34,2 mg, 0,038 mmol, 1 eq.) em N,N- dimetilformamida (0,7 mL), e a reação foi deixada agitar por 45 min. A reação foi purificada por cromatografia de fase reversa (Método A) pa- ra dar #B238 como um sólido branco. Rendimento: 16,1 mg, 0,012 mmol, 33%. LCMS (Protocolo D): m/z 1305,3 [M+H]+, tempo de retenção = 0,92 minuto.
[01145] Etapa 2. Síntese de N-(6-aminohexanoil)-L-valil-N-{4- [({[({[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5- il]acetil}amino)metil]carbamoil}oxi)metil]fenil}-N5-carbamoil-L-ornitinamida (#B239). O composto do título foi preparado em 88% de rendimento a partir de 16,1 mg (0,012 mmol, 1,0 eq.) de #B238 e 20,4 mg (0,24 mmol, 20,0 eq.) de piperidina usando o procedimento descrito para a preparação do composto #B47. LCMS (Protocolo D): m/z 1083,1 [M+H]+, tempo de retenção = 0,67 minuto.
[01146] Etapa 3. Síntese de N-{6-[(bromoacetil)amino]hexanoil}-L- valil-N-{4-[({[({[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)- 4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5-il]acetil}amino)metil]carbamoil}oxi)metil]fenil}-N5-carbamoil-L-ornitinamida (#B237). O composto do título foi preparado em 62% de rendimento a partir de 11,5 mg (0,011 mmol) de #B239, 4 mg (0,017 mmol, 1,5 eq) de 1-[(bromoacetil)oxi]pirrolidina-2,5-diona e 5,7 mg (0,044 mmol, 4,0 eq) de N,N-diisopropiletilamina usando o procedimento descrito para a preparação do composto #B150. LCMS (Protocolo D): m/z 1203,2 [M+H]+, tempo de retenção = 0,77 minuto. 1H RMN (500 MHz, DMSO-d6) δ 9,99 (s, 1 H), 8,45-8,37 (m, 1 H), 8,28-8,20 (m, 1 H), 8,15-8,05 (m, 1 H), 7,86-7,73 (m, 2 H), 7,62-7,54 (m, 2 H), 7,317,22 (m, 2 H), 6,41-6,33 (m, 1 H), 6,30 (d, J = 15,9 Hz, 1 H), 6,11 (dd, J = 11,6 e 1,5 Hz, 1 H), 6,02-5,94 (m, 1 H), 5,86 (dd, J = 11,6 e 7,6 Hz, 1 H), 5,60 (dd, J = 15,9 e 5,6 Hz, 1 H), 5,56-5,48 (m, 1 H), 5,41 (s, 2 H), 5,04 (d, J = 5,4 Hz, 1 H), 4,95 (s, 2 H), 4,43-4,15 (m, 5 H), 3,81 (s, 2 H), 3,69-3,60 (m, 2 H), 3,53-3,45 (m, 1 H), 3,25-3,18 (m, 1 H), 3,092,88 (m, 4 H), 2,73 (d, J = 5,0 Hz, 1 H), 2,57 (d, J = 5,0 Hz, 1 H), 2,342,08 (m, 5 H), 2,03-1,91 (m, 4 H), 1,91-1,74 (m, 4 H), 1,73-1,30 (m, 12 H), 1,29-1,18 (m, 4 H), 1,06 (d, J = 6,4 Hz, 3 H), 0,94 (d, J = 7,3 Hz, 3 H), 0,86 (d, J = 6,6 Hz, 3 H), 0,83 (d, J = 6,9 Hz, 3 H).
[01147] Exemplo A#79
[01148] Preparação de metil [(3R,5S,7R,8R)-8-metoxi-7-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-metoxipent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6- dioxaspiro[2,5]oct-5-il]acetato (#B240).
[01149] Etapa 1. Síntese de metil [(3R,5S,7R,8R)-8-metoxi-7- {(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-metoxipent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6-dioxaspiro[2,5]oct-5-il]acetato (#B240). A uma solução de #B235 (24,2 mg, 0,048 mmol, 1 eq.) em N,N-dimetilformamida (0,5 mL) à temperatura ambiente, foi adicionado Mel (45 μL, 0,7 mmol, 15 eq.) e Ag2O (66,7 mg, 0,29 mmol, 6 eq.), e a reação foi deixada agitar por 23 horas no escuro. Mais MeI (45 μL, 0,7 mmol, 15 eq.) e Ag2O (67 mg, 0,29 mmol, 6 eq.) foi adicionado, e a reação foi agitada por mais 24 horas. A reação foi filtrada sobre celite e purificada por cromatografia de fase reversa (Método A) para dar #B240 como um sólido branco. Rendimento: 12,2 mg, 0,023 mmol, 48%. LCMS (Protocolo D): m/z 536,7 [M+H]+, tempo de retenção = 0,90 minuto. 1H RMN (500 MHz, DMSO- d6) δ 7,75 (d, J = 8,0 Hz, 1 H), 6,36 (d, J = 15,8 Hz, 1 H), 6,16 (d, J = 11,7 Hz, 1 H), 5,75 (dd, J = 11,7 e 8,1 Hz, 1 H), 5,62-5,50 (m, 2 H), 5,10-4,99 (m, 1 H), 4,58-4,51 (m, 1 H), 4,28-4,18 (m, 1 H), 3,70-3,62 (m, 2 H), 3,60 (s, 3 H), 3,55-3,47 (m, 1 H), 3,32 (s, 3 H), 3,14 (s, 3 H), 2,96-2,91 (m, 1 H), 2,70-2,63 (m, 2 H), 2,58-2,52 (m, 1 H), 2,35-2,16 (m, 2 H), 2,06-1,97 (m, 1 H), 1,88-1,75 (m, 2 H), 1,73-1,60 (m, 4 H), 1,18-1,09 (m, 4 H), 1,07 (d, J = 6,4 Hz, 3 H), 0,96 (d, J = 7,3 Hz, 3 H).
[01150] Exemplo A#80
[01151] Preparação de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5-{(3R,4R,5R,7S)-7-[(carbamoilamino)metil]-4-hidroxi-1,6-dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B241). e (2S,3Z)-5- {[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,4R,5R,7S)-4-hidroxi-7-{[(propilcarbamoil)amino]metil}-1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta- 2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en- 2-il acetato (#B242).
[01152] Etapa 1. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-4-hidroxi-7-(isocianatomoetil)-1,6-dioxaspiro[2,5]oct- 5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B243). A uma solução de #NP1 (25,6 mg, 0,048 mmol, 1 eq.) em diclorometano (1 mL) à temperatura ambiente, foi adicionada trietilamina (7,3 mg, 0,072 mmol, 1,5 eq.) seguido por difenilfosforil azida (11,7 μL, 0,053 mmol, 1,1 eq.), e a reação foi deixada agitar por 20 h. A reação foi diluída com diclorometano, lavada com 5% NaHCO3 (aq.) três vezes, seca sobre sulfato de sódio e concentrada in vacuo para dar um óleo amarelo. O óleo foi dissolvido acetonitrila (1 mL) e aquecido para 50°C por 1 hora. A reação foi resfriada para dar #B243 como uma solução em acetonitrila que foi usada sem purificação adicional. Conversão total assumida. LCMS (Protocolo D): m/z 533,6 [M+H]+, tempo de retenção = 0,88 minuto.
[01153] Etapa 2. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3R,4R,5R,7S)-7-[(carbamoilamino)metil]-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B241). A uma solução de #B243 (12,8 mg, 0,024 mmol, 1 eq.) em acetonitrila (0,5 mL) à temperatura ambiente, foi adicionado NH3 (7 M em metanol, 34,3 μL, 0,24 mmol, 10 eq.), e a reação foi deixada agitar por 30 minutos. A reação foi concentrada, diluída com DMSO e purificada por cromatogra- fia de fase reversa (Método A) para dar #B241 como um sólido branco. Rendimento: 6,7 mg, 0,012 mmol, 51%. LCMS (Protocolo D): m/z 550,6 [M+H]+, tempo de retenção = 0,72 minuto. 1H RMN (400 MHz, DMSO-d6) δ 7,79 (d, J = 8,2 Hz, 1 H), 6,41-6,25 (m, 2 H), 6,11 (d, J = 11,7 Hz, 1 H), 6,02-5,94 (m, 1 H), 5,87 (dd, J = 11,7 e 7,4 Hz, 1 H), 5,64 (dd, J = 16,0 e 5,9 Hz, 1 H), 5,57-5,50 (m, 1 H), 5,46 (br s, 1 H), 5,01 (d, J = 5,9 Hz, 1 H), 4,32-4,23 (m, 1 H), 3,88-3,77 (m, 1 H), 3,703,60 (m, 2 H), 3,55-3,46 (m, 1 H), 3,25-3,04 (m, 3 H), 2,75 (d, J = 5,1 Hz, 1 H), 2,60 (d, J = 5,1 Hz, 1 H), 2,35-2,13 (m, 2 H), 1,98 (s, 3 H), 1,88-1,59 (m, 8 H), 1,46-1,37 (m, 1 H), 1,25 (d, J = 6,2 Hz, 3 H), 1,07 (d, J = 6,2 Hz, 3 H), 0,95 (d, J = 7,0 Hz, 3 H).
[01154] Etapa 3. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-4-hidroxi-7-{[(propilcarbamoil)amino]metil}-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B242). A uma solução de #B243 (9 mg, 0,02 mmol, 1 eq.) em acetonitrila (0,4 mL) à temperatura ambiente, foi adicionada n-propilamina (7 μL, 0,085 mmol, 5 eq.), e a reação foi agitada por 10 minutos. A reação foi diluída com DMSO (0,7 mL), concentrada in vacuo e purificada por cromatografia de fase reversa (Método A) para dar #B242 como um sólido branco. Rendimento: 8 mg, 0,014 mmol, 80%. LCMS (Protocolo D): m/z 592,7 [M+H]+, tempo de retenção = 0,80 minuto. 1H RMN (500 MHz, DMSO- de) δ 07,80 (DODd, J = 8,1 Hz, 1 H), 6,41-6,32 (m, 1 H), 6,28 (d, J = 16,0 Hz, 1 H), 6,11 (d, J = 11,7 Hz, 1 H), 6,00-5,93 (m, 1 H), 5,91-5,81 (m, 2 H), 5,62 (dd, J = 16,0 e 5,6 Hz, 1 H), 5,54-5,46 (m, 1 H), 5,02 (d, J = 5,6 Hz, 1 H), 4,31-4,25 (m, 1 H), 3,86-3,77 (m, 1 H), 3,69-3,59 (m, 2 H), 3,53-3,45 (m, 1 H), 3,26-3,08 (m, 3 H), 2,97-2,88 (m, 2 H), 2,75 (d, J = 5,1 Hz, 1 H), 2,60 (d, J = 5,1 Hz, 1 H), 2,35-2,15 (m, 2 H), 1,98 (s, 3 H), 1,88-1,75 (m, 3 H), 1,73-1,60 (m, 4 H), 1,44-1,30 (m, 3 H), 1,25 (d, J = 6,6 Hz, 3 H), 1,07 (d, J = 6,4 Hz, 3 H), 0,95 (d, J = 7,3 Hz, 3 H), 0,82 (app t, J = 7,3 Hz, 3 H).
[01155] Exemplo A#81
[01156] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5- [(3R,4R,5R,7S)-4-hidroxi-7-{[({[(2R)-2-(piridin-2- ildissulfanil)propil]oxi}carbonil)amino]metil}-1,6-dioxaspiro[2,5]oct-5-il]- 3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5- oxopent-3-en-2-il acetato (#B244).
[01157] Etapa 1. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-4-hidroxi-7-{[({[(2R)-2-(piridin-2-ildissulfanil)propil]oxi}carbonil)amino]metil}-1,6-dioxaspiro[2,5]oct-5-il]- 3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5- oxopent-3-en-2-il acetato (#B244). A uma solução de #B147 (8,2 mg, 0,014 mmol, 1 eq.) em diclorometano (0,4 mL) à temperatura ambiente, foi adicionada trietilamina (12,3 μL, 0,088 mmol, 6,3 eq.) seguido por #B221 (9,4 mg, 0,026 mmol, 1,9 eq.) em diclorometano (0,3 mL), e a reação foi agitada por 30 min. 4-N,N-dimetilamino piridina (1 mg, 0,008 mmol, 0,6 eq.) foi adicionada, e a reação foi deixada agitar por 2 horas. A reação foi concentrada, retomada em DMSO (800 uL) e purificada por cromatografia de fase reversa (Método A) para dar #B244 como um sólido branco. Rendimento: 4 mg, 0,005 mmol, 40%. LCMS (Protocolo D): m/z 734,33 [M+H]+, tempo de retenção = 0,91 minuto. 1H RMN (500 MHz, DMSO-d6) δ 8,46-8,40 (m, 1 H), 7,85-7,76 (m, 2 H), 7,36-7,29 (m, 1 H), 7,26-7,20 (m, 1 H), 6,41-6,32 (m, 1 H), 6,25 (d, J = 15,8 Hz, 1 H), 6,11 (d, J = 11,6 Hz, 1 H), 5,87 (dd, J = 11,6 e 7,6 Hz, 1 H), 5,61 (dd, J = 15,8 e 6,0 Hz, 1 H), 5,50-5,43 (m, 1 H), 4,98 (d, J = 6,2 Hz, 1 H), 4,29-4,22 (m, 1 H), 4,10-4,03 (m, 1 H), 4,01-3,85 (m, 2 H), 3,67-3,57 (m, 2 H), 3,52-3,44 (m, 1 H), 3,28-3,21 (m, 1 H), 3,022,93 (m, 1 H), 2,76 (d, J = 5,1 Hz, 1 H), 2,57 (d, J = 5,1 Hz, 1 H), 2,342,13 (m, 2 H), 1,98 (s, 3 H), 1,85-1,53 (m, 9 H), 1,28-1,20 (m, 6 H), 1,05 (d, J = 6,2 Hz, 3 H), 0,93 (d, J = 7,3 Hz, 3 H).
[01158] Exemplo A#82
[01159] Preparação de N-(24-bromo-23-oxo-4,7,10,13,16,19-hexaoxa-22-azatetracosan-1-oil)-L-valil-N-{4-[({[2-({[(3R,5S,7R,8R)-7- {(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi- 1,6-dioxaspiro[2,5]oct-5-il]acetil}amino)etil]carbamoil}oxi)metil]fenil}-N5- carbamoil-L-ornitinamida (#B245).
[01160] Etapa 1. Síntese de terc-butil 1-hidroxi-3,6,9,12,15,18- hexaoxahenicosan-21-oato (#B246). Uma mistura de 3,6,9,12,15- pentaoxaheptadecano-1,17-diol (25 g, 88,7 mmoles, 1 eq.), terc-butil prop-2-enoato (11,3 g, 88,7 mmoles, 1 eq.) e hidróxido de benziltrimeti- lamônio (2,5 mL) foi agitada a 50°C durante a noite. A mistura de rea- ção foi purificada por cromatografia de sílica gel eluindo com etil aceta- to:diclorometano (4%~10%) para gerar #B246 (9,63 g, 25,7%) como um óleo amarelo.
[01161] Etapa 2. Síntese de terc-butil 1-{[(4-metilfenil)sulfonil]oxi}- 3,6,9,12,15,18-hexaoxahenicosan-21-oato (#B247). A uma solução de #B246 (9,63 g, 23,5 mmoles, 1 eq.) e trietilamina (3,56 g, 35,2 mmoles, 1,5 eq.) em diclorometano (150 mL) foi adicionado cloreto 4- metilbenzenossulfonil (6,69 g, 35,2 mmoles, 1,5 eq.) a 0°C, e a solução foi agitada à temperatura ambiente durante a noite. A mistura de reação foi lavada com NaHCO3 aquoso (150 mL), e a fase aquosa foi re-extraída com etil acetato (200 mL x 3). As camadas orgânicas combinadas foram secas sobre sulfato de sódio e concentradas in vacuo, e o resíduo foi purificado por cromatografia de coluna de sílica eluindo com metanol:diclorometano (0,5%~0.8%) para gerar #B247 (9,21 g, 69,7%) como um óleo amarelo.
[01162] Etapa 3. Síntese de terc-butil 1-azido-3,6,9,12,15,18- hexaoxahenicosan-21-oato (#B248). A uma solução de #B247 (13,0 g, 23,0 mmoles, 1 eq.) em acetona/água (150 mL/150 mL), foi adicionada azida de sódio (3,20 g, 49,2 mmoles, 2,1 eq.) e iodeto de sódio (621 mg, 3,45 mmoles, 0,15 eq.), e a reação foi agitada a refluxo durante a noite. A mistura de reação foi extraída com etil acetato (150 mL x 3), e as fases orgânicas foram concentradas in vacuo. O resíduo foi purificado por cromatografia de coluna de sílica eluindo com etil aceta- to:éter de petróleo (12-35%) para gerar #B248 (8,30 g, 83,1%) como um óleo amarelo.
[01163] Etapa 4. Síntese de terc-butil 1-amino-3,6,9,12,15,18- hexaoxahenicosan-21-oato (#B249). Uma suspensão de #B248 (8,30 g, 19,1 mmoles, 1 eq.) e Pd/C (1,0 g) em metanol foi agitada sob balão de hidrogênio à temperatura ambiente durante a noite. A mistura de reação foi filtrada, e o filtrado foi concentrado in vacuo para gerar #B249 (7,80 g, 100%) como um óleo amarelo, que foi diretamente usado na etapa seguinte.
[01164] Etapa 5. Síntese de terc-butil 1-bromo-2-oxo- 6,9,12,15,18,21-hexaoxa-3-azatetracosan-24-oato (#B250). A uma solução de #B249 (5,80 g, 14,1 mmoles, 1 eq.) em diclorometano (300 mL) foram adicionados N,N-diisopropiletilamina (5,50 g, 42,6 mmoles, 3 eq.) e brometo de bromoacetil (4,24 g, 21,3 mmoles, 1,5 eq.) a 0°C, e a reação foi agitada a 0°C por 15 minutos. A mistura de reação foi concentrada até secagem, e o resíduo foi purificado por cromatografia de coluna de sílica eluindo com metanol:diclorometano (0,5-0,8%) para gerar #B250 (5,20 g, 69,3%) como um sólido amarelo.
[01165] Etapa 6. Síntese de ácido 1-bromo-2-oxo-6,9,12,15,18,21- hexaoxa-3-azatetracosan-24-óico (#B251). A uma solução de #B250 (5,20 g, 9,80 mmoles, 1 eq.) em diclorometano (100 mL), foi adicionado ácido trifluoroacético (100 mL) a 0°C, e a solução foi agitada à temperatura ambiente por 3 horas. A mistura de reação foi concentrada in vacuo para gerar #B251 (6,00 g, 100%) como um óleo amarelo.
[01166] Etapa 7. Síntese de pentafluorofenil 1-bromo-2-oxo- 6,9,12,15,18,21-hexaoxa-3-azatetracosan-24-oato (#B252). A uma solução de #B251 (4,65 g, 9,80 mmoles, 1 eq.) e pentafluorofenil trifluo- roacetato (4,12 g, 14,7 mmoles, 1,5 eq.) em diclorometano (150 mL), foi adicionada, em gotas, piridina (4,65 g, 9,80 mmoles, 1,5 eq.) a 0°C, e a solução foi agitada por 30 minutos. A mistura de reação foi lavada com 2 M HCl (150 mL x 2), e a fase aquosa foi extraída com dicloro- metano (150 mL x 2). As camadas orgânicas combinadas foram secas sobre sulfato de sódio e concentradas in vacuo, e o resíduo foi purificado por cromatografia de coluna de sílica eluindo com meta- nol:diclorometano (1,5-2%) para gerar um óleo amarelo, que foi adici-onalmente purificado por HPLC preparativa para gerar #B252 (1,20 g, 19.1%) como um óleo amarelo. 1H RMN (400 MHz, CDCI3) □ 7,06 (br, 1 H), 3,89 (m, 4 H), 3,69-3,59 (m, 22 H), 3,58 (m, 2 H), 2,96 (m, 2 H).
[01167] Etapa 8. Síntese de N-[(9H-fluoren-9-ilmetoxi)carbonil]-L- valil-N-{4-[({[2-({[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5- {[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5- il]acetil}amino)etil]carbamoil}oxi)metil]fenil}-N5-carbamoil-L-ornitinamida (#B253). A uma solução de #B51 (18,5 mg, 0,032 mmol, 1 eq.) em N,N-dimetilformamida (0,8 mL) à temperatura ambiente, foi adicionada N,N-diisopropiletilamina (22,5 μL, 0,128 mmol, 4 eq.) seguido por N- [(9H-fluoren-9-ilmetoxi)carbonil]-L-valil-N5-carbamoil-N-[4-({[(4- nitrofenoxi)carbonil]oxi}metil)fenil]-L-ornitinamida (29,1 mg, 0,038 mmol, 1,2 eq.), e a reação foi agitada por 70 minutos. Mais N-[(9H- fluoren-9-ilmetoxi)carbonil]-L-valil-N5-carbamoil-N-[4-({[(4-nitrofenoxi)carbonil]oxi}metil)fenil]-L-ornitinamida (4,9 mg, 0,006 mmol, 0,2 eq.) foi adicionado, e a reação foi agitada por mais 30 minutos. A reação foi purificada por cromatografia de fase reversa (Método A) para dar #B253 como um sólido branco. Rendimento: 13,1 mg, 0,011 mmol, 34%. LCMS (Protocolo D): m/z 1206,2 [M+H]+, tempo de retenção = 0,91 minuto.
[01168] Etapa 9. Síntese de L-valil-N-{4-[({[2-({[(3R,5S,7R,8R)-7- {(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi- 1,6-dioxaspiro[2,5]oct-5-il]acetil}amino)etil]carbamoil}oxi)metil]fenil}-N5- carbamoil-L-ornitinamida (#B254). O composto do título foi preparado em 76% de rendimento a partir de 13,1 mg (0,011 mmol, 1,0 eq.) de #B253 e 18,7 mg (0,22 mmol, 20,0 eq.) de piperidina usando o procedimento descrito para a preparação do composto #B47. LCMS (Protocolo D): m/z 984,0 [M+H]+, tempo de retenção = 0,67 minuto.
[01169] Etapa 10. Síntese de N-(24-bromo-23-oxo-4,7,10,13,16,19- hexaoxa-22-azatetracosan-1-oil)-L-valil-N-{4-[({[2-({[(3R,5S,7R,8R)-7- {(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi- 1,6-dioxaspiro[2,5]oct-5-il]acetil}amino)etil]carbamoil}oxi)metil]fenil}-N5- carbamoil-L-ornitinamida (#B245). A uma solução de #B254 (8,2 mg, 0,008 mmol, 1 eq.) em N,N-dimetilformamida (0,15 mL) à temperatura ambiente, foi adicionada N,N-diisopropiletilamina (5,7 μL, 0,032 mmol, 4 eq.) seguido por #B252 (7,5 mg, 0,012 mmol, 1,5 eq.) em N,N- dimetilformamida (0,3 mL), e a reação foi deixada agitar à temperatura ambiente por 30 minutos. A reação foi purificada por cromatografia de fase reversa (Método A) para dar #B245 como um sólido branco. Rendimento: 5,4 mg, 0,0038 mmol, 47%. LCMS (Protocolo D): m/z 1440,72 [M+H]+, tempo de retenção = 0,75 minuto. 1H RMN (500 MHz, DMSO-d6) δ 9,98 (s, 1 H), 8,38-8,28 (m, 1 H), 8,17-8,07 (m, 1 H), 7,967,90 (m, 1 H), 7,90-7,84 (m, 1 H), 7,83-7,76 (m, 1 H), 7,64-7,54 (m, 2 H), 7,32-7,23 (m, 2 H), 7,21-7,12 (m, 1 H), 6,41-6,32 (m, 1 H), 6,28 (d, J = 15,8 Hz, 1 H), 6,11 (dd, J = 11,7 e 1,2 Hz, 1 H), 6,02-5,94 (m, 1 H), 5,87 (dd, J = 11,7 e 7,6 Hz, 1 H), 5,60 (dd, J = 15,8 e 5,6 Hz, 1 H), 5,56-5,46 (m, 1 H), 5,41 (s, 2 H), 5,03 (d, J = 5,6 Hz, 1 H), 4,93 (s, 2 H), 4,41-4,34 (m, 1 H), 4,29-4,18 (m, 2 H), 3,85 (s, 2 H), 3,69-3,55 (m, 4 H), 3,54-3,45 (m, 22 H), 3,43-3,39 (m, 2 H), 3,27-3,19 (m, 2 H), 3,162,89 (m, 6 H), 2,74 (d, J = 5,2 Hz, 1 H), 2,58 (d, J = 5,2 Hz, 1 H), 2,422,14 (m, 5 H), 2,01-1,91 (m, 4 H), 1,88-1,75 (m, 3 H), 1,73-1,53 (m, 6 H), 1,52-1,30 (m, 4 H), 1,25 (d, J = 6,4 Hz, 3 H), 1,06 (d, J = 6,1 Hz, 3 H), 0,94 (d, J = 7,3 Hz, 3 H), 0,86 (d, J = 6,9 Hz, 3 H), 0,83 (d, J = 6,9 Hz, 3 H).
[01170] Exemplo A#83
[01171] Preparação de N-(24-bromo-23-oxo-4,7,10,13,16,19-hexaoxa-22-azatetracosan-1-oil)-L-valil-N-[2-(3-{[trans-4-({[(3R,5S,7R,8R)-8-hidroxi-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- hidroxipent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6-dioxaspiro[2,5]oct-5-il]acetil}amino)ciclohexil]oxi}-3-oxopropil)fenil]-L-alaninamida (#B255).
[01172] Etapa 1. Síntese de trans-4-[(terc-butoxicarbonil)amino]ciclohexil (2E)-3-(2-nitrofenil)prop-2-enoato (#B257). A uma solução de ácido (2E)-3-(2-nitrofenil)prop-2-enóico (8,26 g, 55,8 mmoles, 1 eq.) em diclorometano (100 mL), foi adiciona- do #B256 (12 g, 55,8 mmoles, 1 eq.) seguido por 1-etil-3-(3- dimetilaminopropil) carbodiimida-HCl (10,9 g, 55,8 mmoles, 1 eq.), 4- N,N-dimetilamino piridina (680 mg, 5,58 mmoles, 0,1 eq.) e trietilamina (23 mL, 167,7 mmoles, 3 eq.), e a reação foi agitada por 17 horas à temperatura ambiente. A reação foi concentrada e purificada por cro- matografia de coluna flash eluindo com éter de petróleo/etil acetato (4:1) para gerar #B257 (8,8 g, 40%) como um sólido branco.
[01173] Etapa 2. Síntese de trans-4-[(terc-butoxicarbonil)amino]ciclohexil (2E)-3-(2-aminofenil)prop-2-enoato (#B258). A uma solução de #B257 (7,8 g, 20 mmoles, 1 eq.) em etil acetato (150 mL), foi adicionado SnCl2 diidrato (25 g, 0,11 mol, 5,5 eq.), e a reação foi agitada por 16 horas. o pH da solução foi ajustado para pH = 8-9 com NaHCO3 aquoso e filtrado. O bolo de filtro foi lavado com etil acetato/metanol três vezes, e as camadas orgânicas combinadas foram lavadas com salmoura, secas sobre sulfato de sódio e concentradas in vacuo. O resíduo foi purificado por cromatografia de coluna flash eluindo com éter de petróleo/etil acetato (4:1) e etil aceta- to/metanol (20:1) para gerar #B258 (850 mg, 12%) como um sólido amarelo.
[01174] Etapa 3. Síntese de trans-4-[(terc-butoxicarbonil)amino]ciclohexil 3-(2-aminofenil)propanoato (#B259). A uma solução de #B258 (800 mg, 2,2 mmoles, 1 eq.) em etil acetato (10 mL) à temperatura ambiente, foi adicionado Pd/C (1 g), e a mistura foi agitada sob hidrogênio (35 psi) por 30 minutos. A reação foi filtrada e concentrada in vacuo para dar #B259 bruto (500 mg, 63%) como um sólido branco que foi usado sem purificação adicional.
[01175] Etapa 4. Síntese de N-[(9H-fluoren-9-ilmetoxi)carbonil]-L- valil-N-{2-[3-({trans-4-[(terc-butoxicarbonil)amino]ciclohexil}oxi)-3- oxopropil]fenil}-L-alaninamida (#B260). A uma solução de #B259 (400 mg, 1,1 mmol, 1 eq.) em N,N-dimetilformamida (20 mL) à temperatura ambiente, foi adicionado N-[(9H-fluoren-9-ilmetoxi)carbonil]-L-valil-L- alanina (453 mg, 1,1 mmol, 1 eq.), 4-N,N-dimetilamino piridina (12 mg, 0,1 mmol, 0,1 eq.) e hexafluorofosfato de O-(7-azabenzotriazol-1-il)- N,N,N‘,N‘-tetrametilurônio (460 mg, 1,2 mmol, 1,1 eq.), e a reação foi agitada por 3 dias. A mistura de reação foi vertida em água e extraída com etil acetato três vezes. As camadas orgânicas combinadas foram secas sobre sulfato de sódio e concentradas. O resíduo foi purificado por cromatografia de coluna flash eluindo com diclorometano/metanol (20:1 a 10:1) para gerar #B260 (110 mg, 13%) como um sólido branco. 1H RMN (500 MHz, CDCl3) δ 9,18 (s, 1 H), 7,77 (m, 3 H), 7,61 (d, 2 H), 7,40 (m, 4 H), 7,15 (m, 3 H), 6,77 (m, 1 H), 5,47 (d, 1 H), 4,73 (m, 2 H), 4,45 (m, 4 H), 4,24 (m, 1 H), 3,11 (q, 1 H), 2,85 (m, 2 H), 2,69 (m, 2 H), 2,17 (m, 2 H), 1,97 (m, 4 H), 1,65 (m, 1 H), 1,56 (m, 3 H), 1,43 (m, 11 H), 1,25 (m, 4 H), 0,98 (m, 6 H).
[01176] Etapa 5. Síntese de N-[(9H-fluoren-9-ilmetoxi)carbonil]-L- valil-N-(2-{3-[(trans-4-aminociclohexil)oxi]-3-oxopropil}fenil)-L- alaninamida sal de trifluoroacetato (#B261). A #B260 (34,8 mg, 0,046 mmol, 1,0 eq), foi adicionado ácido trifluoroacético pré-resfriado (0,8 mL) a 0°C, e a reação foi deixada agitar por 10 min conforme foi aquecida para temperatura ambiente. A reação foi concentrada, retomada em acetonitrila e reconcentrada três vezes para dar #B261 como uma goma que foi usada na etapa seguinte sem purificação adicional. Presumir conversão total. LCMS (Protocolo D): m/z 655,8 [M+H]+, tempo de retenção = 0,81 minuto.
[01177] Etapa 6. Síntese de N-[(9H-fluoren-9-ilmetoxi)carbonil]-L- valil-N-[2-(3-{[trans-4-({[(3R,5S,7R,8R)-8-hidroxi-7-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-hidroxipent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,6- dioxaspiro[2,5]oct-5-il]acetil}amino)ciclohexil]oxi}-3-oxopropil)fenil]-L- alaninamida (#B262). A uma solução de #B4 (14,1 mg, 0,029 mmol, 1 eq.) em N,N-dimetilformamida (0,2 mL) à temperatura ambiente, foram adicionados N,N-diisopropiletilamina (30,6 μL, 0,17 mmol, 6 eq.) e he- xafluorofosfato de O-(7-azabenzotriazoM-il)-N,N,N‘,N‘-tetrametilurônio (13,6 mg, 0,035 mmol, 1,2 eq.), e a reação foi agitada por cinco minutos. Uma solução de #B261 (35,4 mg, 0,046 mmol, 1,6 eq.) em N,N- dimetilformamida (0,6 mL) foi adicionada, e a reação foi deixada agitar por 30 minutos. A reação foi purificada por cromatografia de fase reversa (Método A) para dar #B262 como um sólido branco. Rendimento: 22,8 mg, 0,02 mmol, 70%. LCMS (Protocolo D): m/z 1131,2 [M+H]+, tempo de retenção = 0,96 minuto.
[01178] Etapa 7. Síntese de L-valil-N-[2-(3-{[trans-4-({[(3R,5S,7R,8R)-8-hidroxi-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- hidroxipent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-1,6-dioxaspiro[2,5]oct-5- il]acetil}amino)ciclohexil]oxi}-3-oxopropil)fenil]-L-alaninamida (#B263). O composto do título foi preparado em 88% de rendimento a partir de 22,8 mg (0,02 mmol, 1,0 eq.) de #B262 e 34,1 mg (0,40 mmol, 20,0 eq.) de piperidina usando o procedimento descrito para a preparação do composto #B47. LCMS (Protocolo D): m/z 908,54 [M+H]+, tempo de retenção = 0,64 minuto.
[01179] Etapa 8. Síntese de N-(24-bromo-23-oxo-4,7,10,13,16,19- hexaoxa-22-azatetracosan-1-oil)-L-valil-N-[2-(3-{[trans-4-({[(3R,5S,7R,8R)-8-hidroxi-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- hidroxipent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-1,6-dioxaspiro[2,5]oct-5-il]acetil}amino)ciclohexil]oxi}-3-oxopropil)fenil]-L-alaninamida (#B255). A uma solução de #B263 (16,1 mg, 0,018 mmol, 1 eq.) em N,N- dimetilformamida (0,2 mL) à temperatura ambiente, foi adicionada N,N- diisopropiletilamina (12,7 μL, 0,072 mmol, 4 eq.) seguido por #B252 (9,4 mg, 0,034 mmol, 1,9 eq.) em N,N-dimetilformamida (0,5 mL), e a reação foi deixada agitar à temperatura ambiente por 15 minutos. Mais #B252 (8,8 mg, 0,014 mmol, 0,75 eq.) em N,N-dimetilformamida (0,3 mL) foi adicionado, e a reação foi agitada por mais 15 minutos. A reação foi purificada por cromatografia de fase reversa (Método A) para dar #B255 como um sólido branco. Rendimento: 14,4 mg, 0,011 mmol, 59%. LCMS (Protocolo D): m/z 1365,75 [M+H]+, tempo de retenção = 0,78 minuto. 1H RMN (500 MHz, DMSO-d6) δ 9,40 (s, 1 H), 8,37-8,29 (m, 1 H), 8,15 (d, J = 7,1 Hz, 1 H), 7,87 (d, J = 8,8 Hz, 1 H), 7,82-7,74 (m, 2 H), 7,29-7,09 (m, 4 H), 6,28 (d, J = 15,9 Hz, 1 H), 5,98 (d, J = 10,5 Hz, 1 H), 5,87 (dd, J = 11,7 e 7,1 Hz, 1 H), 5,60 (dd, J = 15,9 e 5,6 Hz, 1 H), 5,54-5,46 (m, 1 H), 5,22-5,13 (m, 1 H), 5,11 (d, J = 4,7 Hz, 1 H), 5,02 (d, J = 5,1 Hz, 1 H), 4,61-4,52 (m, 1 H), 4,51-4,42 (m, 1 H), 4,30-4,17 (m, 3 H), 3,85 (s, 2 H), 3,69-3,37 (m, 25 H), 3,27-3,19 (m, 3 H), 2,88-2,72 (m, 3 H), 2,57 (d, J = 5,1 Hz, 1 H), 2,42-2,13 (m, 5 H), 2,01-1,91 (m, 2 H), 1,88-1,59 (m, 10 H), 1,53-1,43 (m, 1 H), 1,40-1,16 (m, 7 H), 1,11 (d, J = 6,4 Hz, 3 H), 1,06 (d, J = 6,4 Hz, 3 H), 0,96 (d, J = 7,3 Hz, 3 H), 0,86 (d, J = 6,9 Hz, 3 H), 0,83 (d, J = 6,9 Hz, 3 H).
[01180] Exemplo A#84
[01181] Preparação de metil [(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-metoxi-1,6- dioxaspiro[2,5]oct-5-il] acetato (#B265) e metil [(3R,5S,7R,8R)-7- {(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-hidroxipent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-metoxi-1,6-dioxaspiro[2,5]oct-5-il]acetato (#B264).
[01182] Etapa 1. Síntese de metil [(3R,5S,7R,8R)-7-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-metoxi-1,6- dioxaspiro[2,5]oct-5-il] acetato (#B265). A uma solução de #NP1 (32,9 mg, 0,061 mmol, 1 eq.) em N,N-dimetilformamida (0,7 mL) à temperatura ambiente, foi adicionado Mel (114 μL, 1,83 mmol, 30 eq.) e Ag2O (170 mg, 0,73 mmol, 12 eq.), e a reação foi deixada agitar por 72 horas no escuro. A reação foi filtrada sobre lavagem de celite com N,N- dimetilformamida (0,8 mL) e dividida e duas partes. Uma parte foi levada direta para a etapa dois enquanto a outra foi purificada por cro- matografia de fase reversa (Método A) para dar #B265 como um sólido branco. Rendimento: 4,66 mg, 0,008 mmol, 14%. LCMS (Protocolo D): m/z 564.39 [M+H]+, tempo de retenção = 0,90 minuto. 1H RMN (400 MHz, DMSO-d6) δ 7,79 (d, J = 7,8 Hz, 1 H), 6,41-6,31 (m, 2 H), 6,11 (dd, J = 11,7 e 1,2 Hz, 1 H), 5,87 (dd, J = 11,7 e 7,8 Hz, 1 H), 5,635,51 (m, 2 H), 4,58-4,51 (m, 1 H), 4,28-4,18 (m, 1 H), 3,70-3,57 (m, 5 H), 3,55-3,47 (m, 1 H), 3,33 (s, 3 H), 2,96-2,91 (m, 1 H), 2,71-2,63 (m, 2 H), 2,37-2,15 (m, 2 H), 2,07-1,94 (m, 4 H), 1,88-1,75 (m, 2 H), 1,731,60 (m, 4 H), 1,25 (d, J = 6,2 Hz, 3 H), 1,19-1,11 (m, 1 H), 1,07 (d, J = 6,2 Hz, 3 H), 0,96 (d, J = 7,0 Hz, 3 H).
[01183] Etapa 2. Síntese de metil [(3R,5S,7R,8R)-7-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-hidroxipent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-metoxi-1,6- dioxaspiro[2,5]oct-5-il]acetato (#B264). A uma solução #B265 (20 mg, 0,035 mmol, 1 eq.) em metanol (0,6 mL) à temperatura ambiente, foi adicionado K2CO3 (12,2 mg, 0,088 mmol, 2,5 eq.), e a reação foi deixada agitar por 45 minutos. A reação foi filtrada e lavada com etil acetato. A camada orgânica foi lavada com água e salmoura, seca sobre sulfato de sódio e concentrada. O resíduo foi purificado por cromato- grafia de fase reversa (Método A) para dar #B264 como um sólido branco. Rendimento: 4,2 mg, 0,008 mmol, 23%. LCMS (Protocolo D): m/z 522,40 [M+Na]+, tempo de retenção = 0,81 minuto. 1H RMN (400 MHz, DMSO-d6) δ 7,76 (d, J = 7,8 Hz, 1 H), 6,36 (d, J = 14,4 Hz, 1 H), 5,98 (d, J = 11,7 Hz, 1 H), 5,87 (dd, J = 11,7 e 7,0 Hz, 1 H), 5,63-5,50 (m, 2 H), 5,22-5,08 (m, 2 H), 4,58-4,52 (m, 1 H), 4,28-4,18 (m, 1 H), 3,70-3,57 (m, 5 H), 3,55-3,47 (m, 1 H), 3,32 (s, 3 H), 2,96-2,91 (m, 1 H), 2,71-2,63 (m, 2 H), 2,37-2,16 (m, 2 H), 2,06-1,96 (m, 1 H), 1,891,59 (m, 6 H), 1,20-1,02 (m, 7 H), 0,96 (d, J = 7,4 Hz, 3 H).
[01184] Exemplo A#85
[01185] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,4R,5R,7S)-7-{2-[(4-carbamoilbenzil)amino]-2-oxoetil}-4-hidroxi- 1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetate (#B266).
[01186] Etapa 1. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-{2-[(4-carbamoilbenzil)amino]-2-oxoetil}-4-hidroxi- 1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetate (#B266). A uma solução de #B1 (18,7 mg, 0,03 mmol, 1 eq.) em N,N- dimetilformamida (0,5 mL) à temperatura ambiente, foram adicionados N,N-diisopropiletilamina (21,2 μL, 0,12 mmol, 2 eq.) e 4- (aminometil)benzamida sal de cloridrato (11,2 mg, 0,06 mmol, 2 eq.), e a reação foi agitada por 1 hora. A reação foi purificada por por croma- tografia de fase reversa (Método A) para dar #B266 como um sólido branco. Rendimento: 15,4 mg, 0,023 mmol, 77%. LCMS (Protocolo D): m/z 668,37 [M+Na]+, tempo de retenção = 0,71 minuto. 1H RMN (500 MHz, DMSO-d6) δ 8,50-8,43 (m, 1 H), 7,88 (s, 1 H), 7,82-7,74 (m, 3 H), 7,33-7,25 (m, 3 H), 6,41-6,27 (m, 2 H), 6,10 (d, J = 11,6 Hz, 1 H), 5,87 (dd, J = 11,6 e 7,5 Hz, 1 H), 5,62 (dd, J = 15,8 e 5,5 Hz, 1 H), 5,505,43 (m, 1 H), 5,04 (d, J = 5,4 Hz, 1 H), 4,43-4,20 (m, 3 H), 3,68-3,59 (m, 2 H), 3,53-3,45 (m, 1 H), 3,29-3,23 (m, 1 H), 2,78 (d, J = 5,3 Hz, 1 H), 2,68-2,56 (m, 2 H), 2,35-2,13 (m, 3 H), 1,98 (s, 3 H), 1,90-1,72 (m, 4 H), 1,70 (s, 3 H), 1,66-1,58 (m, 1 H), 1,57-1,49 (m, 1 H), 1,25 (d, J = 6,5 Hz, 3 H), 1,04 (d, J = 6,2 Hz, 3 H), 0,93 (d, J = 7,3 Hz, 3 H).
[01187] Exemplo A#86
[01188] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5-[(3R,4R,5R,7S)-7-{2-[(4-carbamoilfenil)amino]-2-oxoetil}-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B267).
[01189] Etapa 1. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-{2-[(4-carbamoilfenil)amino]-2-oxoetil}-4-hidroxi- 1,6-dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetate (#B267). A uma solução de #NP1 (12,4 mg, 0,023 mmol, 1 eq.) em N,N-dimetilformamida (0,5 mL) à temperatura ambiente, foram adicionados N,N-diisopropiletilamina (20,2 μL, 0,12 mmol, 5 eq.) e hexafluo- rofosfato de O-^-azabenzotriazoM-iO-NXN^N-tetrametHurônio (10,9 mg, 0,028 mmol, 1,2 eq.), e a reação foi agitada por cinco minutos. 4- Aminobenzamida (6,3 mg, 0,046 mmol, 2 eq.) foi adicionada, e a reação foi deixada agitar por 1 hora. A reação foi purificada por cromato- grafia de fase reversa (Método A) para dar #B267 como um sólido branco. Rendimento: 4,5 mg, 0,007 mmol, 30%. LCMS (Protocolo D): m/z 654,37 [M+H]+, tempo de retenção = 0,73 minuto. 1H RMN (500 MHz, DMSO-d6) δ 10,21 (s, 1 H), 7,88-7,75 (m, 3 H), 7,70-7,62 (m, 2 H), 7,23 (s, 1 H), 6,41-6,32 (m, 1 H), 6,26 (d, J = 16,1 Hz, 1 H), 6,10 (d,J = 11,6 Hz, 1 H), 5,87 (dd, J = 11,6 e 7,6 Hz, 1 H), 5,58 (dd, J = 16,1 e 5,3 Hz, 1 H), 5,43-5,34 (m, 1 H), 5,08 (d, J = 5,4 Hz, 1 H), 4,42-4,29 (m, 2 H), 3,70-3,59 (m, 2 H), 3,48-3,40 (m, 1 H), 3,30-3,26 (m, 1 H), 2,81-2,73 (m, 2 H), 2,62 (d, J = 5 Hz, 1 H), 2,31-2,12 (m, 2 H), 1,98 (s, 3 H), 1,96-1,88 (m, 1 H), 1,87-1,74 (m, 2 H), 1,68 (s, 3 H), 1,63-1,50 (m, 2 H), 1,25 (d, J = 6,5 Hz, 3 H), 1,06 (d, J = 6,2 Hz, 3 H), 0,93 (d, J = 7,5 Hz, 3 H).
[01190] Exemplo A#87
[01191] Preparação de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5-{(3R,4R,5R,7S)-7-[(6S,9S)-19-bromo-6-metil-2,5,8,11,18-pentaoxo-9- (propan-2-il)-3,4,7,10,17-pentaazanonadec-1-il]-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B268).
[01192] Etapa 1. Síntese de N-(6-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}hexanoil)-L-valil-N-(4-{9-[(3R,5S,7R,8R)-7- {(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}- 3,6-dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi- 1,6-dioxaspiro[2,5]oct-5-il]-3,7-dioxo-2-oxa-4,6,8-triazanon-1-il}fenil)- N5-carbamoil-L-ornitinamida (#B269). A uma solução de #B243 (19,7 mg, 0,037 mmol, 1 eq.) em acetonitrila (1 mL), foi adicionada N,N- dimetilformamida (0,5 mL), e a acetonitrila foi removida in vacuo. A esta solução, foi adicionada N,N-diisopropiletilamina (32,6 μL, 0,19 mmol, 5 eq.) seguido por uma solução de #B182 (40,5 mg, 0,045 mmol, 1,22 eq.), e a reação foi agitada por 30 minutos. Mais N,N- diisopropiletilamina (32,6 μL, 0,19 mmol, 5 eq.) foi adicionado, e a reação foi agitada por outros 70 minutos. A reação foi purificada por cro- matografia de fase reversa (Método A) para dar #B269 como um sólido branco. Rendimento: 12 mg, 0,009 mmol, 25%. LCMS (Protocolo D): m/z 1320,4 [M+H]+, tempo de retenção = 0,91 minuto.
[01193] Etapa 2. Síntese de N-(6-aminohexanoil)-L-valil-N-(4-{9- [(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4- (acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H-piran-2-il]-3- metilpenta-1,3-dien-1-il}-8-hidroxi-1,6-dioxaspiro[2,5]oct-5-il]-3,7-dioxo- 2-oxa-4,6,8-triazanon-1-il}fenil)-N5-carbamoil-L-ornitinamida sal de acetato (#B270). O composto do título foi preparado em 69% de rendimento a partir de 19,8 mg (0,015 mmol, 1,0 eq.) de #B269 e 25,5 mg (0,3 mmol, 20,0 eq.) de piperidina usando o procedimento descrito para a preparação do composto #B47. LCMS (Protocolo D): m/z 1097,78 [M+H]+, tempo de retenção = 0,64 minuto.
[01194] Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5-{(3R,4R,5R,7S)-7-[(6S,9S)-19-bromo-6-metil-2,5,8,11,18-pentaoxo-9- (propan-2-il)-3,4,7,10,17-pentaazanonadec-1-il]-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B268). O composto do título foi preparado em 64% de rendimento a partir de 12 mg (0,01 mmol, 1 eq.) de #B270, 3,5 mg (0,015 mmol, 1,5 eq) de 1- [(bromoacetil)oxi]pirrolidina-2,5-diona e 5,2 mg (0,04 mmol, 4,0 eq) de N,N-diisopropiletilamina usando o procedimento descrito para a preparação do composto #B150. LCMS (Protocolo D): m/z 1217,43 [M+H]+, tempo de retenção = 0,75 minuto. 1H RMN (500 MHz, DMSO-d6) δ 9,98 (s, 1 H), 8,27-8,20 (m, 1 H), 8,08 (d, J = 7,3 Hz, 1 H), 7,84-7,73 (m, 2 H), 7,62-7,54 (m, 2 H), 7,30-7,23 (m, 2 H), 6,65-6,57 (m, 1 H), 6,41-6,32 (m, 1 H), 6,29 (d, J = 15,9 Hz, 1 H), 6,16-6,07 (m, 2 H), 6,015,93 (m, 1 H), 5,86 (dd, J = 11,5 e 7,6 Hz, 1 H), 5,63 (dd, J = 15,9 e 5,6 Hz, 1 H), 5,55-5,47 (m, 1 H), 5,41 (s, 2 H), 5,01 (d, J = 5,9 Hz, 1 H), 4,94 (s, 1 H), 4,43-4,16 (m, 4 H), 3,81 (s, 2 H), 3,69-3,59 (m, 2 H), 3,54-3,45 (m, 1 H), 3,26-3,10 (m, 3 H), 3,08-2,88 (m, 4 H), 2,74 (d, J = 5,0 Hz, 1 H), 2,60 (d, J = 5,0 Hz, 1 H), 2,35-2,09 (m, 6 H), 2,01-1,92 (m, 4 H), 1,87-1,75 (m, 4 H), 1,74-1,30 (m, 14 H), 1,29-1,19 (m, 4 H), 1,06 (d, J = 6,4 Hz, 3 H), 0,95 (d, J = 7,3 Hz, 3 H), 0,86 (d, J = 6,6 Hz, 3 H), 0,83 (d, J = 6,9 Hz, 3 H).
[01195] Exemplo #A88
[01196] Preparação de (2E)-4-amino-N-[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3S,5S,7S)-7-(2-amino-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]pent-2- enamida (#B271) e (2Z)-4-amino-N-[(2R,3R,5S,6S)-6-{(2E,4E)-5- [(3S,5S,7S)-7-(2-amino-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]pent-2-enamida (#B272)
[01197] Etapa 1. Síntese de (2E)-4-amino-N-[(2R,3R,5S,6S)-6- {(2E,4E)-5-[(3S,5S,7S)-7-(2-amino-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5- il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]pent-2- enamida (#B271) e (2Z)-4-amino-N-[(2R,3R,5S,6S)-6-{(2E,4E)-5- [(3S,5S,7S)-7-(2-amino-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]pent-2- enamida (#B272): A 100 mM de tampão de fosfato de sódio pH 7,4 (3,57 mL), foram adicionados #B129 (7 mg, em 0,23 mL DMSO, 1 eq.), isopropilamina (1,475 mL de uma solução 1 M feita em tampão de fosfato pH 3, gerando uma solução de pH ~7, 100 eq.), piridoxal fosfato (0,295 mL de uma solução 50 mM em tampão de fosfato pH 7,4, 1 eq.), e preparação de enzima ATA-P2-B01 (33 mg, em 0,33 mL de tampão de fosfato pH 7,4, Codexis, lote # D11134, R-seletivo para acetofenona). Após incubação a 30°C, 200 rpm por 19 horas, o pH foi ajustado para ~12 com hidróxido de sódio e a reação foi extraída sete vezes com volume igual de etilacetato. O solvente foi evaporado sob pressão reduzida, o resíduo ressuspenso em 0,25 mL acetonitrila/água 1:1, filtrado e purificado por cromatografia de fase reversa em um total de 10 tiragens (Método I). As frações com tempo de retenção de 10 e 13 min foram coletadas e neutralizadas com hidróxido de amônio antes de liofização para gerar #B271 e #B272, respectivamente, como sólidos brancos.
[01198] #B271; (Rendimento 1,6 mg). HPLC (Protocolo P): tempo de retenção = 6,5 minutos; HRESIMS m/z observou 476,3124 [M+H]+ (previsto para C26H42N3O5 é m/z 476,3124); 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 7,55 (d, J = 8,2 Hz, 1 H), 7,32 (br s, 1 H), 6,77 (br s, 1 H), 6,54 (dd, J = 15,6 e 6,2 Hz, 1 H), 6,27 (d, J = 16,0 Hz, 1 H), 6,21 (dd, J = 15,4 e 1,2 Hz, 1 H), 5,59 (dd, J = 16,0 e 5,5 Hz, 1 H), 5,51 (br t, J = 7,0 Hz,1 H), 4,54 (br q, J = 5,5 Hz, 1 H), 4,30 (m, 1 H), 3,69 (m, 1H), 3,64 (m, 1 H), 3,50 (m, 1 H), 3,42 (m, 1 H), 2,62 (m, 2 H), 2,58-2,52 (m, 1H), 2,34-2,27 (m, 2 H), 2,24-2,17 (m, 2 H), 1,85-1,73 (m, 4 H), 1,70 (s, 3 H), 1,64 (m, 2 H), 1,37 (dd, J = 13,1 e 6,2 Hz, 1 H), 1,07 (d, J = 6,6 Hz, 3 H), 1,05 (d, J = 6,2 Hz, 3 H), 0,96 (d, J = 7,4 Hz, 3 H).
[01199] #B272 (Rendimento 1,1 mg) HPLC (Protocolo P): tempo de retenção = 6,85 minutos; HRESIMS m/z 476,3131 [M+H]+ (previsto para C26H42N3O5 é m/z 476.3124); 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 7,76 (d, J = 7,8 Hz, 1 H), 7,32 (br s, 1 H), 6,77 (br s, 1 H), 6,27 (d, J = 15,6 Hz, 1 H), 5,88 (br d, J = 11,7, 1 H), 5,73 (dd, J = 11,5 e 8,4 Hz, 1 H), 5,59 (dd, J = 16,0 e 5,5 Hz, 1 H), 5,51 (br t, J = 6,6 Hz,1 H), 4,54 (br q, J = 5,5 Hz, 1 H), 4,47 (m, J = 7,0 Hz, 1 H), 4,30 (m, 1 H), 3,65 (m, 2 H), 3,50 (m, 1 H), 2,62 (m, 2 H), 2,55 (m, 1H), 2,34-2,27 (m, 2 H), 2,24-2,18 (m, 2 H), 1,83-1,75 (m, 2 H), 1,70 (s, 3 H), 1,65 (m, 3 H), 1,38 (dd, J = 13,3 e 6,2 Hz, 1 H), 1,24 (br s, 1 H), 1,07 (d, J = 6,2 Hz, 3 H), 1,02 (d, J = 6,6 Hz, 3 H), 0,95 (d, J = 7,4 Hz, 3 H).
[01200] Exemplo #A89
[01201] Preparação de (2E)-4-amino-N-[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3S,5S,7S)-7-(2-amino-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]pent-2- enamida (#B273)
[01202] Etapa 1. Síntese (2E)-4-amino-N-[(2R,3R,5S,6S)-6- {(2E,4E)-5-[(3S,5S,7S)-7-(2-amino-2-oxoetil)-1,6-dioxaspiro[2,5]oct-5- il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]pent-2- enamida (#B273). A #B129 (2,9 mg, em 0,1 mL DMSO, 1 eq.), foi adicionado piridoxal fosfato (0,125 mL de uma solução 50 mM em tampão de fosfato pH 7,4, 1 eq.), e uma mistura de isopropilamina (0,625 mL de uma solução 1 M feita em tampão de fosfato pH 3, dando uma solução de pH ~7, 100 eq.), preparação de enzima ATA-251 (15 mg, em 0,15 mL de tampão de fosfato pH 7,4, Codexis, lote # D11140, S- seletivo para acetofenona) e 100 mM tampão de sódio e fosfato pH 7,4 (1,6 mL) que foi pré-incubado a 45°C, 200 rpm por 1 hora. Após incubação a 37°C, 200 rpm por 22 horas, o pH foi ajustado para ~12 com hidróxido de sódio e a reação foi extraída sete vezes com volume igual de etilacetato. O solvente foi evaporado sob pressão reduzida, o resíduo ressuspenso em 0,25 mL acetonitrila/água 1:1, filtrado e purificado por cromatografia de fase reversa em um total de 4 tiragens (Método J). A fração com tempo de retenção de 9 minutos foi coletada e neutralizada com hidróxido de amônio antes de liofilização para gerar #B273 como um sólido branco. Rendimento: 0,7 mg. HPLC (Protocolo P): tempo de retenção = 6,6 minutos; HRESIMS m/z 476,3126 [M+H]+ (previsto para C26H42N3O5 é m/z 476.3124); 1H RMN (500 MHz, DMSO-d6, mult, J em Hz) δ 7,57 (d, J = 8,3 Hz, 1 H), 7,33 (br s, 1 H), 6,78 (br s, 1 H), 6,54 (dd, J = 15,4 e 6,1 Hz, 1 H), 6,27 (d, J = 15,9 Hz, 1 H), 6,22 (br d, J = 15,4 Hz, 1 H), 5,59 (dd, J = 15,9 e 5,4 Hz, 1 H), 5,51 (br t, J = 7,1 Hz,1 H), 4,54 (q, J = 5,3 Hz, 1 H), 4,30 (m, 1 H), 3,69 (m, 1H), 3,64 (dq, J = 6,8 e 2,2 Hz, 1 H), 3,49 (dt, J = 7 e 2,2 Hz, 1 H), 3,42 (m, 1 H), 2,62 (m, 2 H), 2,58-2,53 (m, 1H), 2,33-2,28 (m, 1 H), 2,23-2,18 (m, 2 H), 1,85-1,74 (m, 4 H), 1,70 (s, 3 H), 1,64 (m, 2 H), 1,37 (dd, J = 13,3 e 6,2 Hz, 1 H), 1,07 (d, J = 6,6 Hz, 3 H), 1,05 (d, J = 6,3 Hz, 3 H), 0,96 (d, J = 7,3 Hz, 3 H).
[01203] Exemplo #A90
[01204] Preparação de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5- {(3R,5S)-7-hidroxi-7-[2-oxo-2-(propilamino)etil]-1,6-dioxaspiro[2,5]oct- 5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro-2H-piran-3- il}amino)-5-oxopent-3-en-2-il acetato (#B274)
[01205] Etapa 1. Síntese de ácido [(3R,7S)-7-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-5-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]ácido (#B174). A uma solução aquosa de #NP2 (4 mg, 0,4 mM, 1 eq.) em 50 mM de tampão MOPS pH 7,5, foram adicionados α-cetoglutarato (0,8 mM concentração final, 2 eq.), ascorbato de sódio (0,08 mM, 0,2 eq.), NH4Fe(II)SO4 (0,04 mM, 0,1 eq.) e Fr9P recombinante da etapa 1 do exemplo #A60 (1,6 μM, 0,004 eq.). Após incubação à temperatura ambiente por 2 horas, a reação foi acidificada para pH ~4-5 com ácido acético e extraída três vezes com volume igual de etilacetato. O solvente foi evaporado sob pressão reduzida, para prover #B174 que foi usado sem purificação adicional. LCMS m/z 536 [M+H]+.
[01206] Etapa 2. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3R,5S,7S)-7-hidroxi-7-[2-oxo-2-(propilamino)etil]-1,6-dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxop ent-3-en-2-il acetato (#B274)
[01207] A #B174 (4,0 mg; 0,0075 mmol) em DMF (0,05M), a 0oC. A isto, foi adicionado HATU (1,4 eq) e a mistura foi agitada por 5 minutos. DIPEA (1 eq), seguido por propilamina (1,5 eq) em DMF foi adicionado e a mistura agitada à temperatura ambiente por 2 horas. A reação foi diluída com acetonitrila e purificada por HPLC de fase reversa (Protocolo K): tempo de retenção = 10,8 minutos; A fração contendo o produto foi imediatamente congelada e liofilizada para gerar #B274 como um sólido branco (2,4 mg; Rendimento 60%). LCMS m/z 577 [M+H]+ 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 7,99 (dd, J = 5,5, 5,5 Hz, 1H), 7,80 (d, J = 8,0 Hz, 1H), 6,36 (m, 1H), 6,21 (d, J = 15,7 Hz, 1H), 6,11 (dd, J = 11,7, 0,9 Hz, 1H), 5,86 (dd, J = 11,6, 7,6 Hz, 1H), 5,53 (dd, 15,7, 6,0 Hz, 1H), 5,47 (m, 1H), 4,65 (m, 1H), 3,65 (m, 1H), 3,64 (m, 1H), 3,48 (m, 1H), 3,05 (m, 1H), 3,01 (m, 1H), 2,47 (m, 2H), 2,40 (m, 2H), 2,29 (m, 1H), 2,19 (m, 1H), 2,11 (m, 1H), 1,98 (s, 3H), 1,80 (m, 3H), 1,68 (s, 3H), 1,64 (m, 1H), 1,41 (m, 2H), 1,29 (m, 1H), 1,25 (d, J = 6,4 Hz, 3H), 1,17 (m, 1H), 1,07 (d, J = 6,7 Hz, 3H), 0,94 (d, J = 7,2 Hz, 3H), 0,84 (dd, J = 7,6, 7,6 Hz, 3H), 13C RMN (100 MHz, DMSO-d6) δ 170,0, 169,7, 164,4, 142,6, 133,9, 133,6, 128,4, 127,3, 122,6, 95,9, 79,7, 74,7, 66,8, 67,7, 54,2, 49,2, 46,0, 45,5, 39,8, 39,7, 37,8, 35,0, 31,4, 28,5, 22,0, 19,5, 17,4, 13,9, 12,6, 12,0, 10,9.
[01208] Exemplo #A91
[01209] Preparação de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5- {(3R,4R,5R,7S)-4,7-dihidroxi-7-[2-oxo-2-(propilamino)etil]-1,6- dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino)-5-oxopent-3-en-2-il acetato (#B275)
[01210] Etapa 1. Síntese de ácido [(3R,7R,8R)-7-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-5,8-dihidroxi- 1,6-dioxaspiro[2,5]oct-5-il]ácido (#B276): A uma solução aquosa de #NP1 (4 mg, 0,4 mM, 1 eq.) em 50 mM de tampão MOPS pH 7,5, foram adicionados α-cetoglutarato (0,8 mM concentração final, 2 eq.), ascorbato de sódio (0,08 mM, 0,2 eq.), NH4Fe(II)SO4 (0,04 mM, 0,1 eq.) e Fr9P recombinante da etapa 1 do exemplo #A60 (1,2 μM, 0,003 eq.). Após incubação à temperatura ambiente por 1 hora e 30 minutos, a reação foi acidificada para pH ~4-5 com ácido acético e extraída três vezes com volume igual de etilacetato. O solvente foi evaporado sob pressão reduzida, e o #B276 cru obtido foi usado sem purificação adi-cional. LCMS m/z 552 [M+H]+.
[01211] Etapa 2. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(3R,4R,5R,7S)-4,7-dihidroxi-7-[2-oxo-2-(propilamino)etil]-1,6- dioxaspiro[2,5]oct-5-il}-3-metilpenta-2,4-dien-1-il]-2,5-dimetiltetrahidro- 2H-piran-3-il}amino) -5-oxopent-3-en-2-il acetato (#B275)
[01212] A #B276 (4,3 mg; 0,0080 mmol) em DMF (0,05M), e a 0oC, foi adicionado HATU (1,4 eq) e deixado agitar por 5 minutos. DIPEA (1 eq), seguido por propilamina (1,5 eq) em DMF foi adicionado, e a reação foi deixada agitar à temperatura ambiente por 1 hora. Um adicional da quantidade de HATU (0,7 eq), DIPEA (1 eq) e propilamina (1 eq) foi adicionado, e a mistura foi agitada por um adicional de 30 minutos. O produto bruto foi diluído com acetonitrila e purificado por HPLC de fase reversa (Protocolo K): tempo de retenção = 8,60 minutos. A fração contendo o produto foi imediatamente congelada e liofilizada para gerar #B275 como um sólido branco (2,3 mg; Rendimento 49%). LCMS m/z 593 [M+H]+ ; 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 8,01 (dd, J = 5,6, 5,6 Hz, 1H), 7,81 (d, J = 8,0 Hz, 1H), 6,36 (m, 1H), 6,24 (d, J = 16,1 Hz, 1H), 6,11 (dd, J = 12,1 Hz, 1H), 5,88 (dd, J = 11,7, 7,7 Hz, 1H), 5,67 (dd, 16,1, 4,4 Hz, 1H), 5,44 (m, 1H), 4,55 (d, 8,7 Hz, 1H),4,33 (m, 1H), 3,65 (m, 1H), 3,64 (m, 1H), 3,48 (m, 1H), 3,33 (m, 1H),3,07 (m, 1H), 3,00 (m, 1H), 2,73 (m, 1H), 2,39 (m, 2H), 2,31 (m, 1H),2,29 (m, 1H), 2,27 (m, 1H), 2,19 (m, 1H), 1,98 (s, 3H), 1,80 (m, 2H),1,69 (s, 3H), 1,64 (m, 1H), 1,49 (m, 1H), 1,41 (m, 2H), 1,25 (d, J = 6,3 Hz, 3H), 1,07 (d, J = 6,3 Hz, 3H), 0,95 (d, J = 7,2 Hz, 3H), 0,84 (m, 3H), 13C RMN (100 MHz, DMSO-d6) δ 170,4, 169,6, 164,7, 143,4, 135,5, 134,0, 128,9, 126,3, 123,4, 95,7, 80,6, 75,4, 71,4, 68,4 (x2), 56,7, 46,7, 46,2, 45,9, 41,3, 40,6, 35,6, 32,1, 29,1, 22,8, 21,4, 20,2, 18,1, 14,6, 12,8, 11,8.
[01213] Exemplo #A92
[01214] Preparação de N-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1- il)hexanoil]-L-valil-N-{4-[({[trans-4-({[(3R,5S,7R,8R)-7-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]acetil}amino)ciclohexil]carbamoil}oxi)metil]fenil}-N5-carbamoil-L-ornitinamida (#B277)
[01215] Etapa 1. Síntese de N-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1- il)hexanoil]-L-valil-N-{4-[({[trans-4-({[(3R,5S,7R,8R)-7-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-8-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]acetil}amino)ciclohexil]carbamoil}oxi)metil]fenil}- N5-carbamoil-L-ornitinamida (#B277). A uma solução de N-[6-(2,5- dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]-L-valil-N5-carbamoil-N-{4- [({[(4-nitrobenzil)oxi]carbonil}oxi)metil]fenil}-L-ornitinamida (MalCValCi- tPABC-PNP, preparado como em Bioconjugado Chem. 2002, 13, 855869, 16,5 mg, 0,022 mmol, 1,1 eq.) e B76 (11,9 mg, 83% de pureza, 0,019 mmol, 1,0 eq.) em N,N-dimetilformamida anidra (1,5 mL), foi adicionada N,N-diisopropiletilamina (30 μL). A mistura de reação foi agitada à temperatura ambiente por 0,5 hora. A mistura de reação foi purificada usando cromatografia de fase reversa (Método B*) para gerar #B277 como um pó branco. Rendimento: 9,3 mg, 40%. HPLC (Protocolo N): tempo de retenção = 9,5 minutos (pureza 94%). LCMS (Protocolo M): m/z 1229,5 [M+H]+.
[01216] Exemplo #A93
[01217] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5- [(3R,4R,5R,7S)-7-{2-[(trans-4-{[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1- il)hexanoil]amino}ciclohexil)amino]-2-oxoetil}-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B278)
[01218] Etapa 1. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-{2-[(trans-4-{[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1- il)hexanoil]amino}ciclohexil)amino]-2-oxoetil}-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B278). Uma solução de ácido 6-maleimidohexanóico (24,2 mg, 0,11 mmol, 5 eq.) e diciclo- hexilearbodiimida (DCC, 49,5 mg, 0,24 mmol, 11 eq.) em N,N- dimetilformamida anidra (1,0 mL) foi agitada à temperatura ambiente por 30 minutos. #B76 (13,0 mg, 83% de pureza, 0,021 mmol, 1,0 eq.) em N,N-dimetilformamida (0,5 mL) foi adicionada e a mistura de reação agitada por 2 horas. A mistura de reação foi purificada usando cromatografia de fase reversa (Método B*) para gerar #B278 como um pó branco. Rendimento: 8,6 mg, 49%. HPLC (Protocolo N): tempo de retenção = 9,6 minutos (pureza 96%). LCMS (Protocolo M): m/z 824,4 [M+H]+.
[01219] Exemplo #A94
[01220] Preparação de (1S,5R)-5-{(1E,3E)-5-[(2S,3S,5R,6R)-5- {[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,5-anidro-1-(carboximatil)-3-C- (clorometil)-2-desoxi-D-eritro-pentitol (#NP5) [PF-06739239]
[01221] Etapa 1. Síntese de (1S,5R)-5-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,5-anidro-1-(carboximatil)-3-C- (clorometil)-2-desoxi-D-eritro-pentitol (#NP5). Uma solução de #NP1 (101 mg, ~70% pura, ~0,19 mmol, 1 eq.) em tetrahidrofurano anidro (1,0 mL) foi misturado com uma solução de cloreto de lítio (61 mg, 1,4 mmol, 7 eq.) em ácido acético anidro (1,0 mL) a 0oC. A solução resultante foi agitada à temperatura ambiente por 1,5 hora. A mistura de reação foi purificada usando cromatografia de fase reversa (Método B*) para gerar #NP5 como um pó branco. Rendimento: 54,6 mg, ~76%. HPLC (Protocolo N): tempo de retenção = 10,5 minutos (pureza 96%). LCMS (Protocolo M): m/z 572,5 [M+H]+. 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 12,00 (br s, D2O permutável), 7,80 (d, J = 8,2, 1H, D2O permutável), 6,36 (dq, J = 6,0, 6,0, 1H), 6,22 (br d, J = 15,6, 1H), 6,11 (d, J = 11,7, 1H), 5,87 (dd, J = 11,7, 7,4, 1H), 5,62 (dd, J = 15,6, 5,4, 1H), 5,47 (br dd, J = 7,0, 7,0, 1H), 5,02 (d, J = 7,0, 1H, D2O permutável), 4,78 (br s, 1H, D2O permutável), 4,27 (m, 1H), 4,09 (dd, 8,1, 6,3, 1H), 3,65 (m, 2H), 3,63 (d, J = 10,9, 1H), 3,50 (m, 1H), 3,46 (d, J = 10,9, 1H), 3,22 (dd, J = 8,6, 7,4, 1H), 2,97 (dd, J = 15,6, 9,0, 1H), 2,60 (dd, J = 15,6, 5,5, 1H), 2,28 (m, 1H), 2,21 (m, 1H), 1,98 (s, 3H), 1,92 (dd, J = 14,7, 6,6, 1H), 1,80 (m, 3H), 1,70 (s, 3H), 1,65 (m, 2H), 1,25 (d, J = 6,6, 3H), 1,07 (d, J = 6,5, 3H), 0,95 (d, J = 7,0, 3H).
[01222] Exemplo #A95
[01223] Preparação de (1S,5R)-5-{(1E,3E)-5-[(2S,3S,5R,6R)-5- {[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,5-anidro-3-C-(clorometil)-2- desoxi-1-[2-oxo-2-(pentafluorofenoxi)etil]-D-eritro-pentitol (#B279)
[01224] Etapa 1. Síntese de (1S,5R)-5-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,5-anidro-3-C-(clorometil)-2- desoxi-1-[2-oxo-2-(pentafluorofenoxi)etil]-D-eritro-pentitol (#B279). A uma solução de #NP5 (18,2 mg, 0,032 mmol, 1 eq.) e HATU (16,8 mg) em N,N-dimetilformamida seca (200 uL), foi adicionada N,N- diisopropiletilamina (20 uL) e a solução foi agitada por 5 min em temperatura ambiente e, em seguida, misturada com pentafluorofenol (25 mg, 0,13 mmol, 4 eq.) em N,N-dimetilformamida (150 ul). A mistura de reação foi agitada por 20 minutos e, em seguida, purificada usando cromatografia de fase reversa (Método B*) para gerar #B279 como um pó branco. Rendimento: 15,1 mg, 74%. HPLC (Protocolo N): tempo de retenção = 14,8 minutos (pureza 94%). LCMS (Protocolo M): m/z 738,3 [M+H]+. 1H RMN (400 MHz, DMSO-d6, mult, J em Hz) δ 7,80 (d, J = 7,8, 1H, D2O permutável), 6,36 (dq, J = 6,0, 6,0, 1H), 6,24 (br d, J = 15,6, 1H), 6,11 (d, J = 11,3, 1H), 5,87 (dd, J = 11,3, 7,4, 1H), 5,66 (dd, J = 15,9, 5,5, 1H), 5,44 (br dd, J = 6,6, 6,6, 1H), 5,16 (d, J = 7,0, 1H, D2O permutável), 4,97 (br s, 1H, D2O permutável), 4,46 (m, 1H), 4,16 (dd, 9,0, 5,8, 1H), 3,70-3,63 (m, 3H), 3,68 (d, J = 10,6, 1H), 3,51 (d, J = 10,9, 1H), 3,49 (m, 1H), 3,27 (dd, J = 9,0, 7,0, 1H), 3,10 (dd, J = 15,6, 4,3, 1H), 2,30 (m, 1H), 2,19 (m, 1H), 2,01 (dd, J = 15,1, 7,0, 1H), 1,97(s, 3H), 1,81 (m, 2H), 1,77 (d, J = 14,8, 1H), 1,70 (s, 3H), 1,64 (m, 1H), 1,25 (d, J = 6,4, 3H), 1,07 (d, J = 6,5, 3H), 0,95 (d, J = 7,0, 3H).
[01225] Exemplo #A96
[01226] Preparação de (1S,5R)-5-{(1E,3E)-5-[(2S,3S,5R,6R)-5- {[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,5-anidro-1-[2-({2-[(bromoacetil)amino]etil}amino)-2-oxoetil]-3-C-(clorometil)-2-desoxi-D- eritro-pentitol (#B280)
[01227] Etapa 1. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-{2-[(2-{[(9H-fluoren-9- ilmetoxi)carbonil]amino}etil)amino]-2-oxoetil}-4-hidroxi-1,6- dioxaspiro[2,5]oct-5-il]-3-metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro- 2H-piran-3-il]amino}-5-oxopent-3-en-2-il acetato (#B281). A uma solução de #NP1 (122,1 mg, ~67% de pureza, 0,22 mmol, 1,2 eq.) e hexa- fluorofosfato de O-(7-azabenzotriazol-1-il)-N,N,N‘,N‘-tetrametilurônio (HATU, 151 mg, 0,30 mmol, 1,7 eq.) em N,N-dimetilformamida (3 mL), foi adicionada N,N-diisopropiletilamina (base de Hunig, 60 uL) e a solução resultante foi agitada à temperatura ambiente por 10 minutos. Bromidrato de N- fluorenilmetiloxicarbonil-1,2-diaminoetano (73 mg, 0,2 mmol, 1 eq.) em N,N-dimetilformamida (0,5 mL) foi, então, adicionada e a solução resultante foi agitada por 10 minutos. A mistura de reação foi filtrada e, em seguida, purificada usando cromatografia de fase reversa (Método F*) para gerar #B281 como um pó branco. Rendimento: 114,5 mg, 64% de rendimento. HPLC (Protocolo N): tempo de retenção = 12,7 minutos (pureza 99%). ESIMS (positivo) m/z 800,7 [M+H]+.
[01228] Etapa 2. Síntese de (1S,5R)-5-{(1E,3E)-5-[(2S,3S,5R,6R)-5- {[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,5-anidro-3-C-(clorometil)-2- desoxi-1-{2-[(2-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}etil)amino]-2- oxoetil}-D-eritro-pentitol (#B282). Uma solução de #B281 (44,5 mg, 0,056 mmol) em tetrahidrofurano anidro (0,4 mL) foi misturada com uma solução de cloreto de lítio (30,0 mg, 0,71 mmol) em ácido acético seco (0,2 mL). O reagente foi agitado à temperatura ambiente por 1,5 hora e, em seguida, purificado usando cromatografia de fase reversa (Método F*) para gerar #B282 como um pó branco. Rendimento: 48,0 mg, 100% de rendimento. HPLC (Protocolo N): tempo de retenção = 16,0 minutos (pureza 96%). ESIMS (positivo) m/z 836,7 [M+H]+.
[01229] Etapa 3. Síntese de (1S,5R)-5-{(1E,3E)-5-[(2S,3S,5R,6R)-5- {[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-1-{2-[(2-aminoetil)amino]-2- oxoetil}-1,5-anidro-3 -C-(clorometil)-2-desoxi-D-eritro-pentitol (#B283). A uma solução de #B282 (48 mg, 0,057 mmol) em DMF (2 mL), foi adicionada piperidina (20 uL). A solução foi agitada à temperatura ambiente por 1 hora e, em seguida, purificada usando cromatografia de fase reversa (Método F*) para gerar #B283 como um pó branco. Rendimento: 26,4 mg, 92% de rendimento. HPLC (Protocolo N): tempo de retenção = 6,8 minutos (pureza 91%). ESIMS (positivo) m/z 614,6 [M+H]+.
[01230] Etapa 4. Síntese de (1S,5R)-5-{(1E,3E)-5-[(2S,3S,5R,6R)-5- {[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,5-anidro-1-[2-({2-[(bromoacetil)amino]etil}amino)-2-oxoetil]-3-C-(clorometil)-2-desoxi-D- eritro-pentitol (#B280). A uma solução de #B283 (9,1 mg, 0,015 mmol) e 1-[(bromoacetil)oxi]pirrolidina-2,5-diona (6,2 mg, 0,024 mmol) em DMF (0,5 mL), foi adicionada N,N’-diisopropiletilamina (base de Hunig, 5.0 uL). A solução resultante foi agitada à temperatura ambiente por 30 minutos e, em seguida, purificada usando cromatografia de fase reversa (Método F*) para gerar #B280 como um pó branco. Rendimento: 5,8 mg, 53% de rendimento. HPLC (Protocolo N): tempo de retenção = 10,0 minutos (pureza 99%). ESIMS (positivo) m/z 736,6 [M+H]+.
[01231] Exemplo #A97
[01232] Preparação de (1S,5R)-5-{(1E,3E)-5-[(2S,3S,5R,6R)-5- {[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,5-anidro-3-C-(clorometil)-2-desoxi-1-(2-metoxi-2-oxoetil)-D-eritro-pentitol (#B284)
[01233] Etapa 1. Síntese de (1S,5R)-5-{(1E,3E)-5-[(2S,3S,5R,6R)-5- {[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,5-anidro-3-C-(clorometil)-2- desoxi-1-(2-metoxi-2-oxoetil)-D-eritro-pentitol (#B284). A uma suspensão de #NP5 (6,2 mg, 0,011 mmol, 1 eq.) e carbonato de potássio (20,0 mg, 0,14 mmol, 12 eq.) em N,N-dimetilformamida (0,5 mL), foi adicionado metil iodeto (20 uL, 0,32 mmol, 29 eq.). A mistura de reação foi agitada a 0 grau por 30 minutos. O sólido foi removido por filtração e o filtrado foi purificado usando cromatografia de fase reversa (Método F*) para gerar #B284 como um pó branco. Rendimento: 5,6 mg, 90% de rendimento. HPLC (Protocolo N): tempo de retenção = 12,7 minutos (pureza 97%). ESIMS (positivo) m/z 586,4 [M+H]+.
[01234] Exemplo #A98
[01235] Preparação de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)-5- {(2S,4S,6S)-4-(clorometil)-4-hidroxi-6-[2-oxo-2-(propilamino)etil]tetrahidro-2H-piran-2-il}-3-metilpenta-2,4-dien-1-il]-2,5- dimetiltetrahidro-2H-piran-3-il} amino)-5-oxopent-3-en-2-il acetato (#B285)
[01236] Etapa 1. Síntese de ácido [(2S,4S,6S)-6-{(1E,3E)-5- [(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-4-(clorometil)- 4-hidroxitetrahidro-2H-piran-2-il]acético (#NP8). Uma solução de #NP2 (70,4 mg, 90% de pureza, 0,19 mmol, 1 eq.) em tetrahidrofurano anidro (1,0 mL) foi misturada com uma solução de cloreto de lítio (50 mg, 1,2 mmol, 6 eq.) em ácido acético seco (1,0 mL) a 0oC. A solução foi agitada à temperatura ambiente por 1,5 hora e, em seguida, a 40 graus por 2 horas. O reagente foi purificado usando cromatografia de fase reversa (Método F*) para gerar #NP8 como um pó branco. Rendimento: 52,0 mg, 72% de rendimento. HPLC (Protocolo N): tempo de retenção = 11,4 minutos (pureza 98%). ESIMS (positivo) m/z 556,2 [M+H]+.
[01237] Etapa 2. Síntese de (2S,3Z)-5-({(2R,3R,5S,6S)-6-[(2E,4E)- 5-{(2S,4S,6S)-4-(clorometil)-4-hidroxi-6-[2-oxo-2-(propilamino)etil]tetrahidro-2H-piran-2-il}-3-metilpenta-2,4-dien-1-il]-2,5- dimetiltetrahidro-2H-piran-3-il} amino)-5-oxopent-3-en-2-il acetato. (#B285). Uma solução de #NP8 (5,0 mg, 0,009 mmol, 1 eq.), hexafluo- rofosfato de O-(7-azabenzotriazol-1-il)-N,N,N‘,N‘-tetrametilurônio (HATU, 5,2 mg, 0,014 mmol, 1,5 eq.), e diisopropiletilamina (base de Hunig, 5,0 uL) em N,N-dimetilformamida anidra (0,5 mL) foi agitada à temperatura ambiente por 10 minutos. Propilamina pura (5,0 uL, 0,08, 9 eq.) foi, então, adicionada e a solução agitada por 1 hora e, em seguida, purificada usando cromatografia de fase reversa (Método F*) para gerar #B285 como um pó branco. Rendimento: 4,2 mg, 90% de rendimento. HPLC (Protocolo N): tempo de retenção = 11,8 minutos (pureza 95%). ESIMS (positivo) m/z 597,4 [M+H]+.
[01238] Exemplo #A99
[01239] Preparação de (1S,5R)-5-{(1E,3E)-5-[(2S,3S,5R,6R)-5- {[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,5-anidro-3-C-(clorometil)-2-desoxi-1-[2-oxo-2-(propilamino)etil]-D-eritro-pentitol (#B286)
[01240] Etapa 1. Síntese de (1S,5R)-5-{(1E,3E)-5-[(2S,3S,5R,6R)-5- {[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,5-anidro-3-C-(clorometil)-2- desoxi-1-[2-oxo-2-(propilamino)etil]-D-eritro-pentitol (#B286). Uma solução de #B63 (18,4 mg, 0,032 mmol, 1 eq.) em tetrahidrofurano anidro (1,0 mL) foi misturada com uma suspensão de cloreto de sódio (50,0 mg) em ácido acético seco (0,5 mL). A solução foi, então, agitada a 50oC por 5 horas e purificada usando cromatografia de fase reversa (Método F*) para gerar #B286 como um pó branco. Rendimento: 17,5 mg, 94% de rendimento. HPLC (Protocolo N): tempo de retenção = 10,9 minutos (pureza 97%). ESIMS (positivo) m/z 613,6 [M+H]+.
[01241] Exemplo #A100
[01242] Preparação de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)-5- [(3R,4R,5R,7S)-7-(2-{[trans-3-(2,5-dioxo-2,5-dihidro-1H-pirrol-1- il)ciclobutil]amino}-2-oxoetil)-4-hidroxi-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5- oxopent-3-en-2-il acetato (#B287) e (1S,5R)-5-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6- dimetiltetrahidro-2H-piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,5-anidro-3- C-(clorometil)-2-desoxi-1-(2-{[trans-3-(2,5-dioxo-2,5-dihidro-1H-pirrol-1- il)ciclobutil]amino}-2-oxoetil)-D-eritro-pentitol (#B288)
[01243] Etapa 1. Síntese de (2S,3Z)-5-{[(2R,3R,5S,6S)-6-{(2E,4E)- 5-[(3R,4R,5R,7S)-7-(2-{[trans-3-(2,5-dioxo-2,5-dihidro-1H-pirrol-1- il)ciclobutil]amino}-2-oxoetil)-4-hidroxi-1,6-dioxaspiro[2,5]oct-5-il]-3- metilpenta-2,4-dien-1-il}-2,5-dimetiltetrahidro-2H-piran-3-il]amino}-5- oxopent-3-en-2-il acetato (#B287). A uma solução de #B73 (32,5 mg, 0,05 mmol, 1 eq.) em 1:2:3 dimetilsulfóxido/bicarbonato de sódio satu- rado/água (6 mL total), foi adicionada N-metoxicarbonilmaleimida (45 mg, 0,29 mmol, 6 eq.). A suspensão foi, então, agitada a 0C por 1 hora. Os produtos foram extraídos com etil acetato (10 mL). A camada orgânica foi seca sobre cloreto de magnésio anidro e, em seguida, evaporada até secagem sob pressão reduzida. O resíduo foi re-dissolvido em diclorometano (2 mL), e trietilamina (90 uL) foi adicionada e, em seguida, agitada a 40 0C por 2 horas. A mistura de reação foi neutralizada com ácido acético e purificada usando cromatografia de fase re-versa (Método F*) para gerar #B287 como um pó branco. Rendimento: 4,3 mg, 11% de rendimento. HPLC (Protocolo N): tempo de retenção = 10,1 minutos (pureza 97%). ESIMS (positivo) m/z 684,4 [M+H]+.
[01244] Etapa 2. Síntese de (1S,5R)-5-{(1E,3E)-5-[(2S,3S,5R,6R)-5- {[(2Z,4S)-4-(acetiloxi)pent-2-enoil]amino}-3,6-dimetiltetrahidro-2H- piran-2-il]-3-metilpenta-1,3-dien-1-il}-1,5-anidro-3-C-(clorometil)-2- desoxi-1-(2-{[trans-3-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)ciclobutil]amino}-2-oxoetil)-D-eritro-pentitol (#B288). Uma solução de #B287 (2,7 mg,) em tetrahidrofurano anidro (200 uL) foi misturada com uma solução de cloreto de lítio (13 mg) em ácido acético seco (200 uL) a 0 grau. A solução foi agitada à temperatura ambiente por 0,5 hora e, em seguida, purificada usando cromatografia de fase reversa (Método F*) para gerar #B288 como um pó branco. Rendimento: 0,8 mg, 26% de rendimento. HPLC (Protocolo N): tempo de retenção = 10,6 minutos (pureza 92%). ESIMS (positivo) m/z 720,7 [M+H]+.
[01245] Procedimentos de Conjugação
[01246] Procedimento de conjugação geral A: Anticorpo Herceptin comercialmente disponível (Genentech Inc) é dializado em Solução Salina Tamponada com Fosfato de Dulbecco (DPBS, Lonza). O anticorpo dializado (5-10 mg/mL) é, então, reagido com PL (3-12) equivalentes de ligante-carga útil (10 mM em dimetil sulfóxido (DMSO)) contendo o N-hidroxissuccinimida éster reativo à temperatura ambiente por 2 horas em tampão de borato 50 mM pH 8,7. Em alguns casos, tampão de borato 50 mM pH 8,7 é substituído por Solução Salina Tamponada com Fosfato de Dulbecco (DPBS, Lonza). Em alguns casos, para melhorar a solubilidade/reatividade do ligante-carga útil, di- metilacetamida (DMA) ou DMSO é adicionada para alcançar 10-15% (v/v) do componente de solvente orgânico na mistura de reação final. A mistura de reação é, então, permutada por tampão em DPBS (pH 7,4) usando Colunas de permuta por tampão GE Healthcare Sephadex G-25 M por instruções do fabricante. O material bruto é purificado por cromatografia por exclusão de tamanho (SEC) usando um sistema a GE AKTA Explorer com uma coluna a GE Superdex 200 e eluente DPBS (pH 7,4). A fração de monômero agrupada de AKTA é, então, concentrada e permutada por tampão em 10mM tampão de succinato de sódio, 5,4% trealose pH 5,1 usando colunas de permuta por tampão GE Healthcare Sephadex G-25 M por instruções do fabricante. O ADC é adicionalmente caracterizado através de cromatografia por exclusão de tamanho (SEC) para pureza e espectrometria de massa em tandem por ionização por eletropulverização de cromatografia líquida (LC-ESI MS) para calcular a razão de droga-anticorpo (carga). A concentração de proteína é determinada através de espectrofotômetro UV.
[01247] Procedimento de conjugação geral B: Anticorpo Herceptin comercialmente disponível (Genentech Inc) ou anticorpos terapêuticos são dializados em 50 mM tampão de fosfato pH 6,8. O anticorpo diali- zado (5-10 mg/mL) é reagido com PL (4-12) equivalentes de ligante- carga útil (5-30 mM em dimetilacetamida (DMA) ou dimetil sulfóxido (DMSO)) contendo o pentafluorofenil éster reativo à temperatura ambiente por 2-20 h em 50 mM tampão de fosfato pH 6,8. Em alguns casos, para melhorar a solubilidade/reatividade do ligante-carga útil, DMA ou DMSO é adicionado para alcançar 10-15% (v/v) do componente de solvente orgânico na mistura de reação final. A mistura de reação é, então, permutada por tampão em DPBS (pH 7,4) usando colunas de permuta por tampão GE Healthcare Sephadex G-25 M por instruções do fabricante. O material bruto é purificado por cromatogra- fia por exclusão de tamanho (SEC) usando um sistema GE AKTA Explorer com uma coluna GE Superdex 200 e eluente DPBS (pH 7,4). A fração de monômero agrupada de AKTA é, então, concentrada e permutada por tampão em 10mM tampão de succinato de sódio, 5,4% trealose pH 5,1 usando colunas de permuta por tampão GE Healthcare Sephadex G-25 M por instruções do fabricante. O ADC é adicionalmente caracterizado através de cromatografia por exclusão de tamanho (SEC) para pureza e espectrometria de massa em tandem por io- nização por eletropulverização de cromatografia líquida (LC-ESI MS) para calcular a razão de droga-anticorpo (carga). A concentração de proteína é determinada através de espectrofotômetro UV.
[01248] Procedimento de conjugação geral C: Anticorpo Herceptin comercialmente disponível (Genentech Inc) ou anticorpos terapêuticos são dializados em Solução Salina Tamponada com Fosfato de Dul- becco (DPBS, Lonza). O anticorpo dializado é reduzido com adição de y(1-7) equivalentes de cloridrato de tris(2-carboxietil)fosfina (TCEP, 5mM em água destilada) e diluído para 15 mg/mL de concentração final de anticorpo usando DPBS, 5mM ácido 2,2’,2’’,2’’’-(Etano-1,2- diildinitrilo)tetraacético (EDTA), pH 7,0 (Tampão A). A reação é incubada a 37°C por 2 horas e, em seguida, resfriada para temperatura ambiente. Conjugação foi realizada por adição de PL (2 a 15) equivalentes de ligante-carga útil (5-10mM em dimetilacetamida (DMA) ou dimetil sulfóxido (DMSO)). Em alguns casos, para melhorar a solubili- dade/reatividade do ligante-carga útil, DMA ou DMSO é adicionado para alcançar 10-15% (v/v) de solvente orgânico total na mistura de reação final, e Tampão A adicionado para alcançar 10 mg/mL de concentração final de anticorpo. A reação é, então, incubada por 2 horas à temperatura ambiente. A mistura de reação é subsequentemente permutada por tampão em DPBS (pH 7,4) usando colunas de permuta por tampão GE Healthcare Sephadex G-25 M por instruções do fabricante. O material bruto é purificado por cromatografia por exclusão de tamanho (SEC) usando um sistema GE AKTA Explorer com uma coluna GE Superdex 200 e eluente DPBS (pH 7,4). A fração de monômero agrupada de AKTA é, então, concentrada se necessário. O ADC é adicio-nalmente caracterizado através de cromatografia por exclusão de tamanho (SEC) para pureza e espectrometria de massa em tandem por ionização por eletropulverização de cromatografia líquida (LC-ESI MS) para calcular a razão de droga-anticorpo (carga). A concentração de proteína é determinada através de espectrofotômetro UV.
[01249] Procedimento de conjugação geral D: O anticorpo terapêutico é dializado em Solução Salina Tamponada com Fosfato de Dul- becco (DPBS, Lonza). O anticorpo dializado é reduzido com adição de y(1-7) equivalentes de cloridrato de tris(2-carboxietil)fosfina (TCEP, 5mM em água destilada) e diluído para 15 mg/mL de concentração final de anticorpo usando DPBS, 5mM ácido 2,2’,2’’,2’’’-(Etano-1,2- diildinitrilo)tetraacético (EDTA), pH 7,0 (Tampão A). A reação é incubada a 37°C por 2 horas e, em seguida, resfriada para temperatura ambiente. A conjugação foi realizada por adição de PL (2 a 15) equivalentes de ligante-carga útil (5-10mM em dimetilacetamida (DMA). Em alguns casos, para melhorar a solubilidade/reatividade do ligante- carga útil, DMA ou DMSO é adicionado para alcançar 10-15% (v/v) de solvente orgânico total na mistura de reação final, e 20X tampão de borato e DPBS é adicionado para alcançar 10 mg/mL de concentração final de anticorpo em tampão de borato 50 mM pH 8,7. A reação é incubada por 3 horas a 37°C ou por 16h à temperatura ambiente. A mistura de reação é subsequentemente permutada por tampão em DPBS (pH 7,4) usando colunas de permuta por tampão GE Healthcare Sephadex G-25 M por instruções do fabricante. O material bruto é purificado por cromatografia por exclusão de tamanho (SEC) usando um sistema GE AKTA Explorer com uma coluna a GE Superdex 200 e eluente DPBS (pH 7,4). A fração de monômero agrupada de AKTA é, então, concentrada se necessário. O ADC é adicionalmente caracterizado através de cromatografia por exclusão de tamanho (SEC) para pureza e espectrometria de massa em tandem por ionização por ele- tropulverização de cromatografia líquida (LC-ESI MS) para calcular a razão de droga-anticorpo (carga). A concentração de proteína é determinada através de espectrofotômetro UV.
[01250] Procedimento de conjugação geral E: O anticorpo terapêu- tico portando resíduos de cisteína extra em relação a anticorpo nativo é dializado em Solução Salina Tamponada com Fosfato de Dulbecco (DPBS, Lonza). O anticorpo dializado é reduzido com adição de 100 equivalentes de cloridrato de tris(2-carboxietil)fosfina (TCEP, 5mM em água destilada) e diluída para 15 mg/mL de concentração final de anticorpo usando DPBS, 5mM ácido 2,2’,2’’,2’’’-(Etano-1,2- diildinitrilo)tetraacético (EDTA), pH 7,0 (Tampão A). A reação é, então, incubada à temperatura ambiente por 2 horas e, em seguida, resfriada para temperatura ambiente. Após a redução, TCEP é removido da mistura de reação usando um dispositivo de ultrafiltração Millipore Amicon Ultra 4 mL 50KD MWCO. A mistura de reação é concentrada para 1/10 do volume original quatro vezes e novamente diluída para o volume original cada vez usando Tampão A. Em alguns casos, a mistura de reação é subsequentemente permutada por tampão em Tampão A usando colunas de permuta por tampão GE Healthcare Sephadex G-25 M por instruções do fabricante. Métodos alternativos, tais como Filtração de Fluxo Tangencial (TFF) ou diálise são também úteis em circunstâncias particulares. Após a redução, os dissulfitos in- ternos/ponte reduzidos do anticorpo são re-oxidados usando 1-1,5mM deidroascorbato (DHA) à temperatura ambiente durante a noite em Tampão A. Após a oxidação, DHA é removido da mistura de reação usando um dispositivo de ultrafiltração Millipore Amicon Ultra 4 mL 50KD MWCO. A mistura de reação é concentrada para 1/10 do volume original quatro vezes e re-diluída para o volume original cada vez usando tampão de borato 50 mM pH 8,7. Em alguns casos, a mistura de reação é subsequentemente permutada por tampão em tampão de borato 50 mM pH 8,7 usando colunas de permuta por tampão GE Healthcare Sephadex G-25 M por instruções do fabricante. Métodos alternativos, tais como TFF ou diálise são também úteis em circunstâncias particulares. A conjugação é realizada por adição de PL (3 a 12) equivalentes de ligante-carga útil (10mM em dimetilacetamida (DMA)). Em alguns casos, para melhorar a solubilidade/reatividade do ligante-carga útil, DMA ou DMSO é adicionado para alcançar 10-15% (v/v) de solvente orgânico total na mistura de reação final, e 20X tampão de borato e DPBS é adicionado para alcançar 5-10 mg/mL de concentração final de anticorpo em tampão de borato 50 mM pH 8,7. A reação é incubada por 3 horas a 37°C ou 16 horas à temperatura ambiente. A mistura de reação é subsequentemente permutada por tampão em DPBS (pH7,4) usando colunas de permuta por tampão GE Healthcare Sephadex G-25 M por instruções do fabricante. O material bruto é purificado por cromatografia por exclusão de tamanho (SEC) usando um sistema GE AKTA Explorer com uma coluna GE Superdex 200 e eluente DPBS (pH 7,4). A fração de monômero agrupada de AKTA é, então, concentrada se necessário. O ADC é adicionalmente caracterizado através de cromatografia por exclusão de tamanho (SEC) para pureza e espectrometria de massa em tandem por ionização por eletropulverização de cromatografia líquida (LC-ESI MS) para calcular a razão de droga-anticorpo (carga). A concentração de proteína é determinada através de espectrofotômetro UV.
[01251] Procedimento de conjugação geral F: O anticorpo terapêutico portando resíduos de cisteína extra em relação a anticorpo nativo é dializado em Solução Salina Tamponada com Fosfato de Dulbecco (DPBS, Lonza). O anticorpo dializado é reduzido com adição de 100 equivalentes de cloridrato de tris(2-carboxietil)fosfina (TCEP, 5mM em água destilada) e diluída para 15 mg/mL de concentração final de anticorpo usando DPBS, 5mM ácido 2,2’,2’’,2’’’-(Etano-1,2- diildinitrilo)tetraacético (EDTA), pH 7,0 (Tampão A). A reação é, então, incubada à temperatura ambiente por 2 horas e, em seguida, resfriada para temperatura ambiente. Após a redução, TCEP é removido da mistura de reação usando um dispositivo de ultrafiltração Millipore Amicon Ultra 4 mL 50KD MWCO. A mistura de reação é concentrada para 1/10 do volume original quatro vezes e novamente diluída para o volume original por vez usando Tampão A. Em alguns casos, a mistura de reação é subsequentemente permutada por tampão em Tampão A usando colunas de permuta por tampão GE Healthcare Sephadex G- 25 M por instruções do fabricante. Métodos alternativos, tais como Filtração de Fluxo Tangencial (TFF) ou diálise são também úteis em circunstâncias particulares. Após a redução, os dissulfitos internos/ponte reduzidos do anticorpo são reoxidados usando 1-1,5mM deidroascor- bato (DHA) à temperatura ambiente durante a noite em Tampão A. Após a oxidação, DHA é removido da mistura de reação usando um dispositivo de ultrafiltração Millipore Amicon Ultra 4mL 50KD MWCO. A mistura de reação é concentrada para 1/10 do volume original quatro vezes e re-diluída para o volume original cada vez usando Tampão A. Em alguns casos, a mistura de reação é subsequentemente permutada por tampão em Tampão A usando colunas de permuta por tampão GE Healthcare Sephadex G-25 M por instruções do fabricante. Métodos alternativos, tais como Filtração de Fluxo Tangencial (TFF) ou diá- lise são também úteis em circunstâncias particulares. A conjugação é realizada por adição de PL (2 a 15) equivalentes de ligante-carga útil (5-10mM em dimetilacetamida (DMA) ou dimetil sulfóxido (DMSO)). Em alguns casos, para melhorar a solubilidade/reatividade do ligante- carga útil, DMA ou DMSO é adicionado para alcançar 10-15% (v/v) de solvente orgânico total na mistura de reação final, e Tampão A adicionado para alcançar 5-10 mg/mL de concentração final de anticorpo. A reação é incubada por 1-2 horas à temperatura ambiente. A mistura de reação é subsequentemente permutada por tampão em DPBS (pH 7,4) usando colunas de permuta por tampão GE Healthcare Sephadex G-25 M por instruções do fabricante. O material bruto é purificado por cromatografia por exclusão de tamanho (SEC) usando um sistema GE AKTA Explorer com uma coluna GE Superdex 200 e eluente DPBS (pH 7,4). A fração de monômero agrupada de AKTA é, então, concentrada se necessário. O ADC é adicionalmente caracterizado através de cromatografia por exclusão de tamanho (SEC) para pureza e espec- trometria de massa em tandem por ionização por eletropulverização de cromatografia líquida (LC-ESI MS) para calcular a razão de droga- anticorpo (carga). A concentração de proteína é determinada através de espectrofotômetro UV.
[01252] Procedimento de conjugação geral G: Reações de conjugação são realizadas na porção superior de um dispositivo de ultrafiltra- ção centrífuga, tal como filtros Amicon Ultra 50k Ultracel (part #UFC805096, GE). 132 mM de uma solução estoque de L-cisteína é preparada em Solução Salina Tamponada com Fosfato de Dulbecco (DPBS, Lonza) 5mM ácido 2,2’,2’’,2’’’-(Etano-1,2- diildinitrilo)tetraacético (EDTA), pH 7,0 (Tampão A). Esta solução (50 uL) é adicionada a uma mistura do respectivo anticorpo mutante portando resíduos de cisteína extra (5 mg) em 950 uL de Tampão A. A concentração final de cisteína na mistura de reação é 6,6 mM. Após deixar a reação descansar à temperatura ambiente por 1,5 hora, o tubo de reação é centrifugado para concentrar o material para aproximadamente 100 uL. A mistura é diluída para 1 mL com Tampão A. Este processo é repetido 4 vezes a fim de remover todo o agente redutor de cisteína. O material resultante é diluído para 1 mL em Tampão A e tratado com 16 uL de uma solução 5 mM do ligante-carga útil de ma- leimida (em dimetil acetamida (DMA) (aproximadamente 5 equivalentes). Após descanso à temperatura ambiente por 1,5 hora, o tubo de reação é centrifugado para concentrar o material para aproximadamente 100 μL. A mistura é diluída para 1 mL com DPBS. A mistura de reação é subsequentemente permutada por tampão em DPBS (pH7,4) usando colunas de permuta por tampão GE Healthcare Sephadex G- 25 M por instruções do fabricante. O material bruto é purificado por cromatografia por exclusão de tamanho (SEC) usando um sistema GE AKTA Explorer com uma coluna GE Superdex 200 e eluente DPBS (pH 7,4). A fração de monômero agrupada de AKTA é, então, concentrada se necessário. O ADC é adicionalmente caracterizado através de cromatografia por exclusão de tamanho (SEC) para pureza e espec- trometria de massa em tandem por ionização por eletropulverização de cromatografia líquida (LC-ESI MS) para calcular a razão de droga- anticorpo (carga). A concentração de proteína é determinada através de espectrofotômetro UV.
[01253] Procedimento de conjugação geral H: O anticorpo terapêutico portando resíduos de glutamina reativos à enzima de transgluta- mina é dializado em Solução Salina Tamponada com Fosfato de Dul- becco (DPBS, Lonza). A conjugação mediada por transglutaminase é realizada misturando 0,5-5,0 mg/mL de glutamina reativa à transglutaminase contendo anticorpo em 25 mM Tris Tampão pH 8,0, 150 mM NaCI, 0,31 mM glutationa reduzida com 5,0 - 20,0-vezes de excesso molar de ligante de amino alquil portando carga útil (5-10mM em dime- tilacetamida (DMA) ou dimetil sulfóxido (DMSO)) e 2% p/v transglutaminase (Ajinomot Activa TI). A reação é, então, incubada de 4-16 horas à temperatura ambiente. A mistura de reação é subsequentemente permutada por tampão em DPBS (pH 7,4) usando colunas de permuta por tampão GE Healthcare Sephadex G-25 M por instruções do fabricante. O material bruto é purificado por cromatografia por exclusão de tamanho (SEC) usando um sistema GE AKTA Explorer com uma coluna GE Superdex 200 e eluente DPBS (pH 7,4). A fração de monômero agrupada de AKTA é, então, concentrada se necessário. O ADC é adicionalmente caracterizado através de cromatografia por exclusão de tamanho (SEC) para pureza e espectrometria de massa em tandem por ionização por eletropulverização de cromatografia líquida (LC-ESI MS) para calcular a razão de droga-anticorpo (carga). A concentração de proteína é determinada através de espectrofotômetro UV.
[01254] Procedimento de conjugação geral I: O mutante de anticorpo terapêutico portando resíduos de cisteína extra em relação a anticorpo nativo é dializado em Solução Salina Tamponada com Fosfato de Dulbecco (DPBS, Lonza). O anticorpo dializado é reduzido com adição de x(25-100) equivalentes de cloridrato de tris(2- carboxietil)fosfina (TCEP, 5mM em água destilada) e diluído para 15 mg/mL de concentração final de anticorpo usando DPBS, 5mM ácido 2,2’,2’’,2’’’-(Etano-1,2-diildinitrilo)tetraacético (EDTA), pH 6,5-7,4 (Tampão A). A reação é, então, incubada à temperatura ambiente por 1-2 horas e, em seguida, resfriada para temperatura ambiente. Após a redução, TCEP é removido da mistura de reação usando um dispositivo de ultrafiltração Millipore Amicon Ultra 4 mL 50KD MWCO. A mistura de reação é concentrada para 1/10 do volume original quatro vezes e novamente diluída para o volume original por vez usando Tampão A. Em alguns casos, a mistura de reação é subsequentemente permutada por tampão em Tampão A usando colunas de permuta por tampão GE Healthcare Sephadex G-25 M por instruções do fabricante. Após a redução, os dissulfitos internos/ponte reduzidos do anticorpo são reoxidados usando 1mM deidroascorbato (DHA) à temperatura ambiente durante a noite em Tampão A. Após a oxidação, DHA é removido da mistura de reação usando um dispositivo de ultrafiltração Millipore Amicon Ultra 4 mL 50KD MWCO. A mistura de reação é concentrada para 1/10 do volume original quatro vezes e re-diluída para o volume original por vez usando Tampão A. Conjugação é realizada por adição de PL (2 a 10) equivalentes de ligante-carga útil (5-10mM em dimetila- cetamida (DMA) ou dimetil sulfóxido (DMSO)). Em alguns casos, para melhorar a solubilidade/reatividade do ligante-carga útil, DMA ou DMSO é adicionado para alcançar 10-15% (v/v) de solvente orgânico total na mistura de reação final, e Tampão A adicionado para alcançar 5-10 mg/mL de concentração final de anticorpo. A reação é incubada por 1-4 h à temperatura ambiente. A mistura de reação é subsequentemente permutada por tampão em DPBS (pH 7,4) usando colunas de permuta por tampão GE Healthcare Sephadex G-25 M por instruções do fabricante. O material bruto é purificado por cromatografia por exclusão de tamanho (SEC) usando um sistema GE AKTA Explorer com uma coluna GE Superdex 200 e eluente DPBS (pH 7,4). A fração de monômero agrupada de AKTA é, então, concentrada se necessário. O ADC é adicionalmente caracterizado através de cromatografia por exclusão de tamanho (SEC) para pureza e espectrometria de massa em tandem por ionização por eletropulverização de cromatografia líquida (LC-ESI MS) para calcular a razão de droga-anticorpo (carga). A concentração de proteína é determinada através de espectrofotômetro UV.
[01255] Condições de SEC e HPLC-ESI MS Usadas para Análise de Conjugados:
[01256] Protocolo 1:
[01257] Protocolo 1 (SEC): Coluna: Coluna: TSK-gel G3000SWxl, 300 x 7,8 mm, 10 μm; Fase Móvel: Solução salina de tampão de fosfato (PBS, 1X), pH 7,4 com 2% acetonitrila; Isocrático; Taxa de fluxo: 1 mL/minuto. Temperatura: temperatura ambiente; Volume de injeção: 5 μL; Instrumento: Agilent 1100 HPLC.
[01258] Protocolo 1b: Coluna: Superdex 200 5/150 GL, 5 x 150 mm, 13 μm; Fase Móvel: Solução salina de tampão de fosfato (PBS, 1X), pH 7,4 com 2% acetonitrila; Isocrático; Taxa de fluxo: 1 mL/minuto. Temperatura: temperatura ambiente; Volume de injeção: 5 μL; Instrumento: Agilent 1100 HPLC.
[01259] Protocolo 2: Protocolo 2 (HPLC): Coluna: Coluna: Agilent Poroshell 300SB-C8, 75 x 2,1 mm, 2,6 μm; Fase Móvel A: 0,1% de ácido fórmico em água (v/v); Fase Móvel B: 0,1% de ácido fórmico em acetonitrila (v/v); Gradiente: 20% B a 45% B por 4 minutos; Taxa de fluxo: 1,0 mL/minuto. Temperatura: 60°C; Detecção: 220 nm; MS (+) faixa 400-2000Da; Volume de injeção: 10 μL; Instrumento: Agilent 1100 LC, Waters MicromassZQ MS. Desconvolução foi realizada usando MaxEnt1. Amostras foram tratadas com 100 vezes de excesso de cloridrato de tris(2-carboxietil)fosfina (TCEP) ou Ditiotreitol (DTT) e incubadas por 15 minutos à temperatura ambiente antes da injeção.
[01260] Protocolo 3: Coluna: Aquity UPLC BEH 200 SEC 1,7um; Fase Móvel: 450mM NaCl; Taxa de fluxo: 0,5 mL/minuto. Temperatura: 35C; Volume de injeção: 10 μL.
[01261] Estudos de Trastuzumab in vitro e in vivo
[01262] É notado que que para os seguintes estudos, trastuzumab na ausência de agentes citotóxicos conjugados não apresenta nenhuma potência significante in vitro ou eficácia in vivo em concentrações de anticorpo equivalentes.
[01263] Procedimento de Ensaio Celular In Vitro
[01264] Células de expressão de alvo (BT474 (câncer de mama), N87 (câncer gástrico), HCC1954 (câncer de mama), MDA-MB-361- DYT2 (câncer de mama)) ou de não expressão (MDA-MB-468) foram semeadas em placas de cultura celular de 96 cavidades por 24 horas antes do tratamento. As células foram tratadas com conjugados de an- ticorpo-droga diluídos 3 vezes em série ou compostos livres (isto é, nenhum conjugado de anticorpo para a droga) em duplicata em 10 concentrações. Viabilidade celular foi determinada por Ensaio MTS de Proliferação Celular CellTiter 96® AQueous One Solution (Promega, Ma-dison WI) 96 horas após tratamento. Viabilidade celular relativa foi de-terminada como porcentagem de controle não tratado. Valores de IC50 foram calculados usando um modelo logístico de quarto parâmetros #203 com XLfit v4,2 (IDBS, Guildford, Surry, UK). Os resultados são mostrados nas Tabelas 4 e 9. A potência various para cima a partir de 0,0002 nm. O teste em outras linhagens celulares é reportado na Tabela 9A. Procedimentos e técnicas similares foram empregados.
[01265] Modelo de Xenoenxerto de Tumor N87 In vivo (Figuras 3 e 4)
[01266] Estudos de eficácia In vivo de conjugados de anticorpo- droga foram realizados com modelos de xenoenxerto expressando alvo usando as linhagens celulares N87. Para esudo de eficácia, 7,5 milhões de células tumorais em 50% matrigel são implantadas subcuta- neamente em camundongos nude de 6-8 semanas até os tamanhos de tumor alcançarem entre 250 e 350 mm3. O doseamento é feito através de injeção de bolus na veia da cauda. Dependendo da resposta do tumor ao tratamento, os animais são injetados com 1-10 mg/kg de conjugados de anticorpo-droga tratados quatro vezes a cada quatro dias. Todos os animais experimentais são monitorados para alteração de peso corporal semanalmente. O volume de tumor é medido duas vezes por semana nos primeiros 50 dias e uma vez por semana a partir daí por um dispositivo de Caliper e calculado com a seguinte fórmula: O volume de tumor = (comprimento x largura2) / 2. Animais são humanamente sacrificados antes de seus vlumes de tumor alcançarem 2500 mm3. O tamanho de tumor é observado diminuir após a primeira semana de tratamento. Os animais podem ser monitorados continuamente para recrescimento de tumor após descontinuação do tratamento.
[01267] Os resultados do teste dos ADC’s 3, 4 e 5 e ADC’s 14 e 15 no modelo de mapeamento in vivo de xenoenxerto de camundongo N87 são mostrados nas Figuras 3 e 4.
[01268] A Tabela 1 provê os Detalhes de preparação para os exemplos #B82-#B108.
[01269] A Tabela 2 provê dados de caracterização para os exem- plos #B82 - #B108.
[01270] A Tabela 3 provê a preparação de Ligantes de Carga Útil #B109 - #B117.
[01271] A Tabela 4 mostra dados de Citotoxicidade in vitro para produtos naturais e análogos sintéticos.
[01272] A Tabela 5 provê os dados de caracterização para Ligantes de Carga Útil #B109 - #B117.
[01273] A Tabela 6 provê a estrutura de ADCs e os ligantes de carga útil usados para prepará-los.
[01274] A Tabela 7 provê Métodos Gerais de preparação de ADCs exemplificados.
[01275] A Tabela 8 provê dados analíticos para ADCs exemplificados.
[01276] A Tabela 9 mostra a Tabela 9: dados de citotoxicidade in vitro para ADCs.Tabelas