WO2023186659A1 - Elektrochemische oxidation von cycloalkanen zu cycloalkanon-verbindungen - Google Patents
Elektrochemische oxidation von cycloalkanen zu cycloalkanon-verbindungen Download PDFInfo
- Publication number
- WO2023186659A1 WO2023186659A1 PCT/EP2023/057342 EP2023057342W WO2023186659A1 WO 2023186659 A1 WO2023186659 A1 WO 2023186659A1 EP 2023057342 W EP2023057342 W EP 2023057342W WO 2023186659 A1 WO2023186659 A1 WO 2023186659A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- reaction medium
- group
- unsubstituted
- alkyl
- particularly preferably
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C25—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES; APPARATUS THEREFOR
- C25B—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES FOR THE PRODUCTION OF COMPOUNDS OR NON-METALS; APPARATUS THEREFOR
- C25B3/00—Electrolytic production of organic compounds
- C25B3/01—Products
- C25B3/07—Oxygen containing compounds
-
- C—CHEMISTRY; METALLURGY
- C25—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES; APPARATUS THEREFOR
- C25B—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES FOR THE PRODUCTION OF COMPOUNDS OR NON-METALS; APPARATUS THEREFOR
- C25B11/00—Electrodes; Manufacture thereof not otherwise provided for
- C25B11/04—Electrodes; Manufacture thereof not otherwise provided for characterised by the material
- C25B11/042—Electrodes formed of a single material
- C25B11/043—Carbon, e.g. diamond or graphene
-
- C—CHEMISTRY; METALLURGY
- C25—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES; APPARATUS THEREFOR
- C25B—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES FOR THE PRODUCTION OF COMPOUNDS OR NON-METALS; APPARATUS THEREFOR
- C25B3/00—Electrolytic production of organic compounds
- C25B3/20—Processes
- C25B3/23—Oxidation
-
- C—CHEMISTRY; METALLURGY
- C25—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES; APPARATUS THEREFOR
- C25B—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES FOR THE PRODUCTION OF COMPOUNDS OR NON-METALS; APPARATUS THEREFOR
- C25B9/00—Cells or assemblies of cells; Constructional parts of cells; Assemblies of constructional parts, e.g. electrode-diaphragm assemblies; Process-related cell features
- C25B9/13—Single electrolytic cells with circulation of an electrolyte
- C25B9/15—Flow-through cells
-
- C—CHEMISTRY; METALLURGY
- C25—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES; APPARATUS THEREFOR
- C25B—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES FOR THE PRODUCTION OF COMPOUNDS OR NON-METALS; APPARATUS THEREFOR
- C25B9/00—Cells or assemblies of cells; Constructional parts of cells; Assemblies of constructional parts, e.g. electrode-diaphragm assemblies; Process-related cell features
- C25B9/17—Cells comprising dimensionally-stable non-movable electrodes; Assemblies of constructional parts thereof
Definitions
- the invention relates to a process for producing unsubstituted or at least monosubstituted cycloalkanones by electrochemical oxidation of unsubstituted or at least monosubstituted saturated cycloaliphatic hydrocarbons by electrochemical oxidation in the presence of an inorganic or organic nitrate salt in an electrolysis cell in a reaction medium in the presence of oxygen.
- Cycloalkanone and cycloalkanol compounds are important intermediates in a variety of industrial manufacturing processes.
- the oxidation of saturated, non-functionalized cycloaliphatic hydrocarbons (and thus non-activated C-H bonds) to corresponding ketones or alcohols requires special reaction conditions in order to selectively convert these inert substances into monofunctional secondary products to be transferred while preserving the ring framework.
- the present invention relates to a process for producing unsubstituted or at least monosubstituted cycloalkanones by electrochemical oxidation of unsubstituted or at least monosubstituted saturated cycloaliphatic hydrocarbons, comprising the process steps
- step (c) electrochemical oxidation of the unsubstituted or at least monosubstituted saturated cycloaliphatic hydrocarbon provided in step (a) in the presence of the organic provided in step (b). Nitrate salt in an electrolysis cell in a reaction medium in the presence of oxygen.
- the process according to the invention is characterized in particular by high selectivity, small amounts of auxiliary chemicals used, the use of electric current as an oxidizing agent and, associated with this, by a reduced amount of waste products.
- atmospheric oxygen can be used to introduce the oxygen function into cycloaliphatic hydrocarbons.
- chemical oxidizing agents such as reactive peroxides, and high-priced catalysts with complex ligand systems can be dispensed with.
- toxic and/or potentially carcinogenic reagents can be reduced or even completely avoided.
- the method developed represents a cost-effective and environmentally friendly alternative to existing syntheses. Due to the simple and safe process conditions, large quantities of the desired compounds can be produced without great effort. Through the present invention, previously cost- and time-intensive processes can be significantly optimized.
- the process according to the invention enables the use of electric current to produce cycloalkanone compounds from unsubstituted cycloalkanes using nitrate salts, which function both as a conductive salt and as an electrochemical mediator. If by-products arise when carrying out the process according to the invention, in particular cycloaliphatic alcohols of the same ring size, this is not a problem because they can be converted into the corresponding ketones by already established subsequent processes.
- the method according to the invention can be carried out under ambient pressure and ambient temperature, which also has an advantageous effect on energy efficiency and thus also environmental compatibility.
- Unsubstituted or at least monosubstituted, saturated cycloaliphatic hydrocarbons that are monocyclic or polycyclic can be used in the process according to the invention.
- Monocyclic or bicyclic cycloaliphatic hydrocarbons are preferred.
- Monocyclic cycloaliphatic hydrocarbons are particularly preferably used in the process according to the invention.
- the monocyclic or polycyclic, in particular monocyclic or bicyclic, saturated cycloaliphatic hydrocarbons used in the process according to the invention can preferably have 5 to 18 C atoms in the ring system.
- These cycloaliphatic hydrocarbons can each be unsubstituted or mono- or poly-substituted. If they are substituted once or multiple times, they are preferably substituted with 1, 2, 3, 4 or 5 substituents, independently of one another, each selected from the group consisting of methyl, phenyl or benzyl.
- phenyl or benzyl substituents themselves can each be unsubstituted or mono- or polysubstituted, with 1, 2 or 3 substituents, independently of one another, each selected from the group consisting of F, CI, Br, and NO2. If the cycloaliphatic hydrocarbons used according to the invention or their substituents have alkyl radicals with more than one carbon atom in the side chain, undesirable side reactions occur at these substituents when carrying out the process according to the invention.
- Particularly preferred in the process according to the invention are monocyclic saturated hydrocarbons with 6 to 12 carbon atoms in the ring, preferably with 8 to 12 carbon atoms in the ring, which are unsubstituted or substituted once or multiple times with 1 as unsubstituted or at least monosubstituted, saturated cycloaliphatic hydrocarbons , 2, 3, 4 or 5 substituents, independently of one another, each selected from the group consisting of methyl, phenyl or benzyl.
- monocyclic saturated hydrocarbons with 8 to 12 carbon atoms in the ring in the process according to the invention, which are unsubstituted or mono- or di- or tri-substituted with a methyl group.
- the saturated monocyclic hydrocarbon is unsubstituted and is selected from the group consisting of cyclohexane, Cycloheptane, cyclooctane, cyclononane, cyclodecane, cycloundecane and cyclododecane, even more preferably selected from the group consisting of cyclooctane, cyclononane, cyclodecane, cycloundecane and cyclododecane, most preferably the hydrocarbon cyclododecane.
- step (b) of the process according to the invention at least one organic nitrate salt is provided.
- This nitrate salt acts both as a conductive salt and as a mediator in the electrochemical oxidation process according to the invention.
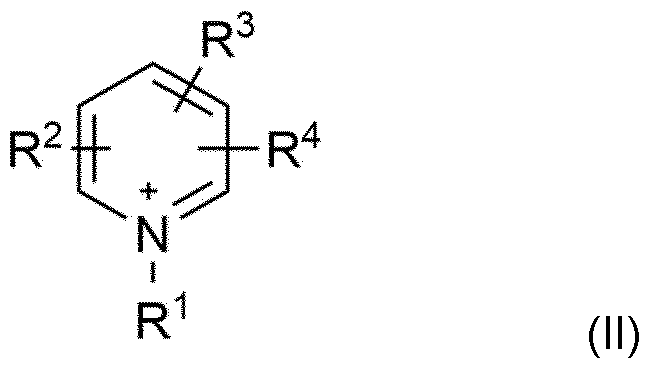
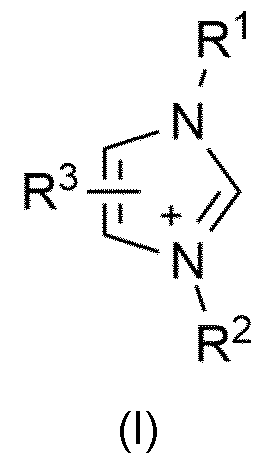
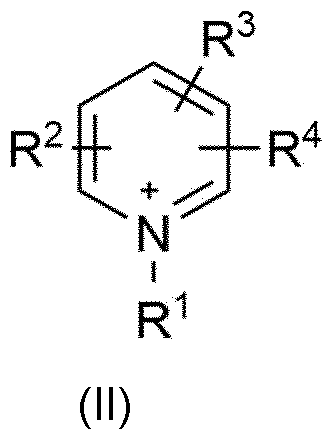
- An organic nitrate of the general formula [cation + ][NO 3 '] is preferably used, the [cation + ] being selected from the group consisting of ammonium ions with the general structure [R 1 R 2 R 3 R 4 N + ] with R 1 , R 2 , R 3 , R 4 , independently of one another, each selected from the group consisting of Ci to Ci6 alkyl, in particular Ci to Cs alkyl, straight-chain or branched, imidazolium cations with the general structure (I)
- ) with R 1 and R 2 independently of one another, each selected from the group consisting of Ci- to Cis-alkyl, straight-chain or branched, in particular Ci- to Cs-alkyl, straight-chain or branched, and R 3 selected from the group consisting of H and Ci- to Cis-alkyl, straight-chain or branched, in particular from the group consisting of H and Ci- to Cs-alkyl, straight-chain or branched,
- Phosphonium ions with the general structure [R 1a R 2a R 3a R 4a P + ] with R 1a , R 2a , R 3a , R 4a , independently of one another, each selected from the group consisting of Ci- to Ci6-alkyl, in particular Ci- to Cs-alkyl, straight chain or branched.
- R 1 and R 2 independently of one another, are each selected from the group consisting of Ci to Cis-alkyl, straight-chain or branched, in particular Ci- to Cs-alkyl, straight-chain or branched and R 3 represents hydrogen.
- Particular preference is given to imidazolium cations of the general formula (I) in which R 1 is methyl and R 2 is ethyl or R 1 is methyl and R 2 is methyl and R 1 is methyl and R 2 is butyl, as well as R 3 each represents hydrogen.
- R 1 is Ci- to Cis-alkyl, straight-chain or branched, in particular for Ci- to Cs -Alkyl, straight chain or branched.
- nitrate salt according to the invention is preferably used, in particular an organic ammonium nitrate salt of the composition [R 1 R 2 R 3 R 4 N + ][NO3'] or an organic phosphonium salt of the composition [R 1a R 2a R 3a R 4a P + ] [NO3'], where an organic Ammonium nitrate salt of the composition [R 1 R 2 R 3 R 4 N + ][NC>3'] is particularly preferred.
- the organic ammonium nitrate salt tetra-n-butyl ammonium nitrate or methyl tri-n-octylammonium nitrate is very particularly preferred.
- the organic phosphonium nitrate salt is most preferably tetra-n-butylphosphonium nitrate or methyltri-n-octylphosphonium nitrate.
- the organic imidazolium nitrate salt is preferably 1-butyl-3-methylimidazolium nitrate.
- Tetra-n-butyl ammonium nitrate or methyl tri-n-octylammonium nitrate is most preferably used as the organic nitrate salt in the process according to the invention.
- the unsubstituted or at least monosubstituted saturated cycloaliphatic hydrocarbon or the organic nitrate salt is introduced and brought into contact with the reaction medium, preferably at least partially or completely dissolved in the reaction medium or mixed with it, and then the other of these two components added.
- the unsubstituted or at least monosubstituted, saturated cycloaliphatic hydrocarbon and the organic nitrate salt are introduced and then brought together with the reaction medium, preferably at least partially or completely dissolved in the reaction medium or mixed with it.
- the unsubstituted or at least monosubstituted, saturated cycloaliphatic hydrocarbon and the inorganic or organic nitrate salt are added to the reaction medium simultaneously or in succession to one another, preferably at least partially or completely dissolved in the reaction medium or with it be mixed.
- the reaction medium used in the process according to the invention is liquid under the conditions under which the process is carried out and is suitable for partially or completely dissolving the components used, ie in particular the unsubstituted or at least monosubstituted, saturated cycloaliphatic hydrocarbon and the inorganic or organic nitrate salt. If at least one of these components is used in liquid form, the reaction medium is preferably easily miscible with this component or these components.
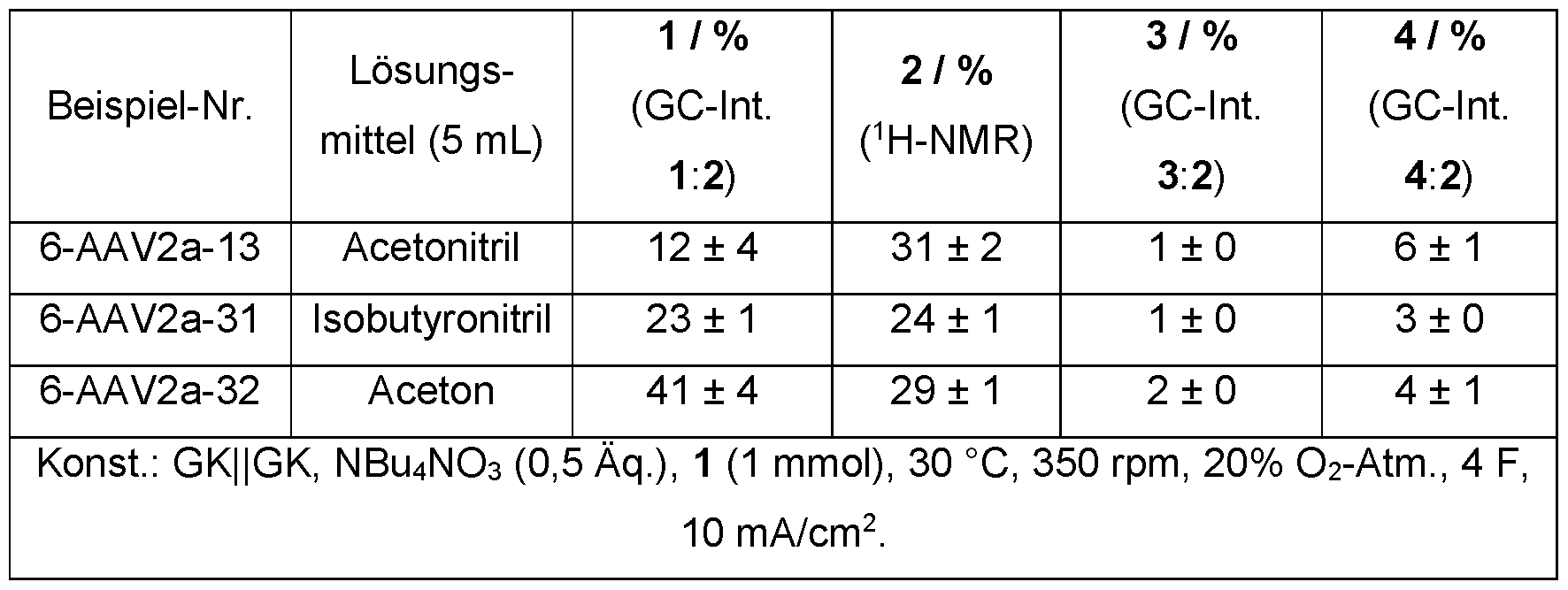
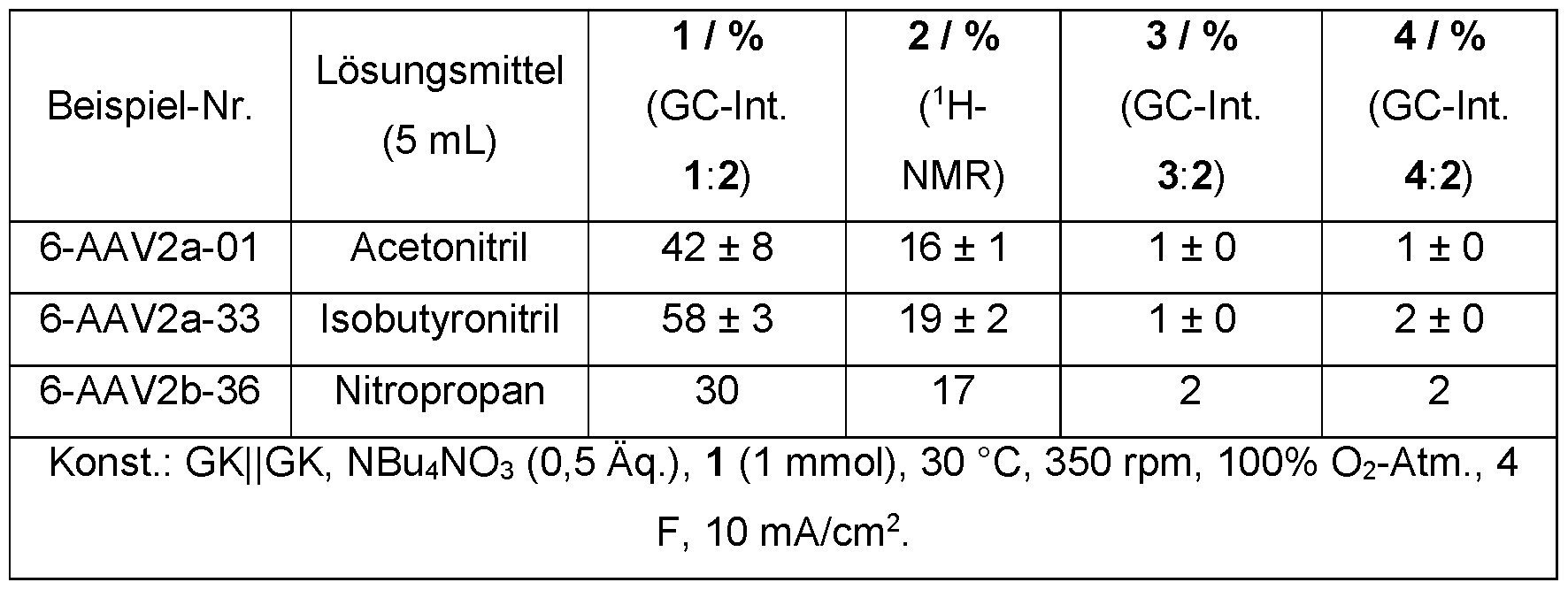
- a polar aprotic reaction medium is preferably used in the process according to the invention for electrochemical oxidation. This can be used in anhydrous form, in dried form or in combination with water.
- the reaction medium advantageously contains water, with aprotic reaction medium in combination with water being preferred.
- the water content in the reaction medium can vary.
- the water content is preferably up to 20% by volume, particularly preferably up to 15% by volume, very particularly preferably up to 10% by volume, even more preferably up to 5% by volume, in each case on the total amount of reaction medium.
- the polar aprotic reaction medium is preferably selected from the group consisting of aliphatic nitriles, aliphatic ketones, cycloaliphatic ketones, dialkyl carbonates, cyclic carbonates, lactones, aliphatic nitroalkanes, dimethyl sulfoxide, esters and ethers or a combination of at least two of these components.
- the reaction medium is particularly preferably selected from the group consisting of acetonitrile, isobutyronitrile, adiponitrile, acetone, dimethyl carbonate, methyl ethyl ketone, 3-pentanone, cyclohexanone, nitromethane, nitropropane, tert-butyl methyl ether, dimethyl sulfoxide, gamma-butyrolactone and epsilon-caprolactone or a combination from at least two of these components.
- the reaction medium is selected from the group consisting of acetonitrile, isobutyronitrile, adiponitrile, dimethyl carbonate and acetone or a combination of at least two of these components.
- the reaction medium is very particularly preferably acetonitrile, isobutyronitrile or adiponitrile in dried or anhydrous form.
- the reaction medium is also very particularly preferred: acetonitrile, isobutyronitrile or adiponitrile, optionally in combination with water.
- the water content is preferably up to 20% by volume, particularly preferably up to 15% by volume, very particularly preferably up to 10% by volume. %, even more preferably up to 5% by volume, based on the total amount of reaction medium.
- solubilizing components it may be advantageous to add further solubilizing components to the reaction medium.
- Suitable advantageous components can be determined through simple preliminary tests on solution behavior.
- Suitable solubilizing components include, for example, primary alcohols, secondary alcohols, monoketones or dialkyl carbonates or mixtures of at least two of these components, possibly in combination with water.
- Aliphatic Ci-6 alcohols can preferably be used in the process according to the invention, with particularly preferred solubilizing components being selected from the group consisting of methanol, ethanol, isopropanol, 2-methyl-2-butanol or mixtures of at least two of these components, if necessary in combination with water.
- dimethyl carbonate as a reaction medium can be particularly advantageous, if necessary in combination with at least one Ci-6 alcohol, in particular selected from the group consisting of methanol, ethanol, isopropanol, 2-methyl-2-butanol, if necessary in combination with water be.
- Ci-6 alcohol in particular selected from the group consisting of methanol, ethanol, isopropanol, 2-methyl-2-butanol, if necessary in combination with water be.
- the water content is preferably up to 20% by volume, particularly preferably up to 15% by volume, very particularly preferably up to 10% by volume more preferably up to 5% by volume, based on the total amount of solubilizing component and water.
- the solubilizing components can preferably be added in amounts of ⁇ 50% by volume, particularly preferably ⁇ 30% by volume and very particularly preferably ⁇ 10% by volume, in each case based on the total amount of reaction medium.
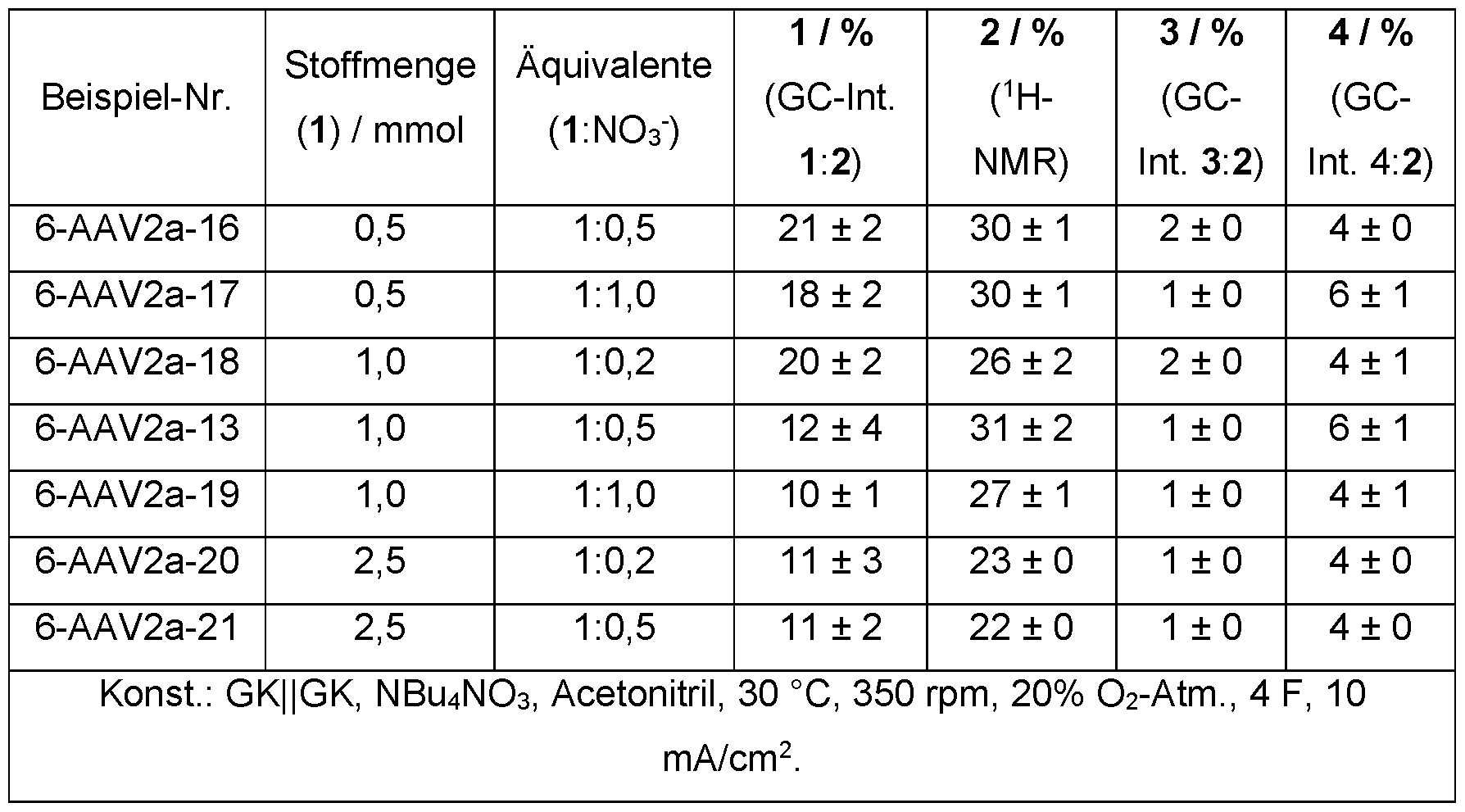
- the organic nitrate salt is preferably used in the process according to the invention in an amount of 0.1 to 2.0, preferably 0.2 to 1.0, particularly preferably 0.3 to 0.8 and very particularly preferably 0.4 to 0.8 equivalents, each based on the amount of unsubstituted or at least monosubstituted, saturated cycloaliphatic hydrocarbon used.
- the electrochemical oxidation of the unsubstituted or at least monosubstituted saturated cycloaliphatic hydrocarbon takes place in the presence of the inorganic or organic nitrate salt in an electrolysis cell in a reaction medium in the presence of oxygen.
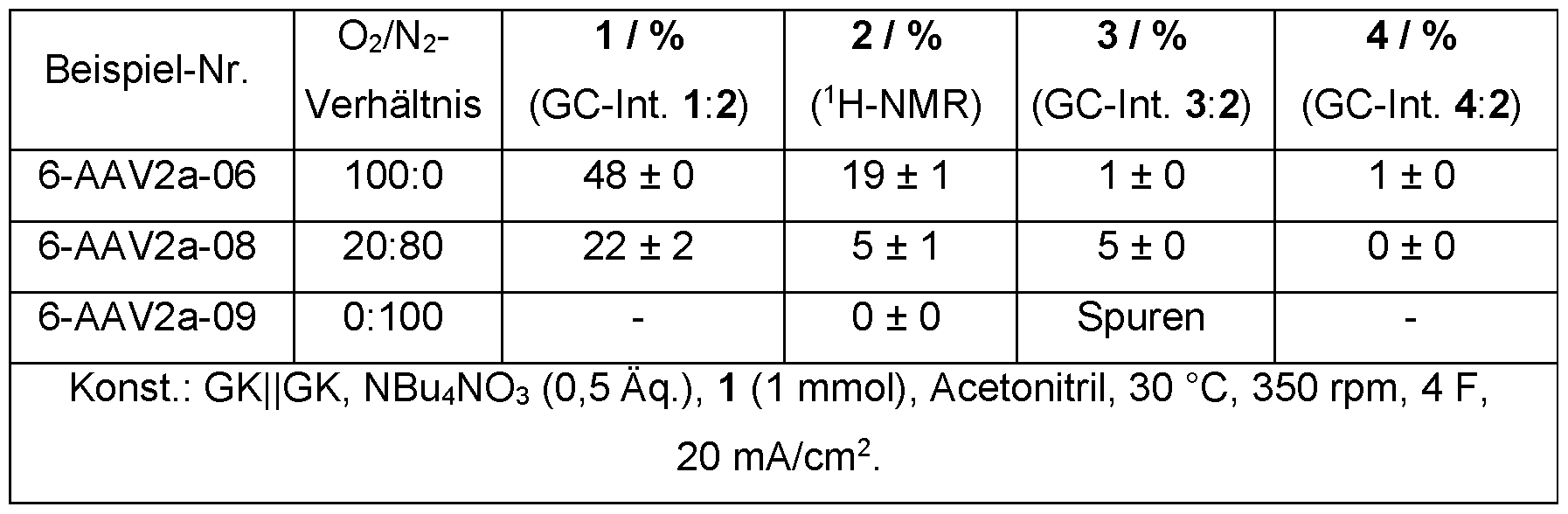
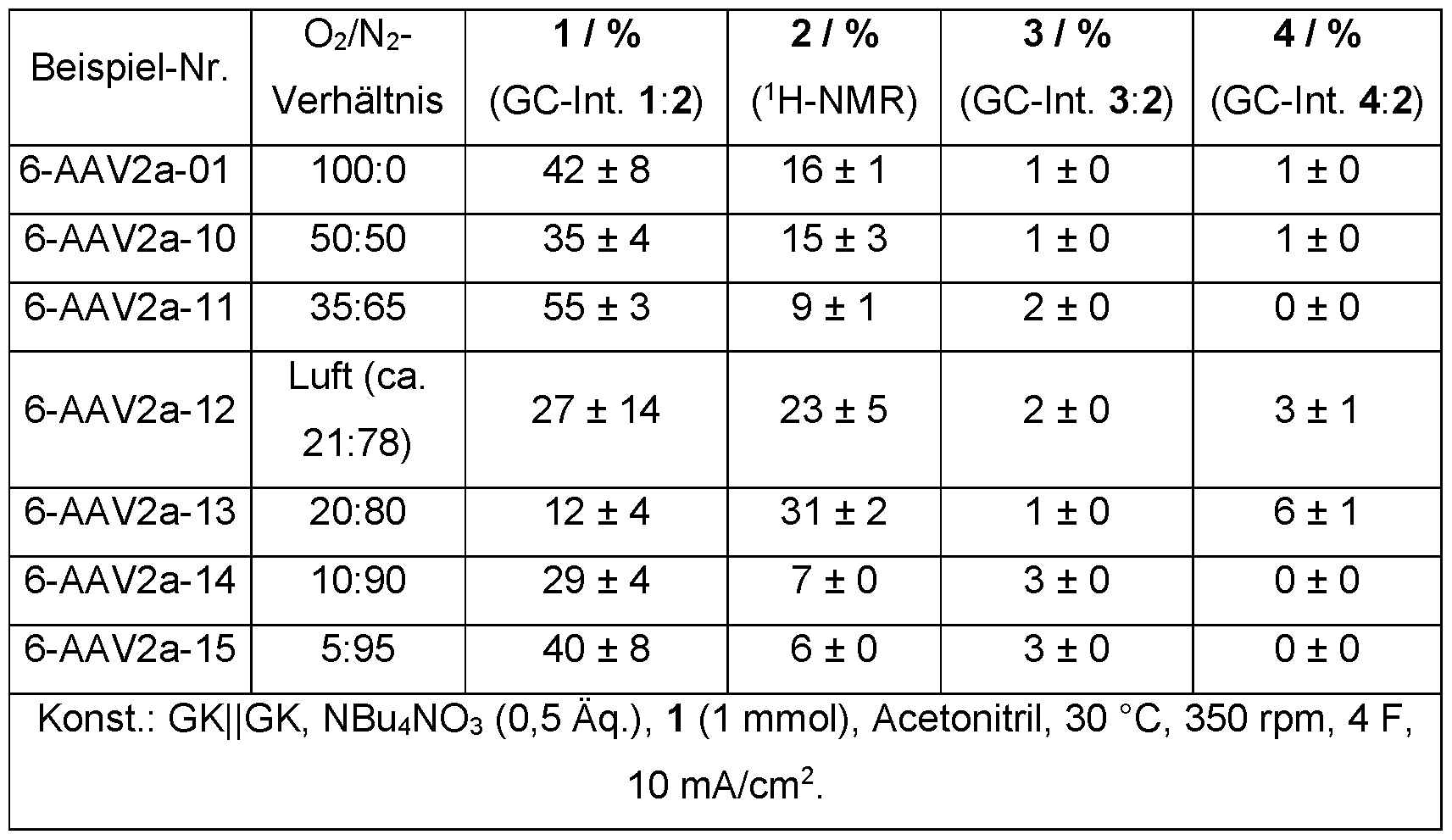
- a gas atmosphere containing oxygen is advantageously provided in spatial connection with the reaction medium.
- the proportion of oxygen in the gas atmosphere can vary.
- the proportion of oxygen in the gas atmosphere is preferably 10 to 100% by volume, particularly preferably 15 to 30% by volume, particularly preferably 15 to 25% by volume, very particularly preferably 18 to 22% by volume.
- the proportion of oxygen in the gas atmosphere can be 10 to 100% by volume, particularly preferably 15 to 100% by volume, particularly preferably 20 to 100% by volume.
- the gas atmosphere is particularly preferably air.
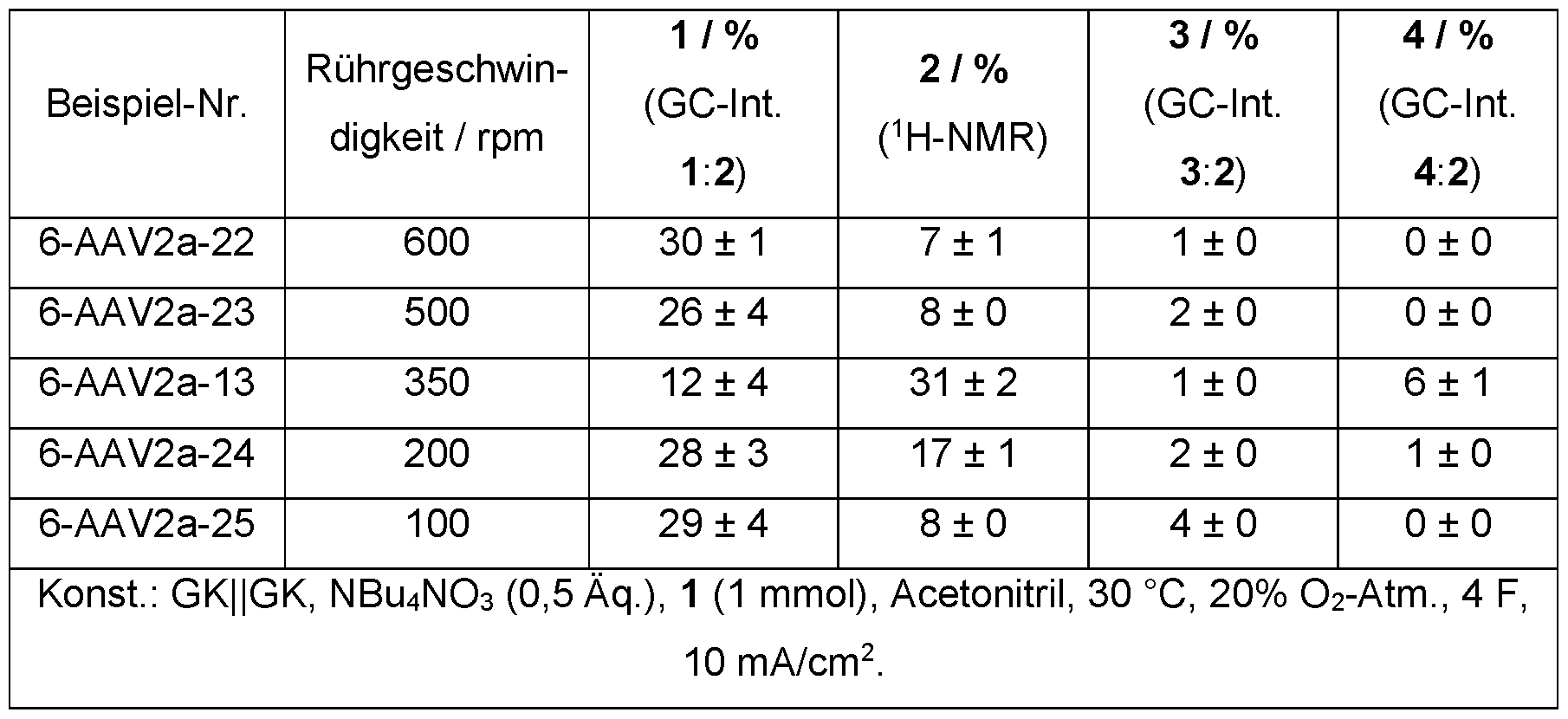
- a gas exchange is advantageously forced between the gas atmosphere and the reaction medium, preferably by introducing a gas atmosphere into the reaction medium or by stirring the liquid phase in the presence of the gas atmosphere.
- the gas exchange between the gas atmosphere and the reaction medium in particular stirring, for example via the geometry of the stirrer or the stirring speed, can be used to control the electrochemical oxidation.
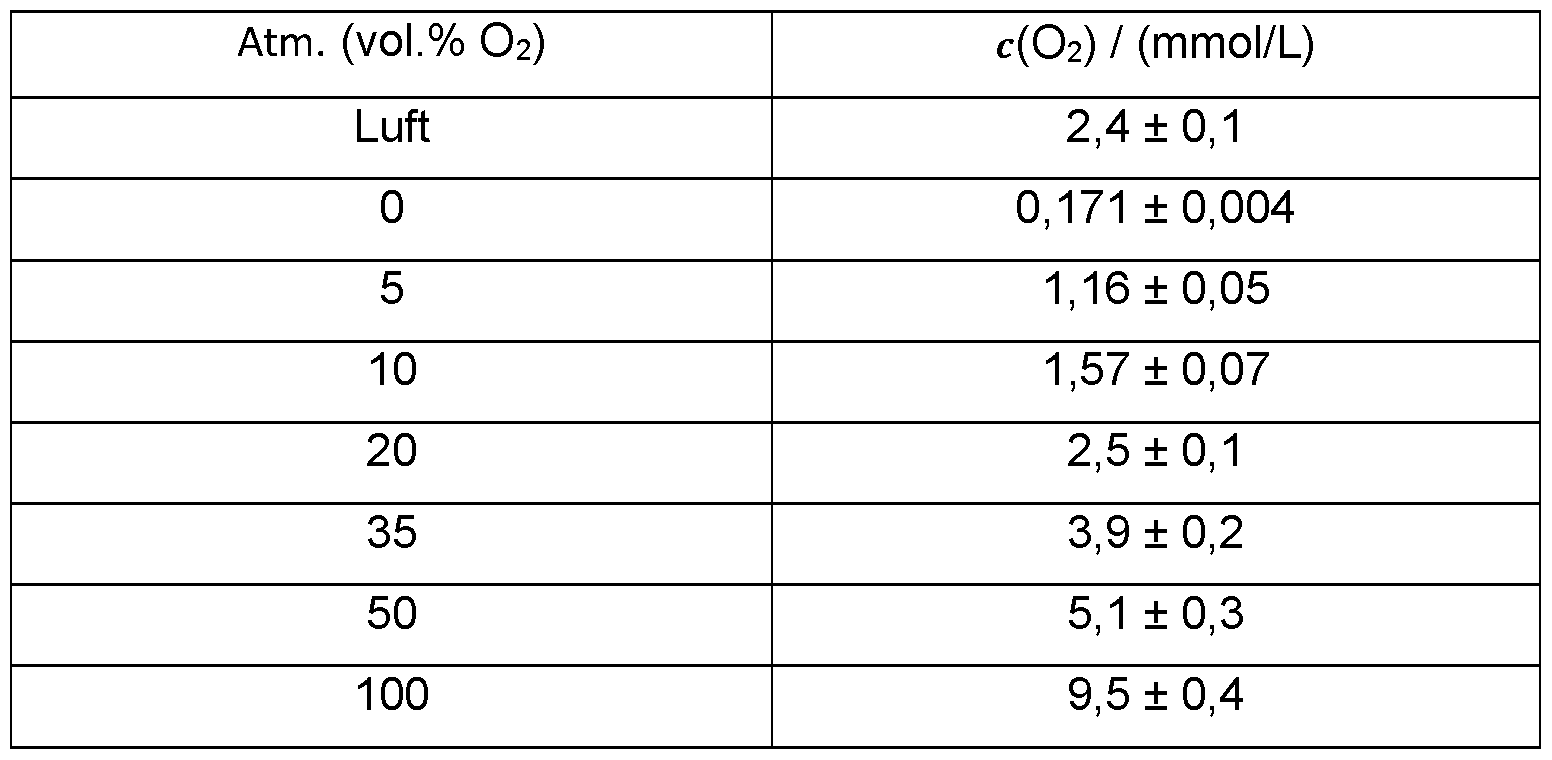
- the amount of oxygen dissolved in the reaction medium is preferably at least 1 mmol/L reaction medium, particularly preferably at least 5 mmol/L reaction medium.
- the amount of oxygen dissolved in the reaction medium is also preferably at least 10 mmol/L reaction medium.
- the process according to the invention for producing unsubstituted or at least monosubstituted cycloalkanones by electrochemical oxidation of unsubstituted or at least monosubstituted saturated cycloaliphatic hydrocarbons by electrochemical oxidation in the presence of an inorganic or organic nitrate salt in a reaction medium in the presence of oxygen can be carried out in both a divided and carry out in an undivided electrolysis cell, with implementation in an undivided electrolysis cell being preferred.
- the undivided electrolysis cell which is preferably used according to the invention has at least two electrodes.
- Anodes and cathodes of common materials can be used here, for example glassy carbon, boron-doped diamond (BDD) or graphite.
- BDD boron-doped diamond
- the use of glassy carbon electrodes is preferred.
- the undivided electrolysis cell preferably has at least one glassy carbon anode or at least one glassy carbon cathode. Both the anode and the cathode are preferably glassy carbon electrodes.
- the distance between the electrodes can vary over a certain range.
- the distance is preferably 0.1 mm to 2.0 cm, particularly preferably 0.1 mm to 1.0 cm, particularly preferably 0.1 mm to 0.5 cm.
- process according to the invention can be carried out batchwise or continuously, preferably in an undivided flow-through electrolysis cell.
- the process according to the invention is preferably carried out with a charge amount of 190 C (2 F) to 970 C (10 F), particularly preferably 320 C to 820 C, very particularly preferably 350 C to 800 C, even more preferably 380 C to 775 C most preferably 380 C to 450 C, in each case based on 1 mmol of unsubstituted or at least monosubstituted saturated cycloaliphatic hydrocarbon.
- the electrochemical oxidation in the process according to the invention preferably takes place at constant current intensity.
- the current density at which the method according to the invention is carried out is preferably at least 5 mA/cm 2 or at least 10 mA/cm 2 or at least 15 mA/cm 2 or at least 20 mA/cm 2 or 20 mA/cm 2 to 50 mA/ cm 2 , whereby the area refers to the geometric area of the electrodes.
- a significant advantage of the method according to the invention is that electric current is used as the oxidizing agent, which is a particularly environmentally friendly agent when it comes from renewable sources, i.e. in particular from biomass, solar thermal energy, geothermal energy, hydropower, wind power or photovoltaics.
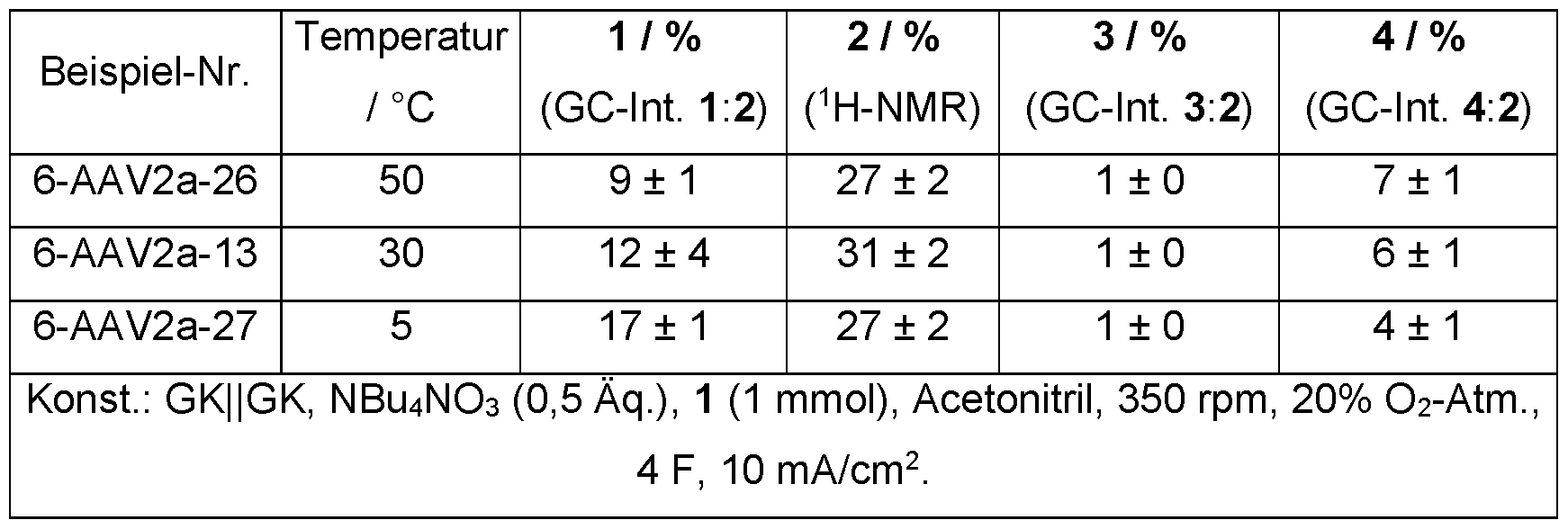
- the process according to the invention can be carried out over a wide temperature range, for example at a temperature in the range from 0 to 60°C, preferably from 5 to 50°C, particularly preferably 10 to 40°C, very particularly preferably 15 to 30°C.
- the process according to the invention can be carried out at increased or reduced pressure. If the process according to the invention is carried out at elevated pressure, a pressure of up to 16 bar is preferred, particularly preferably up to 6 bar.
- the process according to the invention can also preferably be carried out under atmospheric pressure.
- the products produced by the process according to the invention can be isolated or purified by conventional processes known to those skilled in the art, in particular by extraction, crystallization, centrifugation, precipitation, distillation, evaporation or chromatography.
- Analytical grade chemicals were purchased and used from mainstream suppliers (such as TCI, Aldrich, and Acros).
- the oxygen was purchased in 2.5 quality from NIPPON GASES GmbH, Düsseldorf, Germany and used directly.
- NMR spectrometry of 1 H-NMR and 13C-NMR spectra were recorded at 25 °C with a Bruker Avance II 400 (400 MHz, 5 mm BBFO head with z-gradient and ATM, SampleXPress 60 sample changer, Analytician Messtechnik, Düsseldorf, Germany ) recorded.
- the gas introduction was controlled via two mass flow controllers (MFC) model 5850S from Brooks Instrument B.V., Veenendaal, Netherlands.
- MFC mass flow controller
- a regulator was used for the oxygen and nitrogen lines.
- the controllers were controlled using the Smart DDE and Matlab R2017b software.
- the volume flow control was also carried out using a DK800 variable area flowmeter from KR ⁇ HNE Messtechnik GmbH, Duisburg. For all tests carried out, the total volume flow was a constant 20 mL/min, which, limited by the MFCs used, also represents the maximum achievable volume flow.
- the percentage volume flows of the two gases were set using the MFCs and their software.
- the gas bottles were used from the following suppliers: oxygen 2.5 from NIPPON GASES GmbH, Düsseldorf, and nitrogen 4.8 from Nonetheless AG, Weg and nitrogen 5.0 from NIPPON GASES GmbH, Düsseldorf.
- the apparatus was equipped with a gas distributor including an adapter and a Teflon cover for the electrolysis cells.
- the cycloalkane (5.0 mmol) and tetrabutylammonium nitrate (0.5 eq.) were placed in an undivided electrolysis cell (100 mL three-neck round bottom flask, NS29 Teflon stopper with electrode holders, magnetic stirring rod) and dissolved in acetonitrile (25 mL).
- the cell was equipped with glassy carbon electrodes (3 cm ⁇ 1 cm ⁇ 0.3 cm), spaced 0.5 cm apart.
- the immersion area of the electrodes was 1.3 cm 2 . If necessary, oxygen was introduced into the gas space of the reaction vessel via an NS14.5 core olive. It Galvanostatic electrolysis was carried out at a current density of 10 mA/cm 2 at 20 to 30 °C.
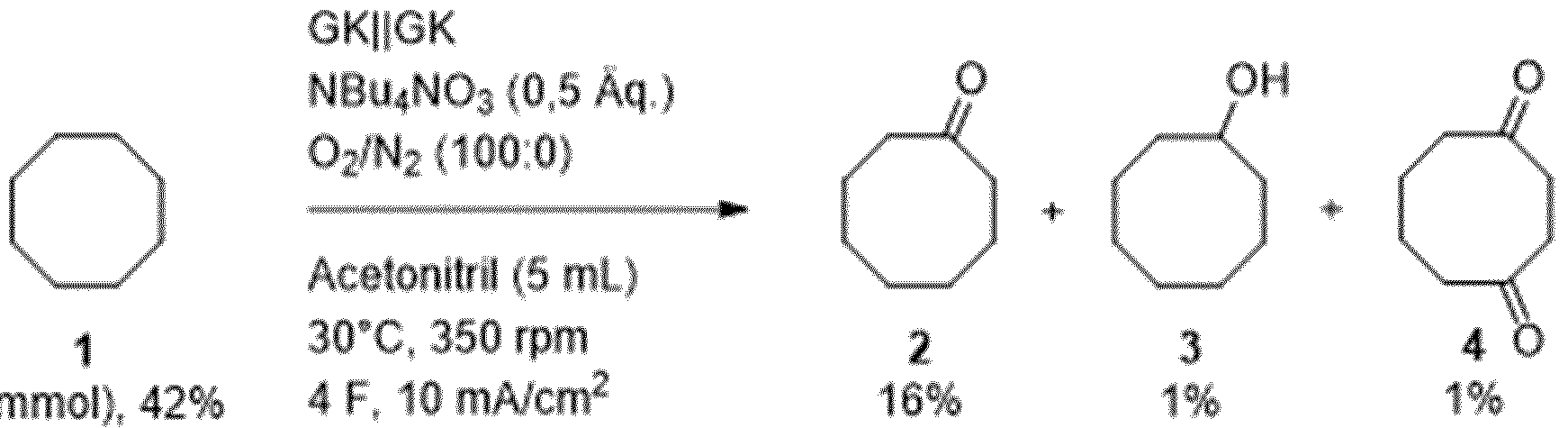
- Cyclooctanone According to AAV1, cyclooctane (0.561 g, 5.0 mmol, 1.0 eq.) was dissolved in acetonitrile (25 mL) and electrolyzed galvanostatically at 30 °C under an oxygen atmosphere with the application of 4 F. After workup according to AAV1, the product was obtained as a colorless liquid (yield: 42%, 0.261 g, 2.07 mmol).
- the electrolyses were carried out in an undivided 5 mL PTFE cell.
- the conductive salt (0.2 to 1.0 eq.) and the substrate (cyclooctane, 0.5-2.5 mmol) were placed in the cell and dissolved in the solvent (5 mL).
- the cell was equipped with a glassy carbon anode and cathode, which had a distance of 0.5 cm (electrode dimension: 7 cm x 1 cm x 0.3 cm, immersion area 1.8 cm 2 ).
- the cells were fixed in a heatable/coolable stainless steel block and supplied with the gas mixture to be examined (100 vol.% O2 to 0 vol.% O2) via an adapter.
- the electrolysis was carried out with constant current intensity, with the current densities (5-60 mA/cm 2 ), temperatures (5-50 ° C), stirring speeds (100-600 rpm) and amounts of charge (4-8 F) being varied. After applying the charge amount, 2 drops of the reaction solution were removed for gas chromatographic analysis. 1,3,5-Trimethoxybenzene is then added to the solution as an NMR standard (1 eq.) and the solvent is removed by distillation (45 ° C, 200 mbar). The yield of the cycloalkanone product was determined using 1 H NMR analysis.
- the electrolyses were carried out in an undivided 5 mL PTFE cell.
- the conductive salt 0.2-1.0 eq.
- the substrate cyclooctane, 0.5-2.5 mmol
- the cell was equipped with a glassy carbon anode and cathode, which had a distance of 0.5 cm (electrode dimension: 7 cm x 1 cm x 0.3 cm, immersion area 1.8 cm 2 ).
- the cells were fixed in a heatable/coolable stainless steel block and supplied with the gas mixture to be examined (100 vol.% O2 to 0 vol.% O2) via an adapter.
- the electrolysis was carried out with constant current strength, with the current densities (5-60 mA/cm 2 ), temperatures (5-50 ° C), stirring speeds (100-600 rpm) and charge quantities (4-8 F) varied.
- 10 mg of 1,3,5-trimethoxybenzene was added to the reaction solution as an internal standard.
- Three drops of the reaction solution were removed for gas chromatographic analysis and quantification of the product. These were eluted using ethyl acetate over approx. 330 mg of 60 M silica gel.
- Approximately 1.5 mL of the filtrate was collected in a GC vial, which was examined for oxidation products using GC-FID and GC-MS.
- the quantification was carried out via a previous calibration of the gas chromatograph.
- Nitrate salt as a mediator/conducting salt
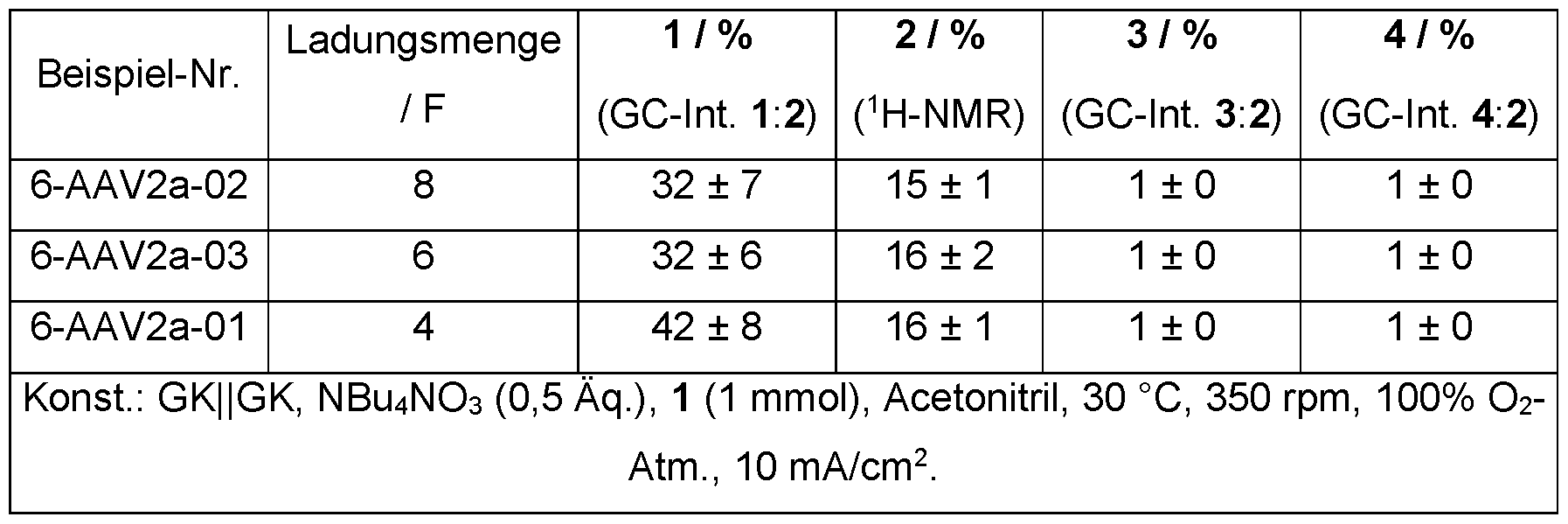
- the amount of charge was examined in a range from 4 F to 8 F (corresponding to 386 C to 772 C for 1 mmol of substrate cyclooctane 1).
- Table 1 Examination of different amounts of charge.
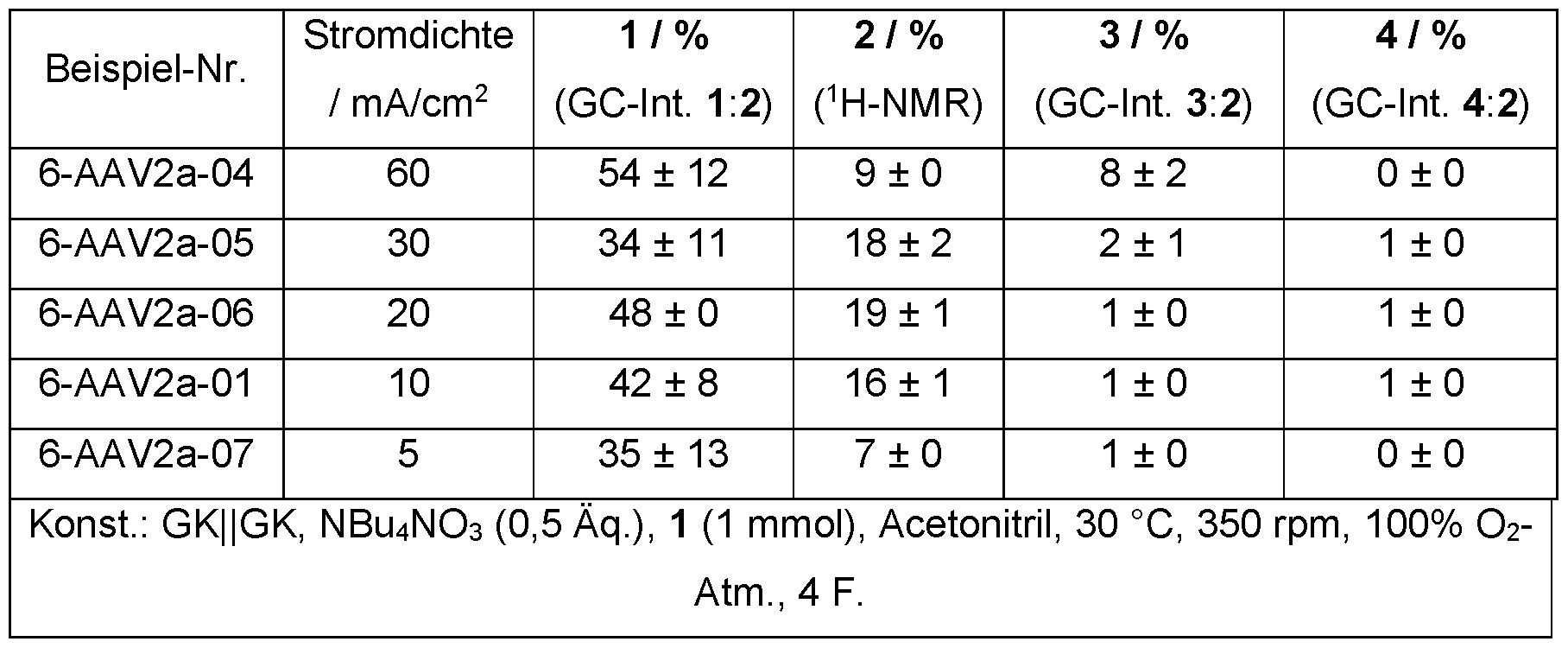
- the current density was varied in the range 5 mA/cm 2 to 60 mA/cm 2 .
- the electrode area in the electrolyte solution was 1.8 cm 2 .
- Example 6e Amount of substance and mediator equivalents To investigate different substrate and mediator concentrations in the solvent (acetonitrile, 5 mL), the amount of substrate and the equivalents of conductive salt/mediator were varied. Table 5: Investigation of different amounts of substances and nitrate equivalents.
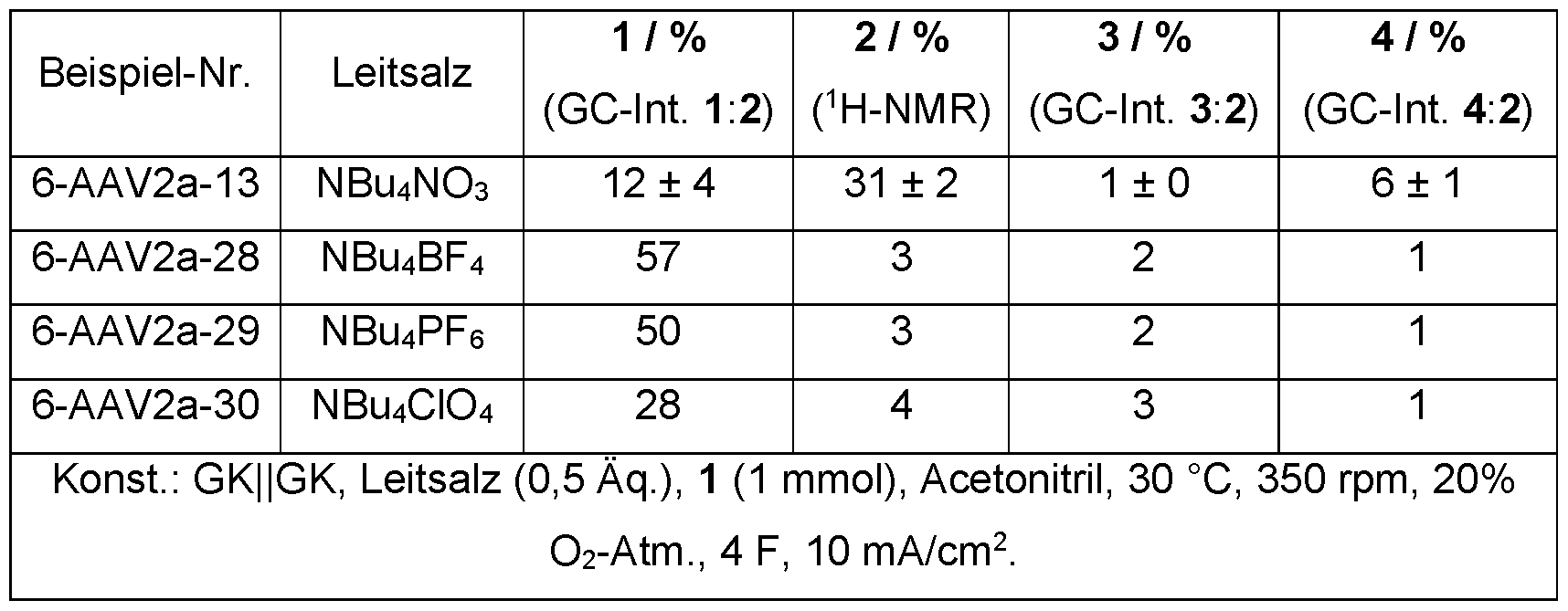
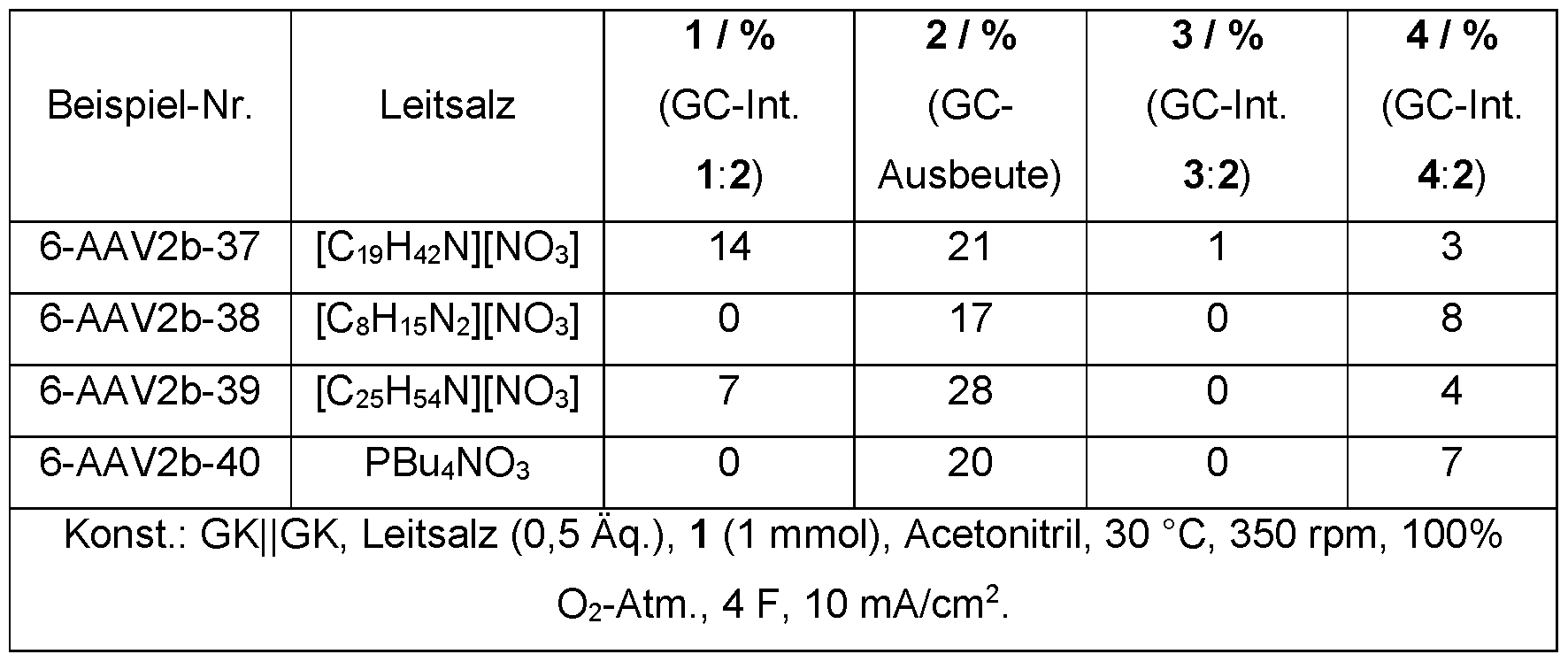
- nitrate as an anion of the conductive salt, also has a mediator effect on the reaction
- the standard tetrabutylammonium nitrate was compared with other common conductive salts.
- the cationic component was left the same.
- the conductive salts that deviate from the standard were only tested once in an electrolysis, which is why no average value was recorded here.
- Table 8 illustrate the dependence of the reaction on the nitrate anion.
- the product 2 forms only to a very small extent with other conductive salt anions.
- 6-AAV2a-09 6-AAV2a-28, 6-AAV2a-29, 6-AAV2a-30
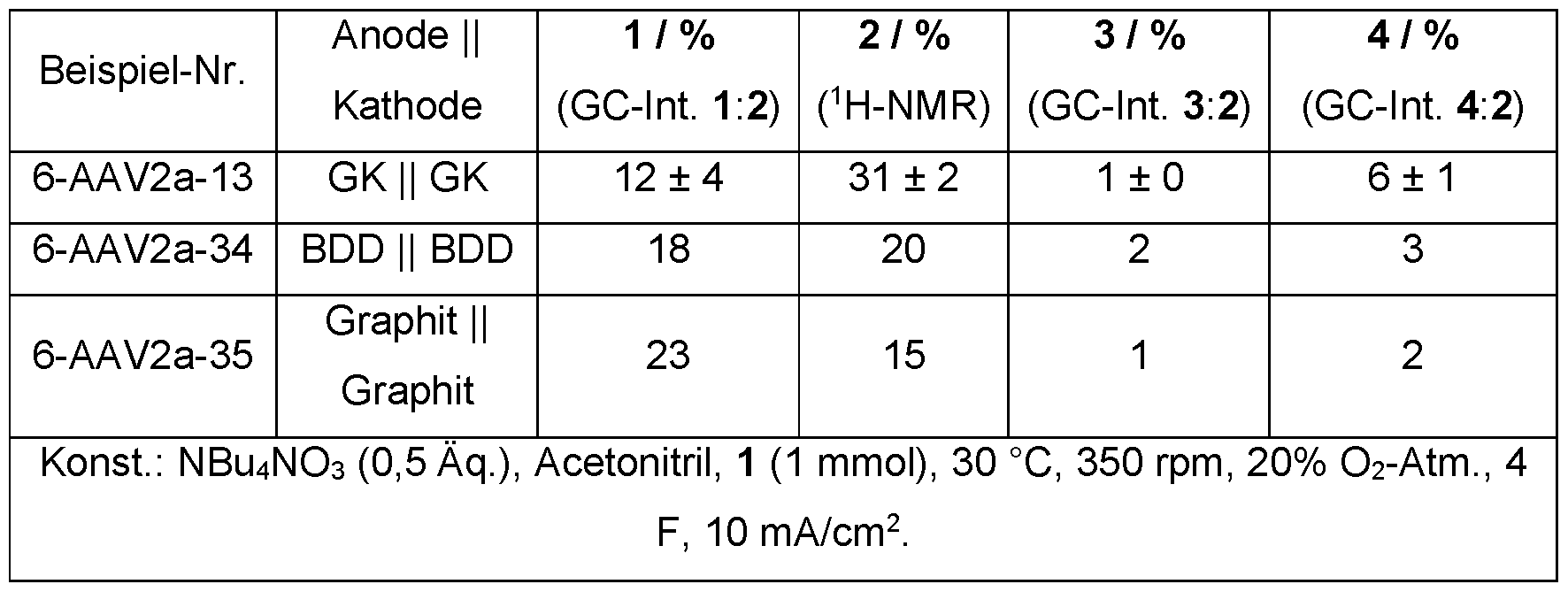
- Electrolysis was carried out on various carbon-based electrode materials.
- the electrode materials that deviate from the GK standard were only tested once in an electrolysis, which is why no average value was recorded here.
- Tetrabutylphosphonium nitrate PBU4NO3
- the anionic nitrate component was left the same.
- the conductive salts that deviate from the standard were only tested once in an electrolysis, which is why no average value was recorded here.
- Table 12 Examination of various cations.
- Example 6I Oxygen solubility in acetonitrile / NBU4NO3 at 25°C and normal pressure:
- Table 13 Dissolved oxygen concentration in MeCN/NBiuNCh depending on the atmospheric oxygen content.
- the cycloalkane (5.0 mmol) and tetrabutylammonium nitrate (0.5 eq.) were dissolved in acetonitrile (25 mL) in an undivided 25 mL glass beaker cell with a gas inlet attachment.
- the cell was equipped with glassy carbon electrodes (7 cm x 1 cm x 0.3 cm) at a distance of 0.5 to 1.0 cm.
- the immersion area of the electrodes was 1.3 cm 2 .
- Galvanostatic electrolysis was carried out at a current density of 10 mA/cm 2 at 20-30 °C.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- Materials Engineering (AREA)
- Metallurgy (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Electrodes For Compound Or Non-Metal Manufacture (AREA)
- Electrolytic Production Of Non-Metals, Compounds, Apparatuses Therefor (AREA)
Abstract
Description

Claims
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US18/850,115 US12571114B2 (en) | 2022-03-28 | 2023-03-22 | Electrochemical oxidation of cycloalkanes to form cycloalkanone compounds |
| JP2024555057A JP7843364B2 (ja) | 2022-03-28 | 2023-03-22 | シクロアルカノン化合物を生成するためのシクロアルカンの電気化学的酸化 |
| CN202380031130.9A CN118974324A (zh) | 2022-03-28 | 2023-03-22 | 电化学氧化环烷烃形成环烷酮化合物 |
| EP23712904.4A EP4499897A1 (de) | 2022-03-28 | 2023-03-22 | Elektrochemische oxidation von cycloalkanen zu cycloalkanon-verbindungen |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP22164767.0 | 2022-03-28 | ||
| EP22164767.0A EP4253603A1 (de) | 2022-03-28 | 2022-03-28 | Elektrochemische oxidation von cycloalkanen zu cycloalkanon-verbindungen |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023186659A1 true WO2023186659A1 (de) | 2023-10-05 |
Family
ID=80978841
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2023/057342 Ceased WO2023186659A1 (de) | 2022-03-28 | 2023-03-22 | Elektrochemische oxidation von cycloalkanen zu cycloalkanon-verbindungen |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US12571114B2 (de) |
| EP (2) | EP4253603A1 (de) |
| JP (1) | JP7843364B2 (de) |
| CN (1) | CN118974324A (de) |
| WO (1) | WO2023186659A1 (de) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN118086927A (zh) * | 2024-01-19 | 2024-05-28 | 清华大学 | 烷烃的氧化方法以及反应装置 |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP4253602A1 (de) | 2022-03-28 | 2023-10-04 | Evonik Operations GmbH | Elektrochemische oxidation von cycloalkenen und cycloalkanen zu alpha,omega-dicarbonsäuren oder ketocarbonsäuren und cycloalkanon-verbindungen |
| EP4253604A1 (de) | 2022-03-28 | 2023-10-04 | Evonik Operations GmbH | Elektrochemische oxidation von fettsäuren und fettsäureestern zu monocarbonsäuren und alpha,omega-dicarbonsäuren |
| EP4253605A1 (de) | 2022-03-28 | 2023-10-04 | Evonik Operations GmbH | Elektrochemische oxidation von cycloalkenen zu alpha,omega-dicarbonsäuren und ketocarbonsäuren |
| EP4632111A3 (de) * | 2024-04-10 | 2026-02-11 | ESy-Labs-GmbH | Verfahren zur herstellung von acetalen von alpha-chlor- oder alpha-brom-aldehyden |
| EP4632110A1 (de) * | 2024-04-10 | 2025-10-15 | ESy-Labs-GmbH | Verfahren zur halogenierung organischer verbindungen |
| CN119082750B (zh) * | 2024-08-30 | 2025-10-17 | 中国科学院上海有机化学研究所 | 一种选择性苄基碳氢键的电化学氧化方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0548141A1 (de) * | 1990-09-13 | 1993-06-30 | Hoechst Aktiengesellschaft | Verfaharen zur herstellung von halogenierten acrylsäuren |
| CN104032327A (zh) * | 2014-06-26 | 2014-09-10 | 天津工业大学 | 一种电化学催化氧化环己烷制备环己醇及环己酮的方法 |
| JP2019099861A (ja) * | 2017-11-30 | 2019-06-24 | 国立研究開発法人産業技術総合研究所 | シクロアルカノール及びシクロアルカノンの製造方法 |
| WO2021260679A1 (en) * | 2020-06-22 | 2021-12-30 | Yeda Research And Development Co. Ltd | Aerobic electrocatalytic oxidation of hydrocarbons |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS59211585A (ja) * | 1983-05-14 | 1984-11-30 | Sugai Kagaku Kogyo Kk | キシレンの電解酸化方法 |
| DE3814498A1 (de) * | 1988-04-29 | 1989-11-09 | Basf Ag | Verfahren zur herstellung von hydroxicarbonsaeureestern |
| JPH03122297A (ja) * | 1989-10-06 | 1991-05-24 | Mitsui Toatsu Chem Inc | カルボン酸ビニルエステルの製造方法 |
| DE19937108A1 (de) * | 1999-08-06 | 2001-02-08 | Basf Ag | Verfahren zur Herstellung von in alpha-Stellung oxidierten Carbonylverbindungen |
| DE19962102A1 (de) * | 1999-12-22 | 2001-06-28 | Basf Ag | Verfahren zur elektrochemischen Oxidation von organischen Verbindungen |
| US6569309B2 (en) * | 2001-07-05 | 2003-05-27 | Asahi Kasei Kabushiki Kaisha | Fuel cell type reactor and method for producing a chemical compound by using the same |
| GB0918616D0 (en) * | 2009-10-23 | 2009-12-09 | 3M Innovative Properties Co | Method of preparing highly fluorinated carboxylic acids and their salts |
| WO2013186094A2 (en) * | 2012-06-15 | 2013-12-19 | Basf Se | Anodic oxidation of organic substrates in the presence of nucleophiles |
| CA2883367A1 (en) * | 2012-09-19 | 2014-03-27 | Liquid Light, Inc. | Electrochemical co-production of chemicals employing the recycling of a hydrogen halide |
| US11851778B2 (en) * | 2017-07-28 | 2023-12-26 | Board Of Trustees Of Michigan State University | Electrochemical reductive carboxylation of unsaturated organic substrates in ionically conductive mediums |
| JP7154595B2 (ja) * | 2019-03-06 | 2022-10-18 | 国立研究開発法人産業技術総合研究所 | 光電気化学反応システムを用いたシクロアルケノンの製造方法 |
| EP3763848A1 (de) * | 2019-07-10 | 2021-01-13 | Technische Universität Berlin | Verfahren zur elektrodicarboxylierung von mindestens einem alken mit kohlendioxid co2 in gegenwart von wasserstoff h2 |
| EP3922758A1 (de) | 2020-06-10 | 2021-12-15 | Evonik Operations GmbH | Verfahren zur elektrochemischen herstellung von alkandicarbonsäuren durch ringöffnende oxidation mittels einer dotierten ni(o)oh schaumelektrode |
| EP3922759A1 (de) * | 2020-06-11 | 2021-12-15 | Minakem | Verfahren zur alpha,beta-desaturierung von verbindungen, die einen carbonylteil enthalten |
| EP4253605A1 (de) | 2022-03-28 | 2023-10-04 | Evonik Operations GmbH | Elektrochemische oxidation von cycloalkenen zu alpha,omega-dicarbonsäuren und ketocarbonsäuren |
| EP4253604A1 (de) | 2022-03-28 | 2023-10-04 | Evonik Operations GmbH | Elektrochemische oxidation von fettsäuren und fettsäureestern zu monocarbonsäuren und alpha,omega-dicarbonsäuren |
| EP4253602A1 (de) | 2022-03-28 | 2023-10-04 | Evonik Operations GmbH | Elektrochemische oxidation von cycloalkenen und cycloalkanen zu alpha,omega-dicarbonsäuren oder ketocarbonsäuren und cycloalkanon-verbindungen |
-
2022
- 2022-03-28 EP EP22164767.0A patent/EP4253603A1/de not_active Ceased
-
2023
- 2023-03-22 JP JP2024555057A patent/JP7843364B2/ja active Active
- 2023-03-22 WO PCT/EP2023/057342 patent/WO2023186659A1/de not_active Ceased
- 2023-03-22 CN CN202380031130.9A patent/CN118974324A/zh active Pending
- 2023-03-22 EP EP23712904.4A patent/EP4499897A1/de active Pending
- 2023-03-22 US US18/850,115 patent/US12571114B2/en active Active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0548141A1 (de) * | 1990-09-13 | 1993-06-30 | Hoechst Aktiengesellschaft | Verfaharen zur herstellung von halogenierten acrylsäuren |
| CN104032327A (zh) * | 2014-06-26 | 2014-09-10 | 天津工业大学 | 一种电化学催化氧化环己烷制备环己醇及环己酮的方法 |
| JP2019099861A (ja) * | 2017-11-30 | 2019-06-24 | 国立研究開発法人産業技術総合研究所 | シクロアルカノール及びシクロアルカノンの製造方法 |
| WO2021260679A1 (en) * | 2020-06-22 | 2021-12-30 | Yeda Research And Development Co. Ltd | Aerobic electrocatalytic oxidation of hydrocarbons |
Non-Patent Citations (5)
| Title |
|---|
| A. KIRSTEG. SCHNAKENBURGF. STECKERA. FISCHERS. R. WALDVOGEL, ANGEW. CHEM. INT. ED., vol. 49, 2010, pages 971 - 975 |
| ANGEW. CHEM., vol. 122, 2010, pages 983 - 987 |
| C. GÜTZB. KLÖCKNERS. R. WALDVOGEL, ORG. PROCESS RES. DEV., vol. 20, 2016, pages 26 - 32 |
| J. AM. CHEM. SOC., no. 139, 2017, pages 7448 - 7551 |
| J. CHEM. COMMUN., 2000, pages 2209 - 2210 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN118086927A (zh) * | 2024-01-19 | 2024-05-28 | 清华大学 | 烷烃的氧化方法以及反应装置 |
Also Published As
| Publication number | Publication date |
|---|---|
| US12571114B2 (en) | 2026-03-10 |
| EP4499897A1 (de) | 2025-02-05 |
| CN118974324A (zh) | 2024-11-15 |
| JP2025510645A (ja) | 2025-04-15 |
| JP7843364B2 (ja) | 2026-04-09 |
| EP4253603A1 (de) | 2023-10-04 |
| US20250215583A1 (en) | 2025-07-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2023186659A1 (de) | Elektrochemische oxidation von cycloalkanen zu cycloalkanon-verbindungen | |
| EP3337801B1 (de) | Verfahren zur herstellung von (4s)-4-(4-cyano-2-methoxyphenyl)-5-ethoxy-2,8-dimethyl-1,4-dihydro-1,6-naphthyridin-3-carboxamid und wiedergewinnung von (4s)-4-(4-cyano-2-methoxyphenyl)-5-ethoxy-2,8-dimethyl-1,4-dihydro-1,6-naphthyridin-3-carboxamid mittels elektrochemischer methoden | |
| EP4499899A1 (de) | ELEKTROCHEMISCHE OXIDATION VON CYCLOALKENEN UND CYCLOALKANEN ZU a,w-DICARBONSÄUREN ODER KETOCARBONSÄUREN UND CYCLOALKANON-VERBINDUNGEN | |
| DE19809532C1 (de) | Verfahren zur Carboxylierung von terminalen Alkinen | |
| WO2023186658A1 (de) | ELEKTROCHEMISCHE OXIDATION VON CYCLOALKENEN ZU α, ω-DICARBONSÄUREN UND KETOCARBONSÄUREN | |
| EP2964813A1 (de) | Elektrochemische kupplung zweier phenole, welche sich in ihrem oxidationspotential unterscheiden | |
| EP3209650A1 (de) | Verfahren zur herstellung von glycerinsäurecarbonat | |
| EP4459011A1 (de) | Elektrochemisches hochdurchsatz-oxidationsverfahren | |
| WO2023186660A1 (de) | Elektrochemische oxidation von fettsäuren und fettsäureestern zu monocarbonsäuren und alpha,omega-dicarbonsäuren | |
| DE69319342T2 (de) | Elektrokatalytische asymmetrische dihydroxylierung von olefinen | |
| EP2964812B1 (de) | Elektrochemische kupplung eines phenols mit einem naphthol | |
| EP0017907B1 (de) | Verfahren zur Herstellung von Cyclohexenderivaten | |
| EP4089202A1 (de) | Anolytfraktion-katalysierte hmf-herstellung | |
| WO2008003620A2 (de) | Elektrochemische herstellung sterisch gehinderter amine | |
| DE102018208510A1 (de) | Salz- und Säuregemisch-katalysierte HMF-Herstellung | |
| EP0702998B1 (de) | Verfahren zur Herstellung von Ketoverbindungen | |
| EP2534281B1 (de) | Verfahren zur herstellung von 4-isopropylcyclohexylmethanol | |
| DE2953189C1 (de) | Verfahren zur Herstellung von Ketonen | |
| EP1413565B1 (de) | Verfahren zur Racematsspaltung von 3-Aminopentannitril | |
| EP0085158B1 (de) | Verfahren zur Herstellung von Cycloalkenonderivaten | |
| Chiba et al. | Electron-Transfer-Induced Intermolecular [2+ 2] Cycloaddition Reactions Assisted by Aromatic “Redox Tag” | |
| Stanciu et al. | ANODIC OXIDATION OF MALIC ACID | |
| DE102010042703A1 (de) | Verfahren zur kontinuierlichen Herstellung von 2-Butanon in heißem Hochdruckwasser mit Elektrolytzusatz |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 23712904 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2024555057 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 18850115 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202380031130.9 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023712904 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2023712904 Country of ref document: EP Effective date: 20241028 |
|
| WWP | Wipo information: published in national office |
Ref document number: 18850115 Country of ref document: US |
|
| WWG | Wipo information: grant in national office |
Ref document number: 18850115 Country of ref document: US |