WO2025117727A1 - Analogs of quinoxaline/quinoline cytotoxins, linker- payloads, protein-drug conjugates, and uses thereof - Google Patents
Analogs of quinoxaline/quinoline cytotoxins, linker- payloads, protein-drug conjugates, and uses thereof Download PDFInfo
- Publication number
- WO2025117727A1 WO2025117727A1 PCT/US2024/057728 US2024057728W WO2025117727A1 WO 2025117727 A1 WO2025117727 A1 WO 2025117727A1 US 2024057728 W US2024057728 W US 2024057728W WO 2025117727 A1 WO2025117727 A1 WO 2025117727A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- antibody
- group
- cancer
- linker
- mmol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Definitions
- the present disclosure relates to analogs of quinoxaline and quinoline antibiotics - these are herein termed “Nupomycins” to differentiate the analogs from those natural counter parts of triostins and echinomycins - linker- payloads, as well as protein-drug conjugates (e.g., antibody -drug conjugates) thereof, pharmaceutical compositions comprising the same, and methods of treating disease therewith.
- Nupomycins analogs of quinoxaline and quinoline antibiotics - these are herein termed “Nupomycins” to differentiate the analogs from those natural counter parts of triostins and echinomycins - linker- payloads, as well as protein-drug conjugates (e.g., antibody -drug conjugates) thereof, pharmaceutical compositions comprising the same, and methods of treating disease therewith.
- Proliferative diseases are characterized by uncontrolled growth and spread of abnormal cells. If the spread is not controlled, it can result in death.
- Abnormal proliferation such as cancer, is caused by both external factors (e.g., tobacco, chemicals, radiation and infectious organisms) and internal factors (inherited mutations, immune system conditions, the mutations that occur from metabolism). These causal factors may act together or in sequence to initiate or promote abnormal proliferation. Cancer is treated by surgery, radiation, chemotherapy, hormones and immunotherapy. However, there is a need for more effective anti-proliferation drugs.
- Quinoxaline antibiotics are of widespread occurrence in nature. They are heterodetic cyclic depsipeptides characterized by the possession of quinoxaline-2- carboxylic acid moieties, and the best-known member of the series is echinomycin which is identical to quinomycin A.
- quinoxalines are powerful antimicrobial agents, cytotoxic to mammalian cells in culture, and display significant inhibitory activity towards a variety of tumors through inhibiting RNA synthesis by specific binding of double-stranded DNA through bisintercalation. Those compounds have a rigid, disulfide -bridged, bicyclic, depsipeptide scaffold, which preorganizes two quinoxaline intercalating units.
- the aromatic groups are oriented in parallel at a distance of 10.5 A, a perfect orientation of two intercalators to interact with two adjacent DNA base pairs.
- Quinoxaline antibiotics reversibly intercalate with the double -helical structure of DNA by interacting with adjacent base pairs and disrupting the structure of DNA and thereby causing cell death (Ross & Bradley, Biochimica et Biophysica Acta (BBA)-Nucleic Acids and Protein Synthesis, 1981, 654(1), 129-134).
- Quiescence is a state of reversible growth arrest in which cells have exited the cell cycle but remain capable of renewed division upon stimulation. Entry into quiescence allows cells to persist in a non-dividing state over extended periods of time and enact mechanisms to protect themselves from damage.
- quiescent cells display some similarities to other non-dividing cell states, such as senescence and terminal differentiation, quiescence possesses unique characteristics and functions. In particular, whereas senescent and terminally differentiated cells arrest permanently and are unable to proliferate further, quiescent cells are defined by their ability to reenter the cell cycle. This broad definition of quiescence encompasses a wide range of diverse cell types in an organism.
- Quiescent cells include tissue-resident adult stem cells, such as hematopoietic, muscle, and neural stem cells, as well as differentiated cells, including fibroblasts, hepatocytes, lymphocytes, and oocytes. Quiescence maintains these cells in a poised state — non-proliferative, but ready to re-enter the cell cycle when confronted with the appropriate stimulus (Oceane Mareseal and Iain M. Cheeseman, Dev Cell. 2020 November 09; 55(3): 259-271).
- Quiescent cancer cells can avoid most chemotherapies and re-enter a proliferative state when conditions are right. This can lead to drug resistance and tumor recurrence.
- Quiescent cancer cells are nonproliferating cells arrested in the GO phase, characterized by ki671ow and p27high. QCCs avoid most chemotherapies, and some treatments could further lead to a higher proportion of QCCs in tumors.
- QCCs are also associated with cancer recurrence since they can re-enter a proliferative state when conditions are favorable.
- the ideal anti-proliferation therapy would enable targeted delivery of highly cytotoxic agents to tumor cells and would leave normal cells unaffected.
- Conventional chemotherapeutic treatment is limited because of the toxic side-effects that arise from effects of the drug on non-cancerous cells.
- Various approaches to targeted drug delivery have been tried, including the use of conjugates of tumor targeted probes (such as antibodies or growth factors) with toxins such as pseudomonas or diphtheria toxins, which arrest the synthesis of proteins and cells.
- the side effects include reaction of the immune system due to non-human components of the conjugates.
- the half-life of the drug conjugates is limited due to elimination from the circulation through renal filtration, and schematic degradation, uptake by the reticuloendothelial system (RES), and accumulation in nontargeted organs and tissues.
- RES reticuloendothelial system
- Another approach uses passive drug carriers such as polymers, liposomes, and polymeric micelles to take advantage of the hyper-permeability of vascular endothelia of tumor tissue.
- Polymeric drugs and macromolecules accumulate within solid tumors due to an enhanced permeability and retention mechanism.
- barriers of using such targeted deliveries include fast clearance of foreign particles from the blood, and technological hindrances in obtaining highly standardized, pharmaceutically acceptable drug delivery systems with the necessary specificity and selectivity for binding tumor cells.
- Protein conjugates such as antibody conjugates, utilize the selective binding of a binding agent to deliver a payload to targets within tissues of subjects.
- the payload can be a therapeutic moiety that is capable of taking action at the target.
- X and Y are independently selected from N and CH; Z is O or NH; m is 1, 2, or 3;
- Ri is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, and aralkyl, each of which is optionally substituted;
- R2 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR;
- R3 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR
- R4 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR;
- R5 is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, aralkyl, each of which is optionally substituted.
- the present disclosure provides a linker-payload compound having a structure according to formula (Ila), (lib), or (lie): or a pharmaceutically acceptable salt thereof.
- X and Y are independently selected from N and CH;
- R2 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR;
- R3 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR;
- R4 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl,
- R5 is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, aralkyl, each of which is optionally substituted;
- LI when present, is a stable or self-immolative linker
- L2 when present, is a linker that is stable or is cleavable by an endosomal/lysosomalenzyme
- L3 when present, is a moiety that modulates the hydrophilicity, physical, and/or chemical properties of the linker-payload compound
- B is a reactive moiety for conjugation to a target protein, antibody, or antigen-binding fragment thereof, comprising at least one member selected from the group consisting of amino, hydroxyl, thiol, a maleimido moiety, a succinyl ester, an activated ester, an
- the present disclosure provides linker-payload compounds having a linker moiety that modulates the hydrophilicity, physical, and/or chemical properties of the linker-payload compounds.
- Non-limiting examples include various sugar or carbohydrate moieties, e.g. mono-, di-, and polysaccharides, cyclic polysaccharides (e.g. cyclodextrins), a quaternary ammonium salt, a polyethylene glycol group, a sulfonic acid group, a phosphonic acid group, and combinations thereof.
- the present disclosure provides a conjugate having the formula having a structure according to Formula (I) or Formula (II):
- BA is an antibody or an antigen-binding fragment thereof
- L is a linker of the formula -L1-L2-L3-B-, wherein
- L is connected to BA through a side chain of an amino acid selected from the group consisting of Gin (-CO-NH-), Lys (-NH-CO-), and Cys (-S-);
- LI when present, is a stable or self-immolative linker
- L2 when present, is a linker that is stable or is cleavable by an endosomal/lysosomalenzyme
- L3 when present, is a moiety that modulates the hydrophilicity, physical, and/or chemical properties of -L-P;
- B is a reactive moiety for conjugation to a target protein, antibody, or antigen-binding fragment thereof, comprising at least one member selected from the group consisting of amino, hydroxyl, thiol, a maleimido moiety, a succinyl ester, an activated ester, an alkylamino; a haloacetamido, -
- P is a moiety having a structure according to Formula (la’), Formula (lb’), or Formula
- X and Y are independently selected from N and CH;
- R2 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR;
- R3 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR;
- R4 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR;
- R5 is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, aralkyl, each of which is optionally substituted; and represents the connecting point of attachment to the linker L; and n is an integer from 1 to 20.
- a conjugate comprising (i) an antibody (BA) or an antigen-binding fragment thereof, (ii) a plurality of payloads, and a linker that covalently connects (i) and (ii), wherein the linker is connected to the BA or antigen-binding fragment thereof through a side chain of an amino acid selected from the group consisting of Gin (-C0-NH-), Lys (-NH-C0-), and Cys (-S-), and has a formula -L1-L2-L3-B-, wherein
- LI when present, is a stable or self-immolative linker
- L2 when present, is a linker that is stable or is cleavable by an endosomal/lysosomalenzyme
- L3 when present, is a moiety that modulates the hydrophilicity, physical, and/or chemical properties of the conjugate
- B is a residue of a reactive moiety for conjugation, comprising at least one member selected from the group consisting of amino, hydroxyl, thiol, a maleimido moiety, a succinyl ester, an activated ester, an alkylamino; a haloacetamido, -N3, ⁇ and the payload has a structure according to Formula (la’), Formula (lb’), or Formula (Ic’):
- X and Y are independently selected from N and CH;
- R2 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR;
- R3 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR;
- R4 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR;
- R5 is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, aralkyl, each of which is optionally substituted; and represents the connecting point of attachment to the linker.
- the present disclosure provides a pharmaceutical composition
- a pharmaceutical composition comprising a conjugate as described herein and one or more pharmaceutically acceptable carriers, excipients or diluents.
- a process of producing a conjugate of Formula (I) or Formula (II) as described herein comprises contacting an antibody or an antigen-binding fragment thereof (BA) with a linker-payload compound (L-P) in the presence of a transglutaminase, wherein L-P has a structure according to formula (Ila), (lib), or (lie) as described herein.
- the present disclosure also provides in another aspect a method of treating a subject suffering from cancer.
- the method comprises administering to the subject a therapeutically effective amount of a compound, composition, conjugate, or pharmaceutical dosage form as described herein.
- a compound, composition, conjugate, or pharmaceutical dosage form as described herein for use in treating a cancer is also provided in the present disclosure.
- the present disclosure provides a use of a therapeutically effective amount of a compound, composition, conjugate, or pharmaceutical dosage form as described herein in the manufacture of a medicament for the treatment of a cancer.
- the present disclosure provides in another aspect a method of selectively killing quiescent cells in a subject.
- The comprises administering to the subject a therapeutically effective amount of a compound, conjugate, or pharmaceutical composition as described herein.
- the present disclosure provides in another aspect a method of selectively killing stem cells in a subject.
- The comprises administering to the subject a therapeutically effective amount of a compound, conjugate, or pharmaceutical composition as described herein.
- the present disclosure provides in another aspect a method of selectively killing resting or naive B- or T- or other immune in a subject.
- The comprises administering to the subject a therapeutically effective amount of a compound, conjugate, or pharmaceutical composition as described herein.
- the present disclosure provides in another aspect a method of selectively killing quiescent cancer cells in a subject preparing for stem cell therapy.
- The comprises administering to the subject a therapeutically effective amount of a compound, conjugate, or pharmaceutical composition as described herein.
- subject is an animal, such as a mammal, including human, such as a patient.
- biological activity refers to the in vivo activities of a compound or physiological responses that result upon in vivo administration of a compound, composition or other mixture.
- Biological activity thus, encompasses therapeutic effects and pharmacokinetic behavior of such compounds, compositions and mixtures. Biological activities can be observed in in vitro systems designed to test for such activities.
- the phrase “specifically binds,” or “binds specifically to,” or the like, means that an antibody or antigen-binding fragment thereof forms a complex with an antigen that is relatively stable under physiologic conditions.
- Specific binding can be characterized by an equilibrium dissociation constant of at least about 1x10-8 M or less (e.g., a smaller KD denotes tighter binding).
- Methods for determining whether two molecules specifically bind are well known in the art and include, for example, equilibrium dialysis, surface plasmon resonance, and the like.
- Antibodies can, for example, be identified by real-time, label free bio-layer interferometry assay on an Octet® HTX biosensor, which bind specifically to a target antigen.
- multi-specific antibodies that bind to one domain in the target antigen and one or more additional antigens or a bi-specific that binds to two different regions of the target antigen are nonetheless considered antibodies that “specifically bind”, as used herein.
- antibodies that bind specifically to the target antigen, but are non-neutralizing also can be used within the scope of the present disclosure to generate antibody-drug conjugates. Such antibodies may function, for example, to deliver a payload to the cells expressing a target antigen.
- antibody means any antigen-binding molecule or molecular complex comprising at least one complementarity determining region (CDR) that specifically binds to or interacts with a particular antigen.
- CDR complementarity determining region
- the term “antibody” includes immunoglobulin molecules comprising four polypeptide chains, two heavy (H) chains and two light (L) chains inter-connected by disulfide bonds, as well as multimers thereof (e.g., IgM).
- Each heavy chain comprises a heavy chain variable region (abbreviated herein as HCVR or VH) and a heavy chain constant region.
- the heavy chain constant region comprises three domains, CHI, CH2 and CH3.
- Each light chain comprises a light chain variable region (abbreviated herein as LCVR or VL) and a light chain constant region.
- the light chain constant region comprises one domain (CL1).
- the VH and VL regions can be further subdivided into regions of hypervariability, termed complementarity determining regions (CDRs), interspersed with regions that are more conserved, termed framework regions (FR).
- CDRs complementarity determining regions
- FR framework regions
- Each VH and VL is composed of three CDRs and four FRs, arranged from amino-terminus to carboxy-terminus in the following order: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4.
- Three CDRs of VH are referred to as HCDR1 , HCDR2, and HCDR3, and three CDRs of VL are referred to as LCDR1, LCDR2 and LCDR3.
- antigen-binding fragment of an antibody means any naturally occurring, enzymatically obtainable, synthetic, or genetically engineered polypeptide or glycoprotein that specifically binds an antigen to form a complex.
- human antibody means antibodies having variable and constant regions derived from human germline immunoglobulin sequences. Human antibodies may nonetheless include amino acid residues not encoded by human germline immunoglobulin sequences (e.g., mutations introduced by random or site-specific mutagenesis in vitro or by somatic mutation in vivo), for example in the CDRs and in particular CDR3.
- human antibody as used herein, is not intended to include antibodies in which CDR sequences derived from the germline of another mammalian species, such as a mouse, have been grafted onto human framework sequences.
- humanized antibody means chimeric antibodies that contain minimal sequence derived from the non-human antibody.
- a humanized antibody is generally a human antibody (recipient antibody) in which residues from one or more CDRs are replaced by residues from one or more CDRs of a non-human antibody (donor antibody).
- the donor antibody can be any suitable non-human antibody, such as a mouse, rat, rabbit, chicken, or non-human primate antibody having a desired specificity, affinity, or biological effect.
- selected framework region residues of the recipient antibody are replaced by the corresponding framework region residues from the donor antibody.
- Humanized antibodies may also comprise residues that are not found in either the recipient antibody or the donor antibody. Such modifications may be made to further refine antibody function. For further details, see Jones et al., Nature, 1986, 321:522-525; Riechmann et al., Nature, 1988, 332:323-329; and Presta, Curr. Op. Struct. Biol., 1992, 2:593-596, each of which is incorporated by reference in its entirety.
- recombinant human antibody means all human antibodies that are prepared, expressed, created or isolated by recombinant means, such as antibodies expressed using a recombinant expression vector transfected into a host cell (described further below), antibodies isolated from a recombinant, combinatorial human antibody library (described further below), antibodies isolated from an animal (e.g., a mouse) that is transgenic for human immunoglobulin genes (see, e.g., Taylor et al. (1992) Nucl. Acids Res. 20:6287-6295) or antibodies prepared, expressed, created or isolated by any other means that involves splicing of human immunoglobulin gene sequences to other DNA sequences.
- nucleic acid molecule having substantial identity to a reference nucleic acid molecule may, in certain instances, encode a polypeptide having the same or substantially similar amino acid sequence as the polypeptide encoded by the reference nucleic acid molecule.
- the phrase “substantial similarity” or “substantially similar” means that two peptide sequences, when optimally aligned, such as by the programs GAP or BESTFIT using default gap weights, share at least 90% sequence identity, even more preferably at least 95%, 98%, or 99% sequence identity. Preferably, residue positions, which are not identical, differ by conservative amino acid substitutions.
- the term “surface plasmon resonance” refers to an optical phenomenon that allows for the analysis of real-time interactions by detection of alterations in protein concentrations within a biosensor matrix, for example using the BIAcoreTM system (Biacore Life Sciences division of GE Healthcare, Piscataway, N.J.).
- KD means the equilibrium dissociation constant of a particular protein-protein interaction (e.g., antibody-antigen interaction). Unless indicated otherwise, the KD values disclosed herein refer to KD values determined by surface plasmon resonance assay at 25° C.
- a “pharmaceutically acceptable salt” is a pharmaceutically acceptable, organic or inorganic acid or base salt of a compound described herein.
- Representative pharmaceutically acceptable salts include, e.g., alkali metal salts, alkali earth salts, ammonium salts, water-soluble and water-insoluble salts, such as the acetate, amsonate (4,4-diaminostilbene-2,2-disulfonate), benzenesulfonate, benzonate, bicarbonate, bisulfate, bitartrate, borate, bromide, butyrate, calcium, calcium edetate, camsylate, carbonate, chloride, citrate, clavulariate, dihydrochloride, edetate, edisylate, estolate, esylate, fiunarate, gluceptate, gluconate, glutamate, glycollylarsanilate, hexafluorophosphate, hexylre
- the terms “treat,” “treating,” or “treatment” refer to the reduction or amelioration of the severity of at least one symptom or indication of the disease, e.g., cancer or hepatitis B infection, due to the administration of a therapeutic agent such as a disclosed antibody to a subject in need thereof.
- a therapeutic agent such as a disclosed antibody to a subject in need thereof.
- the terms include inhibition of progression of disease or of worsening of infection.
- the terms also include positive prognosis of disease, e.g., the subject may be free of infection, the subject may have reduced or no viral titers, the subject may have tumor shrinkage, upon administration of a therapeutic agent such as a disclosed antibody or antibody-drug conjugate.
- the therapeutic agent may be administered at a therapeutic dose to the subject.
- prevent refers to inhibition of manifestation of any symptoms or indications of a disease (e.g., cancer or hepatitis B infection) upon administration of a disclosed antibody or antibody-drug conjugate.
- the term includes prevention of the spread of infection in a subject exposed to the virus or at risk of having hepatitis B infection.
- terapéuticaally effective amount refers to an amount that produces the desired effect for which it is administered. The exact amount will depend on the purpose of the treatment, and will be ascertainable by one skilled in the art using known techniques (see, for example, Lloyd (1999) The Art, Science and Technology of Pharmaceutical Compounding).
- amelioration of the symptoms of a particular disorder by administration of a particular compound or pharmaceutical composition refers to any lessening, whether permanent or temporary, lasting or transient that can be attributed to or associated with administration of the compound or pharmaceutical composition.
- the IC50 refers to an amount, concentration or dosage of a particular test compound that achieves a 50% inhibition of a maximal response in an assay that measures such response.
- moieties are specified by their conventional chemical formulae, written from left to right, they equally encompass the chemically identical moieties that would result from writing the structure from right to left, e.g., -CH2O- is equivalent to -OCH2-.
- alkyl by itself or as part of another substituent, means, unless otherwise stated, a straight (z.e., unbranched) or branched chain saturated hydrocarbon radical.
- alkylene by itself or as part of another substituent means a divalent radical derived from an alkyl.
- an alkyl (or alkylene) group will have from 1 to 24 carbon atoms (i.e., Ci-C24-alkyl), including those groups having 10 or fewer carbon atoms (i.e., Ci-Cio-alkyl).
- a “lower alkyl” or “lower alkylene” is a shorter chain alkyl or alkylene group, generally having six or fewer carbon atoms (i.e., Ci-Ce-alkyl).
- alkyl groups include, but are not limited to, groups such as methyl, ethyl, n-propyl, isopropyl, n- butyl, t-butyl, isobutyl, sec-butyl, homologs and isomers of, for example, n-pentyl, n-hexyl, n-heptyl, n-octyl, and the like.
- alkenyl by itself or as part of another substituent, means, unless otherwise stated, a straight (z'.e., unbranched) or branched chain hydrocarbon radical having one or more carbon-carbon double bonds.
- alkenylene by itself or as part of another substituent means a divalent radical derived from an alkenyl.
- an alkenyl (or alkenylene) group will have from 2 to 24 carbon atoms (i.e., C2-C24-alkenyl), including those groups having 10 or fewer carbon atoms (i.e., C2-Cio-alkenyl).
- a “lower alkenyl” or “lower alkenylene” is a shorter chain alkenyl or alkenylene group, generally having six or fewer carbon atoms (i.e., C2-Ce-alkenyl.
- alkenyl groups include, but are not limited to, vinyl (z'.e., ethenyl), 2-propenyl, crotyl, 2 -isopentenyl, 2-(butadienyl), 2,4-pentadienyl, 3-(l,4- pentadienyl), and the higher homologs and isomers.
- alkynyl by itself or as part of another substituent, means, unless otherwise stated, a straight (i.e., unbranched) or branched chain hydrocarbon radical having one or more carbon-carbon triple bonds, which can include di- and multivalent radicals, having the number of carbon atoms designated i.e., C2-C10 means two to ten carbons in C2-Cio-alkynyl).
- alkynyl groups include, but are not limited to, ethynyl, 1- and 3-propynyl, 3- butynyl, and the higher homologs and isomers.
- alkoxy alkylamino
- alkylthio or thioalkoxy
- heteroalkyl by itself or in combination with another term, means, unless otherwise stated, a straight or branched chain hydrocarbon radical, containing at least one heteroatom in the chain selected from O, N, P, Si and S, and wherein the nitrogen and sulfur atoms may optionally be oxidized, and the nitrogen atom may have an alkyl substituent to fulfill valency and/or may optionally be quaternized.
- the heteroatom(s) O, N, P, Si and S may be placed at any interior position of the heteroalkyl group (i.e., not at the point of attachment to the rest of the molecule).
- -CH 2 -CH N-OCH 3 .
- -CH CH-N(CH 3 )- CH3.
- heteroalkylene by itself or as part of another substituent means a divalent radical derived from heteroalkyl, as exemplified, but not limited by, -CH2-O-CH2-CH2-, -CH2-CH2-O-CH2-CH2-, -CH2-O-CH2-CH2-NH-CH2-, -CH2-CH2-S- CH2-CH2- and -CH2-S-CH2-CH2-NH-CH2-.
- alkylene and heteroalkylene linking groups no orientation of the linking group is implied by the direction in which the formula of the linking group is written. For example, the formula -C(0)2R’- represents both -C(0)2R’- and -R'C(0)2-.
- cycloalkyl and heterocycloalkyl represent, unless otherwise stated, cyclic versions of “alkyl” and “heteroalkyl”, respectively, including bicyclic, tricyclic and bridged bicyclic groups. Additionally, for heterocycloalkyl, a heteroatom can occupy the position at which the heterocycle is attached to the remainder of the molecule.
- cycloalkylene and “heterocycloalkylene” by themselves or as part of another substituent means a divalent radical derived from a cycloalkyl or heterocycloalkyl.
- cycloalkyl examples include Cs-Cio-cycloalkyl, but are not limited to, cyclopentyl, cyclohexyl, 1 -cyclohexenyl, 3-cyclohexenyl, cycloheptyl, norbornanyl, bicyclo(2.2.2)octanyl, and the like.
- heterocycloalkyl examples include C3- Cio-heterocycloalkyl, but are not limited to, l-(l,2,5,6-tetrahydropyridyl), 1-piperidinyl, 2- piperidinyl, 3-piperidinyl, 4-morpholinyl, 3-morpholinyl, tetrahydrofuran-2-yl, tetrahydrofuran-3-yl, tetrahydrothien-2-yl, tetrahydrothien-3-yl, 1-piperazinyl, 2-piperazinyl, 1- or 2-azabicyclo(2.2.2)octanyl, and the like.
- aryl means, unless otherwise stated, a polyunsaturated, aromatic, hydrocarbon substituent which can be a single ring or multiple rings (in some embodiments from 1 to 3 rings) which are fused together or linked covalently.
- Aryl includes C6-C12 aryl rings.
- heteroaryl refers to aryl groups that contain from one to four heteroatoms selected from N, O, and S in the ring(s), wherein the nitrogen and sulfur atoms are optionally oxidized, and the nitrogen atom(s) are optionally quaternized.
- Heteroaryl also includes a 5- to 10-membered ring having one to four heteroatom ring members as described herein.
- a heteroaryl group can be attached to the remainder of the molecule through a carbon or heteroatom.
- arylene and heteroarylene by themselves or as part of another substituent means a divalent radical derived from an aryl or heteroaryl.
- Non-limiting examples of aryl and heteroaryl groups include phenyl, 1 -naphthyl, 2-naphthyl, 4-biphenyl, 1 -pyrrolyl, 2-pyrrolyl, 3-pyrrolyl, 3-pyrazolyl, 2-imidazolyl, 4-imidazolyl, pyrazinyl, 2- oxazolyl, 4-oxazolyl, 5-oxazolyl, 3-isoxazolyl, 4-isoxazolyl, 5-isoxazolyl, 2-thiazolyl, 4- thiazolyl, 5-thiazolyl, 2-furyl, 3-furyl, 2-thienyl, 3-thienyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 2- pyrimidyl, 4-pyrimidyl, 5-benzothiazolyl, purinyl, 2-benzimidazolyl, 5-indolyl, 1 -isoquinolyl, 5
- substituent moieties for cycloalkyl, heterocycloalkyl, cycloalkenyl, and heterocycloalkenyl groups also include substituted and unsubstituted alkyl, substituted and unsubstituted alkenyl, and substituted and unsubstituted alkynyl.
- R’, R", R'” and R" each in some embodiments independently are hydrogen, substituted or unsubstituted heteroalkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocycloalkyl, substituted or unsubstituted aryl (e.g., aryl substituted with 1-3 halogens), substituted or unsubstituted alkyl, alkoxy or thioalkoxy groups, or arylalkyl groups.
- each of the R groups is independently selected as are each R’, R", R’" and R"" groups when more than one of these groups is present.
- R’ and R" are attached to the same nitrogen atom, they can be combined with the nitrogen atom to form a 4-, 5-, 6-, or 7-membered ring.
- -NR’R is meant to include, but not be limited to, 1-pyrrolidinyl and 4-morpholinyl.
- alkyl is meant to include groups including carbon atoms bound to groups other than hydrogen groups, such as haloalkyl e.g., -CF3 and -CH 2 CF3) and acyl e.g., - C(O)CH 3 , -C(O)CF 3 , -C(O)CH 2 OCH 3 , and the like).
- Substituent moieties for aryl and heteroaryl groups are, in some embodiments, selected from deuterium, halo, substituted and unsubstituted alkyl, substituted and unsubstituted alkenyl, and substituted and unsubstituted alkynyl, -OR’, -NR’R", -SR’, - SiR'R"R"', -OC(O)R’, -C(O)R’, -CO 2 R’, -CONR’R", -OC(O)NR’R", -
- Two of the substituent moieties on adjacent atoms of an aryl or heteroaryl ring may optionally form a ring of the formula -Q’-C(O)-(CRR’) q -Q”-, wherein Q’ and Q” are independently -NR-, -O-, -CRR’- or a single bond, and q is an integer of from 0 to 3.
- two of the substituent moieties on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -A-(CH 2 ) r -B-, wherein A and B are independently -CRR’-, -O-, -NR-, -S-, -S(O)-, -S(O) 2 -, -S(O) 2 NR’- or a single bond, and r is an integer of from 1 to 4.
- One of the single bonds of the new ring so formed may optionally be replaced with a double bond.
- two of the substituent moieties on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -(CRR’)s-X’-(CR”R”’)d-, where s and d are independently integers of from 0 to 3, and X’ is -O-, -NR’-, -S-, -S(O)-, -S(O) 2 -, or -S(O) 2 NR’-.
- the substituent moieties R, R’, R" and R’" are, in some embodiments, independently selected from hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocycloalkyl, substituted or unsubstituted aryl, and substituted or unsubstituted heteroaryl.
- halo by itself or as part of another substituent, means, unless otherwise stated, a fluorine, chlorine, bromine, or iodine atom. Additionally, terms such as “haloalkyl,” are meant to include monohaloalkyl and polyhaloalkyl.
- halo(Ci- C alkyl) is meant to include, but not be limited to, trifluoromethyl, 2,2,2-trifluoroethyl, 4- chlorobutyl, 3-bromopropyl, and the like.
- oxo as used herein means an oxygen atom that is double bonded to a carbon atom.
- heteroatom or "ring heteroatom” is meant to include oxygen (O), nitrogen (N), sulfur (S), phosphorus (P), and silicon (Si).
- Some compounds described herein can have asymmetric centers and therefore exist in different enantiomeric and diastereomeric forms.
- a compound as described herein can be in the form of an optical isomer or a diastereomer. Accordingly, the disclosure encompasses compounds and their uses as described herein in the form of their optical isomers, diastereoisomers and mixtures thereof, including a racemic mixture.
- Optical isomers of the compounds of the disclosure can be obtained by known techniques such as asymmetric synthesis, chiral chromatography, simulated moving bed technology or via chemical separation of stereoisomers through the employment of optically active resolving agents.
- stereoisomer means one stereoisomer of a compound that is substantially free of other stereoisomers of that compound.
- a stereomerically pure compound having one chiral center will be substantially free of the opposite enantiomer of the compound.
- a stereomerically pure compound having two chiral centers will be substantially free of other diastereomers of the compound.
- a typical stereomerically pure compound comprises greater than about 80% by weight of one stereoisomer of the compound and less than about 20% by weight of other stereoisomers of the compound, for example greater than about 90% by weight of one stereoisomer of the compound and less than about 10% by weight of the other stereoisomers of the compound, or greater than about 95% by weight of one stereoisomer of the compound and less than about 5% by weight of the other stereoisomers of the compound, or greater than about 97% by weight of one stereoisomer of the compound and less than about 3% by weight of the other stereoisomers of the compound, or greater than about 99% by weight of one stereoisomer of the compound and less than about 1% by weight of the other stereoisomers of the compound.
- the stereoisomer as described above can be viewed as composition comprising two stereoisomers that are present in their respective weight percentages described herein.
- the depicted structure controls. Additionally, if the stereochemistry of a structure or a portion of a structure is not indicated with, for example, bold or dashed lines, the structure or portion of the structure is to be interpreted as encompassing all stereoisomers of it. In some cases, however, where more than one chiral center exists, the structures and names may be represented as single enantiomers to help describe the relative stereochemistry. Those skilled in the art of organic synthesis will know if the compounds are prepared as single enantiomers from the methods used to prepare them.
- the term “compound” is inclusive in that it encompasses a compound or a pharmaceutically acceptable salt, stereoisomer, isotopologue, and/or tautomer thereof.
- a compound includes a pharmaceutically acceptable salt of a tautomer of the compound.
- a compound of includes a pharmaceutically acceptable salt of an isotopologue of the compound.
- X and Y are independently selected from N and CH;
- Ri is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, and aralkyl, each of which is optionally substituted;
- R2 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR;
- R3 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR;
- R4 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR;
- R5 is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, aralkyl, each of which is optionally substituted.
- the compound is of formula (la). In other embodiments, the compound is of formula (lb). In still further embodiments, the compound is of formula (Ic).
- R3 is OH and R2 is hydrogen. In additional embodiments, R3 is OH and R4 is hydrogen.
- Z is O. In other embodiments, Z is NH.
- R5 is selected from the group consisting of alkyl, cycloalkyl, and aralkyl.
- Illustrative embodiments provide for formulae (la), (lb), and (Ic) compounds wherein R5 is selected from the group consisting of isopropyl, isobutyl, cyclopentyl, and cyclohexyl.
- the present disclosure provides a linker-payload compound having a structure according to formula (Ila), (lib), or (lie): or a pharmaceutically acceptable salt thereof.
- X and Y are independently selected from N and CH; Z is O or NH; m is 1, 2, or 3;
- R2 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR;
- R3 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR
- R4 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR;
- Rs is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, aralkyl, each of which is optionally substituted;
- LI when present, is a stable or self-immolative linker
- L2 when present, is a linker that is stable or is cleavable by an endosomal/lysosomalenzyme
- L3 when present, is a moiety that modulates the hydrophilicity, physical, and/or chemical properties of the linker-payload compound
- B is a reactive moiety for conjugation to a target protein, antibody, or antigen-binding fragment thereof, comprising at least one member selected from the group consisting of amino, hydroxyl, thiol, a maleimido moiety, a succinyl ester, an activated ester, an
- R3 is OH and R2 is hydrogen. In additional embodiments, R3 is OH and R4 is hydrogen.
- linkers LI, L2, and L3 are independently and optionally present.
- the linkers in the aggregate comprise generally a linker moiety linked to the payload drug.
- L3 is present and is a polyethylene glycol (PEG) unit, a carbohydrate moiety, or a combination thereof.
- PEG polyethylene glycol
- linkers for use herein may be found, for example, in Antibody- Drug conjugates and Immunotoxins, Phillips, G.
- the linker provided herein is sufficiently stable to exploit the circulating halflife of the antigen binding domain and, at the same time, capable of releasing the payload after antigen-mediated internalization of the conjugate, i.e., of Formula (I) or (II).
- Linkers can be cleavable or non-cleavable.
- Cleavable linkers for use herein include linkers that are cleaved by intracellular metabolism following internalization, e.g., cleavage via hydrolysis, reduction, or enzymatic reaction, such as by an endosomal/lysosomalenzyme.
- Non-cleavable linkers for use herein include linkers that release an attached payload via lysosomal degradation of the antigen binding domain following internalization.
- Illustrative linkers include, but are not limited to, acid-labile linkers, hydrolysis-labile linkers, enzymatically cleavable linkers, reduction labile linkers, self-immolative linkers, and non-cleavable linkers.
- Suitable linkers also include, but are not limited to, those that are or comprise peptides, carbohydrates, glucuronides, polyethylene glycol (PEG) units, hydrazones, mal-caproyl units, dipeptide units, valine-citruline units, and para-aminobenzyl (PAB) units.
- linker molecule or linker technology known in the art can be used within the definitions of LI, L2, and L3.
- a linker is a cleavable linker. In other embodiments, the linker is a non-cleavable linker.
- linkers can comprise or consist of e.g., MC (6-maleimidocaproyl), MP (maleimidopropanoyl), val-cit (valine-citrulline), val-ala (valine-alanine), dipeptide site in protease -cleavable linkers, ala- phe (alanine-phenylalanine), dipeptide site in protease-cleavable linkers, PAB (p- aminobenzyloxycarbonyl), and variants and combinations thereof. Additional examples of linkers that can be used are disclosed, e.g., in U.S. Pat. No. 7,754,681 and in Ducry, Bioconjugate Chem., 2010, 21:5-13, and the references cited therein.
- the linkers are stable in physiological conditions.
- the linkers are cleavable, for instance, able to release at least the payload portion in the presence of an enzyme or at a particular pH range or value.
- a linker comprises an enzyme-cleavable moiety.
- enzyme- cleavable linkers include, but are not limited to, peptide bonds, ester linkages, and hydrazones.
- the L linker comprises a cathepsin-cleavable linker.
- a linker comprises a non-cleavable moiety.
- the linker comprises one or more amino acids. Suitable amino acids include natural, non-natural, standard, non-standard, proteinogenic, non-proteinogenic, and L- or D-a-amino acids.
- the linker comprises alanine, valine, glycine, leucine, isoleucine, methionine, tryptophan, phenylalanine, proline, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, lysine, arginine, histidine, or citrulline, a derivative thereof, or combination thereof.
- one or more side chains of the amino acids is linked to a side chain group, described below.
- the linker comprises valine and citrulline. In some embodiments, the linker comprises lysine, valine, and citrulline. In some embodiments, the linker comprises lysine, valine, and alanine. In some embodiments, the linker comprises valine and alanine.
- the linker comprises a self-immolative group.
- a self- immolative group, a self-immolative linker, or a self-immolative spacer can be any such group known to those of skill in the art.
- a self-immolative linker displays an important role in the cascade mechanism of release of the compound linked. It is defined as a covalent group, which has the role of cleaving two bonds between a protector group and a drug, in the case of drug delivery systems, after a stimulus.
- the stimulus may include enzyme triggers, chemical triggers, such as pH changes, redox systems, 1,4-, 1,6-, 1,8-eliminations, photodegradable triggers, and combinations thereof, among others.
- the self-immolative group is p-aminobenzyl (PAB) or a derivative thereof.
- PAB p-aminobenzyl
- Useful derivatives include p-aminobenzyloxycarbonyl (PABC).
- a linker L3 comprises one or more enhancement groups.
- the enhancement group is linked to the side chain of any amino acid in the linker.
- amino acids for linking enhancement groups include lysine, asparagine, aspartate, glutamine, glutamate, and citrulline.
- the link to the enhancement group can be a direct bond to the amino acid side chain, or the link can be indirect via a spacer and/or reactive group. In one embodiment, spacers and reactive groups include any described herein.
- the enhancement group can be any group that modulates an existing or imparts a beneficial effect to the payload, linker payload, or conjugate including, but not limited to, biological, biochemical, hydrophilicity, synthetic, solubilizing, imaging, detecting, and reactivity effects, and the like.
- the enhancement group is a hydrophilic group.
- the enhancement group is a cyclodextrin.
- the enhancement group is an alkyl, heteroalkyl, alkenyl, heteroalkenyl sulfonic acid, heteroalkenyl taurine, heteroalkenyl phosphoric acid or phosphate, heteroalkenyl amine (e.g., quaternary amine), or heteroalkenyl sugar.
- sugars include, without limitation, monosaccharides, disaccharides, and polysaccharides. Exemplary monosaccharides include glucose, ribose, deoxyribose, xylose, arabinose, mannose, galactose, fructose, and the like.
- sugars include sugar acids such as glucuronic acid, further including conjugated forms such as glucuronides (i.e., via glucuronidation).
- exemplary disaccharides include maltose, sucrose, lactose, lactulose, trehalose, and the like.
- Exemplary polysaccharides include amylose, amylopectin, glycogen, inulin, cellulose, and the like.
- the cyclodextrin can be any cyclodextrin known to those of skill. In some embodiments, the cyclodextrin is alpha cyclodextrin, beta cyclodextrin, or gamma cyclodextrin, or mixtures thereof.
- the cyclodextrin is alpha cyclodextrin. In some embodiments, the cyclodextrin is beta cyclodextrin. In some embodiments, the cyclodextrin is gamma cyclodextrin. In some embodiments, the enhancement group is capable of improving solubility of the remainder of the conjugate. In some embodiments, the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is substituted or non-substituted.
- the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is -(Ofeji-sSChH, -(CH2)n-NH-(CH2)i-sSO3H, -(CH2) n - C(O)NH-(CH 2 )i-5SO 3 H,-(CH2CH2O) m -C(O)NH-(CH2)i-5SO3H, -(CH 2 ) n -N((CH 2 )i- 5 C(O)NH(CH 2 )I-5SO3H)2, -(CH2)n-C(O)N((CH 2 )i-5C(O)NH(CH 2 )i-5SO3H)2, or - (CH 2 CH2O)m-C(O)N((CH2)i-5C(O)NH(CH 2 )i-5SO3H)2, wherein n is 1, 2, 3, 4, or 5, and m is 1, 2, 3, 4, or 5.
- the alkyl or alkenyl sulfonic acid is -(Qfcji-sSChH.
- the heteroalkyl or heteroalkenyl sulfonic acid is -(CH2) n -NH-(CH2)i- 5SO3H, wherein n is 1, 2, 3, 4, or 5.
- the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is -(CH2)n-C(O)NH-(CH2)i-5SO3H, wherein n is 1, 2, 3, 4, or 5.
- the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is - (CH2CH2O)m-C(O)NH-(CH2)i-5SO3H, wherein m is 1, 2, 3, 4, or 5.
- the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is -(CH2) n -N((CH2)i- 5C(O)NH(CH 2 )I-5SO3H) 2 , wherein n is 1, 2, 3, 4, or 5.
- the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is -(CH2) n -C(O)N((CH2)i- 5 C(O)NH(CH2)I-5SO 3 H)2, wherein n is 1, 2, 3, 4, or 5.
- the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is -(CH2CH2O) m -C(O)N((CH2)i- 5C(O)NH(CH 2 )I- 5 SO 3 H)2, wherein m is 1, 2, 3, 4, or 5.
- Moiety B as described herein is a reactive moiety for conjugation to a target protein, antibody, or antigen-binding fragment thereof.
- B comprises a maleimido group (for conjugation with a thiol, e.g., cysteine, of an antigen binding domain), an N-hydroxysuccinimido ester (for conjugation with an amine, e.g., lysine, of an antigen binding domain), or cyclooctynyl group (for conjugation with an antigen binding domain using click chemistry). See, e.g., WO 2020/132658; Ohio et al. Methods Mol. Biol. 2020, 2078:83-87.
- B contains a maleimido group.
- the maleimido group in B reacts with a cysteine residue on an antigen binding domain to form a carbon-sulfur bond.
- B contains an N-hydroxysuccinimido ester group.
- the N-hydroxysuccinimido ester group reacts with a lysine residue on an antigen binding domain to form an amide bond.
- B contains a functional group or moiety that is capable of undergoing a click chemistry reaction (see, e.g., Click Chemistry, Huisgen Proc. Chem. Soc. 1961,357-396; Wang et al. J. Am. Chem. Soc. 2003, 125(11), 3192-3193; and Agard et al. J. Am. Chem. Soc. 2004, 126(46), 15046-15047).
- B contains an alkyne which can react via click chemistry with an azide, such as on a modified antigen binding domain, to form a click chemistry product.
- the alkyne group reacts with an azide.
- the reactive group is an alkyne that is capable of undergoing a 1,3 -cycloaddition reaction with an azide.
- Alkynes that are useful in such embodiments include strained alkynes, e.g., those suitable for strain-promoted alkyneazide cycloadditions (SPAAC), cycloalkynes, e.g., cyclooctynes, benzannulated alkynes, and alkynes capable of undergoing 1,3-cycloaddition reactions with alkynes in the absence of copper catalysts.
- SPAAC strain-promoted alkyneazide cycloadditions
- Alkynes that may be used in such embodiments also include, but are not limited to, dibenzoazacyclooctyne, dibenzocyclooctyne, biarylazacyclooctynone, difluorinated cyclooctyne, substituted, e.g., fluorinated alkynes, aza-cycloalkynes and bicyclo[6.1.0]nonyne.
- alkynes are useful for conjugating antibodies that have been functionalized with azido groups.
- Such functionalized antibodies include antibodies functionalized with azido-polyethylene glycol groups.
- such a functionalized antibody is derived by treating an antibody having at least one glutamine residue, e.g., heavy chain Gln295, with a compound bearing an amino group and an azide group, in the presence of the enzyme transglutaminase.
- glutamine-modified antibody refers to an antibody with at least one covalent linkage from a glutamine side chain to a primary amine compound of the present disclosure.
- the primary amine compound is linked through an amide linkage on the glutamine side chain.
- the glutamine is an endogenous glutamine.
- the glutamine is an endogenous glutamine made reactive by polypeptide engineering (e.g., via amino acid deletion, insertion, substitution, or mutation on the polypeptide).
- the glutamine is polypeptide engineered with an acyl donor glutamine-containing tag (e.g., glutamine- containing peptide tags, Q- tags or TGase recognition tag).
- TGase recognition tag refers to a sequence of amino acids comprising an acceptor glutamine residue and that when incorporated into (e.g., appended to) a polypeptide sequence, under suitable conditions, is recognized by a TGase and leads to cross-linking by the TGase through a reaction between an amino acid side chain within the sequence of amino acids and a reaction partner.
- the recognition tag may be a peptide sequence that is not naturally present in the polypeptide comprising the TGase recognition tag.
- the TGase recognition tag comprises at least one Gin.
- the TGase recognition tag comprises an amino acid sequence XXQX (SEQ ID NO: 1935), wherein X is any amino acid (e.g., conventional amino acid Leu, Ala, Gly, Ser, Vai, Phe, Tyr, His, Arg, Asn, Glu, Asp, Cys, Gin, He, Met, Pro, Thr, Lys, or Trp or nonconventional amino acid).
- X is any amino acid (e.g., conventional amino acid Leu, Ala, Gly, Ser, Vai, Phe, Tyr, His, Arg, Asn, Glu, Asp, Cys, Gin, He, Met, Pro, Thr, Lys, or Trp or nonconventional amino acid).
- the acyl donor glutamine-containing tag comprises an amino acid sequence selected from the group consisting of LLQGG (SEQ ID NO: 1936), LLQG (SEQ ID NO: 1937), LSLSQG (SEQ ID NO: 1938), gGGLLQGG (SEQ ID NO: 1939), gLLQG (SEQ ID NO: 1940), LLQ, gSPLAQSHGG (SEQ ID NO: 1941), gLLQGGG (SEQ ID NO: 1942), gLLQGG (SEQ ID NO: 1943), gLLQ (SEQ ID NO: 1944), LLQLLQGA (SEQ ID NO: 1945), LLQGA (SEQ ID NO: 1946), LLQYQGA (SEQ ID NO: 1947), LLQGSG (SEQ ID NO: 1936), LLQG (SEQ ID NO: 1937), LSLSQG (SEQ ID NO: 1938), gGGLLQGG (SEQ ID NO: 1939), gLLQG (SEQ ID NO: 1940), LLQ
- B is selected from 2-maleimido-l -ethyl, 2-maleimidoacetyl, and 3-maleimidopropanoyl.
- the linker-payload compound is selected from those, and their pharmaceutically acceptable salts, in Table 2. [0101] Table 2. Structures of Linker-Payloads (LP) or a pharmaceutically acceptable salt thereof.
- BA is an antibody or an antigen-binding fragment thereof
- L is a linker of the formula -L1-L2-L3-B-, wherein
- L is connected to BA through a side chain of an amino acid selected from the group consisting of Gin (-CO-NH-), Lys (-NH-CO-), and Cys (-S-);
- LI when present, is a stable or self-immolative linker
- L2 when present, is a linker that is stable or is cleavable by an endosomal/lysosomalenzyme
- L3 when present, is a moiety that modulates the hydrophilicity, physical, and/or chemical properties of -L-P;
- B is a reactive moiety for conjugation to a target protein, antibody, or antigen-binding fragment thereof, comprising at least one member selected from the group consisting of amino, hydroxyl, thiol, a maleimido moiety, a succinyl ester, an activated ester, an alkylamino; a haloacetamido, -
- P is a moiety having a structure according to Formula (la’), Formula (lb’), or Formula
- X and Y are independently selected from N and CH;
- R2 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR;
- R3 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR;
- R4 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR;
- R5 is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, aralkyl, each of which is optionally substituted; and represents the connecting point of attachment to the linker L; and n is an integer from 1 to 20.
- LI is a self-immolative linker.
- An illustrative self-immolative linker is the para-aminobenzyl (PAB) moiety.
- L2 is a linker cleavable by an endosomal/lysosomalenzyme.
- the linker comprises a peptide unit comprising two to four amino acid residues selected from the group consisting of glycine (G), alanine (A), valine (V), phenylalanine (F), proline (P), glutamic acid (E), lysine (K), arginine (R), citrulline (Cit), and combinations thereof.
- Exemplary embodiments include a peptide unit that comprises GGFG, VA, V-Cit, GG, GA, GV, AG, VG, AV, AA, EVA, and EV-Cit.
- the endosomal/lysosomalenzyme is cathepcin B.
- linker that covalently connects (i) and (ii), wherein the linker is connected to the BA or antigen-binding fragment thereof through a side chain of an amino acid selected from the group consisting of Gin (-CO-NH-), Lys (-NH-CO-), and Cys (-S-), and has a formula -Ll- L2-L3-B-, wherein
- LI when present, is a stable or self-immolative linker
- L2 when present, is a linker that is stable or is cleavable by an endosomal/lysosomalenzyme
- L3 when present, is a moiety that modulates the hydrophilicity, physical, and/or chemical properties of the conjugate
- B is a residue of a reactive moiety for conjugation, comprising at least one member selected from the group consisting of amino, hydroxyl, thiol, a maleimido moiety, a succinyl ester, an activated ester, an alkylamino; a haloacetamido, -N3, ⁇ and the payload has a structure according to Formula (la’), Formula (lb’), or Formula (Ic’):
- X and Y are independently selected from N and CH;
- R2 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR;
- R3 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR;
- R4 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR;
- Rs is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, aralkyl, each of which is optionally substituted; and represents the connecting point of attachment to the linker.
- LI is a self-immolative linker of para-aminobenzyl (PAB) moiety.
- L2 is a linker cleavable by an endosomal/lysosomalenzyme, wherein the linker comprises a peptide unit comprising two to four amino acid residues selected from the group consisting of glycine (G), alanine (A), valine (V), phenylalanine (F), proline (P), glutamic acid (E), lysine (K), arginine (R), citrulline (Cit), and combinations thereof.
- the peptide unit comprises GGFG, VA, V-Cit, GG, GA, GV, AG, VG, AV, AA, EVA, and EV-Cit.
- L3 is present and L3 is a polyethylene glycol (PEG) unit, a carbohydrate moiety, or a combination thereof.
- PEG polyethylene glycol
- the BA is an anti-HER2 antibody, an anti-STEAP2 antibody, an anti-MET antibody, an anti-EGFRvIII antibody, an anti-MUC16 antibody, an anti-PRLR antibody, an anti-PSMA antibody, an anti-FGFR2 antibody, an anti-FOLRl antibody, an anti-HER2/HER2 bispecific antibody, an anti HER2/APLP2 bispecific antibody, an anti- MET/MET bispecific antibody, CD33, CD30, CD22, CD79b, Nectin-4, TROP2, BCMA, CD19, Tissue Factor, or an antigen-binding fragment thereof.
- the BA targets a cancer selected from the group consisting of breast cancer, ovarian cancer, prostate cancer, lung cancer, liver cancer, lymphomas, urothelial, cervical, multiple myeloma, gastric, or brain cancer. More specific and illustrative embodiments are described now.
- the BA is an antibody comprising an Fc region modified to enhance binding affinity to FcyR.
- BA is an antibody with one or more mutations selected from F243L, R292P, Y300L, V305I, and P396L.
- BA is an antibody with one or more mutation selected from S239D and I332E.
- BA is an antibody with one or more mutations selected from S239D, I332E, and A330L.
- BA is an antibody with one or more mutations selected from S298A, E333A and K334A.
- BA is an antibody with one or more mutations selected from L234Y, L235Q, G236W, S239M, H268D, D270E, and S298A. In some embodiments, BA is an antibody with one or more mutations selected from D270E, K326D, A330M, and K334E. In some embodiments, BA is an antibody with L234Y, L235Q, G236W, S239M, H268D, D270E, and S298A in one heavy chain and D270E, K326D, A330M, and K334E in the opposing heavy chain.
- BA is an antibody with one or more mutations selected from G236A, S239D, and I332E. In some embodiments, BA is an antibody with one or more mutations selected from M252Y, S254T, and T256E. In some embodiments, BA is an antibody with one or more mutations selected from M428L and N434S. In some embodiments, BA is an antibody with one or more mutations selected from S267E and L328F. In some embodiments, BA is an antibody with one or more mutations selected from N325S and L328F.
- BA is an antibody that comprises a glutamine residue.
- Antibodies comprising glutamine residues can be isolated from natural sources or engineered to comprise one or more glutamine residues. Techniques for engineering glutamine residues into an antibody polypeptide chain (glutaminyl-modified antibodies) are within the skill of the practitioners in the art.
- BA is an N297Q mutant antibody.
- Z is an antibody that has one or more engineered LLQG, LLQGG, LLQLLQG, LLQYQG, LLQGA, LLQGSG, SLLQG, LQG, LLQLQ, LLQLLQ, LLQGR, LLQYQGA, LQGG, LGQG or LLQLLQGA sites. See, e.g., U.S. Patent No. 9,676,871 and U.S. Patent Application Publication No. 2003/0138785.
- the antibody BA is aglycosylated. In other embodiments, the antibody BA is glucosylated.
- BA is an antibody that is a monoclonal antibody, human antibody, humanized antibody, camelised antibody, or chimeric antibody.
- BA is an antibody of any isotype (e.g., IgG, IgE, IgM, IgD, IgA and IgY), class (e.g., IgGl, IgG2, IgG3, IgG4, IgAl and IgA2) or subclass.
- BA has a molecular weight of at least 500, 600, 700, 800, 900, 1000, 10000, 50000 or 100000 Daltons.
- BA can include antibodies, antigen-binding fragments of antibodies, peptides that specifically interact with a particular antigen (e.g., peptibodies), receptor molecules that specifically interact with a particular antigen, proteins comprising a ligand-binding portion of a receptor that specifically binds a particular antigen, antigenbinding scaffolds (e.g., DARPins, HEAT repeat proteins, ARM repeat proteins, tetratricopeptide repeat proteins, and other scaffolds based on naturally occurring repeat proteins, etc., (see, e.g., Boersma and Pluckthun, 2011, Curr. Opin. Biotechnol. 22:849-857, and references cited therein)), and aptamers or portions thereof.
- BA comprises a scFv having binding specificity to a target antigen.
- an antigen-binding domain includes polypeptides that bind a target antigen or a portion thereof with a KD of less than about 500 pM, less than about 400 pM, less than about 300 pM, less than about 200 pM, less than about 100 pM, less than about 90 pM, less than about 80 pM, less than about 70 pM, less than about 60 pM, less than about 50 pM, less than about 40 pM, less than about 30 pM, less than about 20 pM, less than about 10 pM, less than about 5 pM, less than about 4 pM, less than about 2 pM, less than about 1 pM, less than about 0.5 pM, less than about 0.2 pM, less than about 0. 1 pM,
- the framework regions (FRs) of the antibodies or antigenbinding fragment thereof for use in the conjugates provided herein may be identical to the human germline sequences, or may be naturally or artificially modified.
- An amino acid consensus sequence may be defined based on a side-by-side analysis of two or more CDRs.
- CDRs within HCVR and LCVR amino acid sequences are well known in the art and can be used to identify CDRs.
- Exemplary conventions that can be used to identify the boundaries of CDRs include, e.g., the Kabat definition, the Chothia definition, and the AbM definition.
- the Kabat definition is based on sequence variability
- the Chothia definition is based on the location of the structural loop regions
- the AbM definition is a compromise between the Kabat and Chothia approaches. See, e.g., Kabat, "Sequences of Proteins of Immunological Interest," National Institutes of Health, Bethesda, Md. (1991); Al-Lazikani et al., J.
- the antigen-binding domains for use in the conjugates provided herein may comprise or consist of antigen-binding fragments of full antibody molecules.
- Antigen-binding fragments of an antibody may be derived, e.g., from full antibody molecules using any suitable standard techniques such as proteolytic digestion or recombinant genetic engineering techniques involving the manipulation and expression of DNA encoding antibody variable and optionally constant domains.
- DNA is known and/or is readily available from, e.g., commercial sources, DNA libraries (including, e.g., phage-antibody libraries), or can be synthesized.
- the DNA may be sequenced and manipulated chemically or by using molecular biology techniques, for example, to arrange one or more variable and/or constant domains into a suitable configuration, or to introduce codons, create cysteine residues, modify, add or delete amino acids, etc.
- Non-limiting examples of antigen-binding fragments for use in the conjugates provided herein include: (i) Fab fragments; (ii) F(ab’)2 fragments; (iii) Fd fragments; (iv) Fv fragments; (v) single-chain Fv (scFv) molecules; (vi) dAb fragments; and (vii) minimal recognition units consisting of the amino acid residues that mimic the hypervariable region of an antibody (e.g., an isolated complementarity determining region (CDR) such as a CDR3 peptide), or a constrained FR3-CDR3-FR4 peptide.
- CDR complementarity determining region
- an antigen-binding fragment of an antibody includes other engineered molecules, such as domain-specific antibodies, single domain antibodies, domain-deleted antibodies, chimeric antibodies, CDR- grafted antibodies, diabodies, triabodies, tetrabodies, minibodies, nanobodies (e.g., monovalent nanobodies, bivalent nanobodies, etc.), small modular immunopharmaceuticals (SMIPs), and shark variable IgNAR domains.
- SMIPs small modular immunopharmaceuticals
- an antigen-binding fragment of an antibody will comprise at least one variable domain.
- the variable domain may be of any size or amino acid composition and will generally comprise at least one CDR which is adjacent to or in frame with one or more framework sequences.
- the VH and VL domains may be situated relative to one another in any suitable arrangement.
- the variable region may be dimeric and contain VH-VH, VH-VL or VL-VL dimers.
- the antigen-binding fragment of an antibody may contain a monomeric VH or VL domain.
- an antigen-binding fragment of an antibody may contain at least one variable domain covalently linked to at least one constant domain.
- variable and constant domains that may be found within an antigen-binding fragment of an antibody for use in the conjugates provided herein include: (i) VH-CH1 ; (ii) VH-CH2; (iii) VH-CH3; (iv) VH-CH1-CH2; (V) VH-CH1-CH2-CH3; (vi) VH-CH2- CH3; (vii) VH-CL; (viii) VL-CH1; (ix) VL-CH2; (X) VL-CH3; (xi) VL-CH1-CH2; (xii) VL-CH1- CH2-CH3; (xiii) VL-CH2-CH3; and (xiv) VL-CL- In any configuration of variable and constant domains, including any of the exemplary configurations listed
- a hinge region may consist of at least 2 (e.g., 5, 10, 15, 20, 40, 60 or more) amino acids which result in a flexible or semi-flexible linkage between adjacent variable and/or constant domains in a single polypeptide molecule.
- an antigen-binding fragment may comprise a homo-dimer or hetero-dimer (or other multimer) of any of the variable and constant domain configurations listed above in non- covalent association with one another and/or with one or more monomeric VH or VL domain (e.g., by disulfide bond(s)).
- the antigen-binding domains used in the conjugates provided herein may comprise or consist of human antibodies and/or recombinant human antibodies, or antigen-binding fragments thereof.
- the antigen-binding domains used in the conjugates provided herein may comprise or consist of recombinant human antibodies or antigen-binding fragments thereof.
- such recombinant human antibodies have variable and constant regions derived from human germline immunoglobulin sequences.
- such recombinant human antibodies are subjected to in vitro mutagenesis (or, when an animal transgenic for human Ig sequences is used, in vivo somatic mutagenesis) and thus the amino acid sequences of the VH and VL regions of the recombinant antibodies are sequences that, while derived from and related to human germline VH and VL sequences, may not naturally exist within the human antibody germline repertoire in vivo.
- the antigen-binding domains used in the conjugates provided herein also include bispecific antigen-binding molecules, such as bispecific antibodies.
- bispecific formats that can be used in the context of the present disclosure include, without limitation, e.g., scFv-based or diabody bispecific formats, IgG-scFv fusions, dual variable domain (DVD)-Ig, Quadroma, knobs-into-holes, common light chain (e.g., common light chain with knobs-into-holes, etc.), CrossMab, CrossFab, (SEED) body, leucine zipper, Duobody, IgGl/IgG2, dual acting Fab (DAF)-IgG, and Mab 2 bispecific formats (see, e.g., Klein et al. 2012, mAbs 4:6, 1-11, and references cited therein, for a review of the foregoing formats). See also, e.g.,
- bispecific antigen binding molecules may comprise a first antigen-binding domain (also referred to herein as "DI"), and a second antigen-binding domain (also referred to herein as "D2").
- DI first antigen-binding domain
- D2 second antigen-binding domain
- DI and D2 domains of a bispecific antibody are non-competitive with one another. Non-competition between DI and D2 means that, the respective monospecific antigen binding proteins from which DI and D2 were derived do not compete with one another for binding to the target.
- Exemplary antigen-binding protein competition assays are known in the art.
- DI and D2 bind to different (e.g., nonoverlapping, or partially overlapping) epitopes on the target.
- Bispecific antigen-binding molecules may be constructed using the antigen-binding domains of two separate monospecific antibodies.
- a collection of monoclonal monospecific antibodies may be produced using standard methods known in the art. The individual antibodies thus produced may be tested pairwise against one another for cross-competition to the target protein.
- a bispecific antigen-binding molecule can be a single multifunctional polypeptide, or it can be a multimeric complex of two or more polypeptides that are covalently or non-covalently associated with one another. Any antigen binding construct which has the ability to simultaneously bind two separate, non-identical epitopes of the target molecule is regarded as a bispecific antigen-binding molecule.
- Bispecific antigenbinding molecules may be constructed using standard molecular biological techniques (e.g., recombinant DNA and protein expression technology) as will be known to a person of skill in the art.
- bispecific antibodies are also provided wherein one arm of the bispecific antibody binds to an epitope on a first target protein, and the other arm of the bispecific antibody binds to a second epitope on a second target protein.
- bispecific formats that can be used in the context of the present disclosure include, without limitation, e.g., scFv-based or diabody bispecific formats, IgG-scFv fusions, dual variable domain (DVD)-Ig, Quadroma, knobs-into-holes, common light chain (e.g., common light chain with knobs-into-holes, etc.), CrossMab, CrossFab, (SEED)body, leucine zipper, Duobody, IgGl/IgG2, dual acting Fab (DAF)-IgG, and Mab2 bispecific formats (see, e.g., Klein et al.
- Bispecific antibodies can also be constructed using peptide/nucleic acid conjugation, e.g., wherein unnatural amino acids with orthogonal chemical reactivity are used to generate site-specific antibody-oligonucleotide conjugates which then self-assemble into multimeric complexes with defined composition, valency and geometry. (See, e.g., Kazane et al., J. Am. Chem. Soc. (Epub: Dec. 4, 2012)).
- the antigen binding domains for use in the conjugates provided herein also include antibodies comprising variants of any of the HCVR, LCVR, and/or CDR amino acid sequences known in the art.
- variants include variants of any of the HCVR, LCVR, and/or CDR amino acid sequences known in the art having one or more conservative substitutions.
- the antigen binding domains include antibodies or antigen binding fragments thereof having HCVR, LCVR, and/or CDR amino acid sequences with, e.g., 10 or fewer, 8 or fewer, 6 or fewer, 4 or fewer, etc.
- the antigen binding domains include antibodies or antigen binding fragments thereof also include variants having substantial sequence identity to any of the HCVR, LCVR, and/or CDR amino acid sequences known in the art.
- residue positions which are not identical differ by conservative amino acid substitutions.
- a "conservative amino acid substitution” is one in which an amino acid residue is substituted by another amino acid residue having a side chain with similar chemical properties (e.g., charge or hydrophobicity).
- a conservative amino acid substitution will not substantially change the functional properties of a protein.
- the percent sequence identity or degree of similarity may be adjusted upwards to correct for the conservative nature of the substitution. Means for making this adjustment are well-known to those of skill in the art. See, e.g., Pearson (1994) Methods Mol. Biol. 24: 307-331.
- Examples of groups of amino acids that have side chains with similar chemical properties include (1) aliphatic side chains: glycine, alanine, valine, leucine and isoleucine; (2) aliphatic-hydroxyl side chains: serine and threonine; (3) amide-containing side chains: asparagine and glutamine; (4) aromatic side chains: phenylalanine, tyrosine, and tryptophan; (5) basic side chains: lysine, arginine, and histidine; (6) acidic side chains: aspartate and glutamate, and (7) sulfur-containing side chains are cysteine and methionine.
- conservative amino acids substitution groups are: valine-leucine -isoleucine, phenylalanine- tyrosine, lysine-arginine, alanine-valine, glutamate-aspartate, and asparagine-glutamine.
- a conservative replacement is any change having a positive value in the PAM250 log-likelihood matrix disclosed in Gonnet et al. (1992) Science 256: 1443-1445.
- a "moderately conservative" replacement is any change having a nonnegative value in the PAM250 log-likelihood matrix.
- Sequence identity between two different amino acid sequences is typically measured using sequence analysis software. Sequence analysis software matches similar sequences using measures of similarity assigned to various substitutions, deletions and other modifications, including conservative amino acid substitutions.
- GCG software contains programs such as GAP and BESTFIT which can be used with default parameters to determine sequence homology or sequence identity between closely related polypeptides, such as homologous polypeptides from different species of organisms or between a wild type protein and a mutein thereof. See, e.g., GCG Version 6.1. Polypeptide sequences also can be compared using FASTA using default or recommended parameters, a program in GCG Version 6.1.
- FASTA e.g., FASTA2 and FASTA3
- FASTA2 and FASTA3 provides alignments and percent sequence identity of the regions of the best overlap between the query and search sequences (Pearson (2000) supra).
- Another algorithm when comparing a sequence provided herein to a database containing a large number of sequences from different organisms is the computer program BLAST, especially BLASTP or TBLASTN, using default parameters. See, e.g., Altschul et al. (1990) J. Mol. Biol. 215:403-410 and Altschul et al. (1997) Nucleic Acids Res. 25:3389- 402.
- the antigen-binding domains for use in the conjugates provided herein encompass proteins having amino acid sequences that vary from those of the described antibodies but that retain the ability to bind the target proteins.
- Such variant antigen-binding domains comprise one or more additions, deletions, or substitutions of amino acids when compared to parent sequence but exhibit biological activity that is essentially equivalent to that of the described antibodies.
- Two antigen-binding domains are considered bioequivalent if, for example, they are pharmaceutical equivalents or pharmaceutical alternatives whose rate and extent of absorption do not show a significant difference when administered at the same molar dose under similar experimental conditions, either single dose or multiple doses.
- Some antigenbinding domains will be considered equivalents or pharmaceutical alternatives if they are equivalent in the extent of their absorption but not in their rate of absorption and yet may be considered bioequivalent because such differences in the rate of absorption are intentional and are reflected in the labeling, are not essential to the attainment of effective body drug concentrations on, e.g., chronic use, and are considered medically insignificant for the particular drug product studied.
- two antigen-binding domains are bioequivalent if there are no clinically meaningful differences in their safety, purity, and potency.
- two antigen-binding domains are bioequivalent if a patient can be switched one or more times between the reference product and the biological product without an expected increase in the risk of adverse effects, including a clinically significant change in immunogenicity, or diminished effectiveness, as compared to continued therapy without such switching.
- two antigen-binding domains are bioequivalent if they both act by a common mechanism or mechanisms of action for the condition or conditions of use, to the extent that such mechanisms are known.
- Bioequivalence may be demonstrated by in vivo and in vitro methods.
- Bioequivalence measures include, e.g., (a) an in vivo test in humans or other mammals, in which the concentration of the antigen-binding domain or its metabolites is measured in blood, plasma, serum, or other biological fluid as a function of time; (b) an in vitro test that has been correlated with and is reasonably predictive of human in vivo bio availability data; (c) an in vivo test in humans or other mammals in which the appropriate acute pharmacological effect of the antigen-binding domain (or its target) is measured as a function of time; and (d) in a well-controlled clinical trial that establishes safety, efficacy, or bioavailability or bioequivalence of an antigen-binding domain.
- Bioequivalent variants of antigen-binding domains for use in the conjugates provided herein may be constructed by, for example, making various substitutions of residues or sequences or deleting terminal or internal residues or sequences not needed for biological activity.
- cysteine residues not essential for biological activity can be deleted or replaced with other amino acids to prevent formation of unnecessary or incorrect intramolecular disulfide bridges upon renaturation.
- bioequivalent antigenbinding domains may include variants comprising amino acid changes which modify the glycosylation characteristics of the antigen -binding domain, e.g., mutations which eliminate or remove glycosylation.
- the antigen-binding domains for use in the conjugates provided herein bind to a human target protein but not to target protein from other species. In other embodiments, the antigen-binding domains for use in the conjugates provided herein bind to a human target protein and to a target protein from one or more non-human species.
- the antigen-binding domains for use in the conjugates provided herein may bind to a human target protein and may bind or not bind, as the case may be, to one or more of mouse, rat, guinea pig, hamster, gerbil, pig, cat, dog, rabbit, goat, sheep, cow, horse, camel, cynomolgus, marmoset, rhesus or chimpanzee target protein.
- the antigen-binding domains specifically bind human target protein and cynomolgus monkey (e.g., Macacafascicularis) target protein.
- antigen-binding domains for use herein bind human target protein but do not bind, or bind only weakly, to cynomolgus monkey target protein.
- the BA can be linked in the conjugate through an attachment at a particular amino acid within the BA.
- exemplary amino acid attachments that can be used in the context of this embodiment of the disclosure include, e.g., lysine (see, e.g., US 5,208,020; US 2010/0129314; Hollander et al., Bioconjugate Chem., 2008, 19:358-361 ; WO 2005/089808; US 5,714,586; US 2013/0101546; and US 2012/0585592), cysteine (see, e.g., US 2007/0258987; WO 2013/055993; WO 2013/055990; WO 2013/053873; WO 2013/053872; WO 2011/130598; US 2013/0101546; and US 7,750,116), selenocysteine (see, e.g., WO 2008/122039; and Hofer et al., Proc.
- Linkers can also be conjugated to an BA via attachment to carbohydrates (see, e.g., US 2008/0305497, WO 2014/065661, Ryan et al., Food & Agriculture Immunol., 2001, 73:127-130, and Jeger et al., Angew Chem IntEd Engl., 2010, 49:9995-9997).
- BA is bonded to the linker through a lysine residue.
- the antibody or antigen binding molecule is bonded to the linker through a cysteine residue, lysine residue, or glutamine residue.
- the BA is bonded to the linker through a cysteine residue.
- a linker maleimide moiety bonds to an antibody cysteine residue.
- the BA is bonded to the linker through a lysine residue.
- a linker N-hydroxysuccinimide moiety bonds to an antibody lysine residue to form an amide linkage.
- the BA is bonded to the linker through a glutamine residue see, e.g., Jeger et al., Angew Chem Int Ed Engl., 2010, 49:9995-9997 and Dennler et al., Bioconjugate Chem. 2014, 25:569-578).
- Antibodies comprising glutamine residues can be isolated from natural sources or engineered to comprise one or more glutamine residues.
- antibodies or antigen binding molecules are engineered by mutations, for example insertions or deletions to facilitate reaction via transglutaminase.
- antibodies or antigen binding molecules are engineered to remove one or more glycosylation sites.
- antibodies or antigen binding molecules are engineered to add one or more glutamine residues.
- glutamine residues are added within a TGase recognition tag, as described herein.
- Techniques for engineering glutamine residues into an antibody polypeptide chain are within the skill of the practitioners in the art.
- the antibody is aglycosylated.
- BA comprises at least one glutamine residue in at least one polypeptide chain sequence.
- BA comprises two heavy chain polypeptides, each with one Gln295 or Q295 residue.
- BA comprises one or more glutamine residues at a site other than a heavy chain 295. Included herein are antibodies of this section bearing N297Q mutation(s) described herein.
- a glutamine residue is added at the heavy chain C-terminus.
- the glutamine is polypeptide engineered with a glutamine- containing tag (e.g., glutamine-containing peptide tags, Q-tags or TGase recognition tag).
- a glutamine-containing tag e.g., glutamine-containing peptide tags, Q-tags or TGase recognition tag.
- the term “TGase recognition tag” or “Q-Tag” refers to a sequence of amino acids comprising a glutamine residue that when incorporated into (e.g., appended to) a polypeptide sequence, under suitable conditions, is recognized by a transglutaminase (“TGase”) and leads to crosslinking by the TGase through a reaction between an amino acid side chain within the sequence of amino acids and a reactive group.
- the recognition tag may be a peptide sequence that is not naturally present in the polypeptide.
- the TGase recognition tag comprises at least one glutamine.
- the TGase recognition tag comprises an amino acid sequence XXQX, wherein X is any amino acid (e.g., conventional amino acid Leu, Ala, Gly, Ser, Vai, Phe, Tyr, His, Arg, Asn, Glu, Asp, Cys, Gin, He, Met, Pro, Thr, Lys, or Trp or nonconventional amino acid).
- the TGase recognition tag comprises an amino acid sequence selected from the group consisting of LLQGG, LLQG, LSLSQG, GGGLLQGG, GLLQG, LLQ, GSPLAQSHGG, GLLQGGG, GLLQGG, GLLQ, LLQLLQGA, LLQGA, LLQYQGA, LLQGSG, LLQYQG, LLQLLQG, SLLQG, LLQLQ, LLQLLQ, and LLQGR.
- LLQGG amino acid sequence selected from the group consisting of LLQGG, LLQG, LSLSQG, GGGLLQGG, GLLQG, LLQ, GSPLAQSHGG, GLLQGGG, GLLQGG, GLLQ, LLQLLQGA, LLQGA, LLQYQGA, LLQGSG, LLQYQG, LLQLLQG, SLLQG, LLQLQ
- BA includes an antibody heavy chain and further includes a TGase recognition tag at the C-terminus of the antibody heavy chain. In some embodiments, BA includes an antibody heavy chain and further includes a TGase recognition tag at the C- terminus of the antibody heavy chain, wherein the TGase recognition tag is the pentapeptide sequence LLQGA. In some embodiments, BA includes two antibody heavy chains and further includes a TGase recognition tag at the C-terminus of each antibody heavy chain. In some embodiments, BA includes two antibody heavy chains and further includes a TGase recognition tag at the C-terminus of each antibody heavy chain, wherein the TGase recognition tag is the pentapeptide sequence LLQGA.
- the BA can be also modified at one or more glutamine residues via transglutaminase see, e.g., Jeger et al., Angew Chem Int Ed Engl., 2010, 49:9995-9997 and Dennler et al., Bioconjugate Chem. 2014, 25:569-578).
- transglutaminase one or more glutamine residues of an antibody can be coupled to a primary amine compound to provide a moiety capable of reacting with a reactive group on a linker-payload.
- the primary amine compound provides a diene or dienophile.
- the primary amine compound provides a diene or dienophile
- the linkerpayload provides a complementary dienophile or diene, respectively, for conjugation via a Diels-Alder reaction.
- the primary amine compound provides an azido group.
- the primary amine compound provides an azido group
- the linker-payload provides a complementary alkyne, for conjugation via a click reaction.
- the BA comprises a heavy chain and the heavy chain is linked to BA directly or indirectly via a linker. In some embodiments, the BA comprises a light chain and the light chain is linked to BA directly or indirectly via a linker. [0147] In some embodiments, the BA comprises a heavy chain and the C-terminus of the heavy chain is linked to BA directly or indirectly via a linker. In some embodiments, the BA comprises a light chain and the C-terminus of the light chain is linked to BA directly or indirectly via a linker.
- the BA comprises two heavy chains and each of the two heavy chains is linked to BA directly or indirectly via a linker. In some embodiments, the BA comprises two light chains and each of the two light chains is linked to BA directly or indirectly via a linker.
- the BA comprises two heavy chains and C-terminus of each of the two heavy chains is linked to BA directly or indirectly via a linker. In some embodiments, the BA comprises two light chains and C-terminus of each of the two light chains is linked to BA directly or indirectly via a linker.
- the epitope to which the antigen-binding domains bind may consist of a single contiguous sequence of 3 or more (e.g., 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 or more) amino acids of a target protein.
- the relevant epitope may consist of a plurality of non-contiguous amino acids (or amino acid sequences) of the target protein.
- the epitope is located on or near the binding domain of the target protein. In other embodiments, the epitope is located outside of the binding domain of the target protein.
- Various techniques known to persons of ordinary skill in the art can be used to determine the epitope with which the antigen-binding domains used in the ADCs provided herein interact.
- Exemplary techniques that can be used to determine an epitope or binding domain of a particular antigen-binding domain include, e.g. , point mutagenesis (e.g., alanine scanning mutagenesis, arginine scanning mutagenesis, etc.), peptide blots analysis (Reineke, 2004, Methods Mol Biol 248:443-463), protease protection, and peptide cleavage analysis.
- the hydrogen/deuterium exchange method involves deuterium-labeling the protein of interest, followed by binding the antigen-binding domain to the deuterium-labeled protein. Next, the protein/antigen-binding domain complex is transferred to water to allow hydrogendeuterium exchange to occur at all residues except for the residues protected by the antigenbinding domain (which remain deuterium-labeled).
- the target protein After dissociation of the antigen-binding domain, the target protein is subjected to protease cleavage and mass spectrometry analysis, thereby revealing the deuterium-labeled residues which correspond to the specific amino acids with which the antigen-binding domain interacts.
- protease cleavage and mass spectrometry analysis thereby revealing the deuterium-labeled residues which correspond to the specific amino acids with which the antigen-binding domain interacts.
- X-ray crystal structure analysis can also be used to identify the amino acids within a polypeptide with which an antigen-binding domain interacts.
- the antibodies for use in the conjugates described herein are fully human antibodies.
- Methods for generating monoclonal antibodies, including fully human monoclonal antibodies are known in the art. Any such known methods can be used in the context of the present disclosure to make human antibodies that specifically bind to a human protein target.
- high affinity chimeric antibodies to a human protein target are initially isolated having a human variable region and a mouse constant region.
- the antibodies are characterized and selected for desirable characteristics, including affinity, ligand blocking activity, selectivity, epitope, etc.
- mouse constant regions are replaced with a desired human constant region, for example wild-type or modified IgGl or IgG4, to generate a fully human antibody. While the constant region selected may vary according to specific use, high affinity antigen-binding and target specificity characteristics reside in the variable region.
- fully human antibodies are isolated directly from antigen-positive B cells.
- Monoclonal antibodies can be generated by any techniques with which those having ordinary skill in the art will be familiar. Such methods include, but are not limited to, Epstein Barr Virus (EBV) transformation of human peripheral blood cells e.g., containing B lymphocytes), in vitro immunization of human B-cells, fusion of spleen cells from immunized transgenic mice carrying inserted human immunoglobulin genes, isolation from human immunoglobulin V region phage libraries, or other procedures as known in the art and based on the disclosure herein. For example, fully human monoclonal antibodies can be obtained from transgenic mice that have been engineered to produce specific human antibodies in response to antigenic challenge.
- EBV Epstein Barr Virus
- human immunoglobulin transgenes may be mini-gene constructs, or transloci on yeast artificial chromosomes, which undergo B-cell-specific DNA rearrangement and hypermutation in the mouse lymphoid tissue.
- Fully human monoclonal antibodies may be obtained by immunizing the transgenic mice, which may then produce human antibodies specific for a target antigen. Lymphoid cells of the immunized transgenic mice can be used to produce human antibody-secreting hybridomas according to the methods described herein. Polyclonal sera containing fully human antibodies may also be obtained from the blood of the immunized animals.
- Another method for generating human antibodies of the present disclosure includes immortalizing human peripheral blood cells by EBV transformation. See, e.g., U.S. Patent No. 4,464,456.
- Such an immortalized B-cell line (or lymphoblastoid cell line) producing a monoclonal antibody that specifically binds to a target antigen can be identified by immunodetection methods as provided herein, for example, an ELISA, and then isolated by standard cloning techniques.
- the stability of the lymphoblastoid cell line producing an antibody against a target antigen can be improved by fusing the transformed cell line with a murine myeloma to produce a mouse-human hybrid cell line according to methods known in the art (see, e.g., Glasky et al., Hybridoma 8:377-89 (1989)).
- Still another method to generate human monoclonal antibodies is in vitro immunization, which includes priming human splenic B-cells with a target antigen, followed by fusion of primed B-cells with a heterohybrid fusion partner. See, e.g., Boerner et al., 1991 J. Immunol. 147:86-95.
- a B-cell that is producing an antibody against a target antigen is selected and the light chain and heavy chain variable regions are cloned from the B-cell according to molecular biology techniques known in the art (WO 92/02551; U.S. Patent 5,627,052; Babcook et al., Proc. Natl. Acad. Sci. USA 93:7843-48 (1996)) and described herein.
- B-cells from an immunized animal may be isolated from the spleen, lymph node, or peripheral blood sample by selecting a cell that is producing an antibody that specifically binds to a target antigen.
- B-cells may also be isolated from humans, for example, from a peripheral blood sample.
- Methods for detecting single B-cells that are producing an antibody with the desired specificity are well known in the art, for example, by plaque formation, fluorescence-activated cell sorting, in vitro stimulation followed by detection of specific antibody, and the like.
- Methods for selection of specific antibody-producing B-cells include, for example, preparing a single cell suspension of B-cells in soft agar that contains a target antigen. Binding of the specific antibody produced by the B-cell to the antigen results in the formation of a complex, which may be visible as an immune-precipitate.
- the methods for obtaining antibodies of the present disclosure can also adopt various phage display technologies known in the art. See, e.g., Winter et al., 1994 Amu. Rev.
- Human or murine immunoglobulin variable region gene combinatorial libraries may be created in phage vectors that can be screened to select Ig fragments (Fab, Fv, sFv, or multimers thereof) that bind specifically to a target antigen or variant or fragment thereof. See, e.g., U.S. Patent No. 5,223,409; Huse et al., 1989 Science 246:1275-81; Sastry et al., Proc. Natl. Acad. Sci.
- a library containing a plurality of polynucleotide sequences encoding Ig variable region fragments may be inserted into the genome of a filamentous bacteriophage, such as Ml 3 or a variant thereof, in frame with the sequence encoding a phage coat protein.
- a fusion protein may be a fusion of the coat protein with the light chain variable region domain and/or with the heavy chain variable region domain.
- immunoglobulin Fab fragments may also be displayed on a phage particle (see, e.g., U.S. Patent No. 5,698,426).
- Antibody fragments fused to another protein can be also used to enrich phage with antigen. Then, using a random combinatorial library of rearranged heavy (VH) and light (VL) chains from mice immune to the target antigen e.g., HBV sAg, tumor specific antigen), diverse libraries of antibody fragments are displayed on the surface of the phage. These libraries can be screened for complementary variable domains, and the domains purified by, for example, affinity column. See Clackson et al., Nature, V. 352 pp. 624-628 (1991).
- Heavy and light chain immunoglobulin cDNA expression libraries may also be prepared in lambda phage, for example, using XlmmunoZapTM(H) and XImmunoZapTM(L) vectors (Stratagene, La Jolla, California). Briefly, mRNA is isolated from a B-cell population, and used to create heavy and light chain immunoglobulin cDNA expression libraries in the XlmmunoZap(H) and XlmmunoZap(L) vectors. These vectors may be screened individually or co-expressed to form Fab fragments or antibodies (see Huse et al. , supra', see also Sastry et al., supra). Positive plaques may subsequently be converted to a non-lytic plasmid that allows high level expression of monoclonal antibody fragments from E. coll.
- variable regions of a gene expressing a monoclonal antibody of interest are amplified using nucleotide primers.
- primers may be synthesized by one of ordinary skill in the art or may be purchased from commercially available sources. (See, e.g., Stratagene (La Jolla, California), which sells primers for mouse and human variable regions including, among others, primers for Vua, Vub, Vue, Vua, CHI, VL and CL regions.) These primers may be used to amplify heavy or light chain variable regions, which may then be inserted into vectors such as ImmunoZAPTMH or ImmunoZAPTML (Stratagene), respectively.
- vectors may then be introduced into E. coll, yeast, or mammalian-based systems for expression. Large amounts of a single -chain protein containing a fusion of the VH and VL domains may be produced using these methods (see Bird et al., Science 242:423-426, 1988).
- the specific antibody genes may be cloned by isolating and amplifying DNA or mRNA therefrom according to standard procedures as described herein.
- the antibodies produced therefrom may be sequenced and the CDRs identified and the DNA coding for the CDRs may be manipulated as described previously to generate other antibodies according to the disclosure.
- the binding agents of the present disclosure preferably modulate activity of the target antigen in the cell-based assay described herein and/or the in vivo assay described herein and/or bind to one or more of the domains described herein and/or cross-block the binding of one of the antibodies described in this application and/or are cross-blocked from binding the target antigen by one of the antibodies described in this application. Accordingly, such binding agents can be identified using the assays described herein.
- antibodies are generated by first identifying antibodies that bind to one or more of the domains provided herein and/or neutralize in the cell-based and/or in vivo assays described herein and/or cross-block the antibodies described in this application and/or are cross-blocked from binding a target antigen by one of the antibodies described in this application.
- the CDR regions from these antibodies are then used to insert into appropriate biocompatible frameworks to generate binding agents against the target antigen.
- the non-CDR portion of the binding agent may be composed of amino acids or may be a non-protein molecule.
- the assays described herein allow the characterization of binding agents.
- the binding agents of the present disclosure are antibodies as defined herein.
- CDRs complementarity determining regions
- Non-human antibodies of the present disclosure can be, for example, derived from any antibodyproducing animal, such as mouse, rat, rabbit, goat, donkey, or non-human primate (such as monkey (e.g., cynomolgus or rhesus monkey) or ape (e.g., chimpanzee)).
- non-human primate such as monkey (e.g., cynomolgus or rhesus monkey) or ape (e.g., chimpanzee)).
- An antibody from a particular species can be made by, for example, immunizing an animal of that species with the desired immunogen (e.g., HBV sAg, tumor specific antigen or using an artificial system for generating antibodies of that species (e.g., a bacterial or phage display -based system for generating antibodies of a particular species), or by converting an antibody from one species into an antibody from another species by replacing, e.g., the constant region of the antibody with a constant region from the other species, or by replacing one or more amino acid residues of the antibody so that it more closely resembles the sequence of an antibody from the other species.
- the antibody is a chimeric antibody comprising amino acid sequences derived from antibodies from two or more different species.
- Antigen binding proteins may be prepared, and screened for desired properties, by any of a number of conventional techniques. Certain of the techniques involve isolating a nucleic acid encoding a polypeptide chain (or portion thereof) of an antigen binding protein of interest e.g., an anti- HBV sAg antibody, a tumor specific antigen), and manipulating the nucleic acid through recombinant DNA technology.
- the nucleic acid may be fused to another nucleic acid of interest, or altered (e.g., by mutagenesis or other conventional techniques) to add, delete, or substitute one or more amino acid residues, for example.
- the antigen binding proteins may be purified from cells that naturally express them (e.g., an antibody can be purified from a hybridoma that produces it), or produced in recombinant expression systems, using any technique known in the art. See, for example, Monoclonal Antibodies, Hybridomas: A New Dimension in Biological Analyses, Kennet et al. (eds.), Plenum Press, New York (1980); and Antibodies: A Laboratory Manual, Harlow and Land (eds.), Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, (1988).
- Any expression system known in the art can be used to make the recombinant polypeptides of the present disclosure.
- Expression systems are detailed comprehensively above.
- host cells are transformed with a recombinant expression vector that comprises DNA encoding a desired polypeptide.
- the host cells that may be employed are prokaryotes, yeast or higher eukaryotic cells.
- Prokaryotes include gram negative or gram positive organisms, for example E. coli or Bacilli.
- Higher eukaryotic cells include insect cells and established cell lines of mammalian origin. Examples of suitable mammalian host cell lines include the COS-7 line of monkey kidney cells (ATCC CRL 1651) (Gluzman et al.
- an antibody of the present disclosure may have at least one amino acid substitution, providing that the antibody retains binding specificity. Therefore, modifications to the antibody structures are encompassed within the scope of the present disclosure. These may include amino acid substitutions, which may be conservative or nonconservative that do not destroy the target binding capability of an antibody. Conservative amino acid substitutions may encompass non-naturally occurring amino acid residues, which are typically incorporated by chemical peptide synthesis rather than by synthesis in biological systems. These include peptidomimetics and other reversed or inverted forms of amino acid moieties. A conservative amino acid substitution may also involve a substitution of a native amino acid residue with a normative residue such that there is little or no effect on the polarity or charge of the amino acid residue at that position.
- Non-conservative substitutions may involve the exchange of a member of one class of amino acids or amino acid mimetics for a member from another class with different physical properties (e.g., size, polarity, hydrophobicity, charge). Such substituted residues may be introduced into regions of the human antibody that are homologous with non-human antibodies, or into the non-homologous regions of the molecule.
- test variants containing a single amino acid substitution at each desired amino acid residue.
- the variants can then be screened using activity assays known to those skilled in the art.
- Such variants could be used to gather information about suitable variants. For example, if one discovered that a change to a particular amino acid residue resulted in destroyed, undesirably reduced, or unsuitable activity, variants with such a change may be avoided. In other words, based on information gathered from such routine experiments, one skilled in the art can readily determine the amino acids where further substitutions should be avoided either alone or in combination with other mutations.
- a skilled artisan will be able to determine suitable variants of the polypeptide as set forth herein using well-known techniques.
- one skilled in the art may identify suitable areas of the molecule that may be changed without destroying activity by targeting regions not believed to be important for activity.
- even areas that may be important for biological activity or for structure may be subject to conservative amino acid substitutions without destroying the biological activity or without adversely affecting the polypeptide structure.
- One skilled in the art can also analyze the three-dimensional structure and amino acid sequence in relation to that structure in similar polypeptides. In view of such information, one skilled in the art may predict the alignment of amino acid residues of an antibody with respect to its three-dimensional structure. In certain embodiments, one skilled in the art may choose not to make radical changes to amino acid residues predicted to be on the surface of the protein, since such residues may be involved in important interactions with other molecules.
- variants of antibodies include glycosylation variants wherein the number and/or type of glycosylation site has been altered compared to the amino acid sequences of a parent polypeptide.
- variants comprise a greater or a lesser number of N-linked glycosylation sites than the native protein.
- An N-linked glycosylation site is characterized by the sequence: Asn-X-Ser or Asn-X-Thr, wherein the amino acid residue designated as X can be any amino acid residue except proline.
- the substitution of amino acid residues to create this sequence provides a potential new site for the addition of an N-linked carbohydrate chain. Alternatively, substitutions which eliminate this sequence will remove an existing N-linked carbohydrate chain.
- N-linked carbohydrate chains wherein one or more N-linked glycosylation sites (typically those that are naturally occurring) are eliminated and one or more new N- linked sites are created.
- Additional preferred antibody variants include cysteine variants wherein one or more cysteine residues are deleted from or substituted for another amino acid (e.g., serine) as compared to the parent amino acid sequence. Cysteine variants can be useful when antibodies must be refolded into a biologically active conformation such as after the isolation of insoluble inclusion bodies. Cysteine variants generally have fewer cysteine residues than the native protein, and typically have an even number to minimize interactions resulting from unpaired cysteines.
- Desired amino acid substitutions can be determined by those skilled in the art at the time such substitutions are desired.
- preferred amino acid substitutions are those which: (1) reduce susceptibility to proteolysis, (2) reduce susceptibility to oxidation, (3) alter binding affinity for forming protein complexes, (4) alter binding affinities, and/or (4) confer or modify other physiochemical or functional properties on such polypeptides.
- single or multiple amino acid substitutions in certain embodiments, conservative amino acid substitutions
- a conservative amino acid substitution typically may not substantially change the structural characteristics of the parent sequence (e.g., a replacement amino acid should not tend to break a helix that occurs in the parent sequence, or disrupt other types of secondary structure that characterizes the parent sequence).
- a replacement amino acid should not tend to break a helix that occurs in the parent sequence, or disrupt other types of secondary structure that characterizes the parent sequence.
- Examples of art-recognized polypeptide secondary and tertiary structures are described in Proteins, Structures and Molecular Principles (Creighton, Ed., W. H. Freeman and Company, New York (1984)); Introduction to Protein Structure (C. Branden and J. Tooze, eds., Garland Publishing, New York, N.Y. (1991)); and Thornton et al. Nature 354:105 (1991), which are each incorporated herein by reference.
- antibodies of the present disclosure may be chemically bonded with polymers, lipids, or other moieties.
- the binding agents may comprise at least one of the CDRs described herein incorporated into a biocompatible framework structure.
- the biocompatible framework structure comprises a polypeptide or portion thereof that is sufficient to form a conformationally stable structural support, or framework, or scaffold, which is able to display one or more sequences of amino acids that bind to an antigen (e.g. , CDRs, a variable region, etc.) in a localized surface region.
- an antigen e.g. , CDRs, a variable region, etc.
- Such structures can be a naturally occurring polypeptide or polypeptide “fold” (a structural motif), or can have one or more modifications, such as additions, deletions or substitutions of amino acids, relative to a naturally occurring polypeptide or fold.
- These scaffolds can be derived from a polypeptide of any species (or of more than one species), such as a human, other mammal, other vertebrate, invertebrate, plant, bacteria or virus.
- the biocompatible framework structures are based on protein scaffolds or skeletons other than immunoglobulin domains.
- protein scaffolds or skeletons other than immunoglobulin domains.
- those based on fibronectin, ankyrin, lipocalin, neocarzinostain, cytochrome b, CPI zinc finger, PST1, coiled coil, LACI- Dl, Z domain and tendamistat domains may be used See e.g., Nygren and Uhlen, 1997, Curr. Opin. in Struct. Biol., 7, 463-469).
- Humanized antibodies can be produced using techniques known to those skilled in the art (Zhang, W., et al., Molecular Immunology. 42(72):1445-1451, 2005; Hwang W. et al., Methods. 36(7):35-42, 2005; Dall’Acqua WF, et al., Methods 36(1):43-6Q, 2005; and Clark, M., Immunology Today. 27(S):397-402, 2000).
- suitable binding agents include portions of these antibodies, LCDR1, LCDR2, LCDR3, HCDR1, HCDR2, and/or HCDR3.
- the non-CDR portion of the antibody may be a non-protein molecule, wherein the binding agent cross-blocks the binding of an antibody disclosed herein to a target antigen.
- the non- CDR portion of the antibody may be a non-protein molecule in which the antibody exhibits a similar binding pattern to a target antigen in a competition binding assay as that exhibited by at least one of antibodies disclosed herein.
- the non-CDR portion of the antibody may be composed of amino acids, wherein the antibody is a recombinant binding protein or a synthetic peptide, and the recombinant binding protein cross-blocks the binding of an antibody disclosed herein to a target antigen and/or neutralizes a target antigen.
- the non- CDR portion of the antibody may be composed of amino acids, wherein the antibody is a recombinant antibody, and the recombinant antibody exhibits a similar binding pattern to a target antigen in the target epitope competition binding assay (described hereinbelow) as that exhibited by at least one of the antibodies disclosed herein, and/or neutralizes the target antigen.
- an antibody comprises one or more of HCDR1, HCDR2, HCDR3, LCDR1, LCDR2 and LCDR3 as described above, it may be obtained by expression from a host cell containing DNA coding for these sequences.
- a DNA coding for each CDR sequence may be determined on the basis of the amino acid sequence of the CDR and synthesized together with any desired antibody variable region framework and constant region DNA sequences using oligonucleotide synthesis techniques, site-directed mutagenesis and polymerase chain reaction (PCR) techniques as appropriate.
- DNA coding for variable region frameworks and constant regions is widely available to those skilled in the art from genetic sequences databases such as GenBank®.
- the heavy chain and the light chain of the antibody are expressed from a single DNA construct. In some embodiments, the heavy chain and the light chain of the antibody are expressed from two or more separate DNA constructs.
- the DNA encoding an antibody of the present disclosure or fragment thereof may be propagated and expressed according to any of a variety of well-known procedures for nucleic acid excision, ligation, transformation, and transfection using any number of known expression vectors.
- expression of an antibody fragment may be preferred in a prokaryotic host, such as Escherichia coli (see, e.g., Pluckthun et al., 1989 Methods Enzymol. 178:497-515).
- expression of the antibody or a fragment thereof may be preferred in a eukaryotic host cell, including yeast (e.g.
- animal cells including mammalian cells
- plant cells include, but are not limited to, myeloma (such as a mouse NSO line), COS, CHO, or hybridoma cells.
- suitable animal cells include, but are not limited to, myeloma (such as a mouse NSO line), COS, CHO, or hybridoma cells.
- plant cells include tobacco, corn, soybean, and rice cells.
- One or more replicable expression vectors containing DNA encoding an antibody variable and/or constant region may be prepared and used to transform an appropriate cell line, for example, a non-producing myeloma cell line, such as a mouse NSO line or a bacteria, such as E. coli, in which production of the antibody will occur.
- an appropriate cell line for example, a non-producing myeloma cell line, such as a mouse NSO line or a bacteria, such as E. coli, in which production of the antibody will occur.
- the DNA sequence in each vector should include appropriate regulatory sequences, particularly a promoter and leader sequence operatively linked to the variable domain sequence.
- Particular methods for producing antibodies in this way are generally well-known and routinely used. For example, basic molecular biology procedures are described by Maniatis et al.
- DNA sequencing can be performed as described in Sanger et al. (PNAS 74:5463, (1977)) and the Amersham International pic sequencing handbook, and site directed mutagenesis can be carried out according to methods known in the art (Kramer et al., Nucleic Acids Res. 12:9441, (1984); Kunkel Proc. Natl. Acad. Sci. USA 82:488-92 (1985); Kunkel et al., Methods in Enzymol.
- the BA such as antigen-binding domains, for use in the conjugates provided herein encompass proteins having amino acid sequences that vary from those of the described antibodies but that retain the ability to bind the target proteins.
- Such variant antigen-binding domains comprise one or more additions, deletions, or substitutions of amino acids when compared to parent sequence, but exhibit biological activity that is essentially equivalent to that of the described antibodies.
- Two antigen-binding domains are considered bioequivalent if, for example, they are pharmaceutical equivalents or pharmaceutical alternatives whose rate and extent of absorption do not show a significant difference when administered at the same molar dose under similar experimental conditions, either single does or multiple doses.
- Some antigenbinding domains will be considered equivalents or pharmaceutical alternatives if they are equivalent in the extent of their absorption but not in their rate of absorption and yet may be considered bioequivalent because such differences in the rate of absorption are intentional and are reflected in the labeling, are not essential to the attainment of effective body drug concentrations on, e.g., chronic use, and are considered medically insignificant for the particular drug product studied.
- two antigen-binding domains are bioequivalent if there are no clinically meaningful differences in their safety, purity, and potency.
- two antigen-binding domains are bioequivalent if a patient can be switched one or more times between the reference product and the biological product without an expected increase in the risk of adverse effects, including a clinically significant change in immunogenicity, or diminished effectiveness, as compared to continued therapy without such switching.
- two antigen-binding domains are bioequivalent if they both act by a common mechanism or mechanisms of action for the condition or conditions of use, to the extent that such mechanisms are known.
- Bioequivalence may be demonstrated by in vivo and in vitro methods.
- Bioequivalence measures include, e.g., (a) an in vivo test in humans or other mammals, in which the concentration of the antigen-binding domain or its metabolites is measured in blood, plasma, serum, or other biological fluid as a function of time; (b) an in vitro test that has been correlated with and is reasonably predictive of human in vivo bio availability data; (c) an in vivo test in humans or other mammals in which the appropriate acute pharmacological effect of the antigen-binding domain (or its target) is measured as a function of time; and (d) in a well-controlled clinical trial that establishes safety, efficacy, or bioavailability or bioequivalence of an antigen-binding domain.
- Bioequivalent variants of antigen-binding domains for use in the conjugates provided herein may be constructed by, for example, making various substitutions of residues or sequences or deleting terminal or internal residues or sequences not needed for biological activity.
- cysteine residues not essential for biological activity can be deleted or replaced with other amino acids to prevent formation of unnecessary or incorrect intramolecular disulfide bridges upon renaturation.
- bioequivalent antigenbinding domains may include variants comprising amino acid changes which modify the glycosylation characteristics of the antigen -binding domain, e.g., mutations which eliminate or remove glycosylation.
- the BA including antigen-binding domains for use in the conjugates provided herein bind to a human target protein but not to target protein from other species.
- the antigen-binding domains for use in the conjugates provided herein bind to a human target protein and to a target protein from one or more nonhuman species.
- the antigen-binding domains for use in the conjugates provided herein may bind to a human target protein and may bind or not bind, as the case may be, to one or more of mouse, rat, guinea pig, hamster, gerbil, pig, cat, dog, rabbit, goat, sheep, cow, horse, camel, cynomologous, marmoset, rhesus or chimpanzee target protein.
- the antigen-binding domains specifically bind human target protein and cynomolgus monkey (e.g., Macacafascicularis) target protein.
- antigen-binding domains for use herein bind human target protein but do not bind, or bind only weakly, to cynomolgus monkey target protein.
- the present disclosure provides a process of producing a conjugate of Formula (I) or Formula (II) as described herein.
- the process comprises contacting an antibody or an antigen-binding fragment thereof (BA), as described herein, with a linker-payload compound (L-P) in the presence of a transglutaminase, wherein L-P has a structure according to formula (Ila), (lib), or (lie) as described herein.
- the reactive moiety B is selected from the group consisting of an azide, an alkyne, a thiol, a diene, an amino, an active ester, a glutamine-containing peptide tag (Q-tag), and a 1,2, 4, 5 -tetrazine.
- the reactive moiety B is Q-tag.
- the reactive moiety B is an active ester.
- the reactive moiety B is a maleimide.
- the reactive moiety B is a moiety suitable for participation in a Click Reaction, as described herein for click chemistry, or a Diels- Alder reaction.
- moiety B is a diene.
- the reactive moiety B is a substrate of transglutaminase.
- the reactive moiety B is a reactive moiety for conjugation to a target protein, antibody, or antigen-binding fragment thereof, comprising at least one member selected from the group consisting of amino, hydroxyl, thiol, a maleimido moiety, a succinyl ester, an activated ester, an alkylamino; a halo acetamido, -N3,
- the transglutaminase is a bacterial transglutaminase (BTG). In other embodiments, the transglutaminase a native transglutaminase or an engineered transglutaminase.
- the process is carried out with a molar excess of L-P over BA.
- BA is contacted with a 2 to 30-fold, 5 to 25-fold, or 10 to 25-fold molar excess of L-P.
- Exemplary embodiments include the conjugates described in Table 3 below.
- the present disclosure provides a composition comprising a plurality of conjugates of Formula (I), Formula (II), or combinations thereof as described herein.
- the number ratio of payload (P) to BA in the composition is defined as a drugantibody ratio (DAR) of about 0.5 to about 30.0. In additional embodiments, the DAR is about 1 to about 8.
- the present disclosure provides a product that is produced by the process as described herein.
- the present disclosure provides a pharmaceutical composition
- a pharmaceutical composition comprising the compound of formula (la), (lb), (Ic), (Ila), (lib), (lie), or the conjugate of formula (I) or (II), specific examples thereof as disclosed herein (e.g., in Tables 1, 2, and 3), any product produced by a process as described herein, and one or more pharmaceutically acceptable carriers, diluents, and/or excipients.
- the conjugate includes the composition comprising a plurality of conjugates with a defined DAR as described herein.
- the composition further contains, in accordance with accepted practices of pharmaceutical compounding, one or more additional therapeutic agents, pharmaceutically acceptable adjuvants, stabilizers, emulsifiers, preservatives, colorants, buffers, flavor imparting agents.
- additional therapeutic agents pharmaceutically acceptable adjuvants, stabilizers, emulsifiers, preservatives, colorants, buffers, flavor imparting agents.
- the pharmaceutical composition can be administered by any suitable means that results in a concentration of the compound or conjugate in a subject that is effective at treating a disease or condition suitable for treatment with the compounds of the disclosure.
- the compound or conjugate is present in an amount of 1-95% by weight of the total weight of the composition.
- the “therapeutically effective amount” of a compound or conjugate that is administered is governed by such considerations, and is the minimum amount necessary to exert cytotoxic effect. Such amount may be below the amount that is toxic to normal cells, or the subject as a whole.
- the initial therapeutically effective amount of a compound or conjugate of the present disclosure that is administered is in the range of about 0.001 to about 200 mg/kg or about 0.1 to about 20 mg/kg of patient body weight per day, with the typical initial range being about 0.3 to about 15 mg/kg/day.
- Oral unit dosage forms such as tablets and capsules, may contain from about 0.1 mg to about 1000 mg of a compound (or a pharmaceutically acceptable salt, stereoisomer, or tautomer thereof) of the present disclosure.
- such dosage forms contain from about 50 mg to about 500 mg of a compound or conjugate of the present disclosure.
- such dosage forms contain from about 25 mg to about 200 mg of a compound or conjugate of the present disclosure.
- such dosage forms contain from about 10 mg to about 100 mg of a compound or conjugate of the present disclosure.
- such dosage forms contain from about 5 mg to about 50 mg of a compound or conjugate of the present disclosure.
- the compound or conjugate is substantially pure, in that it contains less than about 5%, or less than about 2%, or less than about 1%, or less than about 0.5%, or less than about 0.1%, of other organic small molecules, such as unreacted intermediates or synthesis by-products that are created, for example, in one or more of the steps of a synthesis method.
- the composition is provided in a dosage form that is suitable for oral, parenteral (e.g., intravenously, intramuscularly, subcutaneous, intraarterial), buccal, sublingual, rectal, cutaneous, nasal, vaginal, intranasal, inhalation, transdermal, ocular, intraosseous, otic, or intracranial administration route.
- parenteral e.g., intravenously, intramuscularly, subcutaneous, intraarterial
- buccal sublingual
- rectal cutaneous, nasal, vaginal, intranasal, inhalation, transdermal, ocular, intraosseous, otic, or intracranial administration route.
- the composition dosage form is chosen from tablets, capsules, pills, powders, granulates, suspensions, emulsions, solutions, gels including hydrogels, pastes, patches, ointments, creams, plasters, drenches, osmotic delivery devices, suppositories, enemas, injectables, implants, sprays, and aerosols.
- the pharmaceutical compositions are formulated according to conventional pharmaceutical practice (see, e.g., Remington: The Science and Practice of Pharmacy, 20th edition, 2000, ed. A.R. Gennaro, Lippincott Williams & Wilkins, Philadelphia, and Encyclopedia of Pharmaceutical Technology, eds. J. Swarbrick and J. C. Boylan, 1988-1999, Marcel Dekker, New York).
- compositions can be formulated to release the compound or conjugate immediately upon administration or at any predetermined time or time period after administration (e.g., controlled release formulations).
- controlled release formulations include (i) formulations that create substantially constant concentrations of the agent(s) of the disclosure within the body over an extended period of time; (ii) formulations that after a predetermined lag time create substantially constant concentrations of the agents of the disclosure within the body over an extended period of time; (iii) formulations that sustain the agent(s) action during a predetermined time period by maintaining a relatively constant, effective level of the agent(s) in the body with concomitant minimization of undesirable side effects associated with fluctuations in the plasma level of the agent(s) (sawtooth kinetic pattern); (iv) formulations that localize action of agent(s), e.g., spatial placement of a controlled release composition adjacent to or in the diseased tissue or organ; (v) formulations that achieve convenience of dosing, e.g., administering the composition once per week or
- controlled release is obtained by appropriate selection of various formulation parameters and ingredients, including, e.g., various types of controlled release compositions and coatings.
- the compound is formulated with appropriate excipients into a pharmaceutical composition that, upon administration, releases the compound in a controlled manner. Examples include single or multiple unit tablet or capsule compositions, oil solutions, suspensions, emulsions, microcapsules, molecular complexes, microspheres, nanoparticles, patches, and liposomes.
- a pharmaceutical composition comprising a compound as described herein can be administered parenterally by injection, infusion, or implantation (e.g., intraocular, subcutaneous, intravenous, intramuscular, intraperitoneal) via dosage forms, formulations, or by suitable delivery devices or implants containing conventional, non-toxic pharmaceutically acceptable carriers and adjuvants.
- injection, infusion, or implantation e.g., intraocular, subcutaneous, intravenous, intramuscular, intraperitoneal
- suitable delivery devices or implants containing conventional, non-toxic pharmaceutically acceptable carriers and adjuvants.
- the formulation and preparation of such compositions are well known to those skilled in the art of pharmaceutical formulation.
- Suitable oral compositions as described herein include without limitation tablets, troches, lozenges, aqueous or oily suspensions, dispersible powders or granules, emulsion, hard or soft capsules, syrups or elixirs.
- compositions suitable for single unit dosages that comprise a compound of the disclosure or its pharmaceutically acceptable stereoisomer, salt, or tautomer and a pharmaceutically acceptable carrier.
- compositions of the present disclosure that are suitable for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions.
- liquid formulations of the compounds of the present disclosure contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide pharmaceutically palatable preparations of a compound of the present disclosure.
- a compound or conjugate of the present disclosure in admixture with non-toxic pharmaceutically acceptable excipients is used for the manufacture of tablets.
- excipients include without limitation inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example, corn starch, or alginic acid; binding agents, for example starch, gelatin or acacia, and lubricating agents, for example magnesium stearate, stearic acid or talc.
- the tablets may be uncoated or they may be coated by known coating techniques to delay disintegration and absorption in the gastrointestinal tract and thereby to provide a sustained therapeutic action over a desired time period.
- a time delay material such as glyceryl monostearate or glyceryl distearate may be employed.
- Formulations for oral use may also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, for example peanut oil, liquid paraffin or olive oil.
- a compound or conjugate of the present disclosure is admixed with excipients suitable for maintaining a stable suspension.
- excipients include without limitation are sodium carboxymethylcellulose, methylcellulose, hydroxpropylmethylcellulose, sodium alginate, polyvinylpyrrolidone, gum tragacanth and gum acacia.
- Oral suspensions can also contain dispersing or wetting agents, such as naturally- occurring phosphatide, for example, lecithin, or condensation products of an alkylene oxide with fatty acids, for example polyoxyethylene stearate, or condensation products of ethylene oxide with long chain aliphatic alcohols, for example, heptadecaethyleneoxycetanol, or condensation products of ethylene oxide with partial esters derived from fatty acids and a hexitol such as polyoxyethylene sorbitol monooleate, or condensation products of ethylene oxide with partial esters derived from fatty acids and hexitol anhydrides, for example polyethylene sorbitan monooleate.
- dispersing or wetting agents such as naturally- occurring phosphatide, for example, lecithin, or condensation products of an alkylene oxide with fatty acids, for example polyoxyethylene stearate, or condensation products of ethylene oxide with long chain aliphatic alcohols, for example, heptadecaethyleneoxycetanol,
- the aqueous suspensions may also contain one or more preservatives, for example ethyl, or n-propyl p-hydroxybenzoate, one or more coloring agents, one or more flavoring agents, and one or more sweetening agents, such as sucrose or saccharin.
- preservatives for example ethyl, or n-propyl p-hydroxybenzoate
- coloring agents for example ethyl, or n-propyl p-hydroxybenzoate
- flavoring agents for example ethyl, or n-propyl p-hydroxybenzoate
- sweetening agents such as sucrose or saccharin.
- Oily suspensions may be formulated by suspending a compound or conjugate of the present disclosure in a vegetable oil, for example arachis oil, olive oil, sesame oil or coconut oil, or in a mineral oil such as liquid paraffin.
- the oily suspensions may contain a thickening agent, for example beeswax, hard paraffin or cetyl alcohol.
- Sweetening agents such as those set forth above, and flavoring agents may be added to provide palatable oral preparations. These compositions may be preserved by the addition of an anti-oxidant such as ascorbic acid.
- Dispersible powders and granules suitable for preparation of an aqueous suspension by the addition of water provide a compound or conjugate of the present disclosure in admixture with a dispersing or wetting agent, suspending agent and one or more preservatives.
- a dispersing or wetting agent e.g., glycerol, glycerol, glycerol, glycerol, glycerol, glycerin, glycerin, glycerin, glycerin, glycerin, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, glycerol, glycerol, glycerol, glycerol, glycerol, glycerol, glycerol, glyce
- compositions of the present disclosure may also be in the form of oil- in-water emulsions.
- the oily phase may be a vegetable oil, for example olive oil or arachis oil, or a mineral oil, for example liquid paraffin or mixtures of these.
- Suitable emulsifying agents may be naturally-occurring gums, for example gum acacia or gum tragacanth, naturally-occurring phosphatides, for example soy bean, lecithin, and esters or partial esters derived from fatty acids and hexitol, anhydrides, for example sorbitan monoleate, and condensation reaction products of the said partial esters with ethylene oxide, for example polyoxyethylene sorbitan monoleate.
- the emulsions may also contain sweetening and flavoring agents.
- Syrups and elixirs may be formulated with sweetening agents, for example glycerol, propylene glycol, sorbitol or sucrose. Such formulations may also contain a demulcent, a preservative, and flavoring and coloring agents.
- the pharmaceutical compositions may be in the form of a sterile injectable, an aqueous suspension or an oleaginous suspension. This suspension may be formulated according to the known art using those suitable dispersing or wetting agents and suspending agents which have been mentioned above.
- the sterile injectable preparation may also be sterile injectable solution or suspension in a non-toxic parentally acceptable diluent or solvent, for example as a solution in 1,3-butanediol.
- Suitable vehicles and solvents that may be employed are water, Ringer’ s solution and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil may be employed including synthetic mono-or diglycerides.
- fatty acids such as oleic acid find use in the preparation of injectables.
- a compound or conjugate of the present disclosure can be administered in the form of suppositories for rectal administration of the drug.
- These compositions can be prepared by mixing the drug with a suitable non-irritating excipient which is solid at ordinary temperatures but liquid at the rectal temperature and will therefore melt in the rectum to release the drug.
- suitable non-irritating excipient which is solid at ordinary temperatures but liquid at the rectal temperature and will therefore melt in the rectum to release the drug.
- Such materials are cocoa butter and polyethylene glycols.
- compositions for parenteral administrations are administered in a sterile medium.
- the parenteral formulation can either be a suspension or a solution containing dissolved drug.
- Adjuvants such as local anesthetics, preservatives and buffering agents can also be added to parenteral compositions.
- the composition is especially adapted for administration into or around the eye.
- a composition can be adapted to be used as eye drops, or injected into the eye, e.g., using peribulbar or intravitreal injection.
- Such compositions should be sterile and substantially endotoxin-free, and within an acceptable range of pH.
- a formulation without preservatives is used.
- Formulation of eye medications is known in the art, see, e.g., Ocular Therapeutics and Drug Delivery: A Multi-Disciplinary Approach, Reddy, Ed. (CRC Press 1995); Kaur and Kanwar, Drug Dev Ind Pharm.
- compositions for parenteral use may be provided in unit dosage forms (e.g., in singledose ampoules), or in vials containing several doses and in which a suitable preservative may be added (see below).
- the composition may be in form of a solution, a suspension, an emulsion, an infusion device, or a delivery device for implantation, or it may be presented as a dry powder to be reconstituted with water or another suitable vehicle before use.
- the composition may include suitable parenterally acceptable carriers and/or excipients.
- the active agent(s) may be incorporated into microspheres, microcapsules, nanoparticles, liposomes, or the like for controlled release.
- the composition may include suspending, solubilizing, stabilizing, pH-adjusting agents, tonicity adjusting agents, and/or dispersing agents.
- the pharmaceutical compositions of the disclosure are in a form suitable for sterile injection.
- the active agent(s) are dissolved or suspended in a parenterally acceptable liquid vehicle.
- acceptable vehicles and solvents that may be employed are water, water adjusted to a suitable pH by addition of an appropriate amount of hydrochloric acid, sodium hydroxide or a suitable buffer, 1,3-butanediol, Ringer’s solution, dextrose solution, and isotonic sodium chloride solution.
- the aqueous formulation may also contain one or more preservatives (e.g., methyl, ethyl, or n-propyl p-hydroxybenzoate).
- a dissolution enhancing or solubilizing agent can be added, or the solvent may include 10-60% w/w of propylene glycol.
- compositions can be administered to a subject in a single dose or in multiple doses.
- a compound or conjugate described herein can be administered once a week or for 2, 3, 4, 5, 6, 7, 8, 10, 15, 20, or more weeks.
- specific dosage regimes should be adjusted over time according to the individual need and the professional judgment of a health care provider administering or supervising the administration of the compound.
- the dosage of a compound or conjugate can be increased if the lower dose does not provide sufficient biological activity (e.g., in the treatment of a disease or condition described herein).
- the dosage of the compound or conjugate can be decreased, for example, if the disease or condition is reduced or eliminated, or to reduce undesirable side-effects.
- the present disclosure provides a method of treating a subject suffering from cancer.
- the method comprises administering to the subject a therapeutically effective amount of a compound, conjugate, dosage form, or composition as described herein.
- the compounds or conjugates provided herein are useful, inter alia, for the treatment, prevention and/or amelioration of any disease or disorder associated with or mediated by expression, signaling or activity of the target protein of the antigen-binding domain.
- the compound or conjugate provided herein is used to treat primary and/or metastatic tumors arising in the brain and meninges, oropharynx, lung and bronchial tree, gastrointestinal tract, male and female reproductive tract, muscle, bone, skin and appendages, connective tissue, spleen, immune system, blood forming cells and bone marrow, liver and urinary tract, and special sensory organs such as the eye.
- the compound or conjugate provided herein is used to treat one or more of the following cancers: acute myelogenous leukemia, adult T-cell leukemia, astrocytomas, bladder cancer, breast cancer, PRLR positive (PRLR+) breast cancer, cervical cancer, cholangiocarcinoma, chronic myeloid leukemia, colon cancer, colorectal cancer, endometrial cancer, esophageal cancer, gastric cancer, glioblastomata, head and neck cancer (e.g., head and neck squamous cell carcinoma (HNSCC)), Kaposi’s sarcoma, kidney cancer, leiomyosarcomas, liver cancer, lung cancer (e.g., small cell lung cancer, non-small cell lung cancer (NSCLC)), lymphomas, malignant gliomas, malignant mesothelioma, melanoma, mesothelioma, malignant mesothelioma, MFH/fibrosarcom
- Also provided in an embodiment is a method of selectively killing quiescent cells in a subject.
- the method comprises administering to the subject a therapeutically effective amount of a compound, a conjugate, or a pharmaceutical composition as described herein.
- the quiescent cells are quiescent cancer cells.
- the present disclosure provides a method of selectively killing stem cells in a subject.
- the method comprises administering to the subject a therapeutically effective amount of a compound, a conjugate, or a pharmaceutical composition as described herein.
- the stem cells are hematopoietic stem cells.
- the present disclosure provides a method of selectively killing resting or naive B- or T- or other immune cells in a subject.
- the method comprises administering to the subject a therapeutically effective amount of a compound, a conjugate, or a pharmaceutical composition as described herein.
- the present disclosure provides a method of selectively killing quiescent cancer cells in a subject preparing for stem cell therapy.
- the method comprises administering to the subject a therapeutically effective amount of a compound, a conjugate, or a pharmaceutical composition as described herein.
- the methods described herein further comprise administering to the subject one or more other therapeutic agents, as described in more detail below.
- Combination Therapy
- compositions comprising a compound or conjugate as described herein in combination with one or more additional therapeutically active components, and methods of treatment comprising administering such combinations to a subject.
- the compound or conjugate may be co-formulated with and/or administered in combination with one or more additional therapeutically active component(s) selected from a MET antagonist (e.g., an anti-MET antibody (e.g., onartuzumab, emibetuzumab, and H4H14639D) or small molecule inhibitor of MET), an EGFR antagonist (e.g., an anti-EGFR antibody (e.g., cetuximab or panitumumab) or small molecule inhibitor of EGFR (e.g., gefitinib or erlotinib)), an antagonist of another EGFR family member such as Her2/ErbB2, ErbB3 or ErbB4 (e.g., anti-ErbB2 (e.g., trastuzumab or T-DM1 ⁇ KADCYLA® ⁇ ), anti-ErbB3 or anti-ErbB4 antibody or small molecule inhibitor of ErbB2, ErbB3 or ErbB4 activity
- cytokines such as IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-8, IL-9, IL- 11, IL-12, IL-13, IL-17, IL-18, or to their respective receptors.
- a PD-1 inhibitor such as an anti-PD-1 antibody can be combined with a compound or conjugate as described herein.
- compositions comprising a compound or conjugate as described herein in combination with one or more chemotherapeutic agents.
- chemotherapeutic agents include alkylating agents such as thiotepa and cyclosphosphamide (CytoxanTM); alkyl sulfonates such as busulfan, improsulfan and piposulfan; aziridines such as benzodopa, carboquone, meturedopa, and uredopa; ethylenimines and methylamelamines including altretamine, triethylenemelamine, trietylenephosphoramide, trie thy lene thiopho sphaoramide and trimethylolomelamine; nitrogen mustards such as chlorambucil, chlornaphazine, cholophosphamide, estramustine, ifosfamide, mechlorethamine, mechlorethamine oxide hydrochlor
- paclitaxel TexolTM, Bristol-Myers Squibb Oncology, Princeton, N.J.
- docetaxel TaxotereTM; Aventis Antony, France
- chlorambucil gemcitabine
- 6-thioguanine mercaptopurine
- methotrexate platinum analogs such as cisplatin and carboplatin; vinblastine; platinum; etoposide (VP-16); ifosfamide; mitomycin C; mitoxantrone; vincristine; vinorelbine; navelbine; novantrone; teniposide; daunomycin; aminopterin; xeloda; ibandronate; CPT-11 ; topoisomerase inhibitor RFS 2000; difluoromethylornithine (DMFO); retinoic acid; esperamicins; capecitabine; and pharmaceutically acceptable salts, acids or derivatives of any of the above.
- DMFO difluor
- anti-hormonal agents that act to regulate or inhibit hormone action on tumors
- anti-estrogens including for example tamoxifen, raloxifene, aromatase inhibiting 4(5) -imidazoles, 4-hydroxytamoxifen, trioxifene, keoxifene, LY 117018, onapristone, and toremifene (Fareston); and anti-androgens such as flutamide, nilutamide, bicalutamide, leuprolide, and goserelin; and pharmaceutically acceptable salts, acids or derivatives of any of the above.
- the compound or conjugate may also be administered and/or co-formulated in combination with antivirals, antibiotics, analgesics, corticosteroids, steroids, oxygen, antioxidants, COX inhibitors, cardioprotectants, metal chelators, IFN-gamma, and/or NSAIDs.
- the additional therapeutically active component(s), e.g., any of the agents listed above or derivatives thereof, may be administered just prior to, concurrent with, or shortly after the administration of the compound or conjugate.
- the term "in combination” includes the use of more than one therapy (e.g., one or more prophylactic and/or therapeutic agents). However, the use of the term “in combination” does not restrict the order in which therapies (e.g., prophylactic and/or therapeutic agents) are administered to a subject with a disease or disorder.
- a first therapy (e.g., a conjugate provided herein) can be administered prior to (e.g., 5 minutes, 15 minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4 hours, 6 hours, 12 hours, 24 hours, 48 hours, 72 hours, 96 hours, 1 week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 8 weeks, or 12 weeks before), concomitantly with, or subsequent to (e.g., 5 minutes, 15 minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4 hours, 6 hours, 12 hours, 24 hours, 48 hours, 72 hours, 96 hours, 1 week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 8 weeks, or 12 weeks after) the administration of a second therapy (e.g., a prophylactic or therapeutic agent) to the subject.
- a second therapy e.g., a prophylactic or therapeutic agent
- Administration of the compound or conjugate provided herein and one or more second active agents to a subject can occur simultaneously or sequentially by the same or different routes of administration.
- the suitability of a particular route of administration employed for a particular active agent will depend on the active agent itself (e.g., whether it can be administered orally without decomposing prior to entering the blood stream) and the disease or disorder being treated.
- Linker payload (20) was synthesized from compound 8 as described below.
- the acid (1900 mg, 2.439 mmol) was subjected to coupling with H-Ala- OtBu.HCl (665 mg, 3.659 mmol) in the presence of EEDQ (904 mg, 3.659 mmol) in DCM (9 mL) at ambient temperature in air.
- the reaction reached completion within 30 mins as revealed by UPLC.
- the reaction mixture was concentrated under reduced pressure, redissolved in DMSO (10 mL) and purified by reverse phase chromatography (275 g C18 Aq Isco, 5-75-100% of acetonitrile in water, each containing 0.05% of AcOH). The product eluted in 75% of acetonitrile.
- the reaction reached completion within an hour.
- the reaction mixture was purified by reverse phase chromatography (30 g Cl 8 Aq Isco, 5-50-100% of acetonitrile in water, each containing 0.05% of AcOH).
- the product eluted in 50% of acetonitrile.
- the fractions were combined and analyzed by UPLC and found to be impure.
- the major impurity was the adduct of HATU-unreacted acid along with other minor impurities.
- the fraction was lyophilized. Mass of the product was clearly seen. After lyophilization, the HATU-acid adduct was found to have decomposed giving rise to the acid.
- the material was treated with 25% of TFA in DCM.
- Linker payload 36 was synthesized from Compound 21 as described below.
- Step 1 Fmoc-Ala-Wang resin 21 (6379.6 mg, 4.0 mmol, 0.627 mmol/g loading, 100- 200 mesh) was treated with 20 mL 10% piperidine/DMF in a vessel and gently shaken at ambient temperature. After 15 min, the solution was filtered, and the resin was re-suspended in 20 mL 10% piperidine/DMF and gently shaken. After 5 min, the solution was filtered, and the resin was washed with DMF (3 x 10 mL), DCM (3 x 10 mL), and then DMF (1 x 10 mL).
- Step 2 Resin 22 was treated with 20 mL DBU/piperidine/NMP (2:5:93) in a vessel and gently shaken at ambient temperature. After 15 min, the solution was filtered, and the resin was re-suspended in 20 mL DBU/piperidine/NMP (2:5:93) and gently shaken. After 5 min, the solution was filtered, and the resin was washed with DMF (3 x 10 mL), DCM (3 x 10 mL), and then DMF (1 x 10 mL). A solution of Cbz-OSu (1993.8 mg, 8 mmol) and NMM (0.88 mL, 8 mmol) in 20 mL DMF was added into the vessel and gently shaken overnight.
- Step 3 Resin 23 in a vessel was washed with TFA/TIS/DCM (2:2.5:95.5) solution until a colorless filtrate was observed. Then, the resin was washed DMF (3 x 10 mL) and DCM (3 x 10 mL). A solution of Fmoc-A-Me-Val-OH (2827.4 mg, 8 mmol), DIC (1009.6 mg, 8 mmol), and DMAP (969.4 mg, 8 mmol) in 15 mL DMF/DCM (1:4) was added into the vessel and gently shaken overnight. The solution was filtered, and the resin was washed with DMF (3 x 10 mL) and DCM (3 x 10 mL).
- Step 4 Resin 24 was treated with 20 mL 10% piperidine/DMF in a vessel and gently shaken at ambient temperature.
- Step 5 Resin 25 was treated with 20 mL DBU/piperidine/NMP (2:5:93) in a vessel and gently shaken at ambient temperature. After 15 min, the solution was filtered, and the resin was washed with DMF (3 x 10 mL), DCM (3 x 10 mL), and then DMF (1 x 10 mL). A solution of Fmoc-Ala-OH (2490.7 mg, 8 mmol), DMTMM (2213.8 mg, 8 mmol), and NMM (0.88 mL, 8 mmol) in 20 mL DMF was added into the vessel and gently shaken overnight.
- Step 6 Resin 26 was treated with 20 mL DBU/piperidine/NMP (2:5:93) in a vessel and gently shaken at ambient temperature. After 15 min, the solution was filtered, and the resin was washed with DMF (3 x 10 mL), DCM (3 x 10 mL), and then DMF (1 x 10 mL). A solution of Fmoc-D-Ser(Trt)-OH (4557.2 mg, 8 mmol), HATU (3041.6 mg, 8 mmol), HOAt (1088.8 mg, 8 mmol), and NMM (0.88 mL, 8 mmol) in 20 mL DMF was added into the vessel and gently shaken overnight.
- Step 7 Resin 27 was treated with 20 mL DBU/piperidine/NMP (2:5:93) in a vessel and gently shaken at ambient temperature. After 15 min, the solution was filtered, and the resin was washed with DMF (3 x 10 mL), DCM (3 x 10 mL), and then DMF (1 x 10 mL). A solution of Troc-Cl (1694.8 mg, 8 mmol) and NMM (0.88 mL, 8 mmol) in 20 mL DMF was added into the vessel and gently shaken overnight. The resin was filtered and then washed with DMF (3 x 10 mL) and DCM (3 x 10 mL).
- Step 8 Resin 28 in a vessel was washed with TFA/TIS/DCM (2:2.5:95.5) solution until a colorless filtrate was observed. Then, the resin was washed DMF (3 x 10 mL) and DCM (3 x 10 mL). A solution of Fmoc-A-Me-Val-OH (2827.4 mg, 8 mmol), DIC (1009.6 mg, 8 mmol), and DMAP (969.4 mg, 8 mmol) in 15 mL DMF/DCM (1:4) was added into the vessel and gently shaken overnight. The solution was filtered, and the resin was washed with DMF (3 x 10 mL) and DCM (3 x 10 mL).
- Step 9 Resin 29 was treated with 20 mL 10% piperidine/DMF in a vessel and gently shaken at ambient temperature. After 15 min, the solution was filtered, and the resin was washed with DMF (3 x 10 mL), DCM (3 x 10 mL), and then DMF (1 x 10 mL). A solution of Fmoc-A / -mcthyl-(.S')-2-allylglycinc (2530.1 mg, 7.2 mmol), DMTMM (2762.6 mg, 10 mmol), and NMM (1.1 mL, 10 mmol) in 20 mL DMF was added into the vessel and gently shaken overnight.
- the resin was filtered and then washed with DMF (3 x 10 mL) and DCM (3 x 10 mL). After the resin was dried, a few beads were taken and cleaved with 50% TFA/DCM for checking the complete conversion to product 30 in LCMS.
- the resin was then treated with 20 mL TFA/H2O/DCM (25:5:70) for 30 min. The resin was filtered, and the filtrate was collected. The resin was repeated with 20 mL TFA/H2O/DCM (25:5:70) treatment two more times. The combined filtrate was concentrated and then purified by reverse phase chromatography (Isco EZ Prep Gemini Cl 8 column).
- DMSO-d 6 5 10.10-10.09 (m, 1H), 8.73-8.70 (m, 1H), 8.23-8.22 (m, 1H), 7.95-7.88 (m, 2H), 7.86-7.82 (m, 2H), 7.73-7.71 (m, 1H), 7.64-7.61 (m, 2H), 7.60-7.57 (m, 1H), 7.53-7.46 (m, 2H), 7.45-7.41 (m, 2H), 7.32-7.26 (m, 2H), 7.24-7.18 (m, 2H), 7.17-7.13 (m, 2H), 6.04-5.97 (m, 2H), 5.32-5.25 (m, 4H), 4.84-4.70 (m, 3H), 4.60-4.55 (m, 1H), 4.48-4.34 (m, 4H), 4.22- 4.18 (m, 1H), 3.60-3.46 (m, 42H), 3.11-3.11 (m, 3H), 3.08-3.07 (m, 3H), 2.88-2
- Payload 37 was synthesized from compound 35 as described below.
- the title compound was prepared by reacting 35 (8 mg, 0.009 mmol) with 3- hydroxy quinoline -2 -carboxylic acid (5.1 mg, 0.027 mmol) in the presence of HATU (10.3 mg, 0.027 mmol) and NMM (5.0 mL, 0.045 mmol) in DMF (1 mL) at ambient temperature under argon. Upon completion of the reaction, the reaction mixture was purified by reverse phase chromatography (Isco EZ Prep (Gemini C18 column, 30 x 150 mm, 5-35-100% of acetonitrile in water, each containing 0.05% of AcOH). The product eluted in 60% of acetonitrile.
- Payload 38 was synthesized from compound 19 as described below.
- Payload 39 was synthesized from compound 19 as described below.
- Payload 40 was synthesized from compound 19 as described below.
- the title compound was prepared by reacting 19 (10 mg, 0.011 mmol) with 3- aminoquinoxaline-2 -carboxylic acid (4.2 mg, 0.022 mmol) in the presence of HATU (8.5 mg, 0.022 mmol) and NMM (3.7 pL, 0.034 mmol) in DMF (1 mL) at ambient temperature under argon. Reverse phase chromatography (Isco EZ Prep Gemini Cl 8 column, 30 x 150 mm, 5- 35-100% of acetonitrile in water, each containing 0.05% of AcOH) followed by lyophilization afforded 40 as an off white solid (6.0 mg, 0.006 mmol, 50%).
- Payload 41 was synthesized from compound 19 as described below.
- the title compound was prepared by reacting 19 (10 mg, 0.011 mmol) with 3- chloroquinoxaline-2 -carboxylic acid (4.7 mg, 0.022 mmol) in the presence of HATU (8.5 mg, 0.022 mmol) and NMM (3.7 pL, 0.034 mmol) in DMF (1 mL) at ambient temperature under
- Payload 49 was synthesized from compound 21 as described below.
- Step 1 The compound 42 was synthesized from Fmoc-Ala-Wang resin 21 (1988.1 mg, 1.0 mmol, 0.503 mmol/g loading, 100-200 mesh) as described in the synthesis of compound 30, step 1 to step 6, while replacing the reactant Fmoc-A-Me-Val-OH in the step 3 with Fmoc-A-Me-Ilu-OH. A few beads were taken and cleaved with 50% TFA/DCM for checking the complete conversion to product 42 [Cbz-Ser((?-A-Me-Ilu-A-Me-2-allylglycine- Ala-Fmoc-Ser)-Ala-OH].
- Step 2 Resin 42 was treated with 10 mL DBU/piperidine/NMP (2:5:93) in a vessel and gently shaken at ambient temperature. After 15 min, the solution was filtered, and the resin was washed with DMF (3 x 10 mL), DCM (3 x 10 mL), and then DMF (1 x 10 mL).
- Step 3 Resin 44 was synthesized from resin 43 via the same method as described in the synthesis of compound 30, step 8, while using Fmoc-A-Me-Ilu-OH instead of using Fmoc-A-Me-Val-OH. After the resin was dried, a few beads were taken and cleaved with 50% TFA/DCM for checking the complete conversion to product 44 ⁇ Cbz-Ser[(?-A-Me-Val- A-Me-2-allylglycine-Ala-A-2-carboxyquinoxaline-Ser((?-Fmoc-A-Me-Ilu)]-Ala-OH ⁇ in LCMS. MS (ESI, pos.) calc’d for C64H78N9O15 [1212.56] + ; found (M+H) 1213.15.
- Step 4 Resin 44 was treated with 10 mL 10% piperidine/DMF in a vessel and gently shaken at ambient temperature. After 15 min, the solution was filtered, and the resin was washed with DMF (3 x 10 mL), DCM (3 x 10 mL), and then DMF (1 x 10 mL). A solution of Boc-A-methyl-(S')-2-allylglycine (412.7 mg, 1.8 mmol), DMTMM (553.4 mg, 2 mmol), and NMM (0.22 mL, 2 mmol) in 20 mL DMF was added into the vessel and gently shaken overnight.
- Step 5 Resin 45 was placed in a seal tube and suspended in DCM (100 mL).
- Grubbs- Hoveyda 2 nd generation catalyst (125 mg, 0.2 mmol) was added at ambient temperature and purged with argon before the tube was sealed. The reaction was heated at 40 °C while slowly stirring for 9 days. After the completion of ring-closing metathesis indicated by LCMS, the resin was then treated with 30 mL TFA/H2O/DCM (25:5:70) for 30 min. The resin was filtered, and the filtrate was collected. The resin was repeated with 30 mL TFA/H2O/DCM (25:5:70) treatment two more times.
- Payload 50 was synthesized from compound 48 as described below.
- Compound 50 was synthesized from compound 48 (2.0 mg, 0.0022 mmol) by reacting it with 3-aminoquinoxaline-2 -carboxylic acid (0.53 mg, 0.0028), HATU (1.2 mg, 0.0033 mmol), NMM (0.33 mg, 0.0033 mmol) in DMF (1 mL) as described in the synthesis of compound 38.
- the crude solution was purified by reverse phase chromatography (Isco EZ Prep Gemini C18 column). The product was eluted in 45-55% ACN/H2O (containing 0.1% of formic acid). The fractions were combined and lyophilized to afford the product 38 as a yellow solid (1.7 mg, 0.0016 mmol, 72% yield).
- Payload 51 was synthesized from compound 48 as described below.
- Payload 53 was synthesized from compound 52 as described below.
- Payload 54 was synthesized from compound 52 as described below.
- Compound 54 was synthesized from compound 52 (3.0 mg, 0.0032 mmol) by reacting it with 3-aminoquinoxaline-2 -carboxylic acid (1.2 mg, 0.0063), HATU (2.4 mg, 0.0063 mmol), and NMM (0.64 mg, 0.0063 mmol) in DMF (0.5 mb) as described in the synthesis of compound 50.
- Product 54 was isolated as a yellow solid. (2.2 mg, 0.0020 mmol, 62% yield).
- MS (ESI, pos.) calc’d for C56H70N13O12 [1116.53] + ; found (M+H) 1116.87.
- Payload 55 was synthesized from compound 52 as described below.
- Payload 57 was synthesized from compound 56 as described below.
- Payload 58 was synthesized from compound 56 as described below.
- Payload 59 was synthesized from compound 56 as described below.
- Payload 63 was synthesized from compound 19 as described below.
- Payload 64 was synthesized from compound 56 as described below. [0367] Synthesis of 64: Compound 64 was synthesized from compound 56 (3.0 mg, 0.0031 mmol) by reacting it with 4-hydroxyquinoline-2-carboxylic acid (0.9 mg, 0.0046 mmol), HATU (1.8 mg, 0.0046 mmol), and NMM (0.5 mg, 0.0046 mmol) in DMF (0.5 mL) as described in the synthesis of compound 50. Product 59 was obtained as a yellow solid. (1.6 mg, 0.00 mmol, 45% yield). MS (ESI, pos.) calc’d for C59H74N11O13 [ 1144.55] + ; found (M+H) 1145.00.
- Payload 65 was synthesized from compound 52 as described below.
- the crude solution 67 was added (S)-A-tert-butanesulfinamide (25.5 g, 210.6 mmol) and Ti(OEt)4 (55.2 mL, 263.3 mmol) at rt and stirred overnight.
- the reaction was quenched with 500 mL water and then filtered with celite.
- the celite was rinsed with 200 mL EtOAc three times.
- the organic layer was collected, and the aqueous layer was extracted with 300 mL EtOAc.
- the combined organic layer was dried over Na2SO4 and concentrated under vacuum.
- the crude was purified by silica gel column, and the product was eluted with 0-10% EtOAc/hexane.
- Step 1 Resin 23 (1.5 mmol) in a vessel was washed with TFA/TIS/DCM (2:2.5:95.5) solution until a colorless filtrate was observed. Then, the resin was washed DMF (3 x 20 mL) and DCM (3 x 20 mL). A solution of 73 (629 mg, 1.65 mmol), DIC (340 mg, 2.7 mmol), and DMAP (327 mg, 2.7 mmol) in 20 mL DMF/DCM (1:4) was added into the vessel and gently shaken overnight. The solution was filtered, and the resin was washed with DMF (3 x 20 mL) and DCM (3 x 20 mL).
- Step 2 Resin 74 was treated with 20 mL 10% piperidine/DMF in a vessel and gently shaken at ambient temperature. After 15 min, the solution was filtered, and the resin was washed with DMF (3 x 20 mL), DCM (3 x 20 mL), and then DMF (1 x 20 mL). A solution of Fmoc-A-methyl-(5')-2-allylglycine (949 mg, 2.7 mmol), DMTMM (829 mg, 3.0 mmol), and NMM (0.33 mL, 3.0 mmol) in 20 mL DMF was added into the vessel and gently shaken overnight.
- the resin was filtered and then washed with DMF (3 x 20 mL) and DCM (3 x 20 mL).
- the resin was cleaved with 20 mL TFA/H2O/DCM (25:5:70) and shaken for 1 h.
- the resin was filtered, and the filtrate was collected.
- the resin was again treated with cleavage condition two more times.
- the combined filtrate was concentrated and then purified by RP Cl 8 column.
- Payload 84 was synthesized from intermediate 19 as described below.
- Payload 96 was synthesized from intermediate 85 as described below.
- Payload 97 was synthesized using all the procedural steps described for 96 by replacing l-((tert-butoxycarbonyl)(methyl)amino)cyclobutane-l-carboxylic acid with (5)-2- ((tert-butoxycarbonyl)(methyl)amino)-2-cyclobutylacetic acid.
- Payload 98 was synthesized from intermediate 19 as described below.
- Payload 99 was synthesized from intermediate 98 as described below.
- Payload 105 was synthesized from 21 as described below.
- Payload 107 was synthesized from 106 as described below.
- Payload 109 was synthesized from 108 as described below.
- Payload 119 was synthesized from intermediate 11 as described below.
- the reaction mixture was concentrated under reduced pressure to give faint yellow oil that was redissolved in acetonitrile/water and lyophilized overnight to give white solid.
- the amine was used for the synthesis of the title compound. Accordingly, (4.533 g, 7.661 mmol) of the amine was reacted with 2.174 g (11.491 mmol) of Boc-Ala-OH in THF (50 mL) in the presence of DMTMM (3. 180 g, 11.491 mmol) and NMM (2.543 mL, 22.983 mmol) at rt in air for 15 mins.
- the reaction mixture was concentrated under reduced pressure, redissolved in DMF (5 mL) and purified by reverse phase chromatography using acetonitrile in water each containing 0.05% of AcOH.
- the major isomer (116, cis, 265 mg, 0.224 mmol, 10%) came out in 70% of acetonitrile in water followed by the minor isomer (117, trans, 151 mg, 0.128 mmol, 5%) in 80% of acetonitrile in water.
- Linker 127 was synthesized as described below.
- Linker payload 129 was synthesized from 19 and 127 as described below.
- Linker payload 133 was synthesized from 56 and 127 as described below.
- Linker payload 134 was synthesized from 106 and 7 as described below.
- Linker payload 135 was synthesized from 109 and 7 as described below.
- Linker payload 136 was synthesized from 52 and 7 as described below.
- Linker payload 137 was synthesized from 35 and 127 as described below.
- Linker payload 138 was synthesized from 106 and 127 as described below.
- Linker payload 139 was synthesized from 108 and 127 as described below.
- Payload 141 was synthesized from intermediate 140 which in turn was synthesized as explained for intermediate 35 in example 3 by replacing A-(((9/f-fluoren-9- yl)methoxy)carbonyl)-A-methyl-L-valine with (5 , )-2-((((9/f-fluoren-9- yl)methoxy)carbonyl)(methyl)amino)-2 -cyclopentylacetic acid.
- Payload 142 was synthesized from intermediate 52 as described below.
- Payload 145 was synthesized from intermediate 144 which in turn was synthesized as described for intermediate 104 in example 26 by replacing (2S,3R)-2-((((9H-fluoren-9- yl)methoxy)carbonyl)(methyl)amino)-3,4-dimethylpentanoic acid with (5')-2-((((9/f-fluoren- 9-yl)methoxy)carbonyl)(methyl)amino)-2-cyclopentylacetic acid.
- Payload 146 was synthesized from intermediate 144 as shown below.
- Linker 150 was synthesized as described below.
- the aniline was consumed within 10 mins at the same temperature as revealed by quenching with propylamine.
- Alcohol (827 mg, 2.103 mmol) was added in one portion and reaction mixture was allowed to warm to rt overnight.
- the reaction mixture was concentrated under reduced pressure and the yellow contents were adsorbed onto silica gel and purified by normal phase chromatography (80 g column, hexanes/EtOAc, 2:8). The fractions were combined and concentrated to afford the product as a white solid (1267 mg).
- the product was dissolved in DCM (24 mL) and treated with 7 mL of TFA at room temperature until the Boc group was gone.
- Linker Payload 152 was synthesized from intermediate 151 which in turn was synthesized as explained for intermediate 140 by replacing A-(((9Z7-fluoren-9- yl)methoxy)carbonyl)-A-methyl-L-valine with (S')-2-((((9Z7-fluoren-9- yl)methoxy)carbonyl)(methyl)amino)-2-cyclopentylacetic acid.
- Synthesis of 152 151 (18 mg, 0.0188 mmol), 150 (21 mg, 0.0206 mmol) and HATU were dissolved in DMF at rt in air. NMM (10.4 pL, 0.0938 mmol) was added.
- the reaction reached completion within 10 mins as revealed by UPLC.
- the reaction mixture was purified by reverse phase chromatography (30 g C18Aq, 5-60-100% of acetonitrile in water each containing 0.1% of HCOOH).
- the product came out in 60% of acetonitrile.
- the fractions were combined to afford 18.1 mg of the Boc amine as a white solid which was dissolved in 2 mL of DCM and treated with 0.4 mL of TFA at rt.
- the reaction mixture was concentrated under reduced pressure and the contents were dissolved in DMF and purified by reverse phase chromatography (Isco EZ Prep (Gemini C18 column, 30 x 150 mm, 5-50-100% of acetonitrile in water, each containing 0.1% of HCOOH).
- Linker Payload 153 was synthesized from intermediate 140 as outlined below. [0518] Synthesis of 153: The title compound was synthesized following the procedure described for 152 above. Accordingly, 140 (12.1 mg, 0.0126 mmol) was reacted with 150 (13 mg, 0.0126 mmol) in the presence of HATU (9.6 mg, 0.0252 mmol) and NMM (4.2 pL, 0.0378 mmol) in DMF (0.5 mL) at rt in air.
- Linker Payload 154 was synthesized from intermediate 52 as outlined below. [0522] Synthesis of 154: The title compound was synthesized following the procedure described for 153. Accordingly, 52 (11 mg, 0.0116 mmol) was reacted with 150 (12 mg, 0.0116 mmol) in the presence of HATU (9 mg, 0.0233 mmol) and NMM (6.4 pL, 0.0582 mmol) in DMF (0.6 mL) at rt in air.
- Linker Payload 162 was synthesized from intermediate 52 as outlined below.
- Linker Payload 164 was synthesized from intermediate 19 as outlined below.
- Linker Payload 167 was synthesized from intermediate 125 as outlined below.
- the title compound was synthesized as per the procedure described for 161 where 52 (21 mg, 0.0222 mmol) was reacted with 165 (11 mg, 0.0222 mmol) in the presence of HATU (17 mg, 0.0444 mmol) and NMM (12.3 pL, 0. 1111 mmol) in DMF (0.5 mL) at rt. Work up and chromatography (15.5 g, C18Aq, 5-40-100% of acetonitrile in water each containing 0.1% of HCOOH) followed by lyophilization of the fractions afforded 166 (25.7 mg, 0.0184 mmol, 83%) as a white solid.
- Linker Payload 172 was synthesized from intermediate 147 and 52 as outlined below.
- Linker Payload 177 was synthesized from intermediate 156, 147 and 52 as outlined below. Synthesis of (2R,3S,4S,5R,6S)-2-(acetoxymethyl)-6-(2-amino-4- (hydroxymethyl)phenoxy)tetrahydro-2H-pyran-3,4,5-triyl triacetate (173): To a solution of 156 (1.07 g, 2.14 mmol) in MeOH/THF (24 mL + 24 mL) was added Zn dust (1.27 g, 19.5 mmol) and aq. NH4CI (12 mL) at rt. The reaction was stirred for 1 h for completion. The solution was concentrated and then suspended in MeOH.
- Linker Payload 178 was synthesized from intermediate 140 as outlined below.
- EXAMPLE 56 Bacterial Transglutaminase Conjugation - 1
- Two antibodies an anti-HER2 antibody having variable regions derived from humAb4D5-8 from Carter et al, PNAS, 1992, 89 4285, also known as trastuzumab, and a nonbinding isotype control derived from an immunological antigen having no relation to oncology or infectious diseases, both containing a N297Q mutation, which eliminates N- linked glycosylation of the Fc at this site, were used.
- the mutation allowed the antibodies to be conjugated to a maximum loading of 4 at 295Q and 297Q of the heavy chains.
- the anti- HER2 and Isotype Control antibodies were conjugated at 1-10 mg/mL in PBS pH 7.4.
- Linker payloads were added in a 5 to 25-fold molar excess over antibody and the enzymatic reaction was initiated by addition of 1-12 units of bacterial transglutaminase (Zedira, T1001) per mg antibody and incubated with shaking at 37°C for 4-18 hours.
- the conjugates were purified by size exclusion chromatography and sterile filtered. Protein concentrations were determined by UV spectral analysis. Size-exclusion HPLC established that all conjugates used were >95% monomeric. Yields are reported in Table 1 based on protein contents.
- ADC Benchmark DXd antibody-drug conjugate
- the conjugates were analyzed by LC-MS. Waters QToFs XEVO G2-S and QToFs XEVO G2-XS were used for the analysis.
- DAR PID0 x 0 +PID1 x 1 + . +PIDi x i / Z(PID0+PID1 +PID2. +PIDi)
- DAR Light Chain
- EXAMPLE 59 Characterization of conjugates by High Performance Size Exclusion Liquid Chromatography (SEC-HPLC) and Ultra Performance Size Exclusion Liquid Chromatography (SEC-UPLC).
- conjugates were analyzed by SEC-HPLC or SEC-UPLC.
- EXAMPLE 60 ADC, Payloads, and Linker-payloads Killing Assay Protocol (384-well format)
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Preparation (AREA)
- Peptides Or Proteins (AREA)
Abstract
The disclosure relates to analogs of quinoxaline/quinoline cytotoxic agents, termed"Nupomycins" herein, linker-payloads, and protein-drug conjugates thereof, pharmaceutical compositions comprising same, and methods of treating disease therewith.
Description
ANALOGS OF QUINOXALINE/QUINOLINE CYTOTOXINS, LINKERPAYLOADS, PROTEIN-DRUG CONJUGATES, AND USES THEREOF
[0001] This application claims the benefit of priority to U.S. Provisional Patent Application No. 63/603,966, filed on November 29, 2023 and U.S. Provisional Patent Application No. 63/688,088, filed on August 28, 2024, which applications are incorporated as if fully set forth herein.
FIELD OF INVENTION
[0002] The present disclosure relates to analogs of quinoxaline and quinoline antibiotics - these are herein termed “Nupomycins” to differentiate the analogs from those natural counter parts of triostins and echinomycins - linker- payloads, as well as protein-drug conjugates (e.g., antibody -drug conjugates) thereof, pharmaceutical compositions comprising the same, and methods of treating disease therewith.
BACKGROUND OF THE INVENTION
[0003] Proliferative diseases are characterized by uncontrolled growth and spread of abnormal cells. If the spread is not controlled, it can result in death. Abnormal proliferation, such as cancer, is caused by both external factors (e.g., tobacco, chemicals, radiation and infectious organisms) and internal factors (inherited mutations, immune system conditions, the mutations that occur from metabolism). These causal factors may act together or in sequence to initiate or promote abnormal proliferation. Cancer is treated by surgery, radiation, chemotherapy, hormones and immunotherapy. However, there is a need for more effective anti-proliferation drugs.
[0004] Quinoxaline antibiotics are of widespread occurrence in nature. They are heterodetic cyclic depsipeptides characterized by the possession of quinoxaline-2- carboxylic acid moieties, and the best-known member of the series is echinomycin which is identical to quinomycin A. In general, quinoxalines are powerful antimicrobial agents, cytotoxic to mammalian cells in culture, and display significant inhibitory activity towards a variety of tumors through inhibiting RNA synthesis by specific binding of double-stranded DNA through bisintercalation. Those compounds have a rigid, disulfide -bridged, bicyclic, depsipeptide scaffold, which preorganizes two quinoxaline intercalating units. The aromatic groups are oriented in parallel at a distance of 10.5 A, a perfect orientation of two intercalators to interact with two adjacent DNA base pairs. Quinoxaline antibiotics reversibly
intercalate with the double -helical structure of DNA by interacting with adjacent base pairs and disrupting the structure of DNA and thereby causing cell death (Ross & Bradley, Biochimica et Biophysica Acta (BBA)-Nucleic Acids and Protein Synthesis, 1981, 654(1), 129-134).
[0005] Quiescence is a state of reversible growth arrest in which cells have exited the cell cycle but remain capable of renewed division upon stimulation. Entry into quiescence allows cells to persist in a non-dividing state over extended periods of time and enact mechanisms to protect themselves from damage. Although quiescent cells display some similarities to other non-dividing cell states, such as senescence and terminal differentiation, quiescence possesses unique characteristics and functions. In particular, whereas senescent and terminally differentiated cells arrest permanently and are unable to proliferate further, quiescent cells are defined by their ability to reenter the cell cycle. This broad definition of quiescence encompasses a wide range of diverse cell types in an organism. Quiescent cells include tissue-resident adult stem cells, such as hematopoietic, muscle, and neural stem cells, as well as differentiated cells, including fibroblasts, hepatocytes, lymphocytes, and oocytes. Quiescence maintains these cells in a poised state — non-proliferative, but ready to re-enter the cell cycle when confronted with the appropriate stimulus (Oceane Mareseal and Iain M. Cheeseman, Dev Cell. 2020 November 09; 55(3): 259-271).
[0006] Quiescent cancer cells (QCCs) can avoid most chemotherapies and re-enter a proliferative state when conditions are right. This can lead to drug resistance and tumor recurrence. Quiescent cancer cells (QCCs) are nonproliferating cells arrested in the GO phase, characterized by ki671ow and p27high. QCCs avoid most chemotherapies, and some treatments could further lead to a higher proportion of QCCs in tumors. QCCs are also associated with cancer recurrence since they can re-enter a proliferative state when conditions are favorable. As QCCs lead to drug resistance and tumor recurrence, there is a great need to understand the characteristics of QCCs, decipher the mechanisms that regulate the proliferative-quiescent transition in cancer cells, and develop new strategies to eliminate QCCs residing in solid tumors (Lindell, E., et al., lot. J. Mol. Sci. 2023, 24, 3762). Effectively targeting and killing QCCs would provide another avenue to fight various cancers.
[0007] The ideal anti-proliferation therapy would enable targeted delivery of highly cytotoxic agents to tumor cells and would leave normal cells unaffected. Conventional chemotherapeutic treatment is limited because of the toxic side-effects that arise from effects
of the drug on non-cancerous cells. Various approaches to targeted drug delivery have been tried, including the use of conjugates of tumor targeted probes (such as antibodies or growth factors) with toxins such as pseudomonas or diphtheria toxins, which arrest the synthesis of proteins and cells. However, the side effects include reaction of the immune system due to non-human components of the conjugates. Further, the half-life of the drug conjugates is limited due to elimination from the circulation through renal filtration, and schematic degradation, uptake by the reticuloendothelial system (RES), and accumulation in nontargeted organs and tissues.
[0008] Another approach uses passive drug carriers such as polymers, liposomes, and polymeric micelles to take advantage of the hyper-permeability of vascular endothelia of tumor tissue. Polymeric drugs and macromolecules accumulate within solid tumors due to an enhanced permeability and retention mechanism. However, barriers of using such targeted deliveries include fast clearance of foreign particles from the blood, and technological hindrances in obtaining highly standardized, pharmaceutically acceptable drug delivery systems with the necessary specificity and selectivity for binding tumor cells.
[0009] Protein conjugates, such as antibody conjugates, utilize the selective binding of a binding agent to deliver a payload to targets within tissues of subjects. The payload can be a therapeutic moiety that is capable of taking action at the target.
[0010] Several techniques for conjugating linkers and payloads to antibodies are available. Many conjugates are prepared by non-selective covalent linkage to cysteine or lysine residues in the antibody. This non-selective technique can result in a heterogeneous mixture of products with conjugations at different sites and with different numbers of conjugations per antibody. Thus, there is a need in the art for methods and techniques that provide site- selective antibody conjugation.
[0011] There is a need in the art for additional safe and effective anti-tumor targeting agents that can bind to various antigens to provide enhanced the treatment of diseases such as cancer for use in monotherapy and combination therapies.
[0012] The foregoing discussion is presented solely to provide a better understanding of the nature of the problems confronting the art and should not be construed in any way as an admission as to prior art nor should the citation of any reference herein be construed as an admission that such reference constitutes “prior art” to the instant application.
SUMMARY OF THE DISCLOSURE
[0013] The present disclosure addresses these needs and confers advantages by providing, in one aspect, a compound having a structure according to formula (la), (lb), or (Ic):
 or a pharmaceutically acceptable salt thereof.
or a pharmaceutically acceptable salt thereof.
[0014] In Formulae (la), (lb), and (Ic):
X and Y are independently selected from N and CH; Z is O or NH; m is 1, 2, or 3;
Ri is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, and aralkyl, each of which is optionally substituted;
R2 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR;
R3 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR; R4 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR; and
R5 is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, aralkyl, each of which is optionally substituted. [0015] In another aspect, the present disclosure provides a linker-payload compound having a structure according to formula (Ila), (lib), or (lie):
 or a pharmaceutically acceptable salt thereof.
or a pharmaceutically acceptable salt thereof.
[0016] In Formulae (Ila), (lib), and (lie):
X and Y are independently selected from N and CH;
Z is O or NH; m is 1, 2, or 3;
R2 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR;
R3 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR;
R4 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl,
OR, and NHR; and
R5 is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, aralkyl, each of which is optionally substituted;
LI, when present, is a stable or self-immolative linker;
L2, when present, is a linker that is stable or is cleavable by an endosomal/lysosomalenzyme;
L3, when present, is a moiety that modulates the hydrophilicity, physical, and/or chemical properties of the linker-payload compound; and
B is a reactive moiety for conjugation to a target protein, antibody, or antigen-binding fragment thereof, comprising at least one member selected from the group consisting of amino, hydroxyl, thiol, a maleimido moiety, a succinyl ester, an activated ester, an
UL JU alkylamino; a haloacetamido, -N3, ~
 (where Q is CH or
(where Q is CH or

[0017] In another aspect, the present disclosure provides linker-payload compounds having a linker moiety that modulates the hydrophilicity, physical, and/or chemical properties of the linker-payload compounds. Non-limiting examples include various sugar or carbohydrate moieties, e.g. mono-, di-, and polysaccharides, cyclic polysaccharides (e.g. cyclodextrins), a quaternary ammonium salt, a polyethylene glycol group, a sulfonic acid group, a phosphonic acid group, and combinations thereof.
[0018] In still another aspect, the present disclosure provides a conjugate having the formula having a structure according to Formula (I) or Formula (II):
BA-NH(-L-P)n (I)
BA-S(-L-P)n (II)
[0019] In Formulae (I) and (II):
BA is an antibody or an antigen-binding fragment thereof;
L is a linker of the formula -L1-L2-L3-B-, wherein
L is connected to BA through a side chain of an amino acid selected from the group consisting of Gin (-CO-NH-), Lys (-NH-CO-), and Cys (-S-);
LI, when present, is a stable or self-immolative linker;
L2, when present, is a linker that is stable or is cleavable by an endosomal/lysosomalenzyme;
L3, when present, is a moiety that modulates the hydrophilicity, physical, and/or chemical properties of -L-P; and
B is a reactive moiety for conjugation to a target protein, antibody, or antigen-binding fragment thereof, comprising at least one member selected from the group consisting of amino, hydroxyl, thiol, a maleimido moiety, a succinyl ester, an activated ester, an alkylamino; a haloacetamido, -

(where

P is a moiety having a structure according to Formula (la’), Formula (lb’), or Formula
(Ic’):

X and Y are independently selected from N and CH;
Z is O or NH; m is 1, 2, or 3;
R2 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR;
R3 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR;
R4 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR; and
R5 is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, aralkyl, each of which is optionally substituted; and represents the connecting point of attachment to the linker L; and
n is an integer from 1 to 20.
[0020] In an additional aspect of the present disclosure, there is provided a conjugate comprising (i) an antibody (BA) or an antigen-binding fragment thereof, (ii) a plurality of payloads, and a linker that covalently connects (i) and (ii), wherein the linker is connected to the BA or antigen-binding fragment thereof through a side chain of an amino acid selected from the group consisting of Gin (-C0-NH-), Lys (-NH-C0-), and Cys (-S-), and has a formula -L1-L2-L3-B-, wherein
LI, when present, is a stable or self-immolative linker;
L2, when present, is a linker that is stable or is cleavable by an endosomal/lysosomalenzyme;
L3, when present, is a moiety that modulates the hydrophilicity, physical, and/or chemical properties of the conjugate; and
B is a residue of a reactive moiety for conjugation, comprising at least one member selected from the group consisting of amino, hydroxyl, thiol, a maleimido moiety, a succinyl ester, an activated ester, an alkylamino; a haloacetamido, -N3, ~

 and the payload has a structure according to Formula (la’), Formula (lb’), or Formula (Ic’):
and the payload has a structure according to Formula (la’), Formula (lb’), or Formula (Ic’):


X and Y are independently selected from N and CH;
Z is O or NH; m is 1, 2, or 3;
R2 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR;
R3 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR;
R4 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR; and
R5 is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, aralkyl, each of which is optionally substituted; and represents the connecting point of attachment to the linker.
[0021] In an additional aspect, the present disclosure provides a pharmaceutical composition comprising a conjugate as described herein and one or more pharmaceutically acceptable carriers, excipients or diluents.
[0022] In another aspect of the present disclosure there is provided a process of producing a conjugate of Formula (I) or Formula (II) as described herein. The process comprises contacting an antibody or an antigen-binding fragment thereof (BA) with a linker-payload compound (L-P) in the presence of a transglutaminase, wherein L-P has a structure according to formula (Ila), (lib), or (lie) as described herein.
[0023] The present disclosure also provides in another aspect a method of treating a subject suffering from cancer. The method comprises administering to the subject a therapeutically effective amount of a compound, composition, conjugate, or pharmaceutical dosage form as described herein.
[0024] Also provided in the present disclosure is a compound, composition, conjugate, or pharmaceutical dosage form as described herein for use in treating a cancer. In addition, the present disclosure provides a use of a therapeutically effective amount of a compound, composition, conjugate, or pharmaceutical dosage form as described herein in the manufacture of a medicament for the treatment of a cancer.
[0025] The present disclosure provides in another aspect a method of selectively killing quiescent cells in a subject. The comprises administering to the subject a therapeutically effective amount of a compound, conjugate, or pharmaceutical composition as described herein.
[0026] The present disclosure provides in another aspect a method of selectively killing stem cells in a subject. The comprises administering to the subject a therapeutically effective amount of a compound, conjugate, or pharmaceutical composition as described herein.
[0027] The present disclosure provides in another aspect a method of selectively killing resting or naive B- or T- or other immune in a subject. The comprises administering to the subject a therapeutically effective amount of a compound, conjugate, or pharmaceutical composition as described herein.
[0028] The present disclosure provides in another aspect a method of selectively killing quiescent cancer cells in a subject preparing for stem cell therapy. The comprises administering to the subject a therapeutically effective amount of a compound, conjugate, or pharmaceutical composition as described herein.
DETAILED DESCRIPTION
[0029] Definitions
[0030] To facilitate understanding of the disclosure set forth herein, certain terms are defined below.
[0031] Unless defined otherwise, all technical and scientific terms used herein have the same meaning as is commonly understood by one of ordinary skill in the art. All patents, applications, published applications and other publications are incorporated by reference in their entirety. In the event that there are a plurality of definitions for a term herein, those in this section prevail unless stated otherwise.
[0032] The singular forms "a," "an," and "the" include plural references, unless the context clearly dictates otherwise.
[0033] As used herein "subject" is an animal, such as a mammal, including human, such as a patient.
[0034] As used herein, biological activity refers to the in vivo activities of a compound or physiological responses that result upon in vivo administration of a compound, composition or other mixture. Biological activity, thus, encompasses therapeutic effects and pharmacokinetic behavior of such compounds, compositions and mixtures. Biological activities can be observed in in vitro systems designed to test for such activities.
[0035] The phrase “specifically binds,” or “binds specifically to,” or the like, means that an antibody or antigen-binding fragment thereof forms a complex with an antigen that is relatively stable under physiologic conditions. Specific binding can be characterized by an equilibrium dissociation constant of at least about 1x10-8 M or less (e.g., a smaller KD denotes tighter binding). Methods for determining whether two molecules specifically bind are well known in the art and include, for example, equilibrium dialysis, surface plasmon resonance, and the like. Antibodies can, for example, be identified by real-time, label free bio-layer interferometry assay on an Octet® HTX biosensor, which bind specifically to a target antigen. Moreover, multi-specific antibodies that bind to one domain in the target antigen and one or more additional antigens or a bi-specific that binds to two different regions of the target antigen are nonetheless considered antibodies that “specifically bind”, as used herein. In addition to neutralizing antibodies, antibodies that bind specifically to the target antigen, but are non-neutralizing, also can be used within the scope of the present disclosure to generate antibody-drug conjugates. Such antibodies may function, for example, to deliver a payload to the cells expressing a target antigen.
[0036] The term "antibody," as used herein, means any antigen-binding molecule or molecular complex comprising at least one complementarity determining region (CDR) that specifically binds to or interacts with a particular antigen. The term "antibody" includes immunoglobulin molecules comprising four polypeptide chains, two heavy (H) chains and two light (L) chains inter-connected by disulfide bonds, as well as multimers thereof (e.g., IgM). Each heavy chain comprises a heavy chain variable region (abbreviated herein as HCVR or VH) and a heavy chain constant region. The heavy chain constant region comprises
three domains, CHI, CH2 and CH3. Each light chain comprises a light chain variable region (abbreviated herein as LCVR or VL) and a light chain constant region. The light chain constant region comprises one domain (CL1). The VH and VL regions can be further subdivided into regions of hypervariability, termed complementarity determining regions (CDRs), interspersed with regions that are more conserved, termed framework regions (FR). Each VH and VL is composed of three CDRs and four FRs, arranged from amino-terminus to carboxy-terminus in the following order: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. Three CDRs of VH are referred to as HCDR1 , HCDR2, and HCDR3, and three CDRs of VL are referred to as LCDR1, LCDR2 and LCDR3.
[0037] As used herein, the term “antigen-binding fragment" of an antibody means any naturally occurring, enzymatically obtainable, synthetic, or genetically engineered polypeptide or glycoprotein that specifically binds an antigen to form a complex.
[0038] As used herein, the term "human antibody" means antibodies having variable and constant regions derived from human germline immunoglobulin sequences. Human antibodies may nonetheless include amino acid residues not encoded by human germline immunoglobulin sequences (e.g., mutations introduced by random or site-specific mutagenesis in vitro or by somatic mutation in vivo), for example in the CDRs and in particular CDR3. However, the term "human antibody", as used herein, is not intended to include antibodies in which CDR sequences derived from the germline of another mammalian species, such as a mouse, have been grafted onto human framework sequences.
[0039] As used herein, the term “humanized antibody” means chimeric antibodies that contain minimal sequence derived from the non-human antibody. A humanized antibody is generally a human antibody (recipient antibody) in which residues from one or more CDRs are replaced by residues from one or more CDRs of a non-human antibody (donor antibody). The donor antibody can be any suitable non-human antibody, such as a mouse, rat, rabbit, chicken, or non-human primate antibody having a desired specificity, affinity, or biological effect. In some instances, selected framework region residues of the recipient antibody are replaced by the corresponding framework region residues from the donor antibody.
Humanized antibodies may also comprise residues that are not found in either the recipient antibody or the donor antibody. Such modifications may be made to further refine antibody function. For further details, see Jones et al., Nature, 1986, 321:522-525; Riechmann et al.,
Nature, 1988, 332:323-329; and Presta, Curr. Op. Struct. Biol., 1992, 2:593-596, each of which is incorporated by reference in its entirety.
[0040] As used herein, the term "recombinant human antibody", means all human antibodies that are prepared, expressed, created or isolated by recombinant means, such as antibodies expressed using a recombinant expression vector transfected into a host cell (described further below), antibodies isolated from a recombinant, combinatorial human antibody library (described further below), antibodies isolated from an animal (e.g., a mouse) that is transgenic for human immunoglobulin genes (see, e.g., Taylor et al. (1992) Nucl. Acids Res. 20:6287-6295) or antibodies prepared, expressed, created or isolated by any other means that involves splicing of human immunoglobulin gene sequences to other DNA sequences.
[0041] The term “substantial identity” or “substantially identical,” when referring to a nucleic acid or fragment thereof, indicates that, when optimally aligned with appropriate nucleotide insertions or deletions with another nucleic acid (or its complementary strand), there is nucleotide sequence identity in at least about 90%, and more preferably at least about 95%, 96%, 97%, 98%, or 99% of the nucleotide bases, as measured by any well-known algorithm of sequence identity, such as FASTA, BLAST, or GAP, as discussed in WO 2016/100807 or US 2016/0176953 Al, each of which are incorporated herein by reference in their entirety. A nucleic acid molecule having substantial identity to a reference nucleic acid molecule may, in certain instances, encode a polypeptide having the same or substantially similar amino acid sequence as the polypeptide encoded by the reference nucleic acid molecule.
[0042] As used herein in the context of amino acid sequences, the phrase “substantial similarity” or “substantially similar” means that two peptide sequences, when optimally aligned, such as by the programs GAP or BESTFIT using default gap weights, share at least 90% sequence identity, even more preferably at least 95%, 98%, or 99% sequence identity. Preferably, residue positions, which are not identical, differ by conservative amino acid substitutions.
[0043] As used herein, the term "surface plasmon resonance", refers to an optical phenomenon that allows for the analysis of real-time interactions by detection of alterations in protein concentrations within a biosensor matrix, for example using the BIAcore™ system (Biacore Life Sciences division of GE Healthcare, Piscataway, N.J.).
[0044] As used herein, the term "KD", means the equilibrium dissociation constant of a particular protein-protein interaction (e.g., antibody-antigen interaction). Unless indicated otherwise, the KD values disclosed herein refer to KD values determined by surface plasmon resonance assay at 25° C.
[0045] In this disclosure, a “pharmaceutically acceptable salt” is a pharmaceutically acceptable, organic or inorganic acid or base salt of a compound described herein. Representative pharmaceutically acceptable salts include, e.g., alkali metal salts, alkali earth salts, ammonium salts, water-soluble and water-insoluble salts, such as the acetate, amsonate (4,4-diaminostilbene-2,2-disulfonate), benzenesulfonate, benzonate, bicarbonate, bisulfate, bitartrate, borate, bromide, butyrate, calcium, calcium edetate, camsylate, carbonate, chloride, citrate, clavulariate, dihydrochloride, edetate, edisylate, estolate, esylate, fiunarate, gluceptate, gluconate, glutamate, glycollylarsanilate, hexafluorophosphate, hexylresorcinate, hydrabamine, hydrobromide, hydrochloride, hydroxynaphthoate, iodide, isothionate, lactate, lactobionate, laurate, malate, maleate, mandelate, mesylate, methylbromide, methylnitrate, methylsulfate, mucate, napsylate, nitrate, N-methylglucamine ammonium salt, 3-hydroxy-2 -naphthoate, oleate, oxalate, palmitate, pamoate (l,l-methene-bis-2-hydroxy-3- naphthoate, einbonate), pantothenate, phosphate/diphosphate, picrate, polygalacturonate, propionate, p-toluenesulfonate, salicylate, stearate, subacetate, succinate, sulfate, sulfosaliculate, suramate, tannate, tartrate, teoclate, tosylate, triethiodide, and valerate salts. A pharmaceutically acceptable salt can have more than one charged atom in its structure. In this instance the pharmaceutically acceptable salt can have multiple counterions. Thus, a pharmaceutically acceptable salt can have one or more charged atoms and/or one or more counterions.
[0046] As used herein, the terms “treat,” “treating,” or “treatment” refer to the reduction or amelioration of the severity of at least one symptom or indication of the disease, e.g., cancer or hepatitis B infection, due to the administration of a therapeutic agent such as a disclosed antibody to a subject in need thereof. The terms include inhibition of progression of disease or of worsening of infection. The terms also include positive prognosis of disease, e.g., the subject may be free of infection, the subject may have reduced or no viral titers, the subject may have tumor shrinkage, upon administration of a therapeutic agent such as a disclosed antibody or antibody-drug conjugate. The therapeutic agent may be administered at a therapeutic dose to the subject.
[0047] The terms “prevent,” “preventing,” or “prevention” refer to inhibition of manifestation of any symptoms or indications of a disease (e.g., cancer or hepatitis B infection) upon administration of a disclosed antibody or antibody-drug conjugate. The term includes prevention of the spread of infection in a subject exposed to the virus or at risk of having hepatitis B infection.
[0048] The phrase “therapeutically effective amount” refers to an amount that produces the desired effect for which it is administered. The exact amount will depend on the purpose of the treatment, and will be ascertainable by one skilled in the art using known techniques (see, for example, Lloyd (1999) The Art, Science and Technology of Pharmaceutical Compounding).
[0049] As used herein, amelioration of the symptoms of a particular disorder by administration of a particular compound or pharmaceutical composition refers to any lessening, whether permanent or temporary, lasting or transient that can be attributed to or associated with administration of the compound or pharmaceutical composition.
[0050] As used herein, the IC50 refers to an amount, concentration or dosage of a particular test compound that achieves a 50% inhibition of a maximal response in an assay that measures such response.
[0051] Where moieties are specified by their conventional chemical formulae, written from left to right, they equally encompass the chemically identical moieties that would result from writing the structure from right to left, e.g., -CH2O- is equivalent to -OCH2-.
[0052] The term "alkyl," by itself or as part of another substituent, means, unless otherwise stated, a straight (z.e., unbranched) or branched chain saturated hydrocarbon radical. The term "alkylene" by itself or as part of another substituent means a divalent radical derived from an alkyl. In various embodiments, an alkyl (or alkylene) group will have from 1 to 24 carbon atoms (i.e., Ci-C24-alkyl), including those groups having 10 or fewer carbon atoms (i.e., Ci-Cio-alkyl). A "lower alkyl" or "lower alkylene" is a shorter chain alkyl or alkylene group, generally having six or fewer carbon atoms (i.e., Ci-Ce-alkyl). Examples of alkyl groups include, but are not limited to, groups such as methyl, ethyl, n-propyl, isopropyl, n- butyl, t-butyl, isobutyl, sec-butyl, homologs and isomers of, for example, n-pentyl, n-hexyl, n-heptyl, n-octyl, and the like.
[0053] The term "alkenyl," by itself or as part of another substituent, means, unless otherwise stated, a straight (z'.e., unbranched) or branched chain hydrocarbon radical having one or more carbon-carbon double bonds. The term "alkenylene" by itself or as part of another substituent means a divalent radical derived from an alkenyl. Typically, an alkenyl (or alkenylene) group will have from 2 to 24 carbon atoms (i.e., C2-C24-alkenyl), including those groups having 10 or fewer carbon atoms (i.e., C2-Cio-alkenyl). A "lower alkenyl" or "lower alkenylene" is a shorter chain alkenyl or alkenylene group, generally having six or fewer carbon atoms (i.e., C2-Ce-alkenyl. Examples of alkenyl groups include, but are not limited to, vinyl (z'.e., ethenyl), 2-propenyl, crotyl, 2 -isopentenyl, 2-(butadienyl), 2,4-pentadienyl, 3-(l,4- pentadienyl), and the higher homologs and isomers.
[0054] The term "alkynyl," by itself or as part of another substituent, means, unless otherwise stated, a straight (i.e., unbranched) or branched chain hydrocarbon radical having one or more carbon-carbon triple bonds, which can include di- and multivalent radicals, having the number of carbon atoms designated i.e., C2-C10 means two to ten carbons in C2-Cio-alkynyl). Examples of alkynyl groups include, but are not limited to, ethynyl, 1- and 3-propynyl, 3- butynyl, and the higher homologs and isomers.
[0055] The terms "alkoxy," "alkylamino," and "alkylthio" (or thioalkoxy) are used in their conventional sense and refer to those alkyl groups attached to the remainder of the molecule via an oxygen atom, an amino group, or a sulfur atom, respectively.
[0056] The term "heteroalkyl," by itself or in combination with another term, means, unless otherwise stated, a straight or branched chain hydrocarbon radical, containing at least one heteroatom in the chain selected from O, N, P, Si and S, and wherein the nitrogen and sulfur atoms may optionally be oxidized, and the nitrogen atom may have an alkyl substituent to fulfill valency and/or may optionally be quaternized. The heteroatom(s) O, N, P, Si and S may be placed at any interior position of the heteroalkyl group (i.e., not at the point of attachment to the rest of the molecule). Examples include, but are not limited to, -CH2-CH2- 0-CH3, -CH2-CH2-NH-CH3, -CH2-CH2-N(CH3)-CH3, -CH2-S-CH2-CH3, -CH2-CH2-S(O)- CH3, -CH2-CH2-S(O)2-CH3, -CH=CH-O-CH3. -CH2-CH=N-OCH3. and -CH=CH-N(CH3)- CH3. Up to two heteroatoms may be consecutive, such as, for example, -CH2-NH-OCH3 and -CH2-O-Si(CH3)3- Similarly, the term "heteroalkylene" by itself or as part of another substituent means a divalent radical derived from heteroalkyl, as exemplified, but not limited by, -CH2-O-CH2-CH2-, -CH2-CH2-O-CH2-CH2-, -CH2-O-CH2-CH2-NH-CH2-, -CH2-CH2-S-
CH2-CH2- and -CH2-S-CH2-CH2-NH-CH2-. For alkylene and heteroalkylene linking groups, no orientation of the linking group is implied by the direction in which the formula of the linking group is written. For example, the formula -C(0)2R’- represents both -C(0)2R’- and -R'C(0)2-.
[0057] The terms "cycloalkyl" and "heterocycloalkyl", by themselves or in combination with other terms, represent, unless otherwise stated, cyclic versions of "alkyl" and "heteroalkyl", respectively, including bicyclic, tricyclic and bridged bicyclic groups. Additionally, for heterocycloalkyl, a heteroatom can occupy the position at which the heterocycle is attached to the remainder of the molecule. The terms "cycloalkylene" and "heterocycloalkylene" by themselves or as part of another substituent means a divalent radical derived from a cycloalkyl or heterocycloalkyl. Examples of cycloalkyl include Cs-Cio-cycloalkyl, but are not limited to, cyclopentyl, cyclohexyl, 1 -cyclohexenyl, 3-cyclohexenyl, cycloheptyl, norbornanyl, bicyclo(2.2.2)octanyl, and the like. Examples of heterocycloalkyl include C3- Cio-heterocycloalkyl, but are not limited to, l-(l,2,5,6-tetrahydropyridyl), 1-piperidinyl, 2- piperidinyl, 3-piperidinyl, 4-morpholinyl, 3-morpholinyl, tetrahydrofuran-2-yl, tetrahydrofuran-3-yl, tetrahydrothien-2-yl, tetrahydrothien-3-yl, 1-piperazinyl, 2-piperazinyl, 1- or 2-azabicyclo(2.2.2)octanyl, and the like.
[0058] The term "aryl" means, unless otherwise stated, a polyunsaturated, aromatic, hydrocarbon substituent which can be a single ring or multiple rings (in some embodiments from 1 to 3 rings) which are fused together or linked covalently. “Aryl” includes C6-C12 aryl rings. The term "heteroaryl" refers to aryl groups that contain from one to four heteroatoms selected from N, O, and S in the ring(s), wherein the nitrogen and sulfur atoms are optionally oxidized, and the nitrogen atom(s) are optionally quaternized. “Heteroaryl” also includes a 5- to 10-membered ring having one to four heteroatom ring members as described herein. A heteroaryl group can be attached to the remainder of the molecule through a carbon or heteroatom. The terms "arylene" and "heteroarylene" by themselves or as part of another substituent means a divalent radical derived from an aryl or heteroaryl. Non-limiting examples of aryl and heteroaryl groups include phenyl, 1 -naphthyl, 2-naphthyl, 4-biphenyl, 1 -pyrrolyl, 2-pyrrolyl, 3-pyrrolyl, 3-pyrazolyl, 2-imidazolyl, 4-imidazolyl, pyrazinyl, 2- oxazolyl, 4-oxazolyl, 5-oxazolyl, 3-isoxazolyl, 4-isoxazolyl, 5-isoxazolyl, 2-thiazolyl, 4- thiazolyl, 5-thiazolyl, 2-furyl, 3-furyl, 2-thienyl, 3-thienyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 2- pyrimidyl, 4-pyrimidyl, 5-benzothiazolyl, purinyl, 2-benzimidazolyl, 5-indolyl, 1 -isoquinolyl,
5 -isoquinolyl, 2-quinoxalinyl, 5-quinoxalinyl, 3-quinolyl, and 6-quinolyl. The term "heteroarylium" refers to a heteroaryl group that is positively charged on one or more of the heteroatoms.
[0059] Each of the terms above are meant to include both substituted and unsubstituted forms of the indicated radical. Non-limiting examples of substituent moieties for each type of radical are provided below.
[0060] Substituent moieties for alkyl, heteroalkyl, alkylene, alkenyl, heteroalkylene, heteroalkenyl, alkynyl, cycloalkyl, heterocycloalkyl, cycloalkenyl, and heterocycloalkenyl groups are, in some embodiments, selected from, deuterium, -OR’, =0. =NR’, =N-OR’. - NR’R", -SR’, halo, -SiR'R"R"', -OC(O)R’, -C(O)R’, -CO2R’, -CONR’R", -OC(O)NR’R", - NR"C(O)R', -NR'-C(O)NR"R"', -NR"C(O)2R', -NR-
C(NR'R"R"')=NR"", -NR-C(NR'R")=NR"', -S(O)R’, -S(O)2R’, -S(O)2NR'R", -NRSO2R’, -CN and -NO2 in a number ranging from zero to the number of hydrogen atoms in such radical. In some embodiments, substituent moieties for cycloalkyl, heterocycloalkyl, cycloalkenyl, and heterocycloalkenyl groups also include substituted and unsubstituted alkyl, substituted and unsubstituted alkenyl, and substituted and unsubstituted alkynyl. R’, R", R'” and R"" each in some embodiments independently are hydrogen, substituted or unsubstituted heteroalkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocycloalkyl, substituted or unsubstituted aryl (e.g., aryl substituted with 1-3 halogens), substituted or unsubstituted alkyl, alkoxy or thioalkoxy groups, or arylalkyl groups. When a compound provided herein includes more than one R group, for example, each of the R groups is independently selected as are each R’, R", R’" and R"" groups when more than one of these groups is present. When R’ and R" are attached to the same nitrogen atom, they can be combined with the nitrogen atom to form a 4-, 5-, 6-, or 7-membered ring. For example, -NR’R" is meant to include, but not be limited to, 1-pyrrolidinyl and 4-morpholinyl. From the above discussion of substituent moieties, one of skill in the art will understand that the term "alkyl" is meant to include groups including carbon atoms bound to groups other than hydrogen groups, such as haloalkyl e.g., -CF3 and -CH2CF3) and acyl e.g., - C(O)CH3, -C(O)CF3, -C(O)CH2OCH3, and the like).
[0061] Substituent moieties for aryl and heteroaryl groups are, in some embodiments, selected from deuterium, halo, substituted and unsubstituted alkyl, substituted and unsubstituted alkenyl, and substituted and unsubstituted alkynyl, -OR’, -NR’R", -SR’, -
SiR'R"R"', -OC(O)R’, -C(O)R’, -CO2R’, -CONR’R", -OC(O)NR’R", -
NR"C(O)R', -NR'-C(O)NR"R"', -NR"C(O)2R', -NR-
C(NR'R"R"')=NR"", -NR-C(NR'R")=NR"', -S(O)R', -S(O)2R', -S(O)2NR'R", -NRSO2R’, -CN and -NO2, -R’, -N3, -CH(Ph)2, fluoro(Ci-C4)alkoxy, and fluoro(Ci-C4)alkyl, in a number ranging from zero to the total number of hydrogens on the aromatic ring system; and where R’, R", R'” and R"" are, in some embodiments, independently selected from hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted heteroalkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocycloalkyl, substituted or unsubstituted aryl and substituted or unsubstituted heteroaryl. When a compound provided herein includes more than one R group, for example, each of the R groups is independently selected as are each R’, R", R’" and R"" groups when more than one of these groups is present.
[0062] Two of the substituent moieties on adjacent atoms of an aryl or heteroaryl ring may optionally form a ring of the formula -Q’-C(O)-(CRR’)q-Q”-, wherein Q’ and Q” are independently -NR-, -O-, -CRR’- or a single bond, and q is an integer of from 0 to 3. Alternatively, two of the substituent moieties on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -A-(CH2)r-B-, wherein A and B are independently -CRR’-, -O-, -NR-, -S-, -S(O)-, -S(O)2-, -S(O)2NR’- or a single bond, and r is an integer of from 1 to 4. One of the single bonds of the new ring so formed may optionally be replaced with a double bond. Alternatively, two of the substituent moieties on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -(CRR’)s-X’-(CR”R”’)d-, where s and d are independently integers of from 0 to 3, and X’ is -O-, -NR’-, -S-, -S(O)-, -S(O)2-, or -S(O)2NR’-. The substituent moieties R, R’, R" and R’" are, in some embodiments, independently selected from hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocycloalkyl, substituted or unsubstituted aryl, and substituted or unsubstituted heteroaryl.
[0063] The term "halo," by itself or as part of another substituent, means, unless otherwise stated, a fluorine, chlorine, bromine, or iodine atom. Additionally, terms such as "haloalkyl," are meant to include monohaloalkyl and polyhaloalkyl. For example, the term "halo(Ci- C alkyl" is meant to include, but not be limited to, trifluoromethyl, 2,2,2-trifluoroethyl, 4- chlorobutyl, 3-bromopropyl, and the like.
[0064] The term "oxo" as used herein means an oxygen atom that is double bonded to a carbon atom.
[0065] As used herein, the term "heteroatom" or "ring heteroatom" is meant to include oxygen (O), nitrogen (N), sulfur (S), phosphorus (P), and silicon (Si).
[0066] Some compounds described herein can have asymmetric centers and therefore exist in different enantiomeric and diastereomeric forms. A compound as described herein can be in the form of an optical isomer or a diastereomer. Accordingly, the disclosure encompasses compounds and their uses as described herein in the form of their optical isomers, diastereoisomers and mixtures thereof, including a racemic mixture. Optical isomers of the compounds of the disclosure can be obtained by known techniques such as asymmetric synthesis, chiral chromatography, simulated moving bed technology or via chemical separation of stereoisomers through the employment of optically active resolving agents.
[0067] Unless otherwise indicated, the term “stereoisomer” means one stereoisomer of a compound that is substantially free of other stereoisomers of that compound. Thus, a stereomerically pure compound having one chiral center will be substantially free of the opposite enantiomer of the compound. A stereomerically pure compound having two chiral centers will be substantially free of other diastereomers of the compound. A typical stereomerically pure compound comprises greater than about 80% by weight of one stereoisomer of the compound and less than about 20% by weight of other stereoisomers of the compound, for example greater than about 90% by weight of one stereoisomer of the compound and less than about 10% by weight of the other stereoisomers of the compound, or greater than about 95% by weight of one stereoisomer of the compound and less than about 5% by weight of the other stereoisomers of the compound, or greater than about 97% by weight of one stereoisomer of the compound and less than about 3% by weight of the other stereoisomers of the compound, or greater than about 99% by weight of one stereoisomer of the compound and less than about 1% by weight of the other stereoisomers of the compound. The stereoisomer as described above can be viewed as composition comprising two stereoisomers that are present in their respective weight percentages described herein.
[0068] If there is a discrepancy between a depicted structure and a name given to that structure, then the depicted structure controls. Additionally, if the stereochemistry of a structure or a portion of a structure is not indicated with, for example, bold or dashed lines,
the structure or portion of the structure is to be interpreted as encompassing all stereoisomers of it. In some cases, however, where more than one chiral center exists, the structures and names may be represented as single enantiomers to help describe the relative stereochemistry. Those skilled in the art of organic synthesis will know if the compounds are prepared as single enantiomers from the methods used to prepare them.
[0069] As used herein, and unless otherwise specified to the contrary, the term “compound” is inclusive in that it encompasses a compound or a pharmaceutically acceptable salt, stereoisomer, isotopologue, and/or tautomer thereof. Thus, for instance, a compound includes a pharmaceutically acceptable salt of a tautomer of the compound. Similarly, a compound of includes a pharmaceutically acceptable salt of an isotopologue of the compound.
[0070] Compounds of Formulae (la), (lb), and (Ic)
[0001] In an aspect of the present disclosure, there is provided a compound having a structure according to formula (la), (lb), or (Ic):

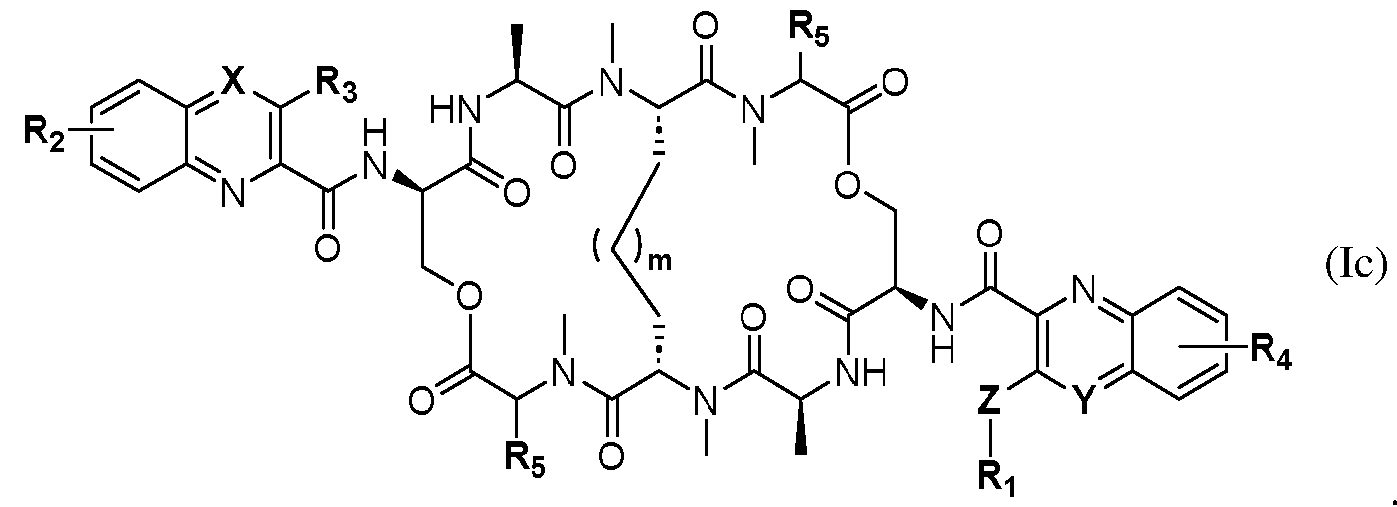
[0002] In formulae (la), (lb), and (Ic):
X and Y are independently selected from N and CH;
Z is O or NH; m is 1, 2, or 3;
Ri is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, and aralkyl, each of which is optionally substituted;
R2 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR;
R3 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR;
R4 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR; and
R5 is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, aralkyl, each of which is optionally substituted.
[0071] In some embodiments, the compound is of formula (la). In other embodiments, the compound is of formula (lb). In still further embodiments, the compound is of formula (Ic).
[0072] In various embodiments:
• X is N and Y is CH;
• X is N and Y is N;
• X is CH and Y is CH; or
• X is CH and Y is N.
[0073] In some embodiments, R3 is OH and R2 is hydrogen. In additional embodiments, R3 is OH and R4 is hydrogen.
[0074] In some embodiments, Z is O. In other embodiments, Z is NH.
[0075] In various embodiments, R5 is selected from the group consisting of alkyl, cycloalkyl, and aralkyl. Illustrative embodiments provide for formulae (la), (lb), and (Ic) compounds wherein R5 is selected from the group consisting of isopropyl, isobutyl, cyclopentyl, and cyclohexyl.
[0076] Specific examples shown in Table 1, and their pharmaceutically acceptable salts, provide additional embodiments of the present disclosure. [0077] Table 1 Structures of Payloads








[0079] In various embodiments, the present disclosure provides a linker-payload compound having a structure according to formula (Ila), (lib), or (lie):
 or a pharmaceutically acceptable salt thereof.
or a pharmaceutically acceptable salt thereof.
[0080] In formulae (Ila), (lib), and (lie):
X and Y are independently selected from N and CH; Z is O or NH; m is 1, 2, or 3;
R2 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR;
R3 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR;
R4 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR; and
Rs is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, aralkyl, each of which is optionally substituted;
LI, when present, is a stable or self-immolative linker;
L2, when present, is a linker that is stable or is cleavable by an endosomal/lysosomalenzyme;
L3, when present, is a moiety that modulates the hydrophilicity, physical, and/or chemical properties of the linker-payload compound; and
B is a reactive moiety for conjugation to a target protein, antibody, or antigen-binding fragment thereof, comprising at least one member selected from the group consisting of amino, hydroxyl, thiol, a maleimido moiety, a succinyl ester, an activated ester, an
0 0 alkylamino; a haloacetamido, -N3, ~
 (where Q is CH or
(where Q is CH or

[0081] In various embodiments:
• X is N and Y is CH;
• X is N and Y is N;
• X is CH and Y is CH; or
• X is CH and Y is N.
[0082] In some embodiments, R3 is OH and R2 is hydrogen. In additional embodiments, R3 is OH and R4 is hydrogen.
[0083] In embodiments, linkers LI, L2, and L3 are independently and optionally present. When present, the linkers in the aggregate comprise generally a linker moiety linked to the payload drug. Thus, in some embodiments, L3 is present and is a polyethylene glycol (PEG) unit, a carbohydrate moiety, or a combination thereof. Further descriptions of moieties and linkers suitable for use as LI, L2, and L3 are as follows.
[0084] In some embodiments, linkers for use herein may be found, for example, in Antibody- Drug conjugates and Immunotoxins, Phillips, G. L., Ed.; Springer Verlag: New York, 2013; Antibody-Drug conjugates, Ducry, L., Ed.; Humana Press, 2013; Antibody-Drug conjugates, Wang, J., Shen, W.-C., and Zaro, J. L., Eds.; Springer International Publishing, 2015. In some embodiments, the linker provided herein is sufficiently stable to exploit the circulating halflife of the antigen binding domain and, at the same time, capable of releasing the payload after antigen-mediated internalization of the conjugate, i.e., of Formula (I) or (II).
[0085] Linkers can be cleavable or non-cleavable. Cleavable linkers for use herein include linkers that are cleaved by intracellular metabolism following internalization, e.g., cleavage via hydrolysis, reduction, or enzymatic reaction, such as by an endosomal/lysosomalenzyme. Non-cleavable linkers for use herein include linkers that release an attached payload via lysosomal degradation of the antigen binding domain following internalization.
[0086] Illustrative linkers include, but are not limited to, acid-labile linkers, hydrolysis-labile linkers, enzymatically cleavable linkers, reduction labile linkers, self-immolative linkers, and non-cleavable linkers. Suitable linkers also include, but are not limited to, those that are or comprise peptides, carbohydrates, glucuronides, polyethylene glycol (PEG) units, hydrazones, mal-caproyl units, dipeptide units, valine-citruline units, and para-aminobenzyl (PAB) units.
[0087] Any linker molecule or linker technology known in the art can be used within the definitions of LI, L2, and L3. In some embodiments, a linker is a cleavable linker. In other embodiments, the linker is a non-cleavable linker. In some embodiments, linkers can comprise or consist of e.g., MC (6-maleimidocaproyl), MP (maleimidopropanoyl), val-cit (valine-citrulline), val-ala (valine-alanine), dipeptide site in protease -cleavable linkers, ala- phe (alanine-phenylalanine), dipeptide site in protease-cleavable linkers, PAB (p- aminobenzyloxycarbonyl), and variants and combinations thereof. Additional examples of linkers that can be used are disclosed, e.g., in U.S. Pat. No. 7,754,681 and in Ducry, Bioconjugate Chem., 2010, 21:5-13, and the references cited therein.
[0088] In some embodiments, the linkers are stable in physiological conditions. In some embodiments, the linkers are cleavable, for instance, able to release at least the payload portion in the presence of an enzyme or at a particular pH range or value. In some embodiments, a linker comprises an enzyme-cleavable moiety. In one embodiment, enzyme-
cleavable linkers include, but are not limited to, peptide bonds, ester linkages, and hydrazones. In some embodiments, the L linker comprises a cathepsin-cleavable linker.
[0089] In other embodiments, a linker comprises a non-cleavable moiety.
[0090] In some embodiments, the linker comprises one or more amino acids. Suitable amino acids include natural, non-natural, standard, non-standard, proteinogenic, non-proteinogenic, and L- or D-a-amino acids. In some embodiments, the linker comprises alanine, valine, glycine, leucine, isoleucine, methionine, tryptophan, phenylalanine, proline, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, lysine, arginine, histidine, or citrulline, a derivative thereof, or combination thereof. In some embodiments, one or more side chains of the amino acids is linked to a side chain group, described below. In some embodiments, the linker comprises valine and citrulline. In some embodiments, the linker comprises lysine, valine, and citrulline. In some embodiments, the linker comprises lysine, valine, and alanine. In some embodiments, the linker comprises valine and alanine.
[0091] In some embodiments, the linker comprises a self-immolative group. A self- immolative group, a self-immolative linker, or a self-immolative spacer can be any such group known to those of skill in the art. A self-immolative linker displays an important role in the cascade mechanism of release of the compound linked. It is defined as a covalent group, which has the role of cleaving two bonds between a protector group and a drug, in the case of drug delivery systems, after a stimulus. The stimulus may include enzyme triggers, chemical triggers, such as pH changes, redox systems, 1,4-, 1,6-, 1,8-eliminations, photodegradable triggers, and combinations thereof, among others. The cascade of reactions of the self-immolative structural construct allows controlled release of a drug (see R.V.Gonzagaetal, et al., Journal of Pharmaceutical Sciences, 109(2020), 3262-3281). In exemplary embodiments, the self-immolative group is p-aminobenzyl (PAB) or a derivative thereof. Useful derivatives include p-aminobenzyloxycarbonyl (PABC). Those of skill in the art will recognize that a self-immolative group is capable of carrying out a chemical reaction which releases the remaining atoms of a linker from a payload.
[0092] In other embodiments, a linker L3 comprises one or more enhancement groups. In some embodiments, the enhancement group is linked to the side chain of any amino acid in the linker. In one embodiment, amino acids for linking enhancement groups include lysine, asparagine, aspartate, glutamine, glutamate, and citrulline. The link to the enhancement group
can be a direct bond to the amino acid side chain, or the link can be indirect via a spacer and/or reactive group. In one embodiment, spacers and reactive groups include any described herein. In some embodiments, the enhancement group can be any group that modulates an existing or imparts a beneficial effect to the payload, linker payload, or conjugate including, but not limited to, biological, biochemical, hydrophilicity, synthetic, solubilizing, imaging, detecting, and reactivity effects, and the like. In some embodiments, the enhancement group is a hydrophilic group. In some embodiments, the enhancement group is a cyclodextrin. In some embodiments, the enhancement group is an alkyl, heteroalkyl, alkenyl, heteroalkenyl sulfonic acid, heteroalkenyl taurine, heteroalkenyl phosphoric acid or phosphate, heteroalkenyl amine (e.g., quaternary amine), or heteroalkenyl sugar. In some embodiments, sugars include, without limitation, monosaccharides, disaccharides, and polysaccharides. Exemplary monosaccharides include glucose, ribose, deoxyribose, xylose, arabinose, mannose, galactose, fructose, and the like. In some embodiments, sugars include sugar acids such as glucuronic acid, further including conjugated forms such as glucuronides (i.e., via glucuronidation). Exemplary disaccharides include maltose, sucrose, lactose, lactulose, trehalose, and the like. Exemplary polysaccharides include amylose, amylopectin, glycogen, inulin, cellulose, and the like. The cyclodextrin can be any cyclodextrin known to those of skill. In some embodiments, the cyclodextrin is alpha cyclodextrin, beta cyclodextrin, or gamma cyclodextrin, or mixtures thereof. In some embodiments, the cyclodextrin is alpha cyclodextrin. In some embodiments, the cyclodextrin is beta cyclodextrin. In some embodiments, the cyclodextrin is gamma cyclodextrin. In some embodiments, the enhancement group is capable of improving solubility of the remainder of the conjugate. In some embodiments, the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is substituted or non-substituted. In some embodiments, the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is -(Ofeji-sSChH, -(CH2)n-NH-(CH2)i-sSO3H, -(CH2)n- C(O)NH-(CH2)i-5SO3H,-(CH2CH2O)m-C(O)NH-(CH2)i-5SO3H, -(CH2)n-N((CH2)i- 5C(O)NH(CH2)I-5SO3H)2, -(CH2)n-C(O)N((CH2)i-5C(O)NH(CH2)i-5SO3H)2, or - (CH2CH2O)m-C(O)N((CH2)i-5C(O)NH(CH2)i-5SO3H)2, wherein n is 1, 2, 3, 4, or 5, and m is 1, 2, 3, 4, or 5. In one embodiment, the alkyl or alkenyl sulfonic acid is -(Qfcji-sSChH. In some embodiments, the heteroalkyl or heteroalkenyl sulfonic acid is -(CH2)n-NH-(CH2)i- 5SO3H, wherein n is 1, 2, 3, 4, or 5. In some embodiments, the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is -(CH2)n-C(O)NH-(CH2)i-5SO3H, wherein n is 1, 2, 3, 4, or 5. In some embodiments, the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is - (CH2CH2O)m-C(O)NH-(CH2)i-5SO3H, wherein m is 1, 2, 3, 4, or 5. In some embodiments,
the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is -(CH2)n-N((CH2)i- 5C(O)NH(CH2)I-5SO3H)2, wherein n is 1, 2, 3, 4, or 5. In some embodiments, the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is -(CH2)n-C(O)N((CH2)i- 5C(O)NH(CH2)I-5SO3H)2, wherein n is 1, 2, 3, 4, or 5. In some embodiments, the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is -(CH2CH2O)m-C(O)N((CH2)i- 5C(O)NH(CH2)I-5SO3H)2, wherein m is 1, 2, 3, 4, or 5.
[0093] Moiety B as described herein is a reactive moiety for conjugation to a target protein, antibody, or antigen-binding fragment thereof. In some embodiments, B comprises a maleimido group (for conjugation with a thiol, e.g., cysteine, of an antigen binding domain), an N-hydroxysuccinimido ester (for conjugation with an amine, e.g., lysine, of an antigen binding domain), or cyclooctynyl group (for conjugation with an antigen binding domain using click chemistry). See, e.g., WO 2020/132658; Ohio et al. Methods Mol. Biol. 2020, 2078:83-87.
[0094] In illustrative embodiments, B contains a maleimido group. In such embodiments, the maleimido group in B reacts with a cysteine residue on an antigen binding domain to form a carbon-sulfur bond.
[0095] In other embodiments, B contains an N-hydroxysuccinimido ester group. In such embodiments, the N-hydroxysuccinimido ester group reacts with a lysine residue on an antigen binding domain to form an amide bond.
[0096] In some embodiments, B contains a functional group or moiety that is capable of undergoing a click chemistry reaction (see, e.g., Click Chemistry, Huisgen Proc. Chem. Soc. 1961,357-396; Wang et al. J. Am. Chem. Soc. 2003, 125(11), 3192-3193; and Agard et al. J. Am. Chem. Soc. 2004, 126(46), 15046-15047). For example, in an embodiment B contains an alkyne which can react via click chemistry with an azide, such as on a modified antigen binding domain, to form a click chemistry product. In some embodiments, the alkyne group reacts with an azide. In an illustrative embodiment, the reactive group is an alkyne that is capable of undergoing a 1,3 -cycloaddition reaction with an azide. Alkynes that are useful in such embodiments include strained alkynes, e.g., those suitable for strain-promoted alkyneazide cycloadditions (SPAAC), cycloalkynes, e.g., cyclooctynes, benzannulated alkynes, and alkynes capable of undergoing 1,3-cycloaddition reactions with alkynes in the absence of copper catalysts. Alkynes that may be used in such embodiments also include, but are not
limited to, dibenzoazacyclooctyne, dibenzocyclooctyne, biarylazacyclooctynone, difluorinated cyclooctyne, substituted, e.g., fluorinated alkynes, aza-cycloalkynes and bicyclo[6.1.0]nonyne. In other embodiments, alkynes are useful for conjugating antibodies that have been functionalized with azido groups. Such functionalized antibodies include antibodies functionalized with azido-polyethylene glycol groups. In some embodiments, such a functionalized antibody is derived by treating an antibody having at least one glutamine residue, e.g., heavy chain Gln295, with a compound bearing an amino group and an azide group, in the presence of the enzyme transglutaminase.
[0097] The term "glutaminyl-modified antibody" refers to an antibody with at least one covalent linkage from a glutamine side chain to a primary amine compound of the present disclosure. In particular embodiments, the primary amine compound is linked through an amide linkage on the glutamine side chain. In certain embodiments, the glutamine is an endogenous glutamine. In other embodiments, the glutamine is an endogenous glutamine made reactive by polypeptide engineering (e.g., via amino acid deletion, insertion, substitution, or mutation on the polypeptide). In additional embodiments, the glutamine is polypeptide engineered with an acyl donor glutamine-containing tag (e.g., glutamine- containing peptide tags, Q- tags or TGase recognition tag).
[0098] The term "TGase recognition tag" refers to a sequence of amino acids comprising an acceptor glutamine residue and that when incorporated into (e.g., appended to) a polypeptide sequence, under suitable conditions, is recognized by a TGase and leads to cross-linking by the TGase through a reaction between an amino acid side chain within the sequence of amino acids and a reaction partner. The recognition tag may be a peptide sequence that is not naturally present in the polypeptide comprising the TGase recognition tag. In some embodiments, the TGase recognition tag comprises at least one Gin. In some embodiments, the TGase recognition tag comprises an amino acid sequence XXQX (SEQ ID NO: 1935), wherein X is any amino acid (e.g., conventional amino acid Leu, Ala, Gly, Ser, Vai, Phe, Tyr, His, Arg, Asn, Glu, Asp, Cys, Gin, He, Met, Pro, Thr, Lys, or Trp or nonconventional amino acid). In some embodiments, the acyl donor glutamine-containing tag comprises an amino acid sequence selected from the group consisting of LLQGG (SEQ ID NO: 1936), LLQG (SEQ ID NO: 1937), LSLSQG (SEQ ID NO: 1938), gGGLLQGG (SEQ ID NO: 1939), gLLQG (SEQ ID NO: 1940), LLQ, gSPLAQSHGG (SEQ ID NO: 1941), gLLQGGG (SEQ ID NO: 1942), gLLQGG (SEQ ID NO: 1943), gLLQ (SEQ ID NO: 1944), LLQLLQGA (SEQ ID
NO: 1945), LLQGA (SEQ ID NO: 1946), LLQYQGA (SEQ ID NO: 1947), LLQGSG (SEQ
ID NO: 1948), LLQYQG (SEQ ID NO: 1949), LLQLLQG (SEQ ID NO: 1950), SLLQG (SEQ
ID NO:1951), LLQLQ (SEQ ID NO:1952), LLQLLQ (SEQ ID NO:1953), and LLQGR (SEQ ID NO: 1954). See for example, WO2012059882, the entire contents of which are incorporated herein.
[0099] In some embodiments, B is selected from 2-maleimido-l -ethyl, 2-maleimidoacetyl, and 3-maleimidopropanoyl.
[0100] In additional embodiments, the linker-payload compound is selected from those, and their pharmaceutically acceptable salts, in Table 2. [0101] Table 2. Structures of Linker-Payloads (LP)








 or a pharmaceutically acceptable salt thereof.
or a pharmaceutically acceptable salt thereof.
[0102] Conjugates
[0103] The present disclosure provides, in additional embodiments, a conjugate having a structure according to Formula (I) or Formula (II):
BA-NH(-L-P)n (I)
BA-S(-L-P)n (II).
[0104] In Formulae (I) and (II):
BA is an antibody or an antigen-binding fragment thereof;
L is a linker of the formula -L1-L2-L3-B-, wherein
L is connected to BA through a side chain of an amino acid selected from the group consisting of Gin (-CO-NH-), Lys (-NH-CO-), and Cys (-S-);
LI, when present, is a stable or self-immolative linker;
L2, when present, is a linker that is stable or is cleavable by an endosomal/lysosomalenzyme;
L3, when present, is a moiety that modulates the hydrophilicity, physical, and/or chemical properties of -L-P; and
B is a reactive moiety for conjugation to a target protein, antibody, or antigen-binding fragment thereof, comprising at least one member selected from the group consisting of amino, hydroxyl, thiol, a maleimido moiety, a succinyl ester, an activated ester, an alkylamino; a haloacetamido, -

(where

P is a moiety having a structure according to Formula (la’), Formula (lb’), or Formula
(Ic’):

X and Y are independently selected from N and CH;
Z is O or NH; m is 1, 2, or 3;
R2 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR;
R3 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR;
R4 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR; and
R5 is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, aralkyl, each of which is optionally substituted; and represents the connecting point of attachment to the linker L; and
n is an integer from 1 to 20.
[0105] In some embodiments, LI is a self-immolative linker. An illustrative self-immolative linker is the para-aminobenzyl (PAB) moiety.
[0106] In additional embodiments, L2 is a linker cleavable by an endosomal/lysosomalenzyme. The linker comprises a peptide unit comprising two to four amino acid residues selected from the group consisting of glycine (G), alanine (A), valine (V), phenylalanine (F), proline (P), glutamic acid (E), lysine (K), arginine (R), citrulline (Cit), and combinations thereof. Exemplary embodiments include a peptide unit that comprises GGFG, VA, V-Cit, GG, GA, GV, AG, VG, AV, AA, EVA, and EV-Cit.
[0107] In additional embodiments, the endosomal/lysosomalenzyme is cathepcin B.
[0108] The present disclosure also provides in embodiments a conjugate comprising
(i) an antibody (BA) or an antigen-binding fragment thereof,
(ii) a plurality of payloads, and a linker that covalently connects (i) and (ii), wherein the linker is connected to the BA or antigen-binding fragment thereof through a side chain of an amino acid selected from the group consisting of Gin (-CO-NH-), Lys (-NH-CO-), and Cys (-S-), and has a formula -Ll- L2-L3-B-, wherein
LI, when present, is a stable or self-immolative linker;
L2, when present, is a linker that is stable or is cleavable by an endosomal/lysosomalenzyme;
L3, when present, is a moiety that modulates the hydrophilicity, physical, and/or chemical properties of the conjugate; and
B is a residue of a reactive moiety for conjugation, comprising at least one member selected from the group consisting of amino, hydroxyl, thiol, a maleimido moiety, a succinyl ester, an activated ester, an alkylamino; a haloacetamido, -N3, ~

 and the payload has a structure according to Formula (la’), Formula (lb’), or Formula (Ic’):
and the payload has a structure according to Formula (la’), Formula (lb’), or Formula (Ic’):

X and Y are independently selected from N and CH;
Z is O or NH; m is 1, 2, or 3;
R2 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR;
R3 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR;
R4 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR; and
Rs is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, aralkyl, each of which is optionally substituted; and represents the connecting point of attachment to the linker.
[0109] In embodiments, LI is a self-immolative linker of para-aminobenzyl (PAB) moiety.
[0110] In further embodiments, L2 is a linker cleavable by an endosomal/lysosomalenzyme, wherein the linker comprises a peptide unit comprising two to four amino acid residues selected from the group consisting of glycine (G), alanine (A), valine (V), phenylalanine (F), proline (P), glutamic acid (E), lysine (K), arginine (R), citrulline (Cit), and combinations thereof. In exemplary embodiments, the peptide unit comprises GGFG, VA, V-Cit, GG, GA, GV, AG, VG, AV, AA, EVA, and EV-Cit.
[0111] In further embodiments, L3 is present and L3 is a polyethylene glycol (PEG) unit, a carbohydrate moiety, or a combination thereof.
[0112] In various embodiments, the BA is an anti-HER2 antibody, an anti-STEAP2 antibody, an anti-MET antibody, an anti-EGFRvIII antibody, an anti-MUC16 antibody, an anti-PRLR antibody, an anti-PSMA antibody, an anti-FGFR2 antibody, an anti-FOLRl antibody, an anti-HER2/HER2 bispecific antibody, an anti HER2/APLP2 bispecific antibody, an anti- MET/MET bispecific antibody, CD33, CD30, CD22, CD79b, Nectin-4, TROP2, BCMA, CD19, Tissue Factor, or an antigen-binding fragment thereof. In accordance with various embodiments, the BA targets a cancer selected from the group consisting of breast cancer, ovarian cancer, prostate cancer, lung cancer, liver cancer, lymphomas, urothelial, cervical, multiple myeloma, gastric, or brain cancer. More specific and illustrative embodiments are described now.
[0113] In some embodiments, the BA is an antibody comprising an Fc region modified to enhance binding affinity to FcyR. In some embodiments, BA is an antibody with one or more mutations selected from F243L, R292P, Y300L, V305I, and P396L. In some embodiments, BA is an antibody with one or more mutation selected from S239D and I332E. In some embodiments, BA is an antibody with one or more mutations selected from S239D, I332E, and A330L. In some embodiments, BA is an antibody with one or more mutations selected from S298A, E333A and K334A. In some embodiments, BA is an antibody with one or more mutations selected from L234Y, L235Q, G236W, S239M, H268D, D270E, and S298A. In some embodiments, BA is an antibody with one or more mutations selected from D270E, K326D, A330M, and K334E. In some embodiments, BA is an antibody with L234Y, L235Q, G236W, S239M, H268D, D270E, and S298A in one heavy chain and D270E, K326D, A330M, and K334E in the opposing heavy chain. In some embodiments, BA is an antibody
with one or more mutations selected from G236A, S239D, and I332E. In some embodiments, BA is an antibody with one or more mutations selected from M252Y, S254T, and T256E. In some embodiments, BA is an antibody with one or more mutations selected from M428L and N434S. In some embodiments, BA is an antibody with one or more mutations selected from S267E and L328F. In some embodiments, BA is an antibody with one or more mutations selected from N325S and L328F.
[0114] In some embodiments, BA is an antibody that comprises a glutamine residue. Antibodies comprising glutamine residues can be isolated from natural sources or engineered to comprise one or more glutamine residues. Techniques for engineering glutamine residues into an antibody polypeptide chain (glutaminyl-modified antibodies) are within the skill of the practitioners in the art. In other embodiments, BA is an N297Q mutant antibody. In further embodiments, Z is an antibody that has one or more engineered LLQG, LLQGG, LLQLLQG, LLQYQG, LLQGA, LLQGSG, SLLQG, LQG, LLQLQ, LLQLLQ, LLQGR, LLQYQGA, LQGG, LGQG or LLQLLQGA sites. See, e.g., U.S. Patent No. 9,676,871 and U.S. Patent Application Publication No. 2003/0138785.
[0115] In some embodiments, the antibody BA is aglycosylated. In other embodiments, the antibody BA is glucosylated.
[0116] In further embodiments, BA is an antibody that is a monoclonal antibody, human antibody, humanized antibody, camelised antibody, or chimeric antibody. In other embodiments, BA is an antibody of any isotype (e.g., IgG, IgE, IgM, IgD, IgA and IgY), class (e.g., IgGl, IgG2, IgG3, IgG4, IgAl and IgA2) or subclass. In some embodiments, BA has a molecular weight of at least 500, 600, 700, 800, 900, 1000, 10000, 50000 or 100000 Daltons.
[0117] In other embodiments, BA can include antibodies, antigen-binding fragments of antibodies, peptides that specifically interact with a particular antigen (e.g., peptibodies), receptor molecules that specifically interact with a particular antigen, proteins comprising a ligand-binding portion of a receptor that specifically binds a particular antigen, antigenbinding scaffolds (e.g., DARPins, HEAT repeat proteins, ARM repeat proteins, tetratricopeptide repeat proteins, and other scaffolds based on naturally occurring repeat proteins, etc., (see, e.g., Boersma and Pluckthun, 2011, Curr. Opin. Biotechnol. 22:849-857,
and references cited therein)), and aptamers or portions thereof. In some embodiments, BA comprises a scFv having binding specificity to a target antigen.
[0118] Methods for determining whether two molecules specifically bind one another are well known in the art and include, for example, equilibrium dialysis, surface plasmon resonance, and the like. For example, an antigen-binding domain, as used herein, includes polypeptides that bind a target antigen or a portion thereof with a KD of less than about 500 pM, less than about 400 pM, less than about 300 pM, less than about 200 pM, less than about 100 pM, less than about 90 pM, less than about 80 pM, less than about 70 pM, less than about 60 pM, less than about 50 pM, less than about 40 pM, less than about 30 pM, less than about 20 pM, less than about 10 pM, less than about 5 pM, less than about 4 pM, less than about 2 pM, less than about 1 pM, less than about 0.5 pM, less than about 0.2 pM, less than about 0. 1 pM, or less than about 0.05 pM, as measured in a surface plasmon resonance assay.
[0119] In some embodiments, the framework regions (FRs) of the antibodies or antigenbinding fragment thereof for use in the conjugates provided herein may be identical to the human germline sequences, or may be naturally or artificially modified. An amino acid consensus sequence may be defined based on a side-by-side analysis of two or more CDRs.
[0120] Methods and techniques for identifying CDRs within HCVR and LCVR amino acid sequences are well known in the art and can be used to identify CDRs. Exemplary conventions that can be used to identify the boundaries of CDRs include, e.g., the Kabat definition, the Chothia definition, and the AbM definition. In general terms, the Kabat definition is based on sequence variability, the Chothia definition is based on the location of the structural loop regions, and the AbM definition is a compromise between the Kabat and Chothia approaches. See, e.g., Kabat, "Sequences of Proteins of Immunological Interest," National Institutes of Health, Bethesda, Md. (1991); Al-Lazikani et al., J. Mol. Biol. 273:927- 948 (1997); and Martin et al., Proc. Natl. Acad. Sci. USA 86:9268-9272 (1989). Public databases are also available for identifying CDR sequences within an antibody.
[0121] The antigen-binding domains for use in the conjugates provided herein may comprise or consist of antigen-binding fragments of full antibody molecules. Antigen-binding fragments of an antibody may be derived, e.g., from full antibody molecules using any suitable standard techniques such as proteolytic digestion or recombinant genetic engineering techniques involving the manipulation and expression of DNA encoding antibody variable
and optionally constant domains. Such DNA is known and/or is readily available from, e.g., commercial sources, DNA libraries (including, e.g., phage-antibody libraries), or can be synthesized. The DNA may be sequenced and manipulated chemically or by using molecular biology techniques, for example, to arrange one or more variable and/or constant domains into a suitable configuration, or to introduce codons, create cysteine residues, modify, add or delete amino acids, etc.
[0122] Non-limiting examples of antigen-binding fragments for use in the conjugates provided herein include: (i) Fab fragments; (ii) F(ab’)2 fragments; (iii) Fd fragments; (iv) Fv fragments; (v) single-chain Fv (scFv) molecules; (vi) dAb fragments; and (vii) minimal recognition units consisting of the amino acid residues that mimic the hypervariable region of an antibody (e.g., an isolated complementarity determining region (CDR) such as a CDR3 peptide), or a constrained FR3-CDR3-FR4 peptide. In other embodiments, an antigen-binding fragment of an antibody includes other engineered molecules, such as domain-specific antibodies, single domain antibodies, domain-deleted antibodies, chimeric antibodies, CDR- grafted antibodies, diabodies, triabodies, tetrabodies, minibodies, nanobodies (e.g., monovalent nanobodies, bivalent nanobodies, etc.), small modular immunopharmaceuticals (SMIPs), and shark variable IgNAR domains.
[0123] In certain embodiments, an antigen-binding fragment of an antibody will comprise at least one variable domain. The variable domain may be of any size or amino acid composition and will generally comprise at least one CDR which is adjacent to or in frame with one or more framework sequences. In antigen-binding fragments having a VH domain associated with a VL domain, the VH and VL domains may be situated relative to one another in any suitable arrangement. For example, the variable region may be dimeric and contain VH-VH, VH-VL or VL-VL dimers. Alternatively, the antigen-binding fragment of an antibody may contain a monomeric VH or VL domain.
[0124] In certain embodiments, an antigen-binding fragment of an antibody may contain at least one variable domain covalently linked to at least one constant domain. Non-limiting, exemplary configurations of variable and constant domains that may be found within an antigen-binding fragment of an antibody for use in the conjugates provided herein include: (i) VH-CH1 ; (ii) VH-CH2; (iii) VH-CH3; (iv) VH-CH1-CH2; (V) VH-CH1-CH2-CH3; (vi) VH-CH2- CH3; (vii) VH-CL; (viii) VL-CH1; (ix) VL-CH2; (X) VL-CH3; (xi) VL-CH1-CH2; (xii) VL-CH1- CH2-CH3; (xiii) VL-CH2-CH3; and (xiv) VL-CL- In any configuration of variable and constant
domains, including any of the exemplary configurations listed above, the variable and constant domains may be either directly linked to one another or may be linked by a full or partial hinge or linker region. A hinge region may consist of at least 2 (e.g., 5, 10, 15, 20, 40, 60 or more) amino acids which result in a flexible or semi-flexible linkage between adjacent variable and/or constant domains in a single polypeptide molecule. In further embodiments, an antigen-binding fragment may comprise a homo-dimer or hetero-dimer (or other multimer) of any of the variable and constant domain configurations listed above in non- covalent association with one another and/or with one or more monomeric VH or VL domain (e.g., by disulfide bond(s)).
[0125] In another embodiment, the antigen-binding domains used in the conjugates provided herein may comprise or consist of human antibodies and/or recombinant human antibodies, or antigen-binding fragments thereof.
[0126] In another embodiment, the antigen-binding domains used in the conjugates provided herein may comprise or consist of recombinant human antibodies or antigen-binding fragments thereof. In some embodiments, such recombinant human antibodies have variable and constant regions derived from human germline immunoglobulin sequences. In certain embodiments, however, such recombinant human antibodies are subjected to in vitro mutagenesis (or, when an animal transgenic for human Ig sequences is used, in vivo somatic mutagenesis) and thus the amino acid sequences of the VH and VL regions of the recombinant antibodies are sequences that, while derived from and related to human germline VH and VL sequences, may not naturally exist within the human antibody germline repertoire in vivo.
[0127] In another embodiment, the antigen-binding domains used in the conjugates provided herein also include bispecific antigen-binding molecules, such as bispecific antibodies.
Methods for making bispecific antibodies are known in the art and may be used to construct bispecific antigen-binding molecules for use herein. Exemplary bispecific formats that can be used in the context of the present disclosure include, without limitation, e.g., scFv-based or diabody bispecific formats, IgG-scFv fusions, dual variable domain (DVD)-Ig, Quadroma, knobs-into-holes, common light chain (e.g., common light chain with knobs-into-holes, etc.), CrossMab, CrossFab, (SEED) body, leucine zipper, Duobody, IgGl/IgG2, dual acting Fab (DAF)-IgG, and Mab2 bispecific formats (see, e.g., Klein et al. 2012, mAbs 4:6, 1-11, and references cited therein, for a review of the foregoing formats). See also, e.g.,
US2018/0134794, which discloses bispecific antigen-binding molecules. Briefly, bispecific
antigen binding molecules may comprise a first antigen-binding domain (also referred to herein as "DI"), and a second antigen-binding domain (also referred to herein as "D2"). The simultaneous binding of the two separate epitopes by the bispecific antigen-binding molecule results in effective ligand blocking with minimal activation of target signaling. In certain embodiments, DI and D2 domains of a bispecific antibody are non-competitive with one another. Non-competition between DI and D2 means that, the respective monospecific antigen binding proteins from which DI and D2 were derived do not compete with one another for binding to the target. Exemplary antigen-binding protein competition assays are known in the art. In certain embodiments, DI and D2 bind to different (e.g., nonoverlapping, or partially overlapping) epitopes on the target. Bispecific antigen-binding molecules may be constructed using the antigen-binding domains of two separate monospecific antibodies. For example, a collection of monoclonal monospecific antibodies may be produced using standard methods known in the art. The individual antibodies thus produced may be tested pairwise against one another for cross-competition to the target protein. If two different antibodies are able to bind to the target at the same time (i.e., do not compete with one another), then the antigen-binding domain from the first antibody and the antigen-binding domain from the second, non-competitive antibody can be engineered into a single bispecific antibody. A bispecific antigen-binding molecule can be a single multifunctional polypeptide, or it can be a multimeric complex of two or more polypeptides that are covalently or non-covalently associated with one another. Any antigen binding construct which has the ability to simultaneously bind two separate, non-identical epitopes of the target molecule is regarded as a bispecific antigen-binding molecule. Bispecific antigenbinding molecules, or variants thereof, may be constructed using standard molecular biological techniques (e.g., recombinant DNA and protein expression technology) as will be known to a person of skill in the art. In another embodiment, bispecific antibodies are also provided wherein one arm of the bispecific antibody binds to an epitope on a first target protein, and the other arm of the bispecific antibody binds to a second epitope on a second target protein. Other exemplary bispecific formats that can be used in the context of the present disclosure include, without limitation, e.g., scFv-based or diabody bispecific formats, IgG-scFv fusions, dual variable domain (DVD)-Ig, Quadroma, knobs-into-holes, common light chain (e.g., common light chain with knobs-into-holes, etc.), CrossMab, CrossFab, (SEED)body, leucine zipper, Duobody, IgGl/IgG2, dual acting Fab (DAF)-IgG, and Mab2 bispecific formats (see, e.g., Klein et al. 2012, mAbs 4:6, 1-11, and references cited therein, for a review of the foregoing formats). Bispecific antibodies can also be constructed using
peptide/nucleic acid conjugation, e.g., wherein unnatural amino acids with orthogonal chemical reactivity are used to generate site-specific antibody-oligonucleotide conjugates which then self-assemble into multimeric complexes with defined composition, valency and geometry. (See, e.g., Kazane et al., J. Am. Chem. Soc. (Epub: Dec. 4, 2012)).
[0128] In another embodiment, the antigen binding domains for use in the conjugates provided herein also include antibodies comprising variants of any of the HCVR, LCVR, and/or CDR amino acid sequences known in the art. In some embodiments, variants include variants of any of the HCVR, LCVR, and/or CDR amino acid sequences known in the art having one or more conservative substitutions. For example, the antigen binding domains include antibodies or antigen binding fragments thereof having HCVR, LCVR, and/or CDR amino acid sequences with, e.g., 10 or fewer, 8 or fewer, 6 or fewer, 4 or fewer, etc. conservative amino acid substitutions relative to any of the HCVR, LCVR, and/or CDR amino acid sequences known in the art. In another embodiment, the antigen binding domains include antibodies or antigen binding fragments thereof also include variants having substantial sequence identity to any of the HCVR, LCVR, and/or CDR amino acid sequences known in the art. In certain embodiments, residue positions which are not identical differ by conservative amino acid substitutions.
[0129] A "conservative amino acid substitution" is one in which an amino acid residue is substituted by another amino acid residue having a side chain with similar chemical properties (e.g., charge or hydrophobicity). In general, a conservative amino acid substitution will not substantially change the functional properties of a protein. In cases where two or more amino acid sequences differ from each other by conservative substitutions, the percent sequence identity or degree of similarity may be adjusted upwards to correct for the conservative nature of the substitution. Means for making this adjustment are well-known to those of skill in the art. See, e.g., Pearson (1994) Methods Mol. Biol. 24: 307-331. Examples of groups of amino acids that have side chains with similar chemical properties include (1) aliphatic side chains: glycine, alanine, valine, leucine and isoleucine; (2) aliphatic-hydroxyl side chains: serine and threonine; (3) amide-containing side chains: asparagine and glutamine; (4) aromatic side chains: phenylalanine, tyrosine, and tryptophan; (5) basic side chains: lysine, arginine, and histidine; (6) acidic side chains: aspartate and glutamate, and (7) sulfur-containing side chains are cysteine and methionine. In some embodiments, conservative amino acids substitution groups are: valine-leucine -isoleucine, phenylalanine-
tyrosine, lysine-arginine, alanine-valine, glutamate-aspartate, and asparagine-glutamine. In another embodiment, a conservative replacement is any change having a positive value in the PAM250 log-likelihood matrix disclosed in Gonnet et al. (1992) Science 256: 1443-1445. A "moderately conservative" replacement is any change having a nonnegative value in the PAM250 log-likelihood matrix.
[0130] Sequence identity between two different amino acid sequences is typically measured using sequence analysis software. Sequence analysis software matches similar sequences using measures of similarity assigned to various substitutions, deletions and other modifications, including conservative amino acid substitutions. For instance, GCG software contains programs such as GAP and BESTFIT which can be used with default parameters to determine sequence homology or sequence identity between closely related polypeptides, such as homologous polypeptides from different species of organisms or between a wild type protein and a mutein thereof. See, e.g., GCG Version 6.1. Polypeptide sequences also can be compared using FASTA using default or recommended parameters, a program in GCG Version 6.1. FASTA (e.g., FASTA2 and FASTA3) provides alignments and percent sequence identity of the regions of the best overlap between the query and search sequences (Pearson (2000) supra). Another algorithm when comparing a sequence provided herein to a database containing a large number of sequences from different organisms is the computer program BLAST, especially BLASTP or TBLASTN, using default parameters. See, e.g., Altschul et al. (1990) J. Mol. Biol. 215:403-410 and Altschul et al. (1997) Nucleic Acids Res. 25:3389- 402.
[0131] The antigen-binding domains for use in the conjugates provided herein encompass proteins having amino acid sequences that vary from those of the described antibodies but that retain the ability to bind the target proteins. Such variant antigen-binding domains comprise one or more additions, deletions, or substitutions of amino acids when compared to parent sequence but exhibit biological activity that is essentially equivalent to that of the described antibodies.
[0132] Two antigen-binding domains are considered bioequivalent if, for example, they are pharmaceutical equivalents or pharmaceutical alternatives whose rate and extent of absorption do not show a significant difference when administered at the same molar dose under similar experimental conditions, either single dose or multiple doses. Some antigenbinding domains will be considered equivalents or pharmaceutical alternatives if they are
equivalent in the extent of their absorption but not in their rate of absorption and yet may be considered bioequivalent because such differences in the rate of absorption are intentional and are reflected in the labeling, are not essential to the attainment of effective body drug concentrations on, e.g., chronic use, and are considered medically insignificant for the particular drug product studied.
[0133] In some embodiments, two antigen-binding domains are bioequivalent if there are no clinically meaningful differences in their safety, purity, and potency.
[0134] In some embodiments, two antigen-binding domains are bioequivalent if a patient can be switched one or more times between the reference product and the biological product without an expected increase in the risk of adverse effects, including a clinically significant change in immunogenicity, or diminished effectiveness, as compared to continued therapy without such switching.
[0135] In some embodiments, two antigen-binding domains are bioequivalent if they both act by a common mechanism or mechanisms of action for the condition or conditions of use, to the extent that such mechanisms are known.
[0136] Bioequivalence may be demonstrated by in vivo and in vitro methods. Bioequivalence measures include, e.g., (a) an in vivo test in humans or other mammals, in which the concentration of the antigen-binding domain or its metabolites is measured in blood, plasma, serum, or other biological fluid as a function of time; (b) an in vitro test that has been correlated with and is reasonably predictive of human in vivo bio availability data; (c) an in vivo test in humans or other mammals in which the appropriate acute pharmacological effect of the antigen-binding domain (or its target) is measured as a function of time; and (d) in a well-controlled clinical trial that establishes safety, efficacy, or bioavailability or bioequivalence of an antigen-binding domain.
[0137] Bioequivalent variants of antigen-binding domains for use in the conjugates provided herein may be constructed by, for example, making various substitutions of residues or sequences or deleting terminal or internal residues or sequences not needed for biological activity. For example, cysteine residues not essential for biological activity can be deleted or replaced with other amino acids to prevent formation of unnecessary or incorrect intramolecular disulfide bridges upon renaturation. In other contexts, bioequivalent antigenbinding domains may include variants comprising amino acid changes which modify the
glycosylation characteristics of the antigen -binding domain, e.g., mutations which eliminate or remove glycosylation.
[0138] In certain embodiments, the antigen-binding domains for use in the conjugates provided herein bind to a human target protein but not to target protein from other species. In other embodiments, the antigen-binding domains for use in the conjugates provided herein bind to a human target protein and to a target protein from one or more non-human species. For example, the antigen-binding domains for use in the conjugates provided herein may bind to a human target protein and may bind or not bind, as the case may be, to one or more of mouse, rat, guinea pig, hamster, gerbil, pig, cat, dog, rabbit, goat, sheep, cow, horse, camel, cynomolgus, marmoset, rhesus or chimpanzee target protein. In some embodiments, the antigen-binding domains specifically bind human target protein and cynomolgus monkey (e.g., Macacafascicularis) target protein. In other embodiments, antigen-binding domains for use herein bind human target protein but do not bind, or bind only weakly, to cynomolgus monkey target protein.
[0139] The BA can be linked in the conjugate through an attachment at a particular amino acid within the BA. Exemplary amino acid attachments that can be used in the context of this embodiment of the disclosure include, e.g., lysine (see, e.g., US 5,208,020; US 2010/0129314; Hollander et al., Bioconjugate Chem., 2008, 19:358-361 ; WO 2005/089808; US 5,714,586; US 2013/0101546; and US 2012/0585592), cysteine (see, e.g., US 2007/0258987; WO 2013/055993; WO 2013/055990; WO 2013/053873; WO 2013/053872; WO 2011/130598; US 2013/0101546; and US 7,750,116), selenocysteine (see, e.g., WO 2008/122039; and Hofer et al., Proc. Natl. Acad. Sci., USA, 2008, 705:12451-12456), formyl glycine (see, e.g., Carrico et al., Nat. Chem. Biol., 2007, 3:321-322; Agarwal et al., Proc. Natl. Acad. Sci., USA, 2013, 110:46-51, and Rabuka et al., Nat. Protocols, 2012, 10: 1052- 1067), non-natural amino acids (see, e.g., WO 2013/068874, and WO 2012/166559), and acidic amino acids (see, e.g., WO 2012/05982). Linkers can also be conjugated to an BA via attachment to carbohydrates (see, e.g., US 2008/0305497, WO 2014/065661, Ryan et al., Food & Agriculture Immunol., 2001, 73:127-130, and Jeger et al., Angew Chem IntEd Engl., 2010, 49:9995-9997).
[0140] In some examples, BA is bonded to the linker through a lysine residue. In some embodiments, the antibody or antigen binding molecule is bonded to the linker through a cysteine residue, lysine residue, or glutamine residue. In certain embodiments, the BA is
bonded to the linker through a cysteine residue. In certain embodiments, a linker maleimide moiety bonds to an antibody cysteine residue. In certain embodiments, the BA is bonded to the linker through a lysine residue. In certain embodiments, a linker N-hydroxysuccinimide moiety bonds to an antibody lysine residue to form an amide linkage.
[0141] In certain embodiments, the BA is bonded to the linker through a glutamine residue see, e.g., Jeger et al., Angew Chem Int Ed Engl., 2010, 49:9995-9997 and Dennler et al., Bioconjugate Chem. 2014, 25:569-578). Antibodies comprising glutamine residues can be isolated from natural sources or engineered to comprise one or more glutamine residues. In certain embodiments, antibodies or antigen binding molecules are engineered by mutations, for example insertions or deletions to facilitate reaction via transglutaminase. In certain embodiments, antibodies or antigen binding molecules are engineered to remove one or more glycosylation sites. In certain embodiments, antibodies or antigen binding molecules are engineered to add one or more glutamine residues. In certain embodiments, glutamine residues are added within a TGase recognition tag, as described herein. Techniques for engineering glutamine residues into an antibody polypeptide chain (glutaminyl-modified antibodies or antigen binding molecules) are within the skill of the practitioners in the art. In certain embodiments, the antibody is aglycosylated.
[0142] In certain embodiments, BA comprises at least one glutamine residue in at least one polypeptide chain sequence. In certain embodiments, BA comprises two heavy chain polypeptides, each with one Gln295 or Q295 residue. In further embodiments, BA comprises one or more glutamine residues at a site other than a heavy chain 295. Included herein are antibodies of this section bearing N297Q mutation(s) described herein. In certain embodiments, a glutamine residue is added at the heavy chain C-terminus.
[0143] In certain embodiments, the glutamine is polypeptide engineered with a glutamine- containing tag (e.g., glutamine-containing peptide tags, Q-tags or TGase recognition tag). The term “TGase recognition tag” or “Q-Tag” refers to a sequence of amino acids comprising a glutamine residue that when incorporated into (e.g., appended to) a polypeptide sequence, under suitable conditions, is recognized by a transglutaminase (“TGase”) and leads to crosslinking by the TGase through a reaction between an amino acid side chain within the sequence of amino acids and a reactive group. The recognition tag may be a peptide sequence that is not naturally present in the polypeptide. In certain embodiments, the TGase recognition tag comprises at least one glutamine. In certain embodiments, the TGase
recognition tag comprises an amino acid sequence XXQX, wherein X is any amino acid (e.g., conventional amino acid Leu, Ala, Gly, Ser, Vai, Phe, Tyr, His, Arg, Asn, Glu, Asp, Cys, Gin, He, Met, Pro, Thr, Lys, or Trp or nonconventional amino acid). In certain embodiments, the TGase recognition tag comprises an amino acid sequence selected from the group consisting of LLQGG, LLQG, LSLSQG, GGGLLQGG, GLLQG, LLQ, GSPLAQSHGG, GLLQGGG, GLLQGG, GLLQ, LLQLLQGA, LLQGA, LLQYQGA, LLQGSG, LLQYQG, LLQLLQG, SLLQG, LLQLQ, LLQLLQ, and LLQGR. See for example, WO2012059882, the entire contents of which are incorporated herein.
[0144] In some embodiments, BA includes an antibody heavy chain and further includes a TGase recognition tag at the C-terminus of the antibody heavy chain. In some embodiments, BA includes an antibody heavy chain and further includes a TGase recognition tag at the C- terminus of the antibody heavy chain, wherein the TGase recognition tag is the pentapeptide sequence LLQGA. In some embodiments, BA includes two antibody heavy chains and further includes a TGase recognition tag at the C-terminus of each antibody heavy chain. In some embodiments, BA includes two antibody heavy chains and further includes a TGase recognition tag at the C-terminus of each antibody heavy chain, wherein the TGase recognition tag is the pentapeptide sequence LLQGA.
[0145] The BA can be also modified at one or more glutamine residues via transglutaminase see, e.g., Jeger et al., Angew Chem Int Ed Engl., 2010, 49:9995-9997 and Dennler et al., Bioconjugate Chem. 2014, 25:569-578). For example, in the presence of transglutaminase, one or more glutamine residues of an antibody can be coupled to a primary amine compound to provide a moiety capable of reacting with a reactive group on a linker-payload. In certain embodiments, the primary amine compound provides a diene or dienophile. In certain embodiments, the primary amine compound provides a diene or dienophile, and the linkerpayload provides a complementary dienophile or diene, respectively, for conjugation via a Diels-Alder reaction. In certain embodiments, the primary amine compound provides an azido group. In certain embodiments, the primary amine compound provides an azido group, and the linker-payload provides a complementary alkyne, for conjugation via a click reaction.
[0146] In some embodiments, the BA comprises a heavy chain and the heavy chain is linked to BA directly or indirectly via a linker. In some embodiments, the BA comprises a light chain and the light chain is linked to BA directly or indirectly via a linker.
[0147] In some embodiments, the BA comprises a heavy chain and the C-terminus of the heavy chain is linked to BA directly or indirectly via a linker. In some embodiments, the BA comprises a light chain and the C-terminus of the light chain is linked to BA directly or indirectly via a linker.
[0148] In some embodiments, the BA comprises two heavy chains and each of the two heavy chains is linked to BA directly or indirectly via a linker. In some embodiments, the BA comprises two light chains and each of the two light chains is linked to BA directly or indirectly via a linker.
[0149] In some embodiments, the BA comprises two heavy chains and C-terminus of each of the two heavy chains is linked to BA directly or indirectly via a linker. In some embodiments, the BA comprises two light chains and C-terminus of each of the two light chains is linked to BA directly or indirectly via a linker.
[0150] The epitope to which the antigen-binding domains bind may consist of a single contiguous sequence of 3 or more (e.g., 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 or more) amino acids of a target protein. Alternatively, the relevant epitope may consist of a plurality of non-contiguous amino acids (or amino acid sequences) of the target protein. In some embodiments, the epitope is located on or near the binding domain of the target protein. In other embodiments, the epitope is located outside of the binding domain of the target protein.
[0151] Various techniques known to persons of ordinary skill in the art can be used to determine the epitope with which the antigen-binding domains used in the ADCs provided herein interact. Exemplary techniques that can be used to determine an epitope or binding domain of a particular antigen-binding domain include, e.g. , point mutagenesis (e.g., alanine scanning mutagenesis, arginine scanning mutagenesis, etc.), peptide blots analysis (Reineke, 2004, Methods Mol Biol 248:443-463), protease protection, and peptide cleavage analysis. In addition, methods such as epitope excision, epitope extraction and chemical modification of antigens can be employed (Tomer, 2000, Protein Science 9:487-496). Another method that can be used to identify the amino acids within a polypeptide with which an antigen-binding domain interacts is hydrogen/deuterium exchange detected by mass spectrometry. In general terms, the hydrogen/deuterium exchange method involves deuterium-labeling the protein of interest, followed by binding the antigen-binding domain to the deuterium-labeled protein.
Next, the protein/antigen-binding domain complex is transferred to water to allow hydrogendeuterium exchange to occur at all residues except for the residues protected by the antigenbinding domain (which remain deuterium-labeled). After dissociation of the antigen-binding domain, the target protein is subjected to protease cleavage and mass spectrometry analysis, thereby revealing the deuterium-labeled residues which correspond to the specific amino acids with which the antigen-binding domain interacts. See, e.g., Ehring (1999) Analytical Biochemistry 267(2):252-259; Engen and Smith (2001) Anal. Chem. 73:256A-265A. X-ray crystal structure analysis can also be used to identify the amino acids within a polypeptide with which an antigen-binding domain interacts.
[0152] In various embodiments, the antibodies for use in the conjugates described herein are fully human antibodies. Methods for generating monoclonal antibodies, including fully human monoclonal antibodies are known in the art. Any such known methods can be used in the context of the present disclosure to make human antibodies that specifically bind to a human protein target.
[0153] Using VELOCIMMUNE™ technology, for example, or any other similar known method for generating fully human monoclonal antibodies, high affinity chimeric antibodies to a human protein target are initially isolated having a human variable region and a mouse constant region. The antibodies are characterized and selected for desirable characteristics, including affinity, ligand blocking activity, selectivity, epitope, etc. If necessary, mouse constant regions are replaced with a desired human constant region, for example wild-type or modified IgGl or IgG4, to generate a fully human antibody. While the constant region selected may vary according to specific use, high affinity antigen-binding and target specificity characteristics reside in the variable region. In certain instances, fully human antibodies are isolated directly from antigen-positive B cells.
[0154] Monoclonal antibodies can be generated by any techniques with which those having ordinary skill in the art will be familiar. Such methods include, but are not limited to, Epstein Barr Virus (EBV) transformation of human peripheral blood cells e.g., containing B lymphocytes), in vitro immunization of human B-cells, fusion of spleen cells from immunized transgenic mice carrying inserted human immunoglobulin genes, isolation from human immunoglobulin V region phage libraries, or other procedures as known in the art and based on the disclosure herein. For example, fully human monoclonal antibodies can be obtained from transgenic mice that have been engineered to produce specific human
antibodies in response to antigenic challenge. Methods for obtaining fully human antibodies from transgenic mice are described, for example, by Green et al. , Nature Genet. 7:13, 1994; Lonberg et al., Nature 368:856, 1994; Taylor et al., Int. Immun. 6:579, 1994; U.S. Patent No. 5,877,397; Bruggemann et al., 1997 Curr. Opin. Biotechnol. 8:455-58; Jakobovits et al., 1995 Ann. N. Y. Acad. Sci. 764:525-35. In this technique, elements of the human heavy and light chain locus are introduced into strains of mice derived from embryonic stem cell lines that contain targeted disruptions of the endogenous heavy chain and light chain loci (see also Bruggemann et al., Curr. Opin. Biotechnol. 8:455-58 (1997)). For example, human immunoglobulin transgenes may be mini-gene constructs, or transloci on yeast artificial chromosomes, which undergo B-cell-specific DNA rearrangement and hypermutation in the mouse lymphoid tissue. Fully human monoclonal antibodies may be obtained by immunizing the transgenic mice, which may then produce human antibodies specific for a target antigen. Lymphoid cells of the immunized transgenic mice can be used to produce human antibody-secreting hybridomas according to the methods described herein. Polyclonal sera containing fully human antibodies may also be obtained from the blood of the immunized animals.
[0155] Another method for generating human antibodies of the present disclosure includes immortalizing human peripheral blood cells by EBV transformation. See, e.g., U.S. Patent No. 4,464,456. Such an immortalized B-cell line (or lymphoblastoid cell line) producing a monoclonal antibody that specifically binds to a target antigen can be identified by immunodetection methods as provided herein, for example, an ELISA, and then isolated by standard cloning techniques. The stability of the lymphoblastoid cell line producing an antibody against a target antigen can be improved by fusing the transformed cell line with a murine myeloma to produce a mouse-human hybrid cell line according to methods known in the art (see, e.g., Glasky et al., Hybridoma 8:377-89 (1989)). Still another method to generate human monoclonal antibodies is in vitro immunization, which includes priming human splenic B-cells with a target antigen, followed by fusion of primed B-cells with a heterohybrid fusion partner. See, e.g., Boerner et al., 1991 J. Immunol. 147:86-95.
[0156] In certain embodiments, a B-cell that is producing an antibody against a target antigen is selected and the light chain and heavy chain variable regions are cloned from the B-cell according to molecular biology techniques known in the art (WO 92/02551; U.S. Patent 5,627,052; Babcook et al., Proc. Natl. Acad. Sci. USA 93:7843-48 (1996)) and described
herein. B-cells from an immunized animal may be isolated from the spleen, lymph node, or peripheral blood sample by selecting a cell that is producing an antibody that specifically binds to a target antigen. B-cells may also be isolated from humans, for example, from a peripheral blood sample.
[0157] Methods for detecting single B-cells that are producing an antibody with the desired specificity are well known in the art, for example, by plaque formation, fluorescence-activated cell sorting, in vitro stimulation followed by detection of specific antibody, and the like. Methods for selection of specific antibody-producing B-cells include, for example, preparing a single cell suspension of B-cells in soft agar that contains a target antigen. Binding of the specific antibody produced by the B-cell to the antigen results in the formation of a complex, which may be visible as an immune-precipitate.
[0158] The methods for obtaining antibodies of the present disclosure can also adopt various phage display technologies known in the art. See, e.g., Winter et al., 1994 Amu. Rev.
Immunol. 12:433-55; Burton et al., 1994 Adv. Immunol. 57:191-280. Human or murine immunoglobulin variable region gene combinatorial libraries may be created in phage vectors that can be screened to select Ig fragments (Fab, Fv, sFv, or multimers thereof) that bind specifically to a target antigen or variant or fragment thereof. See, e.g., U.S. Patent No. 5,223,409; Huse et al., 1989 Science 246:1275-81; Sastry et al., Proc. Natl. Acad. Sci. USA 86:5728-32 (1989); Alting-Mees et al., Strategies in Molecular Biology 3:1-9 (1990); Kang et al., 1991 Proc. Natl. Acad. Sci. USA 88:4363-66; Hoogenboom et al., 1992 J. Molec. Biol. 227:381-388; Schlebusch et al. , 1997 Hybridoma 16:47-52 and references cited therein. For example, a library containing a plurality of polynucleotide sequences encoding Ig variable region fragments may be inserted into the genome of a filamentous bacteriophage, such as Ml 3 or a variant thereof, in frame with the sequence encoding a phage coat protein. A fusion protein may be a fusion of the coat protein with the light chain variable region domain and/or with the heavy chain variable region domain. According to certain embodiments, immunoglobulin Fab fragments may also be displayed on a phage particle (see, e.g., U.S. Patent No. 5,698,426).
[0159] Antibody fragments fused to another protein, such as a minor coat protein, can be also used to enrich phage with antigen. Then, using a random combinatorial library of rearranged heavy (VH) and light (VL) chains from mice immune to the target antigen e.g., HBV sAg, tumor specific antigen), diverse libraries of antibody fragments are displayed on the surface
of the phage. These libraries can be screened for complementary variable domains, and the domains purified by, for example, affinity column. See Clackson et al., Nature, V. 352 pp. 624-628 (1991).
[0160] Heavy and light chain immunoglobulin cDNA expression libraries may also be prepared in lambda phage, for example, using XlmmunoZap™(H) and XImmunoZap™(L) vectors (Stratagene, La Jolla, California). Briefly, mRNA is isolated from a B-cell population, and used to create heavy and light chain immunoglobulin cDNA expression libraries in the XlmmunoZap(H) and XlmmunoZap(L) vectors. These vectors may be screened individually or co-expressed to form Fab fragments or antibodies (see Huse et al. , supra', see also Sastry et al., supra). Positive plaques may subsequently be converted to a non-lytic plasmid that allows high level expression of monoclonal antibody fragments from E. coll.
[0161] In some embodiments, in a hybridoma the variable regions of a gene expressing a monoclonal antibody of interest are amplified using nucleotide primers. These primers may be synthesized by one of ordinary skill in the art or may be purchased from commercially available sources. (See, e.g., Stratagene (La Jolla, California), which sells primers for mouse and human variable regions including, among others, primers for Vua, Vub, Vue, Vua, CHI, VL and CL regions.) These primers may be used to amplify heavy or light chain variable regions, which may then be inserted into vectors such as ImmunoZAP™H or ImmunoZAP™L (Stratagene), respectively. These vectors may then be introduced into E. coll, yeast, or mammalian-based systems for expression. Large amounts of a single -chain protein containing a fusion of the VH and VL domains may be produced using these methods (see Bird et al., Science 242:423-426, 1988).
[0162] Once cells producing antibodies according to the disclosure have been obtained using any of the above-described immunization and other techniques, the specific antibody genes may be cloned by isolating and amplifying DNA or mRNA therefrom according to standard procedures as described herein. The antibodies produced therefrom may be sequenced and the CDRs identified and the DNA coding for the CDRs may be manipulated as described previously to generate other antibodies according to the disclosure.
[0163] The binding agents of the present disclosure preferably modulate activity of the target antigen in the cell-based assay described herein and/or the in vivo assay described herein
and/or bind to one or more of the domains described herein and/or cross-block the binding of one of the antibodies described in this application and/or are cross-blocked from binding the target antigen by one of the antibodies described in this application. Accordingly, such binding agents can be identified using the assays described herein.
[0164] In certain embodiments, antibodies are generated by first identifying antibodies that bind to one or more of the domains provided herein and/or neutralize in the cell-based and/or in vivo assays described herein and/or cross-block the antibodies described in this application and/or are cross-blocked from binding a target antigen by one of the antibodies described in this application. The CDR regions from these antibodies are then used to insert into appropriate biocompatible frameworks to generate binding agents against the target antigen. The non-CDR portion of the binding agent may be composed of amino acids or may be a non-protein molecule. The assays described herein allow the characterization of binding agents. Preferably the binding agents of the present disclosure are antibodies as defined herein.
[0165] Other antibodies according to the disclosure may be obtained by conventional immunization and cell fusion procedures as described herein and known in the art.
[0166] Molecular evolution of the complementarity determining regions (CDRs) in the center of the antibody binding site also has been used to isolate antibodies with increased affinity, for example, antibodies having increased affinity for c-erbB-2, as described by Schier et al., 1996, J. Mol. Biol. 263:551. Accordingly, such techniques are useful in preparing antibodies to a target antigen. Antigen binding proteins directed against a target antigen can be used, for example, in assays to detect the presence of a target antigen, either in vitro or in vivo. The antigen binding proteins also may be employed in purifying a target antigen by immunoaffinity chromatography.
[0167] Although human, partially human, or humanized antibodies will be suitable for many applications, particularly those involving administration of the antibody to a human subject, other types of antigen binding proteins will be suitable for certain applications. Non-human antibodies of the present disclosure can be, for example, derived from any antibodyproducing animal, such as mouse, rat, rabbit, goat, donkey, or non-human primate (such as monkey (e.g., cynomolgus or rhesus monkey) or ape (e.g., chimpanzee)). An antibody from a particular species can be made by, for example, immunizing an animal of that species with
the desired immunogen (e.g., HBV sAg, tumor specific antigen or using an artificial system for generating antibodies of that species (e.g., a bacterial or phage display -based system for generating antibodies of a particular species), or by converting an antibody from one species into an antibody from another species by replacing, e.g., the constant region of the antibody with a constant region from the other species, or by replacing one or more amino acid residues of the antibody so that it more closely resembles the sequence of an antibody from the other species. In some embodiments, the antibody is a chimeric antibody comprising amino acid sequences derived from antibodies from two or more different species.
[0168] Antigen binding proteins may be prepared, and screened for desired properties, by any of a number of conventional techniques. Certain of the techniques involve isolating a nucleic acid encoding a polypeptide chain (or portion thereof) of an antigen binding protein of interest e.g., an anti- HBV sAg antibody, a tumor specific antigen), and manipulating the nucleic acid through recombinant DNA technology. The nucleic acid may be fused to another nucleic acid of interest, or altered (e.g., by mutagenesis or other conventional techniques) to add, delete, or substitute one or more amino acid residues, for example. Furthermore, the antigen binding proteins may be purified from cells that naturally express them (e.g., an antibody can be purified from a hybridoma that produces it), or produced in recombinant expression systems, using any technique known in the art. See, for example, Monoclonal Antibodies, Hybridomas: A New Dimension in Biological Analyses, Kennet et al. (eds.), Plenum Press, New York (1980); and Antibodies: A Laboratory Manual, Harlow and Land (eds.), Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, (1988).
[0169] Any expression system known in the art can be used to make the recombinant polypeptides of the present disclosure. Expression systems are detailed comprehensively above. In general, host cells are transformed with a recombinant expression vector that comprises DNA encoding a desired polypeptide. Among the host cells that may be employed are prokaryotes, yeast or higher eukaryotic cells. Prokaryotes include gram negative or gram positive organisms, for example E. coli or Bacilli. Higher eukaryotic cells include insect cells and established cell lines of mammalian origin. Examples of suitable mammalian host cell lines include the COS-7 line of monkey kidney cells (ATCC CRL 1651) (Gluzman et al. , 1981, Cell 23:175), L cells, 293 cells, C127 cells, 3T3 cells (ATCC CCL 163), Chinese hamster ovary (CHO) cells, HeLa cells, BHK (ATCC CRL 10) cell lines, and the CVI/EBNA cell line derived from the African green monkey kidney cell line CVI (ATCC CCL 70) as
described by McMahan et al., 1991, EMBO J. 10: 2821. Appropriate cloning and expression vectors for use with bacterial, fungal, yeast, and mammalian cellular hosts are described by Pouwels et al. (Cloning Vectors: A Laboratory Manual, Elsevier, New York, 1985).
[0170] It will be appreciated that an antibody of the present disclosure may have at least one amino acid substitution, providing that the antibody retains binding specificity. Therefore, modifications to the antibody structures are encompassed within the scope of the present disclosure. These may include amino acid substitutions, which may be conservative or nonconservative that do not destroy the target binding capability of an antibody. Conservative amino acid substitutions may encompass non-naturally occurring amino acid residues, which are typically incorporated by chemical peptide synthesis rather than by synthesis in biological systems. These include peptidomimetics and other reversed or inverted forms of amino acid moieties. A conservative amino acid substitution may also involve a substitution of a native amino acid residue with a normative residue such that there is little or no effect on the polarity or charge of the amino acid residue at that position.
[0171] Non-conservative substitutions may involve the exchange of a member of one class of amino acids or amino acid mimetics for a member from another class with different physical properties (e.g., size, polarity, hydrophobicity, charge). Such substituted residues may be introduced into regions of the human antibody that are homologous with non-human antibodies, or into the non-homologous regions of the molecule.
[0172] Moreover, one skilled in the art may generate test variants containing a single amino acid substitution at each desired amino acid residue. The variants can then be screened using activity assays known to those skilled in the art. Such variants could be used to gather information about suitable variants. For example, if one discovered that a change to a particular amino acid residue resulted in destroyed, undesirably reduced, or unsuitable activity, variants with such a change may be avoided. In other words, based on information gathered from such routine experiments, one skilled in the art can readily determine the amino acids where further substitutions should be avoided either alone or in combination with other mutations.
[0173] A skilled artisan will be able to determine suitable variants of the polypeptide as set forth herein using well-known techniques. In certain embodiments, one skilled in the art may identify suitable areas of the molecule that may be changed without destroying activity by
targeting regions not believed to be important for activity. In certain embodiments, one can identify residues and portions of the molecules that are conserved among similar polypeptides. In certain embodiments, even areas that may be important for biological activity or for structure may be subject to conservative amino acid substitutions without destroying the biological activity or without adversely affecting the polypeptide structure.
[0174] Additionally, one skilled in the art can review structure-function studies identifying residues in similar polypeptides that are important for activity or structure. In view of such a comparison, one can predict the importance of amino acid residues in a protein that correspond to amino acid residues which are important for activity or structure in similar proteins. One skilled in the art may opt for chemically similar amino acid substitutions for such predicted important amino acid residues.
[0175] One skilled in the art can also analyze the three-dimensional structure and amino acid sequence in relation to that structure in similar polypeptides. In view of such information, one skilled in the art may predict the alignment of amino acid residues of an antibody with respect to its three-dimensional structure. In certain embodiments, one skilled in the art may choose not to make radical changes to amino acid residues predicted to be on the surface of the protein, since such residues may be involved in important interactions with other molecules.
[0176] In certain embodiments, variants of antibodies include glycosylation variants wherein the number and/or type of glycosylation site has been altered compared to the amino acid sequences of a parent polypeptide. In certain embodiments, variants comprise a greater or a lesser number of N-linked glycosylation sites than the native protein. An N-linked glycosylation site is characterized by the sequence: Asn-X-Ser or Asn-X-Thr, wherein the amino acid residue designated as X can be any amino acid residue except proline. The substitution of amino acid residues to create this sequence provides a potential new site for the addition of an N-linked carbohydrate chain. Alternatively, substitutions which eliminate this sequence will remove an existing N-linked carbohydrate chain. Also provided is a rearrangement of N-linked carbohydrate chains wherein one or more N-linked glycosylation sites (typically those that are naturally occurring) are eliminated and one or more new N- linked sites are created. Additional preferred antibody variants include cysteine variants wherein one or more cysteine residues are deleted from or substituted for another amino acid (e.g., serine) as compared to the parent amino acid sequence. Cysteine variants can be useful when antibodies must be refolded into a biologically active conformation such as after the
isolation of insoluble inclusion bodies. Cysteine variants generally have fewer cysteine residues than the native protein, and typically have an even number to minimize interactions resulting from unpaired cysteines.
[0177] Desired amino acid substitutions (whether conservative or non-conservative) can be determined by those skilled in the art at the time such substitutions are desired. According to certain embodiments, preferred amino acid substitutions are those which: (1) reduce susceptibility to proteolysis, (2) reduce susceptibility to oxidation, (3) alter binding affinity for forming protein complexes, (4) alter binding affinities, and/or (4) confer or modify other physiochemical or functional properties on such polypeptides. According to certain embodiments, single or multiple amino acid substitutions (in certain embodiments, conservative amino acid substitutions) may be made in the naturally-occurring sequence (in certain embodiments, in the portion of the polypeptide outside the domain(s) forming intermolecular contacts). In certain embodiments, a conservative amino acid substitution typically may not substantially change the structural characteristics of the parent sequence (e.g., a replacement amino acid should not tend to break a helix that occurs in the parent sequence, or disrupt other types of secondary structure that characterizes the parent sequence). Examples of art-recognized polypeptide secondary and tertiary structures are described in Proteins, Structures and Molecular Principles (Creighton, Ed., W. H. Freeman and Company, New York (1984)); Introduction to Protein Structure (C. Branden and J. Tooze, eds., Garland Publishing, New York, N.Y. (1991)); and Thornton et al. Nature 354:105 (1991), which are each incorporated herein by reference.
[0178] In certain embodiments, antibodies of the present disclosure may be chemically bonded with polymers, lipids, or other moieties.
[0179] The binding agents may comprise at least one of the CDRs described herein incorporated into a biocompatible framework structure. In one example, the biocompatible framework structure comprises a polypeptide or portion thereof that is sufficient to form a conformationally stable structural support, or framework, or scaffold, which is able to display one or more sequences of amino acids that bind to an antigen (e.g. , CDRs, a variable region, etc.) in a localized surface region. Such structures can be a naturally occurring polypeptide or polypeptide “fold” (a structural motif), or can have one or more modifications, such as additions, deletions or substitutions of amino acids, relative to a naturally occurring polypeptide or fold. These scaffolds can be derived from a polypeptide of any species (or of
more than one species), such as a human, other mammal, other vertebrate, invertebrate, plant, bacteria or virus.
[0180] Typically, the biocompatible framework structures are based on protein scaffolds or skeletons other than immunoglobulin domains. For example, those based on fibronectin, ankyrin, lipocalin, neocarzinostain, cytochrome b, CPI zinc finger, PST1, coiled coil, LACI- Dl, Z domain and tendamistat domains may be used See e.g., Nygren and Uhlen, 1997, Curr. Opin. in Struct. Biol., 7, 463-469).
[0181] Humanized antibodies can be produced using techniques known to those skilled in the art (Zhang, W., et al., Molecular Immunology. 42(72):1445-1451, 2005; Hwang W. et al., Methods. 36(7):35-42, 2005; Dall’Acqua WF, et al., Methods 36(1):43-6Q, 2005; and Clark, M., Immunology Today. 27(S):397-402, 2000).
[0182] Additionally, one skilled in the art will recognize that suitable binding agents include portions of these antibodies, LCDR1, LCDR2, LCDR3, HCDR1, HCDR2, and/or HCDR3. The non-CDR portion of the antibody may be a non-protein molecule, wherein the binding agent cross-blocks the binding of an antibody disclosed herein to a target antigen. The non- CDR portion of the antibody may be a non-protein molecule in which the antibody exhibits a similar binding pattern to a target antigen in a competition binding assay as that exhibited by at least one of antibodies disclosed herein. The non-CDR portion of the antibody may be composed of amino acids, wherein the antibody is a recombinant binding protein or a synthetic peptide, and the recombinant binding protein cross-blocks the binding of an antibody disclosed herein to a target antigen and/or neutralizes a target antigen. The non- CDR portion of the antibody may be composed of amino acids, wherein the antibody is a recombinant antibody, and the recombinant antibody exhibits a similar binding pattern to a target antigen in the target epitope competition binding assay (described hereinbelow) as that exhibited by at least one of the antibodies disclosed herein, and/or neutralizes the target antigen.
[0183] Where an antibody comprises one or more of HCDR1, HCDR2, HCDR3, LCDR1, LCDR2 and LCDR3 as described above, it may be obtained by expression from a host cell containing DNA coding for these sequences. A DNA coding for each CDR sequence may be determined on the basis of the amino acid sequence of the CDR and synthesized together with any desired antibody variable region framework and constant region DNA sequences
using oligonucleotide synthesis techniques, site-directed mutagenesis and polymerase chain reaction (PCR) techniques as appropriate. DNA coding for variable region frameworks and constant regions is widely available to those skilled in the art from genetic sequences databases such as GenBank®. In some embodiments, the heavy chain and the light chain of the antibody are expressed from a single DNA construct. In some embodiments, the heavy chain and the light chain of the antibody are expressed from two or more separate DNA constructs.
[0184] Once synthesized, the DNA encoding an antibody of the present disclosure or fragment thereof may be propagated and expressed according to any of a variety of well-known procedures for nucleic acid excision, ligation, transformation, and transfection using any number of known expression vectors. Thus, in certain embodiments expression of an antibody fragment may be preferred in a prokaryotic host, such as Escherichia coli (see, e.g., Pluckthun et al., 1989 Methods Enzymol. 178:497-515). In certain other embodiments, expression of the antibody or a fragment thereof may be preferred in a eukaryotic host cell, including yeast (e.g. , Saccharomyces cerevisiae, Schizosaccharomyces pombe, and Pichia pastoris), animal cells (including mammalian cells) or plant cells. Examples of suitable animal cells include, but are not limited to, myeloma (such as a mouse NSO line), COS, CHO, or hybridoma cells. Examples of plant cells include tobacco, corn, soybean, and rice cells.
[0185] One or more replicable expression vectors containing DNA encoding an antibody variable and/or constant region may be prepared and used to transform an appropriate cell line, for example, a non-producing myeloma cell line, such as a mouse NSO line or a bacteria, such as E. coli, in which production of the antibody will occur. In order to obtain efficient transcription and translation, the DNA sequence in each vector should include appropriate regulatory sequences, particularly a promoter and leader sequence operatively linked to the variable domain sequence. Particular methods for producing antibodies in this way are generally well-known and routinely used. For example, basic molecular biology procedures are described by Maniatis et al. (Molecular Cloning, A Laboratory Manual, 2nd ed., Cold Spring Harbor Laboratory, New York, 1989; see also Maniatis et al, 3rd ed., Cold Spring Harbor Laboratory, New York, (2001)). DNA sequencing can be performed as described in Sanger et al. (PNAS 74:5463, (1977)) and the Amersham International pic sequencing handbook, and site directed mutagenesis can be carried out according to methods
known in the art (Kramer et al., Nucleic Acids Res. 12:9441, (1984); Kunkel Proc. Natl. Acad. Sci. USA 82:488-92 (1985); Kunkel et al., Methods in Enzymol. 154:367-82 (1987); the Anglian Biotechnology Ltd. handbook). Additionally, numerous publications describe techniques suitable for the preparation of antibodies by manipulation of DNA, creation of expression vectors, and transformation and culture of appropriate cells (Mountain A and Adair, J R in Biotechnology and Genetic Engineering Reviews (ed. Tombs, M P, 10, Chapter 1, 1992, Intercept, Andover, UK); “Current Protocols in Molecular Biology”, 1999, F.M. Ausubel (ed.), Wiley Interscience, New York).
[0186] Where it is desired to improve the affinity of antibodies according to the disclosure containing one or more of the above-mentioned CDRs can be obtained by a number of affinity maturation protocols including maintaining the CDRs (Yang et al., J. Mol. Biol., 254, 392-403, 1995), chain shuffling (Marks et al., Bio/Technology, 10, 779-783, 1992), use of mutation strains of E. coli. (Low et al., J. Mol. Biol., 250, 350-368, 1996), DNA shuffling (Patten et al. , Curr. Opin. Biotechnol., 8, 724-733, 1997), phage display (Thompson et al., J. Mol. Biol., 256, 7-88, 1996) and sexual PCR (Crameri, et al., Nature, 391, 288-291, 1998). All of these methods of affinity maturation are discussed by Vaughan et al. (Nature Biotech., 16, 535-539, 1998).
[0187] It will be understood by one skilled in the art that some proteins, such as antibodies, may undergo a variety of posttranslational modifications. The type and extent of these modifications often depends on the host cell line used to express the protein as well as the culture conditions. Such modifications may include variations in glycosylation, methionine oxidation, diketopiperizine formation, aspartate isomerization and asparagine deamidation. A frequent modification is the loss of a carboxy-terminal basic residue (such as lysine or arginine) due to the action of carboxypeptidases (as described in Harris, R.J. Journal of Chromatography 705:129-134, 1995).
[0188] The BA, such as antigen-binding domains, for use in the conjugates provided herein encompass proteins having amino acid sequences that vary from those of the described antibodies but that retain the ability to bind the target proteins. Such variant antigen-binding domains comprise one or more additions, deletions, or substitutions of amino acids when compared to parent sequence, but exhibit biological activity that is essentially equivalent to that of the described antibodies.
[0189] Two antigen-binding domains are considered bioequivalent if, for example, they are pharmaceutical equivalents or pharmaceutical alternatives whose rate and extent of absorption do not show a significant difference when administered at the same molar dose under similar experimental conditions, either single does or multiple doses. Some antigenbinding domains will be considered equivalents or pharmaceutical alternatives if they are equivalent in the extent of their absorption but not in their rate of absorption and yet may be considered bioequivalent because such differences in the rate of absorption are intentional and are reflected in the labeling, are not essential to the attainment of effective body drug concentrations on, e.g., chronic use, and are considered medically insignificant for the particular drug product studied.
[0190] In one embodiment, two antigen-binding domains are bioequivalent if there are no clinically meaningful differences in their safety, purity, and potency.
[0191] In one embodiment, two antigen-binding domains are bioequivalent if a patient can be switched one or more times between the reference product and the biological product without an expected increase in the risk of adverse effects, including a clinically significant change in immunogenicity, or diminished effectiveness, as compared to continued therapy without such switching.
[0192] In one embodiment, two antigen-binding domains are bioequivalent if they both act by a common mechanism or mechanisms of action for the condition or conditions of use, to the extent that such mechanisms are known.
[0193] Bioequivalence may be demonstrated by in vivo and in vitro methods. Bioequivalence measures include, e.g., (a) an in vivo test in humans or other mammals, in which the concentration of the antigen-binding domain or its metabolites is measured in blood, plasma, serum, or other biological fluid as a function of time; (b) an in vitro test that has been correlated with and is reasonably predictive of human in vivo bio availability data; (c) an in vivo test in humans or other mammals in which the appropriate acute pharmacological effect of the antigen-binding domain (or its target) is measured as a function of time; and (d) in a well-controlled clinical trial that establishes safety, efficacy, or bioavailability or bioequivalence of an antigen-binding domain.
[0194] Bioequivalent variants of antigen-binding domains for use in the conjugates provided herein may be constructed by, for example, making various substitutions of residues or
sequences or deleting terminal or internal residues or sequences not needed for biological activity. For example, cysteine residues not essential for biological activity can be deleted or replaced with other amino acids to prevent formation of unnecessary or incorrect intramolecular disulfide bridges upon renaturation. In other contexts, bioequivalent antigenbinding domains may include variants comprising amino acid changes which modify the glycosylation characteristics of the antigen -binding domain, e.g., mutations which eliminate or remove glycosylation.
[0195] In some embodiments, the BA including antigen-binding domains for use in the conjugates provided herein bind to a human target protein but not to target protein from other species. In other embodiments, the antigen-binding domains for use in the conjugates provided herein bind to a human target protein and to a target protein from one or more nonhuman species. For example, the antigen-binding domains for use in the conjugates provided herein may bind to a human target protein and may bind or not bind, as the case may be, to one or more of mouse, rat, guinea pig, hamster, gerbil, pig, cat, dog, rabbit, goat, sheep, cow, horse, camel, cynomologous, marmoset, rhesus or chimpanzee target protein. In one embodiment, the antigen-binding domains specifically bind human target protein and cynomolgus monkey (e.g., Macacafascicularis) target protein. In other embodiments, antigen-binding domains for use herein bind human target protein but do not bind, or bind only weakly, to cynomolgus monkey target protein.
[0196] Process of Producing Conjugates
[0197] In additional embodiments, the present disclosure provides a process of producing a conjugate of Formula (I) or Formula (II) as described herein. The process comprises contacting an antibody or an antigen-binding fragment thereof (BA), as described herein, with a linker-payload compound (L-P) in the presence of a transglutaminase, wherein L-P has a structure according to formula (Ila), (lib), or (lie) as described herein.
[0198] In some embodiments, the reactive moiety B is selected from the group consisting of an azide, an alkyne, a thiol, a diene, an amino, an active ester, a glutamine-containing peptide tag (Q-tag), and a 1,2, 4, 5 -tetrazine. In one embodiment, the reactive moiety B is Q-tag. In another embodiment, the reactive moiety B is an active ester. In yet another embodiment, the reactive moiety B is a maleimide. In still another embodiment, the reactive moiety B is a moiety suitable for participation in a Click Reaction, as described herein for click chemistry,
or a Diels- Alder reaction. In one embodiment, moiety B is a diene. In a further embodiment, the reactive moiety B is a substrate of transglutaminase.
[0199] In additional embodiments, the reactive moiety B is a reactive moiety for conjugation to a target protein, antibody, or antigen-binding fragment thereof, comprising at least one member selected from the group consisting of amino, hydroxyl, thiol, a maleimido moiety, a succinyl ester, an activated ester, an alkylamino; a halo acetamido, -N3,


[0200] In one embodiment, the transglutaminase is a bacterial transglutaminase (BTG). In other embodiments, the transglutaminase a native transglutaminase or an engineered transglutaminase.
[0201] In various embodiments, the process is carried out with a molar excess of L-P over BA. For examples, BA is contacted with a 2 to 30-fold, 5 to 25-fold, or 10 to 25-fold molar excess of L-P.
[0202] Exemplary embodiments include the conjugates described in Table 3 below.
[0203] In some embodiments, the present disclosure provides a composition comprising a plurality of conjugates of Formula (I), Formula (II), or combinations thereof as described herein. The number ratio of payload (P) to BA in the composition is defined as a drugantibody ratio (DAR) of about 0.5 to about 30.0. In additional embodiments, the DAR is about 1 to about 8.
[0204] In additional embodiments, the present disclosure provides a product that is produced by the process as described herein.
[0205] Pharmaceutical Compositions
[0206] In additional embodiments, the present disclosure provides a pharmaceutical composition comprising the compound of formula (la), (lb), (Ic), (Ila), (lib), (lie), or the conjugate of formula (I) or (II), specific examples thereof as disclosed herein (e.g., in Tables 1, 2, and 3), any product produced by a process as described herein, and one or more pharmaceutically acceptable carriers, diluents, and/or excipients. For purposes of these
embodiments, the conjugate includes the composition comprising a plurality of conjugates with a defined DAR as described herein. In some embodiments, the composition further contains, in accordance with accepted practices of pharmaceutical compounding, one or more additional therapeutic agents, pharmaceutically acceptable adjuvants, stabilizers, emulsifiers, preservatives, colorants, buffers, flavor imparting agents. The pharmaceutical composition can be administered by any suitable means that results in a concentration of the compound or conjugate in a subject that is effective at treating a disease or condition suitable for treatment with the compounds of the disclosure.
[0207] In some embodiments, the compound or conjugate is present in an amount of 1-95% by weight of the total weight of the composition. The “therapeutically effective amount” of a compound or conjugate that is administered is governed by such considerations, and is the minimum amount necessary to exert cytotoxic effect. Such amount may be below the amount that is toxic to normal cells, or the subject as a whole. Generally, the initial therapeutically effective amount of a compound or conjugate of the present disclosure that is administered is in the range of about 0.001 to about 200 mg/kg or about 0.1 to about 20 mg/kg of patient body weight per day, with the typical initial range being about 0.3 to about 15 mg/kg/day. Oral unit dosage forms, such as tablets and capsules, may contain from about 0.1 mg to about 1000 mg of a compound (or a pharmaceutically acceptable salt, stereoisomer, or tautomer thereof) of the present disclosure. In another embodiment, such dosage forms contain from about 50 mg to about 500 mg of a compound or conjugate of the present disclosure. In yet another embodiment, such dosage forms contain from about 25 mg to about 200 mg of a compound or conjugate of the present disclosure. In still another embodiment, such dosage forms contain from about 10 mg to about 100 mg of a compound or conjugate of the present disclosure. In a further embodiment, such dosage forms contain from about 5 mg to about 50 mg of a compound or conjugate of the present disclosure.
[0208] In certain embodiments, the compound or conjugate is substantially pure, in that it contains less than about 5%, or less than about 2%, or less than about 1%, or less than about 0.5%, or less than about 0.1%, of other organic small molecules, such as unreacted intermediates or synthesis by-products that are created, for example, in one or more of the steps of a synthesis method.
[0209] In various embodiments, the composition is provided in a dosage form that is suitable for oral, parenteral (e.g., intravenously, intramuscularly, subcutaneous, intraarterial), buccal,
sublingual, rectal, cutaneous, nasal, vaginal, intranasal, inhalation, transdermal, ocular, intraosseous, otic, or intracranial administration route. Thus, in some embodiments, the composition dosage form is chosen from tablets, capsules, pills, powders, granulates, suspensions, emulsions, solutions, gels including hydrogels, pastes, patches, ointments, creams, plasters, drenches, osmotic delivery devices, suppositories, enemas, injectables, implants, sprays, and aerosols. The pharmaceutical compositions are formulated according to conventional pharmaceutical practice (see, e.g., Remington: The Science and Practice of Pharmacy, 20th edition, 2000, ed. A.R. Gennaro, Lippincott Williams & Wilkins, Philadelphia, and Encyclopedia of Pharmaceutical Technology, eds. J. Swarbrick and J. C. Boylan, 1988-1999, Marcel Dekker, New York).
[0210] Pharmaceutical compositions can be formulated to release the compound or conjugate immediately upon administration or at any predetermined time or time period after administration (e.g., controlled release formulations). Examples of controlled release formulations include (i) formulations that create substantially constant concentrations of the agent(s) of the disclosure within the body over an extended period of time; (ii) formulations that after a predetermined lag time create substantially constant concentrations of the agents of the disclosure within the body over an extended period of time; (iii) formulations that sustain the agent(s) action during a predetermined time period by maintaining a relatively constant, effective level of the agent(s) in the body with concomitant minimization of undesirable side effects associated with fluctuations in the plasma level of the agent(s) (sawtooth kinetic pattern); (iv) formulations that localize action of agent(s), e.g., spatial placement of a controlled release composition adjacent to or in the diseased tissue or organ; (v) formulations that achieve convenience of dosing, e.g., administering the composition once per week or once every two weeks; and (vi) formulations that target the action of the agent(s) by using carriers or chemical derivatives to deliver the compound to a particular target cell type. Administration of the compound or conjugate in the form of a controlled release formulation is desirable, in some embodiments, for compounds having a narrow absorption window in the gastrointestinal tract or a relatively short biological half-life.
[0211] In some embodiments, controlled release is obtained by appropriate selection of various formulation parameters and ingredients, including, e.g., various types of controlled release compositions and coatings. In some embodiments, the compound is formulated with appropriate excipients into a pharmaceutical composition that, upon administration, releases
the compound in a controlled manner. Examples include single or multiple unit tablet or capsule compositions, oil solutions, suspensions, emulsions, microcapsules, molecular complexes, microspheres, nanoparticles, patches, and liposomes.
[0212] A pharmaceutical composition comprising a compound as described herein can be administered parenterally by injection, infusion, or implantation (e.g., intraocular, subcutaneous, intravenous, intramuscular, intraperitoneal) via dosage forms, formulations, or by suitable delivery devices or implants containing conventional, non-toxic pharmaceutically acceptable carriers and adjuvants. The formulation and preparation of such compositions are well known to those skilled in the art of pharmaceutical formulation.
[0213] Suitable oral compositions as described herein include without limitation tablets, troches, lozenges, aqueous or oily suspensions, dispersible powders or granules, emulsion, hard or soft capsules, syrups or elixirs.
[0214] In another embodiment, also encompassed are pharmaceutical compositions suitable for single unit dosages that comprise a compound of the disclosure or its pharmaceutically acceptable stereoisomer, salt, or tautomer and a pharmaceutically acceptable carrier.
[0215] The compositions of the present disclosure that are suitable for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions. For instance, liquid formulations of the compounds of the present disclosure contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide pharmaceutically palatable preparations of a compound of the present disclosure.
[0216] For tablet compositions, a compound or conjugate of the present disclosure in admixture with non-toxic pharmaceutically acceptable excipients is used for the manufacture of tablets. Examples of such excipients include without limitation inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example, corn starch, or alginic acid; binding agents, for example starch, gelatin or acacia, and lubricating agents, for example magnesium stearate, stearic acid or talc. The tablets may be uncoated or they may be coated by known coating techniques to delay disintegration and absorption in the gastrointestinal tract and thereby to provide a sustained therapeutic action over a desired time period. For example, a time delay material such as glyceryl monostearate or glyceryl distearate may be employed.
[0217] Formulations for oral use may also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, for example peanut oil, liquid paraffin or olive oil.
[0218] For aqueous suspensions, a compound or conjugate of the present disclosure is admixed with excipients suitable for maintaining a stable suspension. Examples of such excipients include without limitation are sodium carboxymethylcellulose, methylcellulose, hydroxpropylmethylcellulose, sodium alginate, polyvinylpyrrolidone, gum tragacanth and gum acacia.
[0219] Oral suspensions can also contain dispersing or wetting agents, such as naturally- occurring phosphatide, for example, lecithin, or condensation products of an alkylene oxide with fatty acids, for example polyoxyethylene stearate, or condensation products of ethylene oxide with long chain aliphatic alcohols, for example, heptadecaethyleneoxycetanol, or condensation products of ethylene oxide with partial esters derived from fatty acids and a hexitol such as polyoxyethylene sorbitol monooleate, or condensation products of ethylene oxide with partial esters derived from fatty acids and hexitol anhydrides, for example polyethylene sorbitan monooleate. The aqueous suspensions may also contain one or more preservatives, for example ethyl, or n-propyl p-hydroxybenzoate, one or more coloring agents, one or more flavoring agents, and one or more sweetening agents, such as sucrose or saccharin.
[0220] Oily suspensions may be formulated by suspending a compound or conjugate of the present disclosure in a vegetable oil, for example arachis oil, olive oil, sesame oil or coconut oil, or in a mineral oil such as liquid paraffin. The oily suspensions may contain a thickening agent, for example beeswax, hard paraffin or cetyl alcohol.
[0221] Sweetening agents such as those set forth above, and flavoring agents may be added to provide palatable oral preparations. These compositions may be preserved by the addition of an anti-oxidant such as ascorbic acid.
[0222] Dispersible powders and granules suitable for preparation of an aqueous suspension by the addition of water provide a compound or conjugate of the present disclosure in admixture with a dispersing or wetting agent, suspending agent and one or more preservatives. Suitable dispersing or wetting agents and suspending agents are exemplified
by those already mentioned above. Additional excipients, for example sweetening, flavoring and coloring agents, may also be present.
[0223] Pharmaceutical compositions of the present disclosure may also be in the form of oil- in-water emulsions. The oily phase may be a vegetable oil, for example olive oil or arachis oil, or a mineral oil, for example liquid paraffin or mixtures of these. Suitable emulsifying agents may be naturally-occurring gums, for example gum acacia or gum tragacanth, naturally-occurring phosphatides, for example soy bean, lecithin, and esters or partial esters derived from fatty acids and hexitol, anhydrides, for example sorbitan monoleate, and condensation reaction products of the said partial esters with ethylene oxide, for example polyoxyethylene sorbitan monoleate. The emulsions may also contain sweetening and flavoring agents.
[0224] Syrups and elixirs may be formulated with sweetening agents, for example glycerol, propylene glycol, sorbitol or sucrose. Such formulations may also contain a demulcent, a preservative, and flavoring and coloring agents. The pharmaceutical compositions may be in the form of a sterile injectable, an aqueous suspension or an oleaginous suspension. This suspension may be formulated according to the known art using those suitable dispersing or wetting agents and suspending agents which have been mentioned above. The sterile injectable preparation may also be sterile injectable solution or suspension in a non-toxic parentally acceptable diluent or solvent, for example as a solution in 1,3-butanediol. Among the acceptable vehicles and solvents that may be employed are water, Ringer’ s solution and isotonic sodium chloride solution. In addition, sterile, fixed oils are conventionally employed as a solvent or suspending medium. For this purpose, any bland fixed oil may be employed including synthetic mono-or diglycerides. In addition, fatty acids such as oleic acid find use in the preparation of injectables.
[0225] A compound or conjugate of the present disclosure can be administered in the form of suppositories for rectal administration of the drug. These compositions can be prepared by mixing the drug with a suitable non-irritating excipient which is solid at ordinary temperatures but liquid at the rectal temperature and will therefore melt in the rectum to release the drug. Such materials are cocoa butter and polyethylene glycols.
[0226] Compositions for parenteral administrations are administered in a sterile medium. Depending on the vehicle used and concentration the concentration of the drug in the
formulation, the parenteral formulation can either be a suspension or a solution containing dissolved drug. Adjuvants such as local anesthetics, preservatives and buffering agents can also be added to parenteral compositions.
[0227] In some embodiments, the composition is especially adapted for administration into or around the eye. For example, a composition can be adapted to be used as eye drops, or injected into the eye, e.g., using peribulbar or intravitreal injection. Such compositions should be sterile and substantially endotoxin-free, and within an acceptable range of pH. In some embodiments a formulation without preservatives is used. Formulation of eye medications is known in the art, see, e.g., Ocular Therapeutics and Drug Delivery: A Multi-Disciplinary Approach, Reddy, Ed. (CRC Press 1995); Kaur and Kanwar, Drug Dev Ind Pharm. 2002 May; 28(5):473-93; Clinical Ocular Pharmacology, Bartlett et al. (Butterworth-Heinemann; 4th edition (Mar. 15, 2001)); and Ophthalmic Drug Delivery Systems (Drugs and the Pharmaceutical Sciences: a Series of Textbooks and Monographs), Mitra (Marcel Dekker; 2nd Rev&Ex edition (Mar. 1, 2003)).
[0228] Compositions for parenteral use may be provided in unit dosage forms (e.g., in singledose ampoules), or in vials containing several doses and in which a suitable preservative may be added (see below). The composition may be in form of a solution, a suspension, an emulsion, an infusion device, or a delivery device for implantation, or it may be presented as a dry powder to be reconstituted with water or another suitable vehicle before use. Apart from the active agent(s), the composition may include suitable parenterally acceptable carriers and/or excipients. The active agent(s) may be incorporated into microspheres, microcapsules, nanoparticles, liposomes, or the like for controlled release. Furthermore, the composition may include suspending, solubilizing, stabilizing, pH-adjusting agents, tonicity adjusting agents, and/or dispersing agents.
[0229] In some embodiments, the pharmaceutical compositions of the disclosure are in a form suitable for sterile injection. To prepare such a composition, the active agent(s) are dissolved or suspended in a parenterally acceptable liquid vehicle. Among acceptable vehicles and solvents that may be employed are water, water adjusted to a suitable pH by addition of an appropriate amount of hydrochloric acid, sodium hydroxide or a suitable buffer, 1,3-butanediol, Ringer’s solution, dextrose solution, and isotonic sodium chloride solution. The aqueous formulation may also contain one or more preservatives (e.g., methyl, ethyl, or n-propyl p-hydroxybenzoate). In cases where the compound has limited solubility in
water, a dissolution enhancing or solubilizing agent can be added, or the solvent may include 10-60% w/w of propylene glycol.
[0230] The pharmaceutical compositions can be administered to a subject in a single dose or in multiple doses. For example, a compound or conjugate described herein can be administered once a week or for 2, 3, 4, 5, 6, 7, 8, 10, 15, 20, or more weeks. It is to be understood that, for any particular subject, specific dosage regimes should be adjusted over time according to the individual need and the professional judgment of a health care provider administering or supervising the administration of the compound. For example, the dosage of a compound or conjugate can be increased if the lower dose does not provide sufficient biological activity (e.g., in the treatment of a disease or condition described herein). Conversely, the dosage of the compound or conjugate can be decreased, for example, if the disease or condition is reduced or eliminated, or to reduce undesirable side-effects.
[0231] Methods of Treatment
[0232] In additional embodiments, the present disclosure provides a method of treating a subject suffering from cancer. The method comprises administering to the subject a therapeutically effective amount of a compound, conjugate, dosage form, or composition as described herein.
[0233] The compounds or conjugates provided herein are useful, inter alia, for the treatment, prevention and/or amelioration of any disease or disorder associated with or mediated by expression, signaling or activity of the target protein of the antigen-binding domain.
[0234] In some embodiments, the compound or conjugate provided herein is used to treat primary and/or metastatic tumors arising in the brain and meninges, oropharynx, lung and bronchial tree, gastrointestinal tract, male and female reproductive tract, muscle, bone, skin and appendages, connective tissue, spleen, immune system, blood forming cells and bone marrow, liver and urinary tract, and special sensory organs such as the eye. In certain embodiments, the compound or conjugate provided herein is used to treat one or more of the following cancers: acute myelogenous leukemia, adult T-cell leukemia, astrocytomas, bladder cancer, breast cancer, PRLR positive (PRLR+) breast cancer, cervical cancer, cholangiocarcinoma, chronic myeloid leukemia, colon cancer, colorectal cancer, endometrial cancer, esophageal cancer, gastric cancer, glioblastomata, head and neck cancer (e.g., head and neck squamous cell carcinoma (HNSCC)), Kaposi’s sarcoma, kidney cancer,
leiomyosarcomas, liver cancer, lung cancer (e.g., small cell lung cancer, non-small cell lung cancer (NSCLC)), lymphomas, malignant gliomas, malignant mesothelioma, melanoma, mesothelioma, malignant mesothelioma, MFH/fibrosarcoma, multiple myeloma, nasopharyngeal cancer, osteosarcoma, ovarian cancer, pancreatic carcinoma, prostate cancer, castrate-resistant prostate cancer, renal cell carcinoma, residual cancer wherein “residual cancer” means the existence or persistence of one or more cancerous cells in a subject following treatment with an anti-cancer therapy, rhabdomyosarcoma, stomach cancer, synovial sarcoma, thyroid cancer, uterine cancer, and Wilms’ tumor. In some embodiments, the cancer is breast cancer. In some embodiments, the cancer is prostate cancer.
[0235] Also provided in an embodiment is a method of selectively killing quiescent cells in a subject. The method comprises administering to the subject a therapeutically effective amount of a compound, a conjugate, or a pharmaceutical composition as described herein. In some embodiments, the quiescent cells are quiescent cancer cells.
[0236] In another embodiment, the present disclosure provides a method of selectively killing stem cells in a subject. The method comprises administering to the subject a therapeutically effective amount of a compound, a conjugate, or a pharmaceutical composition as described herein. In some embodiments, the stem cells are hematopoietic stem cells.
[0237] In another embodiment, the present disclosure provides a method of selectively killing resting or naive B- or T- or other immune cells in a subject. The method comprises administering to the subject a therapeutically effective amount of a compound, a conjugate, or a pharmaceutical composition as described herein.
[0238] In another embodiment, the present disclosure provides a method of selectively killing quiescent cancer cells in a subject preparing for stem cell therapy. The method comprises administering to the subject a therapeutically effective amount of a compound, a conjugate, or a pharmaceutical composition as described herein.
[0239] In various embodiments, the methods described herein further comprise administering to the subject one or more other therapeutic agents, as described in more detail below.
[0240] Combination Therapy
[0241] Provided herein are compositions comprising a compound or conjugate as described herein in combination with one or more additional therapeutically active components, and methods of treatment comprising administering such combinations to a subject.
[0242] The compound or conjugate may be co-formulated with and/or administered in combination with one or more additional therapeutically active component(s) selected from a MET antagonist (e.g., an anti-MET antibody (e.g., onartuzumab, emibetuzumab, and H4H14639D) or small molecule inhibitor of MET), an EGFR antagonist (e.g., an anti-EGFR antibody (e.g., cetuximab or panitumumab) or small molecule inhibitor of EGFR (e.g., gefitinib or erlotinib)), an antagonist of another EGFR family member such as Her2/ErbB2, ErbB3 or ErbB4 (e.g., anti-ErbB2 (e.g., trastuzumab or T-DM1 {KADCYLA®}), anti-ErbB3 or anti-ErbB4 antibody or small molecule inhibitor of ErbB2, ErbB3 or ErbB4 activity), an antagonist of EGFRvIII (e.g., an anti-EGFRvIII antibody), an IGF1R antagonist (e.g., an anti- IGF1R antibody), a B-raf inhibitor (e.g., vemurafenib, sorafenib, GDC-0879, PLX-4720), a PDGFR-a inhibitor (e.g., an anti-PDGFR-a antibody), a PDGFR-P inhibitor (e.g., an anti- PDGFR-P antibody or small molecule kinase inhibitor such as, e.g., imatinib mesylate or sunitinib malate), a PDGF ligand inhibitor (e.g., anti-PDGF-A, -B, -C, or -D antibody, aptamer, siRNA, etc.), a VEGF antagonist (e.g., a VEGF-Trap such as aflibercept, see, e.g., US 7,087,411 (also referred to herein as a “VEGF-inhibiting fusion protein”), anti- VEGF antibody (e.g., bevacizumab), a small molecule kinase inhibitor of VEGF receptor (e.g., sunitinib, sorafenib or pazopanib)), a DLL4 antagonist (e.g., an anti-DLL4 antibody disclosed in US 2009/0142354 such as REGN421), an Ang2 antagonist (e.g., an anti-Ang2 antibody disclosed in US 2011/0027286 such as H1H685P), a FOLH1 antagonist (e.g., an anti-FOLHl antibody), a STEAP1 or STEAP2 antagonist (e.g., an anti-STEAPl antibody or an anti- STEAP2 antibody), a TMPRSS2 antagonist (e.g., an anti-TMPRSS2 antibody), a MSLN antagonist (e.g., an anti-MSLN antibody), a CA9 antagonist (e.g., an anti-CA9 antibody), a uroplakin antagonist (e.g., an anti-uroplakin (e.g., anti-UPK3A) antibody), a MUC16 antagonist (e.g., an anti-MUC16 antibody), a Tn antigen antagonist (e.g., an anti-Tn antibody), a CLEC12A antagonist (e.g., an anti- CLEC12A antibody), a TNFRSF17 antagonist (e.g., an anti-TNFRSF17 antibody), a LGR5 antagonist (e.g., an anti-LGR5 antibody), a monovalent CD20 antagonist (e.g., a monovalent anti-CD20 antibody such as rituximab), a CD20 x CD3 bispecific antibody, a PD-1 blocking agent (e.g., an anti-PD-1
antibody such as pembrolizumab or nivolumab), etc. Other agents that may be beneficially administered in combination with antibodies provided herein include, e.g., tamoxifen, aromatase inhibitors, and cytokine inhibitors, including small-molecule cytokine inhibitors and antibodies that bind to cytokines such as IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-8, IL-9, IL- 11, IL-12, IL-13, IL-17, IL-18, or to their respective receptors.
[0243] In an exemplary embodiment, a PD-1 inhibitor such as an anti-PD-1 antibody can be combined with a compound or conjugate as described herein.
[0244] In some embodiments, provided herein are pharmaceutical compositions comprising a compound or conjugate as described herein in combination with one or more chemotherapeutic agents. Examples of chemotherapeutic agents include alkylating agents such as thiotepa and cyclosphosphamide (Cytoxan™); alkyl sulfonates such as busulfan, improsulfan and piposulfan; aziridines such as benzodopa, carboquone, meturedopa, and uredopa; ethylenimines and methylamelamines including altretamine, triethylenemelamine, trietylenephosphoramide, trie thy lene thiopho sphaoramide and trimethylolomelamine; nitrogen mustards such as chlorambucil, chlornaphazine, cholophosphamide, estramustine, ifosfamide, mechlorethamine, mechlorethamine oxide hydrochloride, melphalan, novembichin, phenesterine, prednimustine, trofosfamide, uracil mustard; nitrosureas such as carmustine, chlorozotocin, fotemustine, lomustine, nimustine, ranimustine; antibiotics such as aclacinomysins, actinomycin, authramycin, azaserine, bleomycins, cactinomycin, calicheamicin, carabicin, carminomycin, carzinophilin, chromomycins, dactinomycin, daunorubicin, detorubicin, 6-diazo-5-oxo-L-norleucine, doxorubicin, epirubicin, esorubicin, idarubicin, marcellomycin, mitomycins, mycophenolic acid, nogalamycin, olivomycins, peplomycin, potfiromycin, puromycin, quelamycin, rodorubicin, streptonigrin, streptozocin, tubercidin, ubenimex, zinostatin, zorubicin; anti-metabolites such as methotrexate and 5- fluorouracil (5-FU); folic acid analogues such as denopterin, methotrexate, pteropterin, trimetrexate; purine analogs such as fludarabine, 6-mercaptopurine, thiamiprine, thioguanine; pyrimidine analogs such as ancitabine, azacitidine, 6-azauridine, carmofur, cytarabine, dideoxyuridine, doxifluridine, enocitabine, floxuridine; androgens such as calusterone, dromostanolone propionate, epitiostanol, mepitiostane, testolactone; anti-adrenals such as aminoglutethimide, mitotane, trilostane; folic acid replenisher such as frolinic acid; aceglatone; aldophosphamide glycoside; aminolevulinic acid; amsacrine; bestrabucil; bisantrene; edatraxate; defofamine; demecolcine; diaziquone; elfornithine; elliptinium
acetate; etoglucid; gallium nitrate; hydroxyurea; lentinan; lonidamine; mitoguazone; mitoxantrone; mopidamol; nitracrine; pentostatin; phenamet; pirarubicin; podophyllinic acid; 2-ethylhydrazide; procarbazine; PSK™; razoxane; sizofiran; spirogermanium; tenuazonic acid; triaziquone; 2,2’,2”-trichlorotriethylamine; urethan; vindesine; dacarbazine; mannomustine; mitobronitol; mitolactol; pipobroman; gacytosine; arabinoside (“Ara-C”); cyclophosphamide; thiotepa; taxanes, e.g. paclitaxel (Taxol™, Bristol-Myers Squibb Oncology, Princeton, N.J.) and docetaxel (Taxotere™; Aventis Antony, France); chlorambucil; gemcitabine; 6-thioguanine; mercaptopurine; methotrexate; platinum analogs such as cisplatin and carboplatin; vinblastine; platinum; etoposide (VP-16); ifosfamide; mitomycin C; mitoxantrone; vincristine; vinorelbine; navelbine; novantrone; teniposide; daunomycin; aminopterin; xeloda; ibandronate; CPT-11 ; topoisomerase inhibitor RFS 2000; difluoromethylornithine (DMFO); retinoic acid; esperamicins; capecitabine; and pharmaceutically acceptable salts, acids or derivatives of any of the above. Also included in this definition are anti -hormonal agents that act to regulate or inhibit hormone action on tumors such as anti-estrogens including for example tamoxifen, raloxifene, aromatase inhibiting 4(5) -imidazoles, 4-hydroxytamoxifen, trioxifene, keoxifene, LY 117018, onapristone, and toremifene (Fareston); and anti-androgens such as flutamide, nilutamide, bicalutamide, leuprolide, and goserelin; and pharmaceutically acceptable salts, acids or derivatives of any of the above.
[0245] The compound or conjugate may also be administered and/or co-formulated in combination with antivirals, antibiotics, analgesics, corticosteroids, steroids, oxygen, antioxidants, COX inhibitors, cardioprotectants, metal chelators, IFN-gamma, and/or NSAIDs.
[0246] The additional therapeutically active component(s), e.g., any of the agents listed above or derivatives thereof, may be administered just prior to, concurrent with, or shortly after the administration of the compound or conjugate. In some embodiments, provided are pharmaceutical compositions in which a compound or conjugate described herein is coformulated with one or more of the additional therapeutically active component(s) as described herein.
[0247] As used herein, the term "in combination" includes the use of more than one therapy (e.g., one or more prophylactic and/or therapeutic agents). However, the use of the term "in combination" does not restrict the order in which therapies (e.g., prophylactic and/or
therapeutic agents) are administered to a subject with a disease or disorder. A first therapy (e.g., a conjugate provided herein) can be administered prior to (e.g., 5 minutes, 15 minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4 hours, 6 hours, 12 hours, 24 hours, 48 hours, 72 hours, 96 hours, 1 week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 8 weeks, or 12 weeks before), concomitantly with, or subsequent to (e.g., 5 minutes, 15 minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4 hours, 6 hours, 12 hours, 24 hours, 48 hours, 72 hours, 96 hours, 1 week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 8 weeks, or 12 weeks after) the administration of a second therapy (e.g., a prophylactic or therapeutic agent) to the subject. Triple therapy is also contemplated herein.
[0248] Administration of the compound or conjugate provided herein and one or more second active agents to a subject can occur simultaneously or sequentially by the same or different routes of administration. The suitability of a particular route of administration employed for a particular active agent will depend on the active agent itself (e.g., whether it can be administered orally without decomposing prior to entering the blood stream) and the disease or disorder being treated.
[0249] EXAMPLES
[0250] General Procedures. Proton NMR spectra were acquired on a Varian Inova 300 or 500 MHz NMR instrument. Chromatographic purities were determined on an Agilent 1200 Series or 1100 Series LC/MS system with electrospray ionization source and triple -quad ion trap analyzer using a Merck Chromolith RP-18e analytical HPLC column (monolithic, 50 x 2 mm) and the following analytical HPLC method: injection volume 5 pL; flow rate 1 mL/min; 5^95% acetonitrile in water with 0.05% AcOH over 5 mins; Agilent diode array detector at I = 254, 220 or 195 nm; room temperature; or a Waters UPLC/MS-5SQD system using a Kinetex 1.7 mm C18 100A column (50 x 2.1 mm) and the following analytical UPLC method; injection volume 5 p L; flow rate 0.6 mL/min; 10— >-90% acetonitrile (containing 0.05% HCOOH) in water (containing 0.02% HCOOH) over 2.5 mins; full diode array detector; room temperature. Appropriate conjugates were analyzed using a Bruker ultraFleXtreme MALDI-TOF-TOF mass spectrometer. All starting materials and solvents were purchased commercially and used without purification, unless otherwise noted.
[0251] EXAMPLE 1: Linker 7 was synthesized from compound 1 as described below.

[0252] Synthesis of tert-butyl((S)-l-(((S)-l-((4-(hydroxymethyl)phenyl)amino)-l- oxopropan-2-yl)amino)-3-methyl-l-oxobutan-2-yl)carbamate (2): To a mixture of Boc- Val-Ala-OH (2100 mg, 7.283 mmol) and (4-aminophenyl)methanol (987 mg, 8.011 mmol) in DCM/MeOH (9 mL/9 mL) at ambient temperature under argon was added N- ethoxycarbonyl-2-ethoxy-l,2-dihydroquinoline (EEDQ, 2701 mg, 10.925 mmol) and the resulting mixture was allowed to stir overnight. Analysis by UPLC revealed completion of reaction. The reaction mixture was concentrated under reduced pressure, redissolved in DMSO (8 mL) and purified by reverse phase chromatography (275 g C18 Aq Isco, 5-30- 100% of acetonitrile in water, each containing 0.05% of AcOH). The product eluted in 30- 35% of acetonitrile. Fractions were combined and lyophilized to afford 2 as a white solid (2384 mg, 6.059 mmol, 83%). MS (ESI, pos.) calc’d for C20H32N3O5 [394.2]+; found (M+H) 294.1.
[0253] Synthesis of tert-butyl((S)-l-(((S)-l-((4-(chloromethyl)phenyl)amino)-l- oxopropan-2-yl)amino)-3-methyl-l-oxobutan-2-yl)carbamate (3): To a mixture of 2 (299 mg, 0.760 mmol) and benzotriazole (109 mg, 0.912 mmol) in THF (6 mL) at 0 °C under argon was added SOCh (0.065 mL, 0.895 mmol) drop wise. After 10 mins at the same temperature, the reaction mixture was transferred to ambient temperature where it was stirred for an hour. The reaction reached completion within the time as revealed by LCMS. The reaction mixture was slurried in silica gel and purified by normal phase chromatography (40
g column, hexanes/EtOAc). The product eluted in 4:6 hexanes/EtOAc. The fractions were collected and concentrated to afford 3 as a white solid (265 mg, 0.643 mmol, 85%). MS (ESI, pos.) calc’d for C20H31N3O4CI [412.2]+; found (M+H) 412.1.
[0254] Synthesis of benzyl 3-hydroxyquinoline-2-carboxylate (4): To a mixture of 3- hydroxyquinoline -2 -carboxylic acid (800 mg, 4.229 mmol) and NaHCCL (1066 mg, 12.687 mmol) in DMF (12 mL) at ambient temperature was added benzyl bromide (0.8324 mL, 8.458 mmol) via a syringe and the resulting mixture was stirred overnight. Analysis of crude by UPLC revealed desired product along with dibenzylated product and unreacted starting material. The reaction mixture was filtered through a pad of celite and the residue was washed with EtOAc (50 mL). The filtrate was concentrated under reduced pressure and the resulting yellow oil was purified by reverse phase chromatography (275 g C18 Aq Isco, 5-70- 100% of acetonitrile in water, each containing 0.05% AcOH). The desired product eluted in 60% of acetonitrile. The fractions were combined and lyophilized to afford 4 as a white solid (856 mg, 3.065 mmol, 72%). MS (ESI, pos.) calc’d for C17H14NO3 [280.1]+; found (M+H) 280.4.
[0255] Synthesis of benzyl3-((4-((S)-2-((S)-2-((tert-butoxycarbonyl)amino)-3- methylbutanamido)propanamido)benzyl)oxy)quinoline-2-carboxylate (5): A mixture of the phenol (4, 135 mg, 0.483 mmol), acid chloride (3, 223 mg, 0.541 mmol) and K2CO3 (107 mg, 0.774 mmol) in DMF (3.0 mL) was stirred at 70 °C under argon. The reaction reached completion within 90 mins. It was purified by reverse phase chromatography (50 g C18 Aq Isco, 5-60-100% of acetonitrile in water, each containing 0.05% of AcOH). The product eluted in 60% of acetonitrile. The fractions were combined and lyophilized to afford 5 as a white solid (316 mg, 0.483 mmol, 100%). MS (ESI, pos.) calc’d for C37H43N4O7 [655.3]+; found (M+H) 655.8.
[0256] Synthesis of benzyl 3-((4-((34S,37S)-34-isopropyl-2,2,37-trimethyl-4,32,35-trioxo- 3 ,8, 11, 14, 17,20,23,26, 29-nonaoxa-5, 33, 36-triazaoctatriacontan-38- amido)benzyl)oxy)quinoline-2-carboxylate(6): The material 5 (316 mg, 0.4826 mmol) was treated with 25% of TFA in DCM. The reaction reached completion within 30 mins. The reaction mixture was concentrated under reduced pressure, redissolved in acetonitrile/water and lyophilized to afford the product as a faint yellow solid (268 mg, 0.483 mmol, 100%). MS (ESI, pos.) calc’d for C32H35N4O5 [555.2]+; found (M+H) 655.0. The free amine was subjected to the next step. To a stirring solution of the amine (265 mg, 0.478 mmol) and the
acid (284.7 mg, 0.526 mmol) in DMF (3 mL) at ambient temperature under argon were added HATU (272.5 mg, 0.717 mmol) and DIPEA (0.2419 mL, 0.766 mmol) respectively. The reaction reached completion within 90 mins as revealed by LCMS. The reaction mixture was purified by reverse phase chromatography (100 g C18 Aq Isco, 5-60-100% of acetonitrile in water, each containing 0.05% of AcOH). The product eluted in 60% of acetonitrile. The fractions were combined and lyophilized to afford 6 as a faint yellow sticky oil (512 mg, 0.475 mmol, 99%). MS (ESI, pos.) calc’d for CseHsoNsOie [ 1078.5]+; found (M+H) 1078.3.
[0257] Synthesis of 3-((4-((34S,37S)-34-isopropyl-2,2,37-trimethyl-4,32,35-trioxo- 3 ,8, 11, 14, 17,20,23,26, 29-nonaoxa-5, 33, 36-triazaoctatriacontan-38- amido)benzyl)oxy)quinoline-2-carboxylic acid (7): To a stirring solution of 6 (299 mg, 0.277 mmol) in THFilLCTMcOH (0.5 mL each) at ambient temperature under argon was added LiOH (33.2 mg, 1.386 mmol) in one portion. The reaction reached completion within 15 mins as revealed by UPLC with clean reaction profile. The reaction mixture was diluted with DMSO (1.5) and purified by reverse phase chromatography (50 g C18 Aq Isco, 5-30- 100% of acetonitrile in water, each containing 0.05% of AcOH). The product eluted in 30% of AcOH. The fractions were combined and lyophilized to afford 7 as a sticky yellow oil (274 mg, 0.277 mmol, 100%). MS (ESI, pos.) calc’d for C49H73NsOi6Na [1010.5]+; found (M+Na) 1011.2. ’H NMR (CDCl3, 500 MHz): 5 9.02 (s, 1H), 8.01 (d, J=8.0 Hz, 1H), 7.71-7.64 (m, 4H), 7.58-7.50 (m, 3H), 7.41-7.34 (m, 3H), 5.22 (s, 2H), 5.13-5.11 (m, 1H), 4.69-4.66 (m, 1H), 4.29 (t, J=6.5 Hz, 1H), 3.77-3.74 (m, 1H), 3.66-3.48 (m, 35H), 3.27-3.26 (m, 2H), 2.63- 2.47 (m, 2H), 2.18-2.14 (m, 1H), 1.41-1.40 (m, 12H), 0.95-0.92 (m, 6H).
[0258] EXAMPLE 2
[0259] Linker payload N-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-20-(3-((4-((29S,32S)-l- amino-29-isopropyl-32-methyl-27,30-dioxo-3,6,9,12,15,18,21,24-octaoxa-28,31- diazatritriacontan-33-amido)benzyl)oxy)quinoline-2-carboxamido)-ll,24-diisopropyl- 2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa-2,5,12,15,18,25- hexaazabicyclo[12.12.4]triacont-28-en-7-yl)quinoxaline-2-carboxamide (20, M6364)
[0260] Linker payload (20) was synthesized from compound 8 as described below.

[0261] Synthesis of allyl (((9H-fluoren-9-yl)methoxy)carbonyl)-D-serinate (9): Allyl bromide (15.551 mL, 179.950 mmol) was added to a stirring mixture of Fmoc-D-Ser(tBu)- OH (23000 mg, 59.983 mmol) and CS2CO3 (29315 mg, 89.975 mmol) in DMF (25 mL) and acetonitrile (75 mL) at ambient temperature in air. Within 2 h, the reaction reached completion as revealed by TCL as well as UPLC. The reaction mixture was diluted with EtOAc (600 mL) and the organic layer was washed with saturated solution of brine (100 mLX3) followed by washing with water (100 mLXl). The organic layer was dried (MgSCU) and filtered. The filtrate was concentrated under reduced pressure and put into vacuum for 10 mins to afford a colorless oil (28035 mg). MS (ESI, pos.) calc’d for C25H29NO5Na [446.2]+; found (M+Na) 446.3. The crude oil, without purification, was treated with 40% of TFA in
DCM for 3 h at ambient temperature in air. During reaction transesterification with TFA was observed. Upon disappearance of the starting material as revealed by TCL, the reaction mixture was concentrated under reduced pressure. The oil was dissolved in acetonitrile/water (1:1) and the mixture was stirred at rt in air. Upon disappearance of the transesterification product, the reaction mixture was lyophilized to afford 9 (24300 mg) as a white solid. The product was used for the next step without purification. MS (ESI, pos.) calc’d for C2iH2iNO5Na [390.1]+; found (M+Na) 390.2.
[0262] Synthesis of (R)-2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(allyloxy)-3- oxopropyl methyl-L-valinate (10): To a solution of Boc-A-methyl-L-valine (5428 mg, 23.468 mmol) in THF (45 mL) at 0 °C under argon was added triethylamine (4.670 mL, 33.637 mmol) drop wise but fast which was followed by the addition of 2,4,6- trichlorobenzoyl chloride (3.67 mL, 23.468 mmol) drop wise but fast. The reaction mixture turned cloudy white. After exactly 40 mins at the same temperature, solution of the alcohol (5748 mg, 15.645 mmol) and DMAP (573.4 mg, 4.6935 mmol) in THF (15 mL) was added drop wise. The reaction mixture was allowed to warm to ambient temperature overnight. UPLC revealed completion of reaction with clean reaction profile. The reaction mixture was filtered through celite and the residue was washed with EtOAc (100 mL). The filtrate was concentrated under reduced pressure. MS (ESI, pos.) calc’d for C32H4oN2OsNa [603.3]+; found (M+Na) 603.3. The resulting yellow oil was treated with 25% of TFA in DCM. Upon disappearance of the starting material, the rection mixture was concentrated under reduced pressure and the resulting oil was purified by normal phase chromatography (DCM/MeOH). The product eluted in 95:5 combination of DCM/MeOH. The fractions were combined and concentrated under reduced pressure to afford 10 as a white solid (6300 mg, 15.200 mmol, 97%). MS (ESI, pos.) calc’d for C27H33N2O6 [480.2]+; found (M+Na) 480.5.
[0263] Synthesis of (R)-2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(allyloxy)-3- oxopropyl N-((S)-2-((tert-butoxycarbonyl)(methyl)amino)pent-4-enoyl)-N-methyl-L- valinate (11): Note: The acid shown in the scheme above is highly hygroscopic and has to be used accordingly for improved yield. To a mixture of the acid (2028 mg, 8.846 mmol) and DMTMM (3109 mg, 11.234 mmol) in THF (17 mL) at ambient temperature under argon was added the solution of the amine (3374 mg, 7.003 mmol) in THF (10 mL) followed by the addition of NMM (1.40 mL, 12.638 mmol). The reaction reached completion within 30 mins as revealed by UPLC. The reaction mixture was filtered through celite and the residue was
washed with EtOAc (50 mL). The filtrate was concentrated under reduced pressure and the resulting yellow oil was purified by normal phase chromatography (Hexanes/EtOAc). The product eluted in 7:3 hexanes/EtOAc. The fractions were combined and concentrated to afford 11 as a white solid (3460 mg, 5.001 mmol, 71%). MS (ESI, pos.) calc’d for C38H49N3O9Na [714.4]+; found (M+Na) 714.6.
[0264] Synthesis of (6S,9S,13R)-13-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-6- aIIyI-9-isopropyI-2,2,5,8-tetramethyI-4,7,10-trioxo-3,ll-dioxa-5,8-diazatetradecan-14-oic acid (12): To a mixture of 11 (2228 mg, 3.220 mmol) and Pd(PPh3)4 (372 mg, 0.322 mmol) in DCM (15 mL) at ambient temperature under argon was added PhSith (0.397 mL, 3.220 mmol). The reaction reached completion within 7 mins as revealed by UPLC. The reaction mixture was concentrated under reduced pressure, redissolved in DMSO (6 mL) and purified by reverse phase chromatography (100 g C18 Aq Isco, 5-70-100% of acetonitrile in water, each containing 0.05% AcOH). The product eluted in 70% acetonitrile. The fractions were combined and lyophilized to afford 12 as a white solid (1873 mg, 2.874 mmol, 89%). MS (ESI, pos.) calc’d for C35H46N3O9 [652.3]+; found (M+Na) 652.1.
[0265] Synthesis of ((R)-3-(allyloxy)-3-oxo-2-(quinoxaline-2-carboxamido)propyl N-((S)- 2-((tert-butoxycarbonyl)(methyl)amino)pent-4-enoyl)-N-methyl-L-valinate (13): To a solution of 11 (2600 mg, 3.7582 mmol) dissolved in THF (10 mL) was added DBU (0.561 mL, 3.7582 mmol) at 0 °C under argon. After exactly 25 mins at the same temperature, this reaction mixture was added drop wise to a mixture of 2-quinoxalinecarboxylic acid (982 mg, 5.638 mmol), HATU (2143 mg, 5.638 mmol) and NMM (1.24 mL, 11.278 mmol) in THF (12 mL) cooled to 0 °C. After 5 mins at the same temperature, the reaction mixture was transferred to ambient temperature where it was stirred overnight. UPLC revealed formation of product. The reaction mixture was concentrated under reduced pressure and its silica slurry was purified by normal phase chromatography (Hexanes/EtOAc). The product eluted in 6:4 hexanes/EtOAc. The fractions were combined and concentrated to afford 13 as a white solid (1627 mg, 2.601 mmol, 69%). MS (ESI, pos.) calc’d for C32H43N50sNa [648.3]+; found (M+Na) 648.5.
[0266] Synthesis of (R)-2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(allyloxy)-3- oxopropyl N-((S)-2-((tert-butoxycarbonyl)(methyl)amino)pent-4-enoyl)-N-methyl-L- valinate (14): 13 (2268 mg, 3.625 mmol) was treated with 25% of TFA in DCM. After completion of the reaction as revealed by UPLC, the reaction mixture was concentrated under
reduced pressure which was dissolved in acetonitrile/water and lyophilized overnight before subjecting to next step. MS (ESI, pos.) calc’d for C27H36N5O6 [526.4]+; found (M+H) 526.8. The free amine (1789 mg, 3.404 mmol) was reacted with Fmoc-Ala-OH (2119 mg, 6.807 mmol) in the presence of DMTMM (1884 mg, 6.807 mmol) and NMM (1.130 mL, 10.211 mmol) in THF (20 mL) as described for 11. Work up and chromatography by reverse phase chromatography (275 g C18 Aq Isco, 5-75-100% of acetonitrile in water, each containing 0.05% of AcOH) followed by lyophilization afforded 14 as a white solid (2041 mg, 2.4923 mmol, 73%). MS (ESI, pos.) calc’d for C45H51N6O9 [819.4]+; found (M+H) 819.5.
[0267] Synthesis of (R)-3-(((S)-l-(tert-butoxy)-l-oxopropan-2-yl)amino)-3-oxo-2- ( quinoxaline- 2-carboxamido)propyl N-((S)-2-((S)-2-((((9H-fluoren-9- yl)methoxy)carbonyl)amino)-N-methylpropanamido)pent-4-enoyl)-N-methyl-L-valinate (15): The acid counterpart (14, 2000 mg, 2.442 mmol) was treated with phenylsilane (0.301 mL, 2.442 mmol) in DCM (12 mL) in the presence of Pd(PPh3)4 (198 mg, 0.171 mmol) at ambient temperature under argon. The appearance of dark brown color within 6 mins confirmed the disappearance of the acid. The reaction mixture was concentrated under reduced pressure, redissolved in DMSO (10 mL) and purified by reverse phase chromatography (275 g C18 Aq Isco, 5-60-100% of acetonitrile in water, each containing 0.05% of AcOH). The acid eluted in 60% of acetonitrile. The fractions were combined and lyophilized to afford the acid as a white solid. MS (ESI, pos.) calc’d for C42H47N6O9 [779.3]+; found (M+H) 779.9. The acid (1900 mg, 2.439 mmol) was subjected to coupling with H-Ala- OtBu.HCl (665 mg, 3.659 mmol) in the presence of EEDQ (904 mg, 3.659 mmol) in DCM (9 mL) at ambient temperature in air. The reaction reached completion within 30 mins as revealed by UPLC. The reaction mixture was concentrated under reduced pressure, redissolved in DMSO (10 mL) and purified by reverse phase chromatography (275 g C18 Aq Isco, 5-75-100% of acetonitrile in water, each containing 0.05% of AcOH). The product eluted in 75% of acetonitrile. The fractions were combined and lyophilized to afford 15 as a white solid (1677 mg, 1.851 mmol, 76%). MS (ESI, pos.) calc’d for C49H60N7O10 [906.4]+; found (M+H) 906.2.
[0268] Synthesis of (R)-2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3- (((2S,5R,9S,12S,15S)-12-allyl-2-(tert-butoxycarbonyl)-9-isopropyl-10,13-dimethyl- 4,8,1 L14-tetraoxo-5-(quinoxaline-2-carboxainido)-7-oxa-3,10,13-triazahexadecan- 15- yl)amino)-3-oxopropyl N-((S)-2-((tert-butoxycarbonyl)(methyl)amino)pent-4-enoyl)-N-
methyl-L-valinate (16): The solution of the amine counterpart (1666 mg, 1.8388 mmol) in DMF (6 mL) was treated with DBU (0.264 mL, 1.802 mmol) at 0 °C under argon. The reaction mixture was loaded to column within 18 mins and purified by reverse phase chromatography (100 g C18 Aq Isco, 5-40-50-100% of acetonitrile in water, no additive used). The product eluted in 50% of acetonitrile. The fractions were combined and lyophilized to afford free amine as a white solid (1250 mg, 1.828 mmol, 99%). MS (ESI, pos.) calc’d for C34H50N7O8 [684.4]+; found (M+H) 684.2. The free amine (1425 mg, 2.084 mmol) was reacted with 12 (1453 mg, 2.230 mmol) in the presence of DMTMM (865 mg, 3.126 mmol) and NMM (0.416 mL, 3.751 mmol) in THF (9 mL) at ambient temperature as described for 11. Work up and chromatography (100 g C18 Aq Isco, 5-75-85-100% of acetonitrile in water, no additive) followed by lyophilization afforded 16 as a white solid (2745 mg, 2.083 mmol, 100%). MS (ESI, pos.) calc’d for CesFfeNioOieNa [1339.7]+; found (M+Na) 1340.4.
[0269] Synthesis of O-(N-((3S,6S,HS,14S,17R,Z)-17-((((9H-fluoren-9- yl)methoxy)carbonyl)amino)-3-isopropyl-4,12,14-trimethyl-6-(methylamino)-2,5,13,16- tetraoxo-l-oxa-4,12,15-triazacyclooctadec-8-ene-ll-carbonyl)-N-methyl-L-valyl)-N- ( quinoxaline- 2-carbonyl)-D-seryl-L-alanine (17): A solution of Grubbs-Hoveyda 2nd generation catalyst (200 mg, 0.319 mmol) in DCM (10 mL) prepared under argon was added to the solution of 16 (2294 mg, 1.741 mmol) at ambient temperature under argon. After 20 mins at ambient temperature, the reaction mixture was stirred at 44-45 °C on a metallic heating block overnight. Completion of the reaction was confirmed by UPLC. Two peaks corresponding to the product were observed which were hypothesized to be cis/trans. MS (ESI, pos.) calc’d for CevHssNioOieNa [1311.6]+; found (M+Na) 1312.2. The crude reaction mixture was cooled to 0 °C and 8 mL of TFA was added drop wise. The reaction mixture was transferred to ambient temperature where it was stirred for 3 h. After completion of the reaction within the time, it was concentrated under reduced pressure, redissolved in DMSO (4 mL) and purified by reverse phase chromatography (100 g C18 Aq Isco, 5-45-50-100% of acetonitrile in water, no additive used). The product eluted in 45-50% of acetonitrile. The fractions were combined and lyophilized to give impure 17 as a solid (1322 mg, 1.025 mmol, 59%). MS (ESI, pos.) calc’d for C58H73N10O14 [1333 ,5]+; found (M+H) 1333.9.
[0270] Synthesis of (9H-fluoren-9-yl)methyl ((1S,4S,7R,11S,14S,17S,2OR,24S,Z)-11,24- diisopropyl-2,4,12,15,l'7,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-20-(quinoxaline-2-
carboxamido)-9,22-dioxa-2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7- yl)carbamate (18): To a solution of 17 (660 mg, 0.582 mmol) in THF (6 mL) was added DMTMM (322 MG, 1.165 mmol) followed by the addition of NMM (0.193 mL, 1.747 mmol) at ambient temperature under argon. The reaction reached completion within 45 mins as revealed by UPLC. The reaction mixture was concentrated under reduced pressure, redissolved in DMSO (5 mL) and purified by reverse phase chromatography (100 g C18 Aq Isco, 5-60-100% of acetonitrile in water, each containing 0.05% of AcOH). The major product eluted in 60% of acetonitrile. The fractions were combined and lyophilized to afford 18 as a white solid (243 mg, 0.218 mmol, 37%). MS (ESI, pos.) calc’d for C58H71N10O13 [1115.6]+; found (M+H) 1115.9.
[0271] Synthesis of 19: 18 (243 mg, 0.218 mmol) was treated with 5% of piperidine solution in DMF. The reaction reached completion within 10 mins which was purified by reverse phase chromatography (50 g C18 Aq Isco, 5-35-100% of acetonitrile in water, each containing 0.05% of AcOH). The product eluted in 35% of acetonitrile. The fractions were combined and lyophilized to afford 19 as a white solid (158 mg, 0.177 mmol, 81%). MS (ESI, pos.) calc’d for C43H61N10O11 [893.4]+; found (M+H) 893.2.
[0272] Synthesis of N-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-20-(3-((4-((29S,32S)-l-amino- 29-isopropyl-32-methyl-27,30-dioxo-3,6,9,12,15,18,21,24-octaoxa-28,31- diazatritriacontan-33-amido)benzyl)oxy)quinoline-2-carboxamido)-ll,24-diisopropyl- 2,4,12,15,l'7,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa-2,5,12,15,18,25- hexaazabicyclo[12.12.4]triacont-28-en-7-yl)quinoxaline-2-carboxamide (20): To a mixture of the amine (19, 11 mg, 0.0123 mmol) and the acid (7, 18.3 mg, 0.0185 mmol) in DMF (1.5 mL) at ambient temperature under argon was added HATU (9.4 mg, 0.0246 mmol) followed by the addition of NMM (6.8 pL, 0.0616 mmol). The reaction reached completion within an hour. The reaction mixture was purified by reverse phase chromatography (30 g Cl 8 Aq Isco, 5-50-100% of acetonitrile in water, each containing 0.05% of AcOH). The product eluted in 50% of acetonitrile. The fractions were combined and analyzed by UPLC and found to be impure. The major impurity was the adduct of HATU-unreacted acid along with other minor impurities. The fraction was lyophilized. Mass of the product was clearly seen. After lyophilization, the HATU-acid adduct was found to have decomposed giving rise to the acid. The material was treated with 25% of TFA in DCM. After confirming the fall of the Boc group within 15 mins, the reaction mixture was concentrated under reduced pressure,
re-dissoved in DMF (2 mL) and purified by reverse phase chromatography (Isco EZ Prep (Gemini Cl 8 column, 30 x 150 mm, 5-35-100% of acetonitrile in water, each containing 0.05% of AcOH). The product eluted in 35% of acetonitrile. The fractions were combined and lyophilized to afford 20 as a fluffy white solid (16 mg, 0.0091 mmol, 74%). MS (ESI, pos.) calc’d for C87H124N15O24 [ 1762.9] +; found (M+H) 1765.2. 1 H NMR (DMSOe, 500 MHz): 5 9.94 (s, 1H), 9.52 (s, 1H), 8.70-8.61 (m, 1H), 8.20-8.11 (m, 2H), 8.03-7.94 (m, 5H), 7.88-7.81 (m, 4H), 7.63-7.55 (m, 3H), 7.51-7.44 (m, 3H), 6.01-5.99 (m, 2H), 5.34-5.28 (m, 4H), 4.89-4.76 (m, 3H). 4.60-4.47 (m, 3H), 4.40-4.32 (m, 2H), 4.22-4.19 (m, 1H), 3.60-3.48 (m, 42H), 3.10-3.06 (m, 6H), 2.88-2.86 (m, 9H), 2.20-2.17 (m, 2H), 1.99-1.95 (m, 1H), 1.30- 1.23 (m, 10H), 0.96 (d, 1=6.5 Hz,6H), 0.86-0.72 (m, 12H).
[0273] EXAMPLE 3
[0274] Synthesis of linker payload 3-((4-((29S,32S)-l-amino-29-isopropyl-32-methyl- 27,30-dioxo-3,6,9,12,15,18,21,24-octaoxa-28,31-diazatritriacontan-33- amido)benzyl)oxy)-N-((lS,4S,7R,HS,14S,17S,20R,24S,Z)-20-(3-hydroxyquinoline-2- carboxamido)-ll,24-diisopropyl-2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26- octaoxo-9,22-dioxa-2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7- yl)quinoline-2-carboxamide (36, M6409)
[0275] Linker payload 36 was synthesized from Compound 21 as described below.

[0276] Synthesis of O-(N-((S)-2-((S)-2-((5S,8S,12R)-5-allyl-l-(9H-fluoren-9-yl)-8- isopropyl-4,7-dimethyl-3,6,9-trioxo-12-(((2,2,2-trichloroethoxy)carbonyl)amino)-2,10- dioxa-4,7-diazatridecan-13-amido)-N-methylpropanamido)pent-4-enoyl)-N-methyl-L- valyl)-N-((benzyloxy)carbonyl)-D-seryl-L-alanine (30).
[0277] Step 1: Fmoc-Ala-Wang resin 21 (6379.6 mg, 4.0 mmol, 0.627 mmol/g loading, 100- 200 mesh) was treated with 20 mL 10% piperidine/DMF in a vessel and gently shaken at ambient temperature. After 15 min, the solution was filtered, and the resin was re-suspended in 20 mL 10% piperidine/DMF and gently shaken. After 5 min, the solution was filtered, and the resin was washed with DMF (3 x 10 mL), DCM (3 x 10 mL), and then DMF (1 x 10 mL). A solution of Fmoc-Ser(OTrt)OH (4557.2 mg, 8 mmol), HATU (3041.6 mg, 8 mmol), HOAt (1088.8 mg, 8 mmol), and NMM (0.88 mL, 8 mmol) in 10 mL DMF was added into the vessel and gently shaken overnight. The resin was filtered and then washed with DMF (3 x 10 mL) and DCM (3 x 10 mL). After the resin was dried, a few beads were taken and cleaved with 50% TFA/DCM for checking the complete conversion to product 22 (Fmoc-Ser- Ala-OH) in LCMS. MS (ESI, pos.) calc’d for C2iH22N2O6Na [421.14]+; found (M+Na) 421.26.
[0278] Step 2: Resin 22 was treated with 20 mL DBU/piperidine/NMP (2:5:93) in a vessel and gently shaken at ambient temperature. After 15 min, the solution was filtered, and the resin was re-suspended in 20 mL DBU/piperidine/NMP (2:5:93) and gently shaken. After 5 min, the solution was filtered, and the resin was washed with DMF (3 x 10 mL), DCM (3 x 10 mL), and then DMF (1 x 10 mL). A solution of Cbz-OSu (1993.8 mg, 8 mmol) and NMM (0.88 mL, 8 mmol) in 20 mL DMF was added into the vessel and gently shaken overnight. The resin was filtered and then washed with DMF (3 x 10 mL) and DCM (3 x 10 mL). After the resin was dried, a few beads were taken and cleaved with 50% TFA/DCM for checking the complete conversion to product 23 (Cbz-Ser-Ala-OH) in LCMS. MS (ESI, pos.) calc’d for Ci4Hi8N2O6Na [333.11]+; found (M+Na) 333.41.
[0279] Step 3; Resin 23 in a vessel was washed with TFA/TIS/DCM (2:2.5:95.5) solution until a colorless filtrate was observed. Then, the resin was washed DMF (3 x 10 mL) and DCM (3 x 10 mL). A solution of Fmoc-A-Me-Val-OH (2827.4 mg, 8 mmol), DIC (1009.6 mg, 8 mmol), and DMAP (969.4 mg, 8 mmol) in 15 mL DMF/DCM (1:4) was added into the vessel and gently shaken overnight. The solution was filtered, and the resin was washed with DMF (3 x 10 mL) and DCM (3 x 10 mL). After the resin was dried, a few beads were taken and cleaved with 50% TFA/DCM for checking the complete conversion to product 24 [Cbz- Ser(O-Fmoc-A-Me-Val)-Ala-OH] in LCMS. MS (ESI, pos.) calc’d for C^HwN AFNa [668.26]+; found (M+Na) 668.77.
[0280] Step 4: Resin 24 was treated with 20 mL 10% piperidine/DMF in a vessel and gently shaken at ambient temperature. After 15 min, the solution was filtered, and the resin was washed with DMF (3 x 10 mL), DCM (3 x 10 mL), and then DMF (1 x 10 mL). A solution of Fmoc- -mcthyl-CS')-2-allylglycinc (2530.1 mg, 7.2 mmol), DMTMM (2762.6 mg, 10 mmol), and NMM (1.1 mL, 10 mmol) in 20 mL DMF was added into the vessel and gently shaken overnight. The resin was filtered and then washed with DMF (3 x 10 mL) and DCM (3 x 10 mL). After the resin was dried, a few beads were taken and cleaved with 50% TFA/DCM for checking the complete conversion to product 25 [Cbz-Ser((?-A-Me-Val- Fmoc-A-Me-2-allylglycine)-Ala-OH] in LCMS. MS (ESI, pos.) calc’d for C41H49N4O10 [757.35]+; found (M+H) 757.91.
[0281] Step 5: Resin 25 was treated with 20 mL DBU/piperidine/NMP (2:5:93) in a vessel and gently shaken at ambient temperature. After 15 min, the solution was filtered, and the resin was washed with DMF (3 x 10 mL), DCM (3 x 10 mL), and then DMF (1 x 10 mL). A solution of Fmoc-Ala-OH (2490.7 mg, 8 mmol), DMTMM (2213.8 mg, 8 mmol), and NMM (0.88 mL, 8 mmol) in 20 mL DMF was added into the vessel and gently shaken overnight. The resin was filtered and then washed with DMF (3 x 10 mL) and DCM (3 x 10 mL). After the resin was dried, a few beads were taken and cleaved with 50% TFA/DCM for checking the complete conversion to product 26 [Cbz-Ser((?-A-Me-Val-A-Me-2-allylglycine-Fmoc- Ala)-Ala-OH] in LCMS. MS (ESI, pos.) calc’d for C44H53N50iiNa [850.36]+; found (M+Na) 851.00.
[0282] Step 6: Resin 26 was treated with 20 mL DBU/piperidine/NMP (2:5:93) in a vessel and gently shaken at ambient temperature. After 15 min, the solution was filtered, and the resin was washed with DMF (3 x 10 mL), DCM (3 x 10 mL), and then DMF (1 x 10 mL). A solution of Fmoc-D-Ser(Trt)-OH (4557.2 mg, 8 mmol), HATU (3041.6 mg, 8 mmol), HOAt (1088.8 mg, 8 mmol), and NMM (0.88 mL, 8 mmol) in 20 mL DMF was added into the vessel and gently shaken overnight. The resin was filtered and then washed with DMF (3 x 10 mL) and DCM (3 x 10 mL). After the resin was dried, a few beads were taken and cleaved with 50% TFA/DCM for checking the complete conversion to product 27 [Cbz-Ser((?-A-Me- Val-A-Me-2-allylglycine-Ala-Fmoc-Ser)-Ala-OH] in LCMS. MS (ESI, pos.) calc’d for C47H58N6Oi3Na [937.40]+; found (M+Na) 937.98.
[0283] Step 7: Resin 27 was treated with 20 mL DBU/piperidine/NMP (2:5:93) in a vessel and gently shaken at ambient temperature. After 15 min, the solution was filtered, and the
resin was washed with DMF (3 x 10 mL), DCM (3 x 10 mL), and then DMF (1 x 10 mL). A solution of Troc-Cl (1694.8 mg, 8 mmol) and NMM (0.88 mL, 8 mmol) in 20 mL DMF was added into the vessel and gently shaken overnight. The resin was filtered and then washed with DMF (3 x 10 mL) and DCM (3 x 10 mL). After the resin was dried, a few beads were taken and cleaved with 50% TFA/DCM for checking the complete conversion to product 28 [Cbz-Ser(O-A-Me-Val-A-Me-2-allylglycine-Ala-A-Troc-Ser)-Ala-OH] in LCMS. MS (ESI, pos.) calc’d for CssHwChNeOisNa [889.23]+; found (M+Na) 889.82.
[0284] Step 8: Resin 28 in a vessel was washed with TFA/TIS/DCM (2:2.5:95.5) solution until a colorless filtrate was observed. Then, the resin was washed DMF (3 x 10 mL) and DCM (3 x 10 mL). A solution of Fmoc-A-Me-Val-OH (2827.4 mg, 8 mmol), DIC (1009.6 mg, 8 mmol), and DMAP (969.4 mg, 8 mmol) in 15 mL DMF/DCM (1:4) was added into the vessel and gently shaken overnight. The solution was filtered, and the resin was washed with DMF (3 x 10 mL) and DCM (3 x 10 mL). After the resin was dried, a few beads were taken and cleaved with 50% TFA/DCM for checking the complete conversion to product 29 {Cbz- Ser[O-A-Me-Val-A-Me-2-allylglycine-Ala-A-Troc-Ser((?-Fmor-A-Me-Val)]-Ala-OH} in LCMS. MS (ESI, pos.) calc’d for CseHroChNrOieNa [1224.38]+; found (M+Na) 1225.28.
[0285] Step 9: Resin 29 was treated with 20 mL 10% piperidine/DMF in a vessel and gently shaken at ambient temperature. After 15 min, the solution was filtered, and the resin was washed with DMF (3 x 10 mL), DCM (3 x 10 mL), and then DMF (1 x 10 mL). A solution of Fmoc-A/-mcthyl-(.S')-2-allylglycinc (2530.1 mg, 7.2 mmol), DMTMM (2762.6 mg, 10 mmol), and NMM (1.1 mL, 10 mmol) in 20 mL DMF was added into the vessel and gently shaken overnight. The resin was filtered and then washed with DMF (3 x 10 mL) and DCM (3 x 10 mL). After the resin was dried, a few beads were taken and cleaved with 50% TFA/DCM for checking the complete conversion to product 30 in LCMS. The resin was then treated with 20 mL TFA/H2O/DCM (25:5:70) for 30 min. The resin was filtered, and the filtrate was collected. The resin was repeated with 20 mL TFA/H2O/DCM (25:5:70) treatment two more times. The combined filtrate was concentrated and then purified by reverse phase chromatography (Isco EZ Prep Gemini Cl 8 column). The product was eluted in 50-70% ACN/H2O (containing 0.1% of formic acid). The fractions were combined and lyophilized to afford the product as a yellow solid (841 mg, 0.625 mmol, 16% overall yield from step 1). MS (ESI, pos.) calc’d for C62H79C13NsOi7Na [ 1335.45]+; found (M+Na) 1336.53.
[0286] Synthesis of N-((benzyloxy)carbonyl)-O-(N-((3S,6S,llS,14S,17R,Z)-3-isopropyl- 4,12,14-trimethyl-6-(methylamino)-2,5,13,16-tetraoxo-17-(((2,2,2- trichloroethoxy)carbonyl)amino)-l-oxa-4,12,15-triazacyclooctadec-8-ene-ll-carbonyl)- N-methyl-L-valyl)-D-seryl-L-alanine (31): To a solution of 30 (841 mg, 0.625 mmol) in DCM (25 mL) was added Grubbs-Hovey da 2nd generation catalyst (80.3 mg, 0.128 mmol) at ambient temperature. The reaction was heated at 45 °C for 4 days. After the completion of ring-closing metathesis indicated by LCMS, the reaction was cooled to ambient temperature and concentrated in vacuo. The crude was treated with 5% piperidine DMF (3 mL) at ambient temperature for 15 min. The resulting solution was purified by reverse phase chromatography (Isco EZ Prep Gemini Cl 8 column). The product was eluted in 35-40% ACN/H2O (containing 0.1% of formic acid). The fractions were combined and lyophilized to afford product 31 as a yellow solid (175 mg, 0.164 mmol, 26% yield). MS (ESI, pos.) calc’d for C45H67CI3N8O15 [1063.37]+; found (M+H) 1064.16.
[0287] Synthesis of benzyl (2,2,2-trichloroethyl) ((1S,4S,7R,11S,14S,17S,2OR,24S,Z)- 1 L24-diisopropyl-2,4, 12, 15, 17, 25-hexaniethyl-3,6, 10,13,16, 19,23,26-octaoxo-9,22-dioxa-
2.5.12.15.18.25-hexaazabicycIo[12.12.4]triacont-28-ene-7,20-diyI)dicarbamate (32): To a solution of 31 (170 mg, 0.160 mmol) in DMF (3 mL) was added DMTMM (83.9 mg, 0.319 mmol) and NMM (0.035 mL, 0.319 mml) at ambient temperature. The reaction was stirred for 1 h. The crude solution was purified by reverse phase chromatography (Isco EZ Prep Gemini Cl 8 column). The product was eluted in 40-60% ACN/H2O (containing 0.1% of formic acid). The fractions were combined and lyophilized to afford product 32 as a yellow solid (78 mg, 0.0746 mmol, 47% yield). MS (ESI, pos.) calc’d for C45H64CI3N8O14 [1045.36]+; found (M+H) 1046.22.
[0288] Synthesis of benzyl ((lS,4S,7R,HS,14S,17S,20R,24S,Z)-20-amino-ll,24- diisopropyl-2,4,12,15,l'7,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa-
2.5.12.15.18.25-hexaazabicyclo[12.12.4]triacont-28-en-7-yl)carbamate (33): To a solution of 33 (24 mg, 0.0229 mmol) in THF (1 mL) was added Zn dust (10 mg, 0.229 mmol) and 1 M HC1 (1 mL) at ambient temperature and stirred for 16 h. The crude solution was purified by reverse phase chromatography (Isco EZ Prep Gemini Cl 8 column). The product was eluted in 30-40% ACN/H2O (containing 0.1% of formic acid). The fractions were combined and lyophilized to afford the product 33 as a yellow solid (8.0 mg, 0.0092 mmol, 40% yield). MS (ESI, pos.) calc’d for C42H63N8O12 [871.46]+; found (M+H) 872.13.
[0289] Synthesis of benzyl ((lS,4S,7R,HS,14S,17S,20R,24S,Z)-20-(3-hydroxyquinoline- 2-carboxamido)-ll,24-diisopropyl-2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26- octaoxo-9,22-dioxa-2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7- yl)carbamate (34): To a solution of 34 (8.0 mg, 0.0092 mmol) in DMF (0.5 mL) was added HATU (7.0 mg, 0.0184 mmol) and NMM (0.0020 mL, 0.0184 mmol) at ambient temperature. The reaction was stirred for 1 h. The crude solution was purified by reverse phase chromatography (Isco EZ Prep Gemini Cl 8 column). The product was eluted in 50-60% ACN/H2O (containing 0.1% of formic acid). The fractions were combined and lyophilized to afford product 34 as a yellow solid (4.7 mg, 0.0045 mmol, 49% yield). MS (ESI, pos.) calc’d for C52H68N9O14 [1042.49]+; found (M+H) 1043.25.
[0290] Synthesis of N-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-20-amino-ll,24-diisopropyl- 2,4,12,15,T7,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa-2,5,12,15,18,25- hexaazabicyclo[12.12.4]triacont-28-en-7-yl)-3-hydroxyquinoline-2-carboxamide (35): To a solution of 34 (4.7 mg, 0.0045 mmol) in TFA (0.5 mL) was added thioanisole (0.0053 mL, 0.045 mmol) at ambient temperature. The reaction was stirred at 40 °C for 16 h. The crude solution was purified by reverse phase chromatography (Isco EZ Prep Gemini Cl 8 column). The product was eluted in 30-40% ACN/H2O (containing 0.1% of formic acid). The fractions were combined and lyophilized to afford product 35 as a yellow solid (3.0 mg, 0.0033 mmol, 73% yield). MS (ESI, pos.) calc’d for C44H62N9O12 [908.45]+; found (M+H) 909.10.
[0291] Synthesis of 3-((4-((29S,32S)-l-amino-29-isopropyl-32-methyl-27,30-dioxo- 3,6,9,12,15,18,21,24-octaoxa-28,31-diazatritriacontan-33-amido)benzyl)oxy)-N- ((lS,4S,7R,llS,14S,17S,20R,24S,Z)-20-(3-hydroxyquinoline-2-carboxamido)-ll,24- diisopropyl-2,4,12,15,T7,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa- 2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7-yl)quinoline-2-carboxamide (36): To a solution of S in DMF (1 mL) was added linker 7 (3.9 mg, 0.0040 mmol), HATU (1.9 mg, 0.0050 mmol), and NMM (0.0005 mL, 0.0050 mmol). The reaction was stirred at ambient temperature for 1 h. The crude solution was purified by reverse phase chromatography (Isco EZ Prep Gemini Cl 8 column). The product was eluted in 50-60% ACN/H2O (containing 0.1% of formic acid). The fractions were combined and lyophilized to afford the Boc -protected intermediate (2.3 mg, 0.0012 mmol, 37%). MS (ESI, pos) calc’d for C93Hi32Ni4O27Na [1899.93]+; found (M+Na) 1901.18. The Boc-protected intermediate (2.3
mg, 0.0012 mmol) was dissolved in DCM (0.7 mL) and added TFA (0.3 mL) at ambient temperature. The reaction was stirred for 3 h. The crude solution was purified by reverse phase chromatography (Isco EZ Prep (Gemini C18 column). The product was eluted in 40- 50% ACN/H2O (containing 0.1% of formic acid). The fractions were combined and lyophilized to afford product 36 as a yellow solid (1.7 mg, 0.001 mmol, 78% yield). MS (ESI, neg.) calc’d for C88H123N14O25 [1775.88]“; found (M-H) 1776.95. ’H-NMR (500 MHz;
DMSO-d6): 5 10.10-10.09 (m, 1H), 8.73-8.70 (m, 1H), 8.23-8.22 (m, 1H), 7.95-7.88 (m, 2H), 7.86-7.82 (m, 2H), 7.73-7.71 (m, 1H), 7.64-7.61 (m, 2H), 7.60-7.57 (m, 1H), 7.53-7.46 (m, 2H), 7.45-7.41 (m, 2H), 7.32-7.26 (m, 2H), 7.24-7.18 (m, 2H), 7.17-7.13 (m, 2H), 6.04-5.97 (m, 2H), 5.32-5.25 (m, 4H), 4.84-4.70 (m, 3H), 4.60-4.55 (m, 1H), 4.48-4.34 (m, 4H), 4.22- 4.18 (m, 1H), 3.60-3.46 (m, 42H), 3.11-3.11 (m, 3H), 3.08-3.07 (m, 3H), 2.88-2.62 (m, 9H), 2.29-2.19 (m, 2H), 1.97-1.93 (m, 1H), 1.29-1.28 (m, 2H), 1.22 (s, 10H), 1.19 (dd, J = 6.0, 1.1 Hz, 2H), 0.96-0.93 (m, 4H), 0.85-0.84 (m, 3H), 0.82-0.80 (m, 3H), 0.77-0.74 (m, 4H).
[0292] EXAMPLE 4
[0293] Synthesis of N,N'-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-ll,24-diisopropyl- 2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa-2,5,12,15,18,25- hexaazabicyclo[12.12.4]triacont-28-ene-7,20-diyl)bis(3-hydroxyquinoline-2- carboxamide) (37, M6310)
[0294] Payload 37 was synthesized from compound 35 as described below.

[0295] The title compound was prepared by reacting 35 (8 mg, 0.009 mmol) with 3- hydroxy quinoline -2 -carboxylic acid (5.1 mg, 0.027 mmol) in the presence of HATU (10.3 mg, 0.027 mmol) and NMM (5.0 mL, 0.045 mmol) in DMF (1 mL) at ambient temperature under argon. Upon completion of the reaction, the reaction mixture was purified by reverse phase chromatography (Isco EZ Prep (Gemini C18 column, 30 x 150 mm, 5-35-100% of acetonitrile in water, each containing 0.05% of AcOH). The product eluted in 60% of acetonitrile. The fractions were combined and lyophilized to afford 37 (4.7 mg, 0.004 mmol,
49%). MS (ESI, pos.) calc’d for C54H67N10O14 [1079.5]+; found (M+H) 1079.9. ’H NMR (DMSO-d6, 500 MHz): 11.71 (s, 1H), 9.02-8.89 (m, 1H), 8.08 (d, 7=6.0 Hz, 1H, 7.85-7.82 (m, 2H), 7.56 (d, 7=8.5 Hz, 1H), 7.58-7.46 (m, 2H), 6.01 (t, 7=7.0 Hz, 1H), 5.38-5.35 (m, 1H), 4.90-4.81 (m, 3H), 4.64-4.58 (m, 3H), 4.46-4.38 (m, 2H), 3.16 (s, 3H), 2.88 (s, 3H), 2.74-2.70 (m, 2H), 2.20-2.14 (m, 1H), 1.27 (d, 7=7.5 Hz, 3H), 0.94 (d, 7=6.5 Hz, 3H).
[0296] EXAMPLE 5:
[0297] Synthesis of 3-amino-N-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-ll,24-diisopropyl-
2,4,12,15,17,25-hexainethyl-3,6,10, 13,16, 19, 23,26-octaoxo-20-(quinoxaline-2- carboxamido)-9,22-dioxa-2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7- yl)quinoxaline-2-carboxamide (38, M6170)
[0298] Payload 38 was synthesized from compound 19 as described below.

[0299] To a solution of compound 19 (3.0 mg, 0.0034 mmol) in DMF (1 mL) was added 3- aminoquinoxaline-2 -carboxylic acid (1.3 mg, 0.0067), HATU (2.6 mg, 0.0067 mmol), NMM (0.68 mg, 0.0067 mmol) at ambient temperature. The reaction was stirred for 1 h. The crude solution was purified by reverse phase chromatography (Isco EZ Prep Gemini C18 column). The product was eluted in 45-55% ACN/H2O (containing 0.1% of formic acid). The fractions were combined and lyophilized to afford the product 38 as a yellow solid (2.2 mg, 0.0021 mmol, 62% yield). MS (ESI, pos.) calc’d for C52H66N13O12 [1064.50]+; found (M+H) 1065.14. ’H-NMR (500 MHz; DMSO-d6): <5 9.51 (s, 1H), 8.69-8.58 (m, 2H), 8.14 (d, 7 = 7.7 Hz, 1H), 8.04-8.00 (m, 3H), 7.92-7.85 (m, 3H), 7.77-7.71 (m, 2H), 7.54-7.50 (m, 2H), 7.31- 7.28 (m, 1H), 6.00-5.97 (m, 2H), 5.37-5.30 (m, 2H), 4.90-4.81 (m, 4H), 4.61-4.54 (m, 4H), 4.40 (d, 7 = 10.7 Hz, 2H), 3.12 (s, 6H), 2.87 (s, 6H), 2.75-2.63 (m, 2H), 2.43-2.11 (m, 4H), 1.30-1.15 (m, 6H), 0.94-0.89 (m, 6H), 0.83-0.71 (m, 6H).
[0300] EXAMPLE 6:
[0301] Synthesis of N-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-20-(3-hydroxyquinoline-2- carboxamido)-ll,24-diisopropyl-2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26- octaoxo-9,22-dioxa-2,5,12,15,18,25-hexaazabicyc!o[12.12.4]triacont-28-en-7- yl)quinoxaline-2-carboxamide (39, M6169)
[0302] Payload 39 was synthesized from compound 19 as described below.

[0303] The title compound (39) was synthesized from compound 19 (3.0 mg, 0.0034 mmol) by reacting it with 3 -hydroxy quinoline-2-carboxylic acid (1.3 mg, 0.0067), HATU (2.6 mg, 0.0067 mmol), NMM (0.68 mg, 0.0067 mmol) in DMF (1 mL) as described in the synthesis of compound 38. Product 39 was obtained as a yellow solid (2.9 mg, 0.0027 mmol, 81% yield). MS (ESI, pos.): calc’d for C53H66N11O13 [1064.49]+; found (M+H) 1065.06. ’H-NMR (500 MHz; DMSO-d6): 3 11.95-11.60 (m, 1H), 9.55-9.48 (m, 1H), 8.60-8.56 (m, 1H), 8.12- 8.11 (m, 1H), 8.09-7.83 (m, 6H), 7.81-7.60 (m, 3H), 7.53-7.33 (m, 2H), 6.05-5.95 (m, 2H), 5.31 (s, 2H), 4.90-4.78 (m, 4H), 4.65-4.54 (m, 4H), 4.44-4.37 (m, 2H), 3.11-3.09 (m, 6H), 2.84 (s, 6H), 2.78-2.65 (m, 2H), 2.38-2.23 (m, 2H), 2.21-2.08 (m, 2H), 1.28 (s, 6H), 0.90- 0.88 (m, 6H), 0.73 (s, 6H).
[0304] EXAMPLE 7:
[0305] Synthesis of (N-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-20-(3-aminoquinoline-2- carboxamido)-ll,24-diisopropyl-2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26- octaoxo-9,22-dioxa-2,5,12,15,18,25-hexaazabicyc!o[12.12.4]triacont-28-en-7- yl)quinoxaline-2-carboxamide (40, M6253)
[0306] Payload 40 was synthesized from compound 19 as described below.

[0307] The title compound was prepared by reacting 19 (10 mg, 0.011 mmol) with 3- aminoquinoxaline-2 -carboxylic acid (4.2 mg, 0.022 mmol) in the presence of HATU (8.5 mg, 0.022 mmol) and NMM (3.7 pL, 0.034 mmol) in DMF (1 mL) at ambient temperature under argon. Reverse phase chromatography (Isco EZ Prep Gemini Cl 8 column, 30 x 150 mm, 5- 35-100% of acetonitrile in water, each containing 0.05% of AcOH) followed by lyophilization afforded 40 as an off white solid (6.0 mg, 0.006 mmol, 50%). MS (ESI, pos.) calc’d for C53H67N12O12 [ 1063.6]+; found (M+H) 1063.9. ’H NMR (DMSO-d6, 300 MHz): 9.52 (s, 1H), 8.85 (d, 7=9.3 Hz, 1H), 8.60 (d, 7=9.3 Hz, 1H), 8.17-8.14 (m, 1H), 8.04 (t, 7=6.0 Hz, 2H), 7.96-7.88 (m, 4H), 7.61-7.58 (m, 1H), 7.54-7.51 (m, 1H), 7.44 (s, 1H), 7.30- 7.25 (m, 2H), 6.82 (s, 2H), 6.03-5.98 (m, 2H), 5.38-5.31 (m, 2H), 4.91-4.79 (m, 5H), 4.63- 4.51 (m, 5H), 4.43 (m, 2H), 3.21 (s, 3H), 3.19 (s, 3H), 2.88 (s, 6H), 2.27-2.10 (m, 2H), 1.27- 1.23 (m, 6H), 0.97-0.91 (m, 6H), 0.85-0.77 (m, 6H).
[0308] EXAMPLE 8:
[0309] Synthesis of 3-chloro-N-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-ll,24-diisopropyl-
2,4,12,15,17,25-hexamethyI-3,6,10,13,16,19,23,26-octaoxo-20-(quinoxaIine-2- carboxamido)-9,22-dioxa-2,5,12,15,18,25-hexaazabicydo[12.12.4]triacont-28-en-7- yl)quinoxaline-2-carboxamide (41, M6252)
[0310] Payload 41 was synthesized from compound 19 as described below.

[0311] The title compound was prepared by reacting 19 (10 mg, 0.011 mmol) with 3- chloroquinoxaline-2 -carboxylic acid (4.7 mg, 0.022 mmol) in the presence of HATU (8.5 mg, 0.022 mmol) and NMM (3.7 pL, 0.034 mmol) in DMF (1 mL) at ambient temperature under
Il l
argon. Reverse phase chromatography (Isco EZ Prep Gemini Cl 8 column, 30 x 150 mm, 5- 35-100% of acetonitrile in water, each containing 0.05% of AcOH) followed by lyophilization afforded 41 as an off white solid (11 mg, 0.010 mmol, 90%). MS (ESI, pos.) calc’d for C52H64N12O12 [1083.4]+; found (M+H) 1083.9. ’H NMR (DMSO-d6, 300 MHz): 9.52 (s, 1H), 8.91 (d, 7=8.7 Hz, 1H), 8.60 (d, 7=9.3 Hz, 1H), 8.21-8.18 (m, 1H), 8.09-8.01
(m, 4H), 7.98-7.87 (m, 6H), 7.73 (d, 7=5.7 Hz, 1H), 6.02-5.97 (m, 2H), 5.38-5.32 (m, 2H), 4.89-4.79 (m, 5H), 4.62-4.51 (m, 5H), 4.40-4.34 (m, 2H), 3.11 (s, 3H), 3.00 (s, 3H), 2.89 (s, 3H), 2.85 (s, 3H), 2.19-2.12 (m, 2H), 1.30 (d, 7=6.9 Hz, 3H), 1.22 (d, 7=6.9 Hz, 3H), 0.94 (t, 7=6.3 Hz, 6H), 0.84-0.74 (m, 6H). [0312] EXAMPLE 9:
[0313] Synthesis of N,N'-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-ll,24-di((R)-sec-butyl)- 2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa-2,5,12,15,18,25- hexaazabicyclo[12.12.4]triacont-28-ene-7,20-diyl)bis(quinoxaline-2-carboxamide) (49, M6181)
[0314] Payload 49 was synthesized from compound 21 as described below.

[0315] Synthesis of N-((benzyloxy)carbonyl)-O-(N-((3R,6S,9S,14S,17R,Z)-17-((R)-sec- butyl)-6,8,16-trimethyl-14-(methylamino)-4,7,15,18-tetraoxo-3-(quinoxaline-2- carboxamido)-l-oxa-5,8-diazacyclooctadec-ll-ene-9-carbonyl)-N-methyl-L- alloisoleucyl) -D -seryl-L-alanine (46)
[0316] Step 1: The compound 42 was synthesized from Fmoc-Ala-Wang resin 21 (1988.1 mg, 1.0 mmol, 0.503 mmol/g loading, 100-200 mesh) as described in the synthesis of compound 30, step 1 to step 6, while replacing the reactant Fmoc-A-Me-Val-OH in the step 3 with Fmoc-A-Me-Ilu-OH. A few beads were taken and cleaved with 50% TFA/DCM for checking the complete conversion to product 42 [Cbz-Ser((?-A-Me-Ilu-A-Me-2-allylglycine- Ala-Fmoc-Ser)-Ala-OH]. MS (ESI, pos.) calc’d for C48H61N6O13 [929.43]+; found (M+H) 929.84.
[0317] Step 2: Resin 42 was treated with 10 mL DBU/piperidine/NMP (2:5:93) in a vessel and gently shaken at ambient temperature. After 15 min, the solution was filtered, and the resin was washed with DMF (3 x 10 mL), DCM (3 x 10 mL), and then DMF (1 x 10 mL). A solution of quinoxaline -2 -carboxylic acid (348.4 mg, 2 mmol), HATU (760.4 mg, 2 mmol), and NMM (0.22 mL, 2 mmol) in 10 mL DMF was added into the vessel and gently shaken overnight. The resin was filtered and then washed with DMF (3 x 10 mL) and DCM (3 x 10 mL). After the resin was dried, a few beads were taken and cleaved with 50% TFA/DCM for checking the complete conversion to product 43 [Cbz-Ser((?-A-Me-Val-A-Me-2-allylglycine- Ala-A-2-carboxyquinoxaline-Ser)-Ala-OH] in LCMS. MS (ESI, pos.) calc’d for C42H55N8O12 [863.40]+; found (M+H) 863.74.
[0318] Step 3: Resin 44 was synthesized from resin 43 via the same method as described in the synthesis of compound 30, step 8, while using Fmoc-A-Me-Ilu-OH instead of using Fmoc-A-Me-Val-OH. After the resin was dried, a few beads were taken and cleaved with 50% TFA/DCM for checking the complete conversion to product 44 {Cbz-Ser[(?-A-Me-Val- A-Me-2-allylglycine-Ala-A-2-carboxyquinoxaline-Ser((?-Fmoc-A-Me-Ilu)]-Ala-OH} in LCMS. MS (ESI, pos.) calc’d for C64H78N9O15 [1212.56]+; found (M+H) 1213.15.
[0319] Step 4: Resin 44 was treated with 10 mL 10% piperidine/DMF in a vessel and gently shaken at ambient temperature. After 15 min, the solution was filtered, and the resin was washed with DMF (3 x 10 mL), DCM (3 x 10 mL), and then DMF (1 x 10 mL). A solution of Boc-A-methyl-(S')-2-allylglycine (412.7 mg, 1.8 mmol), DMTMM (553.4 mg, 2 mmol), and NMM (0.22 mL, 2 mmol) in 20 mL DMF was added into the vessel and gently shaken overnight. The resin was filtered and then washed with DMF (3 x 10 mL) and DCM (3 x 10 mL). After the resin was dried, a few beads were taken and cleaved with 50% TFA/DCM for checking the complete conversion to product 45 {Cbz-Ser[(?-A-Me-Val-A-Me-2-allylglycine- Ala-A-2-carboxyquinoxaline-Ser((?-A-Me-Ilu-Boc-A-Me-2-allylglycine)]-Ala-OH} in LCMS. MS (ESI, pos.) calc’d for C55H77N10O14 [1101.56]+; found (M+H) 1102.09.
[0320] Step 5: Resin 45 was placed in a seal tube and suspended in DCM (100 mL). Grubbs- Hoveyda 2nd generation catalyst (125 mg, 0.2 mmol) was added at ambient temperature and purged with argon before the tube was sealed. The reaction was heated at 40 °C while slowly stirring for 9 days. After the completion of ring-closing metathesis indicated by LCMS, the resin was then treated with 30 mL TFA/H2O/DCM (25:5:70) for 30 min. The resin was filtered, and the filtrate was collected. The resin was repeated with 30 mL TFA/H2O/DCM
(25:5:70) treatment two more times. The combined filtrate was concentrated and then purified by reverse phase chromatography (Isco EZ Prep Gemini Cl 8 column). The product was eluted in 30-40% ACN/H2O (containing 0.1% of formic acid). The fractions were combined and lyophilized to afford product 46 as a yellow solid (84 mg, 0.0783 mmol, 8% overall yield). MS (ESI, pos.) calc’d for C53H73N10O14 [1073.53]+; found (M+H) 1073.99.
[0321] Synthesis of benzyl ((lS,4S,7R,HS,14S,17S,20R,24S,Z)-ll,24-di((R)-sec-butyl)-
2.4.12.15,T7,25-hexamethyI-3,6,10,13,16,19,23,26-octaoxo-20-(quinoxaIine-2- carboxamido)-9,22-dioxa-2,5,12,15,18,25-hexaazabicycIo[12.12.4]triacont-28-en-7- yl)carbamate (47): To a solution of 46 (84 mg, 0.0783 mmol) in DMF (5 mL) was added DMTMM (43 mg, 0.157 mmol) and NMM (16 mg, 0.157 mmol) at ambient temperature and stirred for 3 h. The crude solution was purified reverse phase chromatography (Isco EZ Prep Gemini Cl 8 column). The product was eluted in 50-60% ACN/H2O (containing 0.1% of formic acid). The fractions were combined and lyophilized to afford product 47 as a yellow solid (46 mg, 0.0429 mmol, 55% yield) MS (ESI, pos.) calc’d for C53H71N10O13 [1055.52]+; found (M+H) 1055.87.
[0322] Synthesis of N-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-20-amino-ll,24-di((R)-sec- butyl)-2,4,12,15,l'7,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa- 2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7-yl)quinoxaline-2-carboxamide (48): To a solution of 47 (46 mg, 0.0429 mmol) in TFA (2.0 mL) was added thioanisole (0.10 mL, 0.885 mmol) at ambient temperature. The reaction was heated to 40 °C and stirred for 16 h. The crude solution was concentrated and then purified by reverse phase chromatography (Isco EZ Prep Gemini Cl 8 column). The product was eluted in 35-40% ACN/H2O (containing 0.1% of formic acid). The fractions were combined and lyophilized to afford product 48 as a yellow solid (16.5 mg, 0.0179 mmol, 40% yield). MS (ESI, pos.) calc’d for C45H65N10O11 [921 ,49]+; found (M+H) 922.09.
[0323] Synthesis of N,N'-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-ll,24-di((R)-sec-butyl)-
2.4.12.15,T7,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa-2,5,12,15,18,25- hexaazabicyclo[12.12.4]triacont-28-ene-7,20-diyl)bis(quinoxaline-2-carboxamide) (49): To a solution of 48 (3.5 mg, 0.0038 mmol) in DMF (1 mL) was added quinoxaline-2- carboxylic acid (1.6 mg, 0.0076 mmol), HATU (2.9 mg, 0.0076 mmol), and NMM (0.77 mg, 0.0076 mmol) at ambient temperature and stirred for 1 h. The crude solution was purified by reverse phase chromatography (Isco EZ Prep Gemini Cl 8 column). The product was eluted
in 50-65% ACN/H2O (containing 0.1% of formic acid). The fractions were combined and lyophilized to afford product 49 as a yellow solid (2.0 mg, 0.0018 mmol, 47% yield). MS (ESI, pos) calc’d for C54H69N12O12 [1077.52]+; found (M+H) 1077.97. ’H-NMR (500 MHz; DMSO-de): 5 9.50 (s, 2H), 8.52 (d, J = 9.3 Hz, 2H), 8.14 (d, J = 8.2 Hz, 2H), 8.03 (d, J = 6.1 Hz, 2H), 7.87-7.82 (m, 6H), 5.99 (t, J = 7.3 Hz, 2H), 5.34 (s, 2H), 4.92 (d, J = 9.9 Hz, 4H), 4.61 (q, J = 6.8 Hz, 2H), 4.53 (dd, J = 11.0, 5.3 Hz, 2H), 4.44 (d, J = 10.4 Hz, 2H), 3.15 (s, 6H), 2.88 (s, 6H), 2.77-2.67 (m, 2H), 2.35-2.29 (m, 2H), 1.97-1.94 (m, 2H), 1.26 (d, J = 7.1 Hz, 8H), 1.02 (dt, J = 13.9, 7.2 Hz, 2H), 0.89 (d, J = 6.5 Hz, 6H), 0.76 (t, J = 7.3 Hz, 6H).
[0324] EXAMPLE 10:
[0325] Synthesis of 3-amino-N-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-ll,24-di((R)-sec- butyl)-2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-20-(quinoxaline-2- carboxamido)-9,22-dioxa-2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7- yl)quinoxaline-2-carboxamide (50, M6410)
[0326] Payload 50 was synthesized from compound 48 as described below.

[0327] Compound 50 was synthesized from compound 48 (2.0 mg, 0.0022 mmol) by reacting it with 3-aminoquinoxaline-2 -carboxylic acid (0.53 mg, 0.0028), HATU (1.2 mg, 0.0033 mmol), NMM (0.33 mg, 0.0033 mmol) in DMF (1 mL) as described in the synthesis of compound 38. The crude solution was purified by reverse phase chromatography (Isco EZ Prep Gemini C18 column). The product was eluted in 45-55% ACN/H2O (containing 0.1% of formic acid). The fractions were combined and lyophilized to afford the product 38 as a yellow solid (1.7 mg, 0.0016 mmol, 72% yield). MS (ESI, pos) calc’d for C54H70N13O12 [1092.53]+; found (M+H) 1093.35. ’H-NMR (300 MHz; DMSO-d6): <5 9.53 (s, 1H), 8.71-8.65 (m, 1H), 8.62-8.57 (m, 1H), 8.53 (d, 7 = 5.5 Hz, 2H), 8.20-8.16 (m, 1H), 8.11-8.06 (m, 2H), 7.92 (d, 7 = 2.1 Hz, 2H), 7.80-7.76 (m, 1H), 7.55 (d, 7 = 5.8 Hz, 2H), 7.33-7.28 (m, 1H), 7.26-7.20 (m, 1H), 6.05-5.96 (m, 2H), 5.33 (dtd, 7 = 4.7, 2.8, 1.0 Hz, 2H), 4.96-4.85 (m, 4H),
4.65-4.61 (m, 2H), 4.55-4.43 (m, 4H), 3.17 (d. 7 = 7.0 Hz, 6H), 2.89 (d, 7 = 7.6 Hz, 6H), 2.76-2.69 (m, 2H), 2.36-2.28 (m, 2H), 2.06-1.93 (m, 2H), 1.28 (dd, 7 = 7.0, 2.9 Hz, 8H), 1.11-1.00 (m, 2H), 0.91 (dt, 7 = 6.5, 3.3 Hz, 6H), 0.79 (td, 7 = 7.1, 2.5 Hz, 6H).
[0328] EXAMPLE 11:
[0329] Synthesis of N-((lS,4S,7R,llS,14S,17S,20R,24S,E)-ll,24-di((R)-sec-butyl)-20-(3- hydroxyquinoline-2-carboxamido)-2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26- octaoxo-9,22-dioxa-2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7- yl)quinoxaline-2-carboxamide (51, M6180)
[0330] Payload 51 was synthesized from compound 48 as described below.

[0331] Compound 51 was synthesized from compound 48 (3.5 mg, 0.0038 mmol) by reacting it with 3-hydroxyquinoline-2-carboxylic acid (1.4 mg, 0.0076), HATU (2.9 mg, 0.0076 mmol), NMM (0.77 mg, 0.0076 mmol) in DMF (1 mb) as described in the synthesis of compound 39. Product 39 was obtained as a yellow solid. (2.0 mg, 0.0018 mmol, 48% yield). MS (ESI, pos) calc’d for C55H70N11O13 [1092.52]+; found (M+H) 1092.98. ’H-NMR (500 MHz; DMSO-d6): 3 11.68 (s, 1H), 9.52 (s, 1H), 8.90 (d, 7 = 9.2 Hz, 1H), 8.56-8.54 (m, 1H), 8.15 (dd, 7 = 8.7, 5.6 Hz, 1H), 8.08 (d, 7 = 6.2 Hz, 1H), 8.03 (d, 7 = 6.3 Hz, 1H), 7.96-7.82 (m, 5H), 7.71 (d, 7 = 8.0 Hz, 1H), 7.52-7.47 (m, 2H), 6.01-5.96 (m, 2H), 5.33 (s, 2H), 4.93- 4.88 (m, 4H), 4.62-4.58 (m, 2H), 4.54-4.43 (m, 4H), 3.16 (d, 7 = 4.4 Hz, 6H), 2.88 (d, 7 = 2.8 Hz, 6H), 2.75-2.62 (m, 2H), 2.35-2.26 (m, 2H), 2.01-1.93 (m, 2H), 1.26 (dt, 7 = 16.0, 8.3 Hz, 8H), 1.08-0.98 (m, 2H), 0.88 (dd, 7 = 11.8, 6.6 Hz, 6H), 0.76 (dt, 7 = 10.9, 7.4 Hz, 6H).
[0332] EXAMPLE 12:
[0333] Synthesis of N,N’-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-ll,24-dicyclopentyl- 2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa-2,5,12,15,18,25-
hexaazabicyclo[12.12.4]triacont-28-ene-7,20-diyl)bis(quinoxaline-2-carboxamide) (53,
M6266)
[0334] Payload 53 was synthesized from compound 52 as described below.

[0335] Synthesis of 52: Compound 52 was synthesized from resin 21 (2.98 g, 1.5 mmol, 0.503 mmol/g loading, 100-200 mesh) as described in the synthesis of compound 48 (Example 9), while replacing the reactant Fmoc-A-Me-Ilu-OH with Fmoc-A/-Mc-2- cyclopentylglycine. The product 52 was isolated as a yellow solid (64 mg, 0.0679 mmol, 4.5% overall yield). MS (ESI, pos.) calc’d for C47H65N10O11 [945.49]+; found (M+H) 945.72.
[0336] Synthesis of 53: Compound 53 was synthesized from compound 52 (3.0 mg, 0.0032 mmol) by reacting it with quinoxaline-2-carboxylic acid (1.1 mg, 0.0063), HATU (2.4 mg, 0.0063 mmol), and NMM (0.64 mg, 0.0063 mmol) in DMF (0.5 mL) as described in the synthesis of compound 49. The product 53 was isolated as a yellow solid (1.8 mg, 0.0016 mmol, 51% yield). MS (ESI, pos.) calc’d for C56H69N12O12 [1101.52]+; found (M+H) 1101.99. ’H-NMR (500 MHz; DMSO-d6): <5 9.51 (s, 2H), 8.59-8.56 (m, 2H), 8.13-8.11 (m, 2H), 8.02 (s, 2H), 7.86 (dt, 7 = 14.6, 7.0 Hz, 6H), 6.02-6.00 (m, 2H), 5.35-5.33 (m, 2H), 4.92- 4.87 (m, 4H), 4.62-4.60 (m, 4H), 4.35 (d, 7 = 10.7 Hz, 2H), 3.13 (s, 6H), 2.86 (s, 6H), 2.77- 2.67 (m, 2H), 2.35-2.26 (m, 4H), 1.72-1.68 (m, 2H), 1.54-1.48 (m, 6H), 1.43-1.30 (m, 8H), 1.25 (d, J = 11.0 Hz, 3H), 1.24 (d, J = 11.0 Hz, 3H).
[0337] EXAMPLE 13:
[0338] Synthesis of 3-amino-N-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-ll,24-dicyclopentyl- 2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-20-(quinoxaline-2- carboxamido)-9,22-dioxa-2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7- yl)quinoxaline-2-carboxamide (54, M6270)
[0339] Payload 54 was synthesized from compound 52 as described below.

[0340] Compound 54 was synthesized from compound 52 (3.0 mg, 0.0032 mmol) by reacting it with 3-aminoquinoxaline-2 -carboxylic acid (1.2 mg, 0.0063), HATU (2.4 mg, 0.0063 mmol), and NMM (0.64 mg, 0.0063 mmol) in DMF (0.5 mb) as described in the synthesis of compound 50. Product 54 was isolated as a yellow solid. (2.2 mg, 0.0020 mmol, 62% yield). MS (ESI, pos.) calc’d for C56H70N13O12 [1116.53]+; found (M+H) 1116.87. ’H-NMR (500 MHz; DMSO-d6): <59.52 (d, J = 6.6 Hz, 1H), 8.69-8.65 (m, 1H), 8.61-8.57 (m, 1H), 8.51-8.49 (m, 2H), 8.17-8.12 (m, 1H), 8.03-8.00 (m, 2H), 7.94-7.91 (m, 2H), 7.55-7.52 (m, 1H), 7.49 (s, 1H), 7.31-7.27 (m, 1H), 6.92-6.88 (m, 2H), 6.06-5.99 (m, 2H), 5.38-5.30 (m, 2H), 4.93- 4.79 (m, 4H), 4.61-4.55 (m, 4H), 4.37-4.33 (m, 2H), 3.14 (s, 6H), 2.90-2.83 (m, 6H), 2.72- 2.62 (m, 2H), 2.43-2.25 (m, 4H), 1.76-1.67 (m, 2H), 1.58-1.49 (m, 6H), 1.43-1.31 (m, 8H), 1.26 (d, 7 = 5.8 Hz, 3H), 1.25 (d, 7 = 5.8 Hz, 3H).
[0341] EXAMPLE 14:
[0342] Synthesis of N-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-ll,24-dicyclopentyl-20-(3- hydroxyquinoIine-2-carboxamido)-2,4,12,15,17,25-hexamethyI-3,6,10,13,16,19,23,26- octaoxo-9,22-dioxa-2,5,12,15,18,25-hexaazabicydo[12.12.4]triacont-28-en-7- yl)quinoxaline-2-carboxamide (55, M6264)
[0343] Payload 55 was synthesized from compound 52 as described below.
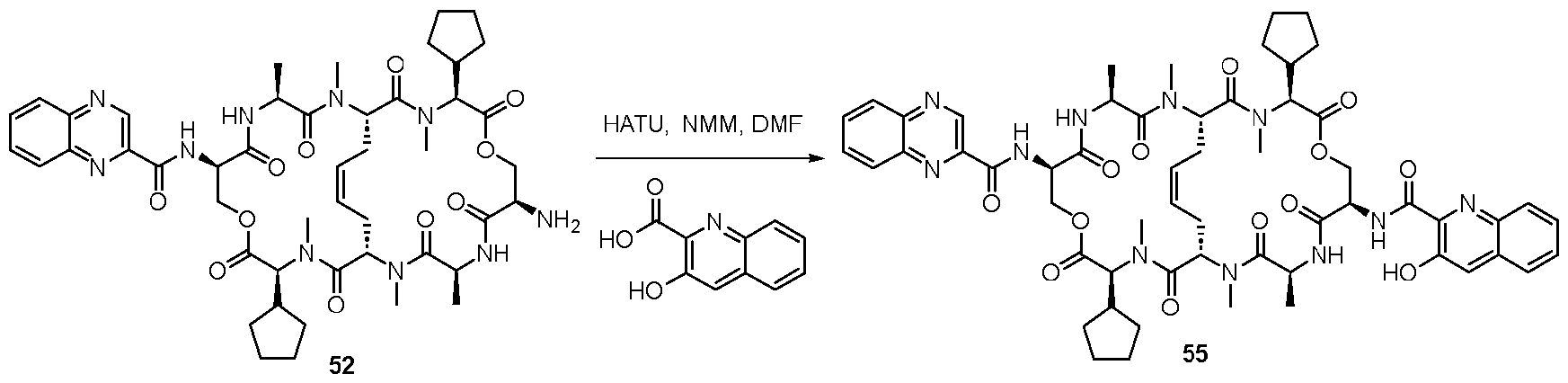
[0344] Compound 55 was synthesized from compound 52 (30 mg, 0.0317 mmol) by reacting it with 3 -hydroxy quinoline-2-carboxylic acid (7.2 mg, 0.0381), HATU (18 mg, 0.0476 mmol), and NMM (4.8 mg, 0.0476 mmol) in DMF (1 mL) as described in the synthesis of
compound 50. Product 54 was obtained as a yellow solid. (24 mg, 0.0215 mmol, 68% yield). MS (ESI, pos.) calc’d for C57H70N11O13 [1116.52]+; found (M+H) 1116.92. 'H-NMR (500 MHz; DMSO-d6): <5 11.70 (s, 1H), 9.54-9.51 (m, 1H), 8.92 (d, J = 9.0 Hz, 1H), 8.59-8.57 (m, 1H), 8.13 (t, J = 7.9 Hz, 1H), 8.10-8.05 (m, 1H), 8.02 (d, 7 = 2.7 Hz, 1H), 7.94-7.89 (m, 2H), 7.87-7.85 (m, 2H), 7.81 (d, 7 = 8.1 Hz, 1H), 7.70 (d, 7 = 8.9 Hz, 1H), 7.51-7.42 (m, 2H), 6.06-6.00 (m, 2H), 5.35 (s, 2H), 4.90 (td, 7 = 10.8, 4.9 Hz, 4H), 4.62-4.57 (m, 4H), 4.40-4.35 (m, 2H), 3.14 (s, 6H), 2.86 (d, 7 = 4.4 Hz, 6H) 2.72-2.66 (m, 2H), 2.37-2.24 (m, 4H), 1.75- 1.66 (m, 2H), 1.56-1.48 (m, 6H), 1.44-1.30 (m, 8H), 1.26 (d. 7 = 7.9 Hz, 3H) ), 1.25 (d, 7 = 7.9 Hz, 3H).
[0345] EXAMPLE 15:
[0346] Synthesis of N,N'-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-ll,24-dicyclohexyl- 2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa-2,5,12,15,18,25- hexaazabicycIo[12.12.4]triacont-28-ene-7,20-diyI)bis(quinoxaIine-2-carboxamide) (57, M6250)
[0347] Payload 57 was synthesized from compound 56 as described below.

[0348] Synthesis of 56. was synthesized from resin 21 (2.98 g, 1.5 mmol, 0.503 mmol/g loading, 100-200 mesh) as described in the synthesis of compound 48 (Example 9), while replacing the reactant Fmoc-A-Me-Ilu-OH with Fmoc-A-Me-2-cyclohexylglycine. The product 56 was isolated as a yellow solid (66 mg, 0.0678 mmol, 4.5% overall yield). MS (ESI, pos.) calc’d for C49H69N10O11 [973.52]+; found (M+H) 973.79.
[0349] Synthesis of 57: Compound 57 was synthesized from compound 56 (3.0 mg, 0.0031 mmol) by reacting it with quinoxaline-2-carboxylic acid (0.8 mg, 0.0046), HATU (1.8 mg, 0.0046 mmol), and NMM (0.47 mg, 0.0046 mmol) in DMF (0.5 mL) as described in the synthesis of compound 53. The product 57 was isolated as a yellow solid (2.2 mg, 0.0019 mmol, 63% yield). MS (ESI, pos.) calc’d for C58H73N12O12 [1129.55]+; found (M+H)
1130.15. 1 H-NMR (500 MHz; DMSO-d6): 3 9.52 (s, 2H), 8.59 (d, J = 8.9 Hz, 2H), 8.14 (s, 2H), 8.03 (d, J = 6.0 Hz, 2H), 7.93 (s, 2H), 7.87 (t, J = 5.9 Hz, 4H), 5.97 (dd, J = 5.8, 0.7 Hz, 2H), 5.33 (d, J = 3.6 Hz, 2H), 4.92 (t, J = 9.4 Hz, 4H), 4.59 (s, 4H), 4.40 (d, J = 10.5 Hz, 2H), 3.13 (s, 6H), 2.87 (s, 6H), 2.72-2.66 (m, 2H), 2.63-2.57 (m, 2H), 2.43-2.28 (m, 2H), 1.85- 1.81 (m, 2H), 1.69-1.66 (m, 2H), 1.60-1.59 (m, 2H), 1.53 (d, 7 = 0.8 Hz, 2H), 1.53-1.52 (m, 2H), 1.42-1.37 (m, 2H), 1.26 (d, 7 = 7.0 Hz, 3H), 1.25 (d, 7 = 7.0 Hz, 3H), 1.15-1.04 (m, 4H), 0.99-0.93 (m, 4H).
[0350] EXAMPLE 16:
[0351] Synthesis of 3-amino-N-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-ll,24-dicyclohexyl- 2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-20-(quinoxaline-2- carboxamido)-9,22-dioxa-2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7- yl)quinoxaline-2-carboxamide (58, M6251)
[0352] Payload 58 was synthesized from compound 56 as described below.

[0353] Compound 58 was synthesized from compound 56 (3.0 mg, 0.0031 mmol) by reacting it with 3-aminoquinoxaline-2 -carboxylic acid (0.95 mg, 0.0046), (1.8 mg, 0.0046 mmol), and NMM (0.47 mg, 0.0046 mmol) in DMF (0.5 mb) as described in the synthesis of compound 50. Product 58 was isolated as a yellow solid. (2.5 mg, 0.0022 mmol, 71% yield). MS (ESI, pos.) calc’d for C58H74N13O12 [1144.56]+; found (M+H) 1145.04. ’H-NMR (500 MHz;
DMSO-d6): 3 9.52 (s, 1H), 8.68-8.66 (m, 1H), 8.57-8.54 (m, 1H), 8. 15 (t, J = 7.0 Hz, 1H), 8.03-8.00 (m, 2H), 7.96-7.87 (m, 4H), 7.78-7.71 (m, 1H), 7.58 (t. 7 = 7.7 Hz, 1H), 7.54-7.50 (m, 2H), 7.32-7.29 (m, 1H), 6.00-5.95 (m, 2H), 5.31 (dd, 7 = 11.8, 2.2 Hz, 2H), 4.94-4.82 (m, 4H), 4.61-4.48 (m, 4H), 4.41 (t, 7 = 9.2 Hz, 2H), 3.15 (s, 6H), 2.87 (d, 7 = 2.2 Hz, 6H), 2.74- 2.62 (m, 2H), 2.46-2.26 (m, 4H), 1.89-1.80 (m, 2H), 1.70-1.66 (m, 2H), 1.61-1.58 (m, 2H), 1.54-1.51 (m, 2H), 1.48-1.47 (m, 2H), 1.42-1.40 (m, 2H), 1.26 (d, 7 = 5.6 Hz, 3H), 1.25 (d, 7 = 5.6 Hz, 3H), 1.13-1.06 (m, 4H), 1.00-0.92 (m, 4H).
[0354] EXAMPLE 17:
[0355] Synthesis of N-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-ll,24-dicyclohexyl-20-(3- hydroxyquinoIine-2-carboxamido)-2,4,12,15,17,25-hexamethyI-3,6,10,13,16,19,23,26- octaoxo-9,22-dioxa-2,5,12,15,18,25-hexaazabicydo[12.12.4]triacont-28-en-7- yl)quinoxaline-2-carboxamide (59, M6256)
[0356] Payload 59 was synthesized from compound 56 as described below.

[0357] Compound 59 was synthesized from compound 56 (20 mg, 0.0206 mmol) by reacting it with 3 -hydroxy quinoline-2-carboxylic acid (5.1 mg, 0.0247), HATU (11 mg, 0.0308 mmol), and NMM (3.1 mg, 0.0308 mmol) in DMF (1 mL) as described in the synthesis of compound 50. Product 54 was obtained as a yellow solid. (11 mg, 0.0096 mmol, 47% yield). MS (ESI, pos.) calc’d for C59H74N11O13 [1144.55]+; found (M+H) 1145.04. ’H-NMR (500 MHz; DMSO-de): <5 11.70 (s, 1H), 9.52 (s, 1H), 8.94-8.92 (m, 1H), 8.59-8.57 (m, 1H), 8.16 (d, 7 = 7.9 Hz, 1H), 8.08 (d, 7 = 6.2 Hz, 1H), 8.03 (d, 7 = 6.1 Hz, 1H), 7.97-7.96 (m, 1H), 7.92-7.86 (m, 2H), 7.86 (s, 1H), 7.83 (d, 7 = 8.2 Hz, 1H), 7.75 (d, 7 = 8.4 Hz, 1H), 7.49 (td, J = 16.8, 9.3 Hz, 2H), 6.01-5.95 (m, 2H), 5.33-5.30 (m, 2H), 4.92 (td, 7 = 11.8, 3.9 Hz, 4H), 4.58 (ddt, 7 = 28.9, 11.6, 5.8 Hz, 4H), 4.46-4.40 (m, 2H), 3.15 (d, 7 = 5.2 Hz, 6H), 2.87 (d, J = 3.7 Hz, 6H), 2.77-2.67 (m, 2H), 2.44-2.26 (m, 4H), 1.86-1.83 (m, 2H), 1.69-1.65 (m, 2H), 1.64-1.58 (m, 2H), 1.53-1.50 (m, 2H), 1.50-1.47 (m, 2H), 1.43-1.38 (m, 2H), 1.26 (d, 7 = 6.8 Hz, 3H), 1.25 (d, 7 = 6.8 Hz, 3H), 1.16-1.05 (m, 4H), 0.98-0.94 (m, 4H).
[0358] EXAMPLE 18
[0359] Synthesis of N-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-20-(3- (aminomethyI)quinoIine-2-carboxamido)-ll,24-diisopropyI-2, 4,12, 15,17, 25-hexamethyI-
3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa-2,5,12,15,18,25- hexaazabicyclo[12.12.4]triacont-28-en-7-yl)quinoxaline-2-carboxamide (63, M6401)
[0360] Payload 63 was synthesized from compound 19 as described below.

[0361] Synthesis of 3-(((tert-butoxycarbonyl)amino)methyl)quinoline-2-carboxylic acid (61): To a solution of l,2-dihydropyrrolo[3,4-b]quinolin-3-one (60, 50 mg, 0.271 mmol) in Water (1 mL) was added IM NaOH (0.54 mL, 0.543 mmol) at ambient temperature. The reaction was heated to 100 °C and stirred for 3 h. The reaction was cooled to ambient temperature and then added BOC2O (0.17 mL, 0.814 mmol). The reaction was stirred at ambient temperature for 1 h. The crude was purified by reverse phase chromatography (Isco EZ Prep Gemini C18 column). The product was eluted in 30-40% ACN/H2O (containing 0.1% of formic acid). The fractions were combined and lyophilized to afford the product as a yellow solid (80 mg, 0.265 mmol, 97% yield). MS (ESI, neg.): calc’d for C16H17N2O4 [301.12]“; found (M-H) 301.19.
[0362] Synthesis of tert-butyl ((2-(((lS,4S,7R,llS,14S,17S,20R,24S,Z)-ll,24-diisopropyl- 2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-20-(quinoxaline-2- carboxamido)-9,22-dioxa-2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7- yl)carbamoyl)quinolin-3-yl)methyl)carbamate (62): To a solution of 19 (4.0 mg, 0.00448 mmol) in DMF (1 mL) was added compound 61 (2.0 mg, 0.00672 mmol), HATU (3.4 mg, 0.00896 mmol), and NMM (0.00099 mL, 0.00896 mmol) at ambient temperature and stirred
for 1 h. The crude was purified by reverse phase chromatography (Isco EZ Prep Gemini C18 column). The product was eluted in 40-50% ACN/H2O (containing 0.1% of formic acid). The fractions were combined and lyophilized to afford the product as a yellow solid (3.8 mg, 0.0032 mmol, 72% yield). MS (ESI, pos.): calc’d for C59H77N12O14 [1177.57]+; found (M+H) 1178.40.
[0363] Synthesis 63: To a solution of compound 62 (3.8 mg, 0.00323 mmol) in DCM (0.7 mL) was added TFA (0.3 mL) at rt ambient temperature and stirred for 3 h. The crude was purified by reverse phase chromatography (Isco EZ Prep Gemini Cl 8 column). The product was eluted in 30-40% ACN/H2O (containing 0.1% of formic acid). The fractions were combined and lyophilized to afford the product as a yellow solid (3.4 mg, 0.00316 mmol, 98% yield). MS (ESI, pos.): calc’d for C54H69N12O12 [1077.52]+; found (M+H) 1078.40. ’H- NMR (300 MHz; DMSO-d6): <5 9.53 (s, 1H), 8.82 (dq, 7 = 3.1, 1.0 Hz, 1H), 8.64-8.61 (m, 1H), 8.52 (s, 1H), 8.36 (d, 7 = 5.0 Hz, 2H), 8.19-8.15 (m, 1H), 8.09-8.05 (m, 2H), 7.98-7.88 (m, 4H), 7.86 (t, 7 = 6.6 Hz, 1H), 7.76-7.70 (m, 1H), 7.60-7.55 (m, 1H), 6.05-5.97 (m, 2H), 5.35-5.30 (m, 2H), 4.88 (td, 7 = 9.4, 5.9 Hz, 4H), 4.64-4.53 (m, 4H), 4.42 (d, 7 = 11.2 Hz, 2H), 4.25-4.22 (m, 2H), 3.13 (s, 6H), 2.89 (d, 7 = 4.0 Hz, 6H), 2.78-2.70 (m, 2H), 2.37-2.14 (m, 4H), 1.27 (d, 7 = 6.8 Hz, 3H), 1.25 (d, 7 = 6.8 Hz, 3H), 0.98 (d, 7 = 6.3, 2.7 Hz, 3H), 0.96 (d, J = 6.3, Hz, 3H), 0.82 (d, J = 6.4 Hz, 3H), 0.80 (d, J = 6.4 Hz, 3H).
[0364] EXAMPLE 19
[0365] Synthesis of N-((lS,4S,7R,llS,14S,17S,20R,24S)-ll,24-dicyclohexyl-20-(4- hydroxyquinoline-2-carboxamido)-2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26- octaoxo-9,22-dioxa-2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7- yl)quinoxaline-2-carboxamide (64, M6255)
[0366] Payload 64 was synthesized from compound 56 as described below.
 [0367] Synthesis of 64: Compound 64 was synthesized from compound 56 (3.0 mg, 0.0031 mmol) by reacting it with 4-hydroxyquinoline-2-carboxylic acid (0.9 mg, 0.0046 mmol), HATU (1.8 mg, 0.0046 mmol), and NMM (0.5 mg, 0.0046 mmol) in DMF (0.5 mL) as described in the synthesis of compound 50. Product 59 was obtained as a yellow solid. (1.6 mg, 0.00 mmol, 45% yield). MS (ESI, pos.) calc’d for C59H74N11O13 [ 1144.55]+; found (M+H) 1145.00. ’H-NMR (500 MHz; DMSO-d6): h 9.50 (q, J = 5.3 Hz, 1H), 8.88-8.83 (m, 1H), 8.59-8.53 (m, 1H), 8.19-8.16 (m, 1H), 8.09-7.99 (m, 4H), 7.99-7.91 (m, 2H), 7.90-7.82 (m, 1H), 7.69-7.48 (m, 2H), 7.28-7.24 (m, 1H), 6.56-6.53 (m, 1H), 6.02-5.94 (m, 2H), 5.35- 5.27 (m, 2H), 4.93-4.83 (m, 3H), 4.81-4.72 (m, 1H), 4.60-4.49 (m, 3H), 4.46-4.34 (m, 3H), 3.19-3.11 (m, 6H), 2.90-2.83 (m, 6H), 2.80-2.63 (m, 2H), 2.42-2.21 (m, 4H), 1.95-1.87 (m, 2H), 1.76-1.37 (m, 9H), 1.31-1.20 (m, 6H), 1.18-0.82 (m, 9H).
[0367] Synthesis of 64: Compound 64 was synthesized from compound 56 (3.0 mg, 0.0031 mmol) by reacting it with 4-hydroxyquinoline-2-carboxylic acid (0.9 mg, 0.0046 mmol), HATU (1.8 mg, 0.0046 mmol), and NMM (0.5 mg, 0.0046 mmol) in DMF (0.5 mL) as described in the synthesis of compound 50. Product 59 was obtained as a yellow solid. (1.6 mg, 0.00 mmol, 45% yield). MS (ESI, pos.) calc’d for C59H74N11O13 [ 1144.55]+; found (M+H) 1145.00. ’H-NMR (500 MHz; DMSO-d6): h 9.50 (q, J = 5.3 Hz, 1H), 8.88-8.83 (m, 1H), 8.59-8.53 (m, 1H), 8.19-8.16 (m, 1H), 8.09-7.99 (m, 4H), 7.99-7.91 (m, 2H), 7.90-7.82 (m, 1H), 7.69-7.48 (m, 2H), 7.28-7.24 (m, 1H), 6.56-6.53 (m, 1H), 6.02-5.94 (m, 2H), 5.35- 5.27 (m, 2H), 4.93-4.83 (m, 3H), 4.81-4.72 (m, 1H), 4.60-4.49 (m, 3H), 4.46-4.34 (m, 3H), 3.19-3.11 (m, 6H), 2.90-2.83 (m, 6H), 2.80-2.63 (m, 2H), 2.42-2.21 (m, 4H), 1.95-1.87 (m, 2H), 1.76-1.37 (m, 9H), 1.31-1.20 (m, 6H), 1.18-0.82 (m, 9H).
[0368] EXAMPLE 20
[0369] Synthesis of N-((lS,4S,7R,llS,14S,17S,20R,24S,E)-ll,24-dicyclopentyl-20-(4- hydroxyquinoline-2-carboxamido)-2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26- octaoxo-9,22-dioxa-2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7- yl)quinoxaline-2-carboxamide (65, M6265)
[0370] Payload 65 was synthesized from compound 52 as described below.

[0371] Synthesis of 65: Compound 56 was synthesized from compound 52 (3 mg, 0.00317 mmol) by reacting it with 3-hydroxyquinoline-2-carboxylic acid (0.72 mg, 0.00381), HATU (1.8 mg, 0.00476 mmol), and NMM (0.48 mg, 0.00476 mmol) in DMF (0.5 mL) as described in the synthesis of compound 50. Product 65 was obtained as a yellow solid (1.8 mg, 0.00161 mmol, 51% yield). MS (ESI, pos.) calc’d for C57H70N11O13 [1116.52]+; found (M+H) 1116.79. ’H-NMR (500 MHz; DMSO-d6): 5 9.52-9.49 (m, 1H), 8.93-8.86 (m, 1H), 8.57-8.49 (m, 1H), 8.46-8.42 (m, 1H), 8.21-8.15 (m, 1H), 8.11-8.04 (m, 1H), 8.03-7.97 (m, 2H), 7.97- 7.90 (m, 2H), 7.76-7.70 (m, 1H), 7.52-7.32 (m, 2H), 7.14-7.06 (m, 1H), 6.29-6.27 (m, 1H),
6.11-6.00 (m, 2H), 5.38-5.27 (m, 2H), 4.95-4.80 (m, 3H), 4.78-4.67 (m, 1H), 4.67-4.54 (m, 3H), 4.48-4.41 (m, 1H), 4.41-4.31 (m, 2H), 3.17-3.11 (m, 6H), 2.90-2.82 (m, 6H), 2.72-2.60 (m, 2H), 2.42-2.21 (m, 4H), 1.75-1.59 (m, 3H), 1.59-1.50 (m, 3H), 1.49-1.31 (m, 6H), 1.31- 1.19 (m, 8H), 1.19-1.05 (m, 2H). [0372] EXAMPLE 21
[0373] Synthesis of N,N’-((1S,4S,7R,11R,14S,17S,2OR,24R,Z)-2,4,12,15,17,25- hexamethyl-ll,24-bis((R)-3-methylbutan-2-yl)-3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa- 2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-ene-7,20-diyl)bis(quinoxaline-2- carboxamide) (83, M6400) [0374] Payload 83 was synthesized from compound 66 as described below.


[0375] Synthesis of(S)-N-((R)-2,3-dimethylbutylidene)-2-methylpropane-2-sulfinamide (68): To a solution of 66 (29.0 g, 105.3 mmol) in 500 mL DCM was added DIBAL-H (115.9 mL, 115.9 mmol, 1.0 M in hexane) at -78 °C and stirred for 1 h. The reaction was quenched with 500 mL aq. NH4CI and stirred at rt for 3 h. The organic layer was collected, and the aqueous layer was extracted with 250 mL Et20 two times. The combined organic layer was passed through Na2SC>4. The crude solution 67 was added (S)-A-tert-butanesulfinamide (25.5 g, 210.6 mmol) and Ti(OEt)4 (55.2 mL, 263.3 mmol) at rt and stirred overnight. The reaction was quenched with 500 mL water and then filtered with celite. The celite was rinsed with 200 mL EtOAc three times. The organic layer was collected, and the aqueous layer was extracted with 300 mL EtOAc. The combined organic layer was dried over Na2SO4 and concentrated under vacuum. The crude was purified by silica gel column, and the product was eluted with 0-10% EtOAc/hexane. Product 68 was obtained as a colorless oil after dried (15.2 g, 74.8 mmol, 71% yield). MS (ESI, pos.) calc’d for C10H22NOS [204.14]+; found (M+H) 204.22.
[0376] Synthesis of(S)-N-((lR,2R)-l-cyano-2,3-dimethylbutyl)-2-methylpropane-2- sulfinamide (69): To a solution of 68 (2.1 g, 10.3 mmol) in 50 mL DCM was added TMSCN (3.89 mL, 40.0 mmol) and Sc(0Tf)3 (8.9 g, 20.7 mmol) at rt. The reaction was stirred for 2 d and then quenched with 50 mL aq. NaHCCL. The organic layer was collected, and the aqueous layer was extracted with EtOAc three times. The combined organic layer was dried over Na2SC>4 and concentrated under vacuum. The crude was purified by silica gel column, and the product was eluted with 40-60% EtOAc/hexane. Two diastereomers were observed by LCMS in 2:1 dr. Major diastereomer 69 was obtained as a white solid after dried (1.05 g, 4.54 mmol, 44% yield). MS (ESI, pos.) calc’d for C11H23N2OS [231.16]+; found (M+H) 231.30.
[0377] Synthesis of(2R,3R )-2-((( ( 9H-fluoren-9-yl)methoxy)carbonyl)amino)-3,4- dimethylpentanoic acid (71 ): To a solution of 69 (2.44 g, 10.59 mmol) in 2 mL THF/water (1:1) was added 20 mL 12 N HC1 at rt. The reaction was stirred at 85 °C overnight. After cooled to rt, the crude solution 70 was diluted with 100 mL H2O neutralized with NaHCCL. To that solution, Fmoc-OSu (7.14 g, 21.2 mmol), NaHCCL (4.45 g, 53.0 mmol), and 100 mL acetone was added at rt and stirred overnight. The reaction was diluted with 100 mL DCM and then quenched with aq. NH4CI. The organic layer was collected and the aqueous layer was extracted with DCM three times. The combined organic layer was dried over Na2SC>4 and concentrated under vacuum. The dried residue was dissolved in 200 mL MTBE and then filtered out the solid. The resulting solution was concentrated under vacuum and then purified by RP Cl 8 column. The product was eluted with 50-55% acctonitrilc/PLO + 0.1% formic acid as the modifier. Product 71 was obtained as a white solid after lyophilized (2.85 g, 7.76 mmol, 73% yield). MS (ESI, pos.) calc’d for C22H2sNO4Na [390.17]+; found (M+Na) 390.33.
[0378] Synthesis of ( 2R, 3R )-2-( ((( 9H-fluoren-9-yl)methoxy )carbonyl )( methyl )amino )-3, 4- dimethylpentanoic acid (73): To the solution of 71 (5.40 g, 14.7 mmol) in 130 mL CHCI3 was added 140 mL TFA and 20 mL 37% aq. formaldehyde at rt. The reaction was stirred overnight. The resulting crude solution 72 was diluted with 50 mL toluene and then dried under vacuum. The dried residue was redissolved in 200 mL CHCI3/TFA (1 :1), and then was added TES (23.5 mL, 147 mmol) at rt. The reaction was stirred overnight. The resulting solution was diluted with 50 mL toluene and then dried under vacuum. The crude was purified by RP C18 column and eluted with 55-60% acctonitrilc/lLC) + 0.1% formic acid as the modifier. Product 73 was obtained as a white solid after lyophilized (2.96 g, 7.76 mmol,
53% yield). The stereochemistry was confirmed by X-ray crystallography. MS (ESI, pos.) calc’d for C23H27NO4Na [404.18]+; found (M+Na) 404.47.
[0379] Synthesis of ((5S,8R,12R)-5-allyl-12-(((benzyloxy)carbonyl)amino)-l-(9H-fluoren-9- yl)-4,7-dimethyl-8-((R)-3-methylbutan-2-yl)-3,6,9-trioxo-2,10-dioxa-4,7-diazatridecan-13- oyl)-L-alanine (75):
[0380] Step 1: Resin 23 (1.5 mmol) in a vessel was washed with TFA/TIS/DCM (2:2.5:95.5) solution until a colorless filtrate was observed. Then, the resin was washed DMF (3 x 20 mL) and DCM (3 x 20 mL). A solution of 73 (629 mg, 1.65 mmol), DIC (340 mg, 2.7 mmol), and DMAP (327 mg, 2.7 mmol) in 20 mL DMF/DCM (1:4) was added into the vessel and gently shaken overnight. The solution was filtered, and the resin was washed with DMF (3 x 20 mL) and DCM (3 x 20 mL). After the resin was dried, a few beads were taken and cleaved with 50% TFA/DCM for checking the complete conversion to product 74 [Cbz-Ser((?-Fmoc-A- Me-3,4-dimethylpentyl)-Ala-OH] in LCMS. MS (ESI, pos.) calc’d for C^H iyNiOyNa [668.26]+; found (M+Na) 668.77.
[0381] Step 2: Resin 74 was treated with 20 mL 10% piperidine/DMF in a vessel and gently shaken at ambient temperature. After 15 min, the solution was filtered, and the resin was washed with DMF (3 x 20 mL), DCM (3 x 20 mL), and then DMF (1 x 20 mL). A solution of Fmoc-A-methyl-(5')-2-allylglycine (949 mg, 2.7 mmol), DMTMM (829 mg, 3.0 mmol), and NMM (0.33 mL, 3.0 mmol) in 20 mL DMF was added into the vessel and gently shaken overnight. The resin was filtered and then washed with DMF (3 x 20 mL) and DCM (3 x 20 mL). The resin was cleaved with 20 mL TFA/H2O/DCM (25:5:70) and shaken for 1 h. The resin was filtered, and the filtrate was collected. The resin was again treated with cleavage condition two more times. The combined filtrate was concentrated and then purified by RP Cl 8 column.
[0382] EXAMPLE 22
[0383] Synthesis of N-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-ll,24-diisopropyl- 2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-20-(quinoxaline-2- carboxamido)-9,22-dioxa-2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7-yl)-3- hydroxyquinoxaline-2-carboxamide (84, M6444)
[0384] Payload 84 was synthesized from intermediate 19 as described below.

[0385] Synthesis of 84: 3.0 mg (0.0034 mmol) of 19 was treated with 3-hydroxy-2- quinoxalinecarboxylic acid (1.92 mg, 0.0101 mmol) in DMF (0.5 mL) in the presence of HATU (3.83 mg, 0.0101 mmol) and NMM (1.1 pL, 0.0101 mmol) at ambient temperature for 5 h. The crude was purified by reverse phase chromatography (Isco EZ Prep Gemini Cl 8 column) using 5-60-100% of acetonitrile in water each containing 0.05% of AcOH. The fractions were combined and lyophilized to afford 84 as a white solid (2.4 mg, 0.0023 mmol, 67%). MS (ESI, pos.) calc’d for C52H65N12O13 [1065 ,47]+; found (M+H) 1066.06. ’H NMR (DMSO-d6, 300 MHz): 89.53 (s, 1H), 8.59 (d, 7=8.7 Hz, 1H), 8.23-8.21 (m, 1H), 8.09-7.89 (m, 7H), 7.63-7.55 (m, 4H), 7.41-7.39 (m, 4H), 6.30 (s, 1H), 6.09-5.98 (m, 2H), 5.38-5.29
(m, 2H), 4.90-4.86 (m, 3H), 4.69-4.44 (m, 7H), 4.28-4.26 (m, 2H), 3.18-3.16 (m, 4H), 2.88- 2.84 (m, 5H), 2.19-2.12 (m, 3H), 1.28-1.22 (m, 5H), 0.99-0.96 (m, 6H), 0.87-0.78 (m, 6H).
[0386] EXAMPLE 23
[0387] Synthesis of N,N'-((1'S,8'R,11'S,14'S,21'R,24'S,Z)-3',11',13',16',24',26'- hexamethyl-2',5',9',12',15',18',22',25'-octaoxo-6',19'-dioxa-3',10',13',16',23',26'- hexaazadispiro[cyclobutane-l,4'-bicyclo[12.12.4]triacontane-17',l"-cyclobutan]-28'-en- 8',21'-diyl)bis(quinoxaline-2-carboxamide) (96, M6563)
[0388] Payload 96 was synthesized from intermediate 85 as described below.

[0389] Synthesis of allyl (quinoxaline-2-carbonyl)-D-serinate (87)'. The title compound was synthesized using the procedure described for compound 13. Accordingly, 12.0 g (28.335 mmol) of 85 was treated with 4.935 g (28.335 mmol) of the acid in the presence of 3.252 mL (22.668 mmol) of DBU, 16.161 g (42.502 mmol) of HATU and 9.410 mL (85.004 mmol) of
NMM in THF (72 mL). After overnight of stirring at ambient temperature, the reaction mixture was filtered through celite to remove the precipitated white solid. The clear filtrate was concentrated under reduced pressure and purified by normal phase chromatography (hexanes/EtOAc). The product eluted in 7:3 combination of the solvents which was not pure. The impure product was treated with 60% of TFA in DCM for 3 hours. The reaction mixture was concentrated under reduced pressure and the contents were redissolved in 30 mL of
acetonitrile and 30 mL of water. The mixture was stirred at rt for 30 mins whereby the transester of TFA converted back to the product. The reaction mixture was extracted in EtOAc. The organic layer was dried (MgSO4), filtered and the filtrate was concentrated under reduced pressure to give yellow oil that was purified by normal phase chromatography (hexanes/EtOAc). The product eluted in 3:7 combination of the solvents. The fractions were concentrated to afford 87 (4.30 g, 14.271 mmol, 50%) as a white solid. MS (ESI, pos.) calc’d for C15H16N3O4 [302.11]+; found (M+H) 302.12.
[0390] Synthesis of (R)-3-(allyloxy)-3-oxo-2-(quinoxaline-2-carboxamido)propyl 1- (methylamino)cyclobutane-l -carboxylate (88): To a suspension of 87 (4.30 g, 14.271 mmol) and l-((tert-butoxycarbonyl)(methyl)amino)cyclobutane-l -carboxylic acid (4.25 g, 18.553 mmol) in DCM (70 mL) at rt/argon was added EDC hydrochloride (6.84 g, 35.679 mmol) followed by the addition of DMAP (0.872 g, 7.136 mmol). All the contents dissolved within 5 minutes giving rise to clear solution that was stirred overnight. Next morning, product resulting from the loss of Boc group was observed as the major along with the hoc containing product. 5 mL of TFA was added that drove the reaction to completion. The reaction mixture was concentrated under reduced pressure and purified by normal phase chromatography (DCM/MeOH). The product eluted in 95:5 combination of the solvents. The fractions were combined and concentrated to afford 88 (2.30 g, 5.576 mmol, 39%) as a colorless oil. MS (ESI, pos.) calc’d for C21H24N4O5 [413. 17]+; found (M+H) 413.44.
[0391] Synthesis of (R)-3-(allyloxy)-3-oxo-2-(quinoxaline-2-carboxamido)propyl 1-((S)-N- methyl-2-(methylamino)pent-4-enamido)cyclobutane-l -carboxylate (89): The title compound was synthesized according to the procedure described for 11. Accordingly 88 (2.23 g, 5.407 mmol) was treated with (S)-2-((tert-butoxycarbonyl)(methyl)amino)pent-4-enoic acid (1.86 g, 8.110 mmol) in THF (30 mL) in the presence of DMTMM (2.39 g, 8.651 mmol) and NMM (1.80 mL, 16.220 mmol) at rt in air. Work up and chromatography (hexanes/EtOAc) gave impure product which, after concentration was treated with 30% of TFA in DCM until all the starting material disappeared. The reaction mixture was concentrated under reduced pressure, redissoved in aceto nitrille and water and lyophilized to afford 89 (2.30 g, 4.393 mmol, 81%) as a white solid. MS (ESI, pos.) calc’d for C27H34N5O6 [524.24]+; found (M+H) 524.39.
[0392] Synthesis of (R)-3-(allyloxy)-3-oxo-2-(quinoxaline-2-carboxamido)propyl l-((S)-2- ((S)-2-((tert-butoxycarbonyl)amino)-N-methylpropanamido)-N-methylpent-4-
enamido)cyclobutane-l -carboxylate (90): 2.23 g (4.259 mmol) of impure 89 was dissolved in THF (30 mL). 2.01 g (10.648 mmol) of Boc-Ala-OH was added followed by the addition of DMTMM (1.95 g, 10.648 mmol) and NMM (1.42 mL, 12.777 mmol). The reaction reached completion within 30 mins. The reaction mixture was filtered through celite and the residue was washed with EtOAc. The filtrate was concentrated under reduced pressure and the resulting yellow oil was dissolved in DMF (12 mL) and purified by reverse phase chromatography (275 g C18Aq) using acetonitrile in water each containing 0.05% of AcOH. The product came out in 70% of acetonitrile. The fractions were combined and lyophilized to afford 90 (1.19 g, 1.752 mmol, 41%) as an off white solid. MS (ESI, pos.) calc’d for C35H46N6O9Na [717.33]; found (M+Na) 717.73.
[0393] Synthesis of (R)-3-(allyloxy)-3-oxo-2-(quinoxaline-2-carboxamido)propyl l-((S)-2- ((S)-2-amino-N-methylpropanamido)-N-methylpent-4-enamido)cyclobutane-l -carboxylate (91): The title compound was prepared by treating 460 mg (0.662 mmol) of 90 with 20% of TFA in DCM until completion of the reaction. The reaction mixture was concentrated under reduced pressure and put into vacuum before using for the next step. The yield was assumed to be quantitative. MS (ESI, pos.) calc’d for C30H38N6O7 [595.28]+; found (M+H) 595.53.
[0394] Synthesis of O-(l-((S)-2-((S)-2-((tert-butoxycarbonyl)amino)-N- methylpropanamido)-N-methylpent-4-enamido)cyclobutane-l-carbonyl)-N-(quinoxaline-2- carbonyl)-D-serine (92): The title compound was prepared according to the procedure described for 12 by treating 731 mg (1.052 mmol) of 90 with 130 pL (1.052 mmol) of phenylsilane in DCM (6 mL) in the presence of Pd(PPh3)4 (61 mg, 0.053 mmol) at rt under argon. Work up and chromatography (100 C18Aq) using acetonitrile in water each containing 0.05% of AcOH followed by lyophilization afforded 92 (511 mg, 0.872 mmol, 83%) as a white solid. MS (ESI, pos.) calc’d for C32H42NeO9Na [677.30]; found (M+Na) 677.67.
[0395] Synthesis of (R)-3-(((S)-l-(((S)-l-((l-(((R)-3-(allyloxy)-3-oxo-2-(quinoxaline-2- carboxamido)propoxy)carbonyl)cyclobutyl)(methyl)amino)-l-oxopent-4-en-2- yl)(methyl)amino)-l-oxopropan-2-yl)amino)-3-oxo-2-(quinoxaline-2-carboxamido)propyl 1- ((S)-2-((S)-2-((tert-butoxycarbonyl)amino)-N-methylpropanamido)-N-methylpent-4- enamido)cyclobutane-l -carboxylate (93): 91 (395 mg, 0.664 mmol) was dissolved in THF (4 mL) to which was added 92 (478 mg, 0.731 mmol). To this homogeneous mixture was added DMTMM (368 mg, 1.328 mmol) followed by the addition of NMM (221 pL, 1.993 mmol) at rt in air. The reaction reached completion within 10 mins with clean reaction profile. 2 mL of
DMF was added and the reaction mixture was directly purified by reverse phase chromatography (100 C18Aq) using acetonitrile in water each containing 0.05% of AcOH. The product came out in 70% of acetonitrile. The fractions were combined and lyophilized to afford 93 (676 mg, 0.549 mmol, 83%) as a white solid. MS (ESI, pos.) calc’d for C62H78Ni2Oi5Na [1253.57]; found (M+Na) 1254.07.
[0396] Synthesis of O-(l-((S)-2-((S)-2-((R)-3-((l-((S)-2-((S)-2-((tert- butoxycarbonyl)amino)-N-methylpropanamido)-N-methylpent-4-enamido)cyclobutane-l- carbonyl)oxy)-2-(quinoxaline-2-carboxamido)propanamido)-N-methylpropanamido)-N- methylpent-4-enamido)cyclobutane-l-carbonyl)-N-(quinoxaline-2-carbonyl)-D-serine (94): The title compound was synthesized according to the procedure described for 12 by treating 676 mg (0.549 mmol) of 93 with phenylsilane (68 pL, 0.549 mmol) in DCM (6 mL) in the presence of Pd(PPh3)4 (32 mg, 0.028 mmol) at rt under argon. Work up and chromatography (C18Aq) using acetonitrile in water each containing 0.05% of AcOH followed by lyophilization afforded 94 (493 mg, 0.414 mmol, 75%) as an off white solid. MS (ESI, pos.) calc’d for Cs^N OisNa [1213.54]; found (M+Na) 1214.02.
[0397] Synthesis of O-(l-((7S,12S,15S,18R,Z)-7-((S)-2-amino-N-methylpropanamido)- N,5 , 13 , 15-tetramethyl-6 , 14, 17,21 -tetraoxo- 18-(quinoxaline-2-carboxamido)-20-oxa-5 ,13,16- triazaspiro [3.17]henicos-9-ene- 12-carboxamido)cyclobutane- 1 -carbonyl)-N-(quinoxaline-2- carbonyl)-D-serine (95): The title compound was prepared according to the procedure described for 17. Accordingly, 493 mg (0.414 mmol) of 94 was dissolved in DCM and Hoveyda-Grubbs 2nd generation catalyst (52 mg, 0.083 mmol) was added at rt under argon. After 10 mins at rt, the reaction mixture was stirred at 45 °C in an aluminum heating block. The reaction reached completion within 3 h. The reaction mixture was cooled to 0 °C and 3.5 mL of TFA was added. The reaction mixture was transferred to rt and allowed to stir until all the ring closing metathesis product disappeared to the product at which time the reaction the reaction mixture was concentrated under reduced pressure. Work and chromatography (50 g Cl 8) using acetonitrile in water each containing 0.05% of AcOH followed by lyophilization of the combined fractions afforded 95 (275 mg, 0.259 mmol, 63%) as an off white solid. MS (ESI, pos.) calc’d for C52H63N12O13 [1063.46]+; found (M+H) 1063.95.
[0398] Synthesis of N,N’-((rS,8’R,ll’S,14’S,2TR,24’S,Z)-3’,i r,13’,16’,24’,26’-hexamethyl- 2’, 5’, 9’, 12’, 15’, 18’,22’,25’-octaoxo-6’, 19’-dioxa-3’, 10’,13’,16’,23’,26’- hexaazadispiro[cyclobutane-l,4’-bicyclo[12. 12.4]triacontane-17’,l”-cyclobutan]-28’-en-8’,21’-
diyl)bis(quinoxaline-2-carboxamide) (96, TCRS-3782, M6563): The title compound was synthesized according to the procedure described for 32. Accordingly, 95 (267 mg, 0.251 mmol) was dissolved in DMF (3 mL). DMTMM (139 mg, 0.502 mmol) was added followed by the addition of NMM (83, 0.753 mmol) at rt in air. Work up and chromatography (50 g C18Aq) using acetonitrile in water each containing 0.05% of AcOH followed by lyophilization of the resulting fractions afforded 96 (86 mg, 0.082 mmol, 33%) as a white solid. MS (ESI, pos.) calc’d for C52H61N12O12 [1045.45]+; found (M+H) 1045.89. ’H-NMR (300 MHz; DMSO-d6): 5 9.53 (s, 2H), 8.33-8.30 (m, 2H), 8.23-8.19 (m, 2H), 8.04-7.94 (m, 8H), 6.02 (br s, 2H), 5.29 (br s, 2H), 4.90-4.85 (m, 3H), 4.59-4.46 (m, 5H), 4.35 (d, J = 10.5 Hz, 2H), 3.21 (s, 5H), 2.93-2.84 (m, 7H), 2.44-2.10 (m, 6H), 2.93-1.87 (m, 8H), 1.31 (d, J=6.9 Hz, 6H).
[0399] EXAMPLE 24
[0400] Synthesis of N,N'-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-ll,24-dicyclobutyl- 2,4,12,15,17,25-hexamethyI-3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa-2,5,12,15,18,25- hexaazabicycIo[12.12.4]triacont-28-ene-7,20-diyI)bis(quinoxaIine-2-carboxamide) (97,
M6597)
[0401] Payload 97 was synthesized using all the procedural steps described for 96 by replacing l-((tert-butoxycarbonyl)(methyl)amino)cyclobutane-l-carboxylic acid with (5)-2- ((tert-butoxycarbonyl)(methyl)amino)-2-cyclobutylacetic acid.

[0402] The title compound was synthesized by treating (2R)-3-[(2S)-2- [[(3S,6S,8Z,l lS,14S,17R)-6-[[(2R)-2-aminopropanoyl]-methyl-amino]-3-cyclobutyl- 4,12,14-trimethyl-2,5,13,16-tetraoxo-17-(quinoxaline-2-carbonylamino)-l-oxa-4,12,15- triazacyclooctadec-8-ene-l l-carbonyl]-methyl-amino]-2-cyclobutyl-acetyl]oxy-2- (quinoxaline-2-carbonylamino)propanoic acid (167 mg, 0.153 mmol) with DMTMM ((85 mg, 0.306 mmol) and NMM (51 p L, 0.459 mmol) in DMF (2 mL). Work up and reverse phase chromatography afforded the compound as a white solid (49 mg, 0.046 mmol, 30%).
MS (ESI, pos.) calc’d for C54H65N12O12 [1073.48]+; found (M+H) 1073.99. 'H-NMR (500 MHz; DMSO-d6): 59.46 (s, 2H), 8.38 (d, J=9.0 Hz, 2H), 8.10 (d, 1=8.0 Hz, 2H), 8.03-8.06 (m, 2H), 7.76-7.58 (m, 6H), 6.12-6.06 (m, 2H), 5.41-5.38 (m, 2H), 5.11 (d, J= 11.0 Hz, 2H), 4.94-4.91 (m, 2H), 4.66-4.63 (m, 4H), 4.35 (d, J=10 Hz, 2H), 3.08 (s, 6H), 2.88 (s, 6H), 2.74- 2.69 (m, 4H), 2.00-1.62 (m, 14H), 1.27 (d, 1=6.5 Hz, 6H).
[0403] EXAMPLE 25
[0404] Synthesis of N,N'-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-ll,24-diisopropyl- 2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa-2,5,12,15,18,25- hexaazabicyclo[12.12.4]triacont-28-ene-7,20-diyl)bis(quinoxaline-2-carboxamide) (98, M6138)
[0405] Payload 98 was synthesized from intermediate 19 as described below.

[0406] Synthesis of 98: To a solution of 19 (40 mg, 0.041 mmol) in DMF (1 mL) was added quinoxaline-2-carboxylic acid (16 mg, 0.076 mmol), HATU (29 mg, 0.076 mmol), and NMM (7.7 mg, 0.076 mmol) at ambient temperature and stirred for 1 h. The crude solution was purified by reverse phase chromatography (Isco EZ Prep Gemini Cl 8 column). The product was eluted in 50-65% ACN/H2O (containing 0.1% of formic acid). The fractions were combined and lyophilized to afford product 98 as a yellow solid (33 mg, 0.031 mmol, 77% yield). MS (ESI, pos) calc’d for C52H65N12O12 [1049.49]+; found (M+H) 1050.52. ’H-NMR (300 MHz; CD3OD): <59.54 (s, 2H), 8.19-8.16 (m, 2H), 7.86 (dd, J = 7.8, 6.2 Hz, 4H), 7.72 (t, J = 7.5 Hz, 2H), 6.12 (t, J = 7.6 Hz, 2H), 5.54-5.50 (m, 2H), 5.08 (d, J = 3.2 Hz, 2H), 4.97 (d, 7 = 10.0 Hz, 2H), 4.79-4.77 (m, 2H), 4.74 (dd, 7 = 7.4, 3.7 Hz, 2H), 4.62 (d, 7 = 11.1 Hz, 2H), 3.34-3.15 (m, 6H), 3.05 (d, 7 = 6.6 Hz, 6H), 2.65-2.61 (m, 2H), 2.57-2.51 (m, 2H), 2.23 (dt, J = 9.9, 6.5 Hz, 2H), 1.41 (d, J = 7.1 Hz, 6H), 1.02 (d, J = 6.4 Hz, 6H), 0.86 (d, J = 6.7 Hz, 6H).
[0407] EXAMPLE 26
[0408] Synthesis of N,N'-((lS,4S,7R,llS,14S,17S,20R,24S)-ll,24-diisopropyl- 2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa-2,5,12,15,18,25- hexaazabicyclo[12.12.4]triacontane-7,20-diyl)bis( quinoxaline- 2-carboxamide) (99, TCRS-3389)
[0409] Payload 99 was synthesized from intermediate 98 as described below.

[0410] Synthesis of 99: To a solution of 98 (20 mg, 0.019) in THF (2 mL) was added Wilkinson’s catalyst (8.8 mg, 0.0095 mmol) at rt under nitrogen. A hydrogen ballon was connected to the reaction vial to replace the nitrogen. The reaction was covered by aluminum foil and stirred overnight. The crude was dried and then purified by Cl 8 column. The product was eluted with 50- 65% ACN/H2O + 0.05% AcOH. Product was isolated as a white solid after lyophilized (3.2 mg, 0.003 mmol, 15% yield). MS (ESI, pos) calc’d for C52IL7N12O12 [ 1051.50]+; found (M+H) 1052.26. 'H-NMR (300 MHz; CDCh): 39.58 (d, J = 3.8 Hz, 2H), 9.12-9.09 (m, 2H), 8.43 (d, 7 = 9.9 Hz, 2H), 8.20-8.10 (m, 4H), 7.98-7.89 (m, 4H), 5.77-5.73 (m, 2H), 5.26-5.05 (m, 6H), 4.49-4.44 (m, 2H), 3.97-3.94 (m, 2H), 3.45-3.41 (m, 6H), 3.03- 2.95 (m, 6H), 2.48-2.35 (m, 4H), 1.76-1.42 (m, 6H), 1.32-1.22 (m, 6H), 1.10-0.98 (m, 12H).
[0411] EXAMPLE 27
[0412] Synthesis of N,N'-((1S,4S,7R,11S,14S,17S,2OR,24S,Z)-2,4,12,15,17,25- hexamethyl-ll,24-bis((R)-3-methylbutan-2-yl)-3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa- 2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-ene-7,20-diyl)bis(quinoxaline-2- carboxamide) (105, M6469)
[0413] Payload 105 was synthesized from 21 as described below.

[0414] Synthesis of (S) -N-((lS,2R)-l-cyano-2,3-dimethylbutyl)-2-methylpropane-2- sulfinamide (100): A solution of Et2AlCN (22.1 mL, 22.1 mmol, 1 M solution in toluene) was added to THF (60 mL) at 0 °C followed by z'PrOH (1.69 mL, 22.1 mmol), and then stirred for 10 min before cooled to -78 °C. To a solution of 68 (3.0 g, 14.7 mmol) in THF (600 mL) was cooled to 0 °C and then added the activated EtOz'PrAlCN solution. The cooling bath was removed, and the reaction was allowed to room temperature overnight. The reaction was quenched with aq. NH4CI. The organic layer was collected, and the aqueous layer was extracted with EtOAc three times. The combined organic layer was dried over Na2SC>4 and concentrated under vacuum. Two diastereomers were observed by LCMS in 95:5 dr. The crude was purified by RP Cl 8 column, and the product was eluted with 30-40% acetonitrile/H2O + 0.1% formic acid as the modifier. Product 100 (a single diastereomer) was obtained as white solid after lyophilized (2.3 g, 10.0 mmol, 69% yield). MS (ESI, pos.) calc’d for C11H23N2OS [231. 16]+; found (M+H) 231.33. [0415] Synthesis of (2S,3R)-2-((((9H-fluoren-9-yl)methoxy)carbonyl)(methyl)amino)-3,4- dimethylpentanoic acid (102): Compound 102 was synthesized from compound 100 (2.3 g, 10.0 mmol) following the same procedure described in the synthesis of compound 73, EXAMPLE 21. Product 102 was isolated as a white solid (1.3 g, 3.4 mmol, 34% yield over 4 steps). MS (ESI, pos.) calc’d for C23H27NO4Na [404.18]+; found (M+Na) 404.41.
[0416] Synthesis of benzyl ((lS,4S,7R,HS,14S,17S,20R,24S,Z)-20-amino-2,4,12,15,17,25- hexamethyl- 11 ,24-bis((R)-3 -methylbutan-2 -yl) -3 ,6 ,10,13,16, 19,23 ,26-octaoxo-9,22-dioxa- 2,5,12,15,18,254iexaazabicyclo[12.12.4]triacont-28-en-7-yl)carbamate (103): Compound 103 was synthesized from resin 21 (1.6 g, 1.0 mmol, 0.627 mmol/g loading, 100-200 mesh) as described in the synthesis of compound 33, EXAMPLE 3, while replacing the reactant Fmoc- N-Me-Val-OH with compound 102. Product 103 was isolated as a white solid (2.7 mg, 0.0029 mmol, 0.29% overall yield). MS (ESI, pos.) calc’d for C^HyiNsOn [927.52]+; found (M+H) 928.14.
[0417] Synthesis of (1S,4S,7R,1 lS,14S,17S,20R,24S,Z)-7 ,20-diamino-2,4,12,15,17,25- hexamethyl-l l,24-bis((R)-3-methylbutan-2-yl)-9,22-dioxa-2,5,12,15,18,25- hexaazabicyclo[12.12.4]triacont-28-en-3,6,10,13,16,19,23,26-octaone (104): To a solution of 103 (2.7 mg, 0.0029 mmol) in TFA (1 mL) was added thioanisole (0.003 mL, 0.029 mmol) at rt. The reaction was heated to 45 °C and stirred overnight. The crude was purified by RP Cl 8 column and the product was eluted with 20-40% aceto nitrilc/PLO + 0.1% formic acid as the modifier. Product 104 was obtained as white solid after lyophilized (1.1 mg, 0.0014 mmol, 48% yield). MS (ESI, pos.) calc’d for CesIfeNsOio [793.48]+; found (M+H) 794.01.
[0418] Synthesis of 105: To a solution of 104 (1.1 mg, 0.0014 mmol) in DMF (0.5 mL) was added quinoxaline -2-carboxylic acid (0.97 mg, 0.0056 mmol), HATU (2.11 mg, 0.0056 mmol), and NMM (0.56 mg, 0.0056 mmol) at ambient temperature under argon. The reaction was stirred at for 1 h. The crude was purified by RP Cl 8 column. The product was eluted with 40-55% acetonitrile/H2O + 0.1% formic acid as the modifier. Product 105 was obtained as white solid after lyophilized (0.9 mg, 0.0008 mmol, 59% yield). MS (ESI, pos.) calc’d for C56H73N12O12 [1105.55]+; found (M+H) 1106.26. ’H-NMR (500 MHz; DMSO-d6): h 9.52- 9.51 (m, 2H), 8.69-8.65 (m, 2H), 8.17-8.13 (m, 2H), 8.09-8.05 (m, 2H), 7.98-7.96 (m, 2H), 7.92-7.87 (m, 4H), 5.99-5.94 (m, 2H), 5.38-5.30 (m, 2H), 5.12-5.09 (m, 2H), 4.92-4.88 (m, 2H), 4.61-4.53 (m, 4H), 4.44-4.39 (m, 2H), 3.11-3.09 (m, 6H), 2.88 (s, 6H), 2.09-2.06 (m, 2H), 2.03-1.97 (m, 4H), 1.70-1.64 (m, 2H), 1.48-1.41 (m, 2H), 0.88-0.81 (m, 6H), 0.77 (d, J = 6.3 Hz, 12H), 0.66-0.60 (m, 6H).
[0419] EXAMPLE 28
[0420] Synthesis of N-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-20-(3-hydroxyquinoline-2- carboxamido)-2,4,12,15,l'7,25-hexamethyl-ll,24-bis((R)-3-methylbutan-2-yl)-
3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa-2,5,12,15,18,25- hexaazabicyclo[12.12.4]triacont-28-en-7-yl)quinoxaline-2-carboxamide (107, M6572)
[0421] Payload 107 was synthesized from 106 as described below.

[0422] Synthesis of N-((lS,4S,7R,l lS,14S,17S,20R,24S,Z)-20-amino-2,4,12,15,17,25- hexamethyl- 11 ,24-bis((R)-3 -methylbutan-2 -yl) -3 ,6 ,10,13,16, 19,23 ,26-octaoxo-9,22-dioxa- 2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7-yl)quinoxaline-2-carboxamide (106): Compound 106 was synthesized from resin 21 (0.6 g, 0.3 mmol, 0.503 mmol/g loading, 100-200 mesh) as described in the synthesis of compound 48, while replacing the reactant Fmoc-N-Me-Ilu-OH with 102. The product 106 was isolated as a yellow solid (19 mg, 0.02 mmol, 6.7% overall yield). MS (ESI, pos.) calc’d for C47H69N10O11 [949.52]+; found (M+H) 949.98.
[0423] Synthesis of 107: To a solution of 106 (3.0 mg, 0.032 mmol) in DMF (1 mL) was added 3-hydroxyquinoline-2-carboxylic acid (0.7 mg, 0.038 mmol), HATU (1.8 mg, 0.0147 mmol), and NMM (0.001 mL, 0.095 mmol) at rt. The reaction was stirred at rt for 30 min. The crude was directly purified by Cl 8 column. The product was eluted with 50-55% ACN/H2O + 0.1% formic acid. The product was obtained as a white solid after lyophilized (1.5 mg, 0.013 mmol, 42% yield). MS (ESI, pos.) calc’d for C57H74N11O13 [ 1120.55]+; found (M+H) 1121.10. ’H-NMR (300 MHz; DMSO-d6): h 9.54 (s, 1H), 8.69-8.63 (m, 1H), 8.34-
8.32 (m, 4H), 8.20-8.16 (m, 1H), 8.14-7.88 (m, 4H), 7.80-7.72 (m, 2H), 7.48-7.47 (m, 1H),
7.32 (d, J = 3.3 Hz, 1H), 5.99 (dd, J = 8.2, 1.0 Hz, 2H), 5.36 (dd, J = 2. 1, 1.0 Hz, 2H), 5. 15- 5.07 (m, 2H), 4.93-4.89 (m, 2H), 4.63-4.45 (m, 6H), 3.15 (d, 7 = 1.5 Hz, 6H), 2.90 (d, 7 = 2.7 Hz, 6H), 2.74 (dd, 7 = 2.4, 1.4 Hz, 2H), 2.29 (t, 7 = 1.6 Hz, 2H), 2.10-2.04 (m, 2H), 1.77-1.67 (m, 2H), 1.29-1.25 (m, 6H), 0.80-0.62 (m, 18H).
[0424] EXAMPLE 29
[0425] Synthesis of N,N'-((1S,4S,7R,11S,14S,17S,2OR,24S,Z)-2,4,12,15,17,25- hexamethyI-ll,24-bis((R)-3-methyIbutan-2-yI)-3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa-
2,5,12,15,18,25-hexaazabicycIo[12.12.4]triacont-28-ene-7,20-diyI)bis(3- hydroxyquinoIine-2-carboxamide) (109, M6627)
[0426] Payload 109 was synthesized from 108 as described below.

[0427] Synthesis of 108: Compound 108 was synthesized from resin 21 (2.4 g, 2.7 mmol, 0.503 mmol/g loading, 100-200 mesh) as described in the synthesis of compound 35, while replacing the reactant Fmoc-A-Me-Ilu-OH with 102. Product 108 was isolated as a yellow solid (6.3 mg, 0.0065 mmol, 0.24% overall yield). MS (ESI, pos.) calc’d for C48H70N9O12 [964.52]+; found (M+H) 965.17.
[0428] Synthesis of 109: Compound 109 was synthesized from compound 108 (1.5 mg, 0.0016 mmol) by reacting it with 3-hydroxyquinoline-2-carboxylic acid (0.35 mg, 0.0019 mmol), HATU (0.89 mg, 0.0023 mmol), and NMM (0.0005 mL, 0.0047 mmol) in DMF (1 mL) as described in the synthesis of compound 107. Product 109 was obtained as a white solid (0.8 mg, 0.0007 mmol, 45% yield). MS (ESI, pos.) calc’d for C58H75N10O14 [1135.55]+; found 1136.28. (M+H) 1121.10. ’H-NMR (300 MHz; DMSO-d6): <5 11.81-11.67 (m, 2H), 9.03-8.93 (m, 2H), 8.16-8.11 (m, 2H), 7.94-7.71 (m, 6H), 7.65-7.40 (m, 4H), 6.04-5.96 (m, 2H), 5.43-5.29 (m, 2H), 5.21-5.02 (m, 2H), 4.97-4.82 (m, 2H), 4.73-4.41 (m, 6H), 3.22-3.14 (m, 6H), 2.90-2.83 (m, 6H), 2.79-2.67 (m, 2H), 2.38-2.25 (m, 2H), 2.16-1.95 (m, 2H), 1.77- 1.61 (m, 2H), 1.36-1.21 (m, 6H), 0.95-0.45 (m, 18H).
[0429] EXAMPLE 30
[0430] Synthesis of N,N'-((lS,4S,7R,llS,14S,17S,20R,24S,E)-ll,24-diisopropyl- 2, 4, 12, 15,17, 25-hexamethyl-3, 6, 10, 13, 16, 19,23, 26-octaoxo-9,22-dioxa- 2,5,12,15,18,25-
hexaazabicyclo[12.12.4]triacont-28-ene-7,20-diyl)bis(3-hydroxyquinoline-2- carboxamide) (119, M6378)
[0431] Payload 119 was synthesized from intermediate 11 as described below.
 [0432] Synthesis of (R)-2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(allyloxy)-3- oxopropyl N-((S)-2-((S)-2-((tert-butoxycarbonyl)amino)-N-methylpropanamido)pent-4- enoyl)-N-methyl-L-valinate (110): 5.30 g (mmol) of 11 was treated with 30% of TFA in DCM until the reaction was completed. The reaction mixture was concentrated under reduced pressure to give faint yellow oil that was redissolved in acetonitrile/water and lyophilized overnight to give white solid. The amine was used for the synthesis of the title compound. Accordingly, (4.533 g, 7.661 mmol) of the amine was reacted with 2.174 g (11.491 mmol) of Boc-Ala-OH in THF (50 mL) in the presence of DMTMM (3. 180 g, 11.491 mmol) and
NMM (2.543 mL, 22.983 mmol) at rt in air for 15 mins. Work up and chromatography (hexanes/EtOAc) as described for 90 afforded 110 (4.90 g, 6.423 mmol, 84%) as a sticky white solid. MS (ESI, pos.) calc’d for C41H55N4O10 [763.38]+; found (M+H) 763.66.
[0432] Synthesis of (R)-2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(allyloxy)-3- oxopropyl N-((S)-2-((S)-2-((tert-butoxycarbonyl)amino)-N-methylpropanamido)pent-4- enoyl)-N-methyl-L-valinate (110): 5.30 g (mmol) of 11 was treated with 30% of TFA in DCM until the reaction was completed. The reaction mixture was concentrated under reduced pressure to give faint yellow oil that was redissolved in acetonitrile/water and lyophilized overnight to give white solid. The amine was used for the synthesis of the title compound. Accordingly, (4.533 g, 7.661 mmol) of the amine was reacted with 2.174 g (11.491 mmol) of Boc-Ala-OH in THF (50 mL) in the presence of DMTMM (3. 180 g, 11.491 mmol) and
NMM (2.543 mL, 22.983 mmol) at rt in air for 15 mins. Work up and chromatography (hexanes/EtOAc) as described for 90 afforded 110 (4.90 g, 6.423 mmol, 84%) as a sticky white solid. MS (ESI, pos.) calc’d for C41H55N4O10 [763.38]+; found (M+H) 763.66.
[0433] Synthesis of N-(((9H-fluoren-9-yl)methoxy)carbonyl)-O-(N-((S)-2-((S)-2-((tert- butoxy carbonyl) amino) -N -methylpropanamido)pent-4-enoyl) -N -methyl -L- valyl) -D -serine
(111): The title compound was prepared as described for 92. Accordingly, 3.20 g (4.195 mmol) of 110 was reacted with 517 microliter (4.195 mmol) of phenylsilane in DCM (12 mL) in the presence of Pd(PPh3)4 (242 mg, 0.210 mmol) at rt under argon for 10 mins. Work up and chromatography using acetonitrile in water each containing 0.05% of AcOH afforded 111 (2.525 g, 3.493 mmol, 83%) as a white solid. MS (ESI, pos.) calc’d for C38H51N4O10 [723.35]+; found (M+H) 723.72.
[0434] Synthesis of (R)-2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(allyloxy)-3- oxopropyl N-((S)-2-((S)-2-amino-N-methylpropanamido)pent-4-enoyl)-N-methyl-L-valinate
(112): 2.45 g of 110 was treated with 30% of TFA in DCM until the reaction reached completion. The reaction mixture was concentrated under reduced pressure to give faint yellow oil that was redissolved in acetonitrile/water and lyophilized overnight to afford 112 as a white solid. The yield was assumed to be quantitative. MS (ESI, pos.) calc’d for C36H47N4O8 [663.33]+; found (M+H) 663.66.
[0435] Synthesis of (5R,9S,12S,15S,18R)-18-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)- 12-allyl-5-((allyloxy)carbonyl)-l-(9H-fluoren-9-yl)-9-isopropyl-10,13,15-trimethyl- 3,8,l l,14,17-pentaoxo-2,7-dioxa-4,10,13,16-tetraazanonadecan-19-yl N-((S)-2-((S)-2-((tert- butoxycarbonyl)amino) -N -methylprop anamido)pent-4-enoy 1) -N -methyl-L- valinate (113): The title compound was prepared according to procedure described for 93. Accordingly, 2.521 g (3.388 mmol) of the acid and 2.312 g (3.488 mmol) of the amine were dissolved in THF (16 mL) at rt in air. DMTMM (1.45 g, 5.240 mmol) was added followed by the addition of NMM (1.15 mL, 10.46 mmol). The reaction reached completion within 10 mins. Work up and chromatography using acetonitrile in water each containing 0.05% of AcOH followed by lyophilization afforded 113 (3.87 g, 2.832 mmol, 81%) as a white solid. MS (ESI, pos.) calc’d for C74H94N8Oi7Na [1389.67]+; found (M+Na) 1390.17.
[0436] Synthesis of N-(((9H-fluoren-9-yl)methoxy)carbonyl)-O-(N-((S)-2-((S)-2-((R)-2- ((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-((N-((S)-2-((S)-2-((tert-
butoxycarbonyl) amino) -N -methylprop anamido)pent-4-enoy 1) -N -methyl-L- valyl)oxy)propanamido)-N-methylpropanamido)pent-4-enoyl)-N-methyl-L-valyl)-D-serine (114): The title compound was synthesized as per the procedure described for 94.
Accordingly, 3.86 (2.822 mmol) of 113 was treated with phenylsilane (348 pL, 2.822 mmol) in DCM (15 mL) in the presence of Pd(PPh3)4 (163 mg, 0.141 mmol) at rt under argon for 10 mins. Work up and chromatography using acetonitrile in water each containing 0.05% of AcOH followed by lyophilization afforded 114 (3.04 g, 2.292 mmol, 81%) as a white solid. MS (ESI, pos.) calc’d for C71H91N8O17 [1327.64]+; found (M+H) 1328.31.
[0437] Synthesis of N-(((9H-fhroren-9-yl)methoxy)carbonyl)-O-(N-((3S,6S,l lS,14S,17R)- 17-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-6-((S)-2-amino-N-methylpropanamido)-3- isopropyl-4,12,14-trimethyl-2,5,13,16-tetraoxo-l-oxa-4,12,15-triazacyclooctadec-8-ene-ll- carbonyl)-N-methyl-L-valyl)-D-serine (115): The title compound was synthesized according to the procedure mentioned for 95. Accordingly, 3.04 g (2.290 mmol) of 114 was heated at 45 °C in the presence of H-G II (143 mg, 0.228 mmol) in DCM (22 mL) for 3 h. After cooling down to rt, the reaction mixture was treated with 5 mL of TFA. Work up and chromatography using acetonitrile in water each containing 0.05% of AcOH followed by lyophilization of the resulting fractions afforded 115 (2.81 g, impure) as a dirty green solid. MS (ESI, pos.) calc’d for C64H79N8O15 [1199.56]+; found (M+H) 1200.13.
[0438] Synthesis of bis((9H-fluoren-9-yl)methyl) ((1S,4S,7R,11S,14S,17S,2OR,24S,Z)-
1 l,24-diisopropyl-2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa-
2.5.12.15.18.25-hexaazabicyclo[12.12.4]triacont-28-ene-7,20-diyl)dicarbamate (116) & bis((9H-fluoren-9-yl)methyl) (( 1 S,4S ,7R, 11 S , 14S , 17S ,20R,24S ,E)- 11 ,24-diisopropyl-
2.4.12.15.17.25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa-2,5,12,15,18,25- hexaazabicyclo[12.12.4]triacont-28-ene-7,20-diyl)dicarbamate (117): The impure material 115 (2.81 g, 2.343 mmol) was stirred in THF (6 mL) in the presence of DMTMM (1.30 g, 4.698 mmol) and NMM (773 pL, 7.031 mmol) at rt in air for 15 mins until all the starting material was consumed. The reaction mixture was concentrated under reduced pressure, redissolved in DMF (5 mL) and purified by reverse phase chromatography using acetonitrile in water each containing 0.05% of AcOH. The major isomer (116, cis, 265 mg, 0.224 mmol, 10%) came out in 70% of acetonitrile in water followed by the minor isomer (117, trans, 151 mg, 0.128 mmol, 5%) in 80% of acetonitrile in water. MS (ESI, pos.) calc’d for 116,
C64H77N8Oi4 [1181.55]+; found (M+H) 1182.27 and MS (ESI, pos.) calc’d for 117, C64H77N8O14 [1181.55]+; found (M+H) 1182.25.
[0439] Synthesis of ( 1 S,4S ,7R, 1 IS , 14S, 17S,20R,24S ,E)-7,20-diamino- 11 ,24-diisopropyl- 2,4,12,15,17,25-hexamethyl-9,22-dioxa-2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28- en-3,6,10,13,16,19,23,26-octaone (118): 42 mg (0.036 mmol) of 117 was treated with 5% of piperidine in DMF at rt in air until all the starting material was consumed. The reaction mixture was purified by reverse phase chromatography using acetonitrile in water each containing 0.05% of AcOH. Lyophilization of the fractions gave 118 (32 mg, impure) as a white solid. MS (ESI, pos.) calc’d for C34H57N8O10 [737.4]+; found (M+H) 737.2.
[0440] Synthesis of 119: 32 mg (0.043 mmol) of 118 was dissolved in DMF (0.5 mL) at rt under argon. 3-Hydroxyquinoline-2-carboxylic acid (25 mg, 0.132 mmol) was added followed by the addition of HATU (50 mg, 0.131 mmol) and NMM (29 pL, 0.264 mmol). After 3 h, the reaction mixture was purified by reverse phase chromatography using acetonitrile in water each containing 0.05% of AcOH. Fractions were combined and lyophilized to afford 119 (6.1 mg, 0.006 mmol, 13%) as a white solid. MS (ESI, pos.) calc’d for C54H67N10O14 [1079.48]+; found (M+H) 1080.20. ’H-NMR (500 MHz; DMSO-d6): <5 10.54 (s, 2H), 9.27 (d, 7=9.0 Hz, 2H), 8.77 (d, 7=7.0 Hz, 2H), 7.78 (d, 7=8.5 Hz, 2H), 7.58 (t, 7=7.0 Hz, 2H), 7.53 (t, 7=7.5 Hz, 2H), 7.46 (s, 2H), 7.19 (d, 7=8.5 Hz, 2H), 5.60-5.58 (m, 2H), 5.37 (d, 7=11.5 Hz, 2H), 5.01-4.98 (m, 2H), 4.86 (d, 7=10.5 Hz, 2H), 4.74 (d, 7=11.5 Hz, 2H), 4.39 (d, 7=6.0 Hz, 2H), 4.19-4.16 (m, 2H), 3.20-3.14 (m, 8H), 2.67 (s, 6H), 1.95- 1.90 (m, 2H), 1.83-1.78 (m, 2H), 1.43 (d, 7=7.0 Hz, 6H), 0.71 (d, 7=7.0 Hz, 6H), 0.66 (d, 7=7.0 Hz, 6H).
[0441] EXAMPLE 31
[0442] Synthesis of 3-((3-(2,2-dimethyl-4-oxo-3,8,ll,14,17,20,23,26,29-nonaoxa-5- azadotriacontan-32-amido)-4-(((2S,3R,4S,5R,6R)-3,4,5-trihydroxy-6- (hydroxymethyl)tetrahydro-2H-pyran-2-yl)oxy)benzyl)oxy)quinoline-2-carboxylic acid (127)
[0443] Linker 127 was synthesized as described below.

[0444] Synthesis of (2R,3S,4S,5R,6S)-2-(acetoxymethyl)-6-(4-(hydroxymethyl)-2- nitrophenoxy)tetrahydro-2H-pyran-3,4,5-triyl triacetate (122): 4-(hydroxymethyl)-2- nitrophenol (120, 3.00 g, 17.737 mmol) and (2R,3S,4S,5R,6R)-2-(acetoxymethyl)-6- bromotetrahydro-2H-pyran-3,4,5-triyl triacetate (121, 7.29 g, 17.737 mmol) were dissolved in acetonitrile (48 mL) at rt under argon. Ag2O (4.52 g, 19.510 mmol) was added and the suspension was stirred for 2 h. The reaction mixture was filtered through celite and the residue was washed with EtOAc. The organic layer was concentrated under reduced pressure and the resulting solid was washed with hexanes followed by Et20 to afford 122 (4.87 g, 9.751 mmol, 55%) as a faint yellow solid. MS (ESI, pos.) calc’d for C21H26NO13 [500.13]+; found (M+H) 500.16.
[0445] Synthesis of allyl 3-hydroxyqumoline-2-carboxylate (123)'. The title compound was prepared by reacting 3 -hydroxy quinoline -2 -carboxylic acid (1.00 g, 5.286 mmol) with ally bromide (686 pL, 7.929 mmol) in the presence of NaHCCh (888 mg, 10.573 mmol) in DMF (15 mL) at rt in air for overnight. The reaction mixture was diluted with EtOAc (200 mL) and the organic layer was washed with saturated solution of NaHCXL (50 mLx3). The organic layer was concentrated under reduced pressure and purified by normal phase chromatography using hexanes/EtOAc. The product came out in 85:15 combination of the solvents. The fractions were combined and concentrated to afford 123 (888 mg, 3.874 mmol, 73%) as a
white solid. Minor bisalkyation was also observed which was not collected. MS (ESI, pos.) calc’d for C13H12NO3 [230.07]+; found (M+H) 230.05.
[0446] Synthesis of (2R,3S,4S,5R,6S)-2-(acetoxymethyl)-6-(4-(((2- ((allyloxy)carbonyl)quinolin-3-yl)oxy)methyl)-2 -nitropheno xy)tetrahydro-2H-pyran-3, 4,5- triyl triacetate (124): 122 (1.27 g, 2.544 mmol) and triphenylphosphine (723 mg, 2.756 mmol) were dissolved in THF at 0 °C under argon. Solution of 123 (486 mg, 2.120 mmol) in THF (4 mL) was added followed by the addition of the solution of DIAD (557 mg, 2.756 mmol) in THF (2 mL). After 5 mins at the same temperature, the reaction mixture was transferred to rt. After 2 h at rt, the reaction mixture was concentrated under reduced pressure and purified by normal phase chromatography using hexanes/EtOAc. The product came out in 4:6 combination of the solvents. The fractions were combined and concentrated under reduced pressure to afford 124 (888 mg, 1.250 mmol, 59%) as a white solid. MS (ESI, pos.) calc’d for C34H35N2O15 [711.20]+; found (M+H) 711.52.
[0447] Synthesis of (2R,3S,4S,5R,6S)-2-(acetoxymethyl)-6-(4-(((2- ((allyloxy)carbonyl)quinolin-3-yl)oxy)methyl)-2-aminophenoxy)tetrahydro-2H-pyran-3,4,5- triyl triacetate (125): 124 (888 mg, 1.250 mmol) was dissolved in THF:MeOH:H2O (5 mL each) at rt in air. CaCL (1.39 g, 12.496 mmol) was added followed by the addition of Fe powder (696 mg, 12.496 mmol). The mixture was stirred at 70 °C for 2 h whereby all the starting material was found to have been consumed. The reaction mixture was filtered through celite and the residue was washed with EtOAc. The filtrate was concentrated under reduced pressure, adsorbed onto silica gel and the slurry was purified by normal phase chromatography using hexanes/EtOAc. The product came out in 3:7 combination of the solvents. The fractions were combined and concentrated to afford 125 (337 mg, 0.495 mmol, 40%) as a white solid. The reaction was found to be time sensitive and monitoring over time was required to get the desired product. MS (ESI, pos.) calc’d for C34H37N2O13 [681.22]+; found (M+H) 681.59.
[0448] Synthesis of (2R,3S,4S,5R,6S)-2-(acetoxymethyl)-6-(4-(((2- ((allyloxy)carbonyl)quinolin-3-yl)oxy)methyl)-2-(2,2-dimethyl-4-oxo- 3,8,l l,14,17,20,23,26,29-nonaoxa-5-azadotriacontan-32-amido)phenoxy)tetrahydro-2H- pyran-3,4,5-triyl triacetate (126): 125 (327 mg, 0.480 mmol) was dissolved in DMF (3 mL) at rt under argon. 2,2-dimethyl-4-oxo-3,8,l l,14,17,20,23,26,29-nonaoxa-5-azadotriacontan-32- oic acid (338 mg, 0.624 mmol) was added followed by the addition of HATU (274 mg, 0.721
mmol), HOAt (65.4 mg, 0.480 mmol) and DIPEA (243 pL, 1.441 mmol). The reaction mixture was stirred overnight and purified by reverse phase chromatography using acetonitrile in water each containing 0.05% of AcOH. The product came out in 55-60% of acetonitrile. The fractions were combined and lyophilized to afford 126 (576 mg, 0.478 mmol, 100%) as a white solid. MS (ESI, pos.) calc’d for CssHsiNsC^Na [1226.52]+; found (M+Na) 1227.11.
[0449] Synthesis of 3-((3-(2,2-dimethyl-4-oxo-3,8,l l,14,17,20,23,26,29-nonaoxa-5- azadotriacontan-32-amido)-4-(((2S,3R,4S,5R,6R)-3,4,5-trihydroxy-6- (hydroxymethyl)tetrahydro-2H-pyran-2-yl)oxy)benzyl)oxy)quinoline-2 -carboxylic acid (127): 126 (576 mg, 0.469 mmol) was dissolved in THF:MeOH:H2O (0.5 mL each) at rt in air. LiOH (112 mg, 4.686 mmol) was added in one portion. The reaction reached completion within 5 mins which was purified by reverse phase chromatography using acetonitrile in water each containing 0.05% of AcOH. The product came out in 35% of acetonitrile. The fractions were combined and lyophilized to afford 127 (356 mg, 0.357 mmol, 76%) as an off white solid. MS (ESI, pos.) calc’d for C47H70N3O20 [996.45]+; found (M+H) 996.95.
[0450] EXAMPLE 32
[0451] Synthesis of N-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-20-(3-((3-(l-amino- 3,6,9,12,15,18,21,24-octaoxaheptacosan-27-amido)-4-(((2S,3R,4S,5S,6R)-3,4,5- trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)oxy)benzyl)oxy)quinoline-2- carboxamido)-ll,24-diisopropyl-2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26- octaoxo-9,22-dioxa-2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7- yl)quinoxaline-2-carboxamide (129, M6577)
[0452] Linker payload 129 was synthesized from 19 and 127 as described below.

[0453] Synthesis of tert-butyl (27-((5-(((2-(((lS,4S,7R,l lS,14S,17S,20R,24S,Z)-l l,24- diisopropyl-2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-20-(quinoxaline-2- carboxamido)-9,22-dioxa-2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7- yl)carbamoyl)quinolin-3-yl)oxy)methyl)-2-(((2S,3R,4S,5R,6R)-3,4,5-trihydroxy-6- (hydroxymethyl)tetrahydro-2H-pyran-2-yl)oxy)phenyl)amino)-27-oxo-3,6,9,12,15,18,21,24- octaoxaheptacosyl)carbamate (128): To a solution of 19 (7.6 mg, 0.0085 mmol) and 127 (8.5 mg, 0.0085 mmol) in DMF (0.6 mL) at rt were added HATU (6.5 mg, 0.017 mmol) and NMM (43 p L, 0.043 mmol) respectively. After 20 mins of stirring, the reaction mixture was purified by reverse phase chromatography (30 g C18Aq, 5-30-40-50-100% of acetonitrile in water each containing 0.05% of AcOH). Product eluted in 50% of acetonitrile. Fractions were combined and lyophilized to afford 128 (11.7 mg, 0.0063 mmol, 73%) as a white solid. MS (ESI, pos.) calc’d for C90H128N13O30 [1870.88]+; found (M+H) 1872.54.
[0454] Synthesis of 129: To solution of 128 (11.7 mg, 0.0063 mmol) in DCM (2 mL) at 0 °C was added 220 pL of TFA. Within 30 mins at the same temperature, the reaction reached completion. The reaction mixture was concentrated under reduced pressure at 20 °C bath temperature. The material was redissolved in acetonitrile/water and lyophilized to afford 129 (8.4 mg, 0.0047 mmol, 76%) as a white solid. MS (ESI, pos.) calc’d for C85H120N13O28 [1770.83]+; found (M+H) 1771.59. 'H-NMR (500 MHz; DMSO-d6): <5 9.53 (s, 1H), 9.21 (br s, 1H), 8.78-8.60 (m, 2H), 8.56-8.54 (m, 1H), 8.28-8.18 (m, 1H), 8.17-7.56 (m, 5H), 7.96-77 (m, 6H), 7.60-7.48 (m, 2H), 7.38-6.88 (m, 2H), 6.22-5.89 (m, 2H), 5.40-5.22 (m, 5H), 4.88- 4.78 (m, 5H), 4.62-4.28 (m, 6H), 4.21-4.01 (m, 4H), 3.80-76 (m, 3H), 3.60-3.11 (m, 35H),
3.23-3.10 (m, 5H), 3.01-2.98 (m, 3H), 2.94-2.82 (m, 5H), 2.77-2.60 (m, 2H), 2.23-2.18 (m, 2H), 1.30-1.20 (m, 10H), 0.99-0.86 (m, 5H), 0.88-0.78 (m, 6H).
[0455] EXAMPLE 33
[0456] Synthesis of N-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-20-(3-((3-(l-amino- 3,6,9,12,15,18,21,24-octaoxaheptacosan-27-amido)-4-(((2S,3R,4S,5S,6R)-3,4,5- trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)oxy)benzyl)oxy)quinoline-2- carboxamido)-ll,24-dicyclopentyl-2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26- octaoxo-9,22-dioxa-2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7- yl)quinoxaline-2-carboxamide (131, M6579) [0457] Linker payload 131 was synthesized from 52 and 127 as described below.

[0458] Synthesis of tert-butyl (27-((5-(((2-(((lS,4S,7R,l lS,14S,17S,20R,24S,Z)-ll,24- dicyclopentyl-2,4, 12,15, 17,25-hexamethyl-3 ,6, 10, 13, 16,19,23 ,26-octaoxo-20-(quinoxaline-2 - carboxamido)-9,22-dioxa-2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7- yl)carbamoyl)quinolin-3-yl)oxy)methyl)-2-(((2S,3R,4S,5R,6R)-3,4,5-trihydroxy-6- (hydroxymethyl)tetrahydro-2H-pyran-2-yl)oxy)phenyl)amino)-27-oxo-3,6,9,12,15,18,21,24- octaoxaheptacosyl)carbamate (130): 52 (121 mg, 0.128 mmol) and 127 (128 mg, 0.128 mmol) were dissolved in DMF (1.5 mb) at rt. HATU (73 mg, 0.192 mmol) and NMM (43 pL, 0.384 mmol) were added. After 20 mins, the reaction mixture was purified by reverse
phase chromatography (30 g C18Aq, 5-35-100% of acetonitrile in water each containing 0.1% of HCOOH). The product came out in 35% of acetonitrile. The fractions were combined and lyophilized to afford 130 (197 mg, 0.102 mmol, 80%) as a white solid. MS (ESI, pos.) calc’d for C94H132N13O30 [1922.91]+; found (M+H) 1925.10.
[0459] Synthesis of 131 : 130 (197 mg, 0.1024 mmol) was dissolved in DCM and cooled to 0 °C. 500 pL of TFA was added. After completion, the reaction mixture was concentrated under reduced pressure. The contents were redissolved in DMF (2 mF) and purified by reverse phase chromatography (30 g C18Aq, using 5-35-50-100% of acetonitrile in water each containing 0.1% of HCOOH). The product came out in 35% of acetonitrile. The fractions were combined and lyophilized to afford 131 (156.2 mg, 0.086 mmol, 84%) as a white solid. MS (ESI, pos.) calc’d for C89H124N13O28 [1823.86]+; found (M+H) 1824.68. 'H- NMR (500 MHz; DMSO-d6): 9.52 (s, 1H), 9.25 (s, 1H), 8.65-8.24 (m, 1H), 8.38 (s, 1H), 8.30 (s, 1H), 8.18-8.16 (m, 1H), 8.04-8.01 (m, 3H), 7.99-7.88 (m, 8H), 7.83-7.80 (m, 8H), 7.54-7.46 (m, 1H), 7.23-7.20 (m, 3H), 6.04 (br s, 1H), 5.34-5.24 (m, 7H), 4.92-4.77 (m, 9H), 4.59-4.53 (m, 16H), 4.38-4.28 (m, 3H), 3,71-3.10 (m, 30H), 2.76-2.58 (m, 6H), 1.73-1.24 (m, 22H).
[0460] EXAMPLE 34
[0461] Synthesis of N-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-20-(3-((3-(l-amino- 3,6,9,12,15,18,21,24-octaoxaheptacosan-27-amido)-4-(((2S,3R,4S,5S,6R)-3,4,5- trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)oxy)benzyl)oxy)quinoline-2- carboxamido)-ll,24-dicyclohexyl-2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26- octaoxo-9,22-dioxa-2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7- yl)quinoxaline-2-carboxamide (133, M6580)
[0462] Linker payload 133 was synthesized from 56 and 127 as described below.

[0463] Synthesis of tert-butyl (27-((5-(((2-(((lS,4S,7R,l lS,14S,17S,20R,24S,Z)-l l,24- dicyclohexyl-2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-20-(quinoxaline-2- carboxamido)-9,22-dioxa-2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7- yl)carbamoyl)quinolin-3-yl)oxy)methyl)-2-(((2S,3R,4S,5R,6R)-3,4,5-trihydroxy-6- (hydroxymethyl)tetrahydro-2H-pyran-2-yl)oxy)phenyl)amino)-27-oxo-3,6,9,12,15,18,21,24- octaoxaheptacosyl)carbamate (132): 56 (8 mg, 0.008 mmol) and 127 (10.6 mg, 0.011 mmol) were dissolved in DMF (0.5 mL) at rt. HATU (9.4 mg, 0.025 mmol) was added followed by the addition of NMM (4.5 p L, 0.041 mmol). After 20 mins, the reaction mixture was purified by reverse phase chromatography (30 g C18Aq, 5-30-40-50-100% of acetonitrile in water each containing 0.05% of AcOH. Product eluted in 50% of acetonitrile. Fractions were combined and lyophilized to afford 132 (11.1 mg, 0.006 mmol, 69%) as a white solid. MS (ESI, pos.) calc’d for C96H136N13O30 [1950.94]+; found (M+H) 1951.64.
[0464] Synthesis of 133: 132 (11 mg, 0.056 mmol) was dissolved in DCM (2 mL) and cooled to 0 °C. 220 pL of TFA was added and the reaction mixture was stirred at the same temperature for 30 mins whereby the reaction was found to have reached completion. The reaction mixture was concentrated under reduced pressure at 20 °C bath temperature. The material was redissolved in acetonitrile/water and lyophilized to afford 133 (7.2 mg, 0.004 mmol, 69%) as a white solid. MS (ESI, pos.) calc’d for C91H128N13O28 [ 1850.89]+; found (M+H) 1851.72. H-NMR (500 MHz; DMSO-d6): <5 9.53 (s, 1H), 9.22 (br s, 1H), 8.68-8.59
(m, 2H), 8.31 (br s, 1H), 8.22 -8.19 (m, 1H), 8.18-7.62 (m, 11H), 7.60-7.46 (m, 2H), 7.30- 7.01 (m, 2H), 6.10-5.90 (m, 2H), 5.48-5.22 (m, 4H), 4.99-4.60 (m, 5H), 4.56-4.22 (m, 7H), 4.20-3.80 (m, 4H), 3.66-3.60 (m, 4H), 3.59-2.89 (m, 34H), 3.20-3.18 (m, 4H), 3.01-2.89 (m, 2H), 2.88-2.76 ( m, 4H), 2.60-2.58 (m, 3H), 2.50-2.44 (m, 2H), 2.01-0.88 (m, 31H).
[0465] EXAMPLE 35
[0466] Synthesis of N-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-20-(3-((4-((29S,32S)-l-amino- 29-isopropyl-32-methyl-27,30-dioxo-3,6,9,12,15,18,21,24-octaoxa-28,31- diazatritriacontan-33-amido)benzyl)oxy)quinoline-2-carboxamido)-2,4,12,15,17,25- hexamethyl-ll,24-bis((R)-3-methylbutan-2-yl)-3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa- 2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7-yl)quinoxaline-2-carboxamide (134, TCRS-3804, M6565)
[0467] Linker payload 134 was synthesized from 106 and 7 as described below.

[0468] Synthesis of 134: Compound 134 was synthesized from 106 (5.0 mg, 0.0053) and 7 (6.2 mg, 0.0063 mmol) as described in the synthesis of compound 36. The crude was injected into Cl 8 column and eluted with 30-45% ACN/H2O + 0.1% formic acid. Product 134 was obtained as a white solid (2.9 mg, 0.0011 mmol, 21% yield over two steps). MS (ESI, pos.) calc’d for C91H132N15O24 [1818.96]+; found 1820.93 (M+H). ’H-NMR (300 MHz; DMSO- d6): 8 10.02 (d, 7 = 6.8 Hz, 1H), 9.55-9.51 (m, 1H), 8.80-8.66 (m, 1H), 8.49-8.40 (m, 2H), 8.28-8.19 (m, 1H), 8.13-8.08 (m, 1H), 8.07-7.81 (m, 8H), 7.66-7.41 (m, 6H), 7.05-6.81 (m, 2H), 6.35-6.31 (m, 1H), 6.01-5.92 (m, 1H), 5.39-5.23 (m, 4H), 5.14-5.03 (m, 2H), 4.95-4.75 (m, 2H), 4.63-4.31 (m, 6H), 4.25-4.18 (m, 1H), 3.63-3.57 (m, 2H), 3.49 (d, 7 = 25.5 Hz, 30H), 3.10 (dd, 7 = 12.3, 7.0 Hz, 6H), 2.94-2.84 (m, 6H), 2.80-2.67 (m, 4H), 2.46-2.26 (m, 6H), 2.10-2.08 (m, 1H), 2.02-1.94 (m, 1H), 1.76-1.64 (m, 1H), 1.35-1.19 (m, 11H), 0.92-0.75 (m, 18H), 0.71-0.58 (m, 6H).
[0469] EXAMPLE 36
[0470] Synthesis of 3-((4-((29S,32S)-l-amino-29-isopropyl-32-methyl-27,30-dioxo- 3,6,9,12,15,18,21,24-octaoxa-28,31-diazatritriacontan-33-amido)benzyI)oxy)-N- ((lS,4S,7R,llS,14S,17S,20R,24S,Z)-20-(3-hydroxyquino!ine-2-carboxamido)- 2,4,12,15,17,25-hexamethyI-ll,24-bis((R)-3-methyIbutan-2-yI)-3,6,10,13,16,19,23,26- octaoxo-9,22-dioxa-2,5,12,15,18,25-hexaazabicydo[12.12.4]triacont-28-en-7- yI)quino!ine-2-carboxamide (135, M6573)
[0471] Linker payload 135 was synthesized from 109 and 7 as described below.

[0472] Synthesis of 135: Compound 135 was synthesized from 109 (6.3 mg, 0.0065) and 7 (7.7 mg, 0.0078 mmol) as described in the synthesis of compound 36. The crude was injected into Cl 8 column and eluted with 40-50% ACN/H2O + 0.1% formic acid. Product 135 was obtained as a white solid (5.9 mg, 0.0032 mmol, 49% yield over two steps). MS (ESI, pos.) calc’d for C92H133N14O25 [1833.96]+; found 1835.78 (M+H). 'H-NMR (300 MHz; DMSO- d6): 8 10.00 (s, 1H), 8.70 (dd, J = 8.8, 0.6 Hz, 1H), 8.24-8.22 (m, 2H), 8.14-8.12 (m, 1H), 8.04 (s, 2H), 7.92-7.80 (m, 8H), 7.66-7.54 (m, 7H), 7.47 (dd, 7 = 7.2, 3.5 Hz, 2H), 6.58 (s, 1H), 6.00-5.95 (m, 2H), 5.36-5.27 (m, 4H), 5.12-5.07 (m, 2H), 4.91-4.81 (m, 2H), 4.62-4.37 (m, 6H), 4.26-4.20 (m, 1H), 3.57 (d, 7 = 4.0 Hz, 2H), 3.53-3.49 (m, 30H), 3.12 (d, 7 = 18.6 Hz, 6H), 2.96 (s, 2H), 2.90 (d, 7 = 2.6 Hz, 6H), 2.77-2.73 (m, 1H), 2.51-2.29 (m, 4H), 2.10- 2.09 (m, 2H), 2.00-1.90 (m, 1H), 1.81-1.58 (m, 1H), 1.33-1.25 (m, 11H), 0.89-0.76 (m, 18H), 0.66-0.62 (m, 6H).
[0473] EXAMPLE 37
[0474] Synthesis of N-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-20-(3-((4-((29S,32S)-l-amino-
29-isopropyI-32-methyI-27,30-dioxo-3,6,9,12,15,18,21,24-octaoxa-28,31- diazatritriacontan-33-amido)benzyI)oxy)quinoIine-2-carboxamido)-ll,24-dicycIopentyI-
2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa-2,5,12,15,18,25- hexaazabicyclo[12.12.4]triacont-28-en-7-yl)quinoxaline-2-carboxamide (136, M6564)
[0475] Linker payload 136 was synthesized from 52 and 7 as described below.

[0476] Synthesis of 136: Compound 136 was synthesized from 52 (6.0 mg, 0.0063) and 7 (7.5 mg, 0.0076 mmol) as described in the synthesis of compound 36. The crude was injected into Cl 8 column and eluted with 30-45% ACN/H2O + 0.1% formic acid. Product 136 was obtained as a white solid (5.2 mg, 0.0029 mmol, 46% yield over two steps). MS (ESI, pos.) calc’d for C91H128N15O24 [1814.93]+; found 1816.71 (M+H). ’H-NMR (500 MHz; DMSO- d6): 8 10.04-9.98 (m, 1H), 9.56-9.51 (m, 1H), 8.82-8.62 (m, 2H), 8.51-8.44 (m, 3H), 8.29- 8.18 (m, 2H), 8.11-7.82 (m, 8H), 7.64-7.44 (m, 4H), 6.09-6.01 (m, 2H), 5.40-5.24 (m, 4H), 4.95-4.73 (m, 4H), 4.64-4.50 (m, 4H), 4.43-4.17 (m, 4H), 3.66-3.19 (m, 32H), 3.19-2.98 (m, 8H), 2.98-2.81 (m, 6H), 2.81-2.63 (m, 4H), 2.44-2.24 (m, 5H), 2.02-1.92 (m, 1H), 1.78-1.71 (m, 1H), 1.55-1.47 (m, 4H), 1.47-1.16 (m, 17H), 1.15-0.97 (m, 4H), 0.91-0.65 (m, 6H).
[0477] EXAMPLE 38
[0478] Synthesis of 3-((3-(l-amino-3,6,9,12,15,18,21,24-octaoxaheptacosan-27-amido)-4- (((2S,3R,4S,5R,6R)-3,4,5-trihydroxy-6-(hydroxymethyI)tetrahydro-2H-pyran-2- yl)oxy)benzyl)oxy)-N-((lS,4S,7R,HS,14S,17S,20R,24S,Z)-20-(3-hydroxyquinoline-2- carboxamido)-ll,24-diisopropyl-2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26- octaoxo-9,22-dioxa-2,5,12,15,18,25-hexaazabicydo[12.12.4]triacont-28-en-7- yI)quino!ine-2-carboxamide (137, M6661)
[0479] Linker payload 137 was synthesized from 35 and 127 as described below.

[0480] Synthesis of 137: Compound 137 was synthesized from 35 (3.0 mg, 0.0033) and 127 (3.9 mg, 0.0040 mmol) as described in the synthesis of compound 131. The crude was injected into Cl 8 column and eluted with 35-45% ACN/H2O + 0.1% formic acid. Product 137 was obtained as a white solid (2.7 mg, 0.0015 mmol, 45% yield over two steps). MS
(ESI, pos.) calc’d for C86H119N12O29 [1783.82]’; found 1784.52 (M-H). 'H-NMR (300 MHz; DMSO-d6): 3 9.33-9.24 (m, 1H), 8.74-8.66 (m, 1H), 8.48-8.42 (m, 2H), 8.37-8.30 (m, 1H), 8.08-7.96 (m, 1H), 7.94-7.81 (m, 4H), 7.80-7.73 (m, 2H), 7.69-7.48 (m, 3H), 7.47-7.35 (m, 2H), 7.27-7.15 (m, 2H), 6.09-5.97 (m, 2H), 5.39-5.21 (m, 5H), 4.89-4.72 (m, 6H), 4.67-3.90 (m, 6H), 3.74-3.66 (m, 4H), 3.53-3.48 (m, 30 H), 3.17-3.06 (m, 12H), 2.87-2.80 (m, 6H),
2.84-2.60 (m, 6H), 2.36-2.16 (m, 6H), 1.29-1.16 (m, 6H), 0.97-0.65 (m, 12H).
[0481] EXAMPLE 39
[0482] Synthesis of N-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-20-(3-((3-(l-amino- 3,6,9,12,15,18,21,24-octaoxaheptacosan-27-amido)-4-(((2S,3R,4S,5R,6R)-3,4,5- trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)oxy)benzyl)oxy)quinoline-2- carboxamido)-2,4,12,15,17,25-hexamethyl-ll,24-bis((R)-3-methylbutan-2-yl)- 3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa-2,5,12,15,18,25- hexaazabicyclo[12.12.4]triacont-28-en-7-yl)quinoxaline-2-carboxamide (138, M6578)
[0483] Linker payload 138 was synthesized from 106 and 127 as described below.

[0484] Synthesis of 138: Compound 138 was synthesized from 106 (6.3 mg, 0.0066) and 127 (7.3 mg, 0.0073 mmol) as described in the synthesis of compound 131. The crude was injected into Cl 8 column and eluted with 35-45% ACN/H2O + 0.1% formic acid. Product 138 was obtained as a white solid (3.6 mg, 0.0020 mmol, 30% yield over two steps). MS (ESI, pos.) calc’d for C89H126N13O28 [1824.88]’; found 1825.54 (M-H). 'H-NMR (300 MHz; DMSO-de): <5 9.55 (s, 1H), 9.22-9.21 (m, 1H), 8.73-8.62 (m, 2H), 8.33 (d, 7 = 5.6 Hz, 1H), 8.21 (dt, 7 = 5.9, 3.1 Hz, 1H), 8.09-7.92 (m, 4H), 7.92-7.79 (m, 2H), 7.77-7.67 (m, 2H), 7.67- 7.50 (m, 2H), 7.30-7.19 (m, 2H), 6.03-5.91 (m, 2H), 5.43-5.21 (m, 5H), 5.19-5.03 (m, 3H), 4.98-4.74 (m, 3H), 4.69-4.29 (m, 6H), 4.28-3.88 (m, 6H), 3.79-3.69 (m, 2H), 3.62-3.44 (m, 30H), 3.13-3.06 (m, 6H), 3.04-2.94 (m, 2H), 2.94-2.84 (m, 6H), 2.82-2.63 (m, 3H), 2.39-2.25 (m, 3H), 2.15-1.95 (m, 3H), 1.77-1.62 (m, 2H), 1.52-1.39 (m, 1H), 1.34-1.12 (m, 8H), 0.87- 0.74 (m, 10H), 0.74-0.56 (m, 8H).
[0485] EXAMPLE 40
[0486] Synthesis of 3-((3-(l-amino-3,6,9,12,15,18,21,24-octaoxaheptacosan-27-amido)-4-
(((2S,3R,4S,5R,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2- yl)oxy)benzyl)oxy)-N-((lS,4S,7R,HS,14S,17S,20R,24S,Z)-20-(3-hydroxyquinoline-2- carboxamido)-2,4,12,15,17,25-hexamethyl-ll,24-bis((R)-3-methylbutan-2-yl)-
3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa-2,5,12,15,18,25- hexaazabicyclo[12.12.4]triacont-28-en-7-yl)quinoline-2-carboxamide (139, M6718)
[0487] Linker payload 139 was synthesized from 108 and 127 as described below.

[0488] Synthesis of 139: Compound 139 was synthesized from 108 (1.8 mg, 0.0019) and 127 (2.2 mg, 0.0022 mmol) as described in the synthesis of compound 131. The crude was injected onto a C18 column and eluted with 35-45% ACN/H2O + 0.1% formic acid. Product 139 was obtained as a white solid (1.1 mg, 0.0006 mmol, 32% yield over two steps). MS (ESI, pos.) calc’d for C90H129N12O29 [1841.90]’; found (M+H) 1843.15. ’H-NMR (500 MHz; DMSO-d6): 3 9.24-9.19 (m, 1H), 8.66-8.60 (m, 1H), 8.57-8.45 (m, 7H), 8.34-8.28 (m, 1H), 8.05-7.98 (m, 1H), 7.92-7.81 (m, 3H), 7.77-7.48 (m, 5H), 7.41-7.14 (m, 6H), 6.91-6.73 (m, 7H), 5.36-5.22 (m, 4H), 5.07-4.99 (m, 2H), 4.90-4.80 (m, 2H), 4.63-4.55 (m, 3H), 4.52-4.39 (m, 2H), 3.74-3.66 (m, 2H), 3.61-3.44 (m, 30H), 3.20-3.06 (m, 6H), 2.94-2.81 (m, 6H), 2.70- 2.58 (m, 4H), 2.40-2.26 (m, 3H), 2.20-2.06 (m, 1H), 1.73-1.66 (m, 1H), 1.30-1.14 (m, 10H), 0.91-0.57 (m, 20H).
[0489] EXAMPLE 41
[0490] Synthesis of 3-Amino-N-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-ll,24-dicyclopentyl-
20-(3-hydroxyquinoIine-2-carboxamido)-2,4,12,15,17,25-hexamethyI-
3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa-2,5,12,15,18,25- hexaazabicyclo[12.12.4]triacont-28-en-7-yl)quinoline-2-carboxamide (141, M6760)
[0491] Payload 141 was synthesized from intermediate 140 which in turn was synthesized as explained for intermediate 35 in example 3 by replacing A-(((9/f-fluoren-9- yl)methoxy)carbonyl)-A-methyl-L-valine with (5,)-2-((((9/f-fluoren-9- yl)methoxy)carbonyl)(methyl)amino)-2 -cyclopentylacetic acid.

[0492] Synthesis of 141: The title compound was synthesized by reacting 140 (7.0 mg, 0.0073 mmol) with 3-aminoquinoline-2-carboxylic acid (2.7 mg, 0.0146 mmol) in the presence of HATU (5.5 mg, 0.0146 mmol) and NMM (4 pL, 0.0365 mmol) in DMF (0.5 mL) at room temperature. Work up and chromatography (Isco EZ Prep (Gemini Cl 8 column, 30 x 150 mm, 5-65-100% of acetonitrile in water, each containing 0. 1% of HCOOH) followed by lyophilization of the fractions afforded 141 (2.5 mg, 0.0022 mmol, 30%) as a yellow solid. MS (ESI, pos.) calc’d for C58H72N11O13 [1130.52]’; found (M+H) 1131.34. ’H-NMR (500 MHz; DMSO-de): 3 11.8 (br, s, 1H), 8.84 (d, 7=9.0 Hz, 1H), 8.08-8.01 (m, 1H), 7.85-7.76 (m, 2H), 7.61 (d, 7=8.5 Hz, 1H), 7.57-7.56 (m, 3H), 7.53-7.45 (m, 3H), 7.32-7.29 (m, 3H), 6.81
(br, s, 2H), 6.06-6.04 (m, 1H), 5.35 (br, s, 1H), 4.90-4.80 (m, 6H), 4.64-4.53 (m, 6H), 4.42- 4.37 (m, 2H), 3.22 (s, 3H), 3.18 (s, 3H), 2.87 (br, s, 6H), 2.60-2.40 (m, 5H), 1.71-1.20 (m, 21H).
[0493] EXAMPLE 42 [0494] Synthesis of N-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-20-(3-aminoquinoline-2- carboxamido)-ll,24-dicyclopentyl-2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26- octaoxo-9,22-dioxa-2,5,12,15,18,25-hexaazabicydo[12.12.4]triacont-28-en-7- yI)quinoxa!ine-2-carboxamide (142, M6769)
[0495] Payload 142 was synthesized from intermediate 52 as described below.

[0496] Synthesis of 142: The title compound was synthesized by reacting 52 (7.2 mg, 0.0076 mmol) with 3-aminoquinoline-2-carboxylic acid (2.15 mg, 0.0114 mmol) in the presence of HATU (4.3 mg, 0.0114 mmol) and NMM (2.5 pL, 0.0229 mmol) in DMF (0.5 mL) at room temperature in air. Work up and reverse phase chromatography (15.5 g C18Aq, 5-60-100% of acetonitrile in water each containing 0.1 % of HCOOH) afforded pure fractions that were combined and lyophilized to afford 142 (7.1 mg, 0.0064 mmol, 84%) as a yellow solid. MS (ESI, pos.) calc’d for C57H71N12O12 [1115.52]’; found (M+H) 1116.34. ’H-NMR (500 MHz; DMSO-de): <5 9.52 (s, 1H), 8.84 (d, 7=9.0 Hz, 1H), 8.56 (d, 7=9.5 Hz, 1H), 8.15 (d, 7=8.5 Hz, 1H), 8.01-7.87 (m, 5H), 7.60 (d, 7=8.0 Hz, 1H), 7.53 (d, 7=8.5 Hz, 1H), 7.45 (s, 1H), 7.29- 7.25 (m, 2H), 7.81 (br, s, 2H), 6.06-6.03 (m, 2H), 5.35 (br, s, 2H), 4.93-4.82 (m, 5H), 4.63- 4.54 (m, 4H), 4.39-4.37 (m, 2H), 3.21 (s, 3H), 3.15 (s, 3H), 2.88-2.87 (m, 6H), 2.86-2.66 (m, 6H), 1.71-1.21 (m, 21H).
[0497] EXAMPLE 43
[0498] Synthesis of N,N'-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-ll,24-dicyclopentyl- 2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa-2,5,12,15,18,25- hexaazabicyclo[12.12.4]triacont-28-ene-7,20-diyl)bis(3-aminoquinoline-2-carboxamide) (145, M6717)
[0499] Payload 145 was synthesized from intermediate 144 which in turn was synthesized as described for intermediate 104 in example 26 by replacing (2S,3R)-2-((((9H-fluoren-9- yl)methoxy)carbonyl)(methyl)amino)-3,4-dimethylpentanoic acid with (5')-2-((((9/f-fluoren- 9-yl)methoxy)carbonyl)(methyl)amino)-2-cyclopentylacetic acid.

[0500] Synthesis of 145: The title compound was synthesized by reacting 144 (41 mg, 0.052 mmol) with 3-aminoquinoline-2-carboxylic acid (29 mg, 0.156 mmol) in the presence of HATU (60 mg, 0. 156 mmol) and NMM (44 pL, 0.260 mmol) in DMF (1.0 mL) at room temperature in air. Work up and reverse phase chromatography (Isco EZ Prep (Gemini Cl 8 column, 30 x 150 mm, 5-55-100% of acetonitrile in water, each containing 0.1% of HCOOH) followed by lyophilization of the pure fractions afforded 145 (11.8 mg, 0.0104 mmol, 20%) as a yellow solid. MS (ESI, pos.) calc’d for C58H72N12O12 [1129.54]"; found (M+H) 1130.36. ’H-NMR (300 MHz; DMSO-d6): <5 8.84 (d, 7=9.3 Hz, 2H), 8.07 (d, 7=6.3 Hz, 2H), 7.64-7.56 (m, 4H), 7.45 (s, 2H), 7.35-7.30 (m, 4H), 6.86-6.81 (m, 4H), 6.09-6.03 (m, 2H), 5.33-5.31
(m, 2H), 4.87-4.64 (m, 4H), 4.63-4.51 (m, 4H), 4.40-4.36 (m, 2H), 3.23 (s, 6H), 2.87 (s, 6H), 2.38-2.30 (m, 5H), 1.66-1.12 (m, 23H).
[0501] EXAMPLE 44
[0502] Synthesis of N,N'-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-ll,24-dicyclopentyl- 2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa-2,5,12,15,18,25- hexaazabicydo[12.12.4]triacont-28-ene-7,20-diyI)bis(3-hydroxyquinoIine-2- carboxamide) (146, M6726)
[0503] Payload 146 was synthesized from intermediate 144 as shown below.

[0504] Synthesis of 146: The title compound was synthesized by reacting 144 (33 mg, 0.042 mmol) with 3-hydroxyquinoline-2-carboxylic acid (24 mg, 0.125 mmol) in the presence of HATU (48 mg, 0.125 mmol) and NMM (28 pL, 0.251 mmol) in DMF (0.5 mL) at room temperature in air. Work up and reverse phase chromatography (Isco EZ Prep (Gemini Cl 8 column, 30 x 150 mm, 5-70-100% of acetonitrile in water, each containing 0.1% of HCOOH) followed by lyophilization of the pure fractions afforded 146 (8.0 mg, 0.0071 mmol, 17%) as a white solid. MS (ESI, pos.) calc’d for C58H71N10O14 [1131.51]"; found (M+H) 1132.33. 'H- NMR (500 MHz; DMSO-d6): <5 11.88 (s, 2H), 8.84 (d, 7=9.0 Hz, 2H), 8.07 (d, 7=6.4 Hz, 2H), 7.64-7.56 (m, 4H), 7.45 (s, 2H), 7.35-7.30 (m, 4H), 6.09-6.03 (m, 2H), 5.33-5.31 (m, 2H),
4.87-4.64 (m, 4H), 4.63-4.51 (m, 4H), 4.40-4.36 (m, 2H), 3.23 (s, 6H), 2.87 (s, 6H), 2.38- 2.30 (m, 5H), 1.66-1.12 (m, 23H).
[0505] EXAMPLE 45
[0506] Linker 150 was synthesized as described below.

[0507] Synthesis of Allyl 3-aminoquinoline-2-carboxylate (147)'. 3-Aminoquinoline-2- carboxylic acid (800 mg, 4.251 mmol) was dissolved in 12 mL of DMF at rt in air. NaHCCL (1.071 g, 12.753 mmol) was added followed by the addition of allyl bromide (735 L, 8.502 mmol) and the reaction mixture was stirred overnight. The reaction mixture was diluted with
EtOAc (150 mL) and the organic layer was washed with saturated solution of NaHCCL (50 mLx3). The organic layer was concentrated under reduced pressure and the residue was purified by normal phase chromatography (hexanes/EtOAc, 8 :2). The fractions were combined and concentrated to afford the desired product (147, 580 mg, 2.541 mmol, 60%) as a white solid. The undesired product was not collected. MS (ESI, pos.) calc’d for C13H13N2O2 [229.09]’; found (M+H) 229.34.
[0508] Synthesis of allyl 3-((((4-((S)-2-((S)-2-amino-3- methylbutanamido )propanamido )benzyl )oxy )carbonyl )amino )quinoline-2-carboxylate (148): 147 (480 mg, 2.103 mmol) was dissolved in DCM (40 mL) and cooled to 0 °C. Solution of triphosgene (437 mg, 1.472 mmol) in DCM (5 mL) was added drop wise followed by the addition of pyridine (542 pL, 6.729 mmol) and solution of DMAP (257 mg, 2.103 mmol) in DCM (5 mL) respectively. The aniline was consumed within 10 mins at the same temperature as revealed by quenching with propylamine. Alcohol (827 mg, 2.103 mmol) was added in one portion and reaction mixture was allowed to warm to rt overnight. The reaction mixture was concentrated under reduced pressure and the yellow contents were adsorbed onto silica gel and purified by normal phase chromatography (80 g column, hexanes/EtOAc, 2:8). The fractions were combined and concentrated to afford the product as a white solid (1267 mg). The product was dissolved in DCM (24 mL) and treated with 7 mL of TFA at room temperature until the Boc group was gone. The contents were concentrated under reduced pressure and the yellow contents were adsorbed onto silica gel and purified by normal phase chromatography (40 g column). The column was first flushed with 80% of EtOAc in hexane. The product was isolated in 9:1 combination of DCM/MeOH. The fractions were combined and concentrated to afford 148 (1.073 g, 1.959 mmol, 93%) as a white solid. MS (ESI, pos.) calc’d for C29H34N5O6 [548.24]’; found (M+H) 548.67.
[0509] Synthesis of allyl 3-((((4-(( 34S, 37S)-34-isopropyl-2, 2,37 -trimethyl-4, 32,35-trioxo- 3, 8, 11, 14, 17,20,23, 26,29-nonaoxa-5, 33, 36-triazaoctatriacontan-38- amido)benzyl)oxy)carbonyl)amino)quinoline-2-carboxylate (149): 148 (211 mg, 0.385 mmol) and t-Boc-N-amido-PEGs-COOH (230 mg, 0.424 mmol) were dissolved in DMF (2 mL) at rt in air. HATU (220 mg, 0.578 mmol) was added followed by the addition of NMM (128 L, 1.156 mmol) respectively. The reaction reached completion within 20 mins which was purified by reverse phase chromatography (50 g C18Aq, 5-55-100% of acetonitrile in water each containing 0.1% of HCOOH). The product came out in 55% of acetonitrile. The
fractions were combined and lyophilized to afford 149 (284 mg, 0.265 mmol, 69%) as a white solid. MS (ESI, pos.) calc’d for C53H79N6O17 [1071.54]"; found (M+H) 1072.38.
[0510] Synthesis of3-(((( 4-( ( 34S,37S )-34-isopropyl-2, 2,37-trimethyl-4, 32, 35-trioxo- 3, 8, 11, 14, 17,20,23, 26,29-nonaoxa-5, 33, 36-triazaoctatriacontan-38- amido)benzyl)oxy)carbonyl)amino)quinoline-2-carboxylic acid (150): 149 (264 mg, 0.246 mmol) was dissolved in DCM (6 mL) at rt under argon. Pd(PPh3)4 (14 mg, 0.012 mmol) was added followed by the drop wise addition of phenylsilane (46 pL, 0.370 mmol). The reaction reached completion within 20 mins. The reaction mixture was concentrated under reduced pressure, the contents were redissolved in DMF (3 mL) and purified by reverse phase chromatography (50 g C18Aq, 5-45-100% of acetonitrile in water each containing 0.1% of HCOOH). The product came out in 45% of acetonitrile. The fractions were combined and lyophilized to afford 150 (181 mg, 0.175 mmol, 71%) as a white solid. MS (ESI, pos.) calc’d for C5oH74N6Oi7Na [1053.51]"; found (M+Na) 1054.23.
[0511] EXAMPLE 46
[0512] Synthesis of 4-((29S,32S)-l-Amino-29-isopropyl-32-methyl-27,30-dioxo- 3,6,9,12,15,18,21,24-octaoxa-28,31-diazatritriacontan-33-amido)benzyl (2- (((lS,4S,7R,llS,14S,17S,20R,24S,Z)-20-(3-aminoquinoline-2-carboxamido)-ll,24- dicyclopentyl-2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa- 2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7-yl)carbamoyl)quinolin-3- yl)carbamate (152, M6743)
[0513] Linker Payload 152 was synthesized from intermediate 151 which in turn was synthesized as explained for intermediate 140 by replacing A-(((9Z7-fluoren-9- yl)methoxy)carbonyl)-A-methyl-L-valine with (S')-2-((((9Z7-fluoren-9- yl)methoxy)carbonyl)(methyl)amino)-2-cyclopentylacetic acid.
 [0514] Synthesis of 152: 151 (18 mg, 0.0188 mmol), 150 (21 mg, 0.0206 mmol) and HATU were dissolved in DMF at rt in air. NMM (10.4 pL, 0.0938 mmol) was added. The reaction reached completion within 10 mins as revealed by UPLC. The reaction mixture was purified by reverse phase chromatography (30 g C18Aq, 5-60-100% of acetonitrile in water each containing 0.1% of HCOOH). The product came out in 60% of acetonitrile. The fractions were combined to afford 18.1 mg of the Boc amine as a white solid which was dissolved in 2 mL of DCM and treated with 0.4 mL of TFA at rt. The reaction mixture was concentrated under reduced pressure and the contents were dissolved in DMF and purified by reverse phase chromatography (Isco EZ Prep (Gemini C18 column, 30 x 150 mm, 5-50-100% of acetonitrile in water, each containing 0.1% of HCOOH). The fractions were combined and lyophilized to afford 152 (13.2 mg, 0.0071 mmol, 77%) a yellow solid. MS (ESI, pos.) calc’d for C93H131N16O25 [1871.94]’; found (M+H) 1872.99. ’H-NMR (500 MHz; DMSO-d6): <5 10.01 (s, 1H), 9.09-9.00 (m, 2H), 8.24-8.23 (m, 1H), 8.05-8.01 (m, 1H), 7.95-7.89 (m, 2H), 7.78-7.72 (m, 2H), 7.65-7.59 (m, 3H), 7.52-7.42 (m, 5H), 7.29-7.10 (m, 2H), 6.82 (br, s, 2H), 6.05-6.02 (m, 1H), 5.34-5.18 (m, 3H), 4.89-4.81 (m, 3H), 4.63-4.53 (m, 3H), 4.41-4.36 (m, 2H), 4.22-4.19 (m, 1H), 3.60-3.37 (m, 68H), 2.38-2.30 (m, 5H), 1.66-1.12 (m, 23H).
[0514] Synthesis of 152: 151 (18 mg, 0.0188 mmol), 150 (21 mg, 0.0206 mmol) and HATU were dissolved in DMF at rt in air. NMM (10.4 pL, 0.0938 mmol) was added. The reaction reached completion within 10 mins as revealed by UPLC. The reaction mixture was purified by reverse phase chromatography (30 g C18Aq, 5-60-100% of acetonitrile in water each containing 0.1% of HCOOH). The product came out in 60% of acetonitrile. The fractions were combined to afford 18.1 mg of the Boc amine as a white solid which was dissolved in 2 mL of DCM and treated with 0.4 mL of TFA at rt. The reaction mixture was concentrated under reduced pressure and the contents were dissolved in DMF and purified by reverse phase chromatography (Isco EZ Prep (Gemini C18 column, 30 x 150 mm, 5-50-100% of acetonitrile in water, each containing 0.1% of HCOOH). The fractions were combined and lyophilized to afford 152 (13.2 mg, 0.0071 mmol, 77%) a yellow solid. MS (ESI, pos.) calc’d for C93H131N16O25 [1871.94]’; found (M+H) 1872.99. ’H-NMR (500 MHz; DMSO-d6): <5 10.01 (s, 1H), 9.09-9.00 (m, 2H), 8.24-8.23 (m, 1H), 8.05-8.01 (m, 1H), 7.95-7.89 (m, 2H), 7.78-7.72 (m, 2H), 7.65-7.59 (m, 3H), 7.52-7.42 (m, 5H), 7.29-7.10 (m, 2H), 6.82 (br, s, 2H), 6.05-6.02 (m, 1H), 5.34-5.18 (m, 3H), 4.89-4.81 (m, 3H), 4.63-4.53 (m, 3H), 4.41-4.36 (m, 2H), 4.22-4.19 (m, 1H), 3.60-3.37 (m, 68H), 2.38-2.30 (m, 5H), 1.66-1.12 (m, 23H).
[0515] EXAMPLE 47
[0516] Synthesis of 4-((29S,32S)-l-amino-29-isopropyl-32-methyl-27,30-dioxo- 3,6,9,12,15,18,21,24-octaoxa-28,31-diazatritriacontan-33-amido)benzyl (2- (((lS,4S,7R,llS,14S,17S,20R,24S,Z)-ll,24-dicyclopentyl-20-(3-hydroxyquinoline-2- carboxamido)-2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa- 2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7-yl)carbamoyl)quinolin-3- yl)carbamate (153, M6762)
[0517] Linker Payload 153 was synthesized from intermediate 140 as outlined below.
 [0518] Synthesis of 153: The title compound was synthesized following the procedure described for 152 above. Accordingly, 140 (12.1 mg, 0.0126 mmol) was reacted with 150 (13 mg, 0.0126 mmol) in the presence of HATU (9.6 mg, 0.0252 mmol) and NMM (4.2 pL, 0.0378 mmol) in DMF (0.5 mL) at rt in air. Work up and chromatography by reverse phase chromatography (15.5 g C18Aq, 5-80-100% of acetonitrile in water each containing 0.1% of HCOOH) followed by lyophilization of the fractions afforded Boc containing amine 14.6 mg which was dissolved in DCM (2 mL) at rt and treated with TFA. After completion of the reaction, work up and chromatography again (15.5 g C18Aq, 5-60-100% of acetonitrile in water each containing 0.1% of HCOOH) followed by lyophilization afforded 153 (10.5 mg, 0.0056 mmol, 44%) as a white solid. MS (ESI, pos.) calc’d for C93H130N15O26 [1872.92]"; found (M+H) 1875.06. ’H-NMR (500 MHz; DMSO-d6): <5 11.88 (s, 1H), 11.44 (s, 1H), 10.01 (s, 1H), 9.09-9.00 (m, 2H), 8.24-8.23 (m, 1H), 8.05-8.01 (m, 1H), 7.95-7.89 (m, 2H), 7.78- 7.72 (m, 2H), 7.65-7.59 (m, 3H), 7.52-7.42 (m, 5H), 7.29-7.10 (m, 2H), 6.05-6.02 (m, 1H), 5.34-5.18 (m, 3H), 4.89-4.81 (m, 3H), 4.63-4.53 (m, 3H), 4.41-4.36 (m, 2H), 4.22-4.19 (m, 1H), 3.60-3.37 (m, 67H), 2.38-2.30 (m, 5H), 1.66-1.12 (m, 23H).
[0518] Synthesis of 153: The title compound was synthesized following the procedure described for 152 above. Accordingly, 140 (12.1 mg, 0.0126 mmol) was reacted with 150 (13 mg, 0.0126 mmol) in the presence of HATU (9.6 mg, 0.0252 mmol) and NMM (4.2 pL, 0.0378 mmol) in DMF (0.5 mL) at rt in air. Work up and chromatography by reverse phase chromatography (15.5 g C18Aq, 5-80-100% of acetonitrile in water each containing 0.1% of HCOOH) followed by lyophilization of the fractions afforded Boc containing amine 14.6 mg which was dissolved in DCM (2 mL) at rt and treated with TFA. After completion of the reaction, work up and chromatography again (15.5 g C18Aq, 5-60-100% of acetonitrile in water each containing 0.1% of HCOOH) followed by lyophilization afforded 153 (10.5 mg, 0.0056 mmol, 44%) as a white solid. MS (ESI, pos.) calc’d for C93H130N15O26 [1872.92]"; found (M+H) 1875.06. ’H-NMR (500 MHz; DMSO-d6): <5 11.88 (s, 1H), 11.44 (s, 1H), 10.01 (s, 1H), 9.09-9.00 (m, 2H), 8.24-8.23 (m, 1H), 8.05-8.01 (m, 1H), 7.95-7.89 (m, 2H), 7.78- 7.72 (m, 2H), 7.65-7.59 (m, 3H), 7.52-7.42 (m, 5H), 7.29-7.10 (m, 2H), 6.05-6.02 (m, 1H), 5.34-5.18 (m, 3H), 4.89-4.81 (m, 3H), 4.63-4.53 (m, 3H), 4.41-4.36 (m, 2H), 4.22-4.19 (m, 1H), 3.60-3.37 (m, 67H), 2.38-2.30 (m, 5H), 1.66-1.12 (m, 23H).
[0519] EXAMPLE 48
[0520] Synthesis of 4-((29S,32S)-l-Amino-29-isopropyl-32-methyl-27,30-dioxo- 3,6,9,12,15,18,21,24-octaoxa-28,31-diazatritriacontan-33-amido)benzyI (2-
(((lS,4S,7R,llS,14S,17S,20R,24S,Z)-ll,24-dicydopentyI-2,4,12,15,17,25-hexamethyI-
3,6,10,13,16,19,23,26-octaoxo-20-(quinoxaIine-2-carboxamido)-9,22-dioxa-
2,5,12,15,18,25-hexaazabicydo[12.12.4]triacont-28-en-7-yI)carbamoyI)quinoIin-3- yl)carbamate (154, M6742)
[0521] Linker Payload 154 was synthesized from intermediate 52 as outlined below.
 [0522] Synthesis of 154: The title compound was synthesized following the procedure described for 153. Accordingly, 52 (11 mg, 0.0116 mmol) was reacted with 150 (12 mg, 0.0116 mmol) in the presence of HATU (9 mg, 0.0233 mmol) and NMM (6.4 pL, 0.0582 mmol) in DMF (0.6 mL) at rt in air. Work up and reverse phase chromatography followed by treatment of the product thereof with TFA in DCM as described above for 153 followed by chromatography again (15.5 g C18Aq, 5-55-100% of acetonitrile in water each containing 0.1% of HCOOH) and lyophilization of the fractions afforded 154 (10.5 mg, 0.0057 mmol, 66%) as a white solid. MS (ESI, pos.) calc’d for C92H128N16O25 [1857.92]"; found (M+H) 1859.16. ’H-NMR (500 MHz; DMSO-d6): <5 11.44 (s, 1H), 10.01 (s, 1H), 9.09-9.00 (m, 2H), 8.24-8.23 (m, 1H), 8.05-8.01 (m, 1H), 7.95-7.89 (m, 2H), 7.78-7.72 (m, 2H), 7.65-7.59 (m,
[0522] Synthesis of 154: The title compound was synthesized following the procedure described for 153. Accordingly, 52 (11 mg, 0.0116 mmol) was reacted with 150 (12 mg, 0.0116 mmol) in the presence of HATU (9 mg, 0.0233 mmol) and NMM (6.4 pL, 0.0582 mmol) in DMF (0.6 mL) at rt in air. Work up and reverse phase chromatography followed by treatment of the product thereof with TFA in DCM as described above for 153 followed by chromatography again (15.5 g C18Aq, 5-55-100% of acetonitrile in water each containing 0.1% of HCOOH) and lyophilization of the fractions afforded 154 (10.5 mg, 0.0057 mmol, 66%) as a white solid. MS (ESI, pos.) calc’d for C92H128N16O25 [1857.92]"; found (M+H) 1859.16. ’H-NMR (500 MHz; DMSO-d6): <5 11.44 (s, 1H), 10.01 (s, 1H), 9.09-9.00 (m, 2H), 8.24-8.23 (m, 1H), 8.05-8.01 (m, 1H), 7.95-7.89 (m, 2H), 7.78-7.72 (m, 2H), 7.65-7.59 (m,
3H), 7.52-7.42 (m, 5H), 7.29-7.10 (m, 2H), 6.05-6.02 (m, 1H), 5.34-5.18 (m, 3H), 4.89-4.81 (m, 3H), 4.63-4.53 (m, 3H), 4.41-4.36 (m, 2H), 4.22-4.19 (m, 1H), 3.60-3.37 (m, 67H), 2.38- 2.30 (m, 5H), 1.66-1.12 (m, 23H).
[0523] EXAMPLE 49 [0524] Linker 160 was synthesized as described below.

[0525] Synthesis of ( 2R,3R,4S,5R,6S )-2-( acetoxymethyl)-6-( 4-(hydroxymethyl)-2- nitrophenoxy)tetrahydro-2H-pyran-3,4,5-triyl triacetate (156)'. The phenol (120, 2.46 g,
14.521 mmol) and the acetobromo-a-D-glucose (155, 5.97 g, 14.521 mmol) were dissolved in acetonitrile (70 mL) at rt under argon. Ag2O (3.63 g, 15.682 mmol) was added in one portion. The resulting heterogeneous mixture was stirred for 2 h whereby UPLC revealed completion of the reaction. The reaction mixture was filtered through celite and the residue was washed with EtOAc (50 mL). The filtrate was concentrated to give orange solid that was suspended in hexanes and filtered. The solid residue was washed with hexanes (100 mLX3) followed by washing with Et20 (100 mLx3). The orange solid 156 (5.95 g, 11.916 mmol, 82%) was collected and used for the next step without chromatographic purification. MS (ESI, pos.) calc’d for C2iH2sNOi3Na [522.13]"; found (M+Na) 522.60.
[0526] Synthesis of (2R,3R,4S,5R,6S)-2-(acetoxymethyl)-6-(4-(((2-
( ( allyloxy )carbonyl)quinolin-3-yl )oxy )methyl )-2-nitrophenoxy)tetrahydro-2H-pyran-3, 4,5- triyl triacetate (157)-. The title compound was synthesized as per the procedure described for
124 where 123 (1.321 g, 5.763 mmol) was treated with 156 (3.454 g, 6.915 mmol) in the presence of DIAD (1.398 g, 6.915 mmol) and PPhs (1.814 g, 6.915 mmol) in THF (30 mL). Work up and chromatography (hexanes/EtOAc, 1:1) afforded 157 (3.371 g, 4.744 mmol, 82%) as white solid. MS (ESI, pos.) calc’d for C34H35N2O15 [711.20]"; found (M+H) 711.72.
[0527] Synthesis of (2R,3R,4S,5R,6S)-2-(acetoxymethyl)-6-(4-(((2- ((allyloxy)carbonyl)quinolin-3-yl)oxy)methyl)-2-aminophenoxy)tetrahydro-2H-pyran-3,4,5- triyl triacetate (158): The title compound was synthesized as per the procedure described for
125 by heating a mixture of 157 (881 mg, 1.240 mmol) with Fe (692 mg, 12.397 mmol) and CaCh (1.376 g, 12.397 mmol) in THF:MeOH:H2O (8 mL each) at 75 °C for 5 h. Work up and chromatography (hexanes/EtOAc, 1 :1) afforded 158 (320 mg, 0.470 mmol, 38%) as white solid. (Note: The reaction works well with Zn/NPLCI in the same combination of solvents at room temperature within 30 mins). MS (ESI, pos.) calc’d for C34H37N2O13 [681.22]"; found (M+H) 681.74.
[0528] Synthesis of (2R,3R,4S,5R,6S)-2-(acetoxymethyl)-6-(4-(((2-
( ( allyloxy )carbonyl)quinolin-3-yl )oxy )methyl)-2-( 2,2-dimethyl-4-oxo-
3, 8, 11, 14, 17,20,23, 26,29-nonaoxa-5-azadotriacontan-32-amido )phenoxy )tetrahydro-2H- pyran-3,4,5-triyl triacetate (159): The title compound was synthesized as per the procedure described for 126 where impure 158 was used for the reaction. Work up and chromatography (50 g C18Aq, 5-55-100% of acetonitrile in water each containing 0.1% of HCOOH) followed
by lyophilization afforded 159 as a white solid. (The yield from this reaction is in general >90%). MS (ESI, pos.) calc’d for C58H82N3O124 [1204.52]’; found (M+H) 1205.15.
[0529] Synthesis of3-((3-( 2, 2-dimethyl-4-oxo-3,8, 11, 14, 17, 20, 23, 26, 29-nonaoxa-5- azadotriacontan-32-amido )-4-( ( ( 2S,3R,4S, 5S,6R )-3,4,5-trihydroxy-6- ( hydroxymethyl)tetrahydro-2H-pyran-2 -yl )oxy)benzyl )oxy)quinoline-2 -carboxylic acid (160)'.
The title compound was synthesized as per the procedure described for 127. Accordingly, 159 (196 mg, 0.163 mmol) was dissolved in THF:MeOH:H2O (0.4 mL each) and LiOH (23 mg, 0.976 mmol) was added in one portion. Work and chromatography (50 g C18Aq, 5-35- 100% of acetonitrile in water each containing 0.1% of HCOOH) followed by lyophilization afforded 160 (121 mg, 0.122 mg, 75%) as a white solid. MS (ESI, pos.) calc’d for C47H70N3O20 [996.45]’; found (M+H) 997.10.
[0530] EXAMPLE 50
[0531] Synthesis of (2S,3S,4S,5R,6S)-6-(2-(l-amino-3,6,9,12,15,18,21,24- octaoxaheptacosan-27-amido)-4-(((2-(((lS,4S,7R,llS,14S,17S,20R,24S,Z)-ll,24- dicyclopentyl-2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-20-(quinoxaline-
2-carboxamido)-9,22-dioxa-2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7- yl)carbamoyl)quinolin-3-yl)oxy)methyl)phenoxy)-3,4,5-trihydroxytetrahydro-2H- pyran-2-carboxylic acid (162, M6731)
[0532] Linker Payload 162 was synthesized from intermediate 52 as outlined below.

[0533] Synthesis of tert-butyl (27-((5-(((2-(((lS,4S, 7R,llS,14S,17S,20R,24S,Z)-ll,24- dicyclopentyl-2,4, 12, 15, 17, 25 -hexamethyl- 3, 6, 10,13, 16, 19,23, 26-octaoxo-20-( quinoxaline-2- carboxamido)-9,22-dioxa-2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7- yl)carbamoyl)quinolin-3-yl)oxy)methyl)-2-(((2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6- (hydroxymethyl)tetrahydro-2H-pyran-2-yl)oxy)phenyl)amino)-27-oxo-3,6,9,12,15,18,21,24- octaoxaheptacosyl)carbamate (161): The title compound was synthesized as per the procedure described for 128 where 52 (13 mg, 0.0138 mmol) was treated with 160 (13.7 mg, 0.138 mmol) in the presence of HATU (10.5 mg, 0.0275 mmol) and NMM (4.6 pL, 0.0413 mmol) in DMF (0.5 mL) at rt in air. Work up and chromatography (15.5 g C18Aq, 5-50- 100% of acetonitrile in water each containing 0.1% of HCOOH) followed by lyophilization afforded 161 (21.2 mg, 0.011 mmol, 80%) as a white solid. MS (ESI, pos.) calc’d for C94H132N13O30 [1921.91]’; found (M+H) 1925.10.
[0534] Synthesis of(2S,3S, 4S, 5R, 6S)-6-(2-(l -amino- 3, 6,9,12,15,18,21,24- octaoxaheptacosan-27-amido)-4-(((2-(((lS,4S, 7R,llS,14S,17S,20R,24S,Z)-ll,24- dicyclopentyl-2,4, 12, 15, 17, 25 -hexamethyl- 3, 6, 10,13, 16, 19,23, 26-octaoxo-20-( quinoxaline- 2- carboxamido)-9,22-dioxa-2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7- yl)carbamoyl ) quinolin- 3 -y I )oxy )methyl )phenoxy)-3,4,5-trihydroxytetrahydro-2H-pyran-2- carboxylic acid (162): 12 mg (0.0062 mmol) of 161 was dissolved in 0.8 mL of MeCN and
0.4 mL of tap water. 15 pL of 1.0 M solution of KBr was added followed by the addition of a
pinch of TEMPO, 20 pL of the saturated solution of NaHCCL and 15 pL of commercial bleach were added respectively. The reaction reached completion within 5 mins as revealed by UPLC. Product peak appeared at the same spot as that of the starting material (SM) peak, however, mass of SM was not observed. The reaction mixture was diluted with 1.2 mL of DMF and loaded into column and purified by reverse phase chromatography (30 g C18Aq, 5- 50-100% of acetonitrile in water each containing 0.1% HCOOH). The fractions were combined and lyophilized to afford white solid (8.2 mg, 68% yield white solid) which was dissolved in 2 mL of DCM and treated with 250 pL of TFA at art in air. After completion, the reaction mixture was concentrated under reduced pressure, the contents were dissolved in DMF (1.2 mL) and purified by reverse phase chromatography (15.5 g C18Aq, 5-40-100% of acetonitrile in water each containing 0.1% of HCOOH). The fractions were combined and lyophilized to afford 162 (6.1 mg, 0.0033 mmol, 78%) as a white solid. MS (ESI, pos.) calc’d for C89H122N13O29 [18836.84]’; found (M+H) 1838.05. ’H-NMR (500 MHz; DMSO-d6): <5 <5 9.52 (s, 1H), 9.25 (s, 1H), 8.65-8.24 (m, 1H), 8.38 (s, 1H), 8.30 (s, 1H), 8.18-8.16 (m, 1H), 8.04-8.01 (m, 3H), 7.99-7.88 (m, 8H), 7.83-7.80 (m, 8H), 7.54-7.46 (m, 1H), 6.04 (br s, 1H), 5.34-5.24 (m, 7H), 4.92-4.77 (m, 9H), 4.59-4.53 (m, 16H), 4.38-4.28 (m, 3H), 3,71-3.10 (m, 30H), 2.76-2.58 (m, 6H), 1.73-1.24 (m, 22H).
[0535] EXAMPLE 51
[0536] Synthesis of (2S,3S,4S,5R,6S)-6-(2-(l-amino-3,6,9,12,15,18,21,24- octaoxaheptacosan-27-amido)-4-(((2-(((lS,4S,7R,llS,14S,17S,20R,24S,Z)-ll,24- diisopropyl-2,4,12,15,17,25-hexamethyl-3,6,10,13,16,19,23,26-octaoxo-20-(quinoxaline-2- carboxamido)-9,22-dioxa-2,5,12,15,18,25-hexaazabicydo[12.12.4]triacont-28-en-7- yI)carbamoyI)quinoIin-3-yI)oxy)methyI)phenoxy)-3,4,5-trihydroxytetrahydro-2H- pyran-2-carboxyIic acid (164, M6719)
[0537] Linker Payload 164 was synthesized from intermediate 19 as outlined below.

[0538] Synthesis of tert-butyl (27-((5-(((2-(((lS,4S, 7R,llS,14S,17S,20R,24S,Z)-ll,24- diisopropyl-2,4, 12, 15,17, 25-hexamethyl-3, 6, 10, 13,16, 19, 23, 26-octaoxo-20-( quinoxaline-2- carboxamido)-9,22-dioxa-2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7- yl)carbamoyl)quinolin-3-yl)oxy)methyl)-2-(((2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6- (hydroxymethyl)tetrahydro-2H-pyran-2-yl)oxy)phenyl)amino)-27-oxo-3,6,9,12,15,18,21,24- octaoxaheptacosyl)carbamate (163): The title compound was synthesized as per the procedure described for 128 where 19 (21 mg, 0.0235 mmol) was treated with 160 (23 mg, 0.0235 mmol) in the presence of HATU (13 mg, 0.035 mmol) and NMM (7.8 pL, 0.071 mmol) in DMF (0.5 mL) at rt in air. Work up and chromatography (30 g C18Aq, 5-45-100% of acetonitrile in water each containing 0.1 % of HCOOH) followed by lyophilization afforded 163 (37 mg, 0.0198 mmol, 84%) as a white solid. MS (ESI, pos.) calc’d for C90H128N13O30 [1870.88]’; found (M+H) 1872.06.
[0539] (2S,3S,4S,5R,6S)-6-(2-(l-amino-3,6,9,12,15,18,21,24-octaoxaheptacosan-27-amido)- 4-(((2-(((lS, 4S, 7R, 11S,14S,17S, 20R, 24S, Z)-ll, 24-diisopropyl-2, 4,12,15,17,25-hexamethyl- 3, 6,10, 13, 16,19, 23, 26-octaoxo-20-( quinoxaline-2-carboxamido )-9, 22-dioxa-2, 5,12, 15, 18,25- hexaazabicyclo[12.12.4]triacont-28-en-7-yl)carbamoyl)quinolin-3-yl)oxy)methyl)phenoxy)- 3,4,5-trihydroxytetrahydro-2H-pyran-2-carboxylic acid (164): The title compound was synthesized as per the procedure described for 162 above. Accordingly, 15 pL of 1.0 M solution of KBr was added followed by the addition of a pinch of TEMPO, 30 pL of the saturated solution of NaHCOs and 35 pL of commercial bleach were added respectively. The
reaction reached completion within 18 mins. Work up and chromatography (50 g, C18Aq, 5- 50-100% of acetonitrile in water each containing 0.1% of HCOOH) followed by lyophilization gave 33.2 mg (0.0176 mmol, 89%) of white solid. 18 mg (0.0095 mmol) of this intermediate was treated with 250 pL of TFA in 2 mL of DCM until all Boc group was gone. Work up and chromatography (15.5 g C18Aq, 5-35-100% of acetonitrile in water each containing 0.1 % of HCOOH) followed by lyophilization of the resulting fractions afforded 164 (14.1 mg, 0.0079 mg, 89%) as a white solid. MS (ESI, pos.) calc’d for C85H118N13O29 [ 1784.81]+; found (M+H) 1787.00. ’H-NMR (300 MHz; DMSO-d6): <5 9.54 (s, 1H), 9.20 (br s, 1H), 8.79-8.59 (m, 2H), 8.55-8.53 (m, 1H), 8.26-8.16 (m, 1H), 8.16-7.54 (m, 5H), 7.94- 7.74 (m, 6H), 7.59-7.46 (m, 2H), 7.37-6.87 (m, 2H), 6.21-5.88 (m, 2H), 5.41-5.21 (m, 5H), 4.78-4.69 (m, 5H), 4.60-4.26 (m, 6H), 4.19-4.0 (m, 4H), 3.70-3.67 (m, 3H), 3.59-3.09 (m, 35H), 3.21-3.09 (m, 5H), 2.99-2.97 (m, 3H), 2.92-2.81 (m, 5H), 2.75-2.50 (m, 2H), 2.22-2.16 (m, 2H), 1.29-1.18 (m, 7H), 0.99-0.86 (m, 5H), 0.88-0.78 (m, 6H).
[0540] EXAMPLE 52
[0541] Synthesis of N-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-ll,24-dicyclopentyl-20-(3-((3- (l-(2,5-dioxo-2,5-dihydro-lH-pyrroI-l-yI)-3,6,9,12,15,18,21,24-octaoxaheptacosan-27- amido)-4-(((2S,3R,4S,5R,6R)-3,4,5-trihydroxy-6-(hydroxymethyI)tetrahydro-2H-pyran- 2-yI)oxy)benzyI)oxy)quinoIine-2-carboxamido)-2,4,12,15,17,25-hexamethyI- 3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa-2,5,12,15,18,25- hexaazabicycIo[12.12.4]triacont-28-en-7-yI)quinoxaIine-2-carboxamide (167,M6794)
[0542] Linker Payload 167 was synthesized from intermediate 125 as outlined below.

[0543] Synthesis of 3 -(( 3-amino-4-( ( ( 2S, 3R, 4S, 5R, 6R)-3, 4, 5-trihydroxy-6-
(hydroxymethyl)tetrahydro-2H-pyran-2-yl)oxy)benzyl)oxy)quinoline-2-carboxylic acid (165): The title compound was prepared by treating 125 (104 mg, 0.0153 mmol) with LiOH (37 mg, 1.528 mmol) in THF:MeOH:H2O (250 pL each) until all the protecting groups were removed. Work up and chromatography (30 g C18Aq, 5-15-100% of acetonitrile in water each containing 0.1% of HCOOH) followed by lyophilization of the fractions afforded 165 (66 mg, 0.140 mmol, 91%) as a white solid. MS (ESI, neg.) calc’d for C23H24N2O9 [471.15]-; found (M+H) 471.08.
[0544] Synthesis ofN-((lS,4S, 7R,llS,14S,17S,20R,24S,Z)-20-(3-((3-amino-4- ((( 2S,3R,4S,5R,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2- yl)oxy)benzyl)oxy)quinoline-2-carboxamido)-ll,24-dicyclopentyl-2,4,12,15,17,25- hexamethyl-3,6,10,13,16,19,23,26-octaoxo-9,22-dioxa-2,5,12,15,18,25- hexaazabicyclo[12.12.4]triacont-28-en-7-yl)quinoxaline-2-carboxamide (166f. The title compound was synthesized as per the procedure described for 161 where 52 (21 mg, 0.0222 mmol) was reacted with 165 (11 mg, 0.0222 mmol) in the presence of HATU (17 mg, 0.0444 mmol) and NMM (12.3 pL, 0. 1111 mmol) in DMF (0.5 mL) at rt. Work up and chromatography (15.5 g, C18Aq, 5-40-100% of acetonitrile in water each containing 0.1% of HCOOH) followed by lyophilization of the fractions afforded 166 (25.7 mg, 0.0184 mmol, 83%) as a white solid. MS (ESI, pos.) calc’d for C70H86N12O19 [ 1399.61 ]+; found (M+H) 1399.54.
[0545] Synthesis ofN-((lS, 4S, 7R, 11S,14S,17S, 20R, 24S, Z)-ll, 24-dicyclopentyl-20-(3-((3-(l- (2, 5-dioxo-2,5-dihydro-lH-pyrrol-l-yl)-3,6,9,12,15,18,21,24-octaoxaheptacosan-27-amido)- 4-( ( ( 2S,3R,4S,5R,6R )-3, 4, 5 -trihydroxy -6-(hydroxymethyl)tetrahydro-2H-pyran-2- yl)oxy )benzyl )oxy )quinoline-2-carboxamido)-2,4, 12, 15, 17,25-hexamethyl-
3.6.10.13.16.19.23.26-octaoxo-9,22-dioxa-2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont- 28-en-7-yl)quinoxaline-2-carboxamide (167)'. The title compound was synthesized as per the procedure described for 159. Accordingly, 166 (19.1 mg, 0.0136 mmol) was reacted with mal-peg8-acid (14 mg, 0.0263 mmol) in the presence of HATU (10 mg, 0.0273 mmol), HOAt (3.7 mg, 0.0273 mmol) and NMM (6.8 pL, 0.0682 mmol) in DMF (0.6 mL) at rt for 2 h. Work up and chromatography (Isco EZ Prep (Gemini C18 column, 30 x 150 mm, 5-50- 100% of acetonitrile in water, each containing 0.1% of HCOOH) followed by lyophilization of the fractions afforded 167 (9.7 mg, 0.0051 mmol, 37%) as a white solid. MS (ESI, pos.) calc’d for C93H123N13O30 [1902.85]+; found (M+H) 1903.66. ’H-NMR (500 MHz; DMSO- d6): <59.53 (s, 1H), 9.20 (s, 1H), 8.62-8.57 (m, 2H), 8.32 (s, 1H), 8.20-8.18 (m, 1H), 8.03-7.93 (m, 5H), 7.83-7.80 (m, 3H), 7.54-7.45 (m, 2H), 7.26-7.20 (m, 2H), 7.01 (s, 2H), 6.05-6.01 (m, 2H), 5.70-5.69 (m, 1H), 5.34-5.25 (m, 4H), 4.94-4.85 (m, 4H), 4.78-4.77 (m, 2H), 4.65- 4.53 (m, 7H), 4.38-4.28 (m, 2H), 4.40-3.40 (m, 49H), 3.21-2.35(m, 10H), 1.73-1.24 (m, 22H).
[0546] EXAMPLE 53
[0547] Synthesis of N-((lS,4S,7R,llS,14S,17S,20R,24S,Z)-20-(3-((29S,32S)-l-amino-29- isopropyI-32-methyI-27,30-dioxo-3,6,9,12,15,18,21,24-octaoxa-28,31-diazatritriacontan- 33-amido)quinoIine-2-carboxamido)-ll,24-dicydopentyI-2,4,12,15,17,25-hexamethyI-
3.6.10.13.16.19.23.26-octaoxo-9,22-dioxa-2,5,12,15,18,25- hexaazabicycIo[12.12.4]triacont-28-en-7-yI)quinoxaIine-2-carboxamide (172, M6805).
[0548] Linker Payload 172 was synthesized from intermediate 147 and 52 as outlined below.

[0549] Synthesis of allyl 3-( ( S )-2-( ( S )-2-( ( tert-butoxycarbonyl )amino )-3- methylbutanamido)propanamido)quinoline-2-carboxylate (169): To a solution of 147 (12 mg, 0.053 mmol) and 168 (15, 0.053 mmol) in DMAc (2 mL) was added HATU (40 mg, 0.11 mmol), HOAt (14 mg, 0.11 mmol), and DIPEA (0.028 mL, 0.16 mmol) at rt. The reaction was stirred for 24 h for completion. The crude was injected into C18 column and eluted with 35-45% ACN/H2O + 0.1% formic acid. Product 169 was obtained as a yellow solid after lyophilization (20 mg, 0.040 mmol, 77% yield). MS (ESI, pos.) calc’d for C26H35N4O6 [499.29]+; found (M+H) 499.19. [0550] Synthesis of allyl 3-((34S,37S)-34-isopropyl-2,2,37-trimethyl-4,32,35-trioxo-
3,8,1 l,14,17,20,23,26,29-nonaoxa-5,33,36-triazaoctatriacontan-38-amido)quinoline-2- carboxylate (170): To a solution of 169 (20 mg, 0.040 mmol) in DCM (0.7 mL) was added TFA (0.3 mL) at rt. The reaction was stirred for 30 min for completion. The crude solution was diluted with toluene (1 mL), and then concentrated to dryness. The resulting solid was dissolved in DMAc (1 mL) followed by the addition of BocNH-PEG8-OH (26 mg, 0.048 mmol), HATU (23 mg, 0.060 mmol), and DIPEA (0.021 mL, 0.12 mmol). The reaction was
stirred at rt overnight for completion. The crude was injected into Cl 8 column and eluted with 45-50% ACN/H2O + 0.1% formic acid. Product 170 was obtained as a clear oil after lyophilization (30 mg, 0.033 mmol, 81% yield). MS (ESI, pos.) calc’d for C45H72N5O15 [922.50]+; found (M+H) 922.92.
[0551] Synthesis of 3-((34S,37S)-34-isopropyl-2,2,37-trimethyl-4,32,35-trioxo- 3,8,1 l,14,17,20,23,26,29-nonaoxa-5,33,36-triazaoctatriacontan-38-amido)quinoline-2- carboxylic acid (171): To a solution of 170 (30 mg, 0.033 mmol) in DCM (2.0 mL) was added Pd(PPh3)4 (11 mg, 0.010 mmol) and phenylsilane (0.02 mL, 0.16 mmol) at rt. The reaction was stirred for 10 min for completion. The crude concentrated and then injected into Cl 8 column and eluted with 30-40% ACN/H2O + 0.1% formic acid. Product 171 was obtained as a clear oil after lyophilization (12 mg, 0.014 mmol, 42% yield). MS (ESI, pos.) calc’d for C42H68N5O15 [882.47]+; found (M+H) 882.33.
[0552] Synthesis of 172: To a solution of 52 (5.0 mg, 0.0053 mmol) in DMAc (1.0 mL) was added 171 (7.0 mg, 0.0079 mmol), HATU (4.0 mg, 0.011 mmol), and NMM (0.0017 mL, 0.016 mmol) at rt. The reaction was stirred for 3 h. The crude concentrated and then injected into Cl 8 column and eluted with 45-55% ACN/H2O + 0.1% formic acid. The coupling product was obtained as a white solid after lyophilization. To that solid was added DCM (0.7 mL) followed by TFA (0.3 mL) at 0 °C. The solution was stirred at 0 °C for 1 h. Then, the solution was diluted with toluene (1 mL) and concentrated to dryness to afford product 172 as a white foam (3.1 mg, 0.0018 mmol, 34% yield over 2 steps). MS (ESI, pos.) calc’d for C84H122N15O23 [ 1708.89]+; found (M+H) 1709.41. ’H-NMR (500 MHz; DMSO-d6): 12.26 (s, 1H), 9.52 (s, 1H), 9.41 (s, 1H), 9.01 (d, 7 = 9.4 Hz, 1H), 8.67 (d, 7 = 6.5 Hz, 1H), 8.57 (d, J = 9.2 Hz, 1H), 8.13 (d, J = 7.6 Hz, 1H), 8.03-8.02 (m, 2H), 7.92 (t, J = 8.0 Hz, 2H), 7.87- 7.83 (m, 2H), 7.76-7.69 (m, 4H), 7.60 ft, 7 = 7.2 Hz, 1H), 7.48 (t, 7 = 7.7 Hz, 1H), 6.04-6.01 (m, 2H), 5.34 (t, 7 = 2.5 Hz, 2H), 4.88 (dd, 7 = 34.9, 10.9 Hz, 4H), 4.60 (t, 7 = 6.9 Hz, 3H), 4.48 (dd, 7 = 8.8, 6.5 Hz, 2H), 4.39-4.33 (m, 3H), 3.58-3.44 (m, 32H), 3.17 (d, 7 = 29.7 Hz, 6H), 2.97 (dd, J = 10.4, 5.3 Hz, 2H), 2.87 (d, J = 4.6 Hz, 6H), 2.79-2.62 (m, 3H), 2.32-2.29 (m, 6H), 2.07-1.98 (m, 2H), 1.73-1.46 (m, 3H), 1.39 (d, 7 = 7.2 Hz, 4H), 1.30-1.22 (m, 12H), 1.18-1.08 (m, 4H), 0.84 (dd, 7 = 16.2, 6.8 Hz, 6H).
[0553] EXAMPLE 54
[0554] Synthesis of 3-(l-amino-3,6,9,12,15,18,21,24-octaoxaheptacosan-27-amido)-4-
(((2S,3R,4S,5R,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2- yl)oxy)benzyl (2-(((lS,4S,7R,HS,14S,17S,20R,24S,Z)-ll,24-dicyclopentyl-2,4,12,15,17,25- hexamethyl-3,6,10,13,16,19,23,26-octaoxo-20-(quinoxaline-2-carboxamido)-9,22-dioxa- 2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7-yl)carbamoyl)quinolin-3- yl)carbamate (177, M6804).
[0555] Linker Payload 177 was synthesized from intermediate 156, 147 and 52 as outlined below. Synthesis of (2R,3S,4S,5R,6S)-2-(acetoxymethyl)-6-(2-amino-4- (hydroxymethyl)phenoxy)tetrahydro-2H-pyran-3,4,5-triyl triacetate (173): To a solution of 156 (1.07 g, 2.14 mmol) in MeOH/THF (24 mL + 24 mL) was added Zn dust (1.27 g, 19.5 mmol) and aq. NH4CI (12 mL) at rt. The reaction was stirred for 1 h for completion. The solution was concentrated and then suspended in MeOH. It was filtered, rinsed with MeOH, and the solution was concentrated to dryness to afford the product 173 as a yellow solid (900 mg, 1.92 mmol, 90% yield). MS (ESI, pos.) calc’d for C21H28NO11 [470.17]+; found (M+H) 470.11.
[0556] Synthesis of (2R,3S,4S,5R,6S)-2-(acetoxymethyl)-6-(2-(2,2-dimethyl-4-oxo-
3.8.11.14.17.20.23.26.29-nonaoxa-5-azadotriacontan-32-amido)-4- (hydroxymethyl)phenoxy)tetrahydro-2H-pyran-3,4,5-triyl triacetate (174): To a solution of
173 (200 mg, 0.426 mmol) in DMAc (3 mL) was added BocNH-PEG8-OH (254 mg, 0.469 mmol), HATU (243 mg, 0.639 mmol), HOAt (87 mg, 0.639 mmol), and DIPEA (0.305 mL, 1.28mmol) at rt. The reaction was stirred for 16 h for completion. The crude was injected into Cl 8 column and eluted with 40-45% ACN/H2O + 0.1% formic acid. Product 174 was obtained as a white solid after lyophilization (240 mg, 0.242 mmol, 57% yield). MS (ESI, pos.) calc’d for C45H73N2O22 [993.47]+; found (M+H) 993.43.
[0557] Synthesis of (2R,3S,4S,5R,6S)-2-(acetoxymethyl)-6-(4-((((2- ((allyloxy)carbonyl)quinolin-3-yl)carbamoyl)oxy)methyl)-2-(2,2-dimethyl-4-oxo-
3.8.11.14.17.20.23.26.29-nonaoxa-5-azadotriacontan-32-amido)phenoxy)tetrahydro-2H- pyran-3,4,5-triyl triacetate (175): The title compound was synthesized as per the procedure described for 148. Accordingly, 147 (29 mg, 0.127 mmol) was treated with triphosgene (34 mg, 0.115 mmol) in the presence of pyridine (32.7 pL, 0.407 mmol) and DMAP (15.5 mg, 0.127 mmol) in DCM (8 mL) at 0 °C under argon for 10 mins before adding the solution of
174 (126 mg, 0.127 mmol) in DCM (1 mL) at the same temperature and allowing the resulting reaction mixture to warm to room temperature overnight. Work up and purification
by reverse phase chromatography (30 g C18Aq, using 5-60-100% of acetonitrile in water each containing 0.1% of HCOOH) followed by lyophilization of the resulting fractions afforded 175 (149 mg, 0.119 mmol, 94%) as a white solid. MS (ESI, pos.) calc’d for C59H83N4O25 [1247.53]+; found (M+H) 1248.24. [0558]

[0559] Synthesis of 3-((((3-(2,2-dimethyl-4-oxo-3,8,ll,14,17,20,23,26,29-nonaoxa-5-
azadotriacontan-32-amido)-4-(((2S,3R,4S,5R,6R)-3,4,5-trihydroxy-6- (hydroxymethyl)tetrahydro-2H-pyran-2-yl)oxy)benzyl)oxy)carbonyl)amino)quinoline-2- carboxylic acid (176): 175 (149 mg, 0.119 mmol) was treated with phenylsilane (14.2 pL, 0.131 mmol) in the presence of Pd(PPh3)4 (7.0 mg, 0.006 mmol) for 10 mins and the resulting reaction mixture was concentrated under reduced pressure. The crude contents were dissolved in THF:MeOH:H2O (1 mL each) at rt/air. K2CO3 (165 mg, 1.195 mmol) was added in one portion. After 30 mins of stirring, the reaction mixture was purified by reverse phase chromatography (50 g C18Aq, 5-35-100% of acetonitrile in water each containing 0.1% of HCOOH). Lyophilization of the resulting combined fraction afforded 176 (76 mg, 0.073 mmol, 61%) as a white solid. MS (ESI, pos.) calc’d for C48H?oN402iNa [1061.45]+; found (M+H) 1062.02.
[0560] Synthesis of 177: 52 (7 mg, 0.007 mmol), 176 (8 mg, 0.007 mmol), HATU (6 mg, 0.015 mmol) were all dissolved in DMF (0.3 mL) at rt/air followed by the addition of NMM (4.1 pL, 0.037 mmol). After 10 mins, the reaction mixture was purified by reverse phase chromatography (15.5 g C18Aq, 5-50-100% of acetonitrile in water each containing 0.1% of HCOOH). The combined fractions were lyophilized to afford the hoc amine as a white solid (12.3 mg). The intermediate was treated with 0.300 mL of TFA in DCM (2 mL) at 0 °C for 15 mins. After complete removal of the protecting group, the reaction mixture was concentrated under reduced pressure and the resulting contents were dissolved in acetonitrile/water (5 mL each) and lyophilized to afford 177 (11.6 mg, 0.006 mmol, 84%) as a white solid. MS (ESI, pos.) calc’d for C90H125N14O29 [ 1864.87]+; found (M+H) 1866.27. ’H-NMR (500 MHz; DMSO-d6): 11.5 (s, 1H), 9.52 (s, 1H), 9.19 (s, 1H), 8.61-8.56 (m, 2H), 8.33 (s, 1H), 8.18-8.16 (m, 1H), 8.02-7.94 (m, 5H), 7.82-7.79 (m, 3H), 7.55-7.44 (m, 2H), 7.25-7.19 (m, 2H), 7.02 (s, 2H), 6.03-6.00 (m, 2H), 5.69-5.67 (m, 1H), 5.33-5.24 (m, 4H), 4.93-4.84 (m, 4H), 4.77-4.76 (m, 2H), 4.64-4.52 (m, 7H), 4.37-4.26 (m, 2H), 4.38-3.35 (m, 49H), 3.21-2.34(m, 10H), 1.73-1.23 (m, 22H).
[0561] EXAMPLE 55
[0562] Linker Payload 178 was synthesized from intermediate 140 as outlined below.

3-(l-amino-3, 6, 9, 12, 15, 18, 21,24-octaoxaheptacosan-27-amido)-4-(((2S, 3R,4S, 5R, 6R)-3,4, 5- trihydroxy-6-( hydroxymethyl)tetrahydro-2H-pyran-2-yl )oxy )benzyl ( 2-
(((lS,4S, 7R,llS,14S,17S,20R,24S,Z)-ll,24-dicyclopentyl-20-(3-hydroxyquinoline-2- carboxamido)-2,4, 12,15,17,25-hexamethyl-3, 6,10,13,16,19, 23, 26-octaoxo-9, 22 -dioxa- 2,5,12,15,18,25-hexaazabicyclo[12.12.4]triacont-28-en-7-yl)carbamoyl)quinolin-3- yl)carbamate 178
Synthesis ofl 78: The title compound was synthesized according to the procedure described for 177 by treating 140 (8 mg, 0.008 mmol) with 176 (8.7 mg, 0.008 mmol) in the presence of NMM (2.8 pL, 0.017 mmol) in DMF (0.2 mL) at rt. Purification by reverse phase chromatography (Isco EZ Prep (Gemini C18 column, 30 x 150 mm, 5-35-100% of acetonitrile in water, each containing 0.1% of HCOOH) followed by lyophilization afforded 8.5 mg of the intermediate that was treated with 300 pL of TFA in DCM (2 mL) at 0 °C for 20 mins. The reaction mixture was concentrated under reduced pressure, the contents were dissolved in acetonitrile/water (5 mL each) and lyophilized to afford 178 (8.5 mg, 0.004 mmol, 51%) as white solid. MS (ESI, pos.) calc’d for C91H126N13O30 [1880.87]"; found (M+H) 1881.49. 'H- NMR (500 MHz; DMSO-d6): S 11.70 (s, 1H), 11.50 (s, 1H), 9.25-9.20 (m, 1H), 8.67-8.61 (m, 1H), 8.58-8.46 (m, 5H), 8.35-8.27 (m, 1H), 8.06-7.99 (m, 1H), 7.93-7.82 (m, 3H), 7.78-7.49 (m, 5H), 7.42-7.15 (m, 5H), 6.92-6.74 (m, 6H), 5.38-5.23 (m, 4H), 5.09-4.98 (m, 2H), 4.92- 4.81 (m, 2H), 4.64-4.54 (m, 3H), 4.53-4.40 (m, 2H), 3.76-3.65 (m, 2H), 3.62-3.45 (m, 30H), 3.21-3.07 (m, 4H), 2.95-2.82 (m, 4H), 2.72-2.59 (m, 3H), 2.42-2.27 (m, 2H), 1.31-1.15 (m, 11H), 0.93-0.57 (m, 26H).
[0563] Additional payload and linker-payload compounds of the present disclosure are prepared in a similar fashion as those disclosed herein.
[0564] Conjugation and Characterization of Antibody-Payload conjugates
[0565] EXAMPLE 56: Bacterial Transglutaminase Conjugation - 1
[0566] Two antibodies, an anti-HER2 antibody having variable regions derived from humAb4D5-8 from Carter et al, PNAS, 1992, 89 4285, also known as trastuzumab, and a nonbinding isotype control derived from an immunological antigen having no relation to oncology or infectious diseases, both containing a N297Q mutation, which eliminates N- linked glycosylation of the Fc at this site, were used. The mutation allowed the antibodies to be conjugated to a maximum loading of 4 at 295Q and 297Q of the heavy chains. The anti- HER2 and Isotype Control antibodies were conjugated at 1-10 mg/mL in PBS pH 7.4. Linker payloads were added in a 5 to 25-fold molar excess over antibody and the enzymatic reaction was initiated by addition of 1-12 units of bacterial transglutaminase (Zedira, T1001) per mg antibody and incubated with shaking at 37°C for 4-18 hours. The conjugates were purified by size exclusion chromatography and sterile filtered. Protein concentrations were determined by UV spectral analysis. Size-exclusion HPLC established that all conjugates used were >95% monomeric. Yields are reported in Table 1 based on protein contents.
[0567] EXAMPLE 57
[0568] Benchmark DXd antibody-drug conjugate (ADC) (Clin Cancer Res. 2016 Oct 15 ;22(20) :5097-5108). The linker payload for the benchmark ADC was purchased from BroadPharma and conjugated to a drug-antibody ratio (DAR) of 4 using the method for interchain disulfide conjugations described above.
[0569] Table 3: Percent yield, purity, and payload to antibody ratios for each of the antibody drug conjugates

 [0570] EXAMPLE 58: Characterization of Antibody-Drug conjugates by Liquid Chromatography-Mass Spectrometry and Calculation of DAR (Drug-to-Antibody Ratio)
[0570] EXAMPLE 58: Characterization of Antibody-Drug conjugates by Liquid Chromatography-Mass Spectrometry and Calculation of DAR (Drug-to-Antibody Ratio)
[0571] To determine the loading of the linker-payloads on the antibody, the conjugates were analyzed by LC-MS. Waters QToFs XEVO G2-S and QToFs XEVO G2-XS were used for the analysis.
[0572] Specifications for the LC analysis are as follows. The conjugates synthesized by transglutaminase (TG) mediated conjugation were analyzed by LC-MS without any further sample preparation step other than dilution. The conjugates were synthesized by cysteine- conjugation, and the sample was reduced by TCEP prior to LC-MS analysis to obtain the molecular ion mass of the light chain and heavy chain separately. The resulting molecular ions, when weighted according to their intensities, corresponded to the loadings listed in Table 3.
[0573] For the assay of the TG conjugates, 50 pg of the conjugate was diluted with milli-Q water to a final concentration of 1 mg/mL. Injections of 5 pL of each sample were made onto LC-MS (Waters Synat G2-Si) and eluted with 0.1 mL/minute of a gradient mobile phase 20- 40% over 25 minutes (Mobile Phase A: 0.1%v/v FA in H2O; Mobile Phase B: 0.1% v/v FA in Acetonitrile). The LC separation was achieved on a Waters Acquity BEH C4 column (1.0 X 50 mM, 1.7 pM) at 80 °C.
[0574] The mass spectrometry charge-distribution spectra were deconvoluted using Masslynx software and the drug to antibody ratio (DAR) was calculated using the following equation. Average DAR calculation of drug (Dn) by distribution peak intensity (PI) for TG conjugations:
DAR = PID0 x 0 +PID1 x 1 + . +PIDi x i / Z(PID0+PID1 +PID2. +PIDi)
(n= 0,1 ,2,3,...,i)
[0575] Average DAR calculation of drug (Dn) by distribution peak intensity (PI) for Cysteine -Maleimide conjugations is as follows:
1) DAR (Light Chain) = [PILO x 0 +PIL1 x 1 + . +PILi x i /
Z(PID0+PID1 +PID2. +PIDi)]*2
2) DAR (Heavy Chain) = [PIHO x 0 +PIH1 x 1 + . +P I Hi x i I
Z(PIHO+PIH1 +PIH2. +PIHi)]*2
3) DAR = DAR (Light Chain) + DAR (Heavy Chain) (n= 0,1 ,2,3,...,i)
(L=Light chain, H= Heavy Chain)
[0576] The results of the average DAR calculation of drug (Dn) by distribution peak intensity (PI) are shown in Table 1.
[0577] For the assay of the cysteine conjugates, 50 pg of the conjugate was incubated with 2 pL of 5 mM TCEP solution for 30 minutes at room temperature and diluted with milli-Q water to a final concentration of 1 mg/mL. Injections of 5 pL of each sample were made onto LC-MS (Waters Synat G2-Si) and eluted with 0.1 mL/minute of a gradient mobile phase 20- 40% over 25 minutes (Mobile Phase A: 0.1%v/v FA in H2O; Mobile Phase B: 0.1% v/v FA in Acetonitrile). The LC separation was achieved on a Waters Acquity BEH C4 column (1.0 X 50 mM, 1.7 pM) at 80 °C.
[0578] The mass spectrometry spectra were deconvoluted using Masslynx software and the drug to antibody ratio (DAR) was calculated using the following equations.
[0579] EXAMPLE 59: Characterization of conjugates by High Performance Size Exclusion Liquid Chromatography (SEC-HPLC) and Ultra Performance Size Exclusion Liquid Chromatography (SEC-UPLC).
[0580] To determine the monomeric purity of the conjugates, the conjugates were analyzed by SEC-HPLC or SEC-UPLC.
[0581] For SEC-HPLC analysis, 50 pg of the conjugate was diluted with PBS to a final concentration of 1 mg/mL. Injections of 10 pL of each sample were made onto SEC-HPLC (Agilent).
[0582] Specifications of the SEC-HPLC analysis are as follows:


[0583] EXAMPLE 60. ADC, Payloads, and Linker-payloads Killing Assay Protocol (384-well format)
[0584] In Vitro Cytotoxicity Assay. In this Example, the ability of various antibody -drug conjugates and naked payloads to kill antigen-expressing tumor cells in vitro was assessed. Data on a group of illustrative compounds are listed in Tables 4 and 5 below.
[0585] Materials
• Corning® 384-well Flat Clear Bottom White Polystyrene TC-treated Microplates, 20 per Bag, with Lid, Sterile, low flange [Corning #3675]
• Nunc 96 well microwell plate, PP, 0.5 mL [ThermoFisher Scientific, cat#267334, VWR #62408-946] - small volume dilution plate
• 96 well deep well plates, 1.1 mL round wells [Axygen Scientific, cat#P-DW-l l-C-S, VWR #47734-788] - large volume dilution plate
• 96 well deep well plates, 2.2 mL round wells [Axygen Scientific, cat#P-DW-20-C-S, VWR #10011-944] - extra-large volume dilution plate
• T75 cm2 flask
• T175 cm2 flask
• Reagent reservoirs, 50 mL, white, individually wrapped [VWR, cat#89094-682]
• Reagent reservoirs, 25 mL, white, polystyrene, 5/bag [VWR, cat# 89094-662]
• Centrifuge conical tube, 50 mL [Starlab E1450-0200]
• Centrifuge conical tube, 15 mL [Fisher 11849650]
• Plate reader [Spectramax I3x]
• Sealing tape, clear polyolefin [Thermo Scientific, cat# 232701]
• McCoy’s Medium 5 A [Life Technologies cat#26600-023]
• RPMI medium 1640 [Gibco cat#A 10491-01]
• Penicillin-Streptomycin L-glutamine Solution 100X [Life technologies, cat# 15140- 122]
• PBS IX without calcium and magnesium salts [Cytiva, cat# SH30028.02]
• TrypLE™ Express [Gibco, cat# 12604-02]
• Fetal Bovine Serum Heat Inactivated [Sigma, cat# F9665]
• Fetal Bovine Serum Heat Inactivated [Cytiva, cat# SV30160.03]
• DMSO Dimethyl sulfoxide, cell culture tested [Sigma, cat#2660-100mL]
• CellTiter-Glo® [Promega, cat# G7573]
• Bovine serum albumin [Sigma, cat#A7906-100g]
• Opti-MEM reduced serum media [Gibco, cat#31985-070]
• Verapamil [Merck Cat # 676777-lOOmg]
[0586] Cell line and culture media
• SKBR3, maintained in McCoy’s 5A, supplemented with 20% Fetal Bovine Serum (Cytiva), 1% Penicillin-Streptomycin.
• NCI-H1975, maintained in RPMI medium 1640, supplemented with 10% Fetal Bovine Serum (Sigma), 1% Penicillin-Streptomycin.
• HCT-15, maintained in RPMI medium 1640, supplemented with 10% Fetal Bovine Serum (Sigma), 1% Penicillin-Streptomycin.
• Jurkat, maintained in RPMI medium 1640, supplemented with 10% Fetal Bovine Serum (Sigma), 1% Penicillin-Streptomycin.
[0587] Procedure and Protocol
• Harvest and seed cells in log phase of growth in assay plates one day prior to adding test articles:
• Aspirate media, rinse cells with 20ml PBS, aspirate PBS
• Add 3-5ml TrypLE to cells depending on whether it is a T75 or a T175 flask, incubate at 37°C, frequently check to see if cells come off the bottom of the flask
• Break up cell clusters to single cells by repeated pipetting.
• Add 6-10ml complete media depending on whether it is a T75 or a T175 flask, spin, resuspend in 3-5mL depending on whether it is a T75 or a T175 flask and count cells
• For SKBR3 and NCI-H1975, spin down cells (250g for 3 minutes), resuspend cells in corresponding growth media. Count cells and dilute to desired concentrations (SKBR3 6.25xl04 cells/ml, NCI-H1975 3.125xl04 cells/ml), seed cells in 20pl media/well to opaque bottom 384-well assay plate with the following density per well and incubate at 37°C 5% CO2 overnight (cell seeded one day before the experiment):
■ SKBR3: 1250/well
■ NCI-H1975: 625/well
• For HCT-15 +- Verapamil, spin down cells (250g for 3 minutes), resuspend cells in corresponding growth media. Count cells and dilute to desired concentrations
(3.125xl04 cells/ml). o Seed 625 cells/well in 20, u L media/well to opaque bottom 96-well assay plate and incubate at 37 °C 5% CO2 overnight, (cell seeded one day before the experiment) o Dilute Verapamil in methanol to 50mg/ml (This stock can be stored at -20°C for long term) o The next day one hour before adding ADC or compound, dilute Verapamil from 50mg/ml stock to 50ug/ml in the growth media. Add I Op L/ well to cells. o Add I Op l/wcll growth media to the non-Verapamil treatment plates. o Return plates to 37° C 5% CO2 incubator.
• For Jurkat, spin down cells (250g for 3 minutes), resuspend cells in corresponding growth media. Count cells and dilute to desired concentrations (6.25xl04 cells/ml), seed cells in 20p I media/well to opaque bottom 96-well assay plate with 1250 cells/well (cell seeded on the same day of the experiment)
• Add test articles (ADC or compound) to the cells.
For Pay loads
• Prepare payload 10 points serial dilution in 100% DMSO at 1 in 4 dilution (e.g. lOpL + 30pL):
• Dilute payloads to 40000nM.
• Add 80p L of 40000nM payload stock (100% DMSO) to the 2nd well of a 96-well plate, add 30ul DMSO to well 3 - 11. Take lOpl from well 2 to well 3, mix thoroughly by pipetting up and down; Take lOpl from well 3 to well 4, mix the same way. . .and so on till well 10, leave the last well (well 11) as blank (containing only DMSO).
• Add 990|iL Opti-Mem with 0.1% BSA from well 2 to well 11 in a 2.2mL deep well dilution plate. Transfer lOpL DMSO serially diluted payload into the deep well plate with Opti-Mem with 0.1% BSA, correspondingly. Mix well. Payloads are now at 1% DMSO.
• Transfer 10 pL of Opti-MEM (0.1% BSA) diluted payload into appropriate wells of the assay plates, the final DMSO concentration is 0.25% in all wells. The payload concentration ranges are lOOnM, 25nM, 6.5nM, . . .0.3815pM and blank (0). Samples are in triplicate or quadruplicate.
For Conjugate (ADC) o Serial dilute ADC/isotype control/naked antibody at 1 in 4 dilution (e.g. I Op L + 30pL): In a 2.2ml deep well dilution plate, add 300pL Opti-Mem with 0.1% BSA to well 3 - 11. Dilute 300pL test article in the 2nd well to 400nM. Take lOOpL from well 2 to well 3, mix thoroughly by pipetting up and down. Take lOOpL from well 3 to well 4, mix the same way. . . and so on till well 10, leaving the last well (well 11) as blank. o Transfer lOpL serial diluted ADC/isotype control/naked into appropriate wells of the assay plates, the final concentration ranges are lOOnM, 25nM, 6.5nM, . . .0.3815pM and blank (0). Samples are in triplicate or quadruplicate. o Incubate plates at 37°C and 5% CO2 for 4 days for the payloads and 7 days for the ADC’s. Equilibrate the plates at RT for 20 minutes o Develop plates by adding 40pL/well of room temperature CellTiter Gio. Seal plates to avoid damage to aperture and to avoid spill of conjugates or payloads (optional).
• Incubate at room on an orbital shaker for 5-10 minutes.
• Relative light units (RLUs) were measured on an Spectramax 13
• The average RLU of untreated well was used as 0% killing (viability 100%).
• Plot % viability in GraphPad Prism, fitting and determining EC50 values by nonlinear regression (curve fit) Log (agonist) vs. response - variable slope (four parameters) over a 10-point dose response curve.
[0588] Table 4A. Cell Killing Data of Payloads


“N/A” stands for “No Activity”, e.g. for the IC50, the value was >1 00 nM; for the percent kill, the “N/A” means that the data is not meaningful or just unreliable for those data points with IC50s >100 nM.
[0589] Table 4B. Cell Killing Data of Payloads


[0590] Table 5. Cell Killing Data of conjugates (ADC) and Their Isotype Controls


* Those ADCs were also tested in NCI H1975 cell line and no measurable activities were found.
** “N/A” stands for No Activity, e.g. for the IC50, the value was >100 nM. For the percent kill, the N/A means that the data is not meaningful or just unreliable for those data points with IC50s >100 nM.
*** ADCs of HER2-162 and Isotype Ctrl- 162 also showed potent activities toward NCI H1975 cell line with an IC50 of 0.6 nM (%kill 97.4) and 0.83 nM (%kill 97.5), respectively.
[0591] EXAMPLE 61. In Vitro Toxicity of M6264 (payload compound # 55) — Killing of Human Primary Cells
[0592] To test the potential toxicity of payload M6264 in quiescent human primary cells, in vitro cytotoxicity assays were performed in human bone marrow (BM) Hematopoietic stem cells (HSC) (Human BM CD34+ cells, StemCell). Briefly, BM HSCs (6000 cells/well) were seeded into 96 well luminescence assay plates (Thermo Fisher Scientific) in growth media (StemSpan SFEM II, StemCell), then were treated with 1:3 serial dilutions of the payload starting from 100 nM. Following a 6-day incubation at 37°C, CellTiter Gio 2.0 was added to each well, plates were mixed for 2 minutes on an orbital shaker, and plates were incubated at room temperature for 10 minutes. Relative light units (RLUs) were measured on an Envision luminometer (PerkinElmer), and IC50 values were calculated using a four-parameter logistic equation over a 10-point dose response curve with GraphPad Prism. The Max % killing was also reported as calculated: Max % kulin aq = 100 X

[0593] M6264 induces killing in BM HSCs with an IC50 value of 4.45 nM and max % kill of 99.4%.
Cell lines + Growth Media:

Reagents:
1. StemSpan SFEM II [STEMCELL, #09655]
2. Opti-MEM I Reduced Serum Medium [Gibco, #31985070]
3. Bovine Serum Albumin solution (BSA) [Sigma-Aldrich, #A8577-50ML]
4. Luminescence assay plates, 96-well, Nunclon Delta-Treated, Flat Bottom [Thermo Fisher Scientific, #136102]
5. CellTiter-Glo 2.0 Cell Viability Assay [Promega, G9243]
6. Dimethylsulfoxide (DMSO), cell culture tested [ATCC, #4-X-5]
7. PlateLoc Thermal Microplate Sealer [Agilent]
8. EnVision Plate Reader [Perkin Elmer].
[0594] As various changes can be made in the above -described subject matter without departing from the scope and spirit of the present invention, it is intended that all subject matter contained in the above description, or defined in the appended claims, be interpreted as descriptive and illustrative of the present invention. Many modifications and variations of the present invention are possible in light of the above teachings. Accordingly, the present description is intended to embrace all such alternatives, modifications, and variances which fall within the scope of the appended claims.
[0595] All patents, applications, publications, test methods, literature, and other materials cited herein are hereby incorporated by reference in their entireties as if fully set forth in this disclosure.
Claims
1. A compound having a structure according to formula (la), (lb), or (Ic):
 or a pharmaceutically acceptable salt thereof, wherein
or a pharmaceutically acceptable salt thereof, wherein
X and Y are independently selected from N and CH;
Z is O or NH; m is 1, 2, or 3;
Ri is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, and aralkyl, each of which is optionally substituted;
R2 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR;
Ra is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR;
R4 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR; and
Rs is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, aralkyl, each of which is optionally substituted.
2. The compound according to claim 1, wherein X is N and Y is CH.
3. The compound according to claim 1, wherein X is N and Y is N.
4. The compound according to claim 1, wherein X is CH and Y is CH.
5. The compound according to claim 1, wherein X is CH and Y is N.
6. The compound according to any of claims 1 to 5, wherein R3 is OH and R2 is hydrogen.
7. The compound according to any of claims 1 to 6, wherein R3 is OH and R4 is hydrogen.
8. The compound according to any of claims 1 to 7, wherein Z is O.
9. The compound according to any of claims 1 to 7, wherein Z is NH.
10. The compound according to any of claims 1 to 9, wherein R5 is selected from the group consisting of alkyl, cycloalkyl, and aralkyl.
11. The compound according to any of claims 1 to 10, wherein R5 is selected from the group consisting of isopropyl, isobutyl, cyclopentyl, and cyclohexyl.
12. The compound according to claim 1, wherein the compound is one selected from the







 or a pharmaceutically acceptable salt thereof.
or a pharmaceutically acceptable salt thereof.
13. A pharmaceutical composition comprising the compound according to any of claims 1 - 12 and one or more pharmaceutically acceptable carriers, diluents, and/or excipients.
14. A pharmaceutical dosage form comprising the compound according to any of claims 1 to 12 or a composition according to claim 13.
15. A linker-payload compound having a structure according to formula (Ila), (Uh), or (lie):

Z is O or NH; m is 1, 2, or 3;
R2 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR; R3 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR;
R4 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR; and
R5 is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, aralkyl, each of which is optionally substituted;
LI, when present, is a stable or self-immolative linker;
L2, when present, is a linker that is stable or is cleavable by an endosomal/lysosomalenzyme;
L3, when present, is a moiety that modulates the hydrophilicity, physical, and/or chemical properties of the linker-payload compound; and
B is a reactive moiety for conjugation to a target protein, antibody, or antigen-binding fragment thereof, comprising at least one member selected from the group consisting of amino, hydroxyl, thiol, a maleimido moiety, a succinyl ester, an activated ester, an
OOO alkylamino; a haloacetamido, -N3, ~
 (where Q is CH or
(where Q is CH or

16. The linker-payload according to claim 15, wherein X is N and Y is CH.
17. The linker-payload according to claim 15, wherein X is N and Y is N.
18. The linker-payload according to claim 15, wherein X is CH and Y is CH.
19. The linker-payload according to claim 15, wherein X is CH and Y is N.
20. The linker-payload according to any of claims 15 - 19, wherein R3 is OH and R2 is hydrogen.
21. The linker-payload according to any of claims 15 - 20, wherein Ra is OH and R4 is hydrogen.
22. The linker-payload according to any of claims 15 - 21, wherein L3 is present and is a polyethylene glycol (PEG) unit, a carbohydrate moiety, or a combination thereof.
23. The linker-payload compound according to claim 15, wherein the compound is:









24. A conjugate having a structure according to Formula (I) or Formula (II): BA-NH(-L-P)n (I)
BA-S(-L-P)n (II) wherein
BA is an antibody or an antigen-binding fragment thereof;
L is a linker of the formula -L1-L2-L3-B-, wherein
L is connected to BA through a side chain of an amino acid selected from the group consisting of Gin (-CO-NH-), Lys (-NH-CO-), and Cys (-S-);
LI, when present, is a stable or self-immolative linker;
L2, when present, is a linker that is stable or is cleavable by an endosomal/lysosomalenzyme;
L3, when present, is a moiety that modulates the hydrophilicity, physical, and/or chemical properties of -L-P; and
B is a reactive moiety for conjugation to a target protein, antibody, or antigen-binding fragment thereof, comprising at least one member selected from the group consisting of amino, hydroxyl, thiol, a maleimido moiety, a succinyl ester, an activated ester, an alkylamino; a haloacetamido, -

(where

P is a moiety having a structure according to Formula (la’), Formula (lb’), or Formula (Ic’):


X and Y are independently selected from N and CH;
Z is O or NH; m is 1, 2, or 3;
R2 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR;
R3 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR;
R4 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR; and
R5 is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, aralkyl, each of which is optionally substituted; and represents the connecting point of attachment to the linker L; and n is an integer from 1 to 20.
25. The conjugate according to claim 24, wherein LI is a self-immolative linker that is para-aminobenzyl (PAB) moiety.
26. The conjugate according to claim 24 or 25, wherein L2 is a linker cleavable by an endosomal/lysosomalenzyme, wherein the linker comprises a peptide unit comprising two to four amino acid residues selected from the group consisting of glycine (G), alanine (A),
valine (V), phenylalanine (F), proline (P), glutamic acid (E), lysine (K), arginine (R), citrulline (Cit), and combinations thereof.
27. The conjugate according to claim 26, wherein the peptide unit comprises GGFG, VA, V-Cit, GG, GA, GV, AG, VG, AV, AA, EVA, and EV-Cit.
28. The conjugate according to claim 24, wherein the endosomal/lysosomalenzyme is cathepcin B.
29. The conjugate according to any of claims 24 to 28, wherein the BA is an anti-HER2 antibody, an anti-STEAP2 antibody, an anti-MET antibody, an anti-EGFRvIII antibody, an anti-MUC16 antibody, an anti-PRLR antibody, an anti-PSMA antibody, an anti-FGFR2 antibody, an anti-FOLRl antibody, an anti-HER2/HER2 bispecific antibody, an anti HER2/APLP2 bispecific antibody, an anti-MET/MET bispecific antibody, CD33, CD30, CD22, CD79b, Nectin-4, TR0P2, BCMA, CD19, Tissue Factor, or an antigen-binding fragment thereof.
30. The conjugate according to any of claims 24 to 29, wherein the BA targets a cancer selected from the group consisting of breast cancer, ovarian cancer, prostate cancer, lung cancer, liver cancer, lymphomas, urothelial, cervical, multiple myeloma, gastric, or brain cancer.
31. A composition comprising a plurality of conjugates of Formula (I), Formula (II), or combinations thereof according to any of claims 24 to 30, wherein the number ratio of P to BA in the composition is a drug-antibody ratio (DAR) of about 0.5 to about 30.0.
32. The composition according to claim 31 having a DAR of about 1 to about 8.
33. A pharmaceutical composition comprising the conjugate according to any of claims 24 to 30 and one or more pharmaceutically acceptable carriers, diluents, and/or excipients.
34. A pharmaceutical composition comprising a therapeutically effective amount of the conjugate of any one of claims 24 to 30, or a pharmaceutically acceptable salt thereof, and one or more other therapeutically active agents in one or more pharmaceutically acceptable carriers, diluents, and/or excipients.
35. A process of producing a conjugate of Formula (I) or Formula (II) according to any of claims 24 to 30, comprising contacting an antibody or an antigen-binding fragment thereof (BA) with a linker-payload compound (E-P) in the presence of a transglutaminase, wherein E-P has a structure according to formula (Ila), (Uh), or (lie):

Z is O or NH; m is 1, 2, or 3;
R2 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR; R3 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR;
R4 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR; and
R5 is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, aralkyl, each of which is optionally substituted;
LI, when present, is a stable or self-immolative linker;
L2, when present, is a linker that is stable or is cleavable by an endosomal/lysosomalenzyme;
L3, when present, is a moiety that modulates the hydrophilicity, physical, and/or chemical properties of the linker-payload compound;
B is a reactive moiety for conjugation to a target protein, antibody, or antigen-binding fragment thereof.
36. The process according to claim 35, wherein the reactive moiety B is selected from the group consisting of an azide, an alkyne, a thiol, a diene, an amino, an active ester, a glutamine-containing peptide tag (Q-tag), and a 1,2,4,5-tetrazine.
37. The process according to claim 36, wherein the reactive moiety B is Q-tag.
38. The process according to claim 36, wherein the reactive moiety B is an active ester.
39. The process according to claim 36, wherein the reactive moiety B is a maleimide.
40. The process according to claim 36, wherein the reactive moiety B is suitable for a Click Reaction or a Diels-Alder reaction.
41. The process according to claim 36, wherein the reactive moiety B is a substrate of transglutaminase.
42. The process according to claim 35, wherein the reactive moiety B is a reactive moiety for conjugation to a target protein, antibody, or antigen-binding fragment thereof, comprising at least one member selected from the group consisting of amino, hydroxyl, thiol, a maleimido moiety, a succinyl ester, an activated ester, an alkylamino; a haloacetamido, -N3,

43. The process according to claim 35, wherein the transglutaminase is bacterial transglutaminase (BTG) or microbial transglutaminase (MTG).
44. The process according to any of claims 35 to 43, wherein the transglutaminase a native transglutaminase or an engineered transglutaminase.
45. The process according to any of claims 35 to 44, wherein BA is contacted with a 2 to 30-fold molar excess of L-P.
46. A product produced by the process according to any of claims 35 to 45.
47. A pharmaceutical composition comprising the product according to claim 46.
48. A method of treating a subject suffering from cancer, comprising administering to the subject a therapeutically effective amount of the compound according to any of claims 1 to
12, the composition according to any of claims 13 and 31 to 33, the pharmaceutical dosage form according to claim 14, or the conjugate according to any of claims 24 to 30.
49. A method of treating a subject suffering from cancer, comprising administering to the subject a therapeutically effective amount of the conjugate having a structure according to Formula (I) or Formula (II):
BA-NH(-L-P)n (I), BA-S(-L-P)n (II), wherein
BA is an antibody or an antigen-binding fragment thereof;
L is a linker of the formula -L1-L2-L3-B-, wherein
L is connected to BA through a side chain of an amino acid selected from the group consisting of Gin (-CO-NH-), Lys (-NH-CO-), and Cys (-S-);
LI, when present, is a stable or self-immolative linker;
L2, when present, is a linker that is stable or is cleavable by an endosomal/lysosomalenzyme;
L3, when present, is a moiety that modulates the hydrophilicity, physical, and/or chemical properties of -L-P; and
B is a reactive moiety for conjugation to a target protein, antibody, or antigen-binding fragment thereof, comprising at least one member selected from the group consisting of amino, hydroxyl, thiol, a maleimido moiety, a succinyl ester, an activated ester, an alkylamino; a haloacetamido, -

(where

P is a moiety having a structure according to Formula (la’), Formula (lb’), or Formula
(Ic’):

Z is O or NH; m is 1, 2, or 3;
R2 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR; R3 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR;
R4 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl -3 alkyl, OR, and NHR; and
R5 is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, aralkyl, each of which is optionally substituted; and represents the connecting point of attachment to the linker L; and
n is an integer from 1 to 20.
50. Use of a therapeutically effective amount of a compound according to any of claims 1 to 12, the composition according to any of claims 13 and 31 to 33, the pharmaceutical dosage form according to claim 14, or the conjugate according to any of claims 24 to 30, in the manufacture of a medicament for the treatment of a cancer.
51. Use according to claim 50, further comprising use of one or more other therapeutic agents in the manufacture of the medicament.
52. A compound according to any of claims 1 to 12, the composition according to any of claims 13 and 31 to 33, the pharmaceutical dosage form according to claim 14, or the conjugate according to any of claims 24 to 30, for use in treating a cancer.
53. The method according to claim 48 or 49, the use according to claim 50 or 51, or the compound, composition, pharmaceutical dosage form, or conjugate according to claim 52, wherein the cancer is selected from the group consisting of myelogenous leukemia, adult T- cell leukemia, astrocytomas, bladder cancer, breast cancer, PRLR positive (PRLR+) breast cancer, cervical cancer, cholangiocarcinoma, chronic myeloid leukemia, colon cancer, colorectal cancer, endometrial cancer, esophageal cancer, gastric cancer, glioblastomata, head and neck cancer (e.g., head and neck squamous cell carcinoma (HNSCC)), Kaposi’s sarcoma, kidney cancer, leiomyosarcomas, liver cancer, lung cancer (e.g., small cell lung cancer, nonsmall cell lung cancer (NSCLC)), lymphomas, malignant gliomas, malignant mesothelioma, melanoma, mesothelioma, malignant mesothelioma, MFH/fibrosarcoma, multiple myeloma, nasopharyngeal cancer, osteosarcoma, ovarian cancer, pancreatic carcinoma, prostate cancer, castrate-resistant prostate cancer, renal cell carcinoma, residual cancer, rhBAomyosarcoma, stomach cancer, synovial sarcoma, thyroid cancer, uterine cancer, and Wilms’ tumor.
54. The method, use, compound, composition, pharmaceutical dosage form, or conjugate according to claim 53, wherein the cancer is breast cancer.
55. The method, use, compound, composition, pharmaceutical dosage form, or conjugate according to claim 53, wherein the cancer is prostate cancer.
56. A conjugate comprising
(i) an antibody (BA) or an antigen-binding fragment thereof,
(ii) a plurality of payloads, and a linker that covalently connects (i) and (ii), wherein the linker is connected to the BA or antigen-binding fragment thereof through a side chain of an amino acid selected from the group consisting of Gin (-CO-NH-), Lys (-NH-CO-), and Cys (-S-), and has a formula -Ll- L2-L3-B-, wherein
LI, when present, is a stable or self-immolative linker;
L2, when present, is a linker that is stable or is cleavable by an endosomal/lysosomalenzyme;
L3, when present, is a moiety that modulates the hydrophilicity, physical, and/or chemical properties of the conjugate; and
B is a residue of a reactive moiety for conjugation, comprising at least one member selected from the group consisting of amino, hydroxyl, thiol, a maleimido moiety, a succinyl ester, an activated ester, an alkylamino; a haloacetamido, -N3, ~

 and the payload has a structure according to Formula (la’), Formula (lb’), or Formula (Ic’):
and the payload has a structure according to Formula (la’), Formula (lb’), or Formula (Ic’):
 or a pharmaceutically acceptable salt thereof, wherein
X and Y are independently selected from N and CH;
or a pharmaceutically acceptable salt thereof, wherein
X and Y are independently selected from N and CH;
Z is O or NH; m is 1, 2, or 3;
R2 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR;
R3 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR;
R4 is selected from the group consisting of hydrogen, OH, NH2, CN, halo, Cl-3 alkyl, OR, and NHR; and
R5 is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heteroalkyl, heteroalkenyl, heterocycloalkyl, aryl, heteroaryl, aralkyl, each of which is optionally substituted; and represents the connecting point of attachment to the linker.
57. The conjugate according to claim 56, wherein LI is a self-immolative linker of paraaminobenzyl (PAB) moiety.
58. The conjugate according to claim 56 or 57, wherein L2 is a linker cleavable by an endosomal/lysosomalenzyme, wherein the linker comprises a peptide unit comprising two to four amino acid residues selected from the group consisting of glycine (G), alanine (A), valine (V), phenylalanine (F), proline (P), glutamic acid (E), lysine (K), arginine (R), citrulline (Cit), and combinations thereof.
59. The conjugate according to claim 58, wherein the peptide unit comprises GGFG, VA, V-Cit, GG, GA, GV, AG, VG, AV, AA, EVA, and EV-Cit.
60. The conjugate according to any of claims 56 to 59, wherein L3 is present and L3 is a polyethylene glycol (PEG) unit, a carbohydrate moiety, or a combination thereof.
61. The conjugate according to any of claims 56 to 60, wherein the BA is an anti-HER2 antibody, an anti-STEAP2 antibody, an anti-MET antibody, an anti-EGFRvIII antibody, an anti-MUC16 antibody, an anti-PRLR antibody, an anti-PSMA antibody, an anti-FGFR2 antibody, an anti-FOLRl antibody, an anti-HER2/HER2 bispecific antibody, an anti HER2/APLP2 bispecific antibody, an anti-MET/MET bispecific antibody, CD33, CD30, CD22, CD79b, Nectin-4, TROP2, BCMA, CD19, Tissue Factor, or an antigen-binding fragment thereof.
62. The conjugate according to any of claims 56 to 60, wherein the BA targets a cancer selected from the group consisting of breast cancer, ovarian cancer, prostate cancer, lung
cancer, liver cancer, lymphomas, urothelial, cervical, multiple myeloma, gastric, or brain cancer.
63. A pharmaceutical composition comprising a conjugate according to any of claims 56 to 62, together with one or more pharmaceutically acceptable carriers, excipients or diluents.
64. A method of selectively killing quiescent cells in a subject comprising administering to the subject a therapeutically effective amount of a compound according to any of claims 1 to 12 and 15 to 23, or a conjugate according to any of claims 24 to 30 and 56 to 62, or a pharmaceutical composition according to any of claims 13, 14, 33, 34, 47, 62, and 63.
65. The method according to claim 64, wherein said quiescent cells are quiescent cancer cells.
66. A method of selectively killing stem cells in a subject comprising administering to the subject a therapeutically effective amount of a compound according to any of claims 1 to 12 and 15 to 23, or a conjugate according to any of claims 24 to 30 and 56 to 62, or a pharmaceutical composition according to any of claims 13, 14, 33, 34, 47, 62, and 63.
67. The method according to claim 66, wherein said stem cells are hematopoietic stem cells.
68. A method of selectively killing resting or naive B- or T- or other immune cells in a subject comprising administering to the subject a therapeutically effective amount of a compound according to any of claims 1 to 12 and 15 to 23, or a conjugate according to any of claims 24 to 30 and 56 to 62, or a pharmaceutical composition according to any of claims 13, 14, 33, 34, 47, 62, and 63.
69. A method of selectively killing quiescent cancer cells in a subject preparing for stem cell therapy comprising administering to the subject a therapeutically effective amount of a compound according to any of claims 1 to 12 and 15 to 23, or a conjugate according to any of claims 24 to 30 and 56 to 62, or a pharmaceutical composition according to any of claims 13, 14, 33, 34, 47, 62, and 63.
70. The method according to any of claims 64 to 69, further comprising administering one or more other therapeutic agents.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363603966P | 2023-11-29 | 2023-11-29 | |
| US63/603,966 | 2023-11-29 | ||
| US202463688088P | 2024-08-28 | 2024-08-28 | |
| US63/688,088 | 2024-08-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025117727A1 true WO2025117727A1 (en) | 2025-06-05 |
Family
ID=94083576
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/057728 Pending WO2025117727A1 (en) | 2023-11-29 | 2024-11-27 | Analogs of quinoxaline/quinoline cytotoxins, linker- payloads, protein-drug conjugates, and uses thereof |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025117727A1 (en) |
Citations (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2013A (en) | 1841-03-24 | Machine foe making pins | ||
| US4464456A (en) | 1982-01-06 | 1984-08-07 | Toray Industries, Inc. | Photosensitive polymer compositions comprising polyether-ester amides |
| WO1992002551A1 (en) | 1990-08-02 | 1992-02-20 | B.R. Centre Limited | Methods for the production of proteins with a desired function |
| US5208020A (en) | 1989-10-25 | 1993-05-04 | Immunogen Inc. | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| US5223409A (en) | 1988-09-02 | 1993-06-29 | Protein Engineering Corp. | Directed evolution of novel binding proteins |
| US5698426A (en) | 1990-09-28 | 1997-12-16 | Ixsys, Incorporated | Surface expression libraries of heteromeric receptors |
| US5714586A (en) | 1995-06-07 | 1998-02-03 | American Cyanamid Company | Methods for the preparation of monomeric calicheamicin derivative/carrier conjugates |
| US5877397A (en) | 1990-08-29 | 1999-03-02 | Genpharm International Inc. | Transgenic non-human animals capable of producing heterologous antibodies of various isotypes |
| US20030138785A1 (en) | 2001-12-21 | 2003-07-24 | Stephan Kopytek | In vivo protein screen based on enzyme-assisted chemically induced dimerization ("CID") |
| WO2005089808A2 (en) | 2004-03-15 | 2005-09-29 | Wyeth | Antibody calicheamicin conjugates |
| US7087411B2 (en) | 1999-06-08 | 2006-08-08 | Regeneron Pharmaceuticals, Inc. | Fusion protein capable of binding VEGF |
| US20070258987A1 (en) | 2000-11-28 | 2007-11-08 | Seattle Genetics, Inc. | Recombinant Anti-Cd30 Antibodies and Uses Thereof |
| WO2008122039A2 (en) | 2007-04-02 | 2008-10-09 | The Government Of The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Selenocysteine mediated hybrid antibody molecules |
| US20080305497A1 (en) | 2007-05-23 | 2008-12-11 | Ventana Medical Systems, Inc. | Polymeric carriers for immunohistochemistry and in situ hybridization |
| US20090142354A1 (en) | 2006-12-14 | 2009-06-04 | Regeneron Pharmaceuticals, Inc. | Human antibodies to human delta like ligand 4 |
| US20100129314A1 (en) | 2008-04-30 | 2010-05-27 | Immunogen Inc. | Potent conjugates and hydrophilic linkers |
| US7750116B1 (en) | 2006-02-18 | 2010-07-06 | Seattle Genetics, Inc. | Antibody drug conjugate metabolites |
| US7754681B2 (en) | 2004-02-23 | 2010-07-13 | Seattle Genetics, Inc. | Heterocyclic self-immolative linkers and conjugates |
| US20110027286A1 (en) | 2009-07-29 | 2011-02-03 | Regeneron Pharmaceuticals, Inc. | High Affinity Human Antibodies to Human Angiopoietin-2 |
| WO2011130598A1 (en) | 2010-04-15 | 2011-10-20 | Spirogen Limited | Pyrrolobenzodiazepines and conjugates thereof |
| WO2012005982A2 (en) | 2010-07-06 | 2012-01-12 | Board Of Supervisors Of Louisiana State University And Agricultural And Mechanical College | Reporter for rna polymerase ii termination |
| WO2012059882A2 (en) | 2010-11-05 | 2012-05-10 | Rinat Neuroscience Corporation | Engineered polypeptide conjugates and methods for making thereof using transglutaminase |
| WO2012166559A1 (en) | 2011-05-27 | 2012-12-06 | Ambrx, Inc. | Compositions containing, methods involving, and uses of non-natural amino acid linked dolastatin derivatives |
| WO2013055990A1 (en) | 2011-10-14 | 2013-04-18 | Seattle Genetics, Inc. | Pyrrolobenzodiazepines and targeted conjugates |
| WO2013055993A1 (en) | 2011-10-14 | 2013-04-18 | Seattle Genetics, Inc. | Pyrrolobenzodiazepines and targeted conjugates |
| WO2013053873A1 (en) | 2011-10-14 | 2013-04-18 | Spirogen Sàrl | Pyrrolobenzodiazepines |
| WO2013053872A1 (en) | 2011-10-14 | 2013-04-18 | Spirogen Sàrl | Synthesis method and intermediates useful in the preparation of pyrrolobenzodiazepines |
| US20130101546A1 (en) | 2011-06-10 | 2013-04-25 | Mersana Therapeutics, Inc. | Protein-Polymer-Drug Conjugates |
| WO2013068874A1 (en) | 2011-11-11 | 2013-05-16 | Pfizer Inc. | Antibody-drug conjugates |
| WO2014065661A1 (en) | 2012-10-23 | 2014-05-01 | Synaffix B.V. | Modified antibody, antibody-conjugate and process for the preparation thereof |
| WO2016100807A2 (en) | 2014-12-19 | 2016-06-23 | Regeneron Pharmaceuticals, Inc. | Human antibodies to influenza hemagglutinin |
| US20180134794A1 (en) | 2016-11-16 | 2018-05-17 | Regeneron Pharmaceuticals, Inc. | Anti-met antibodies, bispecific antigen binding molecules that bind met, and methods of use thereof |
| JP2020094039A (en) * | 2018-11-28 | 2020-06-18 | 岐阜市 | Compound with antitumor effect and production method thereof |
| WO2020132658A2 (en) | 2018-12-21 | 2020-06-25 | Regeneron Pharmaceuticals, Inc. | Tubulysins and protein-tubulysin conjugates |
-
2024
- 2024-11-27 WO PCT/US2024/057728 patent/WO2025117727A1/en active Pending
Patent Citations (37)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2013A (en) | 1841-03-24 | Machine foe making pins | ||
| US4464456A (en) | 1982-01-06 | 1984-08-07 | Toray Industries, Inc. | Photosensitive polymer compositions comprising polyether-ester amides |
| US5223409A (en) | 1988-09-02 | 1993-06-29 | Protein Engineering Corp. | Directed evolution of novel binding proteins |
| US5208020A (en) | 1989-10-25 | 1993-05-04 | Immunogen Inc. | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| WO1992002551A1 (en) | 1990-08-02 | 1992-02-20 | B.R. Centre Limited | Methods for the production of proteins with a desired function |
| US5627052A (en) | 1990-08-02 | 1997-05-06 | B.R. Centre, Ltd. | Methods for the production of proteins with a desired function |
| US5877397A (en) | 1990-08-29 | 1999-03-02 | Genpharm International Inc. | Transgenic non-human animals capable of producing heterologous antibodies of various isotypes |
| US5698426A (en) | 1990-09-28 | 1997-12-16 | Ixsys, Incorporated | Surface expression libraries of heteromeric receptors |
| US5714586A (en) | 1995-06-07 | 1998-02-03 | American Cyanamid Company | Methods for the preparation of monomeric calicheamicin derivative/carrier conjugates |
| US7087411B2 (en) | 1999-06-08 | 2006-08-08 | Regeneron Pharmaceuticals, Inc. | Fusion protein capable of binding VEGF |
| US20070258987A1 (en) | 2000-11-28 | 2007-11-08 | Seattle Genetics, Inc. | Recombinant Anti-Cd30 Antibodies and Uses Thereof |
| US20030138785A1 (en) | 2001-12-21 | 2003-07-24 | Stephan Kopytek | In vivo protein screen based on enzyme-assisted chemically induced dimerization ("CID") |
| US7754681B2 (en) | 2004-02-23 | 2010-07-13 | Seattle Genetics, Inc. | Heterocyclic self-immolative linkers and conjugates |
| WO2005089808A2 (en) | 2004-03-15 | 2005-09-29 | Wyeth | Antibody calicheamicin conjugates |
| US7750116B1 (en) | 2006-02-18 | 2010-07-06 | Seattle Genetics, Inc. | Antibody drug conjugate metabolites |
| US20090142354A1 (en) | 2006-12-14 | 2009-06-04 | Regeneron Pharmaceuticals, Inc. | Human antibodies to human delta like ligand 4 |
| WO2008122039A2 (en) | 2007-04-02 | 2008-10-09 | The Government Of The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Selenocysteine mediated hybrid antibody molecules |
| US20080305497A1 (en) | 2007-05-23 | 2008-12-11 | Ventana Medical Systems, Inc. | Polymeric carriers for immunohistochemistry and in situ hybridization |
| US20100129314A1 (en) | 2008-04-30 | 2010-05-27 | Immunogen Inc. | Potent conjugates and hydrophilic linkers |
| US20110027286A1 (en) | 2009-07-29 | 2011-02-03 | Regeneron Pharmaceuticals, Inc. | High Affinity Human Antibodies to Human Angiopoietin-2 |
| WO2011130598A1 (en) | 2010-04-15 | 2011-10-20 | Spirogen Limited | Pyrrolobenzodiazepines and conjugates thereof |
| WO2012005982A2 (en) | 2010-07-06 | 2012-01-12 | Board Of Supervisors Of Louisiana State University And Agricultural And Mechanical College | Reporter for rna polymerase ii termination |
| WO2012059882A2 (en) | 2010-11-05 | 2012-05-10 | Rinat Neuroscience Corporation | Engineered polypeptide conjugates and methods for making thereof using transglutaminase |
| US9676871B2 (en) | 2010-11-05 | 2017-06-13 | Pfizer Inc. | Engineered polypeptide conjugates and methods for making thereof using transglutaminase |
| WO2012166559A1 (en) | 2011-05-27 | 2012-12-06 | Ambrx, Inc. | Compositions containing, methods involving, and uses of non-natural amino acid linked dolastatin derivatives |
| US20130101546A1 (en) | 2011-06-10 | 2013-04-25 | Mersana Therapeutics, Inc. | Protein-Polymer-Drug Conjugates |
| WO2013055993A1 (en) | 2011-10-14 | 2013-04-18 | Seattle Genetics, Inc. | Pyrrolobenzodiazepines and targeted conjugates |
| WO2013053872A1 (en) | 2011-10-14 | 2013-04-18 | Spirogen Sàrl | Synthesis method and intermediates useful in the preparation of pyrrolobenzodiazepines |
| WO2013053873A1 (en) | 2011-10-14 | 2013-04-18 | Spirogen Sàrl | Pyrrolobenzodiazepines |
| WO2013055990A1 (en) | 2011-10-14 | 2013-04-18 | Seattle Genetics, Inc. | Pyrrolobenzodiazepines and targeted conjugates |
| WO2013068874A1 (en) | 2011-11-11 | 2013-05-16 | Pfizer Inc. | Antibody-drug conjugates |
| WO2014065661A1 (en) | 2012-10-23 | 2014-05-01 | Synaffix B.V. | Modified antibody, antibody-conjugate and process for the preparation thereof |
| WO2016100807A2 (en) | 2014-12-19 | 2016-06-23 | Regeneron Pharmaceuticals, Inc. | Human antibodies to influenza hemagglutinin |
| US20160176953A1 (en) | 2014-12-19 | 2016-06-23 | Regeneron Pharmaceuticals, Inc. | Human Antibodies to Influenza Hemagglutinin |
| US20180134794A1 (en) | 2016-11-16 | 2018-05-17 | Regeneron Pharmaceuticals, Inc. | Anti-met antibodies, bispecific antigen binding molecules that bind met, and methods of use thereof |
| JP2020094039A (en) * | 2018-11-28 | 2020-06-18 | 岐阜市 | Compound with antitumor effect and production method thereof |
| WO2020132658A2 (en) | 2018-12-21 | 2020-06-25 | Regeneron Pharmaceuticals, Inc. | Tubulysins and protein-tubulysin conjugates |
Non-Patent Citations (82)
| Title |
|---|
| "Antibody-Drug conjugates and Immunotoxins", 2013, SPRINGER VERLAG |
| "Antibody-Drug conjugates", 2015, SPRINGER INTERNATIONAL PUBLISHING |
| "Current Protocols in Molecular Biology", 1999, WILEY INTERSCIENCE |
| "Encyclopedia of Pharmaceutical Technology", 1988, COLD SPRING HARBOR LABORATORY PRESS |
| "Huisgen Proc. Chem. Soc.", 1961, article "Click Chemistry", pages: 357 - 396 |
| "Monoclonal Antibodies, Hybridomas: A New Dimension in Biological Analyses", 1980, PLENUM PRESS |
| "Ocular Therapeutics and Drug Delivery: A Multi-Disciplinary Approach", 1995, LIPPINCOTT WILLIAMS & WILKINS |
| AGARD ET AL., J. AM. CHEM. SOC., vol. 126, no. 46, 2004, pages 15046 - 15047 |
| AGARWAL ET AL., PROC. NATL. ACAD. SCI. |
| ALTING-MEES ET AL., STRATEGIES IN MOLECULAR BIOLOGY, vol. 3, 1990, pages 1 - 9 |
| ALTSCHUL ET AL., J. MOL. BIOL., vol. 215, 1990, pages 403 - 410 |
| ALTSCHUL ET AL., NUCLEIC ACIDS RES., vol. 273, 1997, pages 3389 - 402 |
| BABCOOK ET AL., PROC. NATL. ACAD. SCI. USA, vol. 93, 1996, pages 7843 - 48 |
| BARTLETT ET AL.: "Butterworth-Heinemann", CLINICAL OCULAR PHARMACOLOGY, 15 March 2001 (2001-03-15) |
| BIRD ET AL., SCIENCE, vol. 242, 1988, pages 423 - 426 |
| BOERNER ET AL., J. IMMUNOL., vol. 147, 1991, pages 86 - 95 |
| BOERSMAPLUCKTHUN, CURR. OPIN. BIOTECHNOL., vol. 22, 2011, pages 849 - 857 |
| BRUGGEMANN ET AL., CURR. OPIN. BIOTECHNOL., vol. 8, 1997, pages 724 - 733 |
| BURTON ET AL., ADV. IMMUNOL., vol. 57, 1994, pages 191 - 280 |
| CARRICO ET AL., NAT. CHEM. BIOL., vol. 3, 2007, pages 321 - 322 |
| CARTER ET AL., PNAS, vol. 89, 1992, pages 4285 |
| CHIO ET AL., METHODS MOL. BIOL., vol. 2078, 2020, pages 83 - 87 |
| CLACKSON ET AL., NATURE, V., vol. 352, 1991, pages 624 - 628 |
| CLARK, M., IMMUNOLOGY TODAY., vol. 21, no. 8, 2000, pages 397 - 402 |
| CLIN CANCER RES., vol. 22, no. 20, 15 October 2016 (2016-10-15), pages 5097 - 5108 |
| CRAMERI ET AL., NATURE, vol. 391, 1998, pages 288 - 291 |
| DALL' ACQUA WF ET AL., METHODS, vol. 36, no. 1, 2005, pages 43 - 60 |
| DENNLER ET AL., BIOCONJUGATE CHEM., vol. 25, 2014, pages 569 - 578 |
| DUCRY, BIOCONJUGATE CHEM., vol. 21, 2010, pages 5 - 13 |
| EHRING, ANALYTICAL BIOCHEMISTRY, vol. 267, no. 2, 1999, pages 252 - 259 |
| ENGENSMITH, ANAL. CHEM., vol. 73, 2001, pages 256A - 265A |
| GLASKY ET AL., HYBRIDOMA, vol. 8, 1989, pages 377 - 89 |
| GLUZMAN ET AL., CELL, vol. 23, 1981, pages 175 |
| GONNET ET AL., SCIENCE, vol. 256, 1992, pages 1443 - 1445 |
| GREEN ET AL., NATURE GENET., vol. 7, 1994, pages 13 |
| HARRIS, R.J., JOURNAL OF CHROMATOGRAPHY, vol. 705, 1995, pages 129 - 134 |
| HOFER ET AL., PROC. NATL. ACAD. SCI., USA, vol. 105, 2008, pages 12451 - 12456 |
| HOLLANDER ET AL., BIOCONJUGATE CHEM., vol. 19, 2008, pages 358 - 361 |
| HOOGENBOOM ET AL., J. MOLEC. BIOL., vol. 227, 1992, pages 381 - 388 |
| HUSE ET AL., SCIENCE, vol. 246, 1989, pages 1275 - 81 |
| JAKOBOVITS ET AL., ANN. N. Y. ACAD. SCI., vol. 764, 1995, pages 525 - 35 |
| JEGER ET AL., ANGEW CHEM INT ED ENGL., vol. 49, 2010, pages 9995 - 9997 |
| JONES ET AL., NATURE, vol. 321, 1986, pages 522 - 525 |
| KANG ET AL., PROC. NATL. ACAD. SCI. USA, vol. 88, 1991, pages 4363 - 66 |
| KAURKANWAR, DRUG DEV IND PHARM., vol. 28, no. 5, May 2002 (2002-05-01), pages 473 - 93 |
| KAZANE ET AL., J. AM. CHEM. SOC., 4 December 2012 (2012-12-04) |
| KLEIN ET AL., MABS, vol. 4, no. 6, 2012, pages 1 - 11 |
| KOIKE KOTA ET AL: "Design, Synthesis, and Conformation-Activity Study of Unnatural Bridged Bicyclic Depsipeptides as Highly Potent Hypoxia Inducible Factor-1 Inhibitors and Antitumor Agents", JOURNAL OF MEDICINAL CHEMISTRY, vol. 63, no. 8, 23 March 2020 (2020-03-23), US, pages 4022 - 4046, XP093257777, ISSN: 0022-2623, Retrieved from the Internet <URL:https://pubs.acs.org/doi/pdf/10.1021/acs.jmedchem.9b02039> DOI: 10.1021/acs.jmedchem.9b02039 * |
| KRAMER ET AL., NUCLEIC ACIDS RES., vol. 12, 1984, pages 9441 |
| KUNKEL ET AL., METHODS IN ENZYMOL., vol. 154, 1987, pages 367 - 82 |
| KUNKEL, PROC. NATL. ACAD. SCI. USA, vol. 82, 1985, pages 488 - 92 |
| LINDELL, E. ET AL., INT. J. MOL. SCI., vol. 24, 2023, pages 3762 |
| LLOYD, THE ART, SCIENCE AND TECHNOLOGY OF PHARMACEUTICAL COMPOUNDING, 1999 |
| LONBERG ET AL., NATURE, vol. 368, 1994, pages 856 |
| MANIATIS ET AL.: "Molecular Cloning, A Laboratory Manual", 1989, COLD SPRING HARBOR LABORATORY |
| MARKS ET AL., BIO/TECHNOLOGY, vol. 10, 1992, pages 779 - 783 |
| MCMAHAN ET AL., EMBO J., vol. 10, 1991, pages 2821 |
| MITRA: "Ophthalmic Drug Delivery Systems", 1 March 2003, MARCEL DEKKER, article "Drugs and the Pharmaceutical Sciences: a Series of Textbooks and Monographs" |
| NYGRENUHLEN, CURR. OPIN. IN STRUCT. BIOL., vol. 7, 1997, pages 463 - 469 |
| OCÉANE MARESCALIAIN M., CHEESEMAN, DEV CELL., vol. 55, no. 3, 9 November 2020 (2020-11-09), pages 259 - 271 |
| PEARSON, METHODS MOL. BIOL., vol. 24, 1994, pages 307 - 331 |
| PLUCKTHUN ET AL., METHODS ENZYMOL., vol. 178, 1989, pages 497 - 515 |
| PRESTA, CURR. OP. STRUCT. BIOL., vol. 2, 1992, pages 593 - 596 |
| R.V.GONZAGAETAL ET AL., JOURNAL OF PHARMACEUTICAL SCIENCES, vol. 109, 2020, pages 3262 - 3281 |
| RABUKA ET AL., NAT. PROTOCOLS, vol. 10, 2012, pages 1052 - 1067 |
| REINEKE, METHODS MOL BIOL, vol. 248, 2004, pages 443 - 463 |
| RIECHMANN ET AL., NATURE, vol. 332, 1988, pages 323 - 329 |
| ROSSBRADLEY, BIOCHIMICA ET BIOPHYSICA ACTA (BBA)-NUCLEIC ACIDS AND PROTEIN SYNTHESIS, vol. 654, no. 1, 1981, pages 129 - 134 |
| RYAN ET AL., FOOD & AGRICULTURE IMMUNOL., vol. 13, 2001, pages 127 - 130 |
| SANGER ET AL., PNAS, vol. 74, 1977, pages 5463 |
| SASTRY ET AL., PROC. NATL. ACAD. SCI. USA, vol. 86, 1989, pages 5728 - 9272 |
| SCHLEBUSCH ET AL., HYBRIDOMA, vol. 16, 1997, pages 47 - 52 |
| TAYLOR ET AL., INT. IMMUN., vol. 6, 1994, pages 579 |
| TAYLOR ET AL., NUCL. ACIDS RES., vol. 20, 1992, pages 6287 - 6295 |
| THOMPSON ET AL., J. MOL. BIOL., vol. 256, 1996, pages 350 - 368 |
| THORNTON ET AL.: "Introduction to Protein Structure", vol. 354, 1991, GARLAND PUBLISHING, pages: 105 |
| TOMER, PROTEIN SCIENCE, vol. 9, 2000, pages 487 - 496 |
| VAUGHAN ET AL., NATURE BIOTECH., vol. 16, 1998, pages 535 - 539 |
| WANG ET AL., J. AM. CHEM. SOC., vol. 125, no. 11, 2003, pages 3192 - 3193 |
| WINTER ET AL., ANNU. REV. IMMUNOL., vol. 12, 1994, pages 433 - 55 |
| YANG ET AL., J. MOL. BIOL., vol. 254, 1995, pages 392 - 403 |
| ZHANG, W. ET AL., MOLECULAR IMMUNOLOGY., vol. 42, no. 12, 2005, pages 1445 - 1451 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2019253221B2 (en) | PD-L1 binding affimers, and uses related thereto | |
| TWI892268B (en) | Antibody-pyrrolobenzodiazepine derivative complex | |
| CN108697791B (en) | Antibodies that specifically bind to PD-1 and uses thereof | |
| RU2764651C2 (en) | Antibodies to b7-h3 and conjugates of antibody and drug | |
| KR20230038738A (en) | Camptothecin analogues conjugated to glutamine residues in proteins and uses thereof | |
| AU2018357221B2 (en) | Antibodies and antibody-drug conjugates specific for CD123 and uses thereof | |
| TW202102226A (en) | Combination of antibody-pyrrolobenzodiazepine derivative conjugate and PARP inhibitor | |
| AU2018260505A1 (en) | Antibody conjugates comprising toll-like receptor agonist and combination therapies | |
| RS64662B1 (en) | Humanized and affinity matured antibodies to fcrh5 and methods of use | |
| CA3001482A1 (en) | Antibody conjugates comprising toll-like receptor agonist | |
| JP2024502360A (en) | Novel steroid payloads, steroid linkers, containing ADCs, and uses thereof | |
| CA3190569A1 (en) | Anti-fgfr2 antibodies and methods of use thereof | |
| CA3027178A1 (en) | Anti-egfr antibody drug conjugates | |
| TW202027790A (en) | Dc-sign antibody conjugates comprising sting agonists | |
| CN116096413A (en) | Anti-human VISTA antibodies and uses thereof | |
| TW202031298A (en) | Anti-cdh6 antibody-pyrrolobenzodiazepine derivatives conjugate | |
| KR20210061995A (en) | Immune conjugate targeting ADAM9 and methods of using the same | |
| CN118105508A (en) | Drug linker compound and preparation method and use thereof | |
| JP2025508845A (en) | Cytotoxic targeting chimeras for C-C chemokine receptor 2 expressing cells | |
| CN117015381A (en) | Novel steroid payloads, steroid linkers, ADCs containing the same and their uses | |
| WO2025117727A1 (en) | Analogs of quinoxaline/quinoline cytotoxins, linker- payloads, protein-drug conjugates, and uses thereof | |
| AU2023336004A1 (en) | Antibody-drug conjugate including mutant fc region | |
| RU2797559C2 (en) | 3-(5-hydroxy-1-oxoisoindolin-2-yl)piperidine-2,6-dione derivatives and their use in the treatment of diseases associated with zinc finger protein 2 (ikzf2) of the ikaros family | |
| WO2024118785A2 (en) | Tlr7 agonists and antibody-drug-conjugates thereof | |
| WO2025085489A1 (en) | Gspt1-degrading compounds, anti-cd33 antibodies and antibody-drug conjugates and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24828580 Country of ref document: EP Kind code of ref document: A1 |