WO2024148210A1 - Cyclin-dependent kinase 12 modulators and therapeutic uses thereof - Google Patents
Cyclin-dependent kinase 12 modulators and therapeutic uses thereof Download PDFInfo
- Publication number
- WO2024148210A1 WO2024148210A1 PCT/US2024/010390 US2024010390W WO2024148210A1 WO 2024148210 A1 WO2024148210 A1 WO 2024148210A1 US 2024010390 W US2024010390 W US 2024010390W WO 2024148210 A1 WO2024148210 A1 WO 2024148210A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mmol
- compound
- salt
- alkyl
- etoac
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Definitions
- Cyclin-dependent kinase 12 is an important transcription-associated CDK. Cyclin-dependent kinases (CDKs) are a group of serine/threonine protein kinases that play crucial roles in various cellular processes by regulating cell cycle and gene transcription.
- CDK12 shows versatile roles in regulating gene transcription, RNA splicing, translation, DNA damage response, cell cycle progression, and cell proliferation. Recently, increasing evidence demonstrates the important role of CDK12 in various human cancers, illustrating it as both a biomarker of cancer and a potential target for cancer therapy.
- CDK12 also is known to have an important role in DNA repair and homologous recombination, and thus is directly implicated in enabling cancers to survive exposure to poly ADP ribose polymerase (PARP) inhibitors, such as olaparib. Therefore, loss of CDK12 protein or inactivation of CDK12 kinase activity leads to genome instability and renders cells more sensitive to eradication.
- PARP poly ADP ribose polymerase
- CDK12 targets including ATR and histones
- ATR and histones are involved in multiple DNA repair pathways, including mismatch repair.
- defects in mismatch repair have been found to confer sensitivity of cells to cancer immunotherapy.
- modulators of CDK12 could help the immune system eradicate cells expressing aberrant levels of CDK12 in subjects.
- CDK12 also is known to play a role in regulating cell cycle progression and cell proliferation. Specifically, deletion of CDK12 has been shown to prolong the cell cycle. Long-term depletion of CDK12 has many effects including inducing cell accumulation in G2/M phase, inducing the G1/S cell cycle progression defect, and inducing the decreased expression of some crucial DNA replication genes e.g.
- compounds that modulate the CDK12 pathway can used in the treatment of cancer and cancer- related disorders and diseases including breast cancer, ovarian cancer, prostate cancer, and gastric cancer.
- ring A is a 5-7-membered heterocycle comprising 0 or 1 additional ring nitrogen atoms, and ring A can be optionally substituted with 1 to 3 C 1-3 alkyl substituents
- ring B is phenyl or a 6-10-membered heteroaryl comprising 1 or 2 ring nitrogen atoms
- X is CH or N
- n is 0, 1, or 2
- R 1 is halo, CN, SOC 1-3 alkyl, or SO 2 C 1-3 alkyl
- each R 2 is independently halo, CN, OH, –C 0-6 alkylene-N(R N ) 2 , CO 2 R N , COR N , C
- compositions comprising the compounds as disclosed herein.
- methods of treating or preventing a disease or disorder associated with aberrant CDK12 activity in a subject comprising administering to the subject a therapeutically effective amount of a compound as disclosed herein.
- CDK12 modulators such as compounds of Formula (I): , and pharmaceutically acceptable salts thereof wherein ring A, ring B, X, n, R 1 , R 2 , R 3 , R 4 , R 5 , R a , and R N are as described herein.
- ring A is a 5-7-membered heterocycle comprising 0 or 1 additional ring nitrogen atoms.
- ring A can be optionally substituted with 1 to 3 C 1-3 alkyl substituents.
- ring A is pyrrolidine, piperidine, 3-azabicyclo[3.1.0]hexane, or azepane.
- ring A is piperidinine.
- ring A is piperidinine and is substituted with 1 or 2 C 1-3 alkyl substituents.
- ring B can be phenyl or a 6-10-membered heteroaryl comprising 1 or 2 ring nitrogen atoms.
- ring B is phenyl, pyridine, pyrimidine, pyrazine, pyridazine, quinolone, or 4a,5,6,7-tetrahydro-1,6-naphthyridine.
- ring B is phenyl or pyridyl.
- X can be CH or N. In various cases, X is CH. In some cases, X is N.
- the compound has a structure of Formula (Ib): [0017] As disclosed herein, R 1 can be halo, CN SOC 1-3 alkyl, or SO 2 C 1-3 alkyl. In various cases, R 1 is halo, CN or SO 2 C 1-3 alkyl. In some cases, R 1 is F, Cl, CN, or SO 2 CH3. In some cases, R 1 is F. [0018] As disclosed herein, n can be 0, 1, or 2. In various cases, n is 0. In some cases, n is 1 or 2.
- each R 2 can independently be halo, CN, OH, –C 0-6 alkylene-N(R N ) 2 , CO 2 R N , COR N , C 1-6 alkyl, C 1-6 alkoxy, C 1-6 haloalkyl, –C 1-6 hydroxyalkyl, –OC 2-6 hydroxyalkyl, –OC 2-6 alkylene-N(R N ) 2 , CON(R N ) 2 , CON(R N )-4-8-membered heterocycle, C(O)-4-8-membered heterocycle, –[O] 0-1 -4-8 membered heterocycle, or – [O] 0-1 -5-10 membered heteroaryl, wherein each heterocycle and heteroaryl comprises 1, 2, or 3 ring heteroatoms selected from N, O, and S, and the heterocycle or heteroaryl is optionally substituted with 1 or 2 C 1-6 alkyl, and when ring B is a heterocycle or
- each R 2 is independently halo, CN, OH, –C 0-6 alkylene-N(R N ) 2 , C 1-6 alkyl, C 1-6 alkoxy, C 1-6 haloalkyl, –C 1-6 hydroxyalkyl, –OC 2- 6 hydroxyalkyl, –OC 2-6 alkylene-N(R N ) 2 , CON(R N ) 2 , –[O] 0-1 -4-8 membered heterocycle.
- each R 2 is independently Br, CN, OH, C 1-6 haloalkyl, –C 0-6 alkylene-N(R N ) 2 , OC 2-6 hydroxyalkyl, –OC 2-6 alkylene-N(R N ) 2 , or – [O] 0-1 -4-8 membered heterocycle.
- at least one R 2 is oxo.
- R 3 can be H, halo, CN, C 1-3 alkyl, or C 1-3 haloalkyl.
- R 3 is halo or CN.
- R 3 is Cl or CN.
- R 4 can be phenyl or a 5-10-membered heteroaryl comprising 1 to 3 ring atoms independently selected from N, O, and S, and R 4 is optionally substituted with 1, 2, or 3 R a .
- R 4 is indole, 1,7-dihydropyrazolo[1,5-a]pyridine, indazole, benzo[d]imidazole, 4,5,6,7-tetrahydropyrazolo[1,5- a]pyrazine, imidazo[1,2-a]pyridine, pyrazolo[4,3-b]pyridine, [1,2,4]triazolo[4,3-a]pyridine, benzo[d][1,2,3]triazole, phenyl, pyridine, pyrazole, or imidazole.
- R 4 is indole, 1,7-dihydropyrazolo[1,5-a]pyridine, indazole, benzo[d]imidazole, phenyl, pyridine, pyrazole, or imidazole. In some cases, R 4 is substituted with 1, 2, or 3 R a .
- R 5 can be H or C 1-3 alkyl. In various cases, R 5 is H. In some cases, R 5 is C 1-3 alkyl. In some cases, R 5 is CH 3 .
- each R a can independently be halo, OH, CON(R N ) 2 , C 1-6 alkyl, C 1-6 alkoxy, C 1- 6 hydroxyalkyl, CO 2 C 1-6 alkyl, C 0-6 alkylene-N(R N ) 2 , or a 4-6-membered heterocycle comprising 1 or 2 ring atoms selected from N, O, and S.
- each R a is independently halo, C 1-6 alkoxy, C 1-6 alkyl, CO 2 C 1-6 alkyl, C 1-6 hydroxyalkyl, or a 4-6-membered heterocycle comprising 1 or 2 ring atoms selected from N, O, and S.
- each R a is independently F, Cl, C 1-6 alkyl, C 1-6 alkoxy, C 1-6 hydroxyalkyl, or a 4-6-membered heterocycle comprising 1 or 2 ring atoms selected from N, O, and S.
- each R N can independently be H, C 1-6 alkyl, C 1-6 hydroxyalkyl, or C 1-6 alkylene-N(C 1- 3 alkyl) 2 , or two R N groups, together with a nitrogen to which they are each attached, form a 4-10 membered heterocycle comprising 0-2 additional ring heteroatoms independently selected from N, O, and S, and can be optionally substituted with 1 or 2 C 1-6 alkyl.
- each R N is independently H or C 1-6 alkyl.
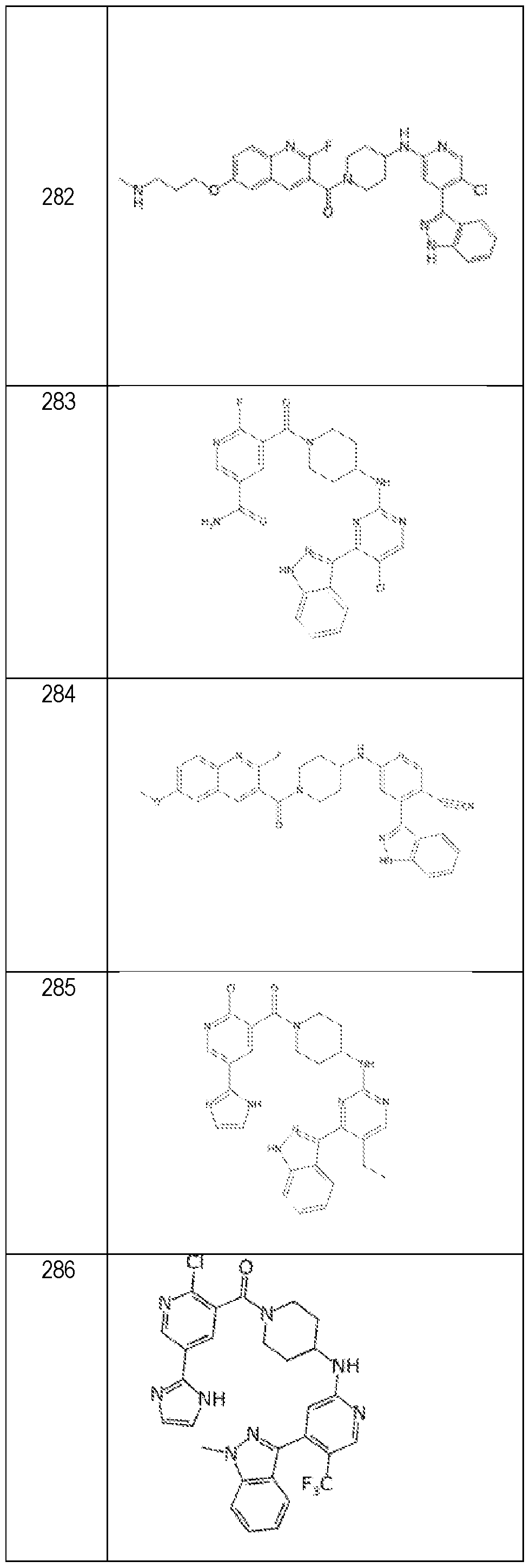
- Compounds as disclosed herein include those as provided in Table A, or a pharmaceutically acceptable salt thereof. Table A.
- a compound as disclosed herein is selected from the group consisting of or a pharmaceutically acceptable salt of any of the foregoing.
- structures depicted herein are also meant to include all isomeric (e.g., enantiomeric, diastereomeric, cis-trans, conformational, and rotational) forms of the structure.
- isomeric e.g., enantiomeric, diastereomeric, cis-trans, conformational, and rotational
- the R and S configurations for each asymmetric center, (Z) and (E) double bond isomers, and (Z) and (E) conformational isomers are included in this disclosure, unless only one of the isomers is specifically indicated.
- stereoisomers refer to compounds that differ in the chirality of one or more stereocenters. Stereoisomers include enantiomers and diastereomers.
- the compounds disclosed herein can exist as a single stereoisomer, or as a mixture of stereoisomers. Stereochemistry of the compounds shown herein indicate a relative stereochemistry, not absolute, unless discussed otherwise.
- a single stereoisomer, diastereomer, or enantiomer refers to a compound that is at least more than 50% of the indicated stereoisomer, diastereomer, or enantiomer, and in some cases, at least 90% or 95% of the indicated stereoisomer, diastereomer, or enantiomer.
- the compounds disclosed herein that have a double bond can exhibit E or Z (not shown) stereochemistry. In some cases, the compounds of Formula (I) exhibit E stereochemistry. In various cases, the compounds of Formula (I) exhibit Z stereochemistry at the double bond.
- the compounds of Formula (I) can have any stereochemical configuration at any sp 3 carbon atoms. In some cases, the compounds of the disclosure are optically pure.
- optically pure refers to the predominant presence of one enantiomer of a compound if multiple stereochemical configurations can exist (e.g., at least 99% enantiomeric excess).
- all tautomeric forms of the compounds of the disclosure are within the scope of the disclosure.
- the compounds of the disclosure are defined herein by their chemical structures and/or chemical names. Where a compound is referred to by both a chemical structure and a chemical name, and the chemical structure and chemical name conflict, the chemical structure is determinative of the compound's identity.
- alkyl refers to straight chained and branched saturated hydrocarbon groups containing one to thirty carbon atoms, for example, one to twenty carbon atoms, or one to ten carbon atoms.
- C n means the alkyl group has “n” carbon atoms.
- C 6 alkyl refers to an alkyl group that has 6 carbon atoms.
- C 1-7 alkyl refers to an alkyl group having a number of carbon atoms encompassing the entire range (i.e., 1 to 6 carbon atoms), as well as all subgroups (e.g., 1-5, 2-5, 3-6, 1, 2, 3, 4, 5, and 6 carbon atoms).
- alkyl groups include, methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl (2- methylpropyl), and t-butyl (1,1-dimethylethyl).
- an alkyl group can be an unsubstituted alkyl group or a substituted alkyl group.
- alkylene refers to a bivalent saturated aliphatic radical.
- Cn means the alkylene group has "n" carbon atoms, e.g., a C 1 alkylene is CH 2 .
- a heterocycle can be a 8-10 membered bicyclic, bridged, fused, or spirocyclic group having 1 or 2 or 3 ring heteroatoms selected from N, O, and S in the bicyclic ring.
- heterocycle groups include piperidine, piperazine, tetrahydrofuran, tetrahydropyran, dihydrofuran, morpholine, oxazepane, thiazole, pyrrole, and pyridine.
- heteroaryl refers to a cyclic aromatic ring having heteroatoms in the ring (e.g., a monocyclic aromatic ring with 5-6 total ring atoms, or a fused bicyclic ring with 10 total ring atoms), and containing one to three heteroatoms selected from nitrogen, oxygen, and sulfur atom in the aromatic ring. Unless otherwise indicated, a heteroaryl group can be unsubstituted or substituted.
- halo refers to refers to a fluoro (F), chloro (Cl), bromo (Br), or iodo (I) group.
- haloalkyl refers to an alkyl group in which one or more of the hydrogen atoms are replaced by halogen. Such groups include but are not limited to, chloromethyl, fluoromethyl, difluoromethyl, trifluoromethyl, 1,1-difluoroethyl, 2-fluoroethyl, 1-chloro-2-fluoromethyl and 2-fluoroisobutyl.
- hydroxyalkyl refers to an alkyl group in which one or more of the hydrogen atoms are replaced by a hydroxyl group (OH).
- OH hydroxyl group
- groups include but are not limited to, hydroxymethyl, hydroxyethyl, and the like.
- a “substituted” functional group is a functional, group having at least one hydrogen radical that is substituted with a non-hydrogen radical (i.e., a substituent).
- non-hydrogen radicals include, but are not limited to, alkyl, cycloalkyl, alkenyl, cycloalkyl, alkynyl, ether, aryl, heteroaryl, heterocycle, hydroxyl, oxy (or oxo), alkoxyl, ester, thioester, acyl, carboxyl, cyano, nitro, amino, sulfhydryl, and halo.
- substituents can be bound to the same carbon or different carbon atoms.
- acid addition salts can be prepared in situ during the final isolation and purification of the compounds.
- acid addition salts can be prepared by 1) reacting the purified compound in its free-base form with a suitable organic or inorganic acid and 2) isolating the salt thus formed.
- acid addition salts might be a more convenient form for use and use of the salt amounts to use of the free basic form.
- Examples of pharmaceutically acceptable, non-toxic acid addition salts are salts of an amino group formed with inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid or by using other methods used in the art such as ion exchange.
- inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid
- organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid or by using other methods used in the art such as ion exchange.
- salts include adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, glycolate, gluconate, glycolate, hemisulfate, heptanoate, hexanoate, hydrochloride, hydrobromide, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2- naphthalenesulfonate, nicotinate, nitrate, oleate, o
- base addition salts can be prepared by 1) reacting the purified compound in its acid form with a suitable organic or inorganic base and 2) isolating the salt thus formed.
- base addition salt might be more convenient and use of the salt form inherently amounts to use of the free acid form.
- Salts derived from appropriate bases include alkali metal (e.g., sodium, lithium, and potassium), alkaline earth metal (e.g., magnesium and calcium), ammonium and N + (C 1 - 4 alkyl) 4 salts. This disclosure also envisions the quaternization of any basic nitrogen-containing groups of the compounds disclosed herein.
- Basic addition salts include pharmaceutically acceptable metal and amine salts. Suitable metal salts include the sodium, potassium, calcium, barium, zinc, magnesium, and aluminum. The sodium and potassium salts are usually preferred. Further pharmaceutically acceptable salts include, when appropriate, nontoxic ammonium, quaternary ammonium, and amine cations formed using counterions such as halide, hydroxide, carboxylate, sulfate, phosphate, nitrate, lower alkyl sulfonate and aryl sulfonate.
- Suitable inorganic base addition salts are prepared from metal bases which include sodium hydride, sodium hydroxide, potassium hydroxide, calcium hydroxide, aluminum hydroxide, lithium hydroxide, magnesium hydroxide, zinc hydroxide and the like.
- Suitable amine base addition salts are prepared from amines which are frequently used in medicinal chemistry because of their low toxicity and acceptability for medical use.
- Ammonia ethylenediamine, N-methyl-glucamine, lysine, arginine, ornithine, choline, N,N'-dibenzylethylenediamine, chloroprocaine, dietanolamine, procaine, N- benzylphenethylamine, diethylamine, piperazine, tris(hydroxymethyl)-aminomethane, tetramethylammonium hydroxide, triethylamine, dibenzylamine, ephenamine, dehydroabietylamine, N-ethylpiperidine, benzylamine, tetramethylammonium, tetraethylammonium, methylamine, dimethylamine, trimethylamine, ethylamine, basic amino acids, dicyclohexylamine and the like.
- compositions that include an effective amount of compounds of the disclosure and one or more pharmaceutically acceptable excipients.
- an "effective amount” includes a “therapeutically effective amount” and a “prophylactically effective amount.”
- therapeutically effective amount refers to an amount effective in treating and/or ameliorating a disease or condition in a subject.
- prophylactically effective amount refers to an amount effective in preventing and/or substantially lessening the chances of a disease or condition in a subject.
- patient and “subject” may be used interchangeably and mean animals, such as dogs, cats, cows, horses, and sheep (i.e., non-human animals) and humans. Particular patients or subjects are mammals (e.g., humans).
- the term “excipient” means any pharmaceutically acceptable additive, carrier, diluent, adjuvant, or other ingredient, other than the active pharmaceutical ingredient (API), suitably selected with respect to the intended form of administration, and consistent with conventional pharmaceutical practices.
- the compounds of the disclosure can be administered alone or as part of a pharmaceutically acceptable composition or formulation.
- the compounds can be administered all at once, as for example, by a bolus injection, multiple times, e.g., by a series of tablets, or delivered substantially uniformly over a period of time, as for example, using transdermal delivery. It is also noted that the dose of the compound can be varied over time.
- the dosage form may also comprise buffering agents.
- Solid compositions of a similar type may also be employed as fillers in soft and hard-filled gelatin capsules using such excipients as lactose or milk sugar as well as high molecular weight polyethylene glycols and the like.
- the solid dosage forms of tablets, dragees, capsules, pills, and granules can be prepared with coatings and shells such as enteric coatings and other coatings well known in the pharmaceutical formulating art. They may optionally contain opacifying agents and can also be of a composition that they release the active ingredient(s) only, or preferentially, in a certain part of the intestinal tract, optionally, in a delayed manner.
- embedding compositions examples include polymeric substances and waxes. Solid compositions of a similar type may also be employed as fillers in soft and hard-filled gelatin capsules using such excipients as lactose or milk sugar as well as high molecular weight polethylene glycols and the like.
- the active compounds can also be in microencapsulated form with one or more excipients as noted above.
- the solid dosage forms of tablets, dragees, capsules, pills, and granules can be prepared with coatings and shells such as enteric coatings, release controlling coatings and other coatings well known in the pharmaceutical formulating art.
- the pharmaceutical compositions may be formulated in a suitable ointment, cream, lotion, or gel, containing the active component suspended or dissolved in one or more carriers, and any needed preservatives or buffers as may be required.
- Carriers for topical administration of the compounds of this disclosure include, but are not limited to, mineral oil, liquid petrolatum, white petrolatum, propylene glycol, polyoxyethylene, polyoxypropylene compound, emulsifying wax and water.
- the pharmaceutical compositions can be formulated in a suitable lotion or cream containing the active components suspended or dissolved in one or more pharmaceutically acceptable carriers.
- Suitable carriers include, but are not limited to, mineral oil, sorbitan monostearate, polysorbate 60, cetyl esters wax, cetearyl alcohol, 2 octyldodecanol, benzyl alcohol and water.
- Ophthalmic formulation, eardrops, and eye drops are also contemplated as being within the scope of this disclosure.
- the present disclosure contemplates the use of transdermal patches, which have the added advantage of providing controlled delivery of a compound to the body. Such dosage forms can be made by dissolving or dispensing the compound in the proper medium. Absorption enhancers can also be used to increase the flux of the compound across the skin.
- Injectable preparations for example, sterile injectable aqueous or oleaginous suspensions may be formulated according to the known art using suitable dispersing or wetting agents and suspending agents.
- the sterile injectable preparation may also be a sterile injectable solution, suspension or emulsion in a nontoxic parenterally acceptable diluent or solvent, for example, as a solution in 1,3-butanediol.
- acceptable vehicles and solvents that may be employed are water, Ringer's solution, U.S.P. and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil can be employed including synthetic mono- or diglycerides.
- fatty acids such as oleic acid are used in the preparation of injectables.
- the injectable formulations can be sterilized, for example, by filtration through a bacterial-retaining filter, or by incorporating sterilizing agents in the form of sterile solid compositions which can be dissolved or dispersed in sterile water or other sterile injectable medium prior to use.
- sterilizing agents in the form of sterile solid compositions which can be dissolved or dispersed in sterile water or other sterile injectable medium prior to use.
- Injectable depot forms are made by forming microencapsule matrices of the compound in biodegradable polymers such as polylactide-polyglycolide. Depending upon the ratio of compound to polymer and the nature of the particular polymer employed, the rate of compound release can be controlled. Examples of other biodegradable polymers include poly(orthoesters) and poly(anhydrides).
- compositions for rectal or vaginal administration are specifically suppositories which can be prepared by mixing the compounds described herein with suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol or a suppository wax which are solid at ambient temperature but liquid at body temperature and therefore melt in the rectum or vaginal cavity and release the active compound.
- suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol or a suppository wax which are solid at ambient temperature but liquid at body temperature and therefore melt in the rectum or vaginal cavity and release the active compound.
- Sterile injectable forms of the compositions described herein may be aqueous or oleaginous suspension. These suspensions may be formulated according to techniques known in the art using suitable dispersing or wetting agents and suspending agents.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally-acceptable diluent or solvent, for example as a solution in 1,3-butanediol.
- a non-toxic parenterally-acceptable diluent or solvent for example as a solution in 1,3-butanediol.
- acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil may be employed including synthetic mono- or di-glycerides.
- Fatty acids, such as oleic acid and its glyceride derivatives are useful in the preparation of injectables, as are natural pharmaceutically acceptable oils, such as olive oil or castor oil, especially in their polyoxyethylated versions.
- the unit dosage form can be the same or different for each dose.
- the compounds of the disclosure can be administered to a subject or patient at dosage levels in the range of about 0.1 to about 3,000 mg per day. For a normal adult human having a body weight of about 70 kg, a dosage in the range of about 0.01 to about 100 mg per kilogram body weight is typically sufficient.
- the specific dosage and dosage range that will be used can potentially depend on a number of factors, including the requirements of the subject or patient, the severity of the condition or disease being treated, and the pharmacological activity of the compound being administered. The determination of dosage ranges and optimal dosages for a particular subject or patient is within the ordinary skill in the art.
- CDK12 Cyclin-dependent kinase 12
- CDK12 Cyclin-dependent kinase 12
- CDC6 Cell division cycle 6
- CDT1 CDT1
- the disclosure provides a method of modulating cyclin-dependent kinase 12 (CDK12) comprising contacting the CDK12 with a therapeutically effective amount of a compound or salt disclosed herein or a formulation thereof, in an amount effective to modulate CDK12 activity.
- the contacting occurs in vitro.
- the contacting occurs in vivo.
- the contacting comprises administering to a subject in need thereof.
- the terms “patient” and “subject” may be used interchangeably and mean animals, such as dogs, cats, cows, horses, and sheep (i.e., non-human animals) and humans.
- the patient is a mammal (e.g., human).
- the subject suffers from cancer.
- the cancer is breast cancer, ovarian cancer, prostate cancer, or gastric cancer.
- the terms “treating”, “treat” or “treatment” and the like can include preventative (e.g., prophylactic) and palliative treatment.
- the disease or disorder is cancer.
- the disease or disorder is breast cancer, ovarian cancer, prostate cancer, or gastric cancer.
- Another aspect of the disclosure provides the use of a compound disclosed herein, a pharmaceutically acceptable salt thereof, or a formulation thereof in the treatment of a disease or disorder associated with aberrant CDK12 activity in a subject, where the treatment increases activity of the subject’s immune system to eradicate cells associated with aberrant CDK12 activity.
- the disease or disorder is cancer.
- the cancer is breast cancer, ovarian cancer, prostate cancer, or gastric cancer.
- R 4 is indole, 1,7- dihydropyrazolo[1,5-a]pyridine, indazole, benzo[d]imidazole, 4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazine, imidazo[1,2-a]pyridine, pyrazolo[4,3-b]pyridine, [1,2,4]triazolo[4,3-a]pyridine, benzo[d][1,2,3]triazole, phenyl, pyridine, pyrazole, or imidazole. 21.
- a pharmaceutical formulation comprising the compound or salt of any one of embodiments 1 to 29 and a pharmaceutically acceptable excipient.
- a method of modulating CDK12 activity comprising contacting the CDK12 with the compound or salt of any one of embodiments 1 to 29 in an amount effective to modulate CDK12 activity.
- 32. A method of treating a disease or disorder associated with aberrant CDK12 activity in a subject, comprising administering to the subject a therapeutically effective amount of the compound or salt of any one of embodiments 1 to 29.
- 33. The method of embodiment 32, wherein the disease or disorder is cancer.
- 34. The method of embodiment 33, wherein the cancer is breast cancer, ovarian cancer, prostate cancer, or gastric cancer. 35.
- Boc-C-6 (2.8 g, yield: 53.6 %) as a yellow solid.
- reaction mixture was stirred at 120 °C for 16 h.
- the LCMS showed the desired MS was detected.
- the reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na 2 SO 4 , filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 85/15 in 15 mins) to afford tert-butyl 4-((5-methyl-4-(1-((2- (trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyrimidin-2-yl)amino)piperidine-1-carboxylate (650.0 mg, yield: 45.2%) as a white solid.
- Boc-C-59 (1.1 g, yield: 57.8%) as yellow solid.
- the compounds of the disclosure were prepared by carboxylic acid / amine condensation reactions, as noted in the below scheme: [0244] The following compounds were synthesized by common carboxylic acid-amine condensation reactions, e.g., conditions (A) or (B) as described below. [0245] (A) To a solution of corresponding carboxylic acid (0.5 mmol) in DMF (5 mL) was added HATU (1.0 mmol) and N, N-Diisopropylethylamine (1.5 mmol) at 25°C. After 20 min, intermediate C (0.55 mmol) in DMF (1 mL) was added. The reaction mixture was stirred at 1 h for 25 °C. The desired mass was detected on LC-MS.
- reaction solution was stirred at rt for 3 h.
- the LCMS showed the desired MS was detected.
- the mixture was concentrated and was purified by prep-HPLC (columns: Gemini 5 um C18150 x 21.2 mm, mobile phase: ACN – H 2 O (0.1% FA), gradient: 50 - 70, 12 min) to afford final compound.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Provided herein are compounds having a structure of Formula (I) and pharmaceutically acceptable salts thereof which can act as modulators of cyclin-dependent kinase 12 (CDK12). Further disclosed herein are methods for treating cancer and cancer-related diseases and disorders, such as breast cancer, ovarian cancer, prostate cancer, and gastric cancer.
Description
CYCLIN-DEPENDENT KINASE 12 MODULATORS AND THERAPEUTIC USES THEREOF FIELD [0001] This disclosure relates to compounds that act as modulators of cyclin-dependent kinase 12 (CDK12), pharmaceutical formulations thereof, and methods of using the compounds to treat diseases and disorders, such as cancer and cancer-related diseases and disorders. BACKGROUND [0002] Cyclin-dependent kinase 12 (CDK12) is an important transcription-associated CDK. Cyclin-dependent kinases (CDKs) are a group of serine/threonine protein kinases that play crucial roles in various cellular processes by regulating cell cycle and gene transcription. It shows versatile roles in regulating gene transcription, RNA splicing, translation, DNA damage response, cell cycle progression, and cell proliferation. Recently, increasing evidence demonstrates the important role of CDK12 in various human cancers, illustrating it as both a biomarker of cancer and a potential target for cancer therapy. [0003] CDK12 also is known to have an important role in DNA repair and homologous recombination, and thus is directly implicated in enabling cancers to survive exposure to poly ADP ribose polymerase (PARP) inhibitors, such as olaparib. Therefore, loss of CDK12 protein or inactivation of CDK12 kinase activity leads to genome instability and renders cells more sensitive to eradication. Several CDK12 targets, including ATR and histones, are involved in multiple DNA repair pathways, including mismatch repair. Importantly, defects in mismatch repair have been found to confer sensitivity of cells to cancer immunotherapy. Accordingly, modulators of CDK12 could help the immune system eradicate cells expressing aberrant levels of CDK12 in subjects. [0004] CDK12 also is known to play a role in regulating cell cycle progression and cell proliferation. Specifically, deletion of CDK12 has been shown to prolong the cell cycle. Long-term depletion of CDK12 has many effects including inducing cell accumulation in G2/M phase, inducing the G1/S cell cycle progression defect, and inducing the decreased expression of some crucial DNA replication genes e.g. TOPBP1 (DNA topoisomerase II binding protein 1), CDC6 (cell division cycle 6) and CDT1 (Cdc10-dependent transcript 1). [0005] As cell cycle is closely related with cell proliferation, aberrant cell cycle progression may result in abnormal cell proliferation. CDK12 is required for multiple steps in mitosis. Thus, CDK12 deficiency has been shown to inhibit cell proliferation and induce apoptosis via the induction of mitotic catastrophe. Additionally, various human cancers characterized by uncontrolled cell proliferation have high levels of CDK12. Accordingly, CDK12 is an attractive target for the treatment of various human cancers. [0006] Thus, compounds that modulate the CDK12 pathway can used in the treatment of cancer and cancer- related disorders and diseases including breast cancer, ovarian cancer, prostate cancer, and gastric cancer.
SUMMARY [0007] Provided herein are compounds having a structure of Formula (I):
 , or pharmaceutically acceptable salts thereof, wherein: ring A is a 5-7-membered heterocycle comprising 0 or 1 additional ring nitrogen atoms, and ring A can be optionally substituted with 1 to 3 C1-3alkyl substituents, ring B is phenyl or a 6-10-membered heteroaryl comprising 1 or 2 ring nitrogen atoms, X is CH or N, n is 0, 1, or 2, R1 is halo, CN, SOC1-3alkyl, or SO2C1-3alkyl, each R2 is independently halo, CN, OH, –C0-6alkylene-N(RN)2, CO2RN, CORN, C1-6alkyl, C1-6alkoxy, C1-6haloalkyl, –C1-6hydroxyalkyl, –OC2-6hydroxyalkyl, –OC2-6alkylene-N(RN)2, CON(RN)2, SO2 C1-6alkyl, CON(RN)-4-8-membered heterocycle, C(O)-4-8-membered heterocycle, –[O]0-1-4-8 membered heterocycle, or –[O]0-1-5-10 membered heteroaryl, wherein each heterocycle and heteroaryl comprises 1, 2, or 3 ring heteroatoms selected from N, O, and S, and the heterocycle or heteroaryl is optionally substituted with 1 or 2 C1-6alkyl, and when ring B is a heterocycle or a 8-10-membered heteroaryl, R2 can be oxo, R3 is H, halo, CN, C1-3alkyl, C1-3haloalkyl, or C(O)N(RN)2, R4 is phenyl or a 5-10-membered heteroaryl comprising 1 to 3 ring atoms independently selected from N, O, and S, and R4 is optionally substituted with 1, 2, or 3 Ra, R5 is H or C1-3alkyl; each Ra is independently halo, OH, C1-6alkyl, C1-6alkoxy, C1-6hydroxyalkyl, CON(RN)2, CO2C1- 6alkyl, C0-6alkylene-N(RN)2, C0-6alkylene-C3-10cycloalkyl, or a 4-6-membered heterocycle comprising 1 or 2 ring atoms selected from N, O, and S, and each RN is independently H, C1-6alkyl, C1-6hydroxyalkyl, or C1-6alkylene- N(C1-3alkyl)2, or two RN groups, together with a nitrogen to which they are each attached, form a 4-10 membered heterocycle comprising 0-2 additional ring heteroatoms independently selected from N, O, and S, and can be optionally substituted with 1 or 2 C1-6alkyl. [0008] Further provided herein are pharmaceutical compositions comprising the compounds as disclosed herein. Also provided are methods of treating or preventing a disease or disorder associated with aberrant CDK12 activity in a subject, comprising administering to the subject a therapeutically effective amount of a compound as disclosed herein. [0009] Further aspects and advantages will be apparent to those of ordinary skill in the art from a review of the following detailed description, taken in conjunction with the drawings. While the compounds and methods disclosed herein are susceptible of cases in various forms, the description hereafter includes specific cases with the understanding that the disclosure is illustrative and is not intended to limit the invention to the specific cases described herein.
DETAILED DESCRIPTION [0010] Provided herein are compounds that can act as CDK12 modulators, such as compounds of Formula (I):
, or pharmaceutically acceptable salts thereof, wherein: ring A is a 5-7-membered heterocycle comprising 0 or 1 additional ring nitrogen atoms, and ring A can be optionally substituted with 1 to 3 C1-3alkyl substituents, ring B is phenyl or a 6-10-membered heteroaryl comprising 1 or 2 ring nitrogen atoms, X is CH or N, n is 0, 1, or 2, R1 is halo, CN, SOC1-3alkyl, or SO2C1-3alkyl, each R2 is independently halo, CN, OH, –C0-6alkylene-N(RN)2, CO2RN, CORN, C1-6alkyl, C1-6alkoxy, C1-6haloalkyl, –C1-6hydroxyalkyl, –OC2-6hydroxyalkyl, –OC2-6alkylene-N(RN)2, CON(RN)2, SO2 C1-6alkyl, CON(RN)-4-8-membered heterocycle, C(O)-4-8-membered heterocycle, –[O]0-1-4-8 membered heterocycle, or –[O]0-1-5-10 membered heteroaryl, wherein each heterocycle and heteroaryl comprises 1, 2, or 3 ring heteroatoms selected from N, O, and S, and the heterocycle or heteroaryl is optionally substituted with 1 or 2 C1-6alkyl, and when ring B is a heterocycle or a 8-10-membered heteroaryl, R2 can be oxo, R3 is H, halo, CN, C1-3alkyl, C1-3haloalkyl, or C(O)N(RN)2, R4 is phenyl or a 5-10-membered heteroaryl comprising 1 to 3 ring atoms independently selected from N, O, and S, and R4 is optionally substituted with 1, 2, or 3 Ra, R5 is H or C1-3alkyl; each Ra is independently halo, OH, C1-6alkyl, C1-6alkoxy, C1-6hydroxyalkyl, CON(RN)2, CO2C1- 6alkyl, C0-6alkylene-N(RN)2, C0-6alkylene-C3-10cycloalkyl, or a 4-6-membered heterocycle comprising 1 or 2 ring atoms selected from N, O, and S, and each RN is independently H, C1-6alkyl, C1-6hydroxyalkyl, or C1-6alkylene- N(C1-3alkyl)2, or two RN groups, together with a nitrogen to which they are each attached, form a 4-10 membered heterocycle comprising 0-2 additional ring heteroatoms independently selected from N, O, and S, and can be optionally substituted with 1 or 2 C1-6alkyl. [0008] Further provided herein are pharmaceutical compositions comprising the compounds as disclosed herein. Also provided are methods of treating or preventing a disease or disorder associated with aberrant CDK12 activity in a subject, comprising administering to the subject a therapeutically effective amount of a compound as disclosed herein. [0009] Further aspects and advantages will be apparent to those of ordinary skill in the art from a review of the following detailed description, taken in conjunction with the drawings. While the compounds and methods disclosed herein are susceptible of cases in various forms, the description hereafter includes specific cases with the understanding that the disclosure is illustrative and is not intended to limit the invention to the specific cases described herein.
DETAILED DESCRIPTION [0010] Provided herein are compounds that can act as CDK12 modulators, such as compounds of Formula (I):
 , and pharmaceutically acceptable salts thereof wherein ring A, ring B, X, n, R1, R2, R3, R4, R5, Ra, and RN are as described herein. Compounds of the Disclosure [0011] Disclosed herein are compounds having a structure of Formula (I):
, and pharmaceutically acceptable salts thereof wherein ring A, ring B, X, n, R1, R2, R3, R4, R5, Ra, and RN are as described herein. Compounds of the Disclosure [0011] Disclosed herein are compounds having a structure of Formula (I):
 , and pharmaceutically acceptable salts thereof wherein: ring A is a 5-7-membered heterocycle comprising 0 or 1 additional ring nitrogen atoms, and ring A can be optionally substituted with 1 to 3 C1-3alkyl substituents; ring B is phenyl or a 6-10-membered heteroaryl comprising 1 or 2 ring nitrogen atoms; X is CH or N; n is 0, 1, or 2; R1 is halo, CN, SOC1-3alkyl, or SO2C1-3alkyl; each R2 is independently halo, CN, OH, –C0-6alkylene-N(RN)2, CO2RN, CORN, C1-6alkyl, C1-6alkoxy, C1-6haloalkyl, –C1-6hydroxyalkyl, –OC2-6hydroxyalkyl, –OC2-6alkylene-N(RN)2, CON(RN)2, SO2C1-6alkyl CON(RN)-4-8- membered heterocycle, C(O)-4-8-membered heterocycle, –[O]0-1-4-8 membered heterocycle, or –[O]0-1- 5-10 membered heteroaryl, wherein each heterocycle and heteroaryl comprises 1, 2, or 3 ring heteroatoms selected from N, O, and S, and the heterocycle or heteroaryl is optionally substituted with 1 or 2 C1-6alkyl, and when ring B is a heterocycle or a 8-10-membered heteroaryl, R2 can be oxo R3 is H, halo, CN, C1-3alkyl, C1-3haloalkyl, C(O)N(RN)2; R4 is phenyl or a 5-10-membered heteroaryl comprising 1 to 3 ring atoms independently selected from N, O, and S, and R4 is optionally substituted with 1, 2, or 3 Ra; R5 is H or C1-3alkyl; each Ra is independently halo, OH, C1-6alkyl, C1-6alkoxy, C1-6hydroxyalkyl, CON(RN)2, CO2C1-6alkyl, C0-6alkylene- N(RN)2, C0-6alkyleneC3-10cycloalkyl, or a 4-6-membered heterocycle comprising 1 or 2 ring atoms selected from N, O, and S; and
each RN is independently H, C1-6alkyl, C1-6hydroxyalkyl, or C1-6alkylene-N(C1-3alkyl)2, or two RN groups, together with a nitrogen to which they are each attached, form a 4-10 membered heterocycle comprising 0-2 additional ring heteroatoms independently selected from N, O, and S, and can be optionally substituted with 1 or 2 C1-6alkyl. [0012] In compounds of Formula (I), ring A is a 5-7-membered heterocycle comprising 0 or 1 additional ring nitrogen atoms. In various cases, ring A can be optionally substituted with 1 to 3 C1-3alkyl substituents. In various cases, ring A is pyrrolidine, piperidine, 3-azabicyclo[3.1.0]hexane, or azepane. In some cases, ring A is piperidinine. In some cases, ring A is piperidinine and is substituted with 1 or 2 C1-3alkyl substituents. [0013] In various cases, the compound has a structure of Formula (Ia):
, and pharmaceutically acceptable salts thereof wherein: ring A is a 5-7-membered heterocycle comprising 0 or 1 additional ring nitrogen atoms, and ring A can be optionally substituted with 1 to 3 C1-3alkyl substituents; ring B is phenyl or a 6-10-membered heteroaryl comprising 1 or 2 ring nitrogen atoms; X is CH or N; n is 0, 1, or 2; R1 is halo, CN, SOC1-3alkyl, or SO2C1-3alkyl; each R2 is independently halo, CN, OH, –C0-6alkylene-N(RN)2, CO2RN, CORN, C1-6alkyl, C1-6alkoxy, C1-6haloalkyl, –C1-6hydroxyalkyl, –OC2-6hydroxyalkyl, –OC2-6alkylene-N(RN)2, CON(RN)2, SO2C1-6alkyl CON(RN)-4-8- membered heterocycle, C(O)-4-8-membered heterocycle, –[O]0-1-4-8 membered heterocycle, or –[O]0-1- 5-10 membered heteroaryl, wherein each heterocycle and heteroaryl comprises 1, 2, or 3 ring heteroatoms selected from N, O, and S, and the heterocycle or heteroaryl is optionally substituted with 1 or 2 C1-6alkyl, and when ring B is a heterocycle or a 8-10-membered heteroaryl, R2 can be oxo R3 is H, halo, CN, C1-3alkyl, C1-3haloalkyl, C(O)N(RN)2; R4 is phenyl or a 5-10-membered heteroaryl comprising 1 to 3 ring atoms independently selected from N, O, and S, and R4 is optionally substituted with 1, 2, or 3 Ra; R5 is H or C1-3alkyl; each Ra is independently halo, OH, C1-6alkyl, C1-6alkoxy, C1-6hydroxyalkyl, CON(RN)2, CO2C1-6alkyl, C0-6alkylene- N(RN)2, C0-6alkyleneC3-10cycloalkyl, or a 4-6-membered heterocycle comprising 1 or 2 ring atoms selected from N, O, and S; and
each RN is independently H, C1-6alkyl, C1-6hydroxyalkyl, or C1-6alkylene-N(C1-3alkyl)2, or two RN groups, together with a nitrogen to which they are each attached, form a 4-10 membered heterocycle comprising 0-2 additional ring heteroatoms independently selected from N, O, and S, and can be optionally substituted with 1 or 2 C1-6alkyl. [0012] In compounds of Formula (I), ring A is a 5-7-membered heterocycle comprising 0 or 1 additional ring nitrogen atoms. In various cases, ring A can be optionally substituted with 1 to 3 C1-3alkyl substituents. In various cases, ring A is pyrrolidine, piperidine, 3-azabicyclo[3.1.0]hexane, or azepane. In some cases, ring A is piperidinine. In some cases, ring A is piperidinine and is substituted with 1 or 2 C1-3alkyl substituents. [0013] In various cases, the compound has a structure of Formula (Ia):
 [0014] In compounds of Formula (I), ring B can be phenyl or a 6-10-membered heteroaryl comprising 1 or 2 ring nitrogen atoms. In various cases, ring B is phenyl, pyridine, pyrimidine, pyrazine, pyridazine, quinolone, or 4a,5,6,7-tetrahydro-1,6-naphthyridine. In some cases, ring B is phenyl or pyridyl. [0015] In various cases, X can be CH or N. In various cases, X is CH. In some cases, X is N. [0016] In various cases, the compound has a structure of Formula (Ib):
[0014] In compounds of Formula (I), ring B can be phenyl or a 6-10-membered heteroaryl comprising 1 or 2 ring nitrogen atoms. In various cases, ring B is phenyl, pyridine, pyrimidine, pyrazine, pyridazine, quinolone, or 4a,5,6,7-tetrahydro-1,6-naphthyridine. In some cases, ring B is phenyl or pyridyl. [0015] In various cases, X can be CH or N. In various cases, X is CH. In some cases, X is N. [0016] In various cases, the compound has a structure of Formula (Ib):
 [0017] As disclosed herein, R1 can be halo, CN SOC1-3alkyl, or SO2C1-3alkyl. In various cases, R1 is halo, CN or SO2C1-3alkyl. In some cases, R1 is F, Cl, CN, or SO2CH3. In some cases, R1 is F. [0018] As disclosed herein, n can be 0, 1, or 2. In various cases, n is 0. In some cases, n is 1 or 2. [0019] As disclosed herein, each R2 can independently be halo, CN, OH, –C0-6alkylene-N(RN)2, CO2RN, CORN, C1-6alkyl, C1-6alkoxy, C1-6haloalkyl, –C1-6hydroxyalkyl, –OC2-6hydroxyalkyl, –OC2-6alkylene-N(RN)2, CON(RN)2, CON(RN)-4-8-membered heterocycle, C(O)-4-8-membered heterocycle, –[O]0-1-4-8 membered heterocycle, or – [O]0-1-5-10 membered heteroaryl, wherein each heterocycle and heteroaryl comprises 1, 2, or 3 ring heteroatoms selected from N, O, and S, and the heterocycle or heteroaryl is optionally substituted with 1 or 2 C1-6alkyl, and when ring B is a heterocycle or a 8-10-membered heteroaryl, R2 can be oxo. In various cases, each R2 is independently halo, CN, OH, –C0-6alkylene-N(RN)2, C1-6alkyl, C1-6alkoxy, C1-6haloalkyl, –C1-6hydroxyalkyl, –OC2-
6hydroxyalkyl, –OC2-6alkylene-N(RN)2, CON(RN)2, –[O]0-1-4-8 membered heterocycle. In some cases, each R2 is independently Br, CN, OH, C1-6haloalkyl, –C0-6alkylene-N(RN)2, OC2-6hydroxyalkyl, –OC2-6alkylene-N(RN)2, or – [O]0-1-4-8 membered heterocycle. In some cases, at least one R2 is oxo. [0020] As disclosed herein, R3 can be H, halo, CN, C1-3alkyl, or C1-3haloalkyl. In various cases, R3 is halo or CN. In some cases, R3 is Cl or CN. [0021] As disclosed herein, R4 can be phenyl or a 5-10-membered heteroaryl comprising 1 to 3 ring atoms independently selected from N, O, and S, and R4 is optionally substituted with 1, 2, or 3 Ra. In various cases, R4 is indole, 1,7-dihydropyrazolo[1,5-a]pyridine, indazole, benzo[d]imidazole, 4,5,6,7-tetrahydropyrazolo[1,5- a]pyrazine, imidazo[1,2-a]pyridine, pyrazolo[4,3-b]pyridine, [1,2,4]triazolo[4,3-a]pyridine, benzo[d][1,2,3]triazole, phenyl, pyridine, pyrazole, or imidazole. In some cases, R4 is indole, 1,7-dihydropyrazolo[1,5-a]pyridine, indazole, benzo[d]imidazole, phenyl, pyridine, pyrazole, or imidazole. In some cases, R4 is substituted with 1, 2, or 3 Ra. [0022] As disclosed herein, R5 can be H or C1-3alkyl. In various cases, R5 is H. In some cases, R5 is C1-3alkyl. In some cases, R5 is CH3. [0023] As disclosed herein, each Ra can independently be halo, OH, CON(RN)2, C1-6alkyl, C1-6alkoxy, C1- 6hydroxyalkyl, CO2C1-6alkyl, C0-6alkylene-N(RN)2, or a 4-6-membered heterocycle comprising 1 or 2 ring atoms selected from N, O, and S. In various cases, each Ra is independently halo, C1-6alkoxy, C1-6alkyl, CO2C1-6alkyl, C1-6hydroxyalkyl, or a 4-6-membered heterocycle comprising 1 or 2 ring atoms selected from N, O, and S. In some cases, each Ra is independently F, Cl, C1-6alkyl, C1-6alkoxy, C1-6hydroxyalkyl, or a 4-6-membered heterocycle comprising 1 or 2 ring atoms selected from N, O, and S. [0024] As disclosed herein, each RN can independently be H, C1-6alkyl, C1-6hydroxyalkyl, or C1-6alkylene-N(C1- 3alkyl)2, or two RN groups, together with a nitrogen to which they are each attached, form a 4-10 membered heterocycle comprising 0-2 additional ring heteroatoms independently selected from N, O, and S, and can be optionally substituted with 1 or 2 C1-6alkyl. In various cases, each RN is independently H or C1-6alkyl. [0025] Compounds as disclosed herein include those as provided in Table A, or a pharmaceutically acceptable salt thereof. Table A.
[0017] As disclosed herein, R1 can be halo, CN SOC1-3alkyl, or SO2C1-3alkyl. In various cases, R1 is halo, CN or SO2C1-3alkyl. In some cases, R1 is F, Cl, CN, or SO2CH3. In some cases, R1 is F. [0018] As disclosed herein, n can be 0, 1, or 2. In various cases, n is 0. In some cases, n is 1 or 2. [0019] As disclosed herein, each R2 can independently be halo, CN, OH, –C0-6alkylene-N(RN)2, CO2RN, CORN, C1-6alkyl, C1-6alkoxy, C1-6haloalkyl, –C1-6hydroxyalkyl, –OC2-6hydroxyalkyl, –OC2-6alkylene-N(RN)2, CON(RN)2, CON(RN)-4-8-membered heterocycle, C(O)-4-8-membered heterocycle, –[O]0-1-4-8 membered heterocycle, or – [O]0-1-5-10 membered heteroaryl, wherein each heterocycle and heteroaryl comprises 1, 2, or 3 ring heteroatoms selected from N, O, and S, and the heterocycle or heteroaryl is optionally substituted with 1 or 2 C1-6alkyl, and when ring B is a heterocycle or a 8-10-membered heteroaryl, R2 can be oxo. In various cases, each R2 is independently halo, CN, OH, –C0-6alkylene-N(RN)2, C1-6alkyl, C1-6alkoxy, C1-6haloalkyl, –C1-6hydroxyalkyl, –OC2-
6hydroxyalkyl, –OC2-6alkylene-N(RN)2, CON(RN)2, –[O]0-1-4-8 membered heterocycle. In some cases, each R2 is independently Br, CN, OH, C1-6haloalkyl, –C0-6alkylene-N(RN)2, OC2-6hydroxyalkyl, –OC2-6alkylene-N(RN)2, or – [O]0-1-4-8 membered heterocycle. In some cases, at least one R2 is oxo. [0020] As disclosed herein, R3 can be H, halo, CN, C1-3alkyl, or C1-3haloalkyl. In various cases, R3 is halo or CN. In some cases, R3 is Cl or CN. [0021] As disclosed herein, R4 can be phenyl or a 5-10-membered heteroaryl comprising 1 to 3 ring atoms independently selected from N, O, and S, and R4 is optionally substituted with 1, 2, or 3 Ra. In various cases, R4 is indole, 1,7-dihydropyrazolo[1,5-a]pyridine, indazole, benzo[d]imidazole, 4,5,6,7-tetrahydropyrazolo[1,5- a]pyrazine, imidazo[1,2-a]pyridine, pyrazolo[4,3-b]pyridine, [1,2,4]triazolo[4,3-a]pyridine, benzo[d][1,2,3]triazole, phenyl, pyridine, pyrazole, or imidazole. In some cases, R4 is indole, 1,7-dihydropyrazolo[1,5-a]pyridine, indazole, benzo[d]imidazole, phenyl, pyridine, pyrazole, or imidazole. In some cases, R4 is substituted with 1, 2, or 3 Ra. [0022] As disclosed herein, R5 can be H or C1-3alkyl. In various cases, R5 is H. In some cases, R5 is C1-3alkyl. In some cases, R5 is CH3. [0023] As disclosed herein, each Ra can independently be halo, OH, CON(RN)2, C1-6alkyl, C1-6alkoxy, C1- 6hydroxyalkyl, CO2C1-6alkyl, C0-6alkylene-N(RN)2, or a 4-6-membered heterocycle comprising 1 or 2 ring atoms selected from N, O, and S. In various cases, each Ra is independently halo, C1-6alkoxy, C1-6alkyl, CO2C1-6alkyl, C1-6hydroxyalkyl, or a 4-6-membered heterocycle comprising 1 or 2 ring atoms selected from N, O, and S. In some cases, each Ra is independently F, Cl, C1-6alkyl, C1-6alkoxy, C1-6hydroxyalkyl, or a 4-6-membered heterocycle comprising 1 or 2 ring atoms selected from N, O, and S. [0024] As disclosed herein, each RN can independently be H, C1-6alkyl, C1-6hydroxyalkyl, or C1-6alkylene-N(C1- 3alkyl)2, or two RN groups, together with a nitrogen to which they are each attached, form a 4-10 membered heterocycle comprising 0-2 additional ring heteroatoms independently selected from N, O, and S, and can be optionally substituted with 1 or 2 C1-6alkyl. In various cases, each RN is independently H or C1-6alkyl. [0025] Compounds as disclosed herein include those as provided in Table A, or a pharmaceutically acceptable salt thereof. Table A.



























































Preparation of 6-(2,5-dichloropyrimidin-4-yl)-1-isopropyl-1,2,3-benzotriazole (B-6)
 [0084] To a solution of 6-bromo-1-isopropyl-1,2,3-benzotriazole (3.6 g, 0.015 mol) in 1,4-dioxane was added 4,4,5,5-tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (5.7 g, 0.023 mol), Pd(dppf)Cl2 (0.55 g, 0.00075 mol) and AcOK (2.9 g, 0.03 mol). The reaction mixture was stirred at 90℃ for 12 h under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed by vacuum. The residue was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 40/60 in 30 mins) to afford 1-isopropyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)-1,2,3-benzotriazole (3.2 g, yield: 61.3 %) as a white solid. LCMS (ESI): calced for C15H22BN3O2 [M+H]+ m/z 288.2, found 288.2. [0085] To a solution of 1-isopropyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,2,3-benzotriazole (3.2 g, 0.011 mol) in 1,4-dioxane / H2O (33 mL) was added 2,4,5-trichloropyrimidine (2.4 g, 0.013 mol), Pd(dppf)Cl2 (0.4 g, 0.00056 mol) and Na2CO3 (2.4 g, 0.022 mol). The reaction mixture was stirred at 80℃ for 6 h under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed by vacuum. The residue was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 50/50 in 30 mins) to afford B-6 (3.1 g, yield: 78.4 %) as a yellow solid. LCMS (ESI): calced for C13H11Cl2N5 [M+H]+ m/z 308.0, found 308.1. Preparation of 5-(2,5-dichloropyrimidin-4-yl)-3-isopropyl-2-methylindazole (B-8)
[0084] To a solution of 6-bromo-1-isopropyl-1,2,3-benzotriazole (3.6 g, 0.015 mol) in 1,4-dioxane was added 4,4,5,5-tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (5.7 g, 0.023 mol), Pd(dppf)Cl2 (0.55 g, 0.00075 mol) and AcOK (2.9 g, 0.03 mol). The reaction mixture was stirred at 90℃ for 12 h under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed by vacuum. The residue was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 40/60 in 30 mins) to afford 1-isopropyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)-1,2,3-benzotriazole (3.2 g, yield: 61.3 %) as a white solid. LCMS (ESI): calced for C15H22BN3O2 [M+H]+ m/z 288.2, found 288.2. [0085] To a solution of 1-isopropyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,2,3-benzotriazole (3.2 g, 0.011 mol) in 1,4-dioxane / H2O (33 mL) was added 2,4,5-trichloropyrimidine (2.4 g, 0.013 mol), Pd(dppf)Cl2 (0.4 g, 0.00056 mol) and Na2CO3 (2.4 g, 0.022 mol). The reaction mixture was stirred at 80℃ for 6 h under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed by vacuum. The residue was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 50/50 in 30 mins) to afford B-6 (3.1 g, yield: 78.4 %) as a yellow solid. LCMS (ESI): calced for C13H11Cl2N5 [M+H]+ m/z 308.0, found 308.1. Preparation of 5-(2,5-dichloropyrimidin-4-yl)-3-isopropyl-2-methylindazole (B-8)
 [0086] To a solution of 5-bromo-3-isopropyl-2-methylindazole (900.0 mg, 3.6 mmol) in 1,4-dioxane (10 mL) was added 4,4,5,5-tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (1083.4 mg, 4.3 mmol), Pd(dppf)Cl2 (130.1 mg, 0.2 mmol) and AcOK (697.9 mg, 7.1 mmol). The reaction mixture was stirred at 90℃ for 12 h under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed by vacuum. The residue was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 40/60 in 30
mins) to afford 3-isopropyl-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)indazole (780.0 mg, yield: 67.2 %) as a yellow solid. LCMS (ESI): calced for C17H25BN2O2 [M+H]+ m/z 301.2, found 301.3. [0087] To a solution of 3-isopropyl-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)indazole (780.0 mg, 2.6 mmol) in 1,4-dioxane / H2O (11 mL )was added 2,4,5-trichloropyrimidine (571.9 mg, 3.1 mmol), Pd(dppf)Cl2 (95.1 mg, 0.1 mmol) and Na2CO3 (550.8 mg, 5.2 mmol). The reaction mixture was stirred at 80℃ for 6 h under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed by vacuum. The residue was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 50/50 in 30 mins) to afford B- 8 (680.0 mg, yield: 73.3 %) as a yellow solid. LCMS (ESI): calced for C15H14Cl2N4 [M+H]+ m/z 321.1, found 321.1. Preparation of 6-(2,5-dichloropyrimidin-4-yl)-1-isopropyl-1H-benzo[d]imidazole (B-9)
[0086] To a solution of 5-bromo-3-isopropyl-2-methylindazole (900.0 mg, 3.6 mmol) in 1,4-dioxane (10 mL) was added 4,4,5,5-tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (1083.4 mg, 4.3 mmol), Pd(dppf)Cl2 (130.1 mg, 0.2 mmol) and AcOK (697.9 mg, 7.1 mmol). The reaction mixture was stirred at 90℃ for 12 h under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed by vacuum. The residue was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 40/60 in 30
mins) to afford 3-isopropyl-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)indazole (780.0 mg, yield: 67.2 %) as a yellow solid. LCMS (ESI): calced for C17H25BN2O2 [M+H]+ m/z 301.2, found 301.3. [0087] To a solution of 3-isopropyl-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)indazole (780.0 mg, 2.6 mmol) in 1,4-dioxane / H2O (11 mL )was added 2,4,5-trichloropyrimidine (571.9 mg, 3.1 mmol), Pd(dppf)Cl2 (95.1 mg, 0.1 mmol) and Na2CO3 (550.8 mg, 5.2 mmol). The reaction mixture was stirred at 80℃ for 6 h under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed by vacuum. The residue was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 50/50 in 30 mins) to afford B- 8 (680.0 mg, yield: 73.3 %) as a yellow solid. LCMS (ESI): calced for C15H14Cl2N4 [M+H]+ m/z 321.1, found 321.1. Preparation of 6-(2,5-dichloropyrimidin-4-yl)-1-isopropyl-1H-benzo[d]imidazole (B-9)
 [0088] To a solution of 4-fluoro-1-isopropyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H- benzo[d]imidazole (1.5 g, 4.9 mmol) in dioxane/H2O=3:1 (30 mL) was added 2,4,5-trichloropyrimidine (901.6 mg, 4.9 mmol), Pd(Dppf)Cl2 (366.0 mg, 0.5 mmol) and Cs2CO3 (432.7 mg, 4.0 mmol) stirred for 12 h at 80 ℃ under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed. Water was added to the solution and extracted with EtOAc. The organic layer was washed with brine, dried over anhydrous sodium sulfate, filtered out and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/00 to 90/10 in 10 mins) afford B-9 (1.1 g, yield: 73.3%) as a white solid. LCMS (ESI): calced for C6H9BrN2 [M+H]+ m/z 325.0, found 324.9. Preparation of 6-(2,5-dichloropyrimidin-4-yl)-1-isopropyl-1H-benzo[d]imidazole (B-10)
[0088] To a solution of 4-fluoro-1-isopropyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H- benzo[d]imidazole (1.5 g, 4.9 mmol) in dioxane/H2O=3:1 (30 mL) was added 2,4,5-trichloropyrimidine (901.6 mg, 4.9 mmol), Pd(Dppf)Cl2 (366.0 mg, 0.5 mmol) and Cs2CO3 (432.7 mg, 4.0 mmol) stirred for 12 h at 80 ℃ under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed. Water was added to the solution and extracted with EtOAc. The organic layer was washed with brine, dried over anhydrous sodium sulfate, filtered out and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/00 to 90/10 in 10 mins) afford B-9 (1.1 g, yield: 73.3%) as a white solid. LCMS (ESI): calced for C6H9BrN2 [M+H]+ m/z 325.0, found 324.9. Preparation of 6-(2,5-dichloropyrimidin-4-yl)-1-isopropyl-1H-benzo[d]imidazole (B-10)
 [0089] To a solution of 6-bromo-1-isopropyl-1H-benzo[d]imidazole (3.0 g, 12.5 mmol) and 4,4,4',4',5,5,5',5'- octamethyl-2,2'-bi(1,3,2-dioxaborolane) (4.8 g, 18.8 mmol) in Dioxane (45 mL) stirred under nitrogen at rt was added KOAc (3.7 g, 37.6 mmol) and Pd(dppf)Cl2 (458.7 mg, 0.63 mmol). The reaction mixture was stirred at 80℃ for 16 h. The mixture was quenched with water and extracted with EtOAc (3 x 70 mL), the organic layer was washed with brine, dried over Na2SO4, filtered out and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 50/50 in 20 mins) to afford 1-isopropyl-6-(4,4,5,5-
tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-benzo[d]imidazole (1.2 g, yield: 33.4%) as a yellow oil. LCMS (ESI): calced for C16H23BN2O2 [M+H]+ m/z 287.2, found 287.2. [0090] A mixture of 1-isopropyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-benzo[d]imidazole (1.2 g, 4.2 mmol), 2,4,5-trichloropyrimidine (764 mg, 4.2 mmol), K2CO3 (1.2 g, 8.4 mmol) and Pd(dppf)Cl2 (307.0 mg, 0.42 mmol) in Dioxane/H2O =4:1 (24 mL) was heated under microwave irradiation at 120°C for 60 min. The crude product was filtered out and concentrated to get the product which was purified by reverse flash chromatography (eluting withH2O(NH3.H2O)/MeCN from 95/5 to 50/50 in 30 mins) to afford B-10 (400.0 mg, yield: 31.1%) as a yellow solid. LCMS (ESI): calced for C14H12Cl2N4 [M+H]+ m/z 307.0, found 306.8. Preparation of 6-(2,5-dichloropyrimidin-4-yl)-1-isopropyl-1H-indazole (B-11)
[0089] To a solution of 6-bromo-1-isopropyl-1H-benzo[d]imidazole (3.0 g, 12.5 mmol) and 4,4,4',4',5,5,5',5'- octamethyl-2,2'-bi(1,3,2-dioxaborolane) (4.8 g, 18.8 mmol) in Dioxane (45 mL) stirred under nitrogen at rt was added KOAc (3.7 g, 37.6 mmol) and Pd(dppf)Cl2 (458.7 mg, 0.63 mmol). The reaction mixture was stirred at 80℃ for 16 h. The mixture was quenched with water and extracted with EtOAc (3 x 70 mL), the organic layer was washed with brine, dried over Na2SO4, filtered out and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 50/50 in 20 mins) to afford 1-isopropyl-6-(4,4,5,5-
tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-benzo[d]imidazole (1.2 g, yield: 33.4%) as a yellow oil. LCMS (ESI): calced for C16H23BN2O2 [M+H]+ m/z 287.2, found 287.2. [0090] A mixture of 1-isopropyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-benzo[d]imidazole (1.2 g, 4.2 mmol), 2,4,5-trichloropyrimidine (764 mg, 4.2 mmol), K2CO3 (1.2 g, 8.4 mmol) and Pd(dppf)Cl2 (307.0 mg, 0.42 mmol) in Dioxane/H2O =4:1 (24 mL) was heated under microwave irradiation at 120°C for 60 min. The crude product was filtered out and concentrated to get the product which was purified by reverse flash chromatography (eluting withH2O(NH3.H2O)/MeCN from 95/5 to 50/50 in 30 mins) to afford B-10 (400.0 mg, yield: 31.1%) as a yellow solid. LCMS (ESI): calced for C14H12Cl2N4 [M+H]+ m/z 307.0, found 306.8. Preparation of 6-(2,5-dichloropyrimidin-4-yl)-1-isopropyl-1H-indazole (B-11)
 [0091] To a solution of 1-isopropyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)indazole (1000.0 mg, 3.49 mmol) in MeCN / H2O (22 mL) was added 2,4,5-trichloropyrimidine (640.9 mg, 3.49 mmol), Pd(PPh3)4 (201.9 g, 0.17 mmol) and Na2CO3 (740.8 mg, 6.99 mmol). The reaction mixture was stirred at 80℃ for 4 h under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed by vacuum. The residue was then quenched water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 50/50 in 30 mins) to afford B-11 (680.0 mg, yield: 54.5 %) as a yellow solid. LCMS (ESI): calced for C14H12Cl2N4 [M+H]+ m/z 307.0, found 307.2.
[0091] To a solution of 1-isopropyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)indazole (1000.0 mg, 3.49 mmol) in MeCN / H2O (22 mL) was added 2,4,5-trichloropyrimidine (640.9 mg, 3.49 mmol), Pd(PPh3)4 (201.9 g, 0.17 mmol) and Na2CO3 (740.8 mg, 6.99 mmol). The reaction mixture was stirred at 80℃ for 4 h under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed by vacuum. The residue was then quenched water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 50/50 in 30 mins) to afford B-11 (680.0 mg, yield: 54.5 %) as a yellow solid. LCMS (ESI): calced for C14H12Cl2N4 [M+H]+ m/z 307.0, found 307.2.
Preparation of tert-butyl 3-(5-chloro-2-(methylsulfonyl)pyrimidin-4-yl)-6,7-dihydropyrazolo[1,5-a]pyrazine- 5(4H)-carboxylate (B-12)
 [0092] A solution of tert-butyl 3-bromo-6,7-dihydropyrazolo[1,5-a]pyrazine-5(4H)-carboxylate (1.5 g, 5.0 mmol), B2pin2 (6.3 g, 26.3 mmol), Pd(dppf)Cl2 (181.0 mg, 0.3 mmol) and AcOK (1.0 g, 9.9 mmol) in 20 mL DMF was charged with N2 and stirred at 100 ℃ for 1 h. The mixture was concentrated and purified by flash chromatography (eluting with PE/EtOAc fron 100/0 to 90/10 in 20 mins) to afford the product tert-butyl 3-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)-6,7-dihydropyrazolo[1,5-a]pyrazine-5(4H)-carboxylate (1.0 g, yield: 57.3%) as a yellow oil. LCMS (ESI) calcd for C17H29BN3O4 [M+H] + m/z 350.2, found 350.0. [0093] A solution of tert-butyl [3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-4H,6H,7H-pyrazolo[1,5- a]pyrazin-5-yl] formate (800.0 mg, 2.28 mmol), 4,5-dichloro-2-(methylsulfanyl)pyrimidine (446.0 mg, 2.28 mmol), Pd(PPh3)4 (132.0 mg, 0.11 mmol) and Cs2CO3 (1042.0 mg, 3.20 mmol) in 15 mL DMF/H2O 10:1 was charged with N2 and stirred at 100℃ for 16 h. The mixture was concentrated and purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 90/10 in 30 mins) to afford the product tert-butyl 3-(5-chloro-2- (methylthio)pyrimidin-4-yl)-6,7-dihydropyrazolo[1,5-a]pyrazine-5(4H)-carboxylate (500.0 mg, yield: 57.5%) as a yellow solid. LCMS (ESI) calcd for C16H21ClN5O2S [M+H] + m/z 382.1, found 382.1. [0094] A solution of tert-butyl {3-[5-chloro-2-(methylsulfanyl)pyrimidin-4-yl]-4H,6H,7H-pyrazolo[1,5-a]pyrazin-5- yl} formate (450.0 mg, 1.17 mg) and m-CPBA (608.0 mg, 3.53 mmol) in 15 mL DCM was stirred at rt for 2 h. The solution was quenched with saturated NaHCO3 solution and then extracted with DCM. The organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product B-12 (400.0 mg, yield: 73.8%) as a yellow solid. LCMS (ESI) calcd for C16H21ClN5O4S [M+H]+ m/z 414.1, found 414.1.
Preparation of 3-isopropyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2- (trimethylsilyl)ethoxy)methyl)-1H-pyrazole (B-13)
[0092] A solution of tert-butyl 3-bromo-6,7-dihydropyrazolo[1,5-a]pyrazine-5(4H)-carboxylate (1.5 g, 5.0 mmol), B2pin2 (6.3 g, 26.3 mmol), Pd(dppf)Cl2 (181.0 mg, 0.3 mmol) and AcOK (1.0 g, 9.9 mmol) in 20 mL DMF was charged with N2 and stirred at 100 ℃ for 1 h. The mixture was concentrated and purified by flash chromatography (eluting with PE/EtOAc fron 100/0 to 90/10 in 20 mins) to afford the product tert-butyl 3-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)-6,7-dihydropyrazolo[1,5-a]pyrazine-5(4H)-carboxylate (1.0 g, yield: 57.3%) as a yellow oil. LCMS (ESI) calcd for C17H29BN3O4 [M+H] + m/z 350.2, found 350.0. [0093] A solution of tert-butyl [3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-4H,6H,7H-pyrazolo[1,5- a]pyrazin-5-yl] formate (800.0 mg, 2.28 mmol), 4,5-dichloro-2-(methylsulfanyl)pyrimidine (446.0 mg, 2.28 mmol), Pd(PPh3)4 (132.0 mg, 0.11 mmol) and Cs2CO3 (1042.0 mg, 3.20 mmol) in 15 mL DMF/H2O 10:1 was charged with N2 and stirred at 100℃ for 16 h. The mixture was concentrated and purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 90/10 in 30 mins) to afford the product tert-butyl 3-(5-chloro-2- (methylthio)pyrimidin-4-yl)-6,7-dihydropyrazolo[1,5-a]pyrazine-5(4H)-carboxylate (500.0 mg, yield: 57.5%) as a yellow solid. LCMS (ESI) calcd for C16H21ClN5O2S [M+H] + m/z 382.1, found 382.1. [0094] A solution of tert-butyl {3-[5-chloro-2-(methylsulfanyl)pyrimidin-4-yl]-4H,6H,7H-pyrazolo[1,5-a]pyrazin-5- yl} formate (450.0 mg, 1.17 mg) and m-CPBA (608.0 mg, 3.53 mmol) in 15 mL DCM was stirred at rt for 2 h. The solution was quenched with saturated NaHCO3 solution and then extracted with DCM. The organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product B-12 (400.0 mg, yield: 73.8%) as a yellow solid. LCMS (ESI) calcd for C16H21ClN5O4S [M+H]+ m/z 414.1, found 414.1.
Preparation of 3-isopropyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2- (trimethylsilyl)ethoxy)methyl)-1H-pyrazole (B-13)
 [0095] To a solution of 4-bromo-3-isopropyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole (1.3 g, 4.0 mmol) in THF (30 mL) was added 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (1.0 g, 6.0 mmol) and n-Bu-Li (0.5 g, 8.0 mol) was added stirred for 1h at -78℃ under N2. LCMS showed the desired MS was detected. The final mixture was quenched with water and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 100/00 to 95/5 in 5 mins) to afford (750.0 mg, yield: 51.0%) as white oil. LCMS (ESI): calced for C18H35BN2O3Si [M+H]+ m/z 367.3, found 367.0. [0096] To a solution of 3-isopropyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2- (trimethylsilyl)ethoxy)methyl)-1H-pyrazole (750.0 mg, 2.0 mmol) in MeCN/H2O=3:1 (15 mL) was added 2,4,5- trichloropyrimidine (374.4 mg, 2.0 mmol), Pd(PPh3)4 (235.9 mg, 0.2 mmol) and Na2CO3 (432.7 mg, 4.0 mmol) stirred for 12 h at 80 ℃ under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed. Water was added to the solution and extracted with EtOAc. The organic layer was washed with brine, dried over anhydrous sodium sulfate, filtered out and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/00 to 90/10 in 10 mins) afford B-13 (390.0 mg, yield: 49.1%) as white solid. LCMS (ESI): calced for C16H24Cl2N4OSi [M+H]+ m/z 387.1, found 387.3. Preparation of 2,5-dichloro-4-phenylpyrimidine (B-16)
[0095] To a solution of 4-bromo-3-isopropyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole (1.3 g, 4.0 mmol) in THF (30 mL) was added 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (1.0 g, 6.0 mmol) and n-Bu-Li (0.5 g, 8.0 mol) was added stirred for 1h at -78℃ under N2. LCMS showed the desired MS was detected. The final mixture was quenched with water and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 100/00 to 95/5 in 5 mins) to afford (750.0 mg, yield: 51.0%) as white oil. LCMS (ESI): calced for C18H35BN2O3Si [M+H]+ m/z 367.3, found 367.0. [0096] To a solution of 3-isopropyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2- (trimethylsilyl)ethoxy)methyl)-1H-pyrazole (750.0 mg, 2.0 mmol) in MeCN/H2O=3:1 (15 mL) was added 2,4,5- trichloropyrimidine (374.4 mg, 2.0 mmol), Pd(PPh3)4 (235.9 mg, 0.2 mmol) and Na2CO3 (432.7 mg, 4.0 mmol) stirred for 12 h at 80 ℃ under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed. Water was added to the solution and extracted with EtOAc. The organic layer was washed with brine, dried over anhydrous sodium sulfate, filtered out and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/00 to 90/10 in 10 mins) afford B-13 (390.0 mg, yield: 49.1%) as white solid. LCMS (ESI): calced for C16H24Cl2N4OSi [M+H]+ m/z 387.1, found 387.3. Preparation of 2,5-dichloro-4-phenylpyrimidine (B-16)
 [0097] To a solution of 2,4,5-trichloropyrimidine (3.1 g, 0.0164 mol) in MeCN / H2O = 10:1 (30 mL) was added phenylboronic acid (2.0 g, 0.0164 mol), Tetrakis(triphenylphosphine)Palladium (1.9 g, 0.0016 mol) and sodium carbonate (3.5 g, 0.0328 mol). The reaction solution was stirred at 80 ℃ for 4 h under N2. The LCMS showed the desired MS was detected. The reaction mixture was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by silica gel
chromatography (eluting with PE / EtOAc from 100/0 to 95/5 in 30 mins) to afford B-16 (2.7 g, yield: 71.9%) as an off-white solid. LCMS (ESI): calcd for C10H6Cl2N2 [M+H] + m/z 224.8, found 224.8. Preparation of 2,5-dichloro-4-(pyridin-3-yl) pyrimidine (B-17)
[0097] To a solution of 2,4,5-trichloropyrimidine (3.1 g, 0.0164 mol) in MeCN / H2O = 10:1 (30 mL) was added phenylboronic acid (2.0 g, 0.0164 mol), Tetrakis(triphenylphosphine)Palladium (1.9 g, 0.0016 mol) and sodium carbonate (3.5 g, 0.0328 mol). The reaction solution was stirred at 80 ℃ for 4 h under N2. The LCMS showed the desired MS was detected. The reaction mixture was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by silica gel
chromatography (eluting with PE / EtOAc from 100/0 to 95/5 in 30 mins) to afford B-16 (2.7 g, yield: 71.9%) as an off-white solid. LCMS (ESI): calcd for C10H6Cl2N2 [M+H] + m/z 224.8, found 224.8. Preparation of 2,5-dichloro-4-(pyridin-3-yl) pyrimidine (B-17)
 [0098] To a solution of 2,4,5-trichloropyrimidine (2.0 g, 10.9 mmol) in dioxane/H2O (40 ml:4 ml) was added pyridin-3-ylboranediol (1.6 g, 13.08 mmol) and Pd(dppf)Cl2 (400 mg, 0.55 mmol) and K2CO3 (3.01 g, 21.8 mmol). The reaction mixture was stirred at 80℃ for 16 h under nitrogen. The solvent was removed by vacumm to get crude product which was purified by flash chromatography (eluting with DCM/MeOH from 100/0 to 95/5 in 30 mins) to afford B-17 (360.0 mg, yield: 13.7 %) as a yellow oil. LCMS (ESI) calcd for C9H5Cl2N3 [M + H] + m/z 226.1, found 226.1. Preparation of 2,5-dichloro-4-(2-methoxyphenyl)pyrimidine (B-18)
[0098] To a solution of 2,4,5-trichloropyrimidine (2.0 g, 10.9 mmol) in dioxane/H2O (40 ml:4 ml) was added pyridin-3-ylboranediol (1.6 g, 13.08 mmol) and Pd(dppf)Cl2 (400 mg, 0.55 mmol) and K2CO3 (3.01 g, 21.8 mmol). The reaction mixture was stirred at 80℃ for 16 h under nitrogen. The solvent was removed by vacumm to get crude product which was purified by flash chromatography (eluting with DCM/MeOH from 100/0 to 95/5 in 30 mins) to afford B-17 (360.0 mg, yield: 13.7 %) as a yellow oil. LCMS (ESI) calcd for C9H5Cl2N3 [M + H] + m/z 226.1, found 226.1. Preparation of 2,5-dichloro-4-(2-methoxyphenyl)pyrimidine (B-18)
 [0099] To a solution of 2,4,5-trichloropyrimidine (2.4 g, 0.0132 mol) in MeCN / H2O = 3:1 (20 mL) were added (2-methoxyphenyl)boronic acid (2.0 g, 0.0132 mol), Tetrakis(triphenylphosphine)Palladium (1.5 g, 0.0013 mol) and sodium carbonate (2.8 g, 0.0264 mol). The reaction solution was stirred at 80 ℃ for 4 h under N2. The LCMS showed the desired MS was detected. The reaction mixture was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by silica gel chromatography (eluting with PE / EtOAc from 100/0 to 95/ 5 in 30 mins) to afford B-18 (2.8 g, yield: 81.2%) as an off-white solid. LCMS (ESI): calcd for C11H8Cl2N2O [M+H] + m/z 255.1, found 254.8.
Preparation of 2,5-dichloro-4-(3-methoxyphenyl)pyrimidine (B-19)
[0099] To a solution of 2,4,5-trichloropyrimidine (2.4 g, 0.0132 mol) in MeCN / H2O = 3:1 (20 mL) were added (2-methoxyphenyl)boronic acid (2.0 g, 0.0132 mol), Tetrakis(triphenylphosphine)Palladium (1.5 g, 0.0013 mol) and sodium carbonate (2.8 g, 0.0264 mol). The reaction solution was stirred at 80 ℃ for 4 h under N2. The LCMS showed the desired MS was detected. The reaction mixture was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by silica gel chromatography (eluting with PE / EtOAc from 100/0 to 95/ 5 in 30 mins) to afford B-18 (2.8 g, yield: 81.2%) as an off-white solid. LCMS (ESI): calcd for C11H8Cl2N2O [M+H] + m/z 255.1, found 254.8.
Preparation of 2,5-dichloro-4-(3-methoxyphenyl)pyrimidine (B-19)
 [0100] To a solution of 2,4,5-trichloropyrimidine (2.4 g, 13.2 mmol) in MeCN / H2O = 3:1 (20 mL) were added (3-methoxyphenyl)boronic acid (2.0 g, 13.2 mmol), Tetrakis(triphenylphosphine)Palladium (1.5 g, 1.3 mmol) and sodium carbonate (2.8 g, 26.4 mmol). The reaction solution was stirred at 80℃ for 4 h under N2. The LCMS showed the desired MS was detected. The reaction mixture was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by silica gel chromatography (eluting with PE / EtOAc from 100/0 to 95/5 in 30 mins) to afford 2 B-19 (1.5 g, yield: 44.2% ) as an off-white solid. LCMS (ESI): calcd for C11H9Cl2N2O [M+H] + m/z 255.1, found 255.0. Preparation of 2,5-dichloro-4-(2-chlorophenyl)pyrimidine (B-21)
[0100] To a solution of 2,4,5-trichloropyrimidine (2.4 g, 13.2 mmol) in MeCN / H2O = 3:1 (20 mL) were added (3-methoxyphenyl)boronic acid (2.0 g, 13.2 mmol), Tetrakis(triphenylphosphine)Palladium (1.5 g, 1.3 mmol) and sodium carbonate (2.8 g, 26.4 mmol). The reaction solution was stirred at 80℃ for 4 h under N2. The LCMS showed the desired MS was detected. The reaction mixture was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by silica gel chromatography (eluting with PE / EtOAc from 100/0 to 95/5 in 30 mins) to afford 2 B-19 (1.5 g, yield: 44.2% ) as an off-white solid. LCMS (ESI): calcd for C11H9Cl2N2O [M+H] + m/z 255.1, found 255.0. Preparation of 2,5-dichloro-4-(2-chlorophenyl)pyrimidine (B-21)
 [0101] To a solution of (2-chlorophenyl)boranediol (2.0 g, 12.8 mmol), 2,4,5-trichloropyrimidine (2.6 g, 14.0 mmol), Pd(PPh3)4 (740.0 mg, 0.6 mmol) and Na2CO3 (2.7 g, 25.6 mmol) in MeCN (15 mL) and H2O (5 mL). The reaction was stirred at 80°C for 4 h under N2. The LCMS showed the desired MS was detected. The reaction mixture was concentrated under reduced pressure to give which was purified by flash silica gel chromatography (eluting with PE/EtOAc from 100/0 to 95/5 in 20 mins) to afford B-21 (1.5 g, yield: 45.3%) as a white solid. LCMS (ESI): calced for C10H5Cl3N2 [M+H]+ m/z 259.0, found 259.0.
[0101] To a solution of (2-chlorophenyl)boranediol (2.0 g, 12.8 mmol), 2,4,5-trichloropyrimidine (2.6 g, 14.0 mmol), Pd(PPh3)4 (740.0 mg, 0.6 mmol) and Na2CO3 (2.7 g, 25.6 mmol) in MeCN (15 mL) and H2O (5 mL). The reaction was stirred at 80°C for 4 h under N2. The LCMS showed the desired MS was detected. The reaction mixture was concentrated under reduced pressure to give which was purified by flash silica gel chromatography (eluting with PE/EtOAc from 100/0 to 95/5 in 20 mins) to afford B-21 (1.5 g, yield: 45.3%) as a white solid. LCMS (ESI): calced for C10H5Cl3N2 [M+H]+ m/z 259.0, found 259.0.
Preparation of 4-{1-[(2-methoxyethyl) trimethyl-$l^{5}-silyl] indazol-3-yl}-2-(methylsulfanyl) pyrimidine-5- carbonitrile (B-22)
 [0102] To a solution of 1-[(2-methoxyethyl)trimethyl-$l^{5}-silyl]-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)indazole (1.2 g, 3.23 mmol) in 1,4-dioxane (20 mL) and was 4-chloro-2-(methylsulfanyl)pyrimidine-5- carbonitrile (500.0 mg, 2.69 mmol), K2CO3 (744.5 mg, 5.39 mmol), Pd(PPh3)4 (155.6 mg, 0.13) and H2O (2 mL). The reaction mixture was stirred at 90℃ for 16 h under N2. The desired mass was detected on LC-MS. The solvent was removed by vacumm to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 95/5 to 85/15 in 10 mins) to afford B-22 (550.0 mg, yield: 50.2 %) as a yellow oil. LCMS (ESI): calced for C19H23N5OSSi [M+H]+ m/z 398.0, found 398.0. Preparation of 3-(2,5-dichloropyrimidin-4-yl)-5-fluoro-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole (B- 23)
[0102] To a solution of 1-[(2-methoxyethyl)trimethyl-$l^{5}-silyl]-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)indazole (1.2 g, 3.23 mmol) in 1,4-dioxane (20 mL) and was 4-chloro-2-(methylsulfanyl)pyrimidine-5- carbonitrile (500.0 mg, 2.69 mmol), K2CO3 (744.5 mg, 5.39 mmol), Pd(PPh3)4 (155.6 mg, 0.13) and H2O (2 mL). The reaction mixture was stirred at 90℃ for 16 h under N2. The desired mass was detected on LC-MS. The solvent was removed by vacumm to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 95/5 to 85/15 in 10 mins) to afford B-22 (550.0 mg, yield: 50.2 %) as a yellow oil. LCMS (ESI): calced for C19H23N5OSSi [M+H]+ m/z 398.0, found 398.0. Preparation of 3-(2,5-dichloropyrimidin-4-yl)-5-fluoro-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole (B- 23)
 [0103] To a solution of (5-fluoro-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)boronic acid (2.1 g, 6.7 mmol) in 1,4-dioxane (50 mL) and H2O (10 mL) was added 2,4,5-trichloropyrimidine (1.3 g, 7.3 mmol), K2CO3 (1.8 g, 13.4 mmol) and Pd(dppf)Cl2 (435.3 mg, 0.6 mmol), then the mixture is stirred for 5 h at 90°C under N2. The desired mass was detected on LC-MS. The mixture was washed with water (100 mL) and extracted with EtOAc (3 x 50 mL), the organic layer was washed with brine, dried over Na2SO4, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 55/45 in 13 mins) to afford B-23 (1.2 g, yield: 38.9 %) as a yellow solid. LCMS (ESI): calcd for C17H19Cl2FN4OSi [M+H]+ m/z 413.1, found 413.3
Preparation of 3-(2-bromo-5-chloropyridin-4-yl)-5,7-difluoro-1-(oxan-3-yl)indazole (B-24)
[0103] To a solution of (5-fluoro-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)boronic acid (2.1 g, 6.7 mmol) in 1,4-dioxane (50 mL) and H2O (10 mL) was added 2,4,5-trichloropyrimidine (1.3 g, 7.3 mmol), K2CO3 (1.8 g, 13.4 mmol) and Pd(dppf)Cl2 (435.3 mg, 0.6 mmol), then the mixture is stirred for 5 h at 90°C under N2. The desired mass was detected on LC-MS. The mixture was washed with water (100 mL) and extracted with EtOAc (3 x 50 mL), the organic layer was washed with brine, dried over Na2SO4, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 55/45 in 13 mins) to afford B-23 (1.2 g, yield: 38.9 %) as a yellow solid. LCMS (ESI): calcd for C17H19Cl2FN4OSi [M+H]+ m/z 413.1, found 413.3
Preparation of 3-(2-bromo-5-chloropyridin-4-yl)-5,7-difluoro-1-(oxan-3-yl)indazole (B-24)
 [0104] A solution of 3-bromo-5,7-difluoro-1-(tetrahydro-2H-pyran-2-yl)-1H-indazole (1.2 g, 3.8 mmol), B2pin2 (6.0 g, 23.5 mmol), Pd(dppf)Cl2 (172.0 mg, 0.23 mmol) and AcOK (1.0 g, 9.4 mmol) in 1,4- dioxane (20 ml) was charged with N2 and stirred at 120 ℃ for 2 h. The mixture was concentrated and purified by flash chromatography (eluting with PE/EtOAc = 100/0 to 90/10 in 10 mins) to afford the product (mixed with B2pin2) as a yellow solid. A solution of 2,4,5-trichloropyrimidine (580.0 mg, 3.2 mmol), [5,7-difluoro-1-(oxan-3-yl)indazol-3- yl]boranediol (1.1 g, 3.8 mmol, fresh made and purified), Bis(triphenylphosphine)palladium(II) chloride (0.2 g, 0.25 mmol) and K2CO3 (0.9 g, 6.4 mmol) in 1,4-dioxane/H2O=10:1 (30 mL) was charged with N2 and stirred at 80 ℃ for 8 h. The mixture was concentrated and purified by flash chromatography (eluting with PE/EtOAc = 100/0 to 90/10 in 30 mins) to afford B-24 (0.8 g, yield: 53.6 %) as a yellow solid. LCMS (ESI) calcd for C16H13Cl2F2N4O [M + H] + ms/z 385.0, found 385.0. Preparation of 3-(2,5-dichloropyrimidin-4-yl)-1-methyl-1H-indazole (B-25)
[0104] A solution of 3-bromo-5,7-difluoro-1-(tetrahydro-2H-pyran-2-yl)-1H-indazole (1.2 g, 3.8 mmol), B2pin2 (6.0 g, 23.5 mmol), Pd(dppf)Cl2 (172.0 mg, 0.23 mmol) and AcOK (1.0 g, 9.4 mmol) in 1,4- dioxane (20 ml) was charged with N2 and stirred at 120 ℃ for 2 h. The mixture was concentrated and purified by flash chromatography (eluting with PE/EtOAc = 100/0 to 90/10 in 10 mins) to afford the product (mixed with B2pin2) as a yellow solid. A solution of 2,4,5-trichloropyrimidine (580.0 mg, 3.2 mmol), [5,7-difluoro-1-(oxan-3-yl)indazol-3- yl]boranediol (1.1 g, 3.8 mmol, fresh made and purified), Bis(triphenylphosphine)palladium(II) chloride (0.2 g, 0.25 mmol) and K2CO3 (0.9 g, 6.4 mmol) in 1,4-dioxane/H2O=10:1 (30 mL) was charged with N2 and stirred at 80 ℃ for 8 h. The mixture was concentrated and purified by flash chromatography (eluting with PE/EtOAc = 100/0 to 90/10 in 30 mins) to afford B-24 (0.8 g, yield: 53.6 %) as a yellow solid. LCMS (ESI) calcd for C16H13Cl2F2N4O [M + H] + ms/z 385.0, found 385.0. Preparation of 3-(2,5-dichloropyrimidin-4-yl)-1-methyl-1H-indazole (B-25)
 [0105] To a solution of 3-bromo-1-methyl-1H-indazole (1.0 g, 4.7 mmol) in 1,4-dioxane (10 mL) was added 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi(1,3,2-dioxaborolane) (2.4 g, 9.5 mmol), AcOK (920.0 mg, 9.5 mmol) and Pd(dppf)Cl2·DCM (190.0 mg, 0.24 mmol). The reaction mixture was stirred under N2 at 90 ℃ for 16 h. The LCMS showed the desired MS was detected. To the mixture was added 2,4,5-trichloropyrimidine (1.2 g, 6.8 mmol), K2CO3 (1.2 g, 9.0 mmol), Pd(dppf)Cl2 (160.0 mg, 0.23 mmol) and H2O (1.0 mL). The reaction mixture was stirred under N2 at 80 ℃ for 16 h under N2. The LCMS showed the desired MS was detected. The mixture was diluted with water and EtOAc and extracted with EtOAc. The organic layers were combined and washed with brine, dried over Na2SO4, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 95/5 to 90/10 in 20 mins) to afford B-25 (800.0 mg, yield: 60.5 %) as a yellow solid. LCMS (ESI): calced for C12H8Cl2N4 [M+H]+ m/z 279.0, found 279.0.
Preparation of 3-(2-chloro-5-(trifluoromethyl)pyrimidin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H- indazole (B-26)
[0105] To a solution of 3-bromo-1-methyl-1H-indazole (1.0 g, 4.7 mmol) in 1,4-dioxane (10 mL) was added 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi(1,3,2-dioxaborolane) (2.4 g, 9.5 mmol), AcOK (920.0 mg, 9.5 mmol) and Pd(dppf)Cl2·DCM (190.0 mg, 0.24 mmol). The reaction mixture was stirred under N2 at 90 ℃ for 16 h. The LCMS showed the desired MS was detected. To the mixture was added 2,4,5-trichloropyrimidine (1.2 g, 6.8 mmol), K2CO3 (1.2 g, 9.0 mmol), Pd(dppf)Cl2 (160.0 mg, 0.23 mmol) and H2O (1.0 mL). The reaction mixture was stirred under N2 at 80 ℃ for 16 h under N2. The LCMS showed the desired MS was detected. The mixture was diluted with water and EtOAc and extracted with EtOAc. The organic layers were combined and washed with brine, dried over Na2SO4, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 95/5 to 90/10 in 20 mins) to afford B-25 (800.0 mg, yield: 60.5 %) as a yellow solid. LCMS (ESI): calced for C12H8Cl2N4 [M+H]+ m/z 279.0, found 279.0.
Preparation of 3-(2-chloro-5-(trifluoromethyl)pyrimidin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H- indazole (B-26)
 [0106] To a solution of 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H- indazole (2.1 g, 0.0056 mol), 2,4-dichloro-5-(trifluoromethyl)pyrimidine (1.5 g, 0.0067 mol), Na2CO3 (0.9 g, 0.0084 mol) and Pd(dppf)Cl2 (205.0 mg,0.0003 mmol) in 1,4-dioxane/H2O=10:1 (20.0 ml) stirred under nitrogen . The reaction mixture was stirred at 80 ℃ for 12 h . The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 97/3 in 15 mins) to afford B-26 (1.4 g, yield: 61.0%) as a white solid. LCMS (ESI): calced for C18H20ClF3N4OSi [M+H]+ m/z 429.1, found 429.1. Preparation of 3-(2-chloro-5-ethylpyrimidin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole (B-27)
[0106] To a solution of 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H- indazole (2.1 g, 0.0056 mol), 2,4-dichloro-5-(trifluoromethyl)pyrimidine (1.5 g, 0.0067 mol), Na2CO3 (0.9 g, 0.0084 mol) and Pd(dppf)Cl2 (205.0 mg,0.0003 mmol) in 1,4-dioxane/H2O=10:1 (20.0 ml) stirred under nitrogen . The reaction mixture was stirred at 80 ℃ for 12 h . The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 97/3 in 15 mins) to afford B-26 (1.4 g, yield: 61.0%) as a white solid. LCMS (ESI): calced for C18H20ClF3N4OSi [M+H]+ m/z 429.1, found 429.1. Preparation of 3-(2-chloro-5-ethylpyrimidin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole (B-27)
 [0107] To a solution of 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H- indazole (1.0 g, 2.7 mmol) in 1,4-dioxane: H2O = 10:1 (10 mL) was added 2,4-dichloro-5-ethylpyrimidine (709.3 mg, 4.0 mmol), K2CO3 (723.6 mg, 5.4 mmol) and Pd(PPh3)4 (156.0 mg, 0.14 mmol). The reaction mixture was stirred under N2 at 90 ℃ for 16 h. The LCMS showed the desired MS was detected. The mixture was quenched with water and extracted with EtOAc, the organic layers were combined and washed with brine, dried over Na2SO4, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 95/5 to 90/10 in 20 mins) to afford B-27 (330.0 mg, yield: 31.8 %) as a white solid. LCMS (ESI): calced for C19H25ClN4OSi [M+H]+ m/z 389.2, found 389.3.
Preparation of 3-(2-chloro-5-methylpyrimidin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole (B-28)
[0107] To a solution of 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H- indazole (1.0 g, 2.7 mmol) in 1,4-dioxane: H2O = 10:1 (10 mL) was added 2,4-dichloro-5-ethylpyrimidine (709.3 mg, 4.0 mmol), K2CO3 (723.6 mg, 5.4 mmol) and Pd(PPh3)4 (156.0 mg, 0.14 mmol). The reaction mixture was stirred under N2 at 90 ℃ for 16 h. The LCMS showed the desired MS was detected. The mixture was quenched with water and extracted with EtOAc, the organic layers were combined and washed with brine, dried over Na2SO4, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 95/5 to 90/10 in 20 mins) to afford B-27 (330.0 mg, yield: 31.8 %) as a white solid. LCMS (ESI): calced for C19H25ClN4OSi [M+H]+ m/z 389.2, found 389.3.
Preparation of 3-(2-chloro-5-methylpyrimidin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole (B-28)
 [0108] To a solution of 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H- indazole (1.1 g, 0.0030 mol), 2,4-dichloro-5-methylpyrimidine (0.8 g, 0.0048 mol), Pd(dppf)Cl2 (150.0 mg, 0.0002 mol) and Na2CO3 (0.7 g, 0.0060 mol) in 1,4-dioxane/H2O = 10:1 (20.0 ml) stirred under nitrogen . The reaction mixture was stirred at 80 ℃ for 16 h. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 95/5 in 15 mins) to afford B-28 (1.0 g, yield: 88.7%) as a white solid. LCMS (ESI): calced for C18H23ClN4OSi [M+H]+ m/z 375.3, found 375.3. Preparation of 3-(2-chloro-5-fluoropyrimidin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole (B-30)
[0108] To a solution of 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H- indazole (1.1 g, 0.0030 mol), 2,4-dichloro-5-methylpyrimidine (0.8 g, 0.0048 mol), Pd(dppf)Cl2 (150.0 mg, 0.0002 mol) and Na2CO3 (0.7 g, 0.0060 mol) in 1,4-dioxane/H2O = 10:1 (20.0 ml) stirred under nitrogen . The reaction mixture was stirred at 80 ℃ for 16 h. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 95/5 in 15 mins) to afford B-28 (1.0 g, yield: 88.7%) as a white solid. LCMS (ESI): calced for C18H23ClN4OSi [M+H]+ m/z 375.3, found 375.3. Preparation of 3-(2-chloro-5-fluoropyrimidin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole (B-30)
 [0109] To a solution of 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H- indazole (1.0 g, 2.7 mmol) , 2,4-dichloro-5-fluoropyrimidine (534.0 mg, 3.2 mmol), Na2CO3 (565.0 mg, 5.3 mmol) and Pd(dppf)Cl2 (195.0 mg, 0.3 mmol) in (MeCN/H2O=3:1)(10.0 ml) stirred under nitrogen at 25℃ . The reaction mixture was stirred at 80 ℃ for 2h. The LCMS showed the desired MS was detected. The reaction mixture was concentrated under reduced pressure to give which was purified by flash silica gel chromatography (eluting with PE/EtOAc from 100/0 to 95/5 in 20 mins) to afford B-30 (530.0 mg, yield: 52.4%) as a white solid. LCMS (ESI): calced for C17H20ClFN4OSi [M+H]+ m/z 379.1, found 379.2.
Preparation of methyl 4-(2,5-dichloropyrimidin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole-3- carboxylate (B-31)
[0109] To a solution of 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H- indazole (1.0 g, 2.7 mmol) , 2,4-dichloro-5-fluoropyrimidine (534.0 mg, 3.2 mmol), Na2CO3 (565.0 mg, 5.3 mmol) and Pd(dppf)Cl2 (195.0 mg, 0.3 mmol) in (MeCN/H2O=3:1)(10.0 ml) stirred under nitrogen at 25℃ . The reaction mixture was stirred at 80 ℃ for 2h. The LCMS showed the desired MS was detected. The reaction mixture was concentrated under reduced pressure to give which was purified by flash silica gel chromatography (eluting with PE/EtOAc from 100/0 to 95/5 in 20 mins) to afford B-30 (530.0 mg, yield: 52.4%) as a white solid. LCMS (ESI): calced for C17H20ClFN4OSi [M+H]+ m/z 379.1, found 379.2.
Preparation of methyl 4-(2,5-dichloropyrimidin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole-3- carboxylate (B-31)
 [0110] To a solution of methyl 4-iodo-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole-3-carboxylate (4.0 g, 11.9 mmol) in 1,4-dioxane was added 4,4,5,5-tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2- dioxaborolane (3.6 g, 14.3 mmol), Pd(dppf)Cl2 (0.4 g, 0.6 mmol) and AcOK (2.3 g, 23.8 mmol). The reaction mixture was stirred at 80 ℃ for 12 h under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed by vacuum. The residue was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 80/20 to 50/50 in 30 mins) to afford methyl 4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole-3-carboxylate (2.5 g, yield: 44.5 %) as a yellow solid. LCMS (ESI): calced for C17H31BN2O5Si [M+H]+ m/z 383.2, found 383.3. [0111] To a solution of methyl 1-[(2-methoxyethyl)trimethyl-$l^{5}-silyl]-4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)pyrazole-3-carboxylate (2.5 g, 6.52 mmol) in MeCN / H2O = 10:1 (55 mL) was added 2,4,5- trichloropyrimidine (1.4 g, 7.82 mmol), Pd(dppf)Cl2 (0.2 g, 0.32 mmol) and Na2CO3 (1.4 g, 12.04 mmol). The reaction mixture was stirred at 80℃ for 4 h under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed by vacuum. The residue was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 50/50 in 30 mins) to afford B-31 (1.9 g, yield: 54.8 %) as a yellow solid. LCMS (ESI): calced for C15H20Cl2N4O3Si [M+H]+ m/z 403.1, found 403.0. Preparation of 2-bromo-5-chloro-4-(3-isopropyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazol-4- yl)pyridine (B-32)
[0110] To a solution of methyl 4-iodo-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole-3-carboxylate (4.0 g, 11.9 mmol) in 1,4-dioxane was added 4,4,5,5-tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2- dioxaborolane (3.6 g, 14.3 mmol), Pd(dppf)Cl2 (0.4 g, 0.6 mmol) and AcOK (2.3 g, 23.8 mmol). The reaction mixture was stirred at 80 ℃ for 12 h under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed by vacuum. The residue was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 80/20 to 50/50 in 30 mins) to afford methyl 4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole-3-carboxylate (2.5 g, yield: 44.5 %) as a yellow solid. LCMS (ESI): calced for C17H31BN2O5Si [M+H]+ m/z 383.2, found 383.3. [0111] To a solution of methyl 1-[(2-methoxyethyl)trimethyl-$l^{5}-silyl]-4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)pyrazole-3-carboxylate (2.5 g, 6.52 mmol) in MeCN / H2O = 10:1 (55 mL) was added 2,4,5- trichloropyrimidine (1.4 g, 7.82 mmol), Pd(dppf)Cl2 (0.2 g, 0.32 mmol) and Na2CO3 (1.4 g, 12.04 mmol). The reaction mixture was stirred at 80℃ for 4 h under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed by vacuum. The residue was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 50/50 in 30 mins) to afford B-31 (1.9 g, yield: 54.8 %) as a yellow solid. LCMS (ESI): calced for C15H20Cl2N4O3Si [M+H]+ m/z 403.1, found 403.0. Preparation of 2-bromo-5-chloro-4-(3-isopropyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazol-4- yl)pyridine (B-32)
 [0112] To a solution of 3-isopropyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2- (trimethylsilyl)ethoxy)methyl)-1H-pyrazole (1.0 g, 2.7 mmol) in dioxane/H2O=5:1 (20 mL) was added 2-bromo-5- chloro-4-iodopyridine (860.0 mg, 2.7 mmol), Pd(Dppf)Cl2 (0.2 g, 0.27 mmol) and K2CO3, (750.0 mg, 5.4 mmol) stirred at 80 ℃ for 5 h. The LCMS showed the desired MS was detected. Water was added to the solution and
extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 100/00 to 80/20 in 10 mins) to afford B-32 (710.0 mg, yield: 59.4%) as white oil. LCMS (ESI): calced for C17H25BrClN3OSi [M+H]+ m/z 430.1, found 430.0. Preparation of 3-(2-bromo-5-chloropyridin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazolo[4,3- b]pyridine (B-36)
[0112] To a solution of 3-isopropyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2- (trimethylsilyl)ethoxy)methyl)-1H-pyrazole (1.0 g, 2.7 mmol) in dioxane/H2O=5:1 (20 mL) was added 2-bromo-5- chloro-4-iodopyridine (860.0 mg, 2.7 mmol), Pd(Dppf)Cl2 (0.2 g, 0.27 mmol) and K2CO3, (750.0 mg, 5.4 mmol) stirred at 80 ℃ for 5 h. The LCMS showed the desired MS was detected. Water was added to the solution and
extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 100/00 to 80/20 in 10 mins) to afford B-32 (710.0 mg, yield: 59.4%) as white oil. LCMS (ESI): calced for C17H25BrClN3OSi [M+H]+ m/z 430.1, found 430.0. Preparation of 3-(2-bromo-5-chloropyridin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazolo[4,3- b]pyridine (B-36)
 [0113] A solution of 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H- pyrazolo[4,3-b]pyridine, 2-bromo-5-chloro-4-iodopyridine (2.8 g, 8.87 mmol), Pd(dppf)2Cl2 (321.6 mg, 0.44 mmol) and K2CO3 (2.4 g, 17.7 mmol) in 15 mL Dioxane/H2O 10:1 was charged with N2 and stirred at 100℃ for 3 h. The desired mass was detected on LC-MS. The mixture was quenched with H2O and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered out and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 50/50 in 20 mins) to afford B-36 (450.0 mg, yield: 11.6%) as a yellow solid. LCMS (ESI): calced for C17H20BrClN4OSi [M+H]+ m/z 439.0, found 438.9. Preparation of 3-(2-bromo-5-chloropyridin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazolo[4,3- c]pyridine (B-37)
[0113] A solution of 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H- pyrazolo[4,3-b]pyridine, 2-bromo-5-chloro-4-iodopyridine (2.8 g, 8.87 mmol), Pd(dppf)2Cl2 (321.6 mg, 0.44 mmol) and K2CO3 (2.4 g, 17.7 mmol) in 15 mL Dioxane/H2O 10:1 was charged with N2 and stirred at 100℃ for 3 h. The desired mass was detected on LC-MS. The mixture was quenched with H2O and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered out and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 50/50 in 20 mins) to afford B-36 (450.0 mg, yield: 11.6%) as a yellow solid. LCMS (ESI): calced for C17H20BrClN4OSi [M+H]+ m/z 439.0, found 438.9. Preparation of 3-(2-bromo-5-chloropyridin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazolo[4,3- c]pyridine (B-37)
 [0114] To a solution of 3-bromo-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazolo[4,3-c]pyridine (1.6 g, 0.0049 mol) in dioxane (20 mL) were added 3-bromo-1-[(2-methoxyethyl)trimethyl-$l^{5}-silyl]pyrazolo[4,3-c]pyridine (2.5 g, 0.0098 mol), Pd(dppf)Cl2 .DCM (0.4 g, 0.0004 mol) and AcOK (1.0 g, 0.0098 mol). The reaction solution was stirred at 90 ℃ for 12 h. The LCMS showed the desired MS was detected. The reaction mixture was concentrated to get (1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazolo[4,3-c]pyridin-3-yl)boronic acid (1.4 g, yield:
67.4%) which was directly used in next step without further purification. LCMS (ESI): calced for C12H20BN3O3Si [M+H]+ m/z 294.0, found 294.0. [0115] To a solution of (1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazolo[4,3-c]pyridin-3-yl)boronic acid (1.4 g, 0.0048 mol) in MeCN/H2O=10:1 (22 ml) were added 2-bromo-5-chloro-4-iodopyridine (2.3 g, 0.0072 mol), Pd(PPh3)4 (550.0 mg, 0.0004 mol), Cs2CO3 (3.1 g, 0.0096 mol). The reaction solution was stirred at 90 ℃ for 8 h. The LCMS showed the desired MS was detected. The reaction mixture was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 90/10 in 20 mins) to afford B-37 (1.4 g, yield: 66.7%) as a yellow solid. LCMS (ESI): calced for C17H20BrClN4OSi [M+H]+ m/z 440.8, found 440.8. Preparation of [3-(2-bromo-5-chloropyridin-4-yl)indol-1-yl]tert-butyl formate (B-38)
[0114] To a solution of 3-bromo-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazolo[4,3-c]pyridine (1.6 g, 0.0049 mol) in dioxane (20 mL) were added 3-bromo-1-[(2-methoxyethyl)trimethyl-$l^{5}-silyl]pyrazolo[4,3-c]pyridine (2.5 g, 0.0098 mol), Pd(dppf)Cl2 .DCM (0.4 g, 0.0004 mol) and AcOK (1.0 g, 0.0098 mol). The reaction solution was stirred at 90 ℃ for 12 h. The LCMS showed the desired MS was detected. The reaction mixture was concentrated to get (1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazolo[4,3-c]pyridin-3-yl)boronic acid (1.4 g, yield:
67.4%) which was directly used in next step without further purification. LCMS (ESI): calced for C12H20BN3O3Si [M+H]+ m/z 294.0, found 294.0. [0115] To a solution of (1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazolo[4,3-c]pyridin-3-yl)boronic acid (1.4 g, 0.0048 mol) in MeCN/H2O=10:1 (22 ml) were added 2-bromo-5-chloro-4-iodopyridine (2.3 g, 0.0072 mol), Pd(PPh3)4 (550.0 mg, 0.0004 mol), Cs2CO3 (3.1 g, 0.0096 mol). The reaction solution was stirred at 90 ℃ for 8 h. The LCMS showed the desired MS was detected. The reaction mixture was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 90/10 in 20 mins) to afford B-37 (1.4 g, yield: 66.7%) as a yellow solid. LCMS (ESI): calced for C17H20BrClN4OSi [M+H]+ m/z 440.8, found 440.8. Preparation of [3-(2-bromo-5-chloropyridin-4-yl)indol-1-yl]tert-butyl formate (B-38)
 [0116] To a solution of tert-butyl [3-(dihydroxyboranyl)indol-1-yl] formate (4.0 g, 15.32 mmol) in 1,4- dioxane:H2O=5:1 (80 mL) was added 2-bromo-5-chloro-4-iodopyridine (5.3 g, 16.85 mmol), Pd(PPh3)4 (885.5 mg, 0.766 mmol) and Potassium carbonate (4.2 g, 30.64 mmol). The reaction was stirred at 80 ℃ for 2 h under N2. The desired mass was detected on LC-MS. The reaction mixture was concentrated and purified by column chromatograph on silica gel (eluting with PE / EtOAc from 100/0 to 90/20 in 20 mins) to afford B-38 (3.4 g, yield: 54.8%) as a white solid. LCMS (ESI): calcd for C18H17BrClN2O2 [M+H]+ ms/z 407.0, found 408.8. Preparation of 6-(2-bromo-5-chloropyridin-4-yl)-4-fluoro-1-isopropyl-1H-benzo[d]imidazole (B-39)
[0116] To a solution of tert-butyl [3-(dihydroxyboranyl)indol-1-yl] formate (4.0 g, 15.32 mmol) in 1,4- dioxane:H2O=5:1 (80 mL) was added 2-bromo-5-chloro-4-iodopyridine (5.3 g, 16.85 mmol), Pd(PPh3)4 (885.5 mg, 0.766 mmol) and Potassium carbonate (4.2 g, 30.64 mmol). The reaction was stirred at 80 ℃ for 2 h under N2. The desired mass was detected on LC-MS. The reaction mixture was concentrated and purified by column chromatograph on silica gel (eluting with PE / EtOAc from 100/0 to 90/20 in 20 mins) to afford B-38 (3.4 g, yield: 54.8%) as a white solid. LCMS (ESI): calcd for C18H17BrClN2O2 [M+H]+ ms/z 407.0, found 408.8. Preparation of 6-(2-bromo-5-chloropyridin-4-yl)-4-fluoro-1-isopropyl-1H-benzo[d]imidazole (B-39)
 [0117] To a solution of 6-bromo-4-fluoro-1-isopropyl-1,3-benzodiazole (4.8 g, 0.0187 mol) in dioxane (75 mL) was added 4,4,5,5-tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (5.7 g, 0.0224 mol), Pd(dppf)Cl2 (1.4 g, 0.0018 mol) and AcOK (3.7 g, 0.0374 mol). The reaction solution was stirred at 90 ℃ for 8 h under N2. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to
get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 80/20 in 20 mins) to afford 4-fluoro-1-isopropyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-benzo[d]imidazole (4.6 g, yield: 74.3%) as a yellow oil. LCMS (ESI): calced for C16H22BFN2O2 [M+H]+ m/z 304.9, found 304.9. [0118] To a solution of 4-fluoro-1-isopropyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3-benzodiazole (2.0 g, 0.0066 mol) in 1,4-dioxane/H2O were added 2-bromo-5-chloro-4-iodopyridine (2.3 g, 0.0072 mol), Pd(PPh3)4 (760.0 mg, 0.0006 mol) and Cs2CO3 (4.3 g, 0.0132 mol). The reaction solution was stirred at 80 ℃ for 8 h under N2. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 80/20 in 20 mins) to afford B-39 (1.7 g, yield: 66.7%) as a yellow solid. LCMS (ESI): calced for C15H12BrClFN3 [M+H]+ m/z 367.8, found 367.8. Preparation of 4-(2-bromo-5-chloropyridin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole (B-40)
[0117] To a solution of 6-bromo-4-fluoro-1-isopropyl-1,3-benzodiazole (4.8 g, 0.0187 mol) in dioxane (75 mL) was added 4,4,5,5-tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (5.7 g, 0.0224 mol), Pd(dppf)Cl2 (1.4 g, 0.0018 mol) and AcOK (3.7 g, 0.0374 mol). The reaction solution was stirred at 90 ℃ for 8 h under N2. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to
get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 80/20 in 20 mins) to afford 4-fluoro-1-isopropyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-benzo[d]imidazole (4.6 g, yield: 74.3%) as a yellow oil. LCMS (ESI): calced for C16H22BFN2O2 [M+H]+ m/z 304.9, found 304.9. [0118] To a solution of 4-fluoro-1-isopropyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3-benzodiazole (2.0 g, 0.0066 mol) in 1,4-dioxane/H2O were added 2-bromo-5-chloro-4-iodopyridine (2.3 g, 0.0072 mol), Pd(PPh3)4 (760.0 mg, 0.0006 mol) and Cs2CO3 (4.3 g, 0.0132 mol). The reaction solution was stirred at 80 ℃ for 8 h under N2. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 80/20 in 20 mins) to afford B-39 (1.7 g, yield: 66.7%) as a yellow solid. LCMS (ESI): calced for C15H12BrClFN3 [M+H]+ m/z 367.8, found 367.8. Preparation of 4-(2-bromo-5-chloropyridin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole (B-40)
 [0119] To a solution of 4-bromo-1H-indazole (28.5 g, 144.0 mmol) in DMF (300 mL) were added NaH (5.2 g, 216.0 mmol, 60% in oil). After 30 min, SEMCl (26.5 g, 159.0 mmol) in DMF was added at 0°C. The reaction was stirred at 25°C for 16 h. The LCMS showed the desired MS was detected. The resulting was then quenched with water, extracted with EtOAc, washed with saturated brine, dried over Na2SO4 and concentrated. The residue was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 70/30 in 30 mins) to afford 4-bromo-1-((2- (trimethylsilyl)ethoxy)methyl)-1H-indazole (40.0 g, yield: 84.3%) as a yellow oil. LCMS (ESI): calced for C13H19BrN2OSi [M+H]+ m/z 329.0, found 328.8. [0120] To a solution of 3-bromo-1-[(2-methoxyethyl)trimethyl-$l^{5}-silyl]indazole (30.0 g, 91.0 mmol) , 4,4,5,5- tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (46.4 g, 182.0 mmol), AcOK (25.9 g, 365.0 mmol) and Pd(dppf)Cl2 (2.0 g, 2.7 mmol) in 1,4-dioxane (200.0 ml) stirred under nitrogen at 25℃ . The reaction mixture was stirred at 90 ℃ for 16 h under N2. The LCMS showed the desired MS was detected. The reaction mixture was concentrated under reduced pressure to give which was purified by flash silica gel chromatography (eluting with PE/EtOAc from 100/0 to 95/5 in 20 mins) to afford 4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole (27.0 g, yield: 78.4%) as a white solid. LCMS (ESI): calced for C19H31BN2O3Si [M+H]+ m/z 375.2, found 375.3. [0121] To a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H- indazole (6.0 g, 16.0 mmol) in 1,4-dioxane:H2O=5:1 (120 mL) was added 2-bromo-5-chloro-4-iodopyridine (5.1 g,
16.0 mmol), Pd(PPh3)4 (0.9 g, 8.0 mmol) and K2CO3 (4.4 g, 32.0 mmol) stirred for 12 h at 80 ℃ under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed. Water was added to the solution and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 100/00 to 85/15 in 10 mins) to afford B-40 (2.7 g, yield: 38.4 %) as white solid. LCMS (ESI): calced for C18H21BrClN3OSi [M+H]+ m/z 438.0, found 438.0. Preparation of 6-chloro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-4-yl)nicotinonitrile (B-41)
[0119] To a solution of 4-bromo-1H-indazole (28.5 g, 144.0 mmol) in DMF (300 mL) were added NaH (5.2 g, 216.0 mmol, 60% in oil). After 30 min, SEMCl (26.5 g, 159.0 mmol) in DMF was added at 0°C. The reaction was stirred at 25°C for 16 h. The LCMS showed the desired MS was detected. The resulting was then quenched with water, extracted with EtOAc, washed with saturated brine, dried over Na2SO4 and concentrated. The residue was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 70/30 in 30 mins) to afford 4-bromo-1-((2- (trimethylsilyl)ethoxy)methyl)-1H-indazole (40.0 g, yield: 84.3%) as a yellow oil. LCMS (ESI): calced for C13H19BrN2OSi [M+H]+ m/z 329.0, found 328.8. [0120] To a solution of 3-bromo-1-[(2-methoxyethyl)trimethyl-$l^{5}-silyl]indazole (30.0 g, 91.0 mmol) , 4,4,5,5- tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (46.4 g, 182.0 mmol), AcOK (25.9 g, 365.0 mmol) and Pd(dppf)Cl2 (2.0 g, 2.7 mmol) in 1,4-dioxane (200.0 ml) stirred under nitrogen at 25℃ . The reaction mixture was stirred at 90 ℃ for 16 h under N2. The LCMS showed the desired MS was detected. The reaction mixture was concentrated under reduced pressure to give which was purified by flash silica gel chromatography (eluting with PE/EtOAc from 100/0 to 95/5 in 20 mins) to afford 4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole (27.0 g, yield: 78.4%) as a white solid. LCMS (ESI): calced for C19H31BN2O3Si [M+H]+ m/z 375.2, found 375.3. [0121] To a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H- indazole (6.0 g, 16.0 mmol) in 1,4-dioxane:H2O=5:1 (120 mL) was added 2-bromo-5-chloro-4-iodopyridine (5.1 g,
16.0 mmol), Pd(PPh3)4 (0.9 g, 8.0 mmol) and K2CO3 (4.4 g, 32.0 mmol) stirred for 12 h at 80 ℃ under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed. Water was added to the solution and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 100/00 to 85/15 in 10 mins) to afford B-40 (2.7 g, yield: 38.4 %) as white solid. LCMS (ESI): calced for C18H21BrClN3OSi [M+H]+ m/z 438.0, found 438.0. Preparation of 6-chloro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-4-yl)nicotinonitrile (B-41)
 [0122] To a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H- indazole (4.8 g, 12.8 mmol) in 1,4-dioxane:H2O=5:1 (100 mL) was added 6-chloro-4-iodopyridine-3-carbonitrile (3.3 g, 12.8 mmol), Pd(PPh3)4 (0.7 g, 0.6 mmol) and K2CO3 (3.5 g, 25.6 mmol) stirred for 12 h at 100 ℃ under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed. Water was added to the solution and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 100/00 to 85/15 in 10 mins) to afford B-41 (1.9 g, yield: 38.2 %) as white solid. LCMS (ESI): calced for C19H21ClN4OSi [M+H]+ m/z 385.1, found 385.0. Preparation of 3-(2-bromo-5-chloropyridin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole (B-42)
[0122] To a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H- indazole (4.8 g, 12.8 mmol) in 1,4-dioxane:H2O=5:1 (100 mL) was added 6-chloro-4-iodopyridine-3-carbonitrile (3.3 g, 12.8 mmol), Pd(PPh3)4 (0.7 g, 0.6 mmol) and K2CO3 (3.5 g, 25.6 mmol) stirred for 12 h at 100 ℃ under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed. Water was added to the solution and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 100/00 to 85/15 in 10 mins) to afford B-41 (1.9 g, yield: 38.2 %) as white solid. LCMS (ESI): calced for C19H21ClN4OSi [M+H]+ m/z 385.1, found 385.0. Preparation of 3-(2-bromo-5-chloropyridin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole (B-42)
 [0123] To a solution of 1H-indazole-3-carboxylic acid (25.0 g, 154.0 mmol) and NBS (27.4 g, 154.0 mmol) in DMF (250.0 ml) stirred under nitrogen at 25℃. The reaction mixture was stirred at 25 ℃ for 1h. The LCMS showed the desired MS was detected. The solution was poured into ice water. The precipitate was collected by
filtration, washed with water, dried over the precipitate to afford 3-bromo-1H-indazole (26.0 g, yield: 86.0%) as a white solid. LCMS (ESI): calced for C7H5BrN2 [M+H]+ m/z 199.0, found 199.0. [0124] To a solution of 3-bromo-1H-indazole (13.0 g, 66.0 mmol) in DMF (80 mL) were added NaH (2.4g, 99.0 mmol, 60% in oil). After 30 min, SEMCl (12.1 g, 72.0 mmol) in DMF was added at 0°C. The reaction was stirred at 25°C for 16 h. The LCMS showed the desired MS was detected. The resulting was then quenched with water, extracted with EtOAc, washed with saturated brine, dried over Na2SO4 and concentrated. The residue was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 70/30 in 30 mins) to afford 3-bromo-1-((2- (trimethylsilyl)ethoxy)methyl)-1H-indazole (20.0 g, yield: 92.5%) as a yellow oil. LCMS (ESI): calced for C13H19BrN2OSi [M+H]+ m/z 329.0, found 329.1. [0125] To a solution of 3-bromo-1-[(2-methoxyethyl)trimethyl-$l^{5}-silyl]indazole (20.0 g, 60.0 mmol) , 4,4,5,5- tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (30.9 g, 120.0 mmol), AcOK (23.9 g, 243.0 mmol) and Pd(dppf)Cl2 (1.34 g, 1.8 mmol) in 1,4-dioxane (200.0 ml). The reaction mixture was stirred at 80 ℃ for 16 h under N2. The LCMS showed the desired MS was detected .The reaction mixture was concentrated under reduced pressure to give which was purified by flash silica gel chromatography (eluting with PE/EtOAc from 100/0 to 95/5 in 20 mins) to afford 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2- (trimethylsilyl)ethoxy)methyl)-1H-indazole (16.0 g, yield: 69.7%) as a white solid. LCMS (ESI): calced for C19H31BN2O3Si [M+H]+ m/z 375.2, found 375.3. [0126] To a solution of 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H- indazole (5.0 g, 0.0133 mol), 2-bromo-5-chloro-4-iodopyridine (4.2 g, 0.0133 mol), Pd(PPh3)4 (1.5 g, 0.0013 mol) and Cs2CO3 (8.7 g, 0.0266 mol) in MeCN and H2O. The reaction solution was stirred at 90 ℃ for 8 h. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 90/10 in 20 mins) to afford B-42 (3.5 g, yield: 58.8%) as a yellow solid. LCMS (ESI): calced for C18H21BrClN3OSi [M+H]+ m/z 440.2, found 440.2. Preparation of 6-chloro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)nicotinonitrile (B-43)
[0123] To a solution of 1H-indazole-3-carboxylic acid (25.0 g, 154.0 mmol) and NBS (27.4 g, 154.0 mmol) in DMF (250.0 ml) stirred under nitrogen at 25℃. The reaction mixture was stirred at 25 ℃ for 1h. The LCMS showed the desired MS was detected. The solution was poured into ice water. The precipitate was collected by
filtration, washed with water, dried over the precipitate to afford 3-bromo-1H-indazole (26.0 g, yield: 86.0%) as a white solid. LCMS (ESI): calced for C7H5BrN2 [M+H]+ m/z 199.0, found 199.0. [0124] To a solution of 3-bromo-1H-indazole (13.0 g, 66.0 mmol) in DMF (80 mL) were added NaH (2.4g, 99.0 mmol, 60% in oil). After 30 min, SEMCl (12.1 g, 72.0 mmol) in DMF was added at 0°C. The reaction was stirred at 25°C for 16 h. The LCMS showed the desired MS was detected. The resulting was then quenched with water, extracted with EtOAc, washed with saturated brine, dried over Na2SO4 and concentrated. The residue was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 70/30 in 30 mins) to afford 3-bromo-1-((2- (trimethylsilyl)ethoxy)methyl)-1H-indazole (20.0 g, yield: 92.5%) as a yellow oil. LCMS (ESI): calced for C13H19BrN2OSi [M+H]+ m/z 329.0, found 329.1. [0125] To a solution of 3-bromo-1-[(2-methoxyethyl)trimethyl-$l^{5}-silyl]indazole (20.0 g, 60.0 mmol) , 4,4,5,5- tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (30.9 g, 120.0 mmol), AcOK (23.9 g, 243.0 mmol) and Pd(dppf)Cl2 (1.34 g, 1.8 mmol) in 1,4-dioxane (200.0 ml). The reaction mixture was stirred at 80 ℃ for 16 h under N2. The LCMS showed the desired MS was detected .The reaction mixture was concentrated under reduced pressure to give which was purified by flash silica gel chromatography (eluting with PE/EtOAc from 100/0 to 95/5 in 20 mins) to afford 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2- (trimethylsilyl)ethoxy)methyl)-1H-indazole (16.0 g, yield: 69.7%) as a white solid. LCMS (ESI): calced for C19H31BN2O3Si [M+H]+ m/z 375.2, found 375.3. [0126] To a solution of 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H- indazole (5.0 g, 0.0133 mol), 2-bromo-5-chloro-4-iodopyridine (4.2 g, 0.0133 mol), Pd(PPh3)4 (1.5 g, 0.0013 mol) and Cs2CO3 (8.7 g, 0.0266 mol) in MeCN and H2O. The reaction solution was stirred at 90 ℃ for 8 h. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 90/10 in 20 mins) to afford B-42 (3.5 g, yield: 58.8%) as a yellow solid. LCMS (ESI): calced for C18H21BrClN3OSi [M+H]+ m/z 440.2, found 440.2. Preparation of 6-chloro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)nicotinonitrile (B-43)
 [0127] To a solution of 6-chloropyridine-3-carbonitrile (5.0 g, 0.0361 mol) in THF (50 mL) were added LDA (21.0 mL, 0.0433 mol 2 M in THF) at -78℃ and stirred 15 min. I2 (10.1 g, 0.0397 mol) in THF (30 mL) was added to the reaction solution and stirred at -78 ℃ for 15 min. The LCMS showed the desired MS was detected. The
reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 95/5 in 20 mins) to afford 6-chloro-4-iodonicotinonitrile (1.8 g, yield: 18.6%) as a yellow solid. [0128] To a solution of 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H- indazole (2.6 g, 0.0068 mol) in MeCN/H2O=10:1 (40 mL) were added 6-chloro-4-iodopyridine-3-carbonitrile (1.8 g, 0.0068 mol), Pd(PPh3)4 (0.8 g, 0.0006 mol) and Cs2CO3 (4.4 g, 0.0136 mol). The reaction solution was stirred at 90 ℃ for 5h. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 90/10 in 20 mins) to afford B-43 (1.6 g, yield: 60.2%) as a yellow oil. LCMS (ESI): calced for C19H21ClN4OSi [M+H]+ m/z 385.0, found 385.0. Preparation of 3-(2-bromo-5-(trifluoromethyl)pyridin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole (B-44)
[0127] To a solution of 6-chloropyridine-3-carbonitrile (5.0 g, 0.0361 mol) in THF (50 mL) were added LDA (21.0 mL, 0.0433 mol 2 M in THF) at -78℃ and stirred 15 min. I2 (10.1 g, 0.0397 mol) in THF (30 mL) was added to the reaction solution and stirred at -78 ℃ for 15 min. The LCMS showed the desired MS was detected. The
reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 95/5 in 20 mins) to afford 6-chloro-4-iodonicotinonitrile (1.8 g, yield: 18.6%) as a yellow solid. [0128] To a solution of 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H- indazole (2.6 g, 0.0068 mol) in MeCN/H2O=10:1 (40 mL) were added 6-chloro-4-iodopyridine-3-carbonitrile (1.8 g, 0.0068 mol), Pd(PPh3)4 (0.8 g, 0.0006 mol) and Cs2CO3 (4.4 g, 0.0136 mol). The reaction solution was stirred at 90 ℃ for 5h. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 90/10 in 20 mins) to afford B-43 (1.6 g, yield: 60.2%) as a yellow oil. LCMS (ESI): calced for C19H21ClN4OSi [M+H]+ m/z 385.0, found 385.0. Preparation of 3-(2-bromo-5-(trifluoromethyl)pyridin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole (B-44)
 [0129] A solution of 2-bromo-4-iodo-5-(trifluoromethyl)pyridine (5.4 g, 15.4 mmol), (1-((2- (trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)boronic acid (5.4 g, 18.5 mmol), Pd(dppf)2Cl2 (563..6 mg, 0.77 mmol) and Cs2CO3 (10.0 g, 30.8 mmol) in 100 mL MeCN/H2O = 10:1 was charged with N2 and stirred at 90℃ for 3 h. The desired mass was detected on LC-MS. The mixture was quenched with H2O and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered out and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 50/50 in 20 mins) to afford B- 44 (2.2 g, yield: 30.3%) as a yellow solid. LCMS (ESI): calced for C19H21BrF3N3OSi [M+H]+ m/z 472.1, found 472.2.
Preparation of 5-chloro-2-methanesulfonyl-4-{[1,2,4]triazolo[4,3-a]pyridin-3-yl}pyrimidine (B-45)
[0129] A solution of 2-bromo-4-iodo-5-(trifluoromethyl)pyridine (5.4 g, 15.4 mmol), (1-((2- (trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)boronic acid (5.4 g, 18.5 mmol), Pd(dppf)2Cl2 (563..6 mg, 0.77 mmol) and Cs2CO3 (10.0 g, 30.8 mmol) in 100 mL MeCN/H2O = 10:1 was charged with N2 and stirred at 90℃ for 3 h. The desired mass was detected on LC-MS. The mixture was quenched with H2O and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered out and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 50/50 in 20 mins) to afford B- 44 (2.2 g, yield: 30.3%) as a yellow solid. LCMS (ESI): calced for C19H21BrF3N3OSi [M+H]+ m/z 472.1, found 472.2.
Preparation of 5-chloro-2-methanesulfonyl-4-{[1,2,4]triazolo[4,3-a]pyridin-3-yl}pyrimidine (B-45)
 [0130] To a solution of 5-chloro-2-(methylsulfanyl)-4-{[1,2,4]triazolo[4,3-a]pyridin-3-yl}pyrimidine (1.4 g, 5.0 mmol) in DCM (30 mL) was added 3-Chloroperoxybenzoic acid (2.6 g, 15.0 mmol) stirred for 5 h at 25 ℃. The solvent was removed. EtOAc was added to the solution and wash with saturated sodium bicarbonate solution. The organic layer was washed with brine, dried over anhydrous sodium sulfate, filtered out and concentrated to get crude product B-45 (1.4 g, yield: 91.5%) was directly used in next step without further purification. LCMS (ESI): calced for C11H8ClN5O2S [M+H]+ m/z 310.0, found 309.8. Preparation of 3-(2-bromo-5-fluoropyridin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole (B-46)
[0130] To a solution of 5-chloro-2-(methylsulfanyl)-4-{[1,2,4]triazolo[4,3-a]pyridin-3-yl}pyrimidine (1.4 g, 5.0 mmol) in DCM (30 mL) was added 3-Chloroperoxybenzoic acid (2.6 g, 15.0 mmol) stirred for 5 h at 25 ℃. The solvent was removed. EtOAc was added to the solution and wash with saturated sodium bicarbonate solution. The organic layer was washed with brine, dried over anhydrous sodium sulfate, filtered out and concentrated to get crude product B-45 (1.4 g, yield: 91.5%) was directly used in next step without further purification. LCMS (ESI): calced for C11H8ClN5O2S [M+H]+ m/z 310.0, found 309.8. Preparation of 3-(2-bromo-5-fluoropyridin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole (B-46)
 [0131] To a solution of 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H- indazole (2.0 g, 5.3 mmol) in 1,4-dioxane/H2O=10:1 (10 mL) was added 2-bromo-5-fluoro-4-iodopyridine (1.9 g, 6.3 mmol), K2CO3 (1.5 g, 11.0 mmol) and Pd(dppf)Cl2 (390.0 mg, 0.54 mmol). The reaction was stirred under N2 at 80 ℃ for 16 h. The mixture was quenched with water and extracted with EtOAc, the organic layers were combined and washed with brine, dried over Na2SO4, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 95/5 to 90/10 in 20 mins) to afford B-46 (1.24 g, yield: 52.8 %) as a yellow solid. LCMS (ESI): calced for C18H21BrFN3OSi [M+H]+ m/z 422.1, found 422.0.
[0131] To a solution of 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H- indazole (2.0 g, 5.3 mmol) in 1,4-dioxane/H2O=10:1 (10 mL) was added 2-bromo-5-fluoro-4-iodopyridine (1.9 g, 6.3 mmol), K2CO3 (1.5 g, 11.0 mmol) and Pd(dppf)Cl2 (390.0 mg, 0.54 mmol). The reaction was stirred under N2 at 80 ℃ for 16 h. The mixture was quenched with water and extracted with EtOAc, the organic layers were combined and washed with brine, dried over Na2SO4, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 95/5 to 90/10 in 20 mins) to afford B-46 (1.24 g, yield: 52.8 %) as a yellow solid. LCMS (ESI): calced for C18H21BrFN3OSi [M+H]+ m/z 422.1, found 422.0.
Preparation of 3-(2-bromo-5-chloropyridin-4-yl)-5,7-difluoro-1-(tetrahydro-2H-pyran-2-yl)-1H-indazole (B- 47)
 [0132] A solution of 2-bromo-5-chloro-4-iodopyridine (1.5 g, 4.7 mmol), [5,7-difluoro-1-(oxan-3-yl)indazol-3- yl]boranediol (1.6 g, 5.6 mmol, fresh made and purified), Bis(triphenylphosphine)palladium(II) chloride (0.4 g, 0.47 mmol) and K2CO3 (1.3 g, 9.4 mmol) in 1,4-dioxane/H2O=10:1 (30 mL) was charged with N2 and stirred at 80 ℃ for 8 h. The LCMS showed the desired MS was detected. The mixture was concentrated and purified by flash chromatography (eluting with PE/EtOAc = 100/0 to 90/10 in 30 mins) to afford B-47 (0.8 g, yield: 39.8 %) as a yellow solid. LCMS (ESI) calcd for C17H14BrClF2N3O [M + H] + ms/z 428.0, found 428.0. Preparation of 3-(2,5-dichloropyrimidin-4-yl)pyrazolo[1,5-a]pyridine (B-48)
[0132] A solution of 2-bromo-5-chloro-4-iodopyridine (1.5 g, 4.7 mmol), [5,7-difluoro-1-(oxan-3-yl)indazol-3- yl]boranediol (1.6 g, 5.6 mmol, fresh made and purified), Bis(triphenylphosphine)palladium(II) chloride (0.4 g, 0.47 mmol) and K2CO3 (1.3 g, 9.4 mmol) in 1,4-dioxane/H2O=10:1 (30 mL) was charged with N2 and stirred at 80 ℃ for 8 h. The LCMS showed the desired MS was detected. The mixture was concentrated and purified by flash chromatography (eluting with PE/EtOAc = 100/0 to 90/10 in 30 mins) to afford B-47 (0.8 g, yield: 39.8 %) as a yellow solid. LCMS (ESI) calcd for C17H14BrClF2N3O [M + H] + ms/z 428.0, found 428.0. Preparation of 3-(2,5-dichloropyrimidin-4-yl)pyrazolo[1,5-a]pyridine (B-48)
 [0133] To a solution of 3-bromopyrazolo[1,5-a]pyridine (1.9 g, 0.0096 mol) in diglyme/H2O (20 mL) were added 4,4,5,5-tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (2.9 g, 0.0114 mol), Palladium diacetate (212.4 mg, 0.0009 mol), PCy3 (507.4 mg, 0.0018 mol) and Potassium acetate (1.8 g, 0.0191 mol). The reaction solution was stirred at 100 ℃ for 2 h. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 90/10 in 20 mins) to afford 3-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)pyrazolo[1,5-a]pyridine (800.0 mg, yield: 33.1%) as a yellow solid. LCMS (ESI): calced for C13H17BN2O2 [M+H]+ m/z 245.2, found 245.2. [0134] To a solution of 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazolo[1,5-a]pyridine (800.0 mg, 3.2774 mmol) in ACN/H2O = 10:1 (10 mL) were added 2,4,5-trichloropyrimidine (1.2 g, 6.5548 mmol), Pd(PPh3)4 (378.7 mg, 0.3277 mmol) and Na2CO3 (694.8 mg, 6.5548 mmol). The reaction solution was stirred at 80 ℃ for 12 h. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with
EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 90/10 in 20 mins) to afford B-48 (600.0 mg, yield: 67.7%) as a yellow solid. LCMS (ESI): calced for C11H6Cl2N4 [M+H]+ m/z 264.9, found 264.9. Preparation of 3-(2-chloro-5-methylpyridin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole (B-49)
[0133] To a solution of 3-bromopyrazolo[1,5-a]pyridine (1.9 g, 0.0096 mol) in diglyme/H2O (20 mL) were added 4,4,5,5-tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (2.9 g, 0.0114 mol), Palladium diacetate (212.4 mg, 0.0009 mol), PCy3 (507.4 mg, 0.0018 mol) and Potassium acetate (1.8 g, 0.0191 mol). The reaction solution was stirred at 100 ℃ for 2 h. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 90/10 in 20 mins) to afford 3-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)pyrazolo[1,5-a]pyridine (800.0 mg, yield: 33.1%) as a yellow solid. LCMS (ESI): calced for C13H17BN2O2 [M+H]+ m/z 245.2, found 245.2. [0134] To a solution of 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazolo[1,5-a]pyridine (800.0 mg, 3.2774 mmol) in ACN/H2O = 10:1 (10 mL) were added 2,4,5-trichloropyrimidine (1.2 g, 6.5548 mmol), Pd(PPh3)4 (378.7 mg, 0.3277 mmol) and Na2CO3 (694.8 mg, 6.5548 mmol). The reaction solution was stirred at 80 ℃ for 12 h. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with
EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 90/10 in 20 mins) to afford B-48 (600.0 mg, yield: 67.7%) as a yellow solid. LCMS (ESI): calced for C11H6Cl2N4 [M+H]+ m/z 264.9, found 264.9. Preparation of 3-(2-chloro-5-methylpyridin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole (B-49)
 [0135] To a solution of 1-[(2-methoxyethyl)trimethyl-$l^{5}-silyl]-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)indazole (1.5 g, 4.0 mmol) , 4-bromo-2-chloro-5-methylpyridine (990.1 mg, 4.8 mmol), Na2CO3 (847.2 mg, 8.0 mmol) and Pd(dppf)Cl2 (292.4 mg, 0.4 mmol) in (1,4-dioxane/H2O=10:1)(15.0 ml) stirred under nitrogen at 25℃ . The reaction mixture was stirred at 80 ℃ for 2 h. The LCMS showed the desired MS was detected .The reaction mixture was concentrated under reduced pressure to give which was purified by flash silica gel chromatography (eluting with PE/EtOAc from 100/0 to 95/5 in 20 mins) to afford B-49 (830.0 mg, yield: 55.9%) as a white solid. LCMS (ESI): calced for C19H24ClN3OSi [M+H]+ m/z 374.1, found 373.8. Preparation of 3-(2-bromo-5-chloropyridin-4-yl)-5-fluoro-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole (B-50)
[0135] To a solution of 1-[(2-methoxyethyl)trimethyl-$l^{5}-silyl]-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)indazole (1.5 g, 4.0 mmol) , 4-bromo-2-chloro-5-methylpyridine (990.1 mg, 4.8 mmol), Na2CO3 (847.2 mg, 8.0 mmol) and Pd(dppf)Cl2 (292.4 mg, 0.4 mmol) in (1,4-dioxane/H2O=10:1)(15.0 ml) stirred under nitrogen at 25℃ . The reaction mixture was stirred at 80 ℃ for 2 h. The LCMS showed the desired MS was detected .The reaction mixture was concentrated under reduced pressure to give which was purified by flash silica gel chromatography (eluting with PE/EtOAc from 100/0 to 95/5 in 20 mins) to afford B-49 (830.0 mg, yield: 55.9%) as a white solid. LCMS (ESI): calced for C19H24ClN3OSi [M+H]+ m/z 374.1, found 373.8. Preparation of 3-(2-bromo-5-chloropyridin-4-yl)-5-fluoro-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole (B-50)
 [0136] To a solution of 3-bromo-5-fluoro-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole (3.9 g, 11.3 mmol) in 1.4-dioxane (50 mL) was added 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi(1,3,2-dioxaborolane) (11.5 g, 45.3 mmol), dry AcOK (2.2 g, 22.6 mmol) and Pd(dppf)Cl2 DCM (915.9 mg, 1.1 mmol), then the mixture is stirred for 5 h at 90°C under N2. The desired mass was detected on LC-MS. The mixture was washed with water and extracted with EtOAc. the organic layer was washed with brine, dried over Na2SO4, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with DCM/MeOH from 100/0 to 95/5 in 8 mins) to afford (5-fluoro-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)boronic acid (2.1 g, yield: 59.7 %) as a yellow solid. LCMS (ESI): calcd for C13H20BFN2O3Si [M+H] + ms/z 311.1, found 311.2.
[0137] To a solution of (5-fluoro-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)boronic acid (2.1 g, 6.7 mmol) in 1.4-dioxane (50 mL) and H2O (10 mL) was added 2-bromo-5-chloro-4-iodopyridine (1,3,2- dioxaborolane) (2.3 g, 7.3 mmol), K2CO3 (1.8 g, 13.4 mmol) and Pd(dppf)Cl2 (435.3 mg, 0.6 mmol), then the mixture is stirred for 5 h at 90°C under N2. The desired mass was detected on LC-MS. The mixture was washed with water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 55/45 in 13 mins) to afford B-50 (1.2 g, yield: 38.9 %) as a yellow solid. LCMS (ESI): calcd for C18H20BrClFN3OSi [M+H] + ms/z 456.0, found 458.1. Preparation of 6-(2-bromo-5-chloropyridin-4-yl)-1-isopropyl-1H-indazole (B-51)
[0136] To a solution of 3-bromo-5-fluoro-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole (3.9 g, 11.3 mmol) in 1.4-dioxane (50 mL) was added 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi(1,3,2-dioxaborolane) (11.5 g, 45.3 mmol), dry AcOK (2.2 g, 22.6 mmol) and Pd(dppf)Cl2 DCM (915.9 mg, 1.1 mmol), then the mixture is stirred for 5 h at 90°C under N2. The desired mass was detected on LC-MS. The mixture was washed with water and extracted with EtOAc. the organic layer was washed with brine, dried over Na2SO4, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with DCM/MeOH from 100/0 to 95/5 in 8 mins) to afford (5-fluoro-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)boronic acid (2.1 g, yield: 59.7 %) as a yellow solid. LCMS (ESI): calcd for C13H20BFN2O3Si [M+H] + ms/z 311.1, found 311.2.
[0137] To a solution of (5-fluoro-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)boronic acid (2.1 g, 6.7 mmol) in 1.4-dioxane (50 mL) and H2O (10 mL) was added 2-bromo-5-chloro-4-iodopyridine (1,3,2- dioxaborolane) (2.3 g, 7.3 mmol), K2CO3 (1.8 g, 13.4 mmol) and Pd(dppf)Cl2 (435.3 mg, 0.6 mmol), then the mixture is stirred for 5 h at 90°C under N2. The desired mass was detected on LC-MS. The mixture was washed with water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 55/45 in 13 mins) to afford B-50 (1.2 g, yield: 38.9 %) as a yellow solid. LCMS (ESI): calcd for C18H20BrClFN3OSi [M+H] + ms/z 456.0, found 458.1. Preparation of 6-(2-bromo-5-chloropyridin-4-yl)-1-isopropyl-1H-indazole (B-51)
 [0138] To a solution of 1-isopropyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)indazole (400.0 mg, 1.40 mmol) in MeCN / H2O was added 2-bromo-5-chloro-4-iodopyridine (444.4 mg, 1.40 mmol), Pd(PPh3)4 (80.8 mg, 0.07 mmol) and Na2CO3 (296.3 mg, 2.80 mmol). The reaction mixture was stirred at 80℃ for 6 h under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed by vacuum. The residue was extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 50/50 in 30 mins) to afford B-51 (310.0 mg, yield: 54.4 %) as a yellow solid. LCMS (ESI) calced for C15H13BrClN3 [M+H]+ m/z 350.0, found 350.1. Preparation of methyl 3-(2,5-dichloropyrimidin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)- 1H-indazole-6- carboxylate (B-52)
[0138] To a solution of 1-isopropyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)indazole (400.0 mg, 1.40 mmol) in MeCN / H2O was added 2-bromo-5-chloro-4-iodopyridine (444.4 mg, 1.40 mmol), Pd(PPh3)4 (80.8 mg, 0.07 mmol) and Na2CO3 (296.3 mg, 2.80 mmol). The reaction mixture was stirred at 80℃ for 6 h under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed by vacuum. The residue was extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 50/50 in 30 mins) to afford B-51 (310.0 mg, yield: 54.4 %) as a yellow solid. LCMS (ESI) calced for C15H13BrClN3 [M+H]+ m/z 350.0, found 350.1. Preparation of methyl 3-(2,5-dichloropyrimidin-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)- 1H-indazole-6- carboxylate (B-52)
 [0139] To a solution of methyl 3-bromo-1-((2-(trimethylsilyl)ethoxy) methyl)-1H-indazole-6-carboxylate (4.4 g, 11.0 mmol) in 1,4-dioxane (66 mL) was added 4,4,5,5-tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2 -dioxaborolan-2- yl)-1,3,2-dioxaborolane (3.5 g, 13.0 mmol), Pd(dppf)Cl2 (0.4 g, 1.0 mmol) and AcOK (2.2 g, 22.0 mmol). The reaction mixture was stirred at 100℃ for 16 h under nitrogen. The LCMS showed the desired MS was detected.
The solvent was removed by vacuum. The residue was extracted with EtOAc (400 mL), the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 60/40 in 20 mins) to afford (6-(methoxycarbonyl)-1-((2- (trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)boronic acid (2.7 g, yield: 47.4%) as a yellow solid. LCMS (ESI): calced for C15H23BN2O5Si [M+H]+ m/z 351.2, found 351.3. [0140] To a solution of (6-(methoxycarbonyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)boronic acid (2.7 g, 6.0 mmol) in 1,4-dioxane (30 mL) and H2O (6 mL) was added 2,4,5-trichloropyrimidine (1.4 g, 7.0 mmol), Pd(dppf)Cl2 (0.2 g, 0.3 mmol) and Na2CO3 (1.3 g, 12.0 mmol). The reaction mixture was stirred at 80 ℃ for 6 h under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed by vacuum. The residue was extracted with EtOAc (200 mL), the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (PE/EtOAc = 100/0 to 50/50) to afford B-52 (1.8 g, yield: 54.8%) as a yellow solid. LCMS (ESI): calced for C19H22Cl2N4O3Si [M+H]+ m/z 453.1, found 453.2. Preparation of 5-chloro-4-(1H-indol-3-yl)-N-(piperidin-4-yl)pyrimidin-2-amine(C-1)
[0139] To a solution of methyl 3-bromo-1-((2-(trimethylsilyl)ethoxy) methyl)-1H-indazole-6-carboxylate (4.4 g, 11.0 mmol) in 1,4-dioxane (66 mL) was added 4,4,5,5-tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2 -dioxaborolan-2- yl)-1,3,2-dioxaborolane (3.5 g, 13.0 mmol), Pd(dppf)Cl2 (0.4 g, 1.0 mmol) and AcOK (2.2 g, 22.0 mmol). The reaction mixture was stirred at 100℃ for 16 h under nitrogen. The LCMS showed the desired MS was detected.
The solvent was removed by vacuum. The residue was extracted with EtOAc (400 mL), the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 60/40 in 20 mins) to afford (6-(methoxycarbonyl)-1-((2- (trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)boronic acid (2.7 g, yield: 47.4%) as a yellow solid. LCMS (ESI): calced for C15H23BN2O5Si [M+H]+ m/z 351.2, found 351.3. [0140] To a solution of (6-(methoxycarbonyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)boronic acid (2.7 g, 6.0 mmol) in 1,4-dioxane (30 mL) and H2O (6 mL) was added 2,4,5-trichloropyrimidine (1.4 g, 7.0 mmol), Pd(dppf)Cl2 (0.2 g, 0.3 mmol) and Na2CO3 (1.3 g, 12.0 mmol). The reaction mixture was stirred at 80 ℃ for 6 h under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed by vacuum. The residue was extracted with EtOAc (200 mL), the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (PE/EtOAc = 100/0 to 50/50) to afford B-52 (1.8 g, yield: 54.8%) as a yellow solid. LCMS (ESI): calced for C19H22Cl2N4O3Si [M+H]+ m/z 453.1, found 453.2. Preparation of 5-chloro-4-(1H-indol-3-yl)-N-(piperidin-4-yl)pyrimidin-2-amine(C-1)
 [0141] To a solution of B-1 (7.5 g, 0.0205 mol) in NMP (150 mL) was added (4-aminopiperidin-1-yl) tert-butyl formate M1 (4.1 g, 0.0205 mol) and N,N-Diisopropylethylamine (5.3 g, 0.041 mol). The reaction was stirred at 80 ℃ for 12 h. The desired mass was detected on LC-MS. Then the mixture was quenched with water, extracted with EtOAc, washed with brine, the combined organic phase were dried over anhydrous Na2SO4, filtered and concentrated to get crude product which was purified by column chromatograph on silica gel (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford di-Boc-C-1 (5.5 g, yield: 48.3 %) as an off-white solid. LCMS (ESI): calcd for C27H35ClN5O4 [M+H] + ms/z 528.2, found 528.6. [0142] To a solution of di-Boc-C-1 (5.5 g, 0.0104 mol) in DCM (100 mL) was added TFA (20 mL) stirred at 25℃ for 5 h. The desired mass was detected on LC-MS. Then the mixture was concentrated, quenched with water, adjusted to PH = 7-8 with NaHCO3(aq.), extracted with EtOAc, the combined organic phase were dried over anhydrous Na2SO4, filtered and concentrated to get crude product which was purified by column chromatograph on silica gel (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-1 (2.4 g, yield: 67.3 %) as an off-white solid. LCMS (ESI): calcd for C17H19ClN5 [M+H] + ms/z 328.1, found 328.2.
Preparation of 5-chloro-4-(1-methyl-1H-indol-3-yl)-N-(piperidin-4-yl)pyrimidin-2-amine hydrochloride (C-2)
[0141] To a solution of B-1 (7.5 g, 0.0205 mol) in NMP (150 mL) was added (4-aminopiperidin-1-yl) tert-butyl formate M1 (4.1 g, 0.0205 mol) and N,N-Diisopropylethylamine (5.3 g, 0.041 mol). The reaction was stirred at 80 ℃ for 12 h. The desired mass was detected on LC-MS. Then the mixture was quenched with water, extracted with EtOAc, washed with brine, the combined organic phase were dried over anhydrous Na2SO4, filtered and concentrated to get crude product which was purified by column chromatograph on silica gel (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford di-Boc-C-1 (5.5 g, yield: 48.3 %) as an off-white solid. LCMS (ESI): calcd for C27H35ClN5O4 [M+H] + ms/z 528.2, found 528.6. [0142] To a solution of di-Boc-C-1 (5.5 g, 0.0104 mol) in DCM (100 mL) was added TFA (20 mL) stirred at 25℃ for 5 h. The desired mass was detected on LC-MS. Then the mixture was concentrated, quenched with water, adjusted to PH = 7-8 with NaHCO3(aq.), extracted with EtOAc, the combined organic phase were dried over anhydrous Na2SO4, filtered and concentrated to get crude product which was purified by column chromatograph on silica gel (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-1 (2.4 g, yield: 67.3 %) as an off-white solid. LCMS (ESI): calcd for C17H19ClN5 [M+H] + ms/z 328.1, found 328.2.
Preparation of 5-chloro-4-(1-methyl-1H-indol-3-yl)-N-(piperidin-4-yl)pyrimidin-2-amine hydrochloride (C-2)
 [0143] To a solution of B-2 (610.0 mg, 2.2 mmol) in NMP (10 mL) was added M1 (441.4 mg, 2.2 mmol) and DIEA (566.9 mg, 4.3 mmol) stirred for 12h at 80 ℃. The LCMS showed the desired MS was detected. Water was added to the solution and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with DCM/MeOH from 100/00 to 95/05 in 10 mins) to afford Boc-C-2 (420.0 mg, yield: 43.2%) as white solid. LCMS (ESI): calced for C23H28ClN5O2 [M+H]+ m/z 442.2, found 442.4. [0144] To a solution of Boc-C-2 (420.0 mg, 0.94 mmol) in HCL1,4-dioxane (4M) (8 mL) was stirred for 3h at 25 ℃. The LCMS showed the desired MS was detected. The solvent was removed. The crude product C-2 (300.0 mg, yield: 92.5%) was directly used in next step without further purification. LCMS (ESI): calced for C18H20ClN5 [M+H]+ m/z 342.1, found 342.3. Preparation of 5-chloro-N-(piperidin-4-yl)-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyrimidin-2-amine (C-3)
[0143] To a solution of B-2 (610.0 mg, 2.2 mmol) in NMP (10 mL) was added M1 (441.4 mg, 2.2 mmol) and DIEA (566.9 mg, 4.3 mmol) stirred for 12h at 80 ℃. The LCMS showed the desired MS was detected. Water was added to the solution and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with DCM/MeOH from 100/00 to 95/05 in 10 mins) to afford Boc-C-2 (420.0 mg, yield: 43.2%) as white solid. LCMS (ESI): calced for C23H28ClN5O2 [M+H]+ m/z 442.2, found 442.4. [0144] To a solution of Boc-C-2 (420.0 mg, 0.94 mmol) in HCL1,4-dioxane (4M) (8 mL) was stirred for 3h at 25 ℃. The LCMS showed the desired MS was detected. The solvent was removed. The crude product C-2 (300.0 mg, yield: 92.5%) was directly used in next step without further purification. LCMS (ESI): calced for C18H20ClN5 [M+H]+ m/z 342.1, found 342.3. Preparation of 5-chloro-N-(piperidin-4-yl)-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyrimidin-2-amine (C-3)
 [0145] To a solution of B-3 (4.3 g, 0.0109 mol), M1 (3.2 g, 0.0159 mol) and DIEA (6.9 g, 0.0538 mol) in NMP (40 ml). The reaction mixture was stirred at 100 ℃ for 12 h. The LCMS showed the desired MS was detected. The reaction mixture was added water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 90/10 in 20 mins) to afford Boc-C-3 (3.5 g, yield: 57.5%) as a white solid. LCMS (ESI): calced for C27H39ClN6O3Si [M+H]+ m/z 559.4, found 559.4. [0146] To a solution of Boc-C-3 (3.5 g, 0.0062 mol) in DCM/TFA=10:1 (50 ml) . The reaction mixture was stirred at 26 ℃ for 6 h. The LCMS showed the desired MS was detected. The reaction mixture was added water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with CH2Cl2/MeOH from 100/0 to 92/8 in
15 mins) to afford C-3 (2.4 g, yield: 84.5%) as a yellow oil. LCMS (ESI): calced for C22H31ClN6OSi [M+H]+ m/z 459.1, found 459.1. Preparation of 5-chloro-4-(1-isopropyl-1H-indol-3-yl)-N-(piperidin-4-yl)pyrimidin-2-amine (C-4)
[0145] To a solution of B-3 (4.3 g, 0.0109 mol), M1 (3.2 g, 0.0159 mol) and DIEA (6.9 g, 0.0538 mol) in NMP (40 ml). The reaction mixture was stirred at 100 ℃ for 12 h. The LCMS showed the desired MS was detected. The reaction mixture was added water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 90/10 in 20 mins) to afford Boc-C-3 (3.5 g, yield: 57.5%) as a white solid. LCMS (ESI): calced for C27H39ClN6O3Si [M+H]+ m/z 559.4, found 559.4. [0146] To a solution of Boc-C-3 (3.5 g, 0.0062 mol) in DCM/TFA=10:1 (50 ml) . The reaction mixture was stirred at 26 ℃ for 6 h. The LCMS showed the desired MS was detected. The reaction mixture was added water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with CH2Cl2/MeOH from 100/0 to 92/8 in
15 mins) to afford C-3 (2.4 g, yield: 84.5%) as a yellow oil. LCMS (ESI): calced for C22H31ClN6OSi [M+H]+ m/z 459.1, found 459.1. Preparation of 5-chloro-4-(1-isopropyl-1H-indol-3-yl)-N-(piperidin-4-yl)pyrimidin-2-amine (C-4)
 [0147] A mixture of B-4 (2.1 g, 6.88 mmol), M-1 (2.06 g, 10.3 mmol), Cs2CO3 (4.4 g, 13.76 mmol), XantPhos (398.8 mg, 0.69 mmol) and Pd2(dba)3 (311.1 mg, 0.34 mmol) in Dioxane (30 mL) was stirred at 100°C for 16 h. The desired mass was detected on LC-MS. Then the mixture was quenched with water, extracted with EtOAc, washed with brine, the combined organic phase were dried over anhydrous Na2SO4, filtered and concentrated to get crude product which was purified by column chromatograph on silica gel (eluting with PE/EtOAc from 100/0 to 75/25 in 20 mins) to afford Boc-C-4 (1.7 g, yield: 52.7%) as a yellow solid. LCMS (ESI): calcd for C25H32ClN5O2 [M+H]+ m/z 470.0, found 470.2. [0148] To a solution of Boc-C-4 (1.7 g, 3.6 mmol) in DCM (20 mL) was added TFA (5 mL). The reaction was stirred at rt for 1 h. The desired mass was detected on LC-MS. Then the mixture was quenched with NaHCO3 (aq.), extracted with EtOAc, washed with brine, the combined organic phase were dried over anhydrous Na2SO4, filtered and concentrated to get crude product C-4 (1.1 g, yield: 82.8%) as a yellow solid. LCMS (ESI): calcd for C20H24ClN5 [M+H]+ m/z 370.2, found 370.0. Preparation of 5-chloro-4-(1H-indol-4-yl)-N-(piperidin-4-yl)pyrimidin-2-amine hydrochloride (C-5)
[0147] A mixture of B-4 (2.1 g, 6.88 mmol), M-1 (2.06 g, 10.3 mmol), Cs2CO3 (4.4 g, 13.76 mmol), XantPhos (398.8 mg, 0.69 mmol) and Pd2(dba)3 (311.1 mg, 0.34 mmol) in Dioxane (30 mL) was stirred at 100°C for 16 h. The desired mass was detected on LC-MS. Then the mixture was quenched with water, extracted with EtOAc, washed with brine, the combined organic phase were dried over anhydrous Na2SO4, filtered and concentrated to get crude product which was purified by column chromatograph on silica gel (eluting with PE/EtOAc from 100/0 to 75/25 in 20 mins) to afford Boc-C-4 (1.7 g, yield: 52.7%) as a yellow solid. LCMS (ESI): calcd for C25H32ClN5O2 [M+H]+ m/z 470.0, found 470.2. [0148] To a solution of Boc-C-4 (1.7 g, 3.6 mmol) in DCM (20 mL) was added TFA (5 mL). The reaction was stirred at rt for 1 h. The desired mass was detected on LC-MS. Then the mixture was quenched with NaHCO3 (aq.), extracted with EtOAc, washed with brine, the combined organic phase were dried over anhydrous Na2SO4, filtered and concentrated to get crude product C-4 (1.1 g, yield: 82.8%) as a yellow solid. LCMS (ESI): calcd for C20H24ClN5 [M+H]+ m/z 370.2, found 370.0. Preparation of 5-chloro-4-(1H-indol-4-yl)-N-(piperidin-4-yl)pyrimidin-2-amine hydrochloride (C-5)
 [0149] To a solution of B-5 (1.1 g, 3.0 mmol) in NMP (10 mL) was added M-1 (721.0 mg, 3.6 mmol) and DIEA (775.4 mg, 6.0 mmol). The reaction mixture was stirred at 100 ℃ for 16 h under N2. The LCMS showed the desired MS was detected. The mixture was quenched with water, extracted with EtOAc, the organic layers were combined and washed with brine, dried over Na2SO4, filtered out and concentrated and obtained 1.2 g crude product. The crude product was dissolved in a solution of HCl in EtOAc (2 M, 20 mL) and stirred at rt for 16 h. The LCMS showed the desired MS was detected. The reaction mixture was filtered. The filter cake was washed
with EtOAc and dried on vacuum to afford C-5 (820.0 mg, yield: 62.0%) as a yellow solid. LCMS (ESI): calced for C17H18ClN5 [M+H]+ m/z 328.1, found 328.0. Preparation of 5-chloro-4-(3-isopropyl-1,2,3-benzotriazol-5-yl)-N-(piperidin-4-yl)pyrimidin-2-amine TFA salt (C-6)
[0149] To a solution of B-5 (1.1 g, 3.0 mmol) in NMP (10 mL) was added M-1 (721.0 mg, 3.6 mmol) and DIEA (775.4 mg, 6.0 mmol). The reaction mixture was stirred at 100 ℃ for 16 h under N2. The LCMS showed the desired MS was detected. The mixture was quenched with water, extracted with EtOAc, the organic layers were combined and washed with brine, dried over Na2SO4, filtered out and concentrated and obtained 1.2 g crude product. The crude product was dissolved in a solution of HCl in EtOAc (2 M, 20 mL) and stirred at rt for 16 h. The LCMS showed the desired MS was detected. The reaction mixture was filtered. The filter cake was washed
with EtOAc and dried on vacuum to afford C-5 (820.0 mg, yield: 62.0%) as a yellow solid. LCMS (ESI): calced for C17H18ClN5 [M+H]+ m/z 328.1, found 328.0. Preparation of 5-chloro-4-(3-isopropyl-1,2,3-benzotriazol-5-yl)-N-(piperidin-4-yl)pyrimidin-2-amine TFA salt (C-6)
 [0150] To a solution of B-6 (3.0 g, 0.0097 mol), M1 (2.3 g, 0.012 mol) in NMP (40 mL) was added DIEA (2.5 g, 0.019 mol). The reaction mixture was stirred at 80℃ for 4 h. The LCMS showed the desired MS was detected. The mixture was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 60/40 in 20 mins) to afford Boc-C-6 (2.8 g, yield: 53.6 %) as a yellow solid. LCMS (ESI): calced for C23H30Cl2N7O2 [M+H]+ m/z 472.2, found 472.4. [0151] To a solution of Boc-C-6 (1.5 g, 0.0032 mol) in TFA/DCM (4mL/20mL) stirred at 25℃ for 4 h. The LCMS showed the desired MS was detected. The solvent was removed by vacuum to afford C-6 (1.2 g, yield: 87.5 %) as a yellow solid. LCMS (ESI): calced for C18H22ClN7 [M+H]+ m/z 486.2, found 372.1. Preparation of 5-chloro-4-(3-isopropyl-2-methylindazol-5-yl)-N-(piperidin-4-yl)pyrimidin-2-amine TFA salt (C-8)
[0150] To a solution of B-6 (3.0 g, 0.0097 mol), M1 (2.3 g, 0.012 mol) in NMP (40 mL) was added DIEA (2.5 g, 0.019 mol). The reaction mixture was stirred at 80℃ for 4 h. The LCMS showed the desired MS was detected. The mixture was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 60/40 in 20 mins) to afford Boc-C-6 (2.8 g, yield: 53.6 %) as a yellow solid. LCMS (ESI): calced for C23H30Cl2N7O2 [M+H]+ m/z 472.2, found 472.4. [0151] To a solution of Boc-C-6 (1.5 g, 0.0032 mol) in TFA/DCM (4mL/20mL) stirred at 25℃ for 4 h. The LCMS showed the desired MS was detected. The solvent was removed by vacuum to afford C-6 (1.2 g, yield: 87.5 %) as a yellow solid. LCMS (ESI): calced for C18H22ClN7 [M+H]+ m/z 486.2, found 372.1. Preparation of 5-chloro-4-(3-isopropyl-2-methylindazol-5-yl)-N-(piperidin-4-yl)pyrimidin-2-amine TFA salt (C-8)
 [0152] To a solution of B-8 (680.0 mg, 2.1 mmol), M1 (511.4 mg, 2.5 mmol) in NMP (20 mL) was added DIEA (547.2 mg, 4.2 mol). The reaction mixture was stirred at 80℃ for 4 h. The LCMS showed the desired MS was detected. The mixture was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 60/40 in 20 mins) to afford Boc-C-8 (460.0 mg, yield: 41.1 %) as a yellow solid. LCMS (ESI): calced for C25H33ClN6O2 [M+H]+ m/z 485.2, found 485.2. [0153] To a solution of Boc-C-8 (460.0 mg, 0.9 mmol) in TFA/DCM (2mL/10mL) stirred at 25℃ for 4 h. The LCMS showed the desired MS was detected. The solvent was removed by vacumm to afford C-8 (390.0 mg, yield: 92.1 %) as a yellow solid. LCMS (ESI): calced for C20H25ClN6 [M+H]+ m/z 499.2, found 384.8.
Preparation of 5-chloro-4-(4-fluoro-1-isopropyl-1H-benzo[d]imidazol-6-yl)-N-(piperidin-4-yl)pyrimidin-2-amine (C- 9)
[0152] To a solution of B-8 (680.0 mg, 2.1 mmol), M1 (511.4 mg, 2.5 mmol) in NMP (20 mL) was added DIEA (547.2 mg, 4.2 mol). The reaction mixture was stirred at 80℃ for 4 h. The LCMS showed the desired MS was detected. The mixture was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 60/40 in 20 mins) to afford Boc-C-8 (460.0 mg, yield: 41.1 %) as a yellow solid. LCMS (ESI): calced for C25H33ClN6O2 [M+H]+ m/z 485.2, found 485.2. [0153] To a solution of Boc-C-8 (460.0 mg, 0.9 mmol) in TFA/DCM (2mL/10mL) stirred at 25℃ for 4 h. The LCMS showed the desired MS was detected. The solvent was removed by vacumm to afford C-8 (390.0 mg, yield: 92.1 %) as a yellow solid. LCMS (ESI): calced for C20H25ClN6 [M+H]+ m/z 499.2, found 384.8.
Preparation of 5-chloro-4-(4-fluoro-1-isopropyl-1H-benzo[d]imidazol-6-yl)-N-(piperidin-4-yl)pyrimidin-2-amine (C- 9)
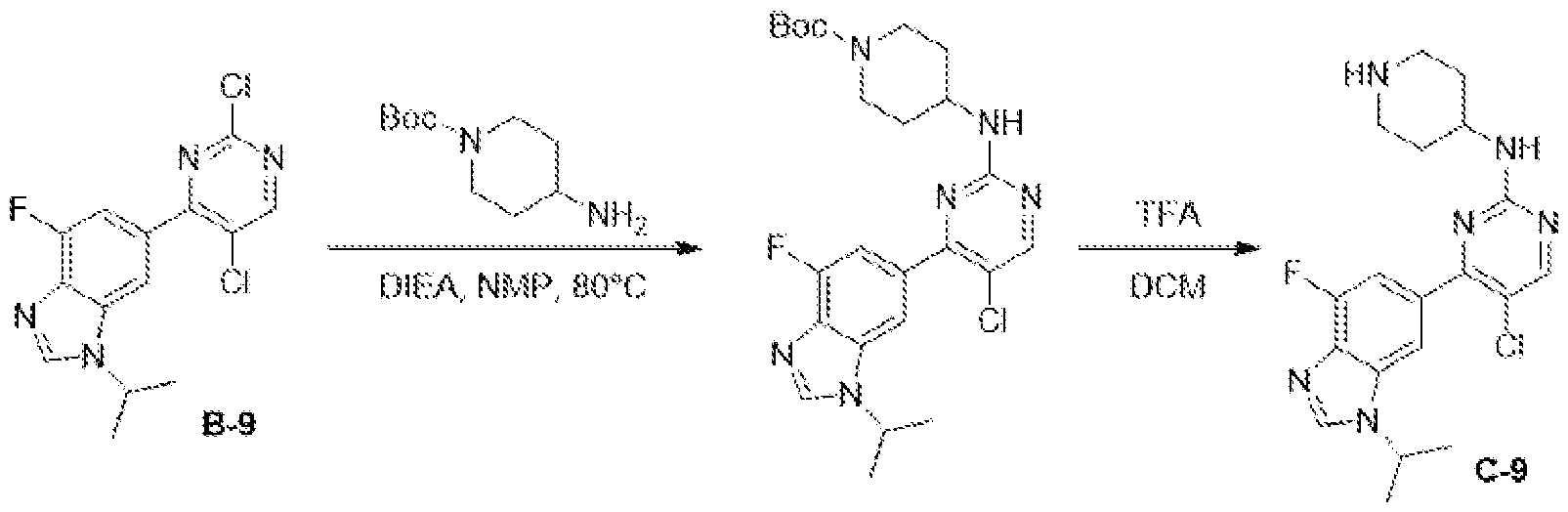 [0154] To a solution of B-9 (1.1 g, 3.4 mmol) in NMP (15 mL) was added M-1 (7403. mg, 3.7 mol) and DIEA (800.0 mg, 6.2 mol) stirred for 12 h at 80 ℃. The LCMS showed the desired MS was detected. Water was added to the solution and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 100/00 to 75/25 in 10 mins) to afford Boc-C-9 (550.0 mg, yield: 35.8%) as a white solid. LCMS (ESI): calced for C24H30ClFN6O2 [M+H]+ m/z 489.2, found 489.4. [0155] To a solution of Boc-C-9 (520 mg, 1.0 mmol) in DCM/TFA=5:1 (10 mL) stirred for 12 h at 25 ℃. The LCMS showed the desired MS was detected. The solution was adjusted to pH 7-8 with saturated sodium carbonate and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with DCM/MeOH from 100/00 to 85/15 in 15 mins) to afford C-9 (360.0 mg, yield: 87.2%) as a white solid. LCMS (ESI): calced for C15H21ClN6 [M+H]+ m/z 389.2, found 389.0. Preparation of 5-chloro-4-(1-isopropyl-1H-indazol-6-yl)-N-(piperidin-4-yl)pyrimidin-2-amine TFA salt (C-11)
[0154] To a solution of B-9 (1.1 g, 3.4 mmol) in NMP (15 mL) was added M-1 (7403. mg, 3.7 mol) and DIEA (800.0 mg, 6.2 mol) stirred for 12 h at 80 ℃. The LCMS showed the desired MS was detected. Water was added to the solution and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 100/00 to 75/25 in 10 mins) to afford Boc-C-9 (550.0 mg, yield: 35.8%) as a white solid. LCMS (ESI): calced for C24H30ClFN6O2 [M+H]+ m/z 489.2, found 489.4. [0155] To a solution of Boc-C-9 (520 mg, 1.0 mmol) in DCM/TFA=5:1 (10 mL) stirred for 12 h at 25 ℃. The LCMS showed the desired MS was detected. The solution was adjusted to pH 7-8 with saturated sodium carbonate and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with DCM/MeOH from 100/00 to 85/15 in 15 mins) to afford C-9 (360.0 mg, yield: 87.2%) as a white solid. LCMS (ESI): calced for C15H21ClN6 [M+H]+ m/z 389.2, found 389.0. Preparation of 5-chloro-4-(1-isopropyl-1H-indazol-6-yl)-N-(piperidin-4-yl)pyrimidin-2-amine TFA salt (C-11)
 [0156] To a solution of B-11 (680.0 mg, 2.21 mmol) in NMP (10 mL) was added M1 (534.7 mg, 2.66 mmol) and DIEA (572.2 mg, 4.43 mmol). The reaction mixture was stirred at 80℃ for 6 h. The LCMS showed the desired MS was detected. The mixture was then quenched water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 60/40 in 20 mins) to afford Boc-C-11 (490.0 mg, yield: 41.3 %) as a yellow solid. LCMS (ESI): calced for C24H31ClN6O2 [M+H]+ m/z 471.2, found 471.5.
[0157] To a solution of Boc-C-11 (490.0 mg, 1.04 mmol) in TFA/DCM (2mL/10mL) stirred at 25℃ for 4 h. The LCMS showed the desired MS was detected. The solvent was removed by vacumm to afford C-10 (350.0 mg, yield: 75.5 %) as a gray solid. LCMS (ESI): calced for C19H23ClN6 [M+H]+ m/z 485.2, found 371.3. Preparation of 5-chloro-4-(3-isopropyl-1H-pyrazol-4-yl)-N-(piperidin-4-yl)pyrimidin-2-amine (C-13)
[0156] To a solution of B-11 (680.0 mg, 2.21 mmol) in NMP (10 mL) was added M1 (534.7 mg, 2.66 mmol) and DIEA (572.2 mg, 4.43 mmol). The reaction mixture was stirred at 80℃ for 6 h. The LCMS showed the desired MS was detected. The mixture was then quenched water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 60/40 in 20 mins) to afford Boc-C-11 (490.0 mg, yield: 41.3 %) as a yellow solid. LCMS (ESI): calced for C24H31ClN6O2 [M+H]+ m/z 471.2, found 471.5.
[0157] To a solution of Boc-C-11 (490.0 mg, 1.04 mmol) in TFA/DCM (2mL/10mL) stirred at 25℃ for 4 h. The LCMS showed the desired MS was detected. The solvent was removed by vacumm to afford C-10 (350.0 mg, yield: 75.5 %) as a gray solid. LCMS (ESI): calced for C19H23ClN6 [M+H]+ m/z 485.2, found 371.3. Preparation of 5-chloro-4-(3-isopropyl-1H-pyrazol-4-yl)-N-(piperidin-4-yl)pyrimidin-2-amine (C-13)
 [0158] To a solution of B-13 (390.0 mg, 1.0 mmol) in NMP (7 mL) was added M1 (303.2 mg, 1.5 mmol) and DIEA (259.5 mg, 2.0 mmol) stirred for 12 h at 80 ℃. The LCMS showed the desired MS was detected. Water was added to the solution and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 100/00 to 90/0 in 10 mins) to afford tert-butyl 4-((5-chloro-4-(3-isopropyl-1-((2- (trimethylsilyl)ethoxy)methyl)-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)piperidine-1-carboxylate (290.0 mg, yield: 52.1%) as yellow solid. LCMS (ESI): calced for C26H43ClN6O3Si [M+H]+ m/z 551.3, found 551.3. [0159] To a solution of tert-butyl 4-((5-chloro-4-(3-isopropyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazol-4- yl)pyrimidin-2-yl)amino)piperidine-1-carboxylate (290.0 mg, 0.5 mmol) in DCM/TFA=5:1 (6 mL) stirred for 12h at 25 ℃. The LCMS showed the desired MS was detected. The filtrate was adjusted to pH 7-8 with saturated sodium carbonate and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with DCM/MeOH from 100/00 to 85/15 in 15 mins) to afford C-13 (140.0 mg, yield: 83.2%) as yellow solid. LCMS (ESI): calced for C15H21ClN6 [M+H]+ m/z 321.2, found 321.0. Preparation of 5-chloro-4-phenyl-N-(piperidin-4-yl)pyrimidin-2-amine (C-16)
[0158] To a solution of B-13 (390.0 mg, 1.0 mmol) in NMP (7 mL) was added M1 (303.2 mg, 1.5 mmol) and DIEA (259.5 mg, 2.0 mmol) stirred for 12 h at 80 ℃. The LCMS showed the desired MS was detected. Water was added to the solution and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 100/00 to 90/0 in 10 mins) to afford tert-butyl 4-((5-chloro-4-(3-isopropyl-1-((2- (trimethylsilyl)ethoxy)methyl)-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)piperidine-1-carboxylate (290.0 mg, yield: 52.1%) as yellow solid. LCMS (ESI): calced for C26H43ClN6O3Si [M+H]+ m/z 551.3, found 551.3. [0159] To a solution of tert-butyl 4-((5-chloro-4-(3-isopropyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazol-4- yl)pyrimidin-2-yl)amino)piperidine-1-carboxylate (290.0 mg, 0.5 mmol) in DCM/TFA=5:1 (6 mL) stirred for 12h at 25 ℃. The LCMS showed the desired MS was detected. The filtrate was adjusted to pH 7-8 with saturated sodium carbonate and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with DCM/MeOH from 100/00 to 85/15 in 15 mins) to afford C-13 (140.0 mg, yield: 83.2%) as yellow solid. LCMS (ESI): calced for C15H21ClN6 [M+H]+ m/z 321.2, found 321.0. Preparation of 5-chloro-4-phenyl-N-(piperidin-4-yl)pyrimidin-2-amine (C-16)
 [0160] To a solution of B-16 (2.0 g, 0.0089 mol) in NMP (20 mL) were added M-1 (1.8 g, 0.0089 mol) and DIEA (2.3 g, 0.0178 mol). The reaction solution was stirred at 80 ℃ for 16 h. LCMS showed the desired MS was detected. The reaction mixture was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by silica gel
chromatography (eluting with PE/EtOAc from 100/0 to 90/10 in 30 mins) to afford Boc-C-16 (2.8 g, yield: 78.6% ) as a yellow solid. LCMS (ESI): calcd for C20H25ClN4O2 [M+H] + m/z 389.0, found 389.0. [0161] Boc-C-16 (2.6 g, 0.0067 mol) was added to DCM/TFA=3:1 (30 mL). The reaction was stirred at rt for 3 h. LCMS showed the desired MS was detected. Then the mixture was concentrated, quenched with water, adjusted to pH = 7 - 8 with NaHCO3, extracted with EtOAc, the combined organic phase was dried over anhydrous Na2SO4. The mixture was concentrated to get crude product C-16 (1.6 g, yield: 80.6%) as yellow solid. LCMS (ESI): calcd for C15H17ClN4 [M+H] + m/z 288.9, found 288.9. Preparation of tert-butyl (4-{[5-chloro-4-(pyridin-3-yl) pyrimidin-2-yl] amino} piperidin-1-yl) formate (C-17)
[0160] To a solution of B-16 (2.0 g, 0.0089 mol) in NMP (20 mL) were added M-1 (1.8 g, 0.0089 mol) and DIEA (2.3 g, 0.0178 mol). The reaction solution was stirred at 80 ℃ for 16 h. LCMS showed the desired MS was detected. The reaction mixture was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by silica gel
chromatography (eluting with PE/EtOAc from 100/0 to 90/10 in 30 mins) to afford Boc-C-16 (2.8 g, yield: 78.6% ) as a yellow solid. LCMS (ESI): calcd for C20H25ClN4O2 [M+H] + m/z 389.0, found 389.0. [0161] Boc-C-16 (2.6 g, 0.0067 mol) was added to DCM/TFA=3:1 (30 mL). The reaction was stirred at rt for 3 h. LCMS showed the desired MS was detected. Then the mixture was concentrated, quenched with water, adjusted to pH = 7 - 8 with NaHCO3, extracted with EtOAc, the combined organic phase was dried over anhydrous Na2SO4. The mixture was concentrated to get crude product C-16 (1.6 g, yield: 80.6%) as yellow solid. LCMS (ESI): calcd for C15H17ClN4 [M+H] + m/z 288.9, found 288.9. Preparation of tert-butyl (4-{[5-chloro-4-(pyridin-3-yl) pyrimidin-2-yl] amino} piperidin-1-yl) formate (C-17)
 [0162] To a solution of B-17 (360 mg, 1.59 mmol) in NMP (5 ml) was added DIEA (411.6 mg, 3.19 mmol) and M1 (384.6 mg, 1.91 mmol). The reaction mixture was stirred at 120℃ for 16 h under nitrogen. The resulting mixture was diluted with water (10 mL) and extracted with EtOAc (10 mL x 3). The combine organic phases were washed with brine, dried over sodium sulfate, concentrated to afford Boc-C-17 (500 mg, yield: 76.31 %) as a yellow oil which was used directly in next step without further purification. LCMS (ESI) calcd for C19H24ClN5O2 [M + H] + ms/z = 390.3, found 390.3. [0163] To a solution of Boc-C-17 (500.0 mg, 1.28 mmol) in HCl-EtOAc (4.0 M, 10 ml). The reaction mixture was stirred at 25℃ for 3 h. The precipitate was collected by filtration, washed with EtOAc to provide C-17 (360.0 mg, 92.3 % yield) as a yellow solid which was used directly in next step without further purification. LCMS (ESI) calcd for C14H16ClN5 [M + H] + ms/z 326.1, found 290.2. Preparation of 5-chloro-4-(2-methoxyphenyl)-N-(piperidin-4-yl)pyrimidin-2-amine (C-18)
[0162] To a solution of B-17 (360 mg, 1.59 mmol) in NMP (5 ml) was added DIEA (411.6 mg, 3.19 mmol) and M1 (384.6 mg, 1.91 mmol). The reaction mixture was stirred at 120℃ for 16 h under nitrogen. The resulting mixture was diluted with water (10 mL) and extracted with EtOAc (10 mL x 3). The combine organic phases were washed with brine, dried over sodium sulfate, concentrated to afford Boc-C-17 (500 mg, yield: 76.31 %) as a yellow oil which was used directly in next step without further purification. LCMS (ESI) calcd for C19H24ClN5O2 [M + H] + ms/z = 390.3, found 390.3. [0163] To a solution of Boc-C-17 (500.0 mg, 1.28 mmol) in HCl-EtOAc (4.0 M, 10 ml). The reaction mixture was stirred at 25℃ for 3 h. The precipitate was collected by filtration, washed with EtOAc to provide C-17 (360.0 mg, 92.3 % yield) as a yellow solid which was used directly in next step without further purification. LCMS (ESI) calcd for C14H16ClN5 [M + H] + ms/z 326.1, found 290.2. Preparation of 5-chloro-4-(2-methoxyphenyl)-N-(piperidin-4-yl)pyrimidin-2-amine (C-18)
 [0164] To a solution of B-18 (2.0 g, 0.0078 mol) in NMP (20 mL) were added M1 (1.6 g, 0.0078 mol) and N,N- Diisopropylethylamine (2.0 g, 0.0156 mol). The reaction solution was stirred at 80 ℃ for 16 h. LCMS showed the desired MS was detected. The reaction mixture was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by silica gel
chromatography (eluting with PE/EtOAc from 100/0 to 90/10 in 30 mins) to afford Boc-C-18 (2.8 g, yield: 83.1% ) as yellow solid. LCMS (ESI): calcd for C21H27ClN4O3 [M+H] + m/z 419.0, found 419.0 [0165] To a solution of Boc-C-18 (2.8 g, 0.0067 mol) in DCM/TFA = 3:1 (24 mL). The reaction was stirred at rt for 3 h. LCMS showed the desired MS was detected. The organic solvent was removed under reduced pressure to get crude product C-18 (1.8 g, yield: 82.1% ) as yellow solid. LCMS (ESI): calcd for C16H19ClN4O [M+H] + m/z 433.1, found 318.9. Preparation of 5-chloro-4-(3-methoxyphenyl)-N-(piperidin-4-yl)pyrimidin-2-amine (C-19)
[0164] To a solution of B-18 (2.0 g, 0.0078 mol) in NMP (20 mL) were added M1 (1.6 g, 0.0078 mol) and N,N- Diisopropylethylamine (2.0 g, 0.0156 mol). The reaction solution was stirred at 80 ℃ for 16 h. LCMS showed the desired MS was detected. The reaction mixture was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by silica gel
chromatography (eluting with PE/EtOAc from 100/0 to 90/10 in 30 mins) to afford Boc-C-18 (2.8 g, yield: 83.1% ) as yellow solid. LCMS (ESI): calcd for C21H27ClN4O3 [M+H] + m/z 419.0, found 419.0 [0165] To a solution of Boc-C-18 (2.8 g, 0.0067 mol) in DCM/TFA = 3:1 (24 mL). The reaction was stirred at rt for 3 h. LCMS showed the desired MS was detected. The organic solvent was removed under reduced pressure to get crude product C-18 (1.8 g, yield: 82.1% ) as yellow solid. LCMS (ESI): calcd for C16H19ClN4O [M+H] + m/z 433.1, found 318.9. Preparation of 5-chloro-4-(3-methoxyphenyl)-N-(piperidin-4-yl)pyrimidin-2-amine (C-19)
 [0166] To a solution of B-19 (1.50 g, 5.9 mmol) in NMP (20 mL) were added M-1 (1.6 g, 7.8 mmol) and N,N- Diisopropylethylamine (2.0 g, 15.6 mmol). The reaction solution was stirred at 80 ℃ for 16 h. LCMS showed the desired MS was detected. The reaction mixture was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by silica gel chromatography (eluting with PE/EtOAc from 100/0 to 90/10 in 30 mins) to afford Boc-C-19 (1.0 g, yield: 37.1%) as a yellow solid. LCMS (ESI): calcd for C21H28ClN4O3 [M+H]+ m/z 419.2, found 419.1. [0167] A solution of Boc-C-19 (1.0 g, 2.4 mmol) was added to DCM/TFA=3:1 (24 mL). The reaction was stirred at rt for 3 h. LCMS showed the desired MS was detected. Then the mixture was concentrated, quenched with water, adjusted to pH = 7 - 8 with NaHCO3, extracted with EtOAc, the combined organic phase was dried over anhydrous Na2SO4. The mixture was concentrated to get crude product C-19 (0.9 g, yield: 89.1% ) as a yellow solid. LCMS (ESI): calcd for C16H20ClN4O [M+H]+ m/z 319.1, found 319.2. Preparation of 5-chloro-4-(2-chlorophenyl)-N-(piperidin-4-yl)pyrimidin-2-amine (C-20)
[0166] To a solution of B-19 (1.50 g, 5.9 mmol) in NMP (20 mL) were added M-1 (1.6 g, 7.8 mmol) and N,N- Diisopropylethylamine (2.0 g, 15.6 mmol). The reaction solution was stirred at 80 ℃ for 16 h. LCMS showed the desired MS was detected. The reaction mixture was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by silica gel chromatography (eluting with PE/EtOAc from 100/0 to 90/10 in 30 mins) to afford Boc-C-19 (1.0 g, yield: 37.1%) as a yellow solid. LCMS (ESI): calcd for C21H28ClN4O3 [M+H]+ m/z 419.2, found 419.1. [0167] A solution of Boc-C-19 (1.0 g, 2.4 mmol) was added to DCM/TFA=3:1 (24 mL). The reaction was stirred at rt for 3 h. LCMS showed the desired MS was detected. Then the mixture was concentrated, quenched with water, adjusted to pH = 7 - 8 with NaHCO3, extracted with EtOAc, the combined organic phase was dried over anhydrous Na2SO4. The mixture was concentrated to get crude product C-19 (0.9 g, yield: 89.1% ) as a yellow solid. LCMS (ESI): calcd for C16H20ClN4O [M+H]+ m/z 319.1, found 319.2. Preparation of 5-chloro-4-(2-chlorophenyl)-N-(piperidin-4-yl)pyrimidin-2-amine (C-20)
 [0168] To a solution of B-21 (1.5 g, 5.8 mmol) in dry NMP (15 mL) was added DIEA (3.7 g, 28.9 mmol) and (4- aminopiperidin-1-yl) tert-butyl formate (2.3 g, 11.6 mmol). The resulting solution was stirred at 80 ℃ for 2 h. The LCMS showed the desired MS was detected. The mixture was quenched with water, extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by silica column chromatography (eluting with PE/EtOAc from 100/0 to 0/100 in 15 mins) to afford
Boc-C-20 (2.3 g, yield: 93.7%) as a white solid. LCMS (ESI): calced for C20H24Cl2N4O2 [M+H]+ m/z 423.1, found 423.2. [0169] To a solution of Boc-C-20 (2.3 g, 5.4 mmol) in HCl-dioxane (50 mL). The reaction was stirred for 2 h at 25℃. The LCMS showed the desired MS was detected. Then the mixture was concentrated, quenched with water, adjusted to pH = 7-8 with NaHCO3, extracted with EtOAc, the combined organic phase was dried over anhydrous Na2SO4. The mixture was concentrated to get crude product which was purified by silica column chromatography (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-20 (1.3 g, yield: 74.0%) as a white solid. LCMS (ESI): calced for C15H16Cl2N4 [M+H]+ m/z 323.1, found 322.8. Preparation of (R)-5-chloro-4-(1H-indol-3-yl)-N-(pyrrolidin-3-yl)pyrimidin-2-amine (C-21)
[0168] To a solution of B-21 (1.5 g, 5.8 mmol) in dry NMP (15 mL) was added DIEA (3.7 g, 28.9 mmol) and (4- aminopiperidin-1-yl) tert-butyl formate (2.3 g, 11.6 mmol). The resulting solution was stirred at 80 ℃ for 2 h. The LCMS showed the desired MS was detected. The mixture was quenched with water, extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by silica column chromatography (eluting with PE/EtOAc from 100/0 to 0/100 in 15 mins) to afford
Boc-C-20 (2.3 g, yield: 93.7%) as a white solid. LCMS (ESI): calced for C20H24Cl2N4O2 [M+H]+ m/z 423.1, found 423.2. [0169] To a solution of Boc-C-20 (2.3 g, 5.4 mmol) in HCl-dioxane (50 mL). The reaction was stirred for 2 h at 25℃. The LCMS showed the desired MS was detected. Then the mixture was concentrated, quenched with water, adjusted to pH = 7-8 with NaHCO3, extracted with EtOAc, the combined organic phase was dried over anhydrous Na2SO4. The mixture was concentrated to get crude product which was purified by silica column chromatography (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-20 (1.3 g, yield: 74.0%) as a white solid. LCMS (ESI): calced for C15H16Cl2N4 [M+H]+ m/z 323.1, found 322.8. Preparation of (R)-5-chloro-4-(1H-indol-3-yl)-N-(pyrrolidin-3-yl)pyrimidin-2-amine (C-21)
 [0170] A solution of B-1 (370.0 mg, 1.01 mmol), tert-butyl (3R)-3-aminopyrrolidine-1-carboxylate (378.0 mg, 2.03 mmol) and DIEA (263.0 mg, 2.03 mmol) in 8 mL NMP was heated at 80 ℃ for 16 h. The LCMS showed the desired MS was detected. The mixture was concentrated and purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 90/10 in 30 mins) to afford di-Boc-C-21 (400.0 mg, yield: 80.8%) as a yellow solid. LCMS (ESI) calcd for C26H32ClN5O4 [M+H-100]+ m/z 414.2, found 414.2. [0171] A solution of di-Boc-C-21 (400.0 mg, 0.77 mmol) in 10 mL HCl-1,4- dioxane was stirred at rt for 2 h. The LCMS showed the desired MS was detected. The residue was partitioned between EtOAc and saturated NaHCO3 solution. The separated organic layer was washed with water, dried over anhydrous Na2SO4 and concentrated under vacuum to afford C-21 (300.0 mg, yield: 90.8%) as a yellow solid. LCMS (ESI) calcd for C16H17ClN5 [M+H]+ m/z 314.1, found 314.1. Preparation of (S)-5-chloro-4-(1H-indol-3-yl)-N-(pyrrolidin-3-yl)pyrimidin-2-amine (C-22)
[0170] A solution of B-1 (370.0 mg, 1.01 mmol), tert-butyl (3R)-3-aminopyrrolidine-1-carboxylate (378.0 mg, 2.03 mmol) and DIEA (263.0 mg, 2.03 mmol) in 8 mL NMP was heated at 80 ℃ for 16 h. The LCMS showed the desired MS was detected. The mixture was concentrated and purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 90/10 in 30 mins) to afford di-Boc-C-21 (400.0 mg, yield: 80.8%) as a yellow solid. LCMS (ESI) calcd for C26H32ClN5O4 [M+H-100]+ m/z 414.2, found 414.2. [0171] A solution of di-Boc-C-21 (400.0 mg, 0.77 mmol) in 10 mL HCl-1,4- dioxane was stirred at rt for 2 h. The LCMS showed the desired MS was detected. The residue was partitioned between EtOAc and saturated NaHCO3 solution. The separated organic layer was washed with water, dried over anhydrous Na2SO4 and concentrated under vacuum to afford C-21 (300.0 mg, yield: 90.8%) as a yellow solid. LCMS (ESI) calcd for C16H17ClN5 [M+H]+ m/z 314.1, found 314.1. Preparation of (S)-5-chloro-4-(1H-indol-3-yl)-N-(pyrrolidin-3-yl)pyrimidin-2-amine (C-22)
 [0172] A solution of B-1 (200.0 mg, 0.55 mmol), tert-butyl (S)-3-aminopyrrolidine-1-carboxylate (153.7 mg, 0.83 mmol) and DIEA (141.9 mg, 1.1 mmol) in 4 mL NMP was heated at 80℃ for 16 h. The LCMS showed the desired MS was detected. The mixture was concentrated and purified by flash chromatography (eluting with
PE/EtOAc = 100/0 to 90/10 in 30 mins) to di-Boc-C-22 (120.0 mg, yield: 42.5%) as a yellow solid. LCMS (ESI): calcd for C26H32ClN5O4 [M+H]+ m/z 514.2, found 514.5. [0173] A solution of di-Boc-C-22 (120.0 mg, 0.23 mmol) in 5 mL HCl-1,4- dioxane was stirred at rt for 2 h. The LCMS showed the desired MS was detected. The residue was partitioned between EtOAc and saturated NaHCO3 solution. The separated organic layer was washed with water, dried over anhydrous Na2SO4 and concentrated under vacuum to afford C-22 (90.0 mg, yield: 94.7%) as a yellow solid. LCMS (ESI) calcd for C16H17ClN5 [M+H]+ m/z 314.1, found 314.0. Preparation of 5-chloro-4-(1H-indol-3-yl)-N-(piperidin-3-yl)pyrimidin-2-amine (C-23)
[0172] A solution of B-1 (200.0 mg, 0.55 mmol), tert-butyl (S)-3-aminopyrrolidine-1-carboxylate (153.7 mg, 0.83 mmol) and DIEA (141.9 mg, 1.1 mmol) in 4 mL NMP was heated at 80℃ for 16 h. The LCMS showed the desired MS was detected. The mixture was concentrated and purified by flash chromatography (eluting with
PE/EtOAc = 100/0 to 90/10 in 30 mins) to di-Boc-C-22 (120.0 mg, yield: 42.5%) as a yellow solid. LCMS (ESI): calcd for C26H32ClN5O4 [M+H]+ m/z 514.2, found 514.5. [0173] A solution of di-Boc-C-22 (120.0 mg, 0.23 mmol) in 5 mL HCl-1,4- dioxane was stirred at rt for 2 h. The LCMS showed the desired MS was detected. The residue was partitioned between EtOAc and saturated NaHCO3 solution. The separated organic layer was washed with water, dried over anhydrous Na2SO4 and concentrated under vacuum to afford C-22 (90.0 mg, yield: 94.7%) as a yellow solid. LCMS (ESI) calcd for C16H17ClN5 [M+H]+ m/z 314.1, found 314.0. Preparation of 5-chloro-4-(1H-indol-3-yl)-N-(piperidin-3-yl)pyrimidin-2-amine (C-23)
 [0174] To a solution of B-1 (150.0 mg, 0.41mmol) in dry NMP (5.0 mL) was added DIEA (265.4 mg, 2.05 mmol) and (3-aminopiperidin-1-yl) tert-butyl formate (90.9 mg, 0.45 mmol). The resulting solution was stirred at 80 ℃ for 2 h. The LCMS showed the desired MS was detected. The mixture was quenched with water, extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by silica column chromatography (eluting with PE/EtOAc from 100/0 to 0/100 in 15 mins) to afford di-Boc-C-23 (150.0 mg, yield: 68.9%) as a white solid. LCMS (ESI): calced for C27H34ClN5O4 [M+H]+ m/z 528.2, found 528.2. [0175] To a solution of di-Boc-C-23 (150.0 mg, 0.23 mmol) in TFA/DCM (5:1)(50 mL). The reaction was stirred for 2 h at 25℃. The LCMS showed the desired MS was detected. Then the mixture was concentrated, quenched with water, adjusted to pH = 7 - 8 with NaHCO3, extracted with EtOAc, the combined organic phase was dried over anhydrous Na2SO4. The mixture was concentrated to get crude product which was purified by silica column chromatography (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-23 (50.0 mg, yield: 53.7%) as a white solid. LCMS (ESI): calced for C15H16Cl2N4 [M+H]+ m/z 328.1, found 327.9.
Preparation of 4-(1H-indazol-3-yl)-2-(piperidin-4-ylamino) pyrimidine-5-carbonitrile (C-26)
[0174] To a solution of B-1 (150.0 mg, 0.41mmol) in dry NMP (5.0 mL) was added DIEA (265.4 mg, 2.05 mmol) and (3-aminopiperidin-1-yl) tert-butyl formate (90.9 mg, 0.45 mmol). The resulting solution was stirred at 80 ℃ for 2 h. The LCMS showed the desired MS was detected. The mixture was quenched with water, extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by silica column chromatography (eluting with PE/EtOAc from 100/0 to 0/100 in 15 mins) to afford di-Boc-C-23 (150.0 mg, yield: 68.9%) as a white solid. LCMS (ESI): calced for C27H34ClN5O4 [M+H]+ m/z 528.2, found 528.2. [0175] To a solution of di-Boc-C-23 (150.0 mg, 0.23 mmol) in TFA/DCM (5:1)(50 mL). The reaction was stirred for 2 h at 25℃. The LCMS showed the desired MS was detected. Then the mixture was concentrated, quenched with water, adjusted to pH = 7 - 8 with NaHCO3, extracted with EtOAc, the combined organic phase was dried over anhydrous Na2SO4. The mixture was concentrated to get crude product which was purified by silica column chromatography (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-23 (50.0 mg, yield: 53.7%) as a white solid. LCMS (ESI): calced for C15H16Cl2N4 [M+H]+ m/z 328.1, found 327.9.
Preparation of 4-(1H-indazol-3-yl)-2-(piperidin-4-ylamino) pyrimidine-5-carbonitrile (C-26)
 [0176] To a solution of B-22 (550.0 mg, 1.38 mmol) in DCM (10 mL) was added m-CPBA (320.0 mg, 0.72 mmol. The reaction mixture was stirred at rt. for 16 h under N2. The desired mass was detected on LC-MS. The mixture was quenched with water, extracted with DCM and washed with brine, dried over anhydrous Na2SO4. The solvent was removed under reduce pressure. The crude product (600.0 mg, yield: 90.9 %) was directly used in next step without further purification. LCMS (ESI): calced for C19H23N5O3SSi [M+H+22]+ m/z 452.0, found 452.1. [0177] To a solution of 2-methanesulfonyl-4-{1-[(2-methoxyethyl) trimethyl-$l^{5}-silyl] indazol-3-yl} pyrimidine- 5-carbonitrile (500.0 mg, 1.16 mmol) in i-PrOH (5 ml) was added M1 (233.7 mg, 1.16 mmol) and DIEA (300.1 mg, 2.32 mmol). The mixture was stirred at 90 ℃ for 2 h. The desired mass was detected on LC-MS. The mixture was quenched with water, extracted with EOAc, washed with brine, dried over anhydrous Na2SO4, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 98/2 to 90/10 in 10 mins) to afford tert-butyl {4-[(5-cyano-4-{1-[(2-methoxyethyl)trimethyl-$l^{5}-silyl]indazol-3- yl}pyrimidin-2-yl)amino]piperidin-1-yl} formate (300.0 mg, yield: 44.5%) as a yellow solid. LCMS (ESI): calced for C28H39N7O3Si [M+H]+ m/z 550.2, found 550.2. [0178] To a solution of tert-butyl {4-[(5-cyano-4-{1-[(2-methoxyethyl) trimethyl-$l^{5}-silyl] indazol-3-yl} pyrimidin-2-yl) amino] piperidin-1-yl} formate (450.0 mg, 0.82 mmol) in DMF (10 mL) was added CsF (621.4 mg, 4.09 mmol). The reaction mixture was stirred at 100 oC for 16 h. The desired mass was detected on LC-MS. The mixture was quenched with water, extracted with EtOAc and concentrated to get the crude product which was added HCl-EtOAc (2 M, 10 ml). The mixture was stirred at rt. for 2 h. The desired mass was detected on LC-MS. The precipitate was collected by filtration, washed with EtOAc to afford C-26 (200.0 mg, yield: 76.6 %) as a yellow solid which was used directly in next step without further purification. LCMS (ESI): calced for C17H17N7 [M+H]+ m/z 320.1, found 320.2.
Preparation of 5-chloro-4-(1H-indol-3-yl)-N-(piperidin-3-yl)pyrimidin-2-amine (C-27)
[0176] To a solution of B-22 (550.0 mg, 1.38 mmol) in DCM (10 mL) was added m-CPBA (320.0 mg, 0.72 mmol. The reaction mixture was stirred at rt. for 16 h under N2. The desired mass was detected on LC-MS. The mixture was quenched with water, extracted with DCM and washed with brine, dried over anhydrous Na2SO4. The solvent was removed under reduce pressure. The crude product (600.0 mg, yield: 90.9 %) was directly used in next step without further purification. LCMS (ESI): calced for C19H23N5O3SSi [M+H+22]+ m/z 452.0, found 452.1. [0177] To a solution of 2-methanesulfonyl-4-{1-[(2-methoxyethyl) trimethyl-$l^{5}-silyl] indazol-3-yl} pyrimidine- 5-carbonitrile (500.0 mg, 1.16 mmol) in i-PrOH (5 ml) was added M1 (233.7 mg, 1.16 mmol) and DIEA (300.1 mg, 2.32 mmol). The mixture was stirred at 90 ℃ for 2 h. The desired mass was detected on LC-MS. The mixture was quenched with water, extracted with EOAc, washed with brine, dried over anhydrous Na2SO4, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 98/2 to 90/10 in 10 mins) to afford tert-butyl {4-[(5-cyano-4-{1-[(2-methoxyethyl)trimethyl-$l^{5}-silyl]indazol-3- yl}pyrimidin-2-yl)amino]piperidin-1-yl} formate (300.0 mg, yield: 44.5%) as a yellow solid. LCMS (ESI): calced for C28H39N7O3Si [M+H]+ m/z 550.2, found 550.2. [0178] To a solution of tert-butyl {4-[(5-cyano-4-{1-[(2-methoxyethyl) trimethyl-$l^{5}-silyl] indazol-3-yl} pyrimidin-2-yl) amino] piperidin-1-yl} formate (450.0 mg, 0.82 mmol) in DMF (10 mL) was added CsF (621.4 mg, 4.09 mmol). The reaction mixture was stirred at 100 oC for 16 h. The desired mass was detected on LC-MS. The mixture was quenched with water, extracted with EtOAc and concentrated to get the crude product which was added HCl-EtOAc (2 M, 10 ml). The mixture was stirred at rt. for 2 h. The desired mass was detected on LC-MS. The precipitate was collected by filtration, washed with EtOAc to afford C-26 (200.0 mg, yield: 76.6 %) as a yellow solid which was used directly in next step without further purification. LCMS (ESI): calced for C17H17N7 [M+H]+ m/z 320.1, found 320.2.
Preparation of 5-chloro-4-(1H-indol-3-yl)-N-(piperidin-3-yl)pyrimidin-2-amine (C-27)
 [0179] To a solution of B-1 (150.0 mg, 0.41mmol) in dry NMP (5.0 mL) was added DIEA (265.4 mg, 2.05 mmol) and (3-aminopiperidin-1-yl) tert-butyl formate (90.9 mg, 0.45 mmol). The resulting solution was stirred at 80 ℃ for 2 h. The LCMS showed the desired MS was detected. The mixture was quenched with water, extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by silica column chromatography (eluting with PE/EtOAc from 100/0 to 0/100 in 15 mins) to afford di-Boc-C-27 (150.0 mg, yield: 68.89%) as a white solid. LCMS (ESI): calced for C27H34ClN5O4 [M+H]+ m/z 528.2, found 528.2. [0180] To a solution of di-Boc-C-27 (150.0 mg, 0.23 mmol) in TFA/DCM=5/1(50 mL). The reaction was stirred for 2 h at 25℃. The LCMS showed the desired MS was detected. Then the mixture was concentrated, quenched with water, adjusted to pH = 7 - 8 with NaHCO3, extracted with EtOAc, the combined organic phase was dried over anhydrous Na2SO4. The mixture was concentrated to get crude productwhich was purified by silica column chromatography (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-27 (50.0 mg, yield: 53.7%) as a white solid. LCMS (ESI): calced for C15H16Cl2N4 [M+H]+ m/z 328.1, found 327.9. Preparation of 5-chloro-4-(5-fluoro-1H-indazol-3-yl)-N-(piperidin-4-yl)pyrimidin-2-amine (C-28)
[0179] To a solution of B-1 (150.0 mg, 0.41mmol) in dry NMP (5.0 mL) was added DIEA (265.4 mg, 2.05 mmol) and (3-aminopiperidin-1-yl) tert-butyl formate (90.9 mg, 0.45 mmol). The resulting solution was stirred at 80 ℃ for 2 h. The LCMS showed the desired MS was detected. The mixture was quenched with water, extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by silica column chromatography (eluting with PE/EtOAc from 100/0 to 0/100 in 15 mins) to afford di-Boc-C-27 (150.0 mg, yield: 68.89%) as a white solid. LCMS (ESI): calced for C27H34ClN5O4 [M+H]+ m/z 528.2, found 528.2. [0180] To a solution of di-Boc-C-27 (150.0 mg, 0.23 mmol) in TFA/DCM=5/1(50 mL). The reaction was stirred for 2 h at 25℃. The LCMS showed the desired MS was detected. Then the mixture was concentrated, quenched with water, adjusted to pH = 7 - 8 with NaHCO3, extracted with EtOAc, the combined organic phase was dried over anhydrous Na2SO4. The mixture was concentrated to get crude productwhich was purified by silica column chromatography (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-27 (50.0 mg, yield: 53.7%) as a white solid. LCMS (ESI): calced for C15H16Cl2N4 [M+H]+ m/z 328.1, found 327.9. Preparation of 5-chloro-4-(5-fluoro-1H-indazol-3-yl)-N-(piperidin-4-yl)pyrimidin-2-amine (C-28)
 [0181] To a solution of B-23 (1.2 g,2.6 mmol) in 1,4-dioxane (20 mL) was added tert-butyl 4-aminopiperidine- 1-carboxylate (1.5 g, 7.8 mmol), Pd2(dba)3 (241.5 mg, 0.2 mmol) and Xantphos (150.4 mg, 0.2 mmol), then the mixture is stirred for 5 h at 100°C under N2. The desired mass was detected on LC-MS. The mixture was washed with water (100 mL) and extracted with EtOAc (3 x 50 mL), the organic layer was washed with brine, dried over Na2SO4, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 95/5 to 45/55 in 14 mins) to afford tert-butyl 4-((5-chloro-4-(5-fluoro-1-((2- (trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyrimidin-2-yl)amino)piperidine-1-carboxylate (723.5 mg, yield: 47.8 %) as a yellow solid. LCMS (ESI): calcd for C27H38ClFN6O3Si [M+H]+ m/z 577.2, found 577.2.
[0182] To a solution of tert-butyl 4-((5-chloro-4-(5-fluoro-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3- yl)pyridin-2-yl)amino)piperidine-1-carboxylate (723.5 mg, 1.2 mmol) in DCM (10 mL) and TFA (10 mL), then the mixture is stirred for 12 h at 25°C. The desired mass was detected on LC-MS. The mixture was washed with sodium carbonate saturated solution (20 mL) and extracted with DCM (3 x 10 mL), the organic layer was washed with brine, dried over Na2SO4, filtered out and concentrated to get C-28 (150.0 mg, yield: 36.1 %) as a yellow solid. LCMS (ESI): calcd for C16H16ClFN6 [M-H]- m/z 345.1, found 344.9. Preparation of tert-butyl 5-chloro-4-(5,7-difluoro-1H-indazol-3-yl)-N-(piperidin-4-yl)pyrimidin-2-amine (C-29)
[0181] To a solution of B-23 (1.2 g,2.6 mmol) in 1,4-dioxane (20 mL) was added tert-butyl 4-aminopiperidine- 1-carboxylate (1.5 g, 7.8 mmol), Pd2(dba)3 (241.5 mg, 0.2 mmol) and Xantphos (150.4 mg, 0.2 mmol), then the mixture is stirred for 5 h at 100°C under N2. The desired mass was detected on LC-MS. The mixture was washed with water (100 mL) and extracted with EtOAc (3 x 50 mL), the organic layer was washed with brine, dried over Na2SO4, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 95/5 to 45/55 in 14 mins) to afford tert-butyl 4-((5-chloro-4-(5-fluoro-1-((2- (trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyrimidin-2-yl)amino)piperidine-1-carboxylate (723.5 mg, yield: 47.8 %) as a yellow solid. LCMS (ESI): calcd for C27H38ClFN6O3Si [M+H]+ m/z 577.2, found 577.2.
[0182] To a solution of tert-butyl 4-((5-chloro-4-(5-fluoro-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3- yl)pyridin-2-yl)amino)piperidine-1-carboxylate (723.5 mg, 1.2 mmol) in DCM (10 mL) and TFA (10 mL), then the mixture is stirred for 12 h at 25°C. The desired mass was detected on LC-MS. The mixture was washed with sodium carbonate saturated solution (20 mL) and extracted with DCM (3 x 10 mL), the organic layer was washed with brine, dried over Na2SO4, filtered out and concentrated to get C-28 (150.0 mg, yield: 36.1 %) as a yellow solid. LCMS (ESI): calcd for C16H16ClFN6 [M-H]- m/z 345.1, found 344.9. Preparation of tert-butyl 5-chloro-4-(5,7-difluoro-1H-indazol-3-yl)-N-(piperidin-4-yl)pyrimidin-2-amine (C-29)
 [0183] A solution of B-24 (0.8 g, 2.0 mmol) and M1 (0.8 g, 4.0 mmol) in NMP (5 mL) was stirred at 80 ℃ for 2 h. The LCMS showed the desired MS was detected. After cooling, the mixture was concentrated and purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 90/10 in 30 mins) to afford tert-butyl 4-((5-chloro-4- (5,7-difluoro-1-(tetrahydro-2H-pyran-2-yl)-1H-indazol-3-yl)pyrimidin-2-yl)amino)piperidine-1-carboxylate (0.6 g, yield: 54.7%) as a yellow solid. LCMS (ESI) calcd for C26H32ClF2N6O3 [M + H] + ms/z 549.2, found 549.2. [0184] A solution of tert-butyl 4-((5-chloro-4-(5,7-difluoro-1-(tetrahydro-2H-pyran-2-yl)-1H-indazol-3- yl)pyrimidin-2-yl)amino)piperidine-1-carboxylate (0.6 g, 1.1 mmol) in 20 mL HCl-1,4-dioxane was stirred at rt for 4 h. The LCMS showed the desired MS was detected. The residue was partitioned between EtOAc and 1M NaOH. The separated organic layer was washed with water, dried over anhydrous Na2SO4 and concentrated under vacuum to afford C-29 (0.20 g, yield: 49.9%). LCMS (ESI) calcd for C16H15ClF2N6 [M + H] + ms/z 365.1, found 365.1. Preparation of 4-(1H-indazol-3-yl)-5-methyl-N-(piperidin-4-yl)pyrimidin-2-amine (C-30)
[0183] A solution of B-24 (0.8 g, 2.0 mmol) and M1 (0.8 g, 4.0 mmol) in NMP (5 mL) was stirred at 80 ℃ for 2 h. The LCMS showed the desired MS was detected. After cooling, the mixture was concentrated and purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 90/10 in 30 mins) to afford tert-butyl 4-((5-chloro-4- (5,7-difluoro-1-(tetrahydro-2H-pyran-2-yl)-1H-indazol-3-yl)pyrimidin-2-yl)amino)piperidine-1-carboxylate (0.6 g, yield: 54.7%) as a yellow solid. LCMS (ESI) calcd for C26H32ClF2N6O3 [M + H] + ms/z 549.2, found 549.2. [0184] A solution of tert-butyl 4-((5-chloro-4-(5,7-difluoro-1-(tetrahydro-2H-pyran-2-yl)-1H-indazol-3- yl)pyrimidin-2-yl)amino)piperidine-1-carboxylate (0.6 g, 1.1 mmol) in 20 mL HCl-1,4-dioxane was stirred at rt for 4 h. The LCMS showed the desired MS was detected. The residue was partitioned between EtOAc and 1M NaOH. The separated organic layer was washed with water, dried over anhydrous Na2SO4 and concentrated under vacuum to afford C-29 (0.20 g, yield: 49.9%). LCMS (ESI) calcd for C16H15ClF2N6 [M + H] + ms/z 365.1, found 365.1. Preparation of 4-(1H-indazol-3-yl)-5-methyl-N-(piperidin-4-yl)pyrimidin-2-amine (C-30)
 [0185] To a solution of B-28 (1.0 g, 0.0026 mmol), M1 (1.1 g, 0.0053 mol) and DIEA (1.7 g, 0.0133 mol) in NMP (15.0 ml). The reaction mixture was stirred at 120 ℃ for 16 h. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash
chromatography (eluting with PE/EtOAc from 100/0 to 85/15 in 15 mins) to afford tert-butyl 4-((5-methyl-4-(1-((2- (trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyrimidin-2-yl)amino)piperidine-1-carboxylate (650.0 mg, yield: 45.2%) as a white solid. LCMS (ESI): calced for C28H42N6O3Si [M+H]+ m/z 539.3, found 539.3. [0186] To a solution of tert-butyl 4-((5-methyl-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyrimidin- 2-yl)amino)piperidine-1-carboxylate (650.0 mg, 0.0012 mol) in DCM/TFA= 5:1 (10.0 ml) . The reaction mixture was stirred at 26 ℃ for 16 h. The LCMS showed the desired MS was detected. The reaction mixture was concentrated, adjusted to 7-8 with Na2CO3 (aq), extracted with EtOAc and concentrated. The residue was purified by flash chromatography (eluting with DCM/MeOH from 100/0 to 90/10 in 15 mins) to afford C-30 (260.0 mg, yield: 70.3%) as a yellow oil. LCMS (ESI): calced for C17H20N6 [M+H]+ m/z 309.1, found 309.1. Preparation of 5-ethyl-4-(1H-indazol-3-yl)-N-(piperidin-4-yl)pyrimidin-2-amine (C-31)
[0185] To a solution of B-28 (1.0 g, 0.0026 mmol), M1 (1.1 g, 0.0053 mol) and DIEA (1.7 g, 0.0133 mol) in NMP (15.0 ml). The reaction mixture was stirred at 120 ℃ for 16 h. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash
chromatography (eluting with PE/EtOAc from 100/0 to 85/15 in 15 mins) to afford tert-butyl 4-((5-methyl-4-(1-((2- (trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyrimidin-2-yl)amino)piperidine-1-carboxylate (650.0 mg, yield: 45.2%) as a white solid. LCMS (ESI): calced for C28H42N6O3Si [M+H]+ m/z 539.3, found 539.3. [0186] To a solution of tert-butyl 4-((5-methyl-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyrimidin- 2-yl)amino)piperidine-1-carboxylate (650.0 mg, 0.0012 mol) in DCM/TFA= 5:1 (10.0 ml) . The reaction mixture was stirred at 26 ℃ for 16 h. The LCMS showed the desired MS was detected. The reaction mixture was concentrated, adjusted to 7-8 with Na2CO3 (aq), extracted with EtOAc and concentrated. The residue was purified by flash chromatography (eluting with DCM/MeOH from 100/0 to 90/10 in 15 mins) to afford C-30 (260.0 mg, yield: 70.3%) as a yellow oil. LCMS (ESI): calced for C17H20N6 [M+H]+ m/z 309.1, found 309.1. Preparation of 5-ethyl-4-(1H-indazol-3-yl)-N-(piperidin-4-yl)pyrimidin-2-amine (C-31)
 [0187] To a solution of B-27 (330.0 mg, 0.85 mmol) in NMP (10 mL) was added M1 (339.8 mg, 1.7 mmol) and DIEA (219.7 mg, 1.7 mmol). The reaction mixture was stirred under nitrogen at 120 ℃ for 24 h. The LCMS showed the desired MS was detected. The mixture was quenched with water and extracted with EtOAc, the organic layers were combined and washed with brine, dried over Na2SO4, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 95/5 to 90/10 in 20 mins) to afford tert-butyl 4-((5-ethyl-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyrimidin-2-yl)amino)piperidine- 1-carboxylate (340.0 mg, yield: 72.5 %) as a yellow solid. LCMS (ESI): calced for C22H27ClN6O2 [M+H]+ m/z 553.3, found 553.6. [0188] To a solution of tert-butyl 4-((5-ethyl-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyrimidin-2- yl)amino)piperidine-1-carboxylate (340.0 mg, 0.62 mmol) in DCM (5 mL) was added TFA (1.0 mL). The reaction mixture was stirred under nitrogen at rt for 16 h. The mixture was treated with saturated aq. NaHCO3, adjusted pH to 7~8 and extracted with DCM. The organic layers were combined and washed with brine, dried over Na2SO4, filtered out and concentrated to get the residue (290.0 mg). To a solution of the residue in THF/H2O=1:1 (5 mL) was added NaOH (48.0 mg, 1.2 mmol). The mixture was stirred at rt for 1 h. The LCMS showed the desired MS was detected. The mixture was adjusted to pH=7~8 with 1M aq. HCl and extracted with EtOAc. The organic layers were combined and washed with brine, dried over Na2SO4, filtered out and concentrated to obtain the crude product C-31 (270.0 mg, yield: 61.7%) which was used for next step without further purification.
Preparation of 4-(1H-indazol-3-yl)-N-(piperidin-4-yl)-5-(trifluoromethyl)pyrimidin-2-amine (C-32)
[0187] To a solution of B-27 (330.0 mg, 0.85 mmol) in NMP (10 mL) was added M1 (339.8 mg, 1.7 mmol) and DIEA (219.7 mg, 1.7 mmol). The reaction mixture was stirred under nitrogen at 120 ℃ for 24 h. The LCMS showed the desired MS was detected. The mixture was quenched with water and extracted with EtOAc, the organic layers were combined and washed with brine, dried over Na2SO4, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 95/5 to 90/10 in 20 mins) to afford tert-butyl 4-((5-ethyl-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyrimidin-2-yl)amino)piperidine- 1-carboxylate (340.0 mg, yield: 72.5 %) as a yellow solid. LCMS (ESI): calced for C22H27ClN6O2 [M+H]+ m/z 553.3, found 553.6. [0188] To a solution of tert-butyl 4-((5-ethyl-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyrimidin-2- yl)amino)piperidine-1-carboxylate (340.0 mg, 0.62 mmol) in DCM (5 mL) was added TFA (1.0 mL). The reaction mixture was stirred under nitrogen at rt for 16 h. The mixture was treated with saturated aq. NaHCO3, adjusted pH to 7~8 and extracted with DCM. The organic layers were combined and washed with brine, dried over Na2SO4, filtered out and concentrated to get the residue (290.0 mg). To a solution of the residue in THF/H2O=1:1 (5 mL) was added NaOH (48.0 mg, 1.2 mmol). The mixture was stirred at rt for 1 h. The LCMS showed the desired MS was detected. The mixture was adjusted to pH=7~8 with 1M aq. HCl and extracted with EtOAc. The organic layers were combined and washed with brine, dried over Na2SO4, filtered out and concentrated to obtain the crude product C-31 (270.0 mg, yield: 61.7%) which was used for next step without further purification.
Preparation of 4-(1H-indazol-3-yl)-N-(piperidin-4-yl)-5-(trifluoromethyl)pyrimidin-2-amine (C-32)
 [0189] To a solution of B-26 (1.4 g, 0.003 mol), M1 (980.0 mg, 0.0049 mol) and DIEA (2.1 g, 0.0162 mol) in DMSO (20.0 ml). The reaction mixture was stirred red at 100 ℃ for 12 h. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 85/15 in 15 mins) to afford tert-butyl 4-((5-(trifluoromethyl)- 4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyrimidin-2-yl)amino)piperidine-1-carboxylate (1.5 g, yield: 77.5%) as a white solid. LCMS (ESI): calced for C28H39F3N6O3Si [M+H]+ m/z 593.3, found 593.3. [0190] To a solution of tert-butyl 4-((5-(trifluoromethyl)-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3- yl)pyrimidin-2-yl)amino)piperidine-1-carboxylate (1.5 g, 0.0042 mol) in DCM/TFA =3:1 (20.0 ml). The reaction mixture was stirred at 26 ℃ for 16 h. The LCMS showed the desired MS was detected. The reaction mixture was concentrated, adjusted to 7-8 with Na2CO3 (aq), extracted with EtOAc and concentrated. The residue was purified by flash chromatography (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-32 (610.0 mg, yield: 66.7%) as a yellow oil. LCMS (ESI): calced for C17H17F3N6 [M+H]+ m/z 363.1, found 363.1. Preparation of 5-chloro-4-(1-methyl-1H-indazol-3-yl)-N-(piperidin-4-yl)pyrimidin-2-amine hydrochloride (C-33)
[0189] To a solution of B-26 (1.4 g, 0.003 mol), M1 (980.0 mg, 0.0049 mol) and DIEA (2.1 g, 0.0162 mol) in DMSO (20.0 ml). The reaction mixture was stirred red at 100 ℃ for 12 h. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 85/15 in 15 mins) to afford tert-butyl 4-((5-(trifluoromethyl)- 4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyrimidin-2-yl)amino)piperidine-1-carboxylate (1.5 g, yield: 77.5%) as a white solid. LCMS (ESI): calced for C28H39F3N6O3Si [M+H]+ m/z 593.3, found 593.3. [0190] To a solution of tert-butyl 4-((5-(trifluoromethyl)-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3- yl)pyrimidin-2-yl)amino)piperidine-1-carboxylate (1.5 g, 0.0042 mol) in DCM/TFA =3:1 (20.0 ml). The reaction mixture was stirred at 26 ℃ for 16 h. The LCMS showed the desired MS was detected. The reaction mixture was concentrated, adjusted to 7-8 with Na2CO3 (aq), extracted with EtOAc and concentrated. The residue was purified by flash chromatography (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-32 (610.0 mg, yield: 66.7%) as a yellow oil. LCMS (ESI): calced for C17H17F3N6 [M+H]+ m/z 363.1, found 363.1. Preparation of 5-chloro-4-(1-methyl-1H-indazol-3-yl)-N-(piperidin-4-yl)pyrimidin-2-amine hydrochloride (C-33)
 [0191] To a solution of B-25 (800.0 mg, 2.9 mmol) in NMP (10 mL) was added M1 (861.1 mg, 4.3 mmol) and DIEA (740.8 mg, 5.7 mmol). The reaction mixture was stirred under N2 at 90 ℃ for 16 h. The LCMS showed the desired MS was detected. The mixture was quenched with water and extracted with EtOAc, the organic layers were combined and washed with brine, dried over Na2SO4, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 95/5 to 90/10 in 20 mins) to afford Boc-C-33 (500.0 mg, yield: 39.4 %) as a yellow solid. LCMS (ESI): calced for C22H27ClN6O2 [M+H]+ m/z 443.2, found 443.4. [0192] Boc-C-33 (500.0 mg, 1.1 mmol) was dissolved in a solution of 1M HCl in EtOAc (10 mL) and stirred at 25 ℃ for 16 h. The LCMS showed the desired MS was detected. The mixture was filtered. The filter cake was
washed with EtOAc and dried on vacuum to afford C-33 (700.0 mg, yield: 90.0%) as a yellow solid. LCMS (ESI): calced for C17H19ClN6 [M+H]+ m/z 343.1, found 343.0. Preparation of (1R,5S,6s)-N-(5-chloro-4-(1H-indazol-3-yl)pyrimidin-2-yl)-3-azabicyclo[3.1.0]hexan-6-amine (C-34)
[0191] To a solution of B-25 (800.0 mg, 2.9 mmol) in NMP (10 mL) was added M1 (861.1 mg, 4.3 mmol) and DIEA (740.8 mg, 5.7 mmol). The reaction mixture was stirred under N2 at 90 ℃ for 16 h. The LCMS showed the desired MS was detected. The mixture was quenched with water and extracted with EtOAc, the organic layers were combined and washed with brine, dried over Na2SO4, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 95/5 to 90/10 in 20 mins) to afford Boc-C-33 (500.0 mg, yield: 39.4 %) as a yellow solid. LCMS (ESI): calced for C22H27ClN6O2 [M+H]+ m/z 443.2, found 443.4. [0192] Boc-C-33 (500.0 mg, 1.1 mmol) was dissolved in a solution of 1M HCl in EtOAc (10 mL) and stirred at 25 ℃ for 16 h. The LCMS showed the desired MS was detected. The mixture was filtered. The filter cake was
washed with EtOAc and dried on vacuum to afford C-33 (700.0 mg, yield: 90.0%) as a yellow solid. LCMS (ESI): calced for C17H19ClN6 [M+H]+ m/z 343.1, found 343.0. Preparation of (1R,5S,6s)-N-(5-chloro-4-(1H-indazol-3-yl)pyrimidin-2-yl)-3-azabicyclo[3.1.0]hexan-6-amine (C-34)
 [0193] To a solution of B-3 (1.5 g, 0.0038 mol), tert-butyl (1R,5S,6s)-6-amino-3-azabicyclo[3.1.0]hexane-3- carboxylate (0.9 g, 0.0045 mol) and DIEA (2.4 g, 0.0189 mol) in DMSO(20 ml). The reaction mixture was stirred at 100 ℃ for 12 h. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 85/15 in 15 mins) to afford tert-butyl (1R,5S,6s)-6-((5-chloro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyrimidin- 2-yl)amino)-3-azabicyclo[3.1.0]hexane-3-carboxylate (1.3 g, yield: 80.9%) as a white solid. LCMS (ESI): calced for C27H37ClN6O3Si [M+H]+ m/z 557.2, found 557.2. [0194] To a solution of tert-butyl (1R,5S,6s)-6-((5-chloro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3- yl)pyrimidin-2-yl)amino)-3-azabicyclo[3.1.0]hexane-3-carboxylate (1.3 g, 0.0023 mol) in DCM/TFA(15 ml/ 3 ml) . The reaction mixture was stirred at 26 ℃ for 16 h. The LCMS showed the desired MS was detected. The reaction mixture was concentrated, adjusted to 7-8 with Na2CO3 (aq), extracted with EtOAc and concentrated. The residue was purified by flash chromatography (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-34 (650.0 mg, yield: 84.7%) as a yellow oil. LCMS (ESI): calced for C16H15ClN6 [M+H]+ m/z 327.0, found 327.0. Preparation of 5-chloro-4-(1Hindol-3-yl)-N-(piperidin-4-yl)pyridin-2-amine(C-36)
[0193] To a solution of B-3 (1.5 g, 0.0038 mol), tert-butyl (1R,5S,6s)-6-amino-3-azabicyclo[3.1.0]hexane-3- carboxylate (0.9 g, 0.0045 mol) and DIEA (2.4 g, 0.0189 mol) in DMSO(20 ml). The reaction mixture was stirred at 100 ℃ for 12 h. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 85/15 in 15 mins) to afford tert-butyl (1R,5S,6s)-6-((5-chloro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyrimidin- 2-yl)amino)-3-azabicyclo[3.1.0]hexane-3-carboxylate (1.3 g, yield: 80.9%) as a white solid. LCMS (ESI): calced for C27H37ClN6O3Si [M+H]+ m/z 557.2, found 557.2. [0194] To a solution of tert-butyl (1R,5S,6s)-6-((5-chloro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3- yl)pyrimidin-2-yl)amino)-3-azabicyclo[3.1.0]hexane-3-carboxylate (1.3 g, 0.0023 mol) in DCM/TFA(15 ml/ 3 ml) . The reaction mixture was stirred at 26 ℃ for 16 h. The LCMS showed the desired MS was detected. The reaction mixture was concentrated, adjusted to 7-8 with Na2CO3 (aq), extracted with EtOAc and concentrated. The residue was purified by flash chromatography (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-34 (650.0 mg, yield: 84.7%) as a yellow oil. LCMS (ESI): calced for C16H15ClN6 [M+H]+ m/z 327.0, found 327.0. Preparation of 5-chloro-4-(1Hindol-3-yl)-N-(piperidin-4-yl)pyridin-2-amine(C-36)
 [0195] To a solution of B-38 (3.4 g, 8.37 mmol) in DMF (40 mL) was added M1 (3.3 g, 16.74 mmol), Pd2(dba)3 (383.4 mg, 0.42 mmol), Xantphos (242.2 mg, 0.42 mmol), and Cs2CO3 (5.5 g, 16.74 mmol). The reaction was stirred at 100 ℃ for 12 h under N2. The desired mass was detected on LC-MS. Then the mixture was quenched with water, extracted with EtOAc, washed with brine, the combined organic phase were dried over anhydrous Na2SO4, filtered and concentrated to get crude product which was purified by column chromatograph on silica gel
(eluting with PE / EtOAc from 100/0 to 90/10 in 20 mins) to afford di-Boc-C-36 (1.6 g, yield: 36.4 %) as a white solid. LCMS (ESI): calcd for C28H36ClN4O4 [M+H] + ms/z 527.2, found 527.2. [0196] To a solution of di-Boc-C-36 (1.6 g, 3.04 mmol) in DCM (40 mL) was added was added TFA (10 mL). The reaction was stirred at 100 ℃ for 12 h under N2. The desired mass was detected on LC-MS. Then the mixture was concentrated, quenched with water, adjusted to PH = 7-8 with NaHCO3(aq.), extracted with EtOAc, the combined organic phase were dried over anhydrous Na2SO4, filtered and concentrated to get crude product which was purified by column chromatograph on silica gel (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-36 (640.0 mg, yield: 64.2 %) as a yellow oil. LCMS (ESI): calcd for C18H20ClN4 [M+H] + ms/z 327.1, found 327.1 Preparation of (S)-5-chloro-4-(1H-indol-3-yl)-N-(pyrrolidin-3-yl)pyridin-2-amine (C-38)
[0195] To a solution of B-38 (3.4 g, 8.37 mmol) in DMF (40 mL) was added M1 (3.3 g, 16.74 mmol), Pd2(dba)3 (383.4 mg, 0.42 mmol), Xantphos (242.2 mg, 0.42 mmol), and Cs2CO3 (5.5 g, 16.74 mmol). The reaction was stirred at 100 ℃ for 12 h under N2. The desired mass was detected on LC-MS. Then the mixture was quenched with water, extracted with EtOAc, washed with brine, the combined organic phase were dried over anhydrous Na2SO4, filtered and concentrated to get crude product which was purified by column chromatograph on silica gel
(eluting with PE / EtOAc from 100/0 to 90/10 in 20 mins) to afford di-Boc-C-36 (1.6 g, yield: 36.4 %) as a white solid. LCMS (ESI): calcd for C28H36ClN4O4 [M+H] + ms/z 527.2, found 527.2. [0196] To a solution of di-Boc-C-36 (1.6 g, 3.04 mmol) in DCM (40 mL) was added was added TFA (10 mL). The reaction was stirred at 100 ℃ for 12 h under N2. The desired mass was detected on LC-MS. Then the mixture was concentrated, quenched with water, adjusted to PH = 7-8 with NaHCO3(aq.), extracted with EtOAc, the combined organic phase were dried over anhydrous Na2SO4, filtered and concentrated to get crude product which was purified by column chromatograph on silica gel (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-36 (640.0 mg, yield: 64.2 %) as a yellow oil. LCMS (ESI): calcd for C18H20ClN4 [M+H] + ms/z 327.1, found 327.1 Preparation of (S)-5-chloro-4-(1H-indol-3-yl)-N-(pyrrolidin-3-yl)pyridin-2-amine (C-38)
 [0197] A solution of B-38 (400.0 mg, 0.98 mmol), (3S)-3-aminopyrrolidin-1-yl tert-butyl formate (274.0 mg, 1.47 mmol), Pd2(dba)3 (90.0 mg, 0.098 mmol), Xantphos (85.0 mg, 0.15 mmol) and Cs2CO3 (638.0 mg, 1.96 mmol) in 10 mL Dioxane was stirred at 80℃ for 2 h. The LCMS showed the desired MS was detected. The solution was quenched with H2O and then extracted with EtOAc (3 × 10 mL). The organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 90/10 in 20 mins) to get di-Boc-C-38 (80.0 mg, yield: 14.2%) as a yellow solid. LCMS (ESI) calcd for C27H34ClN4O4 [M+H]+ m/z 513.2, found 513.2. [0198] A solution of tert-butyl di-Boc-C-38 (80 mg, 0.15 mmol) in 3 mL HCl-1,4-dioxane was stirred at rt for 6 h. The LCMS showed the desired MS was detected. The residue was partitioned between EtOAc and saturated NaHCO3 solution. The separated organic layer was washed with water, dried over anhydrous Na2SO4 and concentrated under vacuum to afford C-38 (50.0 mg, yield: 72.8%). LCMS (ESI) calcd for C17H18ClN4 [M+H]+ m/z 313.1, found 313.0. Preparation of (R)-5-chloro-4-(1H-indol-3-yl)-N-(pyrrolidin-3-yl)pyridin-2-amine (C-39)
[0197] A solution of B-38 (400.0 mg, 0.98 mmol), (3S)-3-aminopyrrolidin-1-yl tert-butyl formate (274.0 mg, 1.47 mmol), Pd2(dba)3 (90.0 mg, 0.098 mmol), Xantphos (85.0 mg, 0.15 mmol) and Cs2CO3 (638.0 mg, 1.96 mmol) in 10 mL Dioxane was stirred at 80℃ for 2 h. The LCMS showed the desired MS was detected. The solution was quenched with H2O and then extracted with EtOAc (3 × 10 mL). The organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 90/10 in 20 mins) to get di-Boc-C-38 (80.0 mg, yield: 14.2%) as a yellow solid. LCMS (ESI) calcd for C27H34ClN4O4 [M+H]+ m/z 513.2, found 513.2. [0198] A solution of tert-butyl di-Boc-C-38 (80 mg, 0.15 mmol) in 3 mL HCl-1,4-dioxane was stirred at rt for 6 h. The LCMS showed the desired MS was detected. The residue was partitioned between EtOAc and saturated NaHCO3 solution. The separated organic layer was washed with water, dried over anhydrous Na2SO4 and concentrated under vacuum to afford C-38 (50.0 mg, yield: 72.8%). LCMS (ESI) calcd for C17H18ClN4 [M+H]+ m/z 313.1, found 313.0. Preparation of (R)-5-chloro-4-(1H-indol-3-yl)-N-(pyrrolidin-3-yl)pyridin-2-amine (C-39)
 [0199] A mixture of B-38 (300.0 mg, 0.74 mmol), tert-butyl (R)-3-aminopyrrolidine-1-carboxylate (165.2 mg, 0.888 mmol), Cs2CO3 (481.0 mg, 1.48 mmol), XantPhos (42.8 mg, 0.074 mmol) and Pd2(dba)3 (33.9 mg, 0.037 mmol) in Dioxane (10 mL) was stirred at 100°C for 16 h. The desired mass was detected on LC-MS. Then the mixture was quenched with water, extracted with EtOAc, washed with brine, the combined organic phase were dried over anhydrous Na2SO4, filtered and concentrated to get crude product which was purified by column chromatograph on silica gel (eluting with PE/EtOAc from 100/0 to 75/25 in 20 mins) to afford di-Boc-C-39 (100.0 mg, yield: 26.4%) as a yellow solid. LCMS (ESI): calced for C27H33ClN4O4 [M+H]+ m/z 513.2, found 513.3. [0200] To a solution of di-Boc-C-39 (100.0 mg, 0.043 mmol) in HCl-Dioxane (4.0 M, 2 mL) was stirred at rt for 16 h. The mixture was concentrated and purified by reverse flash chromatography (eluting with H2O(NH3.H2O)/MeCN from 80/20 to 50/50 in 30 mins) to afford C-39 (50.0 mg, yield: 80.1%) as a white solid. LCMS (ESI): calced for C17H17ClN4 [M+H]+ m/z 313.1, found 313.2. Preparation of 5-chloro-4-(1H-indol-3-yl)-N-(piperidin-3-yl)pyridin-2-amine (C-40)
[0199] A mixture of B-38 (300.0 mg, 0.74 mmol), tert-butyl (R)-3-aminopyrrolidine-1-carboxylate (165.2 mg, 0.888 mmol), Cs2CO3 (481.0 mg, 1.48 mmol), XantPhos (42.8 mg, 0.074 mmol) and Pd2(dba)3 (33.9 mg, 0.037 mmol) in Dioxane (10 mL) was stirred at 100°C for 16 h. The desired mass was detected on LC-MS. Then the mixture was quenched with water, extracted with EtOAc, washed with brine, the combined organic phase were dried over anhydrous Na2SO4, filtered and concentrated to get crude product which was purified by column chromatograph on silica gel (eluting with PE/EtOAc from 100/0 to 75/25 in 20 mins) to afford di-Boc-C-39 (100.0 mg, yield: 26.4%) as a yellow solid. LCMS (ESI): calced for C27H33ClN4O4 [M+H]+ m/z 513.2, found 513.3. [0200] To a solution of di-Boc-C-39 (100.0 mg, 0.043 mmol) in HCl-Dioxane (4.0 M, 2 mL) was stirred at rt for 16 h. The mixture was concentrated and purified by reverse flash chromatography (eluting with H2O(NH3.H2O)/MeCN from 80/20 to 50/50 in 30 mins) to afford C-39 (50.0 mg, yield: 80.1%) as a white solid. LCMS (ESI): calced for C17H17ClN4 [M+H]+ m/z 313.1, found 313.2. Preparation of 5-chloro-4-(1H-indol-3-yl)-N-(piperidin-3-yl)pyridin-2-amine (C-40)
 [0201] To a solution of B-38 (300.0 mg, 0.73 mmol) , (3-aminopiperidin-1-yl) tert-butyl formate (162.5 mg, 0.80 mmol), tris(dibenzylideneacetone)dipalladium (67.2 mg, 0.7 mmol), Caesium carbonateand (478.3 mg, 1.46 mmol), Xantphos (84.9 mg, 0.14 mmol) in 1,4-dioxane (10.0 ml) stirred under nitrogen at 25℃. The reaction mixture was stirred at 80 ℃ for 2 h. The LCMS showed the desired MS was detected. The reaction mixture was concentrated under reduced pressure to give which was purified by flash silica gel chromatography (eluting with PE/EtOAc from 100/0 to 95/5 in 20 mins) to afford di-Boc-C-40 (200.0 mg, yield: 51.4%) as a white solid. LCMS (ESI): calced for C28H35ClN4O4 [M+H]+ m/z 527.2, found 527.2. [0202] To a solution of di-Boc-C-40 (150.0 mg, 0.28 mmol) in TFA/DCM=5/1 (50 mL). The reaction was stirred for 2 h at 25℃. The LCMS showed the desired MS was detected. Then the mixture was concentrated, quenched with water, adjusted to pH = 7 - 8 with NaHCO3, extracted with EtOAc, the combined organic phase was dried over anhydrous Na2SO4. The mixture was concentrated to get crude product which was purified by silica column chromatography (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-40 (30.0 mg, yield: 32.2%) as a white solid. LCMS (ESI): calced for C18H19ClN4 [M+H]+ m/z 327.1, found 327.2.
Preparation of (1R,5S,6s)-N-(5-chloro-4-(1H-indazol-3-yl)pyridin-2-yl)-3-azabicyclo[3.1.0]hexan-6-amine (C-41)
[0201] To a solution of B-38 (300.0 mg, 0.73 mmol) , (3-aminopiperidin-1-yl) tert-butyl formate (162.5 mg, 0.80 mmol), tris(dibenzylideneacetone)dipalladium (67.2 mg, 0.7 mmol), Caesium carbonateand (478.3 mg, 1.46 mmol), Xantphos (84.9 mg, 0.14 mmol) in 1,4-dioxane (10.0 ml) stirred under nitrogen at 25℃. The reaction mixture was stirred at 80 ℃ for 2 h. The LCMS showed the desired MS was detected. The reaction mixture was concentrated under reduced pressure to give which was purified by flash silica gel chromatography (eluting with PE/EtOAc from 100/0 to 95/5 in 20 mins) to afford di-Boc-C-40 (200.0 mg, yield: 51.4%) as a white solid. LCMS (ESI): calced for C28H35ClN4O4 [M+H]+ m/z 527.2, found 527.2. [0202] To a solution of di-Boc-C-40 (150.0 mg, 0.28 mmol) in TFA/DCM=5/1 (50 mL). The reaction was stirred for 2 h at 25℃. The LCMS showed the desired MS was detected. Then the mixture was concentrated, quenched with water, adjusted to pH = 7 - 8 with NaHCO3, extracted with EtOAc, the combined organic phase was dried over anhydrous Na2SO4. The mixture was concentrated to get crude product which was purified by silica column chromatography (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-40 (30.0 mg, yield: 32.2%) as a white solid. LCMS (ESI): calced for C18H19ClN4 [M+H]+ m/z 327.1, found 327.2.
Preparation of (1R,5S,6s)-N-(5-chloro-4-(1H-indazol-3-yl)pyridin-2-yl)-3-azabicyclo[3.1.0]hexan-6-amine (C-41)
 [0203] To a solution of B-42 (1.0 g, 0.0023 mol) in DMF (15 mL) were added (1R,5S,6S)-6-amino-3- azabicyclo[3.1.0]hexan-3-yl tert-butyl formate (0.9 g, 0.0046 mol), Pd2(dba)3 (0.2 g, 0.0002 mol), Xantphos (0.3 g, 0.0004) and Cs2CO3 (1.5 g, 0.0046 mol). The reaction solution was stirred at 80 ℃ for 5 h. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 80/20 in 20 mins) to afford tert-butyl (1R,5S,6s)-6- ((5-chloro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyridin-2-yl)amino)-3-azabicyclo[3.1.0]hexane-3- carboxylate (800.0 g, yield: 60.8%) as a yellow oil. LCMS (ESI): calced for C28H38ClN5O3Si [M+H]+ m/z 556.2, found 556.2. [0204] tert-butyl (1R,5S,6s)-6-((5-chloro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyridin-2- yl)amino)-3-azabicyclo[3.1.0]hexane-3-carboxylate (800.0 mg, 1.4332 mmol) was added to DCM/TFA = 3:1 (10 mL) and stirred at rt for 12 h. The LCMS showed the desired MS was detected. The reaction mixture was concentrated, adjusted to 7-8 with Na2CO3 (aq), extracted with EtOAc and concentrated. The residue was purified by flash chromatography (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-41 (400.0 mg, yield: 83.9%) as a yellow oil. LCMS (ESI): calced for C17H16ClN5 [M+H]+ m/z 326.0, found 326.0. Preparation of 5-chloro-4-(5-fluoro-1H-indazol-3-yl)-N-(piperidin-4-yl)pyridin-2-amine (C-42)
[0203] To a solution of B-42 (1.0 g, 0.0023 mol) in DMF (15 mL) were added (1R,5S,6S)-6-amino-3- azabicyclo[3.1.0]hexan-3-yl tert-butyl formate (0.9 g, 0.0046 mol), Pd2(dba)3 (0.2 g, 0.0002 mol), Xantphos (0.3 g, 0.0004) and Cs2CO3 (1.5 g, 0.0046 mol). The reaction solution was stirred at 80 ℃ for 5 h. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 80/20 in 20 mins) to afford tert-butyl (1R,5S,6s)-6- ((5-chloro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyridin-2-yl)amino)-3-azabicyclo[3.1.0]hexane-3- carboxylate (800.0 g, yield: 60.8%) as a yellow oil. LCMS (ESI): calced for C28H38ClN5O3Si [M+H]+ m/z 556.2, found 556.2. [0204] tert-butyl (1R,5S,6s)-6-((5-chloro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyridin-2- yl)amino)-3-azabicyclo[3.1.0]hexane-3-carboxylate (800.0 mg, 1.4332 mmol) was added to DCM/TFA = 3:1 (10 mL) and stirred at rt for 12 h. The LCMS showed the desired MS was detected. The reaction mixture was concentrated, adjusted to 7-8 with Na2CO3 (aq), extracted with EtOAc and concentrated. The residue was purified by flash chromatography (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-41 (400.0 mg, yield: 83.9%) as a yellow oil. LCMS (ESI): calced for C17H16ClN5 [M+H]+ m/z 326.0, found 326.0. Preparation of 5-chloro-4-(5-fluoro-1H-indazol-3-yl)-N-(piperidin-4-yl)pyridin-2-amine (C-42)
 [0205] To a solution of B-50 (1.2 g, 2.6 mmol) in 1.4-dioxane (20 mL) was added M1 (1.5 g, 7.8 mmol), Pd2(dba)3 (241.5 mg, 0.2 mmol) and Xantphos (150.4 mg, 0.2 mmol), then the mixture is stirred for 5 h at 100°C under N2. The desired mass was detected on LC-MS. The mixture was washed with water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 95/5 to 45/55 in 14 mins) to afford tert-butyl 4-((5-chloro-4-(5-fluoro-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyridin-2-
yl)amino)piperidine-1-carboxylate (723.5 mg, yield: 47.8 %) as a yellow solid. LCMS (ESI): calcd for C28H39ClFN5O3Si [M+H] + ms/z 576.2, found 576.3. [0206] To a solution of tert-butyl 4-((5-chloro-4-(5-fluoro-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3- yl)pyridin-2-yl)amino)piperidine-1-carboxylate (723.5 mg, 1.2 mmol) in DCM (10 mL) and TFA (10 mL), then the mixture is stirred for 12 h at 25°C. The desired mass was detected on LC-MS. The mixture was washed with sodium carbonate saturated solution and extracted with DCM, the organic layer was washed with brine, dried over Na2SO4, filtered out and concentrated to get C-42 (152.5 mg, yield: 35.1 %) as a yellow solid. LCMS (ESI): calcd for C17H17ClFN5 [M+H] + ms/z 346.1, found 346.2. Preparation of 5-chloro-4-(5,7-difluoro-1H-indazol-3-yl)-N-(piperidin-4-yl)pyridin-2-amine (C-43)
[0205] To a solution of B-50 (1.2 g, 2.6 mmol) in 1.4-dioxane (20 mL) was added M1 (1.5 g, 7.8 mmol), Pd2(dba)3 (241.5 mg, 0.2 mmol) and Xantphos (150.4 mg, 0.2 mmol), then the mixture is stirred for 5 h at 100°C under N2. The desired mass was detected on LC-MS. The mixture was washed with water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 95/5 to 45/55 in 14 mins) to afford tert-butyl 4-((5-chloro-4-(5-fluoro-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyridin-2-
yl)amino)piperidine-1-carboxylate (723.5 mg, yield: 47.8 %) as a yellow solid. LCMS (ESI): calcd for C28H39ClFN5O3Si [M+H] + ms/z 576.2, found 576.3. [0206] To a solution of tert-butyl 4-((5-chloro-4-(5-fluoro-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3- yl)pyridin-2-yl)amino)piperidine-1-carboxylate (723.5 mg, 1.2 mmol) in DCM (10 mL) and TFA (10 mL), then the mixture is stirred for 12 h at 25°C. The desired mass was detected on LC-MS. The mixture was washed with sodium carbonate saturated solution and extracted with DCM, the organic layer was washed with brine, dried over Na2SO4, filtered out and concentrated to get C-42 (152.5 mg, yield: 35.1 %) as a yellow solid. LCMS (ESI): calcd for C17H17ClFN5 [M+H] + ms/z 346.1, found 346.2. Preparation of 5-chloro-4-(5,7-difluoro-1H-indazol-3-yl)-N-(piperidin-4-yl)pyridin-2-amine (C-43)
 [0207] A solution of B-47 (800.0 mg, 1.87 mmol), M1 (564.0 mg, 2.80 mmol), Pd2(dba)3 (171.0 mg, 0.19 mmol), Xantphos (162.0 mg, 0.28 mmol) and Cs2CO3 (1.2 g, 3.73 mmol) in DMF (10 mL) was charged with N2 and stirred at 80℃ for 3 h. The LCMS showed the desired MS was detected. The solution was quenched with H2O and then diluted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc 90/10 in 20 mins) to get tert-butyl 4-((5-chloro-4-(5,7-difluoro-1-(tetrahydro-2H-pyran-2-yl)-1H-indazol-3-yl)pyridin-2- yl)amino)piperidine-1-carboxylate (360.0 mg, yield: 31.6%) as a yellow solid. LCMS (ESI) calcd for C27H33ClF2N5O3 [M + H] + ms/z 548.2, found 548.2. [0208] A solution of tert-butyl 4-((5-chloro-4-(5,7-difluoro-1-(tetrahydro-2H-pyran-2-yl)-1H-indazol-3-yl)pyridin- 2-yl)amino)piperidine-1-carboxylate (360.0 mg, 0.66 mmol) in HCl-1,4-dioxane (10 mL) was stirred at rt for 6 h. The LCMS showed the desired MS was detected. The residue was partitioned between EtOAc and saturated NaHCO3 solution. The separated organic layer was washed with water, dried over anhydrous Na2SO4 and concentrated under vacuum to afford C-43 (140.0 mg, yield: 52.8%). LCMS (ESI) calcd for C17H17ClF2N5 [M + H] + ms/z 364.1, found 364.0.
Preparation of 5-chloro-4-(3-isopropyl-1H-pyrazol-4-yl)-N-(piperidin-4-yl)pyridin-2-amine (C-44)
[0207] A solution of B-47 (800.0 mg, 1.87 mmol), M1 (564.0 mg, 2.80 mmol), Pd2(dba)3 (171.0 mg, 0.19 mmol), Xantphos (162.0 mg, 0.28 mmol) and Cs2CO3 (1.2 g, 3.73 mmol) in DMF (10 mL) was charged with N2 and stirred at 80℃ for 3 h. The LCMS showed the desired MS was detected. The solution was quenched with H2O and then diluted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc 90/10 in 20 mins) to get tert-butyl 4-((5-chloro-4-(5,7-difluoro-1-(tetrahydro-2H-pyran-2-yl)-1H-indazol-3-yl)pyridin-2- yl)amino)piperidine-1-carboxylate (360.0 mg, yield: 31.6%) as a yellow solid. LCMS (ESI) calcd for C27H33ClF2N5O3 [M + H] + ms/z 548.2, found 548.2. [0208] A solution of tert-butyl 4-((5-chloro-4-(5,7-difluoro-1-(tetrahydro-2H-pyran-2-yl)-1H-indazol-3-yl)pyridin- 2-yl)amino)piperidine-1-carboxylate (360.0 mg, 0.66 mmol) in HCl-1,4-dioxane (10 mL) was stirred at rt for 6 h. The LCMS showed the desired MS was detected. The residue was partitioned between EtOAc and saturated NaHCO3 solution. The separated organic layer was washed with water, dried over anhydrous Na2SO4 and concentrated under vacuum to afford C-43 (140.0 mg, yield: 52.8%). LCMS (ESI) calcd for C17H17ClF2N5 [M + H] + ms/z 364.1, found 364.0.
Preparation of 5-chloro-4-(3-isopropyl-1H-pyrazol-4-yl)-N-(piperidin-4-yl)pyridin-2-amine (C-44)
 [0209] To a solution of B-32 (710.0 mg, 1.6 mmol) in 1,4-dioxane (15 mL) was added M-1 (397.1 mg, 2.0 mmol), PD2(DBA)3 (150.5 mg, 0.16 mmol),Xantphos (190.2 mg, 0.32 mmol) and Cs2CO3 (1071.3 mg, 3.3 mmol) stirred for 3 h at 100 ℃. The LCMS showed the desired MS was detected. Water was added to the solution and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 100/00 to 75/25 in 10 mins) to afford tert-butyl 4-((5-chloro-4-(3-isopropyl-1-((2- (trimethylsilyl)ethoxy)methyl)-1H-pyrazol-4-yl)pyridin-2-yl)amino)piperidine-1-carboxylate (220.0 mg, yield: 23.2%) as white solid. LCMS (ESI): calced for C27H44ClN5O3Si [M+H]+ m/z 550.3, found 550.3. [0210] To a solution of tert-butyl 4-((5-chloro-4-(3-isopropyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazol-4- yl)pyridin-2-yl)amino)piperidine-1-carboxylate (220 mg, 0.4 mmol) in DCM/TFA=5:1 (5 mL) stirred for 8h at 25 ℃. The LCMS showed the desired MS was detected. The solution was adjusted to pH 7-8 with saturated sodium carbonate and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with DCM/MeOH from 100/00 to 85/15 in 15 mins) to afford C-44 (102.0 mg, yield: 80.0 %) as yellow solid. LCMS (ESI): calced for C16H22ClN5 [M+H]+ m/z 320.2, found 320.0. Preparation of 5-chloro-N-(piperidin-4-yl)-4-(1H-pyrazolo[4,3-b]pyridin-3-yl)pyridin-2-amine (C-48)
[0209] To a solution of B-32 (710.0 mg, 1.6 mmol) in 1,4-dioxane (15 mL) was added M-1 (397.1 mg, 2.0 mmol), PD2(DBA)3 (150.5 mg, 0.16 mmol),Xantphos (190.2 mg, 0.32 mmol) and Cs2CO3 (1071.3 mg, 3.3 mmol) stirred for 3 h at 100 ℃. The LCMS showed the desired MS was detected. Water was added to the solution and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 100/00 to 75/25 in 10 mins) to afford tert-butyl 4-((5-chloro-4-(3-isopropyl-1-((2- (trimethylsilyl)ethoxy)methyl)-1H-pyrazol-4-yl)pyridin-2-yl)amino)piperidine-1-carboxylate (220.0 mg, yield: 23.2%) as white solid. LCMS (ESI): calced for C27H44ClN5O3Si [M+H]+ m/z 550.3, found 550.3. [0210] To a solution of tert-butyl 4-((5-chloro-4-(3-isopropyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazol-4- yl)pyridin-2-yl)amino)piperidine-1-carboxylate (220 mg, 0.4 mmol) in DCM/TFA=5:1 (5 mL) stirred for 8h at 25 ℃. The LCMS showed the desired MS was detected. The solution was adjusted to pH 7-8 with saturated sodium carbonate and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with DCM/MeOH from 100/00 to 85/15 in 15 mins) to afford C-44 (102.0 mg, yield: 80.0 %) as yellow solid. LCMS (ESI): calced for C16H22ClN5 [M+H]+ m/z 320.2, found 320.0. Preparation of 5-chloro-N-(piperidin-4-yl)-4-(1H-pyrazolo[4,3-b]pyridin-3-yl)pyridin-2-amine (C-48)
 [0211] A mixture of B-36 (450.0 mg, 1.03 mmol), M-1 (300.0 mg, 1.5 mmol), Cs2CO3 (669.5 mg, 2.06 mmol), XantPhos (57.8 mg, 0.1 mmol) and Pd2(dba)3 (45.7 mg, 0.05 mmol) in Dioxane (6 mL) was stirred at 100°C for 16 h. The desired mass was detected on LC-MS. Then the mixture was quenched with water, extracted with EtOAc, washed with brine, the combined organic phase were dried over anhydrous Na2SO4, filtered and concentrated to get crude product which was purified by column chromatograph on silica gel (eluting with PE/EtOAc from 100/0 to 75/25 in 20 mins) to afford tert-butyl 4-((5-chloro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-
1H-pyrazolo[4,3-b]pyridin-3-yl)pyridin-2-yl)amino)piperidine-1-carboxylate (330.0 mg, yield: 57.4%) as a yellow solid. LCMS (ESI): calced for C27H39ClN6O3Si [M+H]+ m/z 559.3, found 559.3. [0212] To a solution of tert-butyl 4-((5-chloro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazolo[4,3-b]pyridin- 3-yl)pyridin-2-yl)amino)piperidine-1-carboxylate (330.0 mg, 0.59 mmol) in DCM/TFA=3:1 (6 mL) was stirred at rt for 16 h. Then the mixture was concentrated, adjusted to pH = 7 - 8 with Et3N, purified by reverse flash chromatography (eluting with H2O(NH3.H2O)/MeCN from 80/20 to 50/50 in 30 mins) to afford C-48 (70.0 mg, yield: 36.2%) as a yellow solid. LCMS (ESI): calced for C16H17ClN6 [M+H]+ m/z 329.1, found 329.0. Preparation of 5-chloro-N-(piperidin-4-yl)-4-(1H-pyrazolo[4,3-c]pyridin-3-yl)pyridin-2-amine (C-49)
[0211] A mixture of B-36 (450.0 mg, 1.03 mmol), M-1 (300.0 mg, 1.5 mmol), Cs2CO3 (669.5 mg, 2.06 mmol), XantPhos (57.8 mg, 0.1 mmol) and Pd2(dba)3 (45.7 mg, 0.05 mmol) in Dioxane (6 mL) was stirred at 100°C for 16 h. The desired mass was detected on LC-MS. Then the mixture was quenched with water, extracted with EtOAc, washed with brine, the combined organic phase were dried over anhydrous Na2SO4, filtered and concentrated to get crude product which was purified by column chromatograph on silica gel (eluting with PE/EtOAc from 100/0 to 75/25 in 20 mins) to afford tert-butyl 4-((5-chloro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-
1H-pyrazolo[4,3-b]pyridin-3-yl)pyridin-2-yl)amino)piperidine-1-carboxylate (330.0 mg, yield: 57.4%) as a yellow solid. LCMS (ESI): calced for C27H39ClN6O3Si [M+H]+ m/z 559.3, found 559.3. [0212] To a solution of tert-butyl 4-((5-chloro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazolo[4,3-b]pyridin- 3-yl)pyridin-2-yl)amino)piperidine-1-carboxylate (330.0 mg, 0.59 mmol) in DCM/TFA=3:1 (6 mL) was stirred at rt for 16 h. Then the mixture was concentrated, adjusted to pH = 7 - 8 with Et3N, purified by reverse flash chromatography (eluting with H2O(NH3.H2O)/MeCN from 80/20 to 50/50 in 30 mins) to afford C-48 (70.0 mg, yield: 36.2%) as a yellow solid. LCMS (ESI): calced for C16H17ClN6 [M+H]+ m/z 329.1, found 329.0. Preparation of 5-chloro-N-(piperidin-4-yl)-4-(1H-pyrazolo[4,3-c]pyridin-3-yl)pyridin-2-amine (C-49)
 [0213] To a solution of B-37 (1.4 g, 0.0032 mol) in DMF (15 mL) were added M1 (0.9 g, 0.0048 mol), Pd2(dba)3 (0.3 g, 0.0003 mol), Xantphos (0.4 g, 0.0006 mol) and Cs2CO3 (2.1 g, 0.0064 mol). The reaction solution was stirred at 80 ℃ for 5 h. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with DCM/MeOH from 100/0 to 95/5 in 20 mins) to afford tert-butyl 4-((5-chloro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazolo[4,3- c]pyridin-3-yl)pyridin-2-yl)amino)piperidine-1-carboxylate (900.0 mg, yield: 50.0%) as a yellow oil. LCMS (ESI): calced for C27H39ClN6O3Si [M+H]+ m/z 559.1, found 559.1. [0214] To a solution of tert-butyl 4-((5-chloro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazolo[4,3-c]pyridin- 3-yl)pyridin-2-yl)amino)piperidine-1-carboxylate (900.0 mg, 1.6037 mmol) in DCM/TFA = 3:1 (10 mL). The reaction solution was stirred at rt for 12 h. The LCMS showed the desired MS was detected. The reaction mixture was concentrated, adjusted to 7-8 with Na2CO3 (aq), extracted with EtOAc and concentrated. The residue was purified by flash chromatography (eluting with DCM/MeOH from 100/0 to 80/20 in 20 mins) to afford C-49 (400.0 mg, yield: 74.3%) as a yellow oil. LCMS (ESI): calced for C16H17ClN6 [M+H]+ m/z 326.0, found 326.0. Preparation of 5-fluoro-4-(1H-indazol-3-yl)-N-(piperidin-4-yl)pyrimidin-2-amine (C-50)
[0213] To a solution of B-37 (1.4 g, 0.0032 mol) in DMF (15 mL) were added M1 (0.9 g, 0.0048 mol), Pd2(dba)3 (0.3 g, 0.0003 mol), Xantphos (0.4 g, 0.0006 mol) and Cs2CO3 (2.1 g, 0.0064 mol). The reaction solution was stirred at 80 ℃ for 5 h. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with DCM/MeOH from 100/0 to 95/5 in 20 mins) to afford tert-butyl 4-((5-chloro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazolo[4,3- c]pyridin-3-yl)pyridin-2-yl)amino)piperidine-1-carboxylate (900.0 mg, yield: 50.0%) as a yellow oil. LCMS (ESI): calced for C27H39ClN6O3Si [M+H]+ m/z 559.1, found 559.1. [0214] To a solution of tert-butyl 4-((5-chloro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazolo[4,3-c]pyridin- 3-yl)pyridin-2-yl)amino)piperidine-1-carboxylate (900.0 mg, 1.6037 mmol) in DCM/TFA = 3:1 (10 mL). The reaction solution was stirred at rt for 12 h. The LCMS showed the desired MS was detected. The reaction mixture was concentrated, adjusted to 7-8 with Na2CO3 (aq), extracted with EtOAc and concentrated. The residue was purified by flash chromatography (eluting with DCM/MeOH from 100/0 to 80/20 in 20 mins) to afford C-49 (400.0 mg, yield: 74.3%) as a yellow oil. LCMS (ESI): calced for C16H17ClN6 [M+H]+ m/z 326.0, found 326.0. Preparation of 5-fluoro-4-(1H-indazol-3-yl)-N-(piperidin-4-yl)pyrimidin-2-amine (C-50)
 [0215] To a solution of B-30 (530.0 mg, 1.4 mmol), M-1 (412.2 mg, 2.1 mmol) and DIEA (541.0 mg, 4.2 mmol) in DMSO (10.0 ml) stirred under nitrogen at 25℃. The reaction mixture was stirred at 120 ℃ for 16 h. The LCMS showed the desired MS was detected. The reaction mixture was concentrated under reduced pressure to give which was purified by flash silica gel chromatography (eluting with PE/EtOAc from 100/0 to 95/5 in 20 mins) to afford tert-butyl 4-((5-fluoro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyrimidin-2-yl)amino)piper- idine-1-carboxylate (300.0 mg, yield: 41.8%) as a white solid. LCMS (ESI): calced for C27H39FN6O3Si [M+H]+ m/z 543.3, found 543.2. [0216] To a solution of tert-butyl 4-((5-fluoro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyrimidin-2- yl)amino)piper-idine-1-carboxylate (300.0 mg, 0.6 mmol) in TFA/DCM (5:1)(50 mL). The reaction was stirred for 2 h at 25℃. The LCMS showed the desired MS was detected. Then the mixture was concentrated, quenched with water, adjusted to pH = 7 - 8 with NaHCO3, extracted with EtOAc, the combined organic phase was dried over anhydrous Na2SO4. The mixture was concentrated to get crude product which was purified by silica column chromatography (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-50 (170.0 mg, yield: 69.3%) as a white solid. LCMS (ESI): calced for C16H17FN6 [M+H]+ m/z 313.1, found 313.0. Preparation of 5-chloro-4-(4-fluoro-1-isopropyl-1H-benzo[d]imidazol-6-yl)-N-(piperidin-4-yl)pyridin-2-amine (C-51)
[0215] To a solution of B-30 (530.0 mg, 1.4 mmol), M-1 (412.2 mg, 2.1 mmol) and DIEA (541.0 mg, 4.2 mmol) in DMSO (10.0 ml) stirred under nitrogen at 25℃. The reaction mixture was stirred at 120 ℃ for 16 h. The LCMS showed the desired MS was detected. The reaction mixture was concentrated under reduced pressure to give which was purified by flash silica gel chromatography (eluting with PE/EtOAc from 100/0 to 95/5 in 20 mins) to afford tert-butyl 4-((5-fluoro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyrimidin-2-yl)amino)piper- idine-1-carboxylate (300.0 mg, yield: 41.8%) as a white solid. LCMS (ESI): calced for C27H39FN6O3Si [M+H]+ m/z 543.3, found 543.2. [0216] To a solution of tert-butyl 4-((5-fluoro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyrimidin-2- yl)amino)piper-idine-1-carboxylate (300.0 mg, 0.6 mmol) in TFA/DCM (5:1)(50 mL). The reaction was stirred for 2 h at 25℃. The LCMS showed the desired MS was detected. Then the mixture was concentrated, quenched with water, adjusted to pH = 7 - 8 with NaHCO3, extracted with EtOAc, the combined organic phase was dried over anhydrous Na2SO4. The mixture was concentrated to get crude product which was purified by silica column chromatography (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-50 (170.0 mg, yield: 69.3%) as a white solid. LCMS (ESI): calced for C16H17FN6 [M+H]+ m/z 313.1, found 313.0. Preparation of 5-chloro-4-(4-fluoro-1-isopropyl-1H-benzo[d]imidazol-6-yl)-N-(piperidin-4-yl)pyridin-2-amine (C-51)
 [0217] To a solution of B-39 (1.7 g, 0.0046 mol) in DMF (34 mL) were added M-1 (1.4 g, 0.0069 mol), Pd2(dba)3 (420.0 mg, 0.0004 mol), Xantphos (530.0 mg, 0.0009 mol) and Cs2CO3 (3.0 g, 0.0092 mol). The reaction solution was stirred at 80℃ for 12 h. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with DCM/MeOH from 100/0 to 95/5 in 20 mins) to afford tert-butyl 4-((5-chloro-4-(4-fluoro-1-isopropyl-1H- benzo[d]imidazol-6-yl)pyridin-2-yl)amino)piperidine-1-carboxylate (1.6 g, yield: 67.4%) as a yellow solid. LCMS (ESI): calced for C25H31ClFN5O2 [M+H]+ m/z 488.1, found 488.1. [0218] Tert-butyl 4-((5-chloro-4-(4-fluoro-1-isopropyl-1H-benzo[d]imidazol-6-yl)pyridin-2-yl)amino)piperidine-1- carboxylate (1.6 g, 0.0033 mol) was added to DCM/TFA=5:1 and stirred at rt for 3 h. The LCMS showed the desired MS was detected. Then the mixture was concentrated, quenched with water, adjusted to pH = 7 - 8 with NaHCO3, extracted with EtOAc, the combined organic phase was dried over anhydrous Na2SO4. The mixture was concentrated to get crude product which was purified by flash chromatography (eluting with DCM/MeOH
from 100/0 to 95/5 in 20 mins) to afford C-51 (1.2 g, yield: 87.8%) as a yellow oil. LCMS (ESI): calced for C20H23ClFN5 [M+H]+ m/z 388.1, found 388.1. Preparation of 5-chloro-N-(piperidin-4-yl)-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-4-yl)pyridin-2-amine (C-52)
[0217] To a solution of B-39 (1.7 g, 0.0046 mol) in DMF (34 mL) were added M-1 (1.4 g, 0.0069 mol), Pd2(dba)3 (420.0 mg, 0.0004 mol), Xantphos (530.0 mg, 0.0009 mol) and Cs2CO3 (3.0 g, 0.0092 mol). The reaction solution was stirred at 80℃ for 12 h. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with DCM/MeOH from 100/0 to 95/5 in 20 mins) to afford tert-butyl 4-((5-chloro-4-(4-fluoro-1-isopropyl-1H- benzo[d]imidazol-6-yl)pyridin-2-yl)amino)piperidine-1-carboxylate (1.6 g, yield: 67.4%) as a yellow solid. LCMS (ESI): calced for C25H31ClFN5O2 [M+H]+ m/z 488.1, found 488.1. [0218] Tert-butyl 4-((5-chloro-4-(4-fluoro-1-isopropyl-1H-benzo[d]imidazol-6-yl)pyridin-2-yl)amino)piperidine-1- carboxylate (1.6 g, 0.0033 mol) was added to DCM/TFA=5:1 and stirred at rt for 3 h. The LCMS showed the desired MS was detected. Then the mixture was concentrated, quenched with water, adjusted to pH = 7 - 8 with NaHCO3, extracted with EtOAc, the combined organic phase was dried over anhydrous Na2SO4. The mixture was concentrated to get crude product which was purified by flash chromatography (eluting with DCM/MeOH
from 100/0 to 95/5 in 20 mins) to afford C-51 (1.2 g, yield: 87.8%) as a yellow oil. LCMS (ESI): calced for C20H23ClFN5 [M+H]+ m/z 388.1, found 388.1. Preparation of 5-chloro-N-(piperidin-4-yl)-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-4-yl)pyridin-2-amine (C-52)
 [0219] To a solution of B-40 (2.7 g, 6.1 mmol) in DMF (54 mL) was added M1 (1.4 g, 7.32 mmol), Pd2(dba)3 (0.28 g, 0.3 mmol), Xantphos (0.7 g, 1.22 mmol) and Cs2CO3 (4.0 g, 12.2 mmol) stirred for 6 h at 90 ℃ under nitrogen. The LCMS showed the desired MS was detected. Water was added to the solution and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 100/00 to 75/25 in 15 mins) to afford (1.9 g, yield: 55.7 %) as white solid. LCMS (ESI): calced for C28H40ClN5O3Si [M+H]+ m/z 558.2, found 558.2. [0220] To a solution of of tert-butyl 4-((5-chloro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-4-yl)pyridin- 2-yl)amino)piperidine-1-carboxylate (1.9 g, 3.4 mmol) in DCM/TFA=10:1 (38 mL) stirred for 1 h at 25 ℃. The LCMS showed the desired MS was detected. The filtrate was adjusted to pH 7-8 with saturated sodium carbonate and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with DCM/MeOH from 100/00 to 85/15 in 15 mins) to afford C-52 (1.3 g, yield: 82.7 %) as brown solid. LCMS (ESI): calced for C23H32ClN5OSi [M+H]+ m/z 458.2, found 458.2. Preparation of 6-(piperidin-4-ylamino)-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-4-yl)nicotinonitrile (C-53)
[0219] To a solution of B-40 (2.7 g, 6.1 mmol) in DMF (54 mL) was added M1 (1.4 g, 7.32 mmol), Pd2(dba)3 (0.28 g, 0.3 mmol), Xantphos (0.7 g, 1.22 mmol) and Cs2CO3 (4.0 g, 12.2 mmol) stirred for 6 h at 90 ℃ under nitrogen. The LCMS showed the desired MS was detected. Water was added to the solution and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 100/00 to 75/25 in 15 mins) to afford (1.9 g, yield: 55.7 %) as white solid. LCMS (ESI): calced for C28H40ClN5O3Si [M+H]+ m/z 558.2, found 558.2. [0220] To a solution of of tert-butyl 4-((5-chloro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-4-yl)pyridin- 2-yl)amino)piperidine-1-carboxylate (1.9 g, 3.4 mmol) in DCM/TFA=10:1 (38 mL) stirred for 1 h at 25 ℃. The LCMS showed the desired MS was detected. The filtrate was adjusted to pH 7-8 with saturated sodium carbonate and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with DCM/MeOH from 100/00 to 85/15 in 15 mins) to afford C-52 (1.3 g, yield: 82.7 %) as brown solid. LCMS (ESI): calced for C23H32ClN5OSi [M+H]+ m/z 458.2, found 458.2. Preparation of 6-(piperidin-4-ylamino)-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-4-yl)nicotinonitrile (C-53)
 [0221] To a solution of B-41 (1.9 g, 4.9 mol) in M1 (14.7 g, 73.5) and DIEA (1.27 g, 1.27 mol) stirred for 3 h at 90 ℃. The LCMS showed the desired MS was detected. Water was added to the solution and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 100/00 to 60/40 in 15 mins) to afford tert-butyl 4-((5-cyano-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-4-yl)pyridin-2-yl)amino)
piperidine-1-carboxylate (1.9 g, yield: 70.8 %) as yellow solid. LCMS (ESI): calced for C29H40N6O3Si [M+H]+ m/z 549.3, found 549.3. [0222] To a solution of tert-butyl 4-((5-cyano-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-4-yl)pyridin-2- yl)amino)piperidine-1-carboxylate (1.9 g, 3.4 mmol) in DCM/TFA=10:1 (40 mL) stirred for 1h at 25 ℃. The LCMS showed the desired MS was detected. The filtrate was adjusted to pH 7-8 with saturated sodium carbonate and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with DCM/MeOH from 100/00 to 85/15 in 15 mins) to afford (1.3 g, yield: 83.8 %) as brown solid. LCMS (ESI): calced for C24H32N6OSi [M+H]+ m/z 449.2, found 449.3. Preparation of 5-chloro-N-(piperidin-4-yl)-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyridin-2-amine (C-54)
[0221] To a solution of B-41 (1.9 g, 4.9 mol) in M1 (14.7 g, 73.5) and DIEA (1.27 g, 1.27 mol) stirred for 3 h at 90 ℃. The LCMS showed the desired MS was detected. Water was added to the solution and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with PE/EtOAc from 100/00 to 60/40 in 15 mins) to afford tert-butyl 4-((5-cyano-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-4-yl)pyridin-2-yl)amino)
piperidine-1-carboxylate (1.9 g, yield: 70.8 %) as yellow solid. LCMS (ESI): calced for C29H40N6O3Si [M+H]+ m/z 549.3, found 549.3. [0222] To a solution of tert-butyl 4-((5-cyano-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-4-yl)pyridin-2- yl)amino)piperidine-1-carboxylate (1.9 g, 3.4 mmol) in DCM/TFA=10:1 (40 mL) stirred for 1h at 25 ℃. The LCMS showed the desired MS was detected. The filtrate was adjusted to pH 7-8 with saturated sodium carbonate and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with DCM/MeOH from 100/00 to 85/15 in 15 mins) to afford (1.3 g, yield: 83.8 %) as brown solid. LCMS (ESI): calced for C24H32N6OSi [M+H]+ m/z 449.2, found 449.3. Preparation of 5-chloro-N-(piperidin-4-yl)-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyridin-2-amine (C-54)
 [0223] To a solution of B-42 (3.5 g, 0.008 mol) in DMF (50 mL) were added M1 (2.4 g, 0.012 mol), Xantphos (0.9 g, 0.0016 mol) and Cs2CO3 (5.2 g, 0.016 mol). The reaction solution was stirred at 80 ℃ for 6 h. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 80/20 in 20 mins) to afford (2.9 g, yield: 63.7%) as a yellow oil. CMS (ESI): calced for C28H40ClN5O3Si [M+H]+ m/z 558.4, found 558.4. [0224] tert-butyl 4-((5-chloro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyridin-2- yl)amino)piperidine-1-carboxylate (2.9 g, 0.0052 mol) was added to DCM:TFA=8:1 (45 mL). The reaction solution was stirred at rt for 2 h. The LCMS showed the desired MS was detected. The reaction mixture was concentrated to get crude product which was purified by flash chromatography (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-54 (2.2 g, yield: 90.3%) as a yellow oil. LCMS (ESI): calced for C23H32ClN5OSi [M+H]+ m/z 458.1, found 458.1. Preparation of 6-(piperidin-4-ylamino)-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)nicotinonitrile (C-55)
[0223] To a solution of B-42 (3.5 g, 0.008 mol) in DMF (50 mL) were added M1 (2.4 g, 0.012 mol), Xantphos (0.9 g, 0.0016 mol) and Cs2CO3 (5.2 g, 0.016 mol). The reaction solution was stirred at 80 ℃ for 6 h. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 80/20 in 20 mins) to afford (2.9 g, yield: 63.7%) as a yellow oil. CMS (ESI): calced for C28H40ClN5O3Si [M+H]+ m/z 558.4, found 558.4. [0224] tert-butyl 4-((5-chloro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyridin-2- yl)amino)piperidine-1-carboxylate (2.9 g, 0.0052 mol) was added to DCM:TFA=8:1 (45 mL). The reaction solution was stirred at rt for 2 h. The LCMS showed the desired MS was detected. The reaction mixture was concentrated to get crude product which was purified by flash chromatography (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-54 (2.2 g, yield: 90.3%) as a yellow oil. LCMS (ESI): calced for C23H32ClN5OSi [M+H]+ m/z 458.1, found 458.1. Preparation of 6-(piperidin-4-ylamino)-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)nicotinonitrile (C-55)
 [0225] B-43 (800.0 mg, 2.0728 mmol), M1 (4.2 g, 20.728mol) and Diisopropylethylamine (3.8 g, 4.1456 mmol) was added and stirred at 100℃ for 6 h. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 70/30 in 20 mins) to afford tert-butyl 4-((5-cyano-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3- yl)pyridin-2-yl)amino)piperidine-1-carboxylate (650.0 mg, yield: 55.8%) as a yellow oil. LCMS (ESI): calced for C29H40N6O3Si [M+H]+ m/z 549.2, found 549.2. [0226] To a solution of tert-butyl 4-((5-cyano-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyridin-2- yl)amino)piperidine-1-carboxylate (650.0 mg, 1.1802 mmol) in DCM/TFA = 8:1 (9 mL) and stirred at rt for 2 h. The LCMS showed the desired MS was detected. The reaction mixture was concentrated, adjusted to 7-8 with Na2CO3 (aq), extracted with EtOAc and concentrated. The residue was purified by flash chromatography (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-55 (300.0 mg, yield: 55.4%) as a yellow solid. LCMS (ESI): calced for C24H32N6OSi [M+H]+ m/z 449.2, found 449.2. Preparation of N-(piperidin-4-yl)-5-(trifluoromethyl)-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyridin- 2-amine (C-56)
[0225] B-43 (800.0 mg, 2.0728 mmol), M1 (4.2 g, 20.728mol) and Diisopropylethylamine (3.8 g, 4.1456 mmol) was added and stirred at 100℃ for 6 h. The LCMS showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 70/30 in 20 mins) to afford tert-butyl 4-((5-cyano-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3- yl)pyridin-2-yl)amino)piperidine-1-carboxylate (650.0 mg, yield: 55.8%) as a yellow oil. LCMS (ESI): calced for C29H40N6O3Si [M+H]+ m/z 549.2, found 549.2. [0226] To a solution of tert-butyl 4-((5-cyano-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyridin-2- yl)amino)piperidine-1-carboxylate (650.0 mg, 1.1802 mmol) in DCM/TFA = 8:1 (9 mL) and stirred at rt for 2 h. The LCMS showed the desired MS was detected. The reaction mixture was concentrated, adjusted to 7-8 with Na2CO3 (aq), extracted with EtOAc and concentrated. The residue was purified by flash chromatography (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-55 (300.0 mg, yield: 55.4%) as a yellow solid. LCMS (ESI): calced for C24H32N6OSi [M+H]+ m/z 449.2, found 449.2. Preparation of N-(piperidin-4-yl)-5-(trifluoromethyl)-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyridin- 2-amine (C-56)
 [0227] A mixture of B-44 (2.2 g, 4.67 mmol), M-1 (1.12 g, 5.6 mmol), Cs2CO3 (3.0 g, 9.34 mmol), XantPhos (289.0 mg, 0.5 mmol) and Pd2(dba)3 (210.4 mg, 0.23 mmol) in Dioxane (30 mL) was stirred at 80°C for 16 h. The desired mass was detected on LC-MS. Then the mixture was quenched with water, extracted with EtOAc, washed with brine, the combined organic phase were dried over anhydrous Na2SO4, filtered and concentrated to get crude product which was purified by column chromatograph on silica gel (eluting with PE/EtOAc from 100/0 to 75/25 in 20 mins) to afford tert-butyl 4-((5-(trifluoromethyl)-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3- yl)pyridin-2-yl)amino)piperidine-1-carboxylate (1.3 g, yield: 47.1%) as a yellow solid. LCMS (ESI): calced for C29H40F3N5O3Si [M+H]+ m/z 592.3, found 592.3. [0228] To a solution of tert-butyl 4-((5-(trifluoromethyl)-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3- yl)pyridin-2-yl)amino)piperidine-1-carboxylate (1.3 g, 2.2 mmol) in DCM/TFA=8:1 (20 mL) was stirred at rt for 1 h. Then the mixture was concentrated, adjusted to pH = 7 - 8 with NaHCO3, extracted with EtOAc, washed with brine, the combined organic phase were dried over anhydrous Na2SO4, filtered and concentrated to get crude product which was purified by column chromatograph on silica gel (eluting with DCM/MeOH from 90/10 to 80/20
in 20 mins) to afford C-56 (550.0 mg, yield: 50.9%) as a yellow solid. LCMS (ESI): calced for C24H32F3N5OSi [M+H]+ m/z 492.2, found 492.2. Preparation of 5-methyl-N-(piperidin-4-yl)-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyridin-2-amine (C-57)
[0227] A mixture of B-44 (2.2 g, 4.67 mmol), M-1 (1.12 g, 5.6 mmol), Cs2CO3 (3.0 g, 9.34 mmol), XantPhos (289.0 mg, 0.5 mmol) and Pd2(dba)3 (210.4 mg, 0.23 mmol) in Dioxane (30 mL) was stirred at 80°C for 16 h. The desired mass was detected on LC-MS. Then the mixture was quenched with water, extracted with EtOAc, washed with brine, the combined organic phase were dried over anhydrous Na2SO4, filtered and concentrated to get crude product which was purified by column chromatograph on silica gel (eluting with PE/EtOAc from 100/0 to 75/25 in 20 mins) to afford tert-butyl 4-((5-(trifluoromethyl)-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3- yl)pyridin-2-yl)amino)piperidine-1-carboxylate (1.3 g, yield: 47.1%) as a yellow solid. LCMS (ESI): calced for C29H40F3N5O3Si [M+H]+ m/z 592.3, found 592.3. [0228] To a solution of tert-butyl 4-((5-(trifluoromethyl)-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3- yl)pyridin-2-yl)amino)piperidine-1-carboxylate (1.3 g, 2.2 mmol) in DCM/TFA=8:1 (20 mL) was stirred at rt for 1 h. Then the mixture was concentrated, adjusted to pH = 7 - 8 with NaHCO3, extracted with EtOAc, washed with brine, the combined organic phase were dried over anhydrous Na2SO4, filtered and concentrated to get crude product which was purified by column chromatograph on silica gel (eluting with DCM/MeOH from 90/10 to 80/20
in 20 mins) to afford C-56 (550.0 mg, yield: 50.9%) as a yellow solid. LCMS (ESI): calced for C24H32F3N5OSi [M+H]+ m/z 492.2, found 492.2. Preparation of 5-methyl-N-(piperidin-4-yl)-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyridin-2-amine (C-57)
 [0229] To a solution of B-49 (750.0 mg, 2.0 mmol), M1 (442.8 mg, 2.2 mmol), Cs2CO3 (1.3 g, 4.0 mmol) , Ephos (213.9 mg, 0.4 mmol) and EPhos Pd G4 (183.6 mg, 0.2 mmol) in 1,4-dioxane(10.0 ml) stirred under nitrogen at 25℃ . The reaction mixture was stirred at 80 ℃ for 2 h. The LCMS showed the desired MS was detected .The reaction mixture was concentrated under reduced pressure to give which was purified by flash silica gel chromatography (eluting with PE/EtOAc from 100/0 to 95/5 in 20 mins) to afford tert-butyl 4-((5-methyl- 4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyridin-2-yl)amino)piperidine-1-carboxylate (400.0 mg, yield: 37.0%) as a white solid. LCMS (ESI): calced for C29H43N5O3Si [M+H]+ m/z 538.3, found 538.3. [0230] To a solution of tert-butyl {4-[(4-{1-[(2-methoxyethyl)trimethyl-$l^{5}-silyl]indazol-3-yl}-5-methylpyridin-2- yl)amino]piperidin-1-yl} formate (400.0 mg, 0.63 mmol) in TFA/DCM (5:1)(10 mL). The reaction was stirred for 2 h at 25℃. The LCMS showed the desired MS was detected. The solvent was removed by vacumm, adjusted to 7- 8 with Na2CO3 (aq), extracted with EtOAc and concentrated. The residue was purified by silica column chromatography (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-57 (240.0 mg, yield: 73.7%) as a yellow liquid. LCMS (ESI): calced for C24H35N5OSi [M+H]+ m/z 438.3, found 438.2. Preparation of 4-([1,2,4]triazolo[4,3-a]pyridin-3-yl)-5-chloro-N-(piperidin-4-yl)pyrimidin-2-amine (C-59)
[0229] To a solution of B-49 (750.0 mg, 2.0 mmol), M1 (442.8 mg, 2.2 mmol), Cs2CO3 (1.3 g, 4.0 mmol) , Ephos (213.9 mg, 0.4 mmol) and EPhos Pd G4 (183.6 mg, 0.2 mmol) in 1,4-dioxane(10.0 ml) stirred under nitrogen at 25℃ . The reaction mixture was stirred at 80 ℃ for 2 h. The LCMS showed the desired MS was detected .The reaction mixture was concentrated under reduced pressure to give which was purified by flash silica gel chromatography (eluting with PE/EtOAc from 100/0 to 95/5 in 20 mins) to afford tert-butyl 4-((5-methyl- 4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyridin-2-yl)amino)piperidine-1-carboxylate (400.0 mg, yield: 37.0%) as a white solid. LCMS (ESI): calced for C29H43N5O3Si [M+H]+ m/z 538.3, found 538.3. [0230] To a solution of tert-butyl {4-[(4-{1-[(2-methoxyethyl)trimethyl-$l^{5}-silyl]indazol-3-yl}-5-methylpyridin-2- yl)amino]piperidin-1-yl} formate (400.0 mg, 0.63 mmol) in TFA/DCM (5:1)(10 mL). The reaction was stirred for 2 h at 25℃. The LCMS showed the desired MS was detected. The solvent was removed by vacumm, adjusted to 7- 8 with Na2CO3 (aq), extracted with EtOAc and concentrated. The residue was purified by silica column chromatography (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-57 (240.0 mg, yield: 73.7%) as a yellow liquid. LCMS (ESI): calced for C24H35N5OSi [M+H]+ m/z 438.3, found 438.2. Preparation of 4-([1,2,4]triazolo[4,3-a]pyridin-3-yl)-5-chloro-N-(piperidin-4-yl)pyrimidin-2-amine (C-59)
 [0231] To a solution of B-45 (1.4 g, 4.5 mmol) in NMP (30 mL) was added M-1 (1.1 g, 5.3 mmol) and DIEA (1.1 g, 9.0 mmol) stirred for 12 h at 80 ℃. The LCMS showed the desired MS was detected. Water was added to the solution and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with DCM/MeOH from 100/00 to 95/05 in 10 mins) to afford Boc-C-59 (1.1 g, yield: 57.8%) as yellow solid. LCMS (ESI): calced for C15H16ClN7 [M+H]+ m/z 430.2, found 430.0.
[0232] To a solution of Boc-C-59 (500.0 mg, 1.16 mmol) in HCL-1,4-dioxane (10 mL, 4mol/L) was stirred for 1h at 25 ℃. The LCMS showed the desired MS was detected. The LCMS showed the desired MS was detected. The solvent was removed. The crude product C-59 (360.0 mg, yield: 84.7%) was directly used in next step without further purification. LCMS (ESI): calced for C20H24ClN7O2 [M+H]+ m/z 330.1, found 330.0. Preparation of 5-fluoro-N-(piperidin-4-yl)-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyridin-2-amine (C-60)
[0231] To a solution of B-45 (1.4 g, 4.5 mmol) in NMP (30 mL) was added M-1 (1.1 g, 5.3 mmol) and DIEA (1.1 g, 9.0 mmol) stirred for 12 h at 80 ℃. The LCMS showed the desired MS was detected. Water was added to the solution and extracted with EtOAc. The organic layer was washed with brine, dried anhydrous sodium sulfate, filtered out and concentrated to get the product which was purified by flash chromatography (eluting with DCM/MeOH from 100/00 to 95/05 in 10 mins) to afford Boc-C-59 (1.1 g, yield: 57.8%) as yellow solid. LCMS (ESI): calced for C15H16ClN7 [M+H]+ m/z 430.2, found 430.0.
[0232] To a solution of Boc-C-59 (500.0 mg, 1.16 mmol) in HCL-1,4-dioxane (10 mL, 4mol/L) was stirred for 1h at 25 ℃. The LCMS showed the desired MS was detected. The LCMS showed the desired MS was detected. The solvent was removed. The crude product C-59 (360.0 mg, yield: 84.7%) was directly used in next step without further purification. LCMS (ESI): calced for C20H24ClN7O2 [M+H]+ m/z 330.1, found 330.0. Preparation of 5-fluoro-N-(piperidin-4-yl)-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyridin-2-amine (C-60)
 [0233] To a solution of B-46 (1.24 g, 2.9 mmol) in 1,4-dioxane (20 mL) was added M1 (1.2 g, 5.9 mmol), EPhos Pd G4 (269.2 mg, 0.29 mmol), Ephos (341.4 mg, 0.59 mmol) and Cs2CO3 (1.9 g, 5.9 mmol). The reaction was stirred under nitrogen at 80 ℃ for 16 h under N2. The LCMS showed the desired MS was detected. The mixture was concentrated and purified by flash chromatography (eluting with DCM/MeOH from 98/2 to 95/5 in 20 mins) to afford tert-butyl 4-((5-fluoro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyridin-2- yl)amino)piperidine-1-carboxylate (310.0 mg, yield: 19.5 %) as a yellow oil. LCMS (ESI): calced for C28H40FN5O3Si [M+H]+ m/z 542.3, found 542.2. [0234] To a solution of tert-butyl 4-((5-fluoro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyridin-2- yl)amino)piperidine-1-carboxylate (310.0 mg, 0.57 mmol) in DCM (5 mL) was added TFA (0.5 mL). The reaction mixture was stirred at rt for 5 h. The LCMS showed the desired MS was detected. The mixture was quenched with saturated aq. NaHCO3, adjusted pH to 7~8 and extracted with DCM. The organic layers were combined and washed with brine, dried over Na2SO4, filtered out and concentrated to obtain the crude product C-60 (280.0 mg, yield: 77.6%) which was used for next step without further purification. LCMS (ESI): calced for C23H32FN5OSi [M+H]+ m/z 442.2, found 442.1. Preparation of 5-chloro-N-(piperidin-4-yl)-4-(pyrazolo[1,5-a]pyridin-3-yl)pyrimidin-2-amine (C-61)
[0233] To a solution of B-46 (1.24 g, 2.9 mmol) in 1,4-dioxane (20 mL) was added M1 (1.2 g, 5.9 mmol), EPhos Pd G4 (269.2 mg, 0.29 mmol), Ephos (341.4 mg, 0.59 mmol) and Cs2CO3 (1.9 g, 5.9 mmol). The reaction was stirred under nitrogen at 80 ℃ for 16 h under N2. The LCMS showed the desired MS was detected. The mixture was concentrated and purified by flash chromatography (eluting with DCM/MeOH from 98/2 to 95/5 in 20 mins) to afford tert-butyl 4-((5-fluoro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyridin-2- yl)amino)piperidine-1-carboxylate (310.0 mg, yield: 19.5 %) as a yellow oil. LCMS (ESI): calced for C28H40FN5O3Si [M+H]+ m/z 542.3, found 542.2. [0234] To a solution of tert-butyl 4-((5-fluoro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)pyridin-2- yl)amino)piperidine-1-carboxylate (310.0 mg, 0.57 mmol) in DCM (5 mL) was added TFA (0.5 mL). The reaction mixture was stirred at rt for 5 h. The LCMS showed the desired MS was detected. The mixture was quenched with saturated aq. NaHCO3, adjusted pH to 7~8 and extracted with DCM. The organic layers were combined and washed with brine, dried over Na2SO4, filtered out and concentrated to obtain the crude product C-60 (280.0 mg, yield: 77.6%) which was used for next step without further purification. LCMS (ESI): calced for C23H32FN5OSi [M+H]+ m/z 442.2, found 442.1. Preparation of 5-chloro-N-(piperidin-4-yl)-4-(pyrazolo[1,5-a]pyridin-3-yl)pyrimidin-2-amine (C-61)
 [0235] To a solution of B-48 (600.0 mg, 2.2633 mmol) in NMP (10 mL) were added M1 (683.4 mg, 3.3949 mmol) and DIEA (585.0 mg, 4.5266 mmol). The reaction solution was stirred at 80 ℃ for 14 h. The LCMS
showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 70/30 in 20 mins) to afford Boc-C-61 (650.0 mg, yield: 65.5%) as a yellow solid. LCMS (ESI): calced for C21H25ClN6O2 [M+H]+ m/z 429.1, found 429.1. [0236] To a solution of Boc-C-61 (650.0 mg, 1.5119 mmol) in DCM/TFA = 5:1 (10 mL), and stirred at rt for 2 h. The LCMS showed the desired MS was detected. The reaction mixture was concentrated, adjusted to 7-8 with Na2CO3 (aq), extracted with EtOAc and concentrated. The residue was purified by flash chromatography (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-61 (500.0 mg, yield: 98.6%) as a yellow solid. LCMS (ESI): calced for C16H17ClN6 [M+H]+ m/z 328.8, found 328.8. Preparation of 5-chloro-4-(1-isopropyl-1H-indazol-6-yl)-N-(piperidin-4-yl)pyridin-2-amine TFA salt (C-62)
[0235] To a solution of B-48 (600.0 mg, 2.2633 mmol) in NMP (10 mL) were added M1 (683.4 mg, 3.3949 mmol) and DIEA (585.0 mg, 4.5266 mmol). The reaction solution was stirred at 80 ℃ for 14 h. The LCMS
showed the desired MS was detected. The reaction mixture was added water and extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 70/30 in 20 mins) to afford Boc-C-61 (650.0 mg, yield: 65.5%) as a yellow solid. LCMS (ESI): calced for C21H25ClN6O2 [M+H]+ m/z 429.1, found 429.1. [0236] To a solution of Boc-C-61 (650.0 mg, 1.5119 mmol) in DCM/TFA = 5:1 (10 mL), and stirred at rt for 2 h. The LCMS showed the desired MS was detected. The reaction mixture was concentrated, adjusted to 7-8 with Na2CO3 (aq), extracted with EtOAc and concentrated. The residue was purified by flash chromatography (eluting with DCM/MeOH from 100/0 to 90/10 in 20 mins) to afford C-61 (500.0 mg, yield: 98.6%) as a yellow solid. LCMS (ESI): calced for C16H17ClN6 [M+H]+ m/z 328.8, found 328.8. Preparation of 5-chloro-4-(1-isopropyl-1H-indazol-6-yl)-N-(piperidin-4-yl)pyridin-2-amine TFA salt (C-62)
 [0237] To a solution of B-51 (310.0 mg, 0.88 mmol) in dioxane (5 mL) was added M1 (213.6 mg, 1.06 mmol), Pd2(dba)3 (40.5 mg, 0.04 mmol), Xantphos (51.2 mg, 0.09 mmol) and Cs2CO3 (576.1 mg, 1.77 mmol). The reaction mixture was stirred at 100℃ for 6 h under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed by vacuum. The residue was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 50/50 in 30 mins) to afford Boc-C-62 (170.0 mg, yield: 34.3 %) as a yellow solid. LCMS (ESI): calced for C25H32ClN5O2 [M+H]+ m/z 470.2, found 470.5. [0238] To a solution of Boc-C-62 (170.0 mg, 0.36 mmol) in TFA/DCM (1mL/5mL) stirred at 25℃ for 4 h. The LCMS showed the desired MS was detected. The solvent was removed by vacumm to afford C-62 (110.0 mg, yield: 56.0 %) as a brown solid. LCMS (ESI): calced for C20H24ClN5 [M+H]+ m/z 484.2, found 370.4. Preparation of methyl 4-(5-chloro-2-(piperidin-4-ylamino)pyrimidin-4-yl)-1H-pyrazole-3-carboxylate TFA salt (C- 63)
[0237] To a solution of B-51 (310.0 mg, 0.88 mmol) in dioxane (5 mL) was added M1 (213.6 mg, 1.06 mmol), Pd2(dba)3 (40.5 mg, 0.04 mmol), Xantphos (51.2 mg, 0.09 mmol) and Cs2CO3 (576.1 mg, 1.77 mmol). The reaction mixture was stirred at 100℃ for 6 h under nitrogen. The LCMS showed the desired MS was detected. The solvent was removed by vacuum. The residue was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 50/50 in 30 mins) to afford Boc-C-62 (170.0 mg, yield: 34.3 %) as a yellow solid. LCMS (ESI): calced for C25H32ClN5O2 [M+H]+ m/z 470.2, found 470.5. [0238] To a solution of Boc-C-62 (170.0 mg, 0.36 mmol) in TFA/DCM (1mL/5mL) stirred at 25℃ for 4 h. The LCMS showed the desired MS was detected. The solvent was removed by vacumm to afford C-62 (110.0 mg, yield: 56.0 %) as a brown solid. LCMS (ESI): calced for C20H24ClN5 [M+H]+ m/z 484.2, found 370.4. Preparation of methyl 4-(5-chloro-2-(piperidin-4-ylamino)pyrimidin-4-yl)-1H-pyrazole-3-carboxylate TFA salt (C- 63)
 [0239] To a solution of B-31 (1.9 g, 4.70 mmol), M1 (0.9 g, 4.70 mmol) in NMP (20 mL) was added DIEA (1.2 g, 9.40 mmol). The reaction mixture was stirred at 80℃ for 6 h. The mixture was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 60/40 in 20 mins) to afford tert-butyl 4-((5-chloro-4-(3-(methoxycarbonyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazol-4- yl)pyrimidin-2-yl)amino)piperidine-1-carboxylate (1.4 g, yield: 38.7 %) as a yellow solid. LCMS (ESI): calced for C25H39ClN6O5Si [M+H]+ m/z 567.2, found 567.6. [0240] To a solution of methyl 4-(2-{[1-(tert-butyl-$l^{3}-oxy)piperidin-4-yl]amino}-5-chloropyrimidin-4-yl)-1-[(2- methoxyethyl)trimethyl-$l^{5}-silyl]pyrazole-3-carboxylate (1.4 g, 2.46 mmol) in TFA/DCM (2mL/10mL) stirred at 25℃ for 4 h. The LCMS showed the desired MS was detected. The solvent was removed by vacumm to afford C-63 (0.76 g, yield: 78.9 %) as a yellow solid. LCMS (ESI): calced for C14H17ClN6O2 [M+H]+ m/z 451.1, found 337.2. Preparation of methyl 3-(5-chloro-2-(piperidin-4-ylamino)pyrimidin-4-yl)-1-((2-(trimethylsilyl) ethoxy)methyl)-1H- indazole-6-carboxylate (C-64)
[0239] To a solution of B-31 (1.9 g, 4.70 mmol), M1 (0.9 g, 4.70 mmol) in NMP (20 mL) was added DIEA (1.2 g, 9.40 mmol). The reaction mixture was stirred at 80℃ for 6 h. The mixture was then quenched with water, extracted with EtOAc, the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 60/40 in 20 mins) to afford tert-butyl 4-((5-chloro-4-(3-(methoxycarbonyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazol-4- yl)pyrimidin-2-yl)amino)piperidine-1-carboxylate (1.4 g, yield: 38.7 %) as a yellow solid. LCMS (ESI): calced for C25H39ClN6O5Si [M+H]+ m/z 567.2, found 567.6. [0240] To a solution of methyl 4-(2-{[1-(tert-butyl-$l^{3}-oxy)piperidin-4-yl]amino}-5-chloropyrimidin-4-yl)-1-[(2- methoxyethyl)trimethyl-$l^{5}-silyl]pyrazole-3-carboxylate (1.4 g, 2.46 mmol) in TFA/DCM (2mL/10mL) stirred at 25℃ for 4 h. The LCMS showed the desired MS was detected. The solvent was removed by vacumm to afford C-63 (0.76 g, yield: 78.9 %) as a yellow solid. LCMS (ESI): calced for C14H17ClN6O2 [M+H]+ m/z 451.1, found 337.2. Preparation of methyl 3-(5-chloro-2-(piperidin-4-ylamino)pyrimidin-4-yl)-1-((2-(trimethylsilyl) ethoxy)methyl)-1H- indazole-6-carboxylate (C-64)
 [0241] To a solution of B-52 (1.8 g, 0.004 mol), M-1 (0.97 g, 0.0048 mol) and DIEA (1.03 g, 0.008mol) in NMP (18 mL) was stirred at 80℃ for 6 h. The mixture was extracted with EtOAc (3 x 200 mL), the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 60/40 in 20 mins) to afford methyl 3-(2-((1-(tert- butoxycarbonyl)piperidin-4-yl)amino)-5-chloropyrimidin-4-yl)-1-((2- (trimethylsilyl)ethoxy)methyl)-1H-indazole-6- carboxylate (2.3 g, yield: 80.0%) as a yellow solid. LCMS (ESI): calced for C29H41ClN6O5Si [M+H]+ m/z 617.3, found 617.5. [0242] To a solution of methyl 3-(2-((1-(tert-butoxycarbonyl)piperidin-4-yl)amino)-5-chloropyrimidin-4-yl)-1-((2- (trimethylsilyl)ethoxy)methyl)-1H-indazole-6-carboxylate (2.3 g, 0.003 mol) in DCM (36 mL) was stirred at 25℃ was added TFA (3.6 mL). The reaction mixture was stirred at 25℃ for 4 h. The LCMS showed the desired MS was detected. Then the mixture was concentrated, quenched with water, adjusted to pH = 7 - 8 with NaHCO3, extracted with EtOAc, the combined organic phase was dried over anhydrous Na2SO4. The mixture was concentrated to get crude product which was purified flash chromatography to afford C-64 (1.4 g, yield: 67.6%) as a yellow solid. LCMS (ESI): calced for C24H33ClN6O3Si [M+H]+ m/z 517.2, found 517.2.
[0243] The compounds of the disclosure were prepared by carboxylic acid / amine condensation reactions, as noted in the below scheme:
[0241] To a solution of B-52 (1.8 g, 0.004 mol), M-1 (0.97 g, 0.0048 mol) and DIEA (1.03 g, 0.008mol) in NMP (18 mL) was stirred at 80℃ for 6 h. The mixture was extracted with EtOAc (3 x 200 mL), the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to get crude product which was purified by flash chromatography (eluting with PE/EtOAc from 100/0 to 60/40 in 20 mins) to afford methyl 3-(2-((1-(tert- butoxycarbonyl)piperidin-4-yl)amino)-5-chloropyrimidin-4-yl)-1-((2- (trimethylsilyl)ethoxy)methyl)-1H-indazole-6- carboxylate (2.3 g, yield: 80.0%) as a yellow solid. LCMS (ESI): calced for C29H41ClN6O5Si [M+H]+ m/z 617.3, found 617.5. [0242] To a solution of methyl 3-(2-((1-(tert-butoxycarbonyl)piperidin-4-yl)amino)-5-chloropyrimidin-4-yl)-1-((2- (trimethylsilyl)ethoxy)methyl)-1H-indazole-6-carboxylate (2.3 g, 0.003 mol) in DCM (36 mL) was stirred at 25℃ was added TFA (3.6 mL). The reaction mixture was stirred at 25℃ for 4 h. The LCMS showed the desired MS was detected. Then the mixture was concentrated, quenched with water, adjusted to pH = 7 - 8 with NaHCO3, extracted with EtOAc, the combined organic phase was dried over anhydrous Na2SO4. The mixture was concentrated to get crude product which was purified flash chromatography to afford C-64 (1.4 g, yield: 67.6%) as a yellow solid. LCMS (ESI): calced for C24H33ClN6O3Si [M+H]+ m/z 517.2, found 517.2.
[0243] The compounds of the disclosure were prepared by carboxylic acid / amine condensation reactions, as noted in the below scheme:
 [0244] The following compounds were synthesized by common carboxylic acid-amine condensation reactions, e.g., conditions (A) or (B) as described below. [0245] (A) To a solution of corresponding carboxylic acid (0.5 mmol) in DMF (5 mL) was added HATU (1.0 mmol) and N, N-Diisopropylethylamine (1.5 mmol) at 25℃. After 20 min, intermediate C (0.55 mmol) in DMF (1 mL) was added. The reaction mixture was stirred at 1 h for 25 ℃. The desired mass was detected on LC-MS. The residue was quenched by ice water, extracted with EtOAc. The organic phases were combined, washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by was purified by C18 column (eluting with ACN-H2O (0.1% NH3.H2O) from 5/95 from 80/20 in 30 mins) to afford final compound. [0246] (B) To a solution of corresponding carboxylic acid (0.5 mmol) in DMF (8 mL) were added TCFH (1.0 mmol) and NMI (1.5 mmol). The reaction solution was stirred at rt for 0.5 h and intermediate C (0.55 mmol) was added. The reaction solution was stirred at rt for 3 h. The LCMS showed the desired MS was detected. The mixture was concentrated and was purified by prep-HPLC (columns: Gemini 5 um C18150 x 21.2 mm, mobile phase: ACN – H2O (0.1% FA), gradient: 50 - 70, 12 min) to afford final compound.
[0244] The following compounds were synthesized by common carboxylic acid-amine condensation reactions, e.g., conditions (A) or (B) as described below. [0245] (A) To a solution of corresponding carboxylic acid (0.5 mmol) in DMF (5 mL) was added HATU (1.0 mmol) and N, N-Diisopropylethylamine (1.5 mmol) at 25℃. After 20 min, intermediate C (0.55 mmol) in DMF (1 mL) was added. The reaction mixture was stirred at 1 h for 25 ℃. The desired mass was detected on LC-MS. The residue was quenched by ice water, extracted with EtOAc. The organic phases were combined, washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by was purified by C18 column (eluting with ACN-H2O (0.1% NH3.H2O) from 5/95 from 80/20 in 30 mins) to afford final compound. [0246] (B) To a solution of corresponding carboxylic acid (0.5 mmol) in DMF (8 mL) were added TCFH (1.0 mmol) and NMI (1.5 mmol). The reaction solution was stirred at rt for 0.5 h and intermediate C (0.55 mmol) was added. The reaction solution was stirred at rt for 3 h. The LCMS showed the desired MS was detected. The mixture was concentrated and was purified by prep-HPLC (columns: Gemini 5 um C18150 x 21.2 mm, mobile phase: ACN – H2O (0.1% FA), gradient: 50 - 70, 12 min) to afford final compound.


















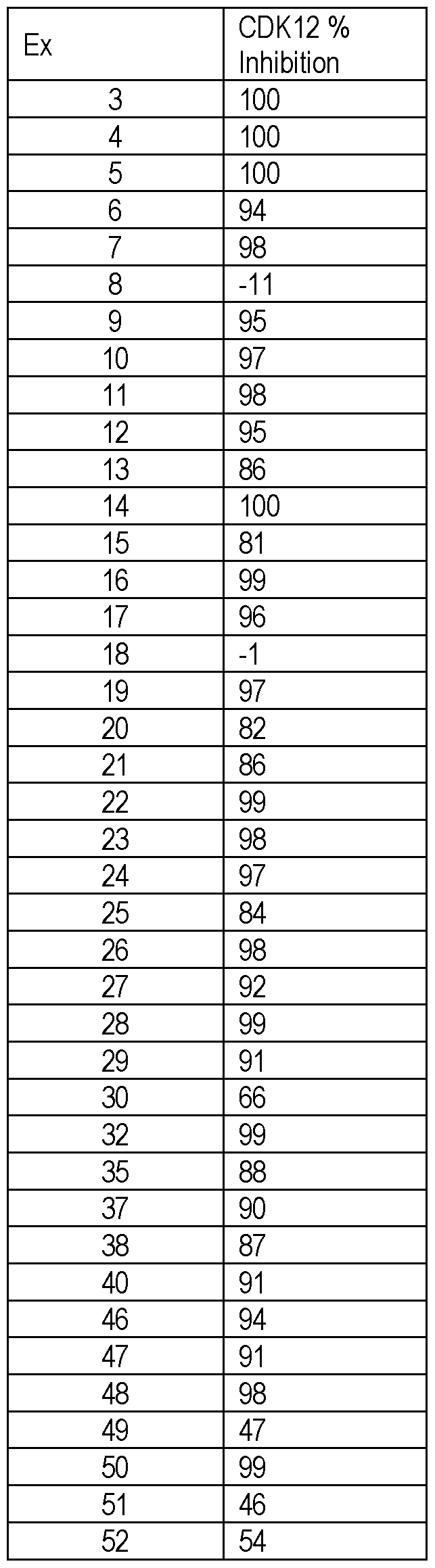





Claims
We Claim: 1. A compound having a structure of Formula (I):
 or a pharmaceutically acceptable salt thereof; wherein: ring A is a 5-7-membered heterocycle comprising 0 or 1 additional ring nitrogen atoms, and ring A can be optionally substituted with 1 to 3 C1-3alkyl substituents; ring B is phenyl or a 6-10-membered heteroaryl comprising 1 or 2 ring nitrogen atoms; X is CH or N; n is 0, 1, or 2; R1 is halo, CN, SOC1-3alkyl, or SO2C1-3alkyl; each R2 is independently halo, CN, OH, –C0-6alkylene-N(RN)2, CO2RN, SO2C1-6alkyl, CORN, C1-6alkyl, C1- 6alkoxy, C1-6haloalkyl, –C1-6hydroxyalkyl, –OC2-6hydroxyalkyl, –OC2-6alkylene-N(RN)2, CON(RN)2, SOsC1-6alkyl, CON(RN)-4-8-membered heterocycle, C(O)-4-8-membered heterocycle, –[O]0-1-4-8 membered heterocycle, or – [O]0-1-5-10 membered heteroaryl, wherein each heterocycle and heteroaryl comprises 1, 2, or 3 ring heteroatoms selected from N, O, and S, and the heterocycle or heteroaryl is optionally substituted with 1 or 2 C1-6alkyl, and when ring B is a heterocycle or a 8-10-membered heteroaryl, R2 can be oxo; R3 is H, halo, CN, C1-3alkyl, C1-3haloalkyl, or C(O)N(RN)2; R4 is phenyl or a 5-10-membered heteroaryl comprising 1 to 3 ring atoms independently selected from N, O, and S, and R4 is optionally substituted with 1, 2, or 3 Ra; R5 is H or C1-3alkyl; each Ra is independently halo, OH, C1-6alkyl, C1-6alkoxy, C1-6hydroxyalkyl, CO2C1-6alkyl, CON(RN)2, C0- 6alkylene-N(RN)2, C0-6alkylene-C3-10cycloalkyl, or a 4-6-membered heterocycle comprising 1 or 2 ring atoms selected from N, O, and S; and each RN is independently H, C1-6alkyl, C1-6hydroxyalkyl, or C1-6alkylene-N(C1-3alkyl)2, or two RN groups, together with a nitrogen to which they are each attached, form a 4-10 membered heterocycle comprising 0-2 additional ring heteroatoms independently selected from N, O, and S, and can be optionally substituted with 1 or 2 C1-6alkyl.
or a pharmaceutically acceptable salt thereof; wherein: ring A is a 5-7-membered heterocycle comprising 0 or 1 additional ring nitrogen atoms, and ring A can be optionally substituted with 1 to 3 C1-3alkyl substituents; ring B is phenyl or a 6-10-membered heteroaryl comprising 1 or 2 ring nitrogen atoms; X is CH or N; n is 0, 1, or 2; R1 is halo, CN, SOC1-3alkyl, or SO2C1-3alkyl; each R2 is independently halo, CN, OH, –C0-6alkylene-N(RN)2, CO2RN, SO2C1-6alkyl, CORN, C1-6alkyl, C1- 6alkoxy, C1-6haloalkyl, –C1-6hydroxyalkyl, –OC2-6hydroxyalkyl, –OC2-6alkylene-N(RN)2, CON(RN)2, SOsC1-6alkyl, CON(RN)-4-8-membered heterocycle, C(O)-4-8-membered heterocycle, –[O]0-1-4-8 membered heterocycle, or – [O]0-1-5-10 membered heteroaryl, wherein each heterocycle and heteroaryl comprises 1, 2, or 3 ring heteroatoms selected from N, O, and S, and the heterocycle or heteroaryl is optionally substituted with 1 or 2 C1-6alkyl, and when ring B is a heterocycle or a 8-10-membered heteroaryl, R2 can be oxo; R3 is H, halo, CN, C1-3alkyl, C1-3haloalkyl, or C(O)N(RN)2; R4 is phenyl or a 5-10-membered heteroaryl comprising 1 to 3 ring atoms independently selected from N, O, and S, and R4 is optionally substituted with 1, 2, or 3 Ra; R5 is H or C1-3alkyl; each Ra is independently halo, OH, C1-6alkyl, C1-6alkoxy, C1-6hydroxyalkyl, CO2C1-6alkyl, CON(RN)2, C0- 6alkylene-N(RN)2, C0-6alkylene-C3-10cycloalkyl, or a 4-6-membered heterocycle comprising 1 or 2 ring atoms selected from N, O, and S; and each RN is independently H, C1-6alkyl, C1-6hydroxyalkyl, or C1-6alkylene-N(C1-3alkyl)2, or two RN groups, together with a nitrogen to which they are each attached, form a 4-10 membered heterocycle comprising 0-2 additional ring heteroatoms independently selected from N, O, and S, and can be optionally substituted with 1 or 2 C1-6alkyl.
2. The compound or salt of claim 1, wherein ring A is pyrrolidine, piperidine, 3- azabicyclo[3.1.0]hexane, or azepane.
3. The compound or salt of claim 2, wherein ring A is piperidinine and is substituted with 1 or 2 C1-3alkyl substituents.
4. The compound or salt of claim 1, having a structure of Formula (Ia):
 5. The compound or salt of any one of claims 1 to 4, wherein ring B is phenyl, pyridine, pyrimidine, pyrazine, pyridazine, quinolone, or 4a,
5. The compound or salt of any one of claims 1 to 4, wherein ring B is phenyl, pyridine, pyrimidine, pyrazine, pyridazine, quinolone, or 4a,
5,6,7-tetrahydro-1,6-naphthyridine.
6. The compound or salt of claim 5, wherein ring B is phenyl or pyridyl.
7. The compound or salt of any one of claims 1 to 6, having a structure of Formula (Ib):

8. The compound or salt of any one of claims 1 to 7, wherein X is CH.
9. The compound or salt of any one of claims 1 to 7, wherein X is N.
10. The compound or salt of any one of claims 1 to 9, wherein R1 is halo, CN, or SO2C1-3alkyl.
11. The compound or salt of claim 10, wherein R1 is F, Cl, CN, or SO2CH3.
12. The compound or salt of claim 11, wherein R1 is F.
13. The compound or salt of any one of claims 1 to 12, wherein n is 0.
14. The compound or salt of any one of claims 1 to 12, wherein n is 1 or 2.
15. The compound or salt of claim 14, wherein each R2 is independently halo, CN, OH, CO2H, COC1-6alkyl, –C0-6alkylene-N(RN)2, C1-6alkyl, C1-6alkoxy, C1-6haloalkyl, –C1-6hydroxyalkyl, –OC2-6hydroxyalkyl, – OC2-6alkylene-N(RN)2, CON(RN)2, –[O]0-1-4-8 membered heterocycle.
16. The compound or salt of claim 14 or 15, wherein each R2 is independently Br, CN, OH, C1- 6haloalkyl, –C0-6alkylene-N(RN)2, OC2-6hydroxyalkyl, –OC2-6alkylene-N(RN)2, or –[O]0-1-4-8 membered heterocycle.
17. The compound or salt of claim 14, wherein at least one R2 is oxo.
18. The compound or salt of any one of claims 1 to 17, wherein R3 is halo or CN.
19. The compound or salt of claim 18, wherein R3 is Cl or CN.
20. The compound or salt of any one of claims 1 to 19, wherein R4 is indole, 1,7- dihydropyrazolo[1,5-a]pyridine, indazole, benzo[d]imidazole, 4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazine, imidazo[1,2-a]pyridine, pyrazolo[4,3-b]pyridine, [1,2,4]triazolo[4,3-a]pyridine, benzo[d][1,2,3]triazole, phenyl, pyridine, pyrazole, or imidazole.
21. The compound or salt of claim 20, wherein R4 is indole, 1,7-dihydropyrazolo[1,5-a]pyridine, indazole, benzo[d]imidazole, pyrazolo[4,3-b]pyridine, phenyl, pyridine, pyrazole, or imidazole.
22. The compound or salt of any one of claims 1 to 21, wherein R4 is substituted with 1, 2, or 3 Ra.
23. The compound or salt of claim 22, wherein each Ra is independently halo, C1-6alkoxy, C1-6alkyl, CO2C1-6alkyl, C1-6hydroxyalkyl, or a 4-6-membered heterocycle comprising 1 or 2 ring atoms selected from N, O, and S.
24. The compound or salt of claim 23, wherein each Ra is independently F, Cl, C1-6alkyl, C1-6alkoxy, C1-6hydroxyalkyl, or a 4-6-membered heterocycle comprising 1 or 2 ring atoms selected from N, O, and S.
25. The compound or salt of any one of claims 1 to 24, wherein R5 is H.
26. The compound or salt of any one of claims 1 to 24, wherein R5 is C1-3alkyl.
27. The compound or salt of any one of claims 1 to 26, wherein each RN is independently H or C1- 6alkyl.
28. A compound, or pharmaceutically acceptable salt thereof, as recited in Table A.
29. The compound or salt of claim 28 selected from
 ,
,

Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CNPCT/CN2023/070607 | 2023-01-05 | ||
| CN2023070607 | 2023-01-05 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024148210A1 true WO2024148210A1 (en) | 2024-07-11 |
Family
ID=89905916
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/010390 Ceased WO2024148210A1 (en) | 2023-01-05 | 2024-01-05 | Cyclin-dependent kinase 12 modulators and therapeutic uses thereof |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2024148210A1 (en) |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007089768A2 (en) * | 2006-01-30 | 2007-08-09 | Exelixis, Inc. | 4-aryl-2-amino-pyrimidines or 4-aryl-2-aminoalkyl-pyrimidines as jak-2 modulators and pharmaceutical compositions containing them |
| KR20120021370A (en) * | 2010-07-28 | 2012-03-09 | 한국과학기술연구원 | 2-Azacyclicamino-4-phenylpyrimidine Derivatives Acting as Iv-β Inhibitors |
| WO2014165065A1 (en) * | 2013-03-13 | 2014-10-09 | Janssen Pharmaceutica Nv | Substituted piperidine compounds and their use as orexin receptor modulators |
| WO2015058163A2 (en) * | 2013-10-18 | 2015-04-23 | Syros Pharmaceuticals, Inc. | Inhibitors of cyclin-dependent kinase 7 (cdk7) |
-
2024
- 2024-01-05 WO PCT/US2024/010390 patent/WO2024148210A1/en not_active Ceased
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007089768A2 (en) * | 2006-01-30 | 2007-08-09 | Exelixis, Inc. | 4-aryl-2-amino-pyrimidines or 4-aryl-2-aminoalkyl-pyrimidines as jak-2 modulators and pharmaceutical compositions containing them |
| KR20120021370A (en) * | 2010-07-28 | 2012-03-09 | 한국과학기술연구원 | 2-Azacyclicamino-4-phenylpyrimidine Derivatives Acting as Iv-β Inhibitors |
| WO2014165065A1 (en) * | 2013-03-13 | 2014-10-09 | Janssen Pharmaceutica Nv | Substituted piperidine compounds and their use as orexin receptor modulators |
| WO2015058163A2 (en) * | 2013-10-18 | 2015-04-23 | Syros Pharmaceuticals, Inc. | Inhibitors of cyclin-dependent kinase 7 (cdk7) |
Non-Patent Citations (1)
| Title |
|---|
| S. M. BERGE ET AL., J. PHARMACEUTICAL SCIENCES, vol. 66, 1977, pages 1 - 19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7460593B2 (en) | Inhibitor of cyclin-dependent kinase 7 (CDK7) | |
| KR102374844B1 (en) | Isoquinolin-3-yl carboxamide and its preparations and uses | |
| CN108024970B (en) | Substituted Heterocyclic Derivatives for Use as CDK Inhibitors | |
| RU2435769C2 (en) | Pyrrolopyridines effective as proteinkinase inhibitors | |
| US9114137B2 (en) | Derivatives of pyrazolophenyl-benzenesulfonamide compounds and use thereof as antitumor agents | |
| US20210317122A1 (en) | Substituted heterocyclic inhibitors of ptpn11 | |
| KR20190012167A (en) | Isoquinolin-3-ylcarboxamide, its preparation and uses | |
| EA028626B1 (en) | Tnf-alpha modulating benzimidazoles | |
| TW201247673A (en) | Imidazo[5,1-f][1,2,4]triazines for the treatment of neurological disorders | |
| AU2014219855A1 (en) | Trk-inhibiting compound | |
| JP2018528193A (en) | Indole derivatives, methods for their preparation and their use in medicine | |
| JP2014051516A (en) | 5-cyano-4-(pyrrolo[2,3b]pyridine-3-yl)-pyrimidine derivatives useful as protein kinase inhibitors | |
| KR20240127910A (en) | Pharmaceutical compositions for treating cancers comprising sos1 inhibitors and anticancer drugs | |
| EA007063B1 (en) | AMINOBENZAMIDE DERIVATIVES AS GLYCOGEN SYNTHASE KINASE 3β INHIBITORS | |
| JP2015503504A (en) | Heteroaryl and uses thereof | |
| CA2799146A1 (en) | Nitrogen-containing heterocyclic compound having inhibitory effect on production of kynurenine | |
| JP2013533318A (en) | Heteroaryl and uses thereof | |
| JP2020502230A (en) | Pyrazolo [3,4-b] pyridine and imidazo [1,5-b] pyridazine as PDE1 inhibitors | |
| WO2021224818A1 (en) | Isoindolone compounds as hpk1 inhibitors | |
| AU2024321427A1 (en) | Pharmaceutical compounds and compositions as c-kit kinase inhibitors | |
| WO2024145505A1 (en) | Pyrimidine carboxamide compounds | |
| TW202421134A (en) | Methods for treating cancer | |
| WO2024148210A1 (en) | Cyclin-dependent kinase 12 modulators and therapeutic uses thereof | |
| JP2025514670A (en) | Pyrido[3,2-d]pyrimidines as HPK1 inhibitors | |
| WO2016139212A1 (en) | Triazolopyridine compounds and methods of use thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24704975 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 24704975 Country of ref document: EP Kind code of ref document: A1 |