WO2016106625A1 - Btk inhibitors - Google Patents
Btk inhibitors Download PDFInfo
- Publication number
- WO2016106625A1 WO2016106625A1 PCT/CN2014/095766 CN2014095766W WO2016106625A1 WO 2016106625 A1 WO2016106625 A1 WO 2016106625A1 CN 2014095766 W CN2014095766 W CN 2014095766W WO 2016106625 A1 WO2016106625 A1 WO 2016106625A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- pyrazin
- amino
- imidazo
- trifluoromethyl
- pyridin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC(C)C(C1)OCC(C(*)(*)OC2([Re])[Re])N1C2=O Chemical compound CC(C)C(C1)OCC(C(*)(*)OC2([Re])[Re])N1C2=O 0.000 description 12
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Definitions
- the present invention relates to Btk inhibitor compounds, to pharmaceutical compositions comprising these compounds and to their use in therapy.
- the present invention relates to the use of Btk inhibitor compounds in the treatment of Bruton’s Tyrosine Kinase (Btk) mediated disorders.
- B lymphocyte activation is key in the generation of adaptive immune responses. Derailed B lymphocyte activation is a hallmark of many autoimmune diseases and modulation of this immune response is therefore of therapeutic interest. Recently the success of B cell therapies in autoimmune diseases has been established. Treatment of rheumatoid arthritis (RA) patients with Rituximab (anti-CD20 therapy) is an accepted clinical therapy by now. More recent clinical trial studies show that treatment with Rituximab also ameliorates disease symptoms in relapsing remitting multiple sclerosis (RRMS) and systemic lupus erythematosus (SLE) patients. This success supports the potential for future therapies in autoimmune diseases targeting B cell immunity.
- RRMS multiple sclerosis
- SLE systemic lupus erythematosus
- Btk Bruton tyrosine kinase
- Btk in the regulation of the production of auto-antibodies in autoimmune diseases.
- regulation of Btk may affect BCR-induced production of pro-inflammatory cytokines and chemokines by B cells, indicating a broad potential for Btk in the treatment of autoimmune diseases.
- Btk inhibitors may also show potential in the treatment of allergic responses [Gilfillan et al, Immunological Reviews 288 (2009) pp149-169] .
- Btk is also reported to be implicated in RANKL-induced osteoclast differentiation [Shinohara et al, Cell 132 (2008) pp794-806] and therefore may also be of interest for the treatment of bone resorption disorders.
- B cell malignancies Other diseases with an important role for dysfunctional B cells are B cell malignancies. Indeed anti-CD20 therapy is used effectively in the clinic for the treatment of follicular lymphoma, diffuse large B-cell lymphoma and chronic lymphocytic leukemia [Lim et al, Haematologica, 95 (2010) pp135-143] .
- the reported role for Btk in the regulation of proliferation and apoptosis of B cells indicates there is potential for Btk inhibitors in the treatment of B cell lymphomas as well. Inhibition of Btk seems to be relevant in particular for B cell lymphomas due to chronic active BCR signaling [Davis et al, Nature, 463 (2010) pp88-94] .
- Btk inhibitor compounds Some classes of Btk inhibitor compounds have been described as kinase inhibitors, e.g. Imidazo [1, 5-f] [1, 2, 4] triazine compounds have been described in WO2005097800 and WO2007064993. Imidazo [1, 5-a] pyrazine compounds have been described in WO2005037836 and WO2001019828 as IGF-1R enzyme inhibitors.
- Btk inhibitors are not selective over Src-family kinases. With dramatic adverse effects reported for knockouts of Src-family kinases, especially for double and triple knockouts, this is seen as prohibitive for the development of Btk inhibitors that are not selective over the Src-family kinases.
- Lyn-deficient mice exhibit autoimmunity mimicking the phenotype of human lupus nephritis.
- Fyn-deficient mice also show pronounced neurological defects.
- Lyn knockout mice also show an allergic-like phenotype, indicating Lyn as a broad negative regulator of the IgE-mediated allergic response by controlling mast cell responsiveness and allergy-associated traits [Odom et al, J. Exp. Med., 199 (2004) pp1491-1502] .
- Lyn knock-out mice develop severe splenomegaly (myeloid expansion) and disseminated monocyte/macrophage tumors [Harder et al, Immunity, 15 (2001) pp603-615] . These observations are in line with hyperresponsive B cells, mast cells and myeloid cells, and increased Ig levels observed in Lyn-deficient mice.
- Female Src knockout mice are infertile due to reduced follicle development and ovulation [Roby et al, Endocrine, 26 (2005) pp169-176] .
- the double knockouts Src -/- Fyn -/- and Src -/- Yes -/- show a severe phenotype with effects on movement and breathing.
- an inhibitor that inhibits multiple or all kinases of the Src-family kinases simultaneously may cause serious adverse effects.
- the present invention provides compounds which inhibit Btk activity, their use for treatment of Btk mediated diseases and disorders, in particular autoimmune diseases and inflammatory diseases, as well as pharmaceutical compositions comprising such compounds and pharmaceutical carriers.
- alkyl refers to an aliphatic hydrocarbon group having one of its hydrogen atoms replaced with a bond having the specified number of carbon atoms.
- an alkyl group contains, for example, from 1 to 6 carbon atoms (1-6C) alkyl or from 1 to 4 carbon atoms (1-4C) alkyl or from 1 to 3 carbon atoms (1-3C) alkyl.
- an alkyl group is linear. In another embodiment, an alkyl group is branched.
- Non-limiting examples of alkyl groups include methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, isobutyl, tert-butyl, n-pentyl, neopentyl, isopentyl, n-hexyl, isohexyl and neohexyl.
- (1-3C) alkylamino refers to an alkyl group having 1 to 3 carbon atoms which is linked by an amino group to the compound having formula I.
- Non-limiting examples include methylamino, ethylamino and propylamino.
- Alkoxy refers to an alkyl-O-group represented by a linear or branched alkyl group of indicated number of carbon atoms attached through an oxygen bridge; for example " (1-6C) Alkoxy” includes -OCH 3 , -OCH 2 CH 3 , -OCH (CH 3 ) 2 , -O (CH 2 ) 5 CH 3 , and the like.
- alkyl groups are unsubstituted or substituted with 1 to 3 substituents on each carbon atom.
- an effective amount refers to an amount of the compound of Formula I and/or an additional therapeutic agent, or a composition thereof, that is effective in producing the desired therapeutic, ameliorative, inhibitory or preventative effect when administered to a subject suffering from a BTK-mediated disease or disorder.
- an effective amount can refer to each individual agent or to the combination as a whole, wherein the amounts of all agents administered are together effective, but wherein the component agent of the combination may not be present individually in an effective amount.
- halogen refers to fluorine, chlorine, bromine or iodine. Fluorine, chlorine or bromine being preferred halogens; fluorine being more preferred.
- cycloalkyl refers to a saturated mono-or multicyclic ring system containing the specified number of ring carbon atoms, and no heteroatom.
- (C 3-6 ) cycloalkyl or (3-6C) cycloalkyl refers to a saturated ring having from 3 to 6 ring carbon atoms.
- monocyclic cycloalkyls include cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl.
- the cycloalkyl is cyclopropyl.
- cycloalkylmethylene refers to a cycloakyl group as defined above, linked to a methyl group, wherein two of the hydrogen atoms of the methyl group have been replaced with a bond such that the methyl group links the cycloalkyl group to the compound having formula I.
- Cycloalkoxy refers to a cycloalkyl-O-group represented by a cycloalkyl group of indicated number of carbon atoms attached through an oxygen bridge to the compound of the invention; for example “ (3-6C) cycloalkoxy” includes –O-cyclopropyl, -O-cyclobutyl, --O-cyclopentyl, or –O-cyclohexyl.
- C 0 as employed in expressions such as “ (C 0-6 ) alkylene” means a direct covalent bond; or when employed in expressions such as “ (C 0-6 ) alkyl” means hydrogen.
- an integer defining the presence of a certain number of atoms in a group is equal to zero, it means that the atoms adjacent thereto are connected directly by a bond; for example, in the structure wherein s is an integer equal to zero, 1 or 2, the structure is when s is zero; or it means that the indicated atom is absent; for example -S (O) 0 -means -S-.
- heterocycloalkyl described as containing from “1 to 4 heteroatoms” means the heterocycloalkyl can contain 1, 2, 3 or 4 heteroatoms.
- variable definition on each occurrence is independent of its definition at every other occurrence. Also, combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
- variable definitions containing terms having repeated terms e.g., (CRiRj) r , where r is the integer 2, Ri is a defined variable, and Rj is a defined variable
- the value of Ri may differ in each instance in which it occurs, and the value of Rj may differ in each instance in which it occurs.
- Ri and Rj are independently selected from the group consisting of methyl, ethyl, propyl and butyl, then (CRiRj) 2 can be
- X a -X b shall have the same meaning as the term "X a-b “ or “ (a-bX) ” , wherein X is any atom and a and b are any integers.
- C 1 -C 4 shall have the same meaning as “C 1-4 " or “ (1-4C) ” .
- a x shall have the same meaning, and be interchangeable with, “AX” , wherein “A” is any atom and “x” or “X” are any integer.
- R 1 shall have the same meaning, and be interchangeable with, "R1".
- the attachment point is at the last group.
- (C 1-3 ) alkoxycarbonyl refers to, e.g. and the term (C 1-4 ) alkylcarbonyloxy refers to, e.g.
- purified refers to the physical state of a compound after the compound has been isolated through a synthetic process (e.g., from a reaction mixture) , from a natural source, or a combination thereof.
- purified also refers to the physical state of a compound after the compound has been obtained from a purification process or processes described herein or well-known to the skilled artisan (e.g., chromatography, recrystallization, and the like) , in sufficient purity to be characterizable by standard analytical techniques described herein or well-known to the skilled artisan.
- substituted means that one or more hydrogens on the designated atom/atoms is/are replaced with a selection from the indicated group, provided that the designated atom’s normal valency under the existing circumstances is not exceeded, and that the substitution results in a stable compound. Combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
- Stable compound or “stable structure” is defined as a compound or structure that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and formulation into an efficacious therapeutic agent.
- a “subject” is a human or non-human mammal.
- a subject is a human.
- the subject is a chimpanzee.
- the attachment point is at the last group, unless otherwise specified on the substituent group by a dash. A dash on the substituent group would then represent the point of attachment.
- the present invention provides Btk inhibitor compounds according to Formula I or pharmaceutically acceptable salts thereof
- R 11 is independently selected from the group consisting of:
- B 1 is N or C (R 7 ) ;
- B 2 is N or C (R 8 ) ;
- R 7 is H, halogen, (1-3C) alkyl, (1-6C) alkoxy , (3-6C) cycloalkoxy, amino, or (1-3C) alkylamino;
- R 8 is H, halogen, (1-3C) alkyl, (1-6C) alkoxy , (3-6C) cycloalkoxy, amino, or (1-3C) alkylamino;
- R 9 is H, halogen, (1-3C) alkyl, (1-6C) alkoxy , (3-6C) cycloalkoxy, amino, or (1-3C) alkylamino;
- R 10 is H, halogen, (1-3C) alkyl, (1-6C) alkoxy , (3-6C) cycloalkoxy, amino, or (1-3C) alkylamino;
- heteroaromatic ring L is selected from the group consisting of:
- R 5 is H, cyano, (1-4C) alkyl, (3-6C) cycloalkyl, or (3-6C) cycloalkoxy,
- R 5 may optionally be substituted with one, two or three halogens
- ring M is selected from the group consisting of:
- T is C (R e ) 2 , O, NR e , or a bond;
- U is C (R d ) 2 , O, or NR d ;
- V is CH 2 or O
- each R c is independently selected from H, fluoro, methyl or trifluoromethyl
- each R d is independently selected from H, (1-3C) alkyl, (1-3C) alkoxy, cyclopropyl or cyclopropylmethylene;
- each R e is independently selected from H or (1-6C) alkyl
- T is C (R e ) 2 .
- the invention relates to a compound according to Formula I wherein the ring M is selected from the group consisting of
- the invention relates to a compound according to Formula I wherein the heteroaromatic ring L is
- B 1 is CH
- B 2 is CH.
- the invention in another aspect relates to a compound according to Formula I wherein R 5 is selected from the group consisting of hydrogen, CN, cyclopropyl, (1-4C) alkyl, and (3-6C) cycloalkoxy; wherein the alkyl may optionally be substituted with one, two or three halogen.
- the (3-6C) cycloalkoxy is cyclopropoxy.
- the invention relates to a compound according to Formula I wherein R 5 is selected from the group consisting of hydrogen, methyl, cyclopropyl, cyclopropoxyl, and trifluoromethyl.
- the invention also relates to those compounds wherein all specific definitions for B 1 , B 2 , Q, T, U, V, R 5 , R 7 , R 8 , R 9 , R 10 , R 11 , R c , R d , R e , and all substituent groups in the various aspects of the inventions defined here above occur in any combination within the definition of the Btk inhibitor compounds of Formula I or pharmaceutically acceptable salts thereof.
- Non-limiting examples of the compounds of the present invention include:
- the compounds of this invention include the salts, solvates, hydrates or prodrugs of the compounds.
- the use of the terms “salt” , “solvate” , “hydrate” , “prodrug” and the like, is intended to equally apply to the salt, solvate, hydrate and prodrug of enantiomers, stereoisomers, rotamers, tautomers, positional isomers, or racemates of the inventive compounds.
- the Btk inhibitor compounds of the present invention which can be in the form of a free base, may be isolated from the reaction mixture in the form of a pharmaceutically acceptable salt.
- the compounds of Formula I can form salts which are also within the scope of this invention.
- Reference to a compound of Formula I herein is understood to include reference to pharmaceutically acceptable salts thereof, unless otherwise indicated.
- pharmaceutically acceptable salt (s) or “salt” , as employed herein, denotes acidic salts formed with inorganic and/or organic acids, as well as basic salts formed with inorganic and/or organic bases.
- salts of the compounds of Formula I may be formed, for example, by reacting a compound of Formula I with an amount of acid or base, such as an equivalent amount, in a medium such as one in which the salt precipitates or in an aqueous medium followed by lyophilization.
- Exemplary acid addition salts include acetates, ascorbates, benzoates, benzenesulfonates, bisulfates, borates, butyrates, citrates, camphorates, camphorsulfonates, fumarates, hydrochlorides, hydrobromides, hydroiodides, lactates, maleates, methanesulfonates, naphthalenesulfonates, nitrates, oxalates, phosphates, propionates, salicylates, succinates, sulfates, tartarates, thiocyanates, toluenesulfonates (also known as tosylates, ) and the like.
- Exemplary basic salts include ammonium salts, alkali metal salts such as sodium, lithium, and potassium salts, alkaline earth metal salts such as calcium and magnesium salts, salts with organic bases (for example, organic amines) such as dicyclohexylamines, t-butyl amines, and salts with amino acids such as arginine, lysine and the like.
- alkali metal salts such as sodium, lithium, and potassium salts
- alkaline earth metal salts such as calcium and magnesium salts
- salts with organic bases for example, organic amines
- organic amines such as dicyclohexylamines, t-butyl amines
- salts with amino acids such as arginine, lysine and the like.
- Basic nitrogen-containing groups may be quarternized with agents such as lower alkyl halides (e.g., methyl, ethyl, and butyl chlorides, bromides and iodides) , dialkyl sulfates (e.g., dimethyl, diethyl, and dibutyl sulfates) , long chain halides (e.g., decyl, lauryl, and stearyl chlorides, bromides and iodides) , aralkyl halides (e.g., benzyl and phenethyl bromides) , and others.
- agents such as lower alkyl halides (e.g., methyl, ethyl, and butyl chlorides, bromides and iodides) , dialkyl sulfates (e.g., dimethyl, diethyl, and dibutyl sulfates) , long chain halides (e.g
- the Btk inhibitor compounds of the present invention may exist as amorphous forms or crystalline forms.
- the compounds of Formula I may have the ability to crystallize in more than one form, a characteristic known as polymorphism, and it is understood that such polymorphic forms (“polymorphs”) are within the scope of Formula I.
- Polymorphism generally can occur as a response to changes in temperature or pressure or both and can also result from variations in the crystallization process.
- Polymorphs can be distinguished by various physical characteristics known in the art such as x-ray diffraction patterns, solubility and melting point.
- the compounds having Formula I or the pharmaceutically acceptable salts may form hydrates or solvates. It is known to those of skill in the art that charged compounds form hydrated species when lyophilized with water, or form solvated species when concentrated in a solution with an appropriate organic solvent.
- the compounds of this invention include the hydrates or solvates of the compounds listed.
- One or more compounds of the invention having Formula I or the pharmaceutically acceptable salts or solvates thereof may exist in unsolvated as well as solvated forms with pharmaceutically acceptable solvents such as water, ethanol, and the like, and it is intended that the invention embrace both solvated and unsolvated forms.
- “Solvate” means a physical association of a compound of this invention with one or more solvent molecules. This physical association involves varying degrees of ionic and covalent bonding, including hydrogen bonding. In certain instances the solvate will be capable of isolation, for example when one or more solvent molecules are incorporated in the crystal lattice of the crystalline solid. "Solvate” encompasses both solution-phase and isolatable solvates. Non-limiting examples of suitable solvates include ethanolates, methanolates, and the like.
- “Hydrate” is a solvate wherein the solvent molecule is H 2 O.
- solvates Preparation of solvates is generally known.
- M. Caira et al, J. Pharmaceutical Sci., 93 (3) , 601-611 (2004) describe the preparation of the solvates of the antifungal fluconazole in ethyl acetate as well as from water.
- Similar preparations of solvates, hemisolvate, hydrates and the like are described by E. C. van Tonder et al, AAPS PharmSciTech., 5(1) , article 12 (2004) ; and A. L. Bingham et al, Chem. Commun. 603-604 (2001) .
- a typical, non-limiting, process involves dissolving the inventive compound in desired amounts of the desired solvent (organic or water or mixtures thereof) at a higher than ambient temperature, and cooling the solution at a rate sufficient to form crystals which are then isolated by standard methods.
- Analytical techniques such as, for example IR spectroscopy, show the presence of the solvent (or water) in the crystals as a solvate (or hydrate) .
- the compounds of Formula I may contain asymmetric or chiral centers, and, therefore, exist in different stereoisomeric forms. It is intended that all stereoisomeric forms of the compounds of Formula I, as well as mixtures thereof, including racemic mixtures, form part of the present invention.
- the present invention embraces all geometric and positional isomers. For example, if a compound of Formula I incorporates a double bond or a fused ring, both the cis-and trans-forms, as well as mixtures, are embraced within the scope of the invention.
- Such stereoisomeric forms also include enantiomers and diastereoisomers, etc.
- chiral compounds For chiral compounds, methods for asymmetric synthesis whereby the pure stereoisomers are obtained are well known in the art, e.g. synthesis with chiral induction, synthesis starting from chiral intermediates, enantioselective enzymatic conversions, separation of stereoisomers using chromatography on chiral media. Such methods are described in Chirality in Industry (edited by A.N. Collins, G.N. Sheldrake and J. Crosby, 1992; John Wiley) . Likewise methods for synthesis of geometrical isomers are also well known in the art.
- Diastereomeric mixtures can be separated into their individual diastereomers on the basis of their physical chemical differences by methods well known to those skilled in the art, such as, for example, by chromatography and/or fractional crystallization.
- Enantiomers can be separated by converting the enantiomeric mixture into a diastereomeric mixture by reaction with an appropriate optically active compound (e.g. chiral auxiliary such as a chiral alcohol or Mosher’s acid chloride) , separating the diastereomers and converting (e.g. hydrolyzing) the individual diastereomers to the corresponding pure enantiomers.
- an appropriate optically active compound e.g. chiral auxiliary such as a chiral alcohol or Mosher’s acid chloride
- some of the compounds of Formula I may be atropisomers (e.g. substituted biaryls) and are considered as part of this invention.
- Enantiomers can also be separated by use of chiral HPLC column.
- All stereoisomers for example, geometric isomers, optical isomers and the like
- of the present compounds including those of the salts, solvates, esters and prodrugs of the compounds as well as the salts, solvates and esters of the prodrugs
- those which may exist due to asymmetric carbons on various substituents including enantiomeric forms (which may exist even in the absence of asymmetric carbons) , rotameric forms, atropisomers, and diastereomeric forms, are contemplated within the scope of this invention, as are positional isomers.
- Individual stereoisomers of the compounds of the invention may, for example, be substantially free of other isomers, or may be admixed, for example, as racemates or with all other, or other selected, stereoisomers.
- the chiral centers of the present invention can have the S or R configuration as defined by the IUPAC 1974 Recommendations.
- the use of the terms "salt” , “solvate” , “ester” , “prodrug” and the like, is intended to equally apply to the salt, solvate, ester and prodrug of enantiomers, stereoisomers, rotamers, tautomers, positional isomers, racemates or prodrugs of the inventive compounds.
- prodrugs means a compound (e.g, a drug precursor) that is transformed in vivo to yield a compound of Formula I or a pharmaceutically acceptable salt, hydrate or solvate of the compound. The transformation may occur by various mechanisms (e.g. by metabolic or chemical processes) , such as, for example, through hydrolysis in blood.
- prodrugs are provided by T. Higuchi and W. Stella, “Pro-drugs as Novel Delivery Systems, ” Vol. 14 of the A.C.S. Symposium Series, and in Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987.
- the atoms may exhibit their natural isotopic abundances, or one or more of the atoms may be artificially enriched in a particular isotope having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number predominantly found in nature.
- the present invention is meant to include all suitable isotopic variations of the compounds of generic Formula I.
- different isotopic forms of hydrogen (H) include protium ( 1 H) and deuterium ( 2 H) .
- Protium is the predominant hydrogen isotope found in nature. Enriching for deuterium may afford certain therapeutic advantages, such as increasing in vivo half-life or reducing dosage requirements, or may provide a compound useful as a standard for characterization of biological samples.
- Isotopically-enriched compounds within generic Formula I can be prepared without undue experimentation by conventional techniques well known to those skilled in the art or by processes analogous to those described in the Schemes and Examples herein using appropriate isotopically-enriched reagents and/or intermediates.
- Certain isotopically-labelled compounds of Formula I are useful in compound and/or substrate tissue distribution assays. Tritiated (i.e., 3 H) and carbon-14 (i.e., 14 C) isotopes are particularly preferred for their ease of preparation and detectability. Further, substitution with heavier isotopes such as deuterium (i.e., 2 H) may afford certain therapeutic advantages resulting from greater metabolic stability (e.g., increased in vivo half-life or reduced dosage requirements) and hence may be preferred in some circumstances.
- Isotopically labelled compounds of Formula I can generally be prepared by following procedures analogous to those disclosed in the Schemes and/or in the Examples herinbelow, by substituting an appropriate isotopically labeled reagent for a non-isotopically labeled reagent.
- the compounds having Formula I and pharmaceutical compositions thereof can be used to treat or prevent a variety of conditions, diseases or disorders mediated by Bruton's Tyrosine kinase (Btk) .
- Btk-mediated conditions, diseases or disorders include, but are not limited to: (1) arthritis, including rheumatoid arthritis, juvenile arthritis, psoriatic arthritis and osteoarthritis; (2) asthma and other obstructive airways diseases, including chronic asthma, late asthma, airway hyper-responsiveness, bronchitis, bronchial asthma, allergic asthma, intrinsic asthma, extrinsic asthma, dust asthma, adult respiratory distress syndrome, recurrent airway obstruction, and chronic obstruction pulmonary disease including emphysema; (3) autoimmune diseases or disorders, including those designated as single organ or single cell-type autoimmune disorders, for example Hashimoto's thyroiditis, autoimmune hemolytic anemia, autoimmune atrophic gastritis of pernicious anemia, autoimmune encephalomyelitis, autoimmune orchit
- B-ALL marginal zone B cell lymphoma, chronic lymphocytic leukemia, diffuse large B cell lymphoma, Burkitt lymphoma, mediastinal large B-cell lymphoma) , Hodgkin lymphoma, NK and T cell lymphomas; TEL-Syk and ITK-Syk fusion driven tumors, myelomas including multiple myeloma, myeloproliferative disorders kidney cancer, lung cancer, muscle cancer, bone cancer, bladder cancer, brain cancer, melanoma including oral and metastatic melanoma, Kaposi's sarcoma, proliferative diabetic retinopathy, and angiogenic-associated disorders including solid tumors, and pancreatic cancer; (5) diabetes, including Type I diabetes and complications from diabetes; (6) eye diseases, disorders or conditions including autoimmune diseases of the eye, keratoconjunctivitis, vernal conjunctivitis, uveitis including uveitis associated with Behcet
- the invention thus provides compounds of Formula I and salts thereof for use in therapy, and particularly in the treatment of disorders, diseases and conditions mediated by inappropriate Btk activity.
- the inappropriate Btk activity referred to herein is any Btk activity that deviates from the normal Btk activity expected in a particular mammalian subject.
- Inappropriate Btk activity may take the form of, for instance, an abnormal increase in activity, or an aberration in the timing and or control of Btk activity.
- Such inappropriate activity may result then, for example, from overexpression or mutation of the protein kinase leading to inappropriate or uncontrolled activation.
- the present invention provides for the use of a compound of Formula I, or a pharmaceutically acceptable salt thereof for the manufacture of a medicament for the treatment of a Btk-mediated disorder.
- the present invention provides methods of regulating, modulating, or inhibiting Btk for the prevention and/or treatment of disorders related to unregulated or inappropriate Btk activity.
- the present invention provides a method for treating a subject suffering from a disorder mediated by Btk, which comprises administering to said subject a compound of Formula I or a pharmaceutically acceptable salt thereof in an amount effective to treat the Btk-mediated disorder.
- a further aspect of the invention resides in the use of a compound of Formula I or a pharmaceutically acceptable salt thereof for the manufacture of a medicament to be used for the treatment of chronic B cell disorders in which T cells play a prominent role.
- Btk mediated diseases, conditions and disorders mean any disease, condition or disorder in which B cells, mast cells, myeloid cells or osteoclasts play a central role.
- diseases include but are not limited to, immune, autoimmune and inflammatory diseases, allergies, infectious diseases, bone resorption disorders and proliferative diseases.
- Immune, autoimmune and inflammatory diseases that may be treated or prevented with the compounds of the present invention include rheumatic diseases (e.g. rheumatoid arthritis, psoriatic arthritis, infectious arthritis, progressive chronic arthritis, deforming arthritis, osteoarthritis, traumatic arthritis, gouty arthritis, Reiter’s syndrome, polychondritis, acute synovitis and spondylitis) , glomerulonephritis (with or without nephrotic syndrome) , Goodpasture's syndrome, (and associated glomerulonephritis and pulmonary hemorrhage) , atherosclerosis, autoimmune hematologic disorders (e.g.
- hemolytic anemia aplasic anemia, idiopathic thrombocytopenia, chronic idiopathic thrombocytopenic purpura (ITP) , and neutropenia
- ITP chronic idiopathic thrombocytopenic purpura
- neutropenia autoimmune gastritis
- autoimmune inflammatory bowel diseases e.g.
- ulcerative colitis and Crohn’s disease irritable bowel syndrome
- host versus graft disease allograft rejection, chronic thyroiditis
- Graves’ disease Sjorgren's disease, scleroderma, diabetes (type I and type II) , active hepatitis (acute and chronic) , pancreatitis, primary billiary cirrhosis, myasthenia gravis, multiple sclerosis, systemic lupus erythematosis, psoriasis, atopic dermatitis, dermatomyositis, contact dermatitis, eczema, skin sunburns, vasculitis (e.g.
- Behcet’s disease chronic renal insufficiency
- Stevens-Johnson syndrome inflammatory pain, idiopathic sprue, cachexia, sarcoidosis, Guillain-Barré syndrome, uveitis, conjunctivitis, kerato conjunctivitis, otitis media, periodontal disease
- Addison's disease Parkinson's disease, Alzheimer's disease, diabetes, septic shock, myasthenia gravis, pulmonary interstitial fibrosis, asthma, bronchitis, rhinitis, sinusitis, pneumoconiosis, pulmonary insufficiency syndrome, pulmonary emphysema, pulmonary fibrosis, silicosis, chronic inflammatory pulmonary disease (e.g. chronic obstructive pulmonary disease) and other inflammatory or obstructive disease on airways.
- chronic inflammatory pulmonary disease e.g. chronic obstructive pulmonary disease
- Allergies that may be treated or prevented include, among others, allergies to foods, food additives, insect poisons, dust mites, pollen, animal materials and contact allergans, type I hypersensitivity allergic asthma, allergic rhinitis, allergic conjunctivitis.
- Infectious diseases that may be treated or prevented include, among others, sepsis, septic shock, endotoxic shock, sepsis by Gram-negative bacteria, shigellosis, meningitis, cerebral malaria, pneumonia, tuberculosis, viral myocarditis, viral hepatitis (hepatitis A, hepatitis B and hepatitis C) , HIV infection, retinitis caused by cytomegalovirus, influenza, herpes, treatment of infections associated with severe burns, myalgias caused by infections, cachexia secondary to infections, and veterinary viral infections such as lentivirus, caprine arthritic virus, visna-maedi virus, feline immunodeficiency virus, bovine immunodeficiency virus or canine immunodeficiency virus.
- Bone resorption disorders that may be treated or prevented include, among others, osteoporosis, osteoarthritis, traumatic arthritis, gouty arthritis and bone disorders related with multiple myeloma.
- Proliferative diseases that may be treated or prevented include, among others, non-Hodgkin lymphoma (in particular the subtypes diffuse large B-cell lymphoma (DLBCL) and mantle cell lymphoma (MCL) ) , B cell chronic lymphocytic leukemia and acute lymphoblastic leukemia (ALL) with mature B cell, ALL in particular.
- non-Hodgkin lymphoma in particular the subtypes diffuse large B-cell lymphoma (DLBCL) and mantle cell lymphoma (MCL)
- ALL acute lymphoblastic leukemia
- the compounds of Formula I or pharmaceutically acceptable salts may be used for the treatment of B cell lymphomas resulting from chronic active B cell receptor signaling.
- Yet another aspect of the present invention provides a method for treating diseases caused by or associated with Fc receptor signaling cascades, including FceRI and/or FcgRI-mediated degranulation as a therapeutic approach towards the treatment or prevention of diseases characterized by, caused by and/or associated with the release or synthesis of chemical mediators of such Fc receptor signaling cascades or degranulation.
- Fc receptor signaling cascades including FceRI and/or FcgRI-mediated degranulation
- Btk is known to play a critical role inimmunotyrosine-based activation motif (ITAM) singaling, B cell receptor signaling, T cell receptor signaling and is an essential component of integrin beta (1) , beta (2) , and beta (3) signaling in neutrophils.
- ITAM inimmunotyrosine-based activation motif
- compounds of the present invention can be used to regulate Fc receptor, ITAM, B cell receptor and integrin signaling cascades, as well as the cellular responses elicited through these signaling cascades.
- cellular responses include respiratory burst, cellular adhesion, cellular degranulation, cell spreading, cell migration, phagocytosis, calcium ion flux, platelet aggregation and cell maturation.
- compositions in which at least one compound of Formula I or a pharmaceutically acceptable salt thereof is administered in combination with at least one other active agent.
- the other active agent is an anti-inflammatory agent, an immunosuppressant agent, or a chemotherapeutic agent.
- Anti-inflammatory agents include but are not limited to NSAIDs, non-specific and COX-2 specific cyclooxgenase enzyme inhibitors, gold compounds, corticosteroids, methotrexate, tumor necrosis factor receptor (TNF) receptors antagonists, immunosuppressants and methotrexate.
- NSAIDs include, but are not limited to, ibuprofen, flurbiprofen, naproxen and naproxen sodium, diclofenac, combinations of diclofenac sodium and misoprostol, sulindac, oxaprozin, diflunisal, piroxicam, indomethacin, etodolac, fenoprofen calcium, ketoprofen, sodium nabumetone, sulfasalazine, tolmetin sodium, and hydroxychloroquine.
- NSAIDs also include COX-2 specific inhibitors such as celecoxib, valdecoxib, lumiracoxib and/or etoricoxib.
- the anti-inflammatory agent is a salicylate.
- Salicylates include by are not limited to acetylsalicylic acid or aspirin, sodium salicylate, and choline and magnesium salicylates.
- the anti-inflammatory agent may also be a corticosteroid.
- the corticosteroid may be cortisone, dexamethasone, methylprednisolone, prednisolone, prednisolone sodium phosphate, or prednisone.
- the anti-inflammatory agent is a gold compound such as gold sodium thiomalate or auranofin.
- the invention also includes embodiments in which the anti-inflammatory agent is a metabolic inhibitor such as a dihydrofolate reductase inhibitor, such as methotrexate or a dihydroorotate dehydrogenase inhibitor, such as leflunomide.
- a metabolic inhibitor such as a dihydrofolate reductase inhibitor, such as methotrexate or a dihydroorotate dehydrogenase inhibitor, such as leflunomide.
- At least one anti-inflammatory agent is an anti-C5 monoclonal antibody (such as eculizumab or pexelizumab) , a TNF antagonist, such as entanercept, or infliximab, which is an anti-TNF alpha monoclonal antibody.
- an anti-C5 monoclonal antibody such as eculizumab or pexelizumab
- TNF antagonist such as entanercept, or infliximab
- Still other embodiments of the invention pertain to combinations in which at least one active agent is an immunosuppressant agent, such as an immunosuppressant compound chosen from methotrexate, leflunomide, cyclosporine, tacrolimus, azathioprine, and mycophenolate mofetil.
- an immunosuppressant agent such as an immunosuppressant compound chosen from methotrexate, leflunomide, cyclosporine, tacrolimus, azathioprine, and mycophenolate mofetil.
- B-cells and B-cell precursors expressing BTK have been implicated in the pathology of B-cell malignancies, including, but not limited to, B-cell lymphoma, lymphoma (including Hodgkin's and non-Hodgkin's lymphoma) , hairy cell lymphoma, multiple myeloma, chronic and acute myelogenous leukemia and chronic and acute lymphocytic leukemia.
- BTK has been shown to be an inhibitor of the Fas/APO-1 (CD-95) death inducing signaling complex (DISC) in B-lineage lymphoid cells.
- DISC Fas/APO-1
- the fate of leukemia/lymphoma cells may reside in the balance between the opposing proapoptotic effects of caspases activated by DISC and an upstream anti-apoptotic regulatory mechanism involving BTK and/or its substrates (Vassilev et al., J. Biol. Chem. 1998, 274, 1646-1656) .
- BTK inhibitors are useful as chemosensitizing agents, and, thus, are useful in combination with other chemotherapeutic agents, in particular, drugs that induce apoptosis.
- chemotherapeutic agents include topoisomerase I inhibitors (camptothecin or topotecan) , topoisomerase II inhibitors (e.g. daunomycin and etoposide) , alkylating agents (e.g. cyclophosphamide, melphalan and BCNU) , tubulin directed agents (e.g. taxol and vinblastine) , and biological agents (e.g. antibodies such as anti CD20 antibody, IDEC 8, immunotoxins, and cytokines) .
- topoisomerase I inhibitors camptothecin or topotecan
- topoisomerase II inhibitors e.g. daunomycin and etoposide
- alkylating agents e.g. cyclophosphamide, melphalan and BC
- Btk activity has also been associated with some leukemias expressing the bcr-abl fusion gene resulting from translocation of parts of chromosome 9 and 22. This abnormality is commonly observed in chronic myelogenous leukemia. Btk is constitutively phosphorylated by the bcr-abl kinase which initiates downstream survival signals which circumvents apoptosis in bcr-abl cells. (N. Feldhahn et al. J. Exp. Med. 2005 201 (11) : 1837-1852) .
- the compound (s) of Formula I and the other pharmaceutically active agent (s) may be administered together or separately and, when administered separately this may occur simultaneously or sequentially in any order.
- the amounts of the compound (s) of Formula I and the other pharmaceutically active agent (s) and the relative timings of administration will be selected in order to achieve the desired combined therapeutic effect.
- a compound of Formula I may be combined with one or more other active agents such as: (1) TNF- ⁇ inhibitors such as infliximab etanercept adalimumab certolizumab pegol and golimumab (2) non-selective COX-I/COX-2 inhibitors (such as piroxicam, diclofenac, propionic acids such as naproxen, flubiprofen, fenoprofen, ketoprofen and ibuprofen, fenamates such as mefenamic acid, indomethacin, sulindac, etodolac, azapropazone, pyrazolones such as phenylbutazone, salicylates such as aspirin) ; (3) COX-2 inhibitors (such as meloxicam, cele
- the present invention also provides for "triple combination" therapy, comprising a compound of Formula I or a pharmaceutically acceptable salt thereof together with beta 2 -adrenoreceptor agonist and an anti-inflammatory corticosteroid.
- this combination is for treatment and/or prophylaxis of asthma, COPD or allergic rhinitis.
- the beta 2 -adrenoreceptor agonist and/or the anti-inflammatory corticosteroid can be as described above and/or as described in WO 03/030939 A1.
- triple combination is a compound of Formula I or a pharmaceutically acceptable salt thereof in combination with the components of (salmeterol xinafoate and fluticasone propionate) , (budesonide and formoterol fumarate) , or (mometasone furoate and formoterol) .
- a compound of Formula I may be combined with one or more of an anticancer agents.
- an anticancer agents can be found in Cancer Principles and Practice of Oncology by V.T. Devita and S. Hellman (editors) , 6 th edition (February 15, 2001) , Lippincott Williams &Wilkins Publishers. A person of ordinary skill in the art would be able to discern which combinations of agents would be useful based on the particular characteristics of the drugs and the cancer involved.
- Such anti-cancer agents include, but are not limited to, the following: (1) estrogen receptor modulator such as diethylstibestral, tamoxifen, raloxifene, idoxifene, LY353381, LY117081, toremifene, fluoxymestero, and SH646; (2) other hormonal agents including aromatase inhibitors (e.g., aminoglutethimide, tetrazole anastrozole, letrozole and exemestane) , luteinizing hormone release hormone (LHRH) analogues, ketoconazole, goserelin acetate, leuprolide, megestrol acetate and mifepristone; (3) androgen receptor modulator such as finasteride and other 5 ⁇ -reductase inhibitors, nilutamide, flutamide, bicalutamide, liarozole, and abiraterone acetate; (4) retinoid receptor modulator such
- Cytotoxic/cytostatic agents refer to compounds which cause cell death or inhibit cell proliferation primarily by interfering directly with the cell’s functioning or inhibit or interfere with cell mytosis, including alkylating agents, tumor necrosis factors, intercalators, hypoxia activatable compounds, microtubule inhibitors/microtubule-stabilizing agents, inhibitors of mitotic kinesins, inhibitors of histone deacetylase, inhibitors of kinases involved in mitotic progression, antimetabolites; biological response modifiers; hormonal/anti-hormonal therapeutic agents, haematopoietic growth factors, monoclonal antibody targeted therapeutic agents, topoisomerase inhibitors, proteasome inhibitors and ubiquitin ligase inhibitors.
- cytotoxic agents include, but are not limited to, sertenef, cachectin, chlorambucil, cyclophosphamide, ifosfamide, mechlorethamine, melphalan, uracil mustard, thiotepa, busulfan, carmustine, lomustine, streptozocin, tasonermin, lonidamine, carboplatin, altretamine, dacarbazine, procarbazine, prednimustine, dibromodulcitol, ranimustine, fotemustine, nedaplatin, oxaliplatin, temozolomide, heptaplatin, estramustine, improsulfan tosilate, trofosfamide, nimustine, dibrospidium chloride, pumitepa, lobaplatin, satraplatin, profiromycin, cisplatin, irofulven, dexifos
- hypoxia activatable compound is tirapazamine.
- proteasome inhibitors include but are not limited to lactacystin and bortezomib.
- microtubule inhibitors/microtubule-stabilising agents include vincristine, vinblastine, vindesine, vinzolidine, vinorelbine, vindesine sulfate, 3’ , 4’ -didehydro-4’ -deoxy-8’ -norvincaleukoblastine, podophyllotoxins (e.g., etoposide (VP-16) and teniposide (VM-26) ) , paclitaxel, docetaxol, rhizoxin, dolastatin, mivobulin isethionate, auristatin, cemadotin, RPR109881, BMS184476, vinflunine, cryptophycin, anhydrovinblastine, N, N-dimethyl-L-valyl-L-valyl-N-methyl-L-valyl-L-prolyl-L-proline-t-butylamide, TDX258, the epothilones (see
- topoisomerase inhibitors are topotecan, hycaptamine, irinotecan, rubitecan, 6-ethoxypropionyl-3’ , 4’ -O-exo-benzylidene-chartreusin, lurtotecan, 7- [2-(N-isopropylamino) ethyl] - (20S) camptothecin, BNP1350, BNPI1100, BN80915, BN80942, etoposide phosphate, teniposide, sobuzoxane, 2’ -dimethylamino-2’ -deoxy-etoposide, GL331, N-[2- (dimethylamino) ethyl] -9-hydroxy-5, 6-dimethyl-6H-pyrido [4, 3-b] carbazole-1-carboxamide, asulacrine, 2, 3- (methylenedioxy) -5-methyl-7-hydroxy-8-methoxybenzo [c]
- inhibitors of mitotic kinesins include, but are not limited to inhibitors of KSP, inhibitors of MKLP1, inhibitors of CENP-E, inhibitors of MCAK, inhibitors of Kif14, inhibitors of Mphosph1 and inhibitors of Rab6-KIFL.
- histone deacetylase inhibitors include, but are not limited to, vorinostat, trichostatin A, oxamflatin, PXD101, MG98, valproic acid and scriptaid.
- “Inhibitors of kinases involved in mitotic progression” include, but are not limited to, inhibitors of aurora kinase, inhibitors of Polo-like kinases (PLK; in particular inhibitors of PLK-1) , inhibitors of bub-1 and inhibitors of bub-R1.
- PLK Polo-like kinases
- An example of an “aurora kinase inhibitor” is VX-680.
- Antiproliferative agents includes antisense RNA and DNA oligonucleotides such as G3139, ODN698, RVASKRAS, GEM231, and INX3001, and antimetabolites such as enocitabine, carmofur, tegafur, pentostatin, doxifluridine, trimetrexate, fludarabine, capecitabine, galocitabine, cytarabine ocfosfate, fosteabine sodium hydrate, raltitrexed, paltitrexid, emitefur, tiazofurin, decitabine, nolatrexed, pemetrexed, nelzarabine, 2’ -deoxy-2’ -methylidenecytidine, 2’ -fluoromethylene-2’ -deoxycytidine, N6- [4-deoxy-4- [N2- [2, 4-tetradecadienoyl] glycylamino] -L-
- Non-limiting examples of suitable agents used in cancer therapy include, but are not limited to, abarelix; aldesleukin; alemtuzumab; alitretinoin; allopurinol; altretamine; amifostine; anastrozole; arsenic trioxide; asparaginase; azacitidine; bendamustine; bevacuzimab; bexarotene; bleomycin; bortezomib; busulfan; calusterone; capecitabine; carboplatin; carmustine; cetuximab; chlorambucil; cisplatin; cladribine; clofarabine; cyclophosphamide; cytarabine; dacarbazine; dactinomycin, actinomycin D; dalteparin; darbepoetin alfa; dasatinib; daunorubicin; degarelix; den
- the other therapeutic ingredient (s) may be used in the form of salts, for example as alkali metal or amine salts or as acid addition salts, or prodrugs, or as esters, for example lower alkyl esters, or as solvates, for example hydrates, to optimise the activity and/or stability and/or physical characteristics, such as solubility, of the therapeutic ingredient. It will be clear also that, where appropriate, the therapeutic ingredients may be used in optically pure form.
- compositions comprising a combination as defined above together with a pharmaceutically acceptable diluent, carrier or excipient represent a further aspect of the invention.
- These combinations are of particular interest in respiratory diseases and are conveniently adapted for inhaled or intranasal delivery.
- the individual compounds of such combinations may be administered either sequentially or simultaneously in separate or combined pharmaceutical compositions.
- the individual compounds will be administered simultaneously in a combined pharmaceutical composition.
- Appropriate doses of known therapeutic agents will be readily appreciated by those skilled in the art.
- the invention further provides a pharmaceutical composition which comprises a compound of Formula I and salts, solvates and physiological functional derivatives thereof, and one or more pharmaceutically acceptable carriers, diluents, or excipients.
- a pharmaceutical composition which comprises a compound of Formula I and salts, solvates and physiological functional derivatives thereof, and one or more pharmaceutically acceptable carriers, diluents, or excipients.
- the compounds of the Formula I and salts, solvates and physiological functional derivatives thereof, are as described above.
- the carrier (s) , diluent (s) or excipient (s) must be acceptable in the sense of being compatible with the other ingredients of the formulation and not deleterious to the recipient thereof.
- a process for the preparation of a pharmaceutical composition including admixing a compound of the Formula I, or salts, solvates and physiological functional derivatives thereof, with one or more pharmaceutically acceptable carriers, diluents or excipients.
- compositions of the present invention may be presented in unit dose forms containing a predetermined amount of active ingredient per unit dose.
- a unit may contain, for example, 5 ⁇ g to 1 g, preferably 1 mg to 700 mg, more preferably 5 mg to 100 mg of a compound of the Formula I, depending on the condition being treated, the route of administration and the age, weight and condition of the patient.
- Such unit doses may therefore be administered more than once a day.
- Preferred unit dosage compositions are those containing a daily dose or sub-dose (for administration more than once a day) , as herein above recited, or an appropriate fraction thereof, of an active ingredient.
- such pharmaceutical compositions may be prepared by any of the methods well known in the pharmacy art.
- compositions of the present invention may be adapted for administration by any appropriate route, for example by the oral (including buccal or sublingual) , rectal, topical, inhaled, nasal, ocular, sublingual, subcutaneous, local or parenteral (including intravenous and intramuscular) route, and the like, all in unit dosage forms for administration.
- Such compositions may be prepared by any method known in the art of pharmacy, for example by bringing into association the active ingredient with the carrier (s) or excipient (s) .
- Dosage forms include tablets, troches, dispersions, suspensions, solutions, capsules, creams, ointments, aerosols, and the like.
- the present invention provides a pharmaceutical composition adapted for administration by the oral route, for treating, for example, rheumatoid arthritis.
- the present invention provides a pharmaceutical composition adapted for administration by the nasal route, for treating, for example, allergic rhinitis.
- the present invention provides a pharmaceutical composition adapted for administration by the inhaled route, for treating, for example, asthma, Chronic Obstructive Pulmonary disease (COPD) or Acute Respiratory Distress Syndrome (ARDS) .
- COPD Chronic Obstructive Pulmonary disease
- ARDS Acute Respiratory Distress Syndrome
- the present invention provides a pharmaceutical composition adapted for administration by the ocular route, for treating, diseases of the eye, for example, conjunctivitis.
- the present invention provides a pharmaceutical composition adapted for administration by the parenteral (including intravenous) route, for treating, for example, cancer.
- the pharmaceutical composition of the invention may be presented in unit-dose or multi-dose containers, e.g. injection liquids in predetermined amounts, for example in sealed vials and ampoules, and may also be stored in a freeze dried (lyophilized) condition requiring only the addition of sterile liquid carrier, e.g. water, prior to use.
- sterile liquid carrier e.g. water
- the active agent may be compressed into solid dosage units, such as pills, tablets, or be processed into capsules or suppositories.
- the active agent can be applied as a fluid composition, e.g. as an injection preparation, in the form of a solution, suspension, emulsion, or as a spray, e.g. a nasal spray.
- solid dosage units For making solid dosage units, the use of conventional additives such as fillers, colorants, polymeric binders and the like is contemplated. In general any pharmaceutically acceptable additive which does not interfere with the function of the active compounds can be used. Suitable carriers with which the active agent of the invention can be administered as solid compositions include lactose, starch, cellulose derivatives and the like, or mixtures thereof, used in suitable amounts. For parenteral administration, aqueous suspensions, isotonic saline solutions and sterile injectable solutions may be used, containing pharmaceutically acceptable dispersing agents and/or wetting agents, such as propylene glycol or butylene glycol.
- compositions of the present invention which are adapted for oral administration may be presented as discrete units such as capsules or tablets; powders or granules; solutions or suspensions in aqueous or non-aqueous liquids; edible foams or whips; or oil-in-water liquid emulsions or water-in-oil liquid emulsions.
- the active drug component can be combined with an oral, non-toxic pharmaceutically acceptable inert carrier such as ethanol, glycerol, water and the like.
- an oral, non-toxic pharmaceutically acceptable inert carrier such as ethanol, glycerol, water and the like.
- Powders are prepared by comminuting the compound to a suitable fine size and mixing with a similarly comminuted pharmaceutical carrier such as an edible carbohydrate, as, for example, starch or mannitol. Flavoring, preservative, dispersing and coloring agent can also be present.
- Capsules are made by preparing a powder mixture, as described above, and filling formed gelatin sheaths.
- Glidants and lubricants such as colloidal silica, talc, magnesium stearate, calcium stearate or solid polyethylene glycol can be added to the powder mixture before the filling operation.
- a disintegrating or solubilizing agent such as agar-agar, calcium carbonate or sodium carbonate can also be added to improve the availability of the medicament when the capsule is ingested.
- suitable binders include starch, gelatin, natural sugars such as glucose or beta-lactose, corn sweeteners, natural and synthetic gums such as acacia, tragacanth or sodium alginate, carboxymethylcellulose, polyethylene glycol, waxes and the like.
- Lubricants used in these dosage forms include sodium oleate, sodium stearate, magnesium stearate, sodium benzoate, sodium acetate, sodium chloride and the like.
- Disintegrators include, without limitation, starch, methyl cellulose, agar, bentonite, xanthan gum and the like.
- Tablets are formulated, for example, by preparing a powder mixture, granulating or slugging, adding a lubricant and disintegrant and pressing into tablets.
- a powder mixture is prepared by mixing the compound, suitably comminuted, with a diluent or base as described above, and optionally, with a binder such as carboxymethylcellulose, an aliginate, gelatin, or polyvinyl pyrrolidone, a solution retardant such as paraffin, a resorption accelerator such as a quaternary salt and/or an absorption agent such as bentonite, kaolin or dicalcium phosphate.
- a binder such as carboxymethylcellulose, an aliginate, gelatin, or polyvinyl pyrrolidone
- a solution retardant such as paraffin
- a resorption accelerator such as a quaternary salt
- an absorption agent such as bentonite, kaolin or dicalcium phosphate.
- the powder mixture can be granulated by wetting with a binder such as syrup, starch paste, acadia mucilage or solutions of cellulosic or polymeric materials and forcing through a screen.
- a binder such as syrup, starch paste, acadia mucilage or solutions of cellulosic or polymeric materials and forcing through a screen.
- the powder mixture can be run through the tablet machine and the result is imperfectly formed slugs broken into granules.
- the granules can be lubricated to prevent sticking to the tablet forming dies by means of the addition of stearic acid, a stearate salt, talc or mineral oil.
- the lubricated mixture is then compressed into tablets.
- the compounds of the present invention can also be combined with a free flowing inert carrier and compressed into tablets directly without going through the granulating or slugging steps.
- a clear or opaque protective coating consisting of a sealing coat of shellac, a coating of
- Oral fluids such as solution, syrups and elixirs can be prepared in dosage unit form so that a given quantity contains a predetermined amount of the compound.
- Syrups can be prepared by dissolving the compound in a suitably flavored aqueous solution, while elixirs are prepared through the use of a non-toxic alcoholic vehicle.
- Suspensions can be formulated by dispersing the compound in a non-toxic vehicle.
- Solubilizers and emulsifiers such as ethoxylated isostearyl alcohols and polyoxy ethylene sorbitol ethers, preservatives, flavor additive such as peppermint oil or natural sweeteners or saccharin or other artificial sweeteners, and the like can also be added.
- dosage unit compositions for oral administration can be microencapsulated.
- the formulation can also be prepared to prolong or sustain the release, for example, by coating or embedding particulate material in polymers, wax or the like.
- the compounds of Formula I, and salts, solvates and physiological functional derivatives thereof, can also be administered in the form of liposome delivery systems, such as small unilamellar vesicles, large unilamellar vesicles and multilamellar vesicles.
- liposomes can be formed from a variety of phospholipids, such as cholesterol, stearylamine or phosphatidylcholines.
- the compounds of Formula I and salts, solvates and physiological functional derivatives thereof may also be delivered by the use of monoclonal antibodies as individual carriers to which the compound molecules are coupled.
- the compounds may also be coupled with soluble polymers as targetable drug carriers.
- Such polymers can include polyvinylpyrrolidone, pyran copolymer, polyhydroxypropylmethacrylamide-phenol, polyhydroxyethylaspartamidephenol, or polyethyleneoxidepolylysine substituted with palmitoyl residues.
- the compounds may be coupled to a class of biodegradable polymers useful in achieving controlled release of a drug, for example, polylactic acid, polyepsilon caprolactone, polyhydroxy butyric acid, polyorthoesters, polyacetals, polydihydropyrans, polycyanoacrylates and cross-linked or amphipathic block copolymers of hydrogels.
- a class of biodegradable polymers useful in achieving controlled release of a drug, for example, polylactic acid, polyepsilon caprolactone, polyhydroxy butyric acid, polyorthoesters, polyacetals, polydihydropyrans, polycyanoacrylates and cross-linked or amphipathic block copolymers of hydrogels.
- Dosage forms for inhaled administration may conveniently be formulated as aerosols or dry powders.
- the compound or salt of Formula I is in a particle-size-reduced form, and more preferably the size-reduced form is obtained or obtainable by micronisation.
- the preferable particle size of the size-reduced (e.g. micronised) compound or salt or solvate is defined by a D50 value of about 0.5 to about 10 microns (for example as measured using laser diffraction) .
- Aerosol formulations can comprise a solution or fine suspension of the active substance in a pharmaceutically acceptable aqueous or non-aqueous solvent. Aerosol formulations can be presented in single or multidose quantities in sterile form in a sealed container, which can take the form of a cartridge or refill for use with an atomising device or inhaler. Alternatively the sealed container may be a unitary dispensing device such as a single dose nasal inhaler or an aerosol dispenser fitted with a metering valve (metered dose inhaler) which is intended for disposal once the contents of the container have been exhausted.
- a metering valve metered dose inhaler
- the dosage form comprises an aerosol dispenser
- it preferably contains a suitable propellant under pressure such as compressed air, carbon dioxide or an organic propellant such as a hydrofluorocarbon (HFC) .
- suitable HFC propellants include 1, 1, 1, 2, 3, 3, 3-heptafluoropropane and 1, 1, 1, 2-tetrafluoroethane.
- the aerosol dosage forms can also take the form of a pump-atomiser.
- the pressurised aerosol may contain a solution or a suspension of the active compound. This may require the incorporation of additional excipients e.g. co-solvents and/or surfactants to improve the dispersion characteristics and homogeneity of suspension formulations. Solution formulations may also require the addition of co-solvents such as ethanol.
- Other excipient modifiers may also be incorporated to improve, for example, the stability and/or taste and/or fine particle mass characteristics (amount and/or profile) of the formulation.
- the pharmaceutical composition is a dry powder inhalable composition.
- a dry powder inhalable composition can comprise a powder base such as lactose, glucose, trehalose, mannitol or starch, the compound of Formula I or salt or solvate thereof (preferably in particle-size-reduced form, e.g. in micronised form) , and optionally a performance modifier such as L-leucine or another amino acid, and/or metals salts of stearic acid such as magnesium or calcium stearate.
- the dry powder inhalable composition comprises a dry powder blend of lactose and the compound of Formula I or salt thereof.
- the lactose is preferably lactose hydrate e.g. lactose monohydrate and/or is preferably inhalation-grade and/or fine-grade lactose.
- the particle size of the lactose is defined by 90% or more (by weight or by volume) of the lactose particles being less than 1000 microns (micrometres) (e.g. 10-1000 microns e.g. 30-1000 microns) in diameter, and/or 50% or more of the lactose particles being less than 500 microns (e.g. 10-500 microns) in diameter. More preferably, the particle size of the lactose is defined by 90% or more of the lactose particles being less than 300 microns (e.g.
- the particle size of the lactose is defined by 90% or more of the lactose particles being less than 100-200 microns in diameter, and/or 50% or more of the lactose particles being less than 40-70 microns in diameter. It is preferable that about 3 to about 30% (e.g. about 10%) (by weight or by volume) of the particles are less than 50 microns or less than 20 microns in diameter.
- a suitable inhalation-grade lactose is E9334 lactose (10% fines) (Borculo Domo Ingredients, Hanzeplein 25, 8017 J D Zwolle, Netherlands) .
- a pharmaceutical composition for inhaled administration can be incorporated into a plurality of sealed dose containers (e.g. containing the dry powder composition) mounted longitudinally in a strip or ribbon inside a suitable inhalation device.
- the container is rupturable or peel-openable on demand and the dose of e.g. the dry powder composition can be administered by inhalation via the device such as the device (GlaxoSmithKline) .
- Dosage forms for ocular administration may be formulated as solutions or suspensions with excipients suitable for ophthalmic use.
- Dosage forms for nasal administration may conveniently be formulated as aerosols, solutions, drops, gels or dry powders.
- compositions adapted for administration by inhalation include fine particle dusts or mists, which may be generated by means of various types of metered, dose pressurized aerosols, nebulizers or insufflators.
- the compound of Formula I or a pharmaceutically acceptable salt or solvate thereof may be formulated as a fluid formulation for delivery from a fluid dispenser.
- a fluid dispenser may have, for example, a dispensing nozzle or dispensing orifice through which a metered dose of the fluid formulation is dispensed upon the application of a user-applied force to a pump mechanism of the fluid dispenser.
- Such fluid dispensers are generally provided with a reservoir of multiple metered doses of the fluid formulation, the doses being dispensable upon sequential pump actuations.
- the dispensing nozzle or orifice may be configured for insertion into the nostrils of the user for spray dispensing of the fluid formulation into the nasal cavity.
- a fluid dispenser of the aforementioned type is described and illustrated in WO-A-2005/044354, the entire content of which is hereby incorporated herein by reference.
- the dispenser has a housing which houses a fluid discharge device having a compression pump mounted on a container for containing a fluid formulation.
- the housing has at least one finger-operable side lever which is movable inwardly with respect to the housing to cam the container upwardly in the housing to cause the pump to compress and pump a metered dose of the formulation out of a pump stem through a nasal nozzle of the housing.
- a particularly preferred fluid dispenser is of the general type illustrated in FIGS. 30-40 of WO-A-2005/044354.
- the invention further includes a pharmaceutical composition of a compound of Formula I or pharmaceutically acceptable salts thereof, as hereinbefore described, in combination with packaging material suitable for said composition, said packaging material including instructions for the use of the composition for the use as hereinbefore described.
- the compound of the present invention when administered in combination with other therapeutic agents normally administered by the inhaled, intravenous, oral or intranasal route, that the resultant pharmaceutical composition may be administered by the same routes.
- compositions may include other agents conventional in the art having regard to the type of formulation in question, for example those suitable for oral administration may include flavoring agents.
- a therapeutically effective amount of a compound of the present invention will depend upon a number of factors including, for example, the age and weight of the animal, the precise condition requiring treatment and its severity, the particular compound having Formula I, the nature of the formulation, and the route of administration, and will ultimately be at the discretion of the attendant physician or veterinarian.
- an effective amount of a compound of Formula I for the treatment of diseases or conditions associated with inappropriate Btk activity will generally be in the range of 5 ⁇ g to 100 mg/kg body weight of recipient (mammal) per day and more usually in the range of 5 ⁇ g to 10 mg/kg body weight per day.
- This amount may be given in a single dose per day or more usually in a number (such as two, three, four, five or six) of sub-doses per day such that the total daily dose is the same.
- An effective amount of a salt or solvate, thereof, may be determined as a proportion of the effective amount of the compound of Formula I per se.
- a dosage for humans preferably contains 0.0001-25 mg of a compound of Formula I or pharmaceutically acceptable salts thereof per kg body weight.
- the desired dose may be presented as one dose or as multiple subdoses administered at appropriate intervals throughout the day, or, in case of female recipients, as doses to be administered at appropriate daily intervals throughout the menstrual cycle.
- the dosage as well as the regimen of administration may differ between a female and a male recipient.
- the compounds of the present invention can be prepared by methods well known in the art of organic chemistry. See, for example, J. March, ‘Advanced Organic Chemistry’ 4 th Edition, John Wiley and Sons. During synthetic sequences it may be necessary and/or desirable to protect sensitive or reactive groups on any of the molecules concerned. This is achieved by means of conventional protecting groups, such as those described in T. W. Greene and P.G.M. Wutts ‘Protective Groups in Organic Synthesis’ 3 rd Edition, John Wiley and Sons, 1999. The protective groups are optionally removed at a convenient subsequent stage using methods well known in the art.
- the products of the reactions are optionally isolated and purified, if desired, using conventional techniques, but not limited to, filtration, distillation, crystallization, chromatography and the like. Such materials are optionally characterized using conventional means, including physical constants and spectral data.
- the compounds of Formula I can be prepared by the general synthetic routes shown in the schemes below.
- Reduction of 3-chloropyrazine-2-carbonitrile (II) can be accomplished by hydrogenation in the presence of a suitable catalyst system and solvent, for example Raney-Nickel ethanol to provide (3-chloropyrazin-2-yl) methanamine (III) . This amine can then be reacted with the acid (IV) .
- a suitable catalyst system and solvent for example Raney-Nickel ethanol
- the reaction of IV can be carried out in a solvent such as DMF, THF or DCM in the presence of a base such as DIPEA, N-methylmorpholine, 4-DMAP or triethylamine and in the presence of a coupling reagent such as PyBOP, TBTU, EDCI or HATU to form N- ( (3-chloropyrazin-2-yl) methyl) amide (V) .
- a coupling reagent such as PyBOP, TBTU, EDCI or HATU
- Cyclization chloropyrazine (V) can be performed using condensation reagents like phosphorousoxychloride under heating conditions to provide the 8-chloroimidazo [1, 5-a] pyrazine derivatives VI.
- bromination can be accomplished using bromine or N-bromosuccinimide in a suitable solvent like DCM or DMF at appropriate temperature to obtain compounds of formula VII.
- 8-Aminoimidazo [1, 5-a] pyrazine derivatives (VIII) can be prepared from compounds VII using ammonia (gas) in isopropanol at elevated temperature in a pressure vessel (>4 atm) or with primary amine (such as dimethoxybenzylamine) under heating.
- Compounds of formula I can be prepared from compounds of formula VIII using an appropriate boronic acid or pinacol ester (IX) , in the presence of a suitable palladium catalyst system, for example bis (diphenylphosphino) ferrocene palladium (II) chloride complex or tetrakis (triphenylphosphine) palladium (0) in the presence of an inorganic base like potassium carbonate, cesium carbonate or potassium phosphate in a suitable solvent system like combinations of dioxane and water.
- a suitable palladium catalyst system for example bis (diphenylphosphino) ferrocene palladium (II) chloride complex or tetrakis (triphenylphosphine) palladium (0) in the presence of an inorganic base like potassium carbonate, cesium carbonate or potassium phosphate in a suitable solvent system like combinations of dioxane and water.
- Palladium catalysts and conditions to form either the pinacol esters or to couple the boronic acids or pinacol esters with the 1-bromoimidazo [1, 5-a] pyrazin-8-amine are well known to the skilled organic chemist –see, for example, Ei-ichi Negishi (Editor) , Armin de Meijere (Associate Editor) , Handbook of Organopalladium Chemistry for Organic Synthesis, John Wiley and Sons, 2002.
- the acid intermediates IV are commercially available or can be readily prepared using methods well known to the skilled organic chemist.
- Mass Spectrometry Electron Spray spectra were recorded on the Applied Biosystems API-165 single quad mass spectrometer in alternating positive and negative ion mode using Flow Injection. The mass range was 120-2000 Da. and scanned with a step rate of 0.2 Da. and the capillary voltage was set to 5000 V. N 2 gas was used for nebulisation.
- solvent B MeCN-0.1% TFA
- solvent A H 2 O-0.1% TFA
- solvent B MeCN-0.1% TFA
- Step 1 (3R, 6S) -1-benzyl 3-methyl 6- ( ( (tert-butyldiphenylsilyl) oxy) methyl) piperidine-1, 3- dicarboxylate
- Step 2 (3R, 6S) -1- ( (benzyloxy) carbonyl) -6- ( ( (tert-butyldiphenylsilyl) oxy) methyl) piperidine-3- carboxylic acid
- Step 3 (2S, 5R) -benzyl 2- ( ( (tert-butyldiphenylsilyl) oxy) methyl) -5- ( ( (3-chloropyrazin-2- yl) methyl) carbamoyl) piperidine-1-carboxylate
- Step 4 (2S, 5R) -benzyl 2- ( ( (tert-butyldiphenylsilyl) oxy) methyl) -5- (8-chloroimidazo [1, 5- a] pyrazin-3-yl) piperidine-1-carboxylate
- Step 5 (2S, 5R) -benzyl 5- (1-bromo-8- chloroimidazo [1, 5-a] pyrazin-3-yl) -2- ( ( (tert-butyldiphenylsilyl) oxy) methyl) piperidine-1- carboxylate
- Step 6 (2S, 5R) -benzyl 5- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3- yl) -2- ( ( (tert-butyldiphenylsilyl) oxy) methyl) piperidine-1-carboxylate
- Step 7 (2S, 5R) -benzyl 5- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3- yl) -2- (hydroxymethyl) piperidine-1-carboxylate
- Step 8 (2S, 5R) -benzyl 5- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3- yl) -2-formylpiperidine-1-carboxylate
- Step 9 (2S, 5R) -benzyl 5- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3- yl) -2- ( (isopropyl (2-methoxy-2-oxoethyl) amino) methyl) piperidine-1-carboxylate
- Step 9 (7R, 9aS) -7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -2-isopropylhexahydro-1H- pyrido [1, 2-a] pyrazin-4 (6H) -one
- Step 1 (6R, 8aS) -6- (1-Bromo-8- ( (2, 4-dimethoxybenzyl) amino) -5-fluoroimidazo [1, 5-a] pyrazin-3- yl) tetrahydro-1H-oxazolo [3, 4-a] pyridin-3 (5H) -one
- Step 2 (6R, 8aS) -6- (8-Amino-1-bromo-5-fluoroimidazo [1, 5-a] pyrazin-3-yl) tetrahydro-1H- oxazolo [3, 4-a] pyridin-3 (5H) -one
- Step 1 (2R, 5R) -tert-butyl 2- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -5- (hydroxymethyl) morpholine-4-carboxylate
- Step 2 (2R, 5S) -tert-butyl 2- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -5-formylmorpholine-4- carboxylate
- Step3 (2R, 5R) -tert-butyl 2- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -5- (3-methoxy-3-oxoprop-1-en- 1-yl) morpholine-4-carboxylate
- Step4 (2R, 5R) -tert-butyl 2- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -5- (3-methoxy-3- oxopropyl) morpholine-4-carboxylate
- Step 5 (2R, 5R) -2- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -5- (3-methoxy-3-oxopropyl) morpholin- 4-ium chloride
- Step 6 (3R, 8aR) -3- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) tetrahydro-1H-pyrrolo [2, 1- c] [1, 4] oxazin-6 (7H) -one
- Step 7 (3R, 8aR) -3- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) tetrahydro-1H-pyrrolo [2, 1- c] [1, 4] oxazin-6 (7H) -one
- Step 8 (3R, 8aR) -3- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) tetrahydro-1H-pyrrolo [2, 1- c] [1, 4] oxazin-6 (7H) -one
- Step 2 (2R, 5S) -tert-butyl 2- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -5- ( ( (tert- butyldiphenylsilyl) oxy) methyl) morpholine-4-carboxylate
- Step 3 (2R, 5S) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3- yl) -5- ( ( (tert-butyldiphenylsilyl) oxy) methyl) morpholine-4-carboxylate
- Step 4 (2R, 5R) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3- yl) -5- (hydroxymethyl) morpholine-4-carboxylate
- Step 5 (2R, 5S) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3- yl) -5-formylmorpholine-4-carboxylate
- Step 6 (2R, 5R) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3- yl) -5- ( (isopropyl (2-methoxy-2-oxoethyl) amino) methyl) morpholine-4-carboxylate
- Step 7 (3R, 9aR) -3- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -8- isopropylhexahydropyrazino [2, 1-c] [1, 4] oxazin-6 (1H) -one
- Step 2 (2R, 5R) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3- yl) -5- ( (cyclopropyl (2-methoxy-2-oxoethyl) amino) methyl) morpholine-4-carboxylate
- Step 3 (3R, 9aR) -3- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -8- cyclopropylhexahydropyrazino [2, 1-c] [1, 4] oxazin-6 (1H) -one
- Step 2 (2R, 5R) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3- yl) -5- ( (tert-butyl (2-methoxy-2-oxoethyl) amino) methyl) morpholine-4-carboxylate
- Step 3 (3R, 9aR) -3- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -8- (tert- butyl) hexahydropyrazino [2, 1-c] [1, 4] oxazin-6 (1H) -one
- Step 1 (2R, 5S) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) -5-fluoroimidazo [1, 5- a] pyrazin-3-yl) -5- ( ( (tert-butyldiphenylsilyl) oxy) methyl) morpholine-4-carboxylate
- Step 2 (2R, 5R) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) -5-fluoroimidazo [1, 5- a] pyrazin-3-yl) -5- (hydroxymethyl) morpholine-4-carboxylate
- Step 3 (2R, 5S) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) -5-fluoroimidazo [1, 5- a] pyrazin-3-yl) -5-formylmorpholine-4-carboxylate
- Step 4 (2R, 5R) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) -5-fluoroimidazo [1, 5- a] pyrazin-3-yl) -5- ( (isopropyl (2-methoxy-2-oxoethyl) amino) methyl) morpholine-4-carboxylate
- Step 5 (3R, 9aR) -3- (8-amino-1-bromo-5-fluoroimidazo [1, 5-a] pyrazin-3-yl) -8 - isopropylhexahydropyrazino [2, 1-c] [1, 4] oxazin-6 (1H) -one
- Step 1 methyl 6-formylnicotinate
- Step 2 methyl 6- (4-methoxy-4-oxobutanoyl) nicotinate
- Step 3 methyl 6- (1, 1-difluoro-4-methoxy-4-oxobutyl) nicotinate
- Step 4 methyl 6- (1, 1-difluoro-4-methoxy-4-oxobutyl) piperidine-3-carboxylate
- Step 5 methyl 9, 9-difluoro-6-oxooctahydro-1H-quinolizine-3-carboxylate
- Step 6 9, 9-difluoro-6-oxooctahydro-1H-quinolizine-3-carboxylic acid
- Step 7 N- ( (3-chloropyrazin-2-yl) methyl) -9, 9-difluoro-6-oxooctahydro-1H-quinolizine-3- carboxamide
- Step 8 7- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-quinolizin-4 (6H) -one
- Step 9 7- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-quinolizin- 4 (6H) -one
- Step 10 7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-quinolizin- 4 (6H) -one
- Step 1 (3R, 6R) -1-benzyl 3-methyl 6-formylpiperidine-1, 3-dicarboxylate
- Step 2 (3R, 6R) -1-benzyl 3-methyl 6- ( (S) -2, 2, 2-trifluoro-1- ( (trimethylsilyl) oxy) ethyl) piperidine- 1, 3-dicarboxylate
- Step 3 (3R, 6R) -1-benzyl 3-methyl 6- ( (S) -2, 2, 2-trifluoro-1-hydroxyethyl) piperidine-1, 3- dicarboxylate
- Step 4 (3R, 6R) -1-benzyl 3-methyl 6- (2, 2, 2-trifluoroacetyl) piperidine-1, 3-dicarboxylate
- Step 5 (3R, 6R) -1-benzyl 3-methyl 6- ( (E) -4-ethoxy-1, 1, 1-trifluoro-4-oxobut-2-en-2- yl) piperidine-1, 3-dicarboxylate
- Step 6 (1S, 6R, 8aR) -methyl 3-oxo-1- (trifluoromethyl) octahydroindolizine-6-carboxylate
- Step 7 (1S, 6R, 8aR) -3-oxo-1- (trifluoromethyl) octahydroindolizine-6-carboxylic acid
- Step 8 (1S, 6R, 8aR) -N- ( (3-chloropyrazin-2-yl) methyl) -3-oxo-1- (trifluoromethyl) octahydroindolizine-6-carboxamide
- Step 9 (1S, 6R, 8aR) -6- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1- (trifluoromethyl) hexahydroindolizin-3 (2H) -one
- Step 10 (1S, 6R, 8aR) -6- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1- (trifluoromethyl) hexahydroindolizin-3 (2H) -one
- Step 11 (1S, 6R, 8aR) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1- (trifluoromethyl) hexahydroindolizin-3 (2H) -one
- Step 12 (single) (1S, 6R, 8aR) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1-(trifluoromethyl) hexahydroindolizin-3 (2H) -one
- Step 1 (3R, 6S) -1-benzyl 3-methyl 6-formylpiperidine-1, 3-dicarboxylate
- Step 2 (3R, 6S) -1-benzyl 3-methyl 6- ( (R) -2, 2, 2-trifluoro-1- ( (trimethylsilyl) oxy) ethyl) piperidine- 1, 3-dicarboxylate
- Step 3 (3R, 6S) -1-benzyl 3-methyl 6- ( (R) -2, 2, 2-trifluoro-1-hydroxyethyl) piperidine-1, 3- dicarboxylate
- Step 4 (3R, 6S) -1-benzyl 3-methyl 6- (2, 2, 2-trifluoroacetyl) piperidine-1, 3-dicarboxylate
- Step 5 (3R, 6S) -1-benzyl 3-methyl 6- ( (Z) -4-ethoxy-1, 1, 1-trifluoro-4-oxobut-2-en-2- yl) piperidine-1, 3-dicarboxylate
- Step 6 (1R, 6R, 8aS) -methyl 3-oxo-1- (trifluoromethyl) octahydroindolizine-6-carboxylate
- Step 7 (1R, 6R, 8aS) -3-oxo-1- (trifluoromethyl) octahydroindolizine-6-carboxylic acid
- Step 8 (1R, 6R, 8aS) -N- ( (3-chloropyrazin-2-yl) methyl) -3-oxo-1- (trifluoromethyl) octahydroindolizine-6-carboxamide
- Step 9 (1R, 6R, 8aS) -6- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1- (trifluoromethyl) hexahydroindolizin-3 (2H) -one
- Step 10 (1R, 6R, 8aS) -6- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1- (trifluoromethyl) hexahydroindolizin-3 (2H) -one
- Step 11 (1R, 6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1- (trifluoromethyl) hexahydroindolizin-3 (2H) -one
- Step 12 (two iosmers) (1R, 6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1- (trifluoromethyl) hexahydroindolizin-3 (2H) -one
- Step 1 (3R, 6S) -1-benzyl 3-methyl 6- (1-hydroxyethyl) piperidine-1, 3-dicarboxylate
- Step 6 (6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1-methyltetrahydro-1H- oxazolo [3, 4-a] pyridin-3 (5H) -one
- Step 7 (1S, 6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1-methyltetrahydro-1H- oxazolo [3, 4-a] pyridin-3 (5H) -one
- Step 1 (2R, 5R) -tert-butyl 2- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -5- (hydroxymethyl) morpholine-4-carboxylate
- Step 2 (2R, 5R) -tert-butyl 5- ( (2- (tert-butoxy) -2-oxoethoxy) methyl) -2- (8-chloroimidazo [1, 5- a] pyrazin-3-yl) morpholine-4-carboxylate
- Step 3 (2R, 5R) -tert-butyl 2- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -5- ( (2- (tert-butoxy) - 2-oxoethoxy) methyl) morpholine-4-carboxylate
- Step4 (2R, 5R) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3- yl) -5- ( (2- (tert-butoxy) -2-oxoethoxy) methyl) morpholine-4-carboxylate
- Step5 2- ( ( (3R, 6R) -6- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3- yl) morpholin-3-yl) methoxy) acetic acid
- Step6 (7R, 9aR) -7- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3- yl) tetrahydro-1H- [1, 4] oxazino [3, 4-c] [1, 4] oxazin-4 (3H) -one
- Step 1 (6R, 8aS) -N-methoxy-N-methyl-3-oxooctahydroindolizine-6-carboxamide
- Step 4 (6R, 8aS) -6- ( ( (tert-butyldimethylsilyl) oxy) methyl) -2, 2-dimethylhexahydroindolizin- 3 (2H) -one
- Step 6 (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizine-6-carboxylic acid
- Step 7 (6R, 8aS) -N- ( (3-chloropyrazin-2-yl) methyl) -2, 2-dimethyl-3-oxooctahydroindolizine-6- carboxamide
- Step 8 (6R, 8aS) -N- ( (3-chloropyrazin-2-yl) methyl) -2, 2-dimethyl-3-oxooctahydroindolizine-6- carboxamide
- Step 9 (6R, 8aS) -6- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -2, 2- dimethylhexahydroindolizin-3 (2H) -one
- Step 10 (6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -2, 2- dimethylhexahydroindolizin-3 (2H) -one
- Step 1 (6R, 8aS) -6- (8-amino-1-bromo-5-fluoro-6-methoxy-5, 6-dihydroimidazo [1, 5-a] pyrazin-3- yl) -2, 2-dimethylhexahydroindolizin-3 (2H) -one
- SELECTFLUOR (412 mg, 1.163 mmol) was added to a stirred, cooled 0 °C (6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -2, 2-dimethylhexahydroindolizin-3 (2H) -one (400 mg, 1.057 mmol) inMeCN (12 ml) and MeOH (12.00 ml) and the mixture was stirred at room temperature for Overnight.
- Step 2 (6R, 8aS) -6- (8-amino-1-bromo-5-fluoroimidazo [1, 5-a] pyrazin-3-yl) -2, 2- dimethylhexahydroindolizin-3 (2H) -one
- Step 1 methyl 2- (5-bromopyridin-2-yl) -2-methylpropanoate
- Step 4 3- (5-bromopyridin-2-yl) -1-diazo-3-methylbutan-2-one
- Step 5 methyl 3- (5-bromopyridin-2-yl) -3-methylbutanoate
- Step 6 methyl 6- (4-methoxy-2-methyl-4-oxobutan-2-yl) nicotinate
- Step 7 (trans) methyl 1, 1-dimethyl-3-oxooctahydroindolizine-6-carboxylate
- Step 8 (trans) 1, 1-dimethyl-3-oxooctahydroindolizine-6-carboxylic acid
- Step 9 (trans) N- ( (3-chloropyrazin-2-yl) methyl) -1, 1-dimethyl-3-oxooctahydroindolizine-6- carboxamide
- Step 10 (trans) 6- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1, 1-dimethylhexahydroindolizin-3 (2H) - one
- Step 11 (trans) 6- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1, 1- dimethylhexahydroindolizin-3 (2H) -one
- Step 12 (trans) 6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1- dimethylhexahydroindolizin-3 (2H) -one
- Step 13 (6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1- dimethylhexahydroindolizin-3 (2H) -one
- Step 1 (6R, 8aS) -6- (8-amino-1-bromo-5-fluoro-6-methoxy-5, 6-dihydroimidazo [1, 5-a] pyrazin-3- yl) -1, 1-dimethylhexahydroindolizin-3 (2H) -one
- Step 2 (6R, 8aS) -6- (8-amino-1-bromo-5-fluoroimidazo [1, 5-a] pyrazin-3-yl) -1, 1- dimethylhexahydroindolizin-3 (2H) -one
- Step 1 (6R, 8aS) -6- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -2, 2-dimethylhexahydroindolizin- 3 (2H) -one
- Step 3 (6R, 8aS) -6- (1-bromo-8-chloro-5-methylimidazo [1, 5-a] pyrazin-3-yl) -2, 2- dimethylhexahydroindolizin-3 (2H) -one
- NBS 118 mg, 0.661 mmol
- 6S, 8aR 8-chloro-5-methyl-imidazo [1, 5-a] pyrazin-3-yl) -1, 1-dimethylhexahydroindolizin-3 (2H) -one (200 mg, 0.601 mmol) in acetonitrile (10 mL) and the mixture was stirred at 25 °C overnight.
- Step 4 (6R, 8aS) -6- (8-amino-1-bromo-5-methylimidazo [1, 5-a] pyrazin-3-yl) -2, 2- dimethylhexahydroindolizin-3 (2H) -one
- Step 2 methyl 6- (2- ( (2, 4-dimethoxybenzyl) amino) ethyl) nicotinate
- Step 3 methyl 6- (2- ( (2, 4-dimethoxybenzyl) (phenoxycarbonyl) amino) ethyl) nicotinate
- Step 4 Methyl 6- (2- ( (2, 4-dimethoxybenzyl) (phenoxycarbonyl) amino) ethyl) piperidine-3- carboxylate
- Step 5 methyl 2- (2, 4-dimethoxybenzyl) -1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7- carboxylate
- Step 6 (trans) methyl 1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7-carboxylate
- Step 7 (trans) methyl 2-ethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7-carboxylate
- Step 8 (trans) 2-ethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7-carboxylic acid
- Step 9 (trans) N- ( (3-chloropyrazin-2-yl) methyl) -2-ethyl-1-oxooctahydro-1H-pyrido [1, 2- c] pyrimidine-7-carboxamide
- Step 10 (trans) 7- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -2-ethyloctahydro-1H-pyrido [1, 2- c] pyrimidin-1-one
- Step 11 (trans) 7- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -2-ethyloctahydro-1H- pyrido [1, 2-c] pyrimidin-1-one
- Step 12 (trans) 7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -2-ethyloctahydro-1H- pyrido [1, 2-c] pyrimidin-1-one
- Step 13 (trans, single, isomer 1 &isomer 2) 7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -2- ethyloctahydro-1H-pyrido [1, 2-c] pyrimidin-1-one
- Step 1 (2R, 5S) -tert-butyl 5- ( ( (tert-butyldiphenylsilyl) oxy) methyl) -2- (5, 8-dichloroimidazo [1, 5- a] pyrazin-3-yl) morpholine-4-carboxylate
- Step 2 (2R, 5S) -tert-butyl 2- (1-bromo-8-chloro-5- (2-hydroxypropan-2-yl) imidazo [1, 5-a] pyrazin- 3-yl) -5-methylmorpholine-4-carboxylate
- Step 3 ( (2R, 5S) -tert-butyl 2- (1-bromo-5-chloro-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5- a] pyrazin-3-yl) -5- ( ( (tert-butyldiphenylsilyl) oxy) methyl) morpholine-4-carboxylate
- Step 4 (2R, 5R) -tert-butyl 2- (1-bromo-5-chloro-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5- a] pyrazin-3-yl) -5- (hydroxymethyl) morpholine-4-carboxylate
- Step 5 (2R, 5S) -tert-butyl 2- (1-bromo-5-chloro-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5- a] pyrazin-3-yl) -5-formylmorpholine-4-carboxylate
- Step 6 (2R, 5R) -tert-butyl 2- (1-bromo-5-chloro-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5- a] pyrazin-3-yl) -5- ( (E) -3-methoxy-3-oxoprop-1-en-1-yl) morpholine-4-carboxylate
- the mixture was warmed to room temperature and stirred for 1 h.
- the reaction mixture was quenched with pH 5 phosphate buffer, extracted with ethyl acetate (2x20 mL) .
- the combined organic layers were washed with brine, dried over sodium sulfate, filtered and concentrated.
- Step 7 (2R, 5R) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3- yl) -5- (3-methoxy-3-oxopropyl) morpholine-4-carboxylate
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Diabetes (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Neurology (AREA)
- Physical Education & Sports Medicine (AREA)
- Hematology (AREA)
- Epidemiology (AREA)
- Obesity (AREA)
- Endocrinology (AREA)
- Emergency Medicine (AREA)
- Immunology (AREA)
- Rheumatology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Pulmonology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Provided are Bruton's Tyrosine Kinase (Btk) inhibitor compounds according to Formula I or pharmaceutically acceptable salts thereof, pharmaceutical compositions comprising these compounds and their use in therapy. In particular, the use of Btk inhibitor compounds of Formula I in the treatment of Btk mediated disorders is provided.
Description
The present invention relates to Btk inhibitor compounds, to pharmaceutical compositions comprising these compounds and to their use in therapy. In particular, the present invention relates to the use of Btk inhibitor compounds in the treatment of Bruton’s Tyrosine Kinase (Btk) mediated disorders.
B lymphocyte activation is key in the generation of adaptive immune responses. Derailed B lymphocyte activation is a hallmark of many autoimmune diseases and modulation of this immune response is therefore of therapeutic interest. Recently the success of B cell therapies in autoimmune diseases has been established. Treatment of rheumatoid arthritis (RA) patients with Rituximab (anti-CD20 therapy) is an accepted clinical therapy by now. More recent clinical trial studies show that treatment with Rituximab also ameliorates disease symptoms in relapsing remitting multiple sclerosis (RRMS) and systemic lupus erythematosus (SLE) patients. This success supports the potential for future therapies in autoimmune diseases targeting B cell immunity.
Bruton tyrosine kinase (Btk) is a Tec family non-receptor protein kinase, expressed in B cells and myeloid cells. The function of Btk in signaling pathways activated by the engagement of the B cell receptor (BCR) and FcεR1 on mast cells is well established. In addition, a function for Btk as a downstream target in Toll-like receptor signaling was suggested. Functional mutations in Btk in human results in the primary immunodeficiency disease called XLA which is characterized by a defect in B cell development with a block between pro-and pre-B cell stage. This results in an almost complete absence of B lymphocytes in human causing a pronounced reduction of serum immunoglobulin of all classes. These finding support the key role for Btk in the regulation of the production of auto-antibodies in autoimmune diseases. In addition, regulation of Btk may affect BCR-induced production of pro-inflammatory cytokines and chemokines by B cells, indicating a broad potential for Btk in the treatment of autoimmune diseases.
With the regulatory role reported for Btk in FcεR-mediated mast cell activation, Btk inhibitors may also show potential in the treatment of allergic responses [Gilfillan et al, Immunological Reviews 288 (2009) pp149-169] .
Furthermore, Btk is also reported to be implicated in RANKL-induced osteoclast differentiation [Shinohara et al, Cell 132 (2008) pp794-806] and therefore may also be of interest for the treatment of bone resorption disorders.
Other diseases with an important role for dysfunctional B cells are B cell malignancies. Indeed anti-CD20 therapy is used effectively in the clinic for the treatment of follicular lymphoma, diffuse large B-cell lymphoma and chronic lymphocytic leukemia [Lim et al, Haematologica, 95 (2010) pp135-143] . The reported role for Btk in the regulation of proliferation and apoptosis of B cells indicates there is potential for Btk inhibitors in the treatment of B cell lymphomas as well. Inhibition of Btk seems to be relevant in particular for B cell lymphomas due to chronic active BCR signaling [Davis et al, Nature, 463 (2010) pp88-94] .
Some classes of Btk inhibitor compounds have been described as kinase inhibitors, e.g. Imidazo [1, 5-f] [1, 2, 4] triazine compounds have been described in WO2005097800 and WO2007064993. Imidazo [1, 5-a] pyrazine compounds have been described in WO2005037836 and WO2001019828 as IGF-1R enzyme inhibitors.
Some of the Btk inhibitors reported are not selective over Src-family kinases. With dramatic adverse effects reported for knockouts of Src-family kinases, especially for double and triple knockouts, this is seen as prohibitive for the development of Btk inhibitors that are not selective over the Src-family kinases.
Both Lyn-deficient and Fyn-deficient mice exhibit autoimmunity mimicking the phenotype of human lupus nephritis. In addition, Fyn-deficient mice also show pronounced neurological defects. Lyn knockout mice also show an allergic-like phenotype, indicating Lyn as a broad negative regulator of the IgE-mediated allergic response by controlling mast cell responsiveness and allergy-associated traits [Odom et al, J. Exp. Med., 199 (2004) pp1491-1502] . Furthermore, aged Lyn knock-out mice develop severe splenomegaly (myeloid expansion) and disseminated monocyte/macrophage tumors [Harder et al, Immunity, 15 (2001) pp603-615] . These observations are in line with hyperresponsive B cells, mast cells and myeloid cells, and increased Ig levels observed in Lyn-deficient mice. Female Src knockout mice are infertile due to reduced follicle development and ovulation [Roby et al, Endocrine, 26 (2005) pp169-176] . The double knockouts Src-/-Fyn-/-and Src-/-Yes-/-show a severe phenotype with effects on movement and breathing. The triple knockouts Src-/-Fyn-/-Yes-/-die at day 9.5 [Klinghoffer et al, EMBO J., 18 (1999) pp2459-2471] . For the double knockout Src-/-Hck-/-, two thirds of the mice die at birth, with surviving mice developing osteopetrosis, extramedullary hematopoiseis, anemia, leukopenia [Lowell et al, Blood, 87 (1996) pp1780-1792] .
Hence, an inhibitor that inhibits multiple or all kinases of the Src-family kinases simultaneously may cause serious adverse effects.
SUMMARY OF THE INVENTION
The present invention provides compounds which inhibit Btk activity, their use for treatment of Btk mediated diseases and disorders, in particular autoimmune diseases and inflammatory diseases, as well as pharmaceutical compositions comprising such compounds and pharmaceutical carriers.
Definitions
The terms used herein have their ordinary meaning and the meaning of such terms is independent at each occurrence thereof. That notwithstanding, and except where stated otherwise, the following definitions apply throughout the specification and claims. Chemical names, common names, and chemical structures may be used interchangeably to describe the same structure. These definitions apply regardless of whether a term is used by itself or in combination with other terms, unless otherwise indicated. Hence, the definition of "alkyl" applies to "alkyl" as well as the "alkyl" portions of "hydroxyalkyl, " "fluoroalkyl, " "alkoxy" , etc.
As used herein, and throughout this disclosure, the following terms, unless otherwise indicated, shall be understood to have the following meanings:
The term "alkyl, ” as used herein, refers to an aliphatic hydrocarbon group having one of its hydrogen atoms replaced with a bond having the specified number of carbon atoms. In different embodiments, an alkyl group contains, for example, from 1 to 6 carbon atoms (1-6C) alkyl or from 1 to 4 carbon atoms (1-4C) alkyl or from 1 to 3 carbon atoms (1-3C) alkyl. In one embodiment, an alkyl group is linear. In another embodiment, an alkyl group is branched. Non-limiting examples of alkyl groups include methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, isobutyl, tert-butyl, n-pentyl, neopentyl, isopentyl, n-hexyl, isohexyl and neohexyl.
The term “ (1-3C) alkylamino, as used herein, refers to an alkyl group having 1 to 3 carbon atoms which is linked by an amino group to the compound having formula I. Non-limiting examples include methylamino, ethylamino and propylamino.
"Alkoxy" refers to an alkyl-O-group represented by a linear or branched alkyl group of indicated number of carbon atoms attached through an oxygen bridge; for example " (1-6C) Alkoxy" includes -OCH3, -OCH2CH3, -OCH (CH3) 2, -O (CH2) 5CH3, and the like.
Unless otherwise specifically noted as only “unsubstituted” or only “substituted” , alkyl groups are unsubstituted or substituted with 1 to 3 substituents on each carbon atom.
The term “amount effective” or "effective amount" as used herein, refers to an amount of the compound of Formula I and/or an additional therapeutic agent, or a composition thereof, that is effective in producing the desired therapeutic, ameliorative, inhibitory or preventative effect when administered to a subject suffering from a BTK-mediated disease or disorder. In the combination therapies of the present invention, an effective amount can refer to each individual agent or to the combination as a whole, wherein the amounts of all agents administered are together effective, but wherein the component agent of the combination may not be present individually in an effective amount.
The term “halogen” , as used herein, refers to fluorine, chlorine, bromine or iodine. Fluorine, chlorine or bromine being preferred halogens; fluorine being more preferred.
The term "cycloalkyl, " as used herein, refers to a saturated mono-or multicyclic ring system containing the specified number of ring carbon atoms, and no heteroatom. In a like manner the term " (C3-6) cycloalkyl" or (3-6C) cycloalkyl” refers to a saturated ring having from 3 to 6 ring carbon atoms. Non-limiting examples of monocyclic cycloalkyls include cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl. In one embodiment, the cycloalkyl is cyclopropyl.
The term “cycloalkylmethylene” , as used herein, refers to a cycloakyl group as defined above, linked to a methyl group, wherein two of the hydrogen atoms of the methyl group have been replaced with a bond such that the methyl group links the cycloalkyl group to the compound having formula I.
"Cycloalkoxy" refers to a cycloalkyl-O-group represented by a cycloalkyl group of indicated number of carbon atoms attached through an oxygen bridge to the compound of the invention; for example " (3-6C) cycloalkoxy" includes –O-cyclopropyl, -O-cyclobutyl, --O-cyclopentyl, or –O-cyclohexyl.
The term “C0” as employed in expressions such as “ (C0-6) alkylene” means a direct covalent bond; or when employed in expressions such as " (C0-6) alkyl" means hydrogen. Similarly, when an integer defining the presence of a certain number of atoms in a group is equal to zero, it means that the atoms adjacent thereto are connected directly by a bond; for example, in the structure wherein s is an integer equal to zero, 1 or 2, the structure is
wherein s is an integer equal to zero, 1 or 2, the structure is when s is zero; or it means that the indicated atom is absent; for example -S (O) 0-means -S-.
when s is zero; or it means that the indicated atom is absent; for example -S (O) 0-means -S-.
Unless expressly stated to the contrary, all ranges cited herein are inclusive. For example, a heterocycloalkyl described as containing from "1 to 4 heteroatoms" means the heterocycloalkyl can contain 1, 2, 3 or 4 heteroatoms.
When any variable occurs more than one time in any constituent or in any formula depicting and describing compounds of the invention, its definition on each occurrence is independent of its definition at every other occurrence. Also, combinations of substituents and/or variables are permissible only if such combinations result in stable compounds. For variable definitions containing terms having repeated terms, e.g., (CRiRj) r, where r is the integer 2, Ri is a defined variable, and Rj is a defined variable, the value of Ri may differ in each instance in which it occurs, and the value of Rj may differ in each instance in which it occurs. For example, if Ri and Rj are independently selected from the group consisting of methyl, ethyl, propyl and butyl, then (CRiRj) 2 can be

As used herein, the term "Xa-Xb" , shall have the same meaning as the term "Xa-b" or “ (a-bX) ” , wherein X is any atom and a and b are any integers. For example, "C1-C4" shall have the same meaning as "C1-4" or “ (1-4C) ” . Additionally, when referring to a functional group generically, "Ax" shall have the same meaning, and be interchangeable with, "AX" , wherein "A" is any atom and "x" or "X" are any integer. For example, "R1" shall have the same meaning, and be interchangeable with, "R1".
In the above definitions with multifunctional groups, the attachment point is at the last group. For example, the term (C1-3) alkoxycarbonyl refers to, e.g.  and the term (C1-4) alkylcarbonyloxy refers to, e.g.
and the term (C1-4) alkylcarbonyloxy refers to, e.g. 
The term "purified" as used herein, refers to the physical state of a compound after the compound has been isolated through a synthetic process (e.g., from a reaction mixture) , from a natural source, or a combination thereof. The term "purified" also refers to the physical state of a compound after the compound has been obtained from a purification process or processes described herein or well-known to the skilled artisan (e.g., chromatography,
recrystallization, and the like) , in sufficient purity to be characterizable by standard analytical techniques described herein or well-known to the skilled artisan.
The term “substituted” , as used herein, means that one or more hydrogens on the designated atom/atoms is/are replaced with a selection from the indicated group, provided that the designated atom’s normal valency under the existing circumstances is not exceeded, and that the substitution results in a stable compound. Combinations of substituents and/or variables are permissible only if such combinations result in stable compounds. “Stable compound” or “stable structure” is defined as a compound or structure that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and formulation into an efficacious therapeutic agent.
The term “optionally substituted” means that a compound may or may not be substituted with the specified groups, radicals or moieties.
A “subject” is a human or non-human mammal. In one embodiment, a subject is a human. In another embodiment, the subject is a chimpanzee.
In the above definitions with multifunctional groups, the attachment point is at the last group, unless otherwise specified on the substituent group by a dash. A dash on the substituent group would then represent the point of attachment.
It should be noted that any carbon as well as heteroatom with unsatisfied valences in the text, schemes, examples and tables herein is assumed to have the sufficient number of hydrogen atom (s) to satisfy the valences.
Compounds of the Invention
The present invention provides Btk inhibitor compounds according to Formula I or pharmaceutically acceptable salts thereof

wherein:
R11 is independently selected from the group consisting of:
a) H,
b) halogen,
c) cyano,
d) (1-6C) alkyl,
wherein in aromatic ring K
B1 is N or C (R7) ;
B2 is N or C (R8) ;
R7 is H, halogen, (1-3C) alkyl, (1-6C) alkoxy , (3-6C) cycloalkoxy, amino, or (1-3C) alkylamino;
R8 is H, halogen, (1-3C) alkyl, (1-6C) alkoxy , (3-6C) cycloalkoxy, amino, or (1-3C) alkylamino;
R9 is H, halogen, (1-3C) alkyl, (1-6C) alkoxy , (3-6C) cycloalkoxy, amino, or (1-3C) alkylamino;
R10 is H, halogen, (1-3C) alkyl, (1-6C) alkoxy , (3-6C) cycloalkoxy, amino, or (1-3C) alkylamino;
wherein heteroaromatic ring L is selected from the group consisting of:

R5 is H, cyano, (1-4C) alkyl, (3-6C) cycloalkyl, or (3-6C) cycloalkoxy,
wherein R5 may optionally be substituted with one, two or three halogens;
wherein ring M is selected from the group consisting of:

Q is C=O or CH2;
T is C (Re) 2, O, NRe, or a bond;
U is C (Rd) 2, O, or NRd;
V is CH2 or O;
each Rc is independently selected from H, fluoro, methyl or trifluoromethyl;
each Rd is independently selected from H, (1-3C) alkyl, (1-3C) alkoxy, cyclopropyl or cyclopropylmethylene;
each Re is independently selected from H or (1-6C) alkyl; and
with the proviso that:
when Q is CH2, then T is C (Re) 2.
In one aspect the invention relates to a compound according to Formula I wherein the ring M is selected from the group consisting of

In another aspect the invention relates to a compound according to Formula I wherein the heteroaromatic ring L is

B1 is CH; and
B2 is CH.
In another aspect the invention relates to a compound according to Formula I wherein R5 is selected from the group consisting of hydrogen, CN, cyclopropyl, (1-4C) alkyl, and (3-6C) cycloalkoxy; wherein the alkyl may optionally be substituted with one, two or three halogen. In a preferred embodiment, the (3-6C) cycloalkoxy is cyclopropoxy.
In a further aspect the invention relates to a compound according to Formula I wherein R5 is selected from the group consisting of hydrogen, methyl, cyclopropyl, cyclopropoxyl, and trifluoromethyl.
The invention also relates to those compounds wherein all specific definitions for B1, B2, Q, T, U, V, R5, R7, R8, R9, R10, R11, Rc, Rd, Re, and all substituent groups in the various aspects of the inventions defined here above occur in any combination within the definition of the Btk inhibitor compounds of Formula I or pharmaceutically acceptable salts thereof.
Non-limiting examples of the compounds of the present invention include:
4- (8-amino-5-fluoro-3- ( (3R, 9aR) -8-isopropyl-6-oxooctahydropyrazino [2, 1-c] [1, 4] oxazin-3-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-ethoxy-5-fluoro-N- (4- (trifluoromethyl) pyridin-2-yl) benzamide;
4- (8-amino-5-chloro-3- ( (6R, 8aS) -3-oxooctahydroindolizin-6-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-fluoro-5-methoxy-N- (1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl) benzamide;
4- (8-amino-5-fluoro-3- ( (6R, 8aS) -3-oxooctahydroindolizin-6-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-fluoro-5-methoxy-N- (1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl) benzamide;
4- (8-amino-3- ( (3S, 9aR) -9, 9-difluorooctahydro-2H-quinolizin-3-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-ethoxy-N- (4- (trifluoromethyl) pyridin-2-yl) benzamide;
4- (8-amino-3- ( (3R, 9aR) -9, 9-difluorooctahydro-2H-quinolizin-3-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-ethoxy-N- (4- (trifluoromethyl) pyridin-2-yl) benzamide;
cis 4- (8-amino-3- (9, 9-difluorooctahydro-2H-quinolizin-3-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-ethoxy-N- (4- (trifluoromethyl) pyridin-2-yl) benzamide;
4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- (1-tert-butyl-1H-1, 2, 3-triazol-4-yl) -5-ethoxy-2-fluorobenzamide
4- {8-amino-5-chloro-3- [ (3R, 8aR) -6-oxohexahydro-1H-pyrrolo [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-fluoro-5-methoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (3R, 8aR) -6-oxohexahydro-1H-pyrrolo [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-fluoro-5-methoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-5-fluoro-3- [ (3R, 8aR) -6-oxohexahydro-1H-pyrrolo [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-fluoro-5-methoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N-[4- (difluoromethyl) pyridin-2-yl] -3-ethoxybenzamide;
4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N-[4- (difluoromethyl) pyridin-2-yl] -3-methoxybenzamide;
4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4-(difluoromethyl) pyridin-2-yl] -3-methoxybenzamide;
4- {8-amino-5-chloro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N-[4- (difluoromethyl) pyridin-2-yl] -3-ethoxybenzamide;
4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N-[4- (1, 1-difluoroethyl) pyridin-2-yl] -3-ethoxybenzamide;
4- {8-amino-5-chloro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N-[4- (1, 1-difluoroethyl) pyridin-2-yl] -3-ethoxybenzamide;
4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-propoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-propoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-fluoro-5-methoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-fluoro-5-methoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-5-cyano-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-5-fluoro-3- [ (6R) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-(cyclopropyloxy) -N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-5-chloro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-5-chloro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N-pyridin-2-ylbenzamide;
4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N-pyridin-2-ylbenzamide;
4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- (4-cyanopyridin-2-yl) -3-ethoxy-5-fluorobenzamide;
4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [6- (trifluoromethyl) pyrimidin-4-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -2-chloro-5-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -2-chloro-5-ethoxy-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -5-ethoxy-2, 3-difluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide;
4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -2-chloro-5-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -2-chloro-5-ethoxy-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide;
4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -2-chloro-5-ethoxy-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide;
4- {8-amino-5-chloro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -2-chloro-5-ethoxy-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide;
4- {8-amino-5-fluoro-3- [ (3R, 9aR) -8- (1-methylethyl) -6-oxooctahydropyrazino [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -2-chloro-5-ethoxy-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide;
4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -2-chloro-5-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-5-chloro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -2-chloro-5-ethoxy-N-pyridin-2-ylbenzamide;
4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -2-chloro-5-ethoxy-N-pyridin-2-ylbenzamide;
4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N-pyridin-2-ylbenzamide;
4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -5-ethoxy-2-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
5- {8-amino-5-chloro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -4-methyl-N- [4- (trifluoromethyl) pyridin-2-yl] pyridine-2-carboxamide;
4- {8-amino-3- [ (6S, 8aR) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (6S, 8aR) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (6S, 8aR) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (7S, 9aR) -octahydropyrido [2, 1-c] [1, 4] oxazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (3R, 9aR) -9, 9-difluoro-6-oxooctahydro-2H-quinolizin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (3R, 9aR) -9, 9-difluoro-6-oxooctahydro-2H-quinolizin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (7R, 9aS) -octahydropyrido [2, 1-c] [1, 4] oxazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (6S, 8aR) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (3S, 9aS) -9, 9-difluoro-6-oxooctahydro-2H-quinolizin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (3S, 9aS) -9, 9-difluoro-6-oxooctahydro-2H-quinolizin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (3R, 8aR) -6-oxohexahydro-1H-pyrrolo [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (1S, 6R, 8aR) -3-oxo-1- (trifluoromethyl) octahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (3R, 9aS) -9, 9-difluoro-6-oxooctahydro-2H-quinolizin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (3R, 9aS) -9, 9-difluoro-6-oxooctahydro-2H-quinolizin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (3S, 9aR) -9, 9-difluoro-6-oxooctahydro-2H-quinolizin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (3S, 9aR) -9, 9-difluoro-6-oxooctahydro-2H-quinolizin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-fluoro-5-methoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (1S, 6R, 8aS) -1-methyl-3-oxotetrahydro-1H- [1, 3] oxazolo [4, 3-c] [1, 4] oxazin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (1R, 6R, 8aS) -1-methyl-3-oxotetrahydro-1H- [1, 3] oxazolo [4, 3-c] [1, 4] oxazin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (3R, 9aR) -6-oxohexahydro-1H- [1, 4] oxazino [3, 4-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] -5-fluoroimidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] -5-fluoroimidazo [1, 5-a] pyrazin-1-yl} -3-fluoro-5-methoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizin-6-yl] -5-fluoroimidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (1R, 6R, 8aS) -3-oxo-1- (trifluoromethyl) octahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (1R, 6R, 8aS) -3-oxo-1- (trifluoromethyl) octahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -5-ethoxy-2-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizin-6-yl] -5-methylimidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxotetrahydro-1H- [1, 3] oxazolo [4, 3-c] [1, 4] oxazin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizin-6-yl] -5-methylimidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -5-ethoxy-2-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
5- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -4-ethoxy-N-[4- (trifluoromethyl) pyridin-2-yl] pyridine-2-carboxamide;
4- {8-amino-3- [ (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizin-6-yl] -5-fluoroimidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizin-6-yl] -5-fluoroimidazo [1, 5-a] pyrazin-1-yl} -3-fluoro-5-methoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N-(4-cyclopropylpyridin-2-yl) -3-ethoxy-5-fluorobenzamide;
4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- (4-cyclopropylpyridin-2-yl) -3-ethoxy-5-fluorobenzamide;
4- {8-amino-3- [ (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -5-ethoxy-2-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -5-ethoxy-2-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizin-6-yl] -5-fluoroimidazo [1, 5-a] pyrazin-1-yl} -5-ethoxy-2-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizin-6-yl] -5-fluoroimidazo [1, 5-a] pyrazin-1-yl} -5-ethoxy-2-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide;
4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -5-ethoxy-2-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- (4-cyclopropylpyridin-2-yl) -3-ethoxy-5-fluorobenzamide;
4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (difluoromethyl) pyridin-2-yl] -3-ethoxy-5-fluorobenzamide;
4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (1, 1-difluoroethyl) pyridin-2-yl] -3-ethoxy-5-fluorobenzamide;
4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- (4-cyclopropylpyridin-2-yl) -3-ethoxy-5-fluorobenzamide;
4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- (4-cyclopropylpyridin-2-yl) -5-ethoxy-2-fluorobenzamide;
4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- (4-cyclopropylpyridin-2-yl) -5-ethoxy-2-fluorobenzamide;
4- [8-amino-3- (2-ethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidin-7-yl) imidazo [1, 5-a] pyrazin-1-yl] -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- [8-amino-3- (2-ethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidin-7-yl) imidazo [1, 5-a] pyrazin-1-yl] -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- [8-amino-3- (1, 1-dimethyl-3-oxooctahydroindolizin-6-yl) imidazo [1, 5-a] pyrazin-1-yl] -3-ethoxy-5-fluoro-N-pyridin-2-ylbenzamide;
4- [8-amino-3- (1-oxohexahydro-3H-pyrido [1, 2-c] [1, 3] oxazin-7-yl) imidazo [1, 5-a] pyrazin-1-yl] -3-ethoxy-5-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- [8-amino-3- (1-oxohexahydro-3H-pyrido [1, 2-c] [1, 3] oxazin-7-yl) imidazo [1, 5-a] pyrazin-1-yl] -3-ethoxy-5-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4-amino-5- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N-[4- (trifluoromethyl) pyridin-2-yl] pyridine-2-carboxamide;
5- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -6-methoxy-N- [4- (trifluoromethyl) pyridin-2-yl] pyridine-2-carboxamide;
5- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4-(trifluoromethyl) pyridin-2-yl] pyrimidine-2-carboxamide;
5- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -6-(ethylamino) -N- [4- (trifluoromethyl) pyridin-2-yl] pyridine-2-carboxamide;
4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-chloro-5-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-chloro-5-ethoxy-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide;
4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-chloro-5-ethoxy-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide;
4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-chloro-N- [4- (difluoromethyl) pyridin-2-yl] -5-ethoxybenzamide;
4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-chloro-5-ethoxy-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-chloro-5-methoxy-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide;
4- {8-amino-3- [ (3R, 8aR) -6-oxohexahydro-1H-pyrrolo [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (3R, 9aR) -8- (1-methylethyl) -6-oxooctahydropyrazino [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (3R, 9aR) -8- (1-methylethyl) -6-oxooctahydropyrazino [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide;
4- {8-amino-3- [ (3R, 9aR) -8- (1-methylethyl) -6-oxooctahydropyrazino [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (3R, 9aR) -8-cyclopropyl-6-oxooctahydropyrazino [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (3R, 9aR) -8-cyclopropyl-6-oxooctahydropyrazino [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (3R, 9aR) -8-tert-butyl-6-oxooctahydropyrazino [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (3R, 9aR) -8-tert-butyl-6-oxooctahydropyrazino [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-5-chloro-3- [ (3R, 9aR) -8- (1-methylethyl) -6-oxooctahydropyrazino [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-5-fluoro-3- [ (3R, 9aR) -8- (1-methylethyl) -6-oxooctahydropyrazino [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-5-fluoro-3- [ (3R, 9aR) -8- (1-methylethyl) -6-oxooctahydropyrazino [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxohexahydro [1, 3] oxazolo [3, 4-a] pyridin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-5-chloro-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (7R, 9aR) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (7R, 9aR) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N-pyridin-2-ylbenzamide;
4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N-pyridin-2-ylbenzamide;
4- {8-amino-5-chloro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N-pyridin-2-ylbenzamide;
4- {8-amino-5-chloro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-chloro-5-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide;
4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N-pyridin-2-ylbenzamide;
4- [8-amino-3- (octahydropyrido [2, 1-c] [1, 4] oxazin-7-yl) imidazo [1, 5-a] pyrazin-1-yl] -3-ethoxy-5-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- [8-amino-3- (octahydropyrido [2, 1-c] [1, 4] oxazin-7-yl) imidazo [1, 5-a] pyrazin-1-yl] -3-ethoxy-5-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide;
4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N-pyridin-2-ylbenzamide;
4- {8-amino-5-chloro-3- [ (2R, 6R, 8aS) -2- (cyclopropylmethyl) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-5-chloro-3- [ (2R, 6R, 8aS) -2- (cyclopropylmethyl) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (3R, 7R, 9aS) -3-methyl-4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide;
4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-methoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] -5-fluoroimidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N-pyridin-2-ylbenzamide;
4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] -5-fluoroimidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide;
4- [8-amino-3- (2, 3, 3-trimethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidin-7-yl) imidazo [1, 5-a] pyrazin-1-yl] -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (1, 1-difluoroethyl) pyridin-2-yl] -3-ethoxybenzamide;
4- [8-amino-3- (2, 3, 3-trimethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidin-7-yl) imidazo [1, 5-a] pyrazin-1-yl] -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -5-ethoxy-2-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide;
4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (difluoromethyl) pyridin-2-yl] -3-ethoxybenzamide;
4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- (4-cyclopropylpyridin-2-yl) -3-ethoxybenzamide;
4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide;
4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (cyclopropyloxy) pyridin-2-yl] -3-ethoxy-5-fluorobenzamide;
4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N-[4- (cyclopropyloxy) pyridin-2-yl] -3-ethoxybenzamide;
4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-fluoro-5-methoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (7R, 9aS) -3, 3-dimethyl-4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-3- [ (3S, 7R, 9aS) -3-methyl-4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide;
4- {8-amino-5-chloro-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide;
4- {8-amino-5-chloro-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N-pyridin-2-ylbenzamide;
4- [8-amino-3- (3, 3-dimethyl-1-oxohexahydro-3H-pyrido [1, 2-c] [1, 3] oxazin-7-yl) imidazo [1, 5-a] pyrazin-1-yl] -N- [4- (difluoromethyl) pyridin-2-yl] -3-ethoxy-5-fluorobenzamide;
4- {8-amino-5-chloro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N-(4-cyanopyridin-2-yl) -3-ethoxy-5-fluorobenzamide;
4- [8-amino-3- (3, 3-dimethyl-1-oxohexahydro-3H-pyrido [1, 2-c] [1, 3] oxazin-7-yl) imidazo [1, 5-a] pyrazin-1-yl] -N- [4- (difluoromethyl) pyridin-2-yl] -3-ethoxy-5-fluorobenzamide; and
4- (8-amino-3- (2- (2-methoxyethyl) -4-oxooctahydro-1H-pyrido [1, 2-a] pyrazin-7-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-ethoxy-N- (4- (trifluoromethyl) pyridin-2-yl) benzamide.
The compounds of this invention include the salts, solvates, hydrates or prodrugs of the compounds. The use of the terms "salt" , "solvate" , “hydrate” , "prodrug" and the like, is intended to equally apply to the salt, solvate, hydrate and prodrug of enantiomers, stereoisomers, rotamers, tautomers, positional isomers, or racemates of the inventive compounds.
Salts
The Btk inhibitor compounds of the present invention, which can be in the form of a free base, may be isolated from the reaction mixture in the form of a pharmaceutically acceptable salt.
The compounds of Formula I can form salts which are also within the scope of this invention. Reference to a compound of Formula I herein is understood to include reference to pharmaceutically acceptable salts thereof, unless otherwise indicated. The term "pharmaceutically acceptable salt (s) "or “salt” , as employed herein, denotes acidic salts formed with inorganic and/or organic acids, as well as basic salts formed with inorganic and/or organic bases. In addition, when a compound of Formula I contains both a basic moiety, such as, but not limited to a pyridine or imidazole, and an acidic moiety, such as, but not limited to a carboxylic acid, zwitterions ("inner salts") may be formed and are included within the term "salt (s) " as used herein. Such acidic and basic salts used within the scope of the invention are pharmaceutically acceptable (i.e., non-toxic, physiologically acceptable) salts. Salts of the compounds of Formula I may be formed, for example, by reacting a compound of Formula I with an amount of acid or base, such as an equivalent amount, in a medium such as one in which the salt precipitates or in an aqueous medium followed by lyophilization.
Exemplary acid addition salts include acetates, ascorbates, benzoates, benzenesulfonates, bisulfates, borates, butyrates, citrates, camphorates, camphorsulfonates, fumarates, hydrochlorides, hydrobromides, hydroiodides, lactates, maleates, methanesulfonates, naphthalenesulfonates, nitrates, oxalates, phosphates, propionates, salicylates, succinates, sulfates, tartarates, thiocyanates, toluenesulfonates (also known as tosylates, ) and the like.
Additionally, acids which are generally considered suitable for the formation of pharmaceutically useful salts from basic pharmaceutical compounds are discussed, for example, by P. Stahl et al, Camille G. (eds. ) Handbook of Pharmaceutical Salts. Properties, Selection and Use. (2002) Zurich: Wiley-VCH; S. Berge et al, Journal of Pharmaceutical Sciences (1977) 66 (1) 1-19; P. Gould, International J. of Pharmaceutics (1986) 33 201-217; Anderson et al, The Practice of Medicinal Chemistry (1996) , Academic Press, New York; and in The Orange Book (Food &Drug Administration, Washington, D.C. on their website) . These disclosures are incorporated herein by reference.
Exemplary basic salts include ammonium salts, alkali metal salts such as sodium, lithium, and potassium salts, alkaline earth metal salts such as calcium and magnesium salts, salts with organic bases (for example, organic amines) such as dicyclohexylamines, t-butyl amines, and salts with amino acids such as arginine, lysine and the like. Basic nitrogen-containing groups may be quarternized with agents such as lower alkyl halides (e.g., methyl, ethyl, and butyl chlorides, bromides and iodides) , dialkyl sulfates (e.g., dimethyl, diethyl, and dibutyl sulfates) , long chain halides (e.g., decyl, lauryl, and stearyl chlorides, bromides and iodides) , aralkyl halides (e.g., benzyl and phenethyl bromides) , and others.
Crystals
The Btk inhibitor compounds of the present invention may exist as amorphous forms or crystalline forms.
The compounds of Formula I may have the ability to crystallize in more than one form, a characteristic known as polymorphism, and it is understood that such polymorphic forms ("polymorphs") are within the scope of Formula I. Polymorphism generally can occur as a response to changes in temperature or pressure or both and can also result from variations in the crystallization process. Polymorphs can be distinguished by various physical characteristics known in the art such as x-ray diffraction patterns, solubility and melting point.
Solvates
The compounds having Formula I or the pharmaceutically acceptable salts may form hydrates or solvates. It is known to those of skill in the art that charged compounds form hydrated species when lyophilized with water, or form solvated species when concentrated in a solution with an appropriate organic solvent. The compounds of this invention include the hydrates or solvates of the compounds listed.
One or more compounds of the invention having Formula I or the pharmaceutically acceptable salts or solvates thereof may exist in unsolvated as well as solvated forms with pharmaceutically acceptable solvents such as water, ethanol, and the like, and it is
intended that the invention embrace both solvated and unsolvated forms. "Solvate" means a physical association of a compound of this invention with one or more solvent molecules. This physical association involves varying degrees of ionic and covalent bonding, including hydrogen bonding. In certain instances the solvate will be capable of isolation, for example when one or more solvent molecules are incorporated in the crystal lattice of the crystalline solid. "Solvate" encompasses both solution-phase and isolatable solvates. Non-limiting examples of suitable solvates include ethanolates, methanolates, and the like. "Hydrate" is a solvate wherein the solvent molecule is H2O.
Preparation of solvates is generally known. Thus, for example, M. Caira et al, J. Pharmaceutical Sci., 93 (3) , 601-611 (2004) describe the preparation of the solvates of the antifungal fluconazole in ethyl acetate as well as from water. Similar preparations of solvates, hemisolvate, hydrates and the like are described by E. C. van Tonder et al, AAPS PharmSciTech., 5(1) , article 12 (2004) ; and A. L. Bingham et al, Chem. Commun. 603-604 (2001) . A typical, non-limiting, process involves dissolving the inventive compound in desired amounts of the desired solvent (organic or water or mixtures thereof) at a higher than ambient temperature, and cooling the solution at a rate sufficient to form crystals which are then isolated by standard methods. Analytical techniques such as, for example IR spectroscopy, show the presence of the solvent (or water) in the crystals as a solvate (or hydrate) .
Optical Isomers
The compounds of Formula I may contain asymmetric or chiral centers, and, therefore, exist in different stereoisomeric forms. It is intended that all stereoisomeric forms of the compounds of Formula I, as well as mixtures thereof, including racemic mixtures, form part of the present invention. In addition, the present invention embraces all geometric and positional isomers. For example, if a compound of Formula I incorporates a double bond or a fused ring, both the cis-and trans-forms, as well as mixtures, are embraced within the scope of the invention. Such stereoisomeric forms also include enantiomers and diastereoisomers, etc.
For chiral compounds, methods for asymmetric synthesis whereby the pure stereoisomers are obtained are well known in the art, e.g. synthesis with chiral induction, synthesis starting from chiral intermediates, enantioselective enzymatic conversions, separation of stereoisomers using chromatography on chiral media. Such methods are described in Chirality in Industry (edited by A.N. Collins, G.N. Sheldrake and J. Crosby, 1992; John Wiley) . Likewise methods for synthesis of geometrical isomers are also well known in the art.
Diastereomeric mixtures can be separated into their individual diastereomers on the basis of their physical chemical differences by methods well known to those skilled in the art,
such as, for example, by chromatography and/or fractional crystallization. Enantiomers can be separated by converting the enantiomeric mixture into a diastereomeric mixture by reaction with an appropriate optically active compound (e.g. chiral auxiliary such as a chiral alcohol or Mosher’s acid chloride) , separating the diastereomers and converting (e.g. hydrolyzing) the individual diastereomers to the corresponding pure enantiomers. Also, some of the compounds of Formula I may be atropisomers (e.g. substituted biaryls) and are considered as part of this invention. Enantiomers can also be separated by use of chiral HPLC column.
It is also possible that the compounds of Formula I may exist in different tautomeric forms, and all such forms are embraced within the scope of the invention. Also, for example, all keto-enol and imine-enamine forms of the compounds are included in the invention.
All stereoisomers (for example, geometric isomers, optical isomers and the like) of the present compounds (including those of the salts, solvates, esters and prodrugs of the compounds as well as the salts, solvates and esters of the prodrugs) , such as those which may exist due to asymmetric carbons on various substituents, including enantiomeric forms (which may exist even in the absence of asymmetric carbons) , rotameric forms, atropisomers, and diastereomeric forms, are contemplated within the scope of this invention, as are positional isomers. Individual stereoisomers of the compounds of the invention may, for example, be substantially free of other isomers, or may be admixed, for example, as racemates or with all other, or other selected, stereoisomers. The chiral centers of the present invention can have the S or R configuration as defined by the IUPAC 1974 Recommendations. The use of the terms "salt" , "solvate" , “ester” , "prodrug" and the like, is intended to equally apply to the salt, solvate, ester and prodrug of enantiomers, stereoisomers, rotamers, tautomers, positional isomers, racemates or prodrugs of the inventive compounds.
Prodrugs
A discussion of prodrugs is provided in T. Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems (1987) 14 of the A.C.S. Symposium Series, and in Bioreversible Carriers in Drug Design, (1987) Edward B. Roche, ed., American Pharmaceutical Association and Pergamon Press. The term “prodrug” means a compound (e.g, a drug precursor) that is transformed in vivo to yield a compound of Formula I or a pharmaceutically acceptable salt, hydrate or solvate of the compound. The transformation may occur by various mechanisms (e.g. by metabolic or chemical processes) , such as, for example, through hydrolysis in blood. A discussion of the use of prodrugs is provided by T. Higuchi and W. Stella, “Pro-drugs as Novel Delivery Systems, ” Vol. 14 of the A.C.S. Symposium Series, and in Bioreversible Carriers in
Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987.
Isotopes
In the compounds of Formula I, the atoms may exhibit their natural isotopic abundances, or one or more of the atoms may be artificially enriched in a particular isotope having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number predominantly found in nature. The present invention is meant to include all suitable isotopic variations of the compounds of generic Formula I. For example, different isotopic forms of hydrogen (H) include protium (1H) and deuterium (2H) . Protium is the predominant hydrogen isotope found in nature. Enriching for deuterium may afford certain therapeutic advantages, such as increasing in vivo half-life or reducing dosage requirements, or may provide a compound useful as a standard for characterization of biological samples. Isotopically-enriched compounds within generic Formula I can be prepared without undue experimentation by conventional techniques well known to those skilled in the art or by processes analogous to those described in the Schemes and Examples herein using appropriate isotopically-enriched reagents and/or intermediates.
Certain isotopically-labelled compounds of Formula I (e.g. those labeled with 3H and 14C) are useful in compound and/or substrate tissue distribution assays. Tritiated (i.e., 3H) and carbon-14 (i.e., 14C) isotopes are particularly preferred for their ease of preparation and detectability. Further, substitution with heavier isotopes such as deuterium (i.e., 2H) may afford certain therapeutic advantages resulting from greater metabolic stability (e.g., increased in vivo half-life or reduced dosage requirements) and hence may be preferred in some circumstances. Isotopically labelled compounds of Formula I can generally be prepared by following procedures analogous to those disclosed in the Schemes and/or in the Examples herinbelow, by substituting an appropriate isotopically labeled reagent for a non-isotopically labeled reagent.
Utilities
The compounds having Formula I and pharmaceutical compositions thereof can be used to treat or prevent a variety of conditions, diseases or disorders mediated by Bruton's Tyrosine kinase (Btk) . Such Btk-mediated conditions, diseases or disorders include, but are not limited to: (1) arthritis, including rheumatoid arthritis, juvenile arthritis, psoriatic arthritis and osteoarthritis; (2) asthma and other obstructive airways diseases, including chronic asthma, late asthma, airway hyper-responsiveness, bronchitis, bronchial asthma, allergic asthma, intrinsic asthma, extrinsic asthma, dust asthma, adult respiratory distress syndrome, recurrent airway obstruction, and chronic obstruction pulmonary disease including emphysema; (3) autoimmune
diseases or disorders, including those designated as single organ or single cell-type autoimmune disorders, for example Hashimoto's thyroiditis, autoimmune hemolytic anemia, autoimmune atrophic gastritis of pernicious anemia, autoimmune encephalomyelitis, autoimmune orchitis, Goodpasture's disease, autoimmune thrombocytopenia including idiopathic thrombopenic purpura, sympathetic ophthalmia, myasthenia gravis, Graves' disease, primary biliary cirrhosis, chronic aggressive hepatitis, ulcerative colitis and membranous glomerulopathy, those designated as involving systemic autoimmune disorder, for example systemic lupus erythematosis, immune thrombocytopenic purpura, rheumatoid arthritis, Sjogren's syndrome, Reiter's syndrome, polymyositis-dermatomyositis, systemic sclerosis, polyarteritis nodosa, multiple sclerosis and bullous pemphigoid, and additional autoimmune diseases, which can be B-cell (humoral) based or T-cell based, including Cogan's syndrome, ankylosing spondylitis, Wegener's granulomatosis, autoimmune alopecia, Type I or juvenile onset diabetes, and thyroiditis; (4) cancers or tumors, including alimentary/gastrointestinal tract cancer, colon cancer, liver cancer, skin cancer including mast cell tumor and squamous cell carcinoma, breast and mammary cancer, ovarian cancer, prostate cancer, lymphoma and leukemia (including but not limited to acute myelogenous leukemia, chronic myelogenous leukemia, mantle cell lymphoma, NHL B cell lymphomas (e.g. precursor B-ALL, marginal zone B cell lymphoma, chronic lymphocytic leukemia, diffuse large B cell lymphoma, Burkitt lymphoma, mediastinal large B-cell lymphoma) , Hodgkin lymphoma, NK and T cell lymphomas; TEL-Syk and ITK-Syk fusion driven tumors, myelomas including multiple myeloma, myeloproliferative disorders kidney cancer, lung cancer, muscle cancer, bone cancer, bladder cancer, brain cancer, melanoma including oral and metastatic melanoma, Kaposi's sarcoma, proliferative diabetic retinopathy, and angiogenic-associated disorders including solid tumors, and pancreatic cancer; (5) diabetes, including Type I diabetes and complications from diabetes; (6) eye diseases, disorders or conditions including autoimmune diseases of the eye, keratoconjunctivitis, vernal conjunctivitis, uveitis including uveitis associated with Behcet's disease and lens-induced uveitis, keratitis, herpetic keratitis, conical keratitis, corneal epithelial dystrophy, keratoleukoma, ocular premphigus, Mooren's ulcer, scleritis, Grave's ophthalmopathy, Vogt-Koyanagi-Harada syndrome, keratoconjunctivitis sicca (dry eye) , phlyctenule, iridocyclitis, sarcoidosis, endocrine ophthalmopathy, sympathetic ophthalmitis, allergic conjunctivitis, and ocular neovascularization; (7) intestinal inflammations, allergies or conditions including Crohn's disease and/or ulcerative colitis, inflammatory bowel disease, coeliac diseases, proctitis, eosinophilic gastroenteritis, and mastocytosis; (8) neurodegenerative diseases including motor neuron disease, Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis, Huntington's disease, cerebral
ischemia, or neurodegenerative disease caused by traumatic injury, strike, glutamate neurotoxicity or hypoxia; ischemic/reperfusion injury in stroke, myocardial ischemica, renal ischemia, heart attacks, cardiac hypertrophy, atherosclerosis and arteriosclerosis, organ hypoxia; (9) platelet aggregation and diseases associated with or caused by platelet activation, such as arteriosclerosis, thrombosis, intimal hyperplasia and restenosis following vascular injury; (10) conditions associated with cardiovascular diseases, including restenosis, acute coronary syndrome, myocardial infarction, unstable angina, refractory angina, occlusive coronary thrombus occurring post-thrombolytic therapy or post-coronary angioplasty, a thrombotically mediated cerebrovascular syndrome, embolic stroke, thrombotic stroke, transient ischemic attacks, venous thrombosis, deep venous thrombosis, pulmonary embolus, coagulopathy, disseminated intravascular coagulation, thrombotic thrombocytopenic purpura, thromboangiitis obliterans, thrombotic disease associated with heparin-induced thrombocytopenia, thrombotic complications associated with extracorporeal circulation, thrombotic complications associated with instrumentation such as cardiac or other intravascular catheterization, intra-aortic balloon pump, coronary stent or cardiac valve, conditions requiring the fitting of prosthetic devices, and the like; (11) skin diseases, conditions or disorders including atopic dermatitis, eczema, psoriasis, scleroderma, pruritus and other pruritic conditions; (12) allergic reactions including anaphylaxis, allergic rhinitis, allergic dermatitis, allergic urticaria, angioedema, allergic asthma, or allergic reaction to insect bites, food, drugs, or pollen; (13) transplant rejection, including pancreas islet transplant rejection, bone marrow transplant rejection, graft-versus-host disease, organ and cell transplant rejection such as bone marrow, cartilage, cornea, heart, intervertebral disc, islet, kidney, limb, liver, lung, muscle, myoblast, nerve, pancreas, skin, small intestine, or trachea, and xeno transplantation; and (14) low grade scarring including scleroderma, increased fibrosis, keloids, post-surgical scars, pulmonary fibrosis, vascular spasms, migraine, reperfusion injury, and post-myocardial infarction.
The invention thus provides compounds of Formula I and salts thereof for use in therapy, and particularly in the treatment of disorders, diseases and conditions mediated by inappropriate Btk activity.
The inappropriate Btk activity referred to herein is any Btk activity that deviates from the normal Btk activity expected in a particular mammalian subject. Inappropriate Btk activity may take the form of, for instance, an abnormal increase in activity, or an aberration in the timing and or control of Btk activity. Such inappropriate activity may result then, for example, from overexpression or mutation of the protein kinase leading to inappropriate or uncontrolled activation.
In one embodiment, the present invention provides for the use of a compound of Formula I, or a pharmaceutically acceptable salt thereof for the manufacture of a medicament for the treatment of a Btk-mediated disorder.
In another embodiment, the present invention provides methods of regulating, modulating, or inhibiting Btk for the prevention and/or treatment of disorders related to unregulated or inappropriate Btk activity.
In a further embodiment, the present invention provides a method for treating a subject suffering from a disorder mediated by Btk, which comprises administering to said subject a compound of Formula I or a pharmaceutically acceptable salt thereof in an amount effective to treat the Btk-mediated disorder.
A further aspect of the invention resides in the use of a compound of Formula I or a pharmaceutically acceptable salt thereof for the manufacture of a medicament to be used for the treatment of chronic B cell disorders in which T cells play a prominent role.
Thus, the compounds according to the invention may be used in therapies to treat or prevent Bruton’s Tyrosine Kinase (Btk) mediated diseases, conditions and disorders. Btk mediated diseases, conditions and disorders as used herein, mean any disease, condition or disorder in which B cells, mast cells, myeloid cells or osteoclasts play a central role. These diseases include but are not limited to, immune, autoimmune and inflammatory diseases, allergies, infectious diseases, bone resorption disorders and proliferative diseases.
Immune, autoimmune and inflammatory diseases that may be treated or prevented with the compounds of the present invention include rheumatic diseases (e.g. rheumatoid arthritis, psoriatic arthritis, infectious arthritis, progressive chronic arthritis, deforming arthritis, osteoarthritis, traumatic arthritis, gouty arthritis, Reiter’s syndrome, polychondritis, acute synovitis and spondylitis) , glomerulonephritis (with or without nephrotic syndrome) , Goodpasture's syndrome, (and associated glomerulonephritis and pulmonary hemorrhage) , atherosclerosis, autoimmune hematologic disorders (e.g. hemolytic anemia, aplasic anemia, idiopathic thrombocytopenia, chronic idiopathic thrombocytopenic purpura (ITP) , and neutropenia) , autoimmune gastritis, and autoimmune inflammatory bowel diseases (e.g. ulcerative colitis and Crohn’s disease) , irritable bowel syndrome, host versus graft disease, allograft rejection, chronic thyroiditis, Graves’ disease, Sjorgren's disease, scleroderma, diabetes (type I and type II) , active hepatitis (acute and chronic) , pancreatitis, primary billiary cirrhosis, myasthenia gravis, multiple sclerosis, systemic lupus erythematosis, psoriasis, atopic dermatitis, dermatomyositis, contact dermatitis, eczema, skin sunburns, vasculitis (e.g. Behcet’s disease) , ANCA-associated and other vasculitudes, chronic renal insufficiency, Stevens-Johnson
syndrome, inflammatory pain, idiopathic sprue, cachexia, sarcoidosis, Guillain-Barré syndrome, uveitis, conjunctivitis, kerato conjunctivitis, otitis media, periodontal disease, Addison's disease, Parkinson's disease, Alzheimer's disease, diabetes, septic shock, myasthenia gravis, pulmonary interstitial fibrosis, asthma, bronchitis, rhinitis, sinusitis, pneumoconiosis, pulmonary insufficiency syndrome, pulmonary emphysema, pulmonary fibrosis, silicosis, chronic inflammatory pulmonary disease (e.g. chronic obstructive pulmonary disease) and other inflammatory or obstructive disease on airways.
Allergies that may be treated or prevented include, among others, allergies to foods, food additives, insect poisons, dust mites, pollen, animal materials and contact allergans, type I hypersensitivity allergic asthma, allergic rhinitis, allergic conjunctivitis.
Infectious diseases that may be treated or prevented include, among others, sepsis, septic shock, endotoxic shock, sepsis by Gram-negative bacteria, shigellosis, meningitis, cerebral malaria, pneumonia, tuberculosis, viral myocarditis, viral hepatitis (hepatitis A, hepatitis B and hepatitis C) , HIV infection, retinitis caused by cytomegalovirus, influenza, herpes, treatment of infections associated with severe burns, myalgias caused by infections, cachexia secondary to infections, and veterinary viral infections such as lentivirus, caprine arthritic virus, visna-maedi virus, feline immunodeficiency virus, bovine immunodeficiency virus or canine immunodeficiency virus.
Bone resorption disorders that may be treated or prevented include, among others, osteoporosis, osteoarthritis, traumatic arthritis, gouty arthritis and bone disorders related with multiple myeloma.
Proliferative diseases that may be treated or prevented include, among others, non-Hodgkin lymphoma (in particular the subtypes diffuse large B-cell lymphoma (DLBCL) and mantle cell lymphoma (MCL) ) , B cell chronic lymphocytic leukemia and acute lymphoblastic leukemia (ALL) with mature B cell, ALL in particular.
In particular the compounds of Formula I or pharmaceutically acceptable salts may be used for the treatment of B cell lymphomas resulting from chronic active B cell receptor signaling.
Yet another aspect of the present invention provides a method for treating diseases caused by or associated with Fc receptor signaling cascades, including FceRI and/or FcgRI-mediated degranulation as a therapeutic approach towards the treatment or prevention of diseases characterized by, caused by and/or associated with the release or synthesis of chemical mediators of such Fc receptor signaling cascades or degranulation. In addition, Btk is known to play a critical role inimmunotyrosine-based activation motif (ITAM) singaling, B cell receptor
signaling, T cell receptor signaling and is an essential component of integrin beta (1) , beta (2) , and beta (3) signaling in neutrophils. Thus, compounds of the present invention can be used to regulate Fc receptor, ITAM, B cell receptor and integrin signaling cascades, as well as the cellular responses elicited through these signaling cascades. Non-limiting examples of cellular responses that may be regulated or inhibited include respiratory burst, cellular adhesion, cellular degranulation, cell spreading, cell migration, phagocytosis, calcium ion flux, platelet aggregation and cell maturation.
Combination Therapy
Included herein are methods of treatment and/or pharmaceutical compositions in which at least one compound of Formula I or a pharmaceutically acceptable salt thereof is administered in combination with at least one other active agent. The other active agent is an anti-inflammatory agent, an immunosuppressant agent, or a chemotherapeutic agent. Anti-inflammatory agents include but are not limited to NSAIDs, non-specific and COX-2 specific cyclooxgenase enzyme inhibitors, gold compounds, corticosteroids, methotrexate, tumor necrosis factor receptor (TNF) receptors antagonists, immunosuppressants and methotrexate.
Examples of NSAIDs include, but are not limited to, ibuprofen, flurbiprofen, naproxen and naproxen sodium, diclofenac, combinations of diclofenac sodium and misoprostol, sulindac, oxaprozin, diflunisal, piroxicam, indomethacin, etodolac, fenoprofen calcium, ketoprofen, sodium nabumetone, sulfasalazine, tolmetin sodium, and hydroxychloroquine. Examples of NSAIDs also include COX-2 specific inhibitors such as celecoxib, valdecoxib, lumiracoxib and/or etoricoxib.
In some embodiments, the anti-inflammatory agent is a salicylate. Salicylates include by are not limited to acetylsalicylic acid or aspirin, sodium salicylate, and choline and magnesium salicylates.
The anti-inflammatory agent may also be a corticosteroid. For example, the corticosteroid may be cortisone, dexamethasone, methylprednisolone, prednisolone, prednisolone sodium phosphate, or prednisone.
In additional embodiments the anti-inflammatory agent is a gold compound such as gold sodium thiomalate or auranofin.
The invention also includes embodiments in which the anti-inflammatory agent is a metabolic inhibitor such as a dihydrofolate reductase inhibitor, such as methotrexate or a dihydroorotate dehydrogenase inhibitor, such as leflunomide.
Other embodiments of the invention pertain to combinations in which at least one anti-inflammatory agent is an anti-C5 monoclonal antibody (such as eculizumab or pexelizumab) ,
a TNF antagonist, such as entanercept, or infliximab, which is an anti-TNF alpha monoclonal antibody.
Still other embodiments of the invention pertain to combinations in which at least one active agent is an immunosuppressant agent, such as an immunosuppressant compound chosen from methotrexate, leflunomide, cyclosporine, tacrolimus, azathioprine, and mycophenolate mofetil.
B-cells and B-cell precursors expressing BTK have been implicated in the pathology of B-cell malignancies, including, but not limited to, B-cell lymphoma, lymphoma (including Hodgkin's and non-Hodgkin's lymphoma) , hairy cell lymphoma, multiple myeloma, chronic and acute myelogenous leukemia and chronic and acute lymphocytic leukemia.
BTK has been shown to be an inhibitor of the Fas/APO-1 (CD-95) death inducing signaling complex (DISC) in B-lineage lymphoid cells. The fate of leukemia/lymphoma cells may reside in the balance between the opposing proapoptotic effects of caspases activated by DISC and an upstream anti-apoptotic regulatory mechanism involving BTK and/or its substrates (Vassilev et al., J. Biol. Chem. 1998, 274, 1646-1656) .
It has also been discovered that BTK inhibitors are useful as chemosensitizing agents, and, thus, are useful in combination with other chemotherapeutic agents, in particular, drugs that induce apoptosis. Examples of other chemotherapeutic agents that can be used in combination with chemosensitizing BTK inhibitors include topoisomerase I inhibitors (camptothecin or topotecan) , topoisomerase II inhibitors (e.g. daunomycin and etoposide) , alkylating agents (e.g. cyclophosphamide, melphalan and BCNU) , tubulin directed agents (e.g. taxol and vinblastine) , and biological agents (e.g. antibodies such as anti CD20 antibody, IDEC 8, immunotoxins, and cytokines) .
Btk activity has also been associated with some leukemias expressing the bcr-abl fusion gene resulting from translocation of parts of chromosome 9 and 22. This abnormality is commonly observed in chronic myelogenous leukemia. Btk is constitutively phosphorylated by the bcr-abl kinase which initiates downstream survival signals which circumvents apoptosis in bcr-abl cells. (N. Feldhahn et al. J. Exp. Med. 2005 201 (11) : 1837-1852) .
The compound (s) of Formula I and the other pharmaceutically active agent (s) may be administered together or separately and, when administered separately this may occur simultaneously or sequentially in any order. The amounts of the compound (s) of Formula I and the other pharmaceutically active agent (s) and the relative timings of administration will be selected in order to achieve the desired combined therapeutic effect.
For the treatment of the inflammatory diseases, rheumatoid arthritis, psoriasis, inflammatory bowel disease, COPD, asthma and allergic rhinitis a compound of Formula I may be combined with one or more other active agents such as: (1) TNF-α inhibitors such as infliximab etanercept
etanercept adalimumab
adalimumab certolizumab pegol
certolizumab pegol  and golimumab
and golimumab (2) non-selective COX-I/COX-2 inhibitors (such as piroxicam, diclofenac, propionic acids such as naproxen, flubiprofen, fenoprofen, ketoprofen and ibuprofen, fenamates such as mefenamic acid, indomethacin, sulindac, etodolac, azapropazone, pyrazolones such as phenylbutazone, salicylates such as aspirin) ; (3) COX-2 inhibitors (such as meloxicam, celecoxib, rofecoxib, valdecoxib and etoricoxib) ; (4) other agents for treatment of rheumatoid arthritis including methotrexate, leflunomide, sulfasalazine, azathioprine, cyclosporin, tacrolimus, penicillamine, bucillamine, actarit, mizoribine, lobenzarit, ciclesonide, hydroxychloroquine, d-penicillamine, aurothiomalate, auranofin or parenteral or oral gold, cyclophosphamide, Lymphostat-B, BAFF/APRIL inhibitors and CTLA-4-Ig or mimetics thereof; (5) leukotriene biosynthesis inhibitor, 5-lipoxygenase (5-LO) inhibitor or 5-lipoxygenase activating protein (FLAP) antagonist such as zileuton; (6) LTD4 receptor antagonist such as zafirlukast, montelukast and pranlukast; (7) PDE4 inhibitor such as roflumilast, cilomilast, AWD-12-281 (Elbion) , and PD-168787 (Pfizer) ; (8) antihistaminic H1 receptor antagonists such as cetirizine, levocetirizine, loratadine, desloratadine, fexofenadine, astemizole, azelastine, levocabastine, olopatidine, methapyrilene and chlorpheniramine; (9) α1-and α2-adrenoceptor agonist vasoconstrictor sympathomimetic agent, such as propylhexedrine, phenylephrine, phenylpropanolamine, pseudoephedrine, naphazoline hydrochloride, oxymetazoline hydrochloride, tetrahydrozoline hydrochloride, xylometazoline hydrochloride, and ethylnorepinephrine hydrochloride; (10) anticholinergic agents such as ipratropium bromide, tiotropium bromide, oxitropium bromide, aclindinium bromide, glycopyrrolate, (R, R) -glycopyrrolate, pirenzepine, and telenzepine; (11) β-adrenoceptor agonists such as metaproterenol, isoproterenol, isoprenaline, albuterol, formoterol (particularly the fumarate salt) , salmeterol (particularly the xinafoate salt) , terbutaline, orciprenaline, bitolterol mesylate, fenoterol, and pirbuterol, or methylxanthanines including theophylline and aminophylline, sodium cromoglycate; (12) insulin-like growth factor type I (IGF-l) mimetic; (13) glucocorticosteroids, especially inhaled glucocorticoid with reduced systemic side effects, such as prednisone, prednisolone, flunisolide, triamcinolone acetonide, beclomethasone dipropionate, budesonide, fluticasone propionate, ciclesonide and mometasone furoate; (14) kinase inhibitors such as inhibitors of the Janus Kinases (JAK 1 and/or JAK2 and/or JAK 3 and/or TYK2) , p38 MAPK and IKK2; (15) B-cell targeting biologics such as rituximab
(2) non-selective COX-I/COX-2 inhibitors (such as piroxicam, diclofenac, propionic acids such as naproxen, flubiprofen, fenoprofen, ketoprofen and ibuprofen, fenamates such as mefenamic acid, indomethacin, sulindac, etodolac, azapropazone, pyrazolones such as phenylbutazone, salicylates such as aspirin) ; (3) COX-2 inhibitors (such as meloxicam, celecoxib, rofecoxib, valdecoxib and etoricoxib) ; (4) other agents for treatment of rheumatoid arthritis including methotrexate, leflunomide, sulfasalazine, azathioprine, cyclosporin, tacrolimus, penicillamine, bucillamine, actarit, mizoribine, lobenzarit, ciclesonide, hydroxychloroquine, d-penicillamine, aurothiomalate, auranofin or parenteral or oral gold, cyclophosphamide, Lymphostat-B, BAFF/APRIL inhibitors and CTLA-4-Ig or mimetics thereof; (5) leukotriene biosynthesis inhibitor, 5-lipoxygenase (5-LO) inhibitor or 5-lipoxygenase activating protein (FLAP) antagonist such as zileuton; (6) LTD4 receptor antagonist such as zafirlukast, montelukast and pranlukast; (7) PDE4 inhibitor such as roflumilast, cilomilast, AWD-12-281 (Elbion) , and PD-168787 (Pfizer) ; (8) antihistaminic H1 receptor antagonists such as cetirizine, levocetirizine, loratadine, desloratadine, fexofenadine, astemizole, azelastine, levocabastine, olopatidine, methapyrilene and chlorpheniramine; (9) α1-and α2-adrenoceptor agonist vasoconstrictor sympathomimetic agent, such as propylhexedrine, phenylephrine, phenylpropanolamine, pseudoephedrine, naphazoline hydrochloride, oxymetazoline hydrochloride, tetrahydrozoline hydrochloride, xylometazoline hydrochloride, and ethylnorepinephrine hydrochloride; (10) anticholinergic agents such as ipratropium bromide, tiotropium bromide, oxitropium bromide, aclindinium bromide, glycopyrrolate, (R, R) -glycopyrrolate, pirenzepine, and telenzepine; (11) β-adrenoceptor agonists such as metaproterenol, isoproterenol, isoprenaline, albuterol, formoterol (particularly the fumarate salt) , salmeterol (particularly the xinafoate salt) , terbutaline, orciprenaline, bitolterol mesylate, fenoterol, and pirbuterol, or methylxanthanines including theophylline and aminophylline, sodium cromoglycate; (12) insulin-like growth factor type I (IGF-l) mimetic; (13) glucocorticosteroids, especially inhaled glucocorticoid with reduced systemic side effects, such as prednisone, prednisolone, flunisolide, triamcinolone acetonide, beclomethasone dipropionate, budesonide, fluticasone propionate, ciclesonide and mometasone furoate; (14) kinase inhibitors such as inhibitors of the Janus Kinases (JAK 1 and/or JAK2 and/or JAK 3 and/or TYK2) , p38 MAPK and IKK2; (15) B-cell targeting biologics such as rituximab (16) selective
costimulation modulators such as abatacept (Orencia) ; (17) interleukin inhibitors, such as IL-1 inhibitor anakinra (Kineret) and IL-6 inhibitor tocilizumab (Actemra) .
(16) selective
costimulation modulators such as abatacept (Orencia) ; (17) interleukin inhibitors, such as IL-1 inhibitor anakinra (Kineret) and IL-6 inhibitor tocilizumab (Actemra) .
The present invention also provides for "triple combination" therapy, comprising a compound of Formula I or a pharmaceutically acceptable salt thereof together with beta2-adrenoreceptor agonist and an anti-inflammatory corticosteroid. Preferably this combination is for treatment and/or prophylaxis of asthma, COPD or allergic rhinitis. The beta2-adrenoreceptor agonist and/or the anti-inflammatory corticosteroid can be as described above and/or as described in WO 03/030939 A1. Representative examples of such a "triple" combination are a compound of Formula I or a pharmaceutically acceptable salt thereof in combination with the components of (salmeterol xinafoate and fluticasone propionate) ,
(salmeterol xinafoate and fluticasone propionate) ,  (budesonide and formoterol fumarate) , or
(budesonide and formoterol fumarate) , or (mometasone furoate and formoterol) .
(mometasone furoate and formoterol) .
For the treatment of cancer a compound of Formula I may be combined with one or more of an anticancer agents. Examples of such agents can be found in Cancer Principles and Practice of Oncology by V.T. Devita and S. Hellman (editors) , 6th edition (February 15, 2001) , Lippincott Williams &Wilkins Publishers. A person of ordinary skill in the art would be able to discern which combinations of agents would be useful based on the particular characteristics of the drugs and the cancer involved. Such anti-cancer agents include, but are not limited to, the following: (1) estrogen receptor modulator such as diethylstibestral, tamoxifen, raloxifene, idoxifene, LY353381, LY117081, toremifene, fluoxymestero, and SH646; (2) other hormonal agents including aromatase inhibitors (e.g., aminoglutethimide, tetrazole anastrozole, letrozole and exemestane) , luteinizing hormone release hormone (LHRH) analogues, ketoconazole, goserelin acetate, leuprolide, megestrol acetate and mifepristone; (3) androgen receptor modulator such as finasteride and other 5α-reductase inhibitors, nilutamide, flutamide, bicalutamide, liarozole, and abiraterone acetate; (4) retinoid receptor modulator such as bexarotene, tretinoin, 13-cis-retinoic acid, 9-cis-retinoic acid, α-difluoromethylornithine, ILX23-7553, trans-N- (4’ -hydroxyphenyl) retinamide, and N-4-carboxyphenyl retinamide; (5) antiproliferative agent such asantisense RNA and DNA oligonucleotides such as G3139, ODN698, RVASKRAS, GEM231, and INX3001, and antimetabolites such as enocitabine, carmofur, tegafur, pentostatin, doxifluridine, trimetrexate, fludarabine, capecitabine, galocitabine, cytarabine ocfosfate, fosteabine sodium hydrate, raltitrexed, paltitrexid, emitefur, tiazofurin, decitabine, nolatrexed, pemetrexed, nelzarabine, 2’ -deoxy-2’ -methylidenecytidine, 2’ -fluoromethylene-2’ -deoxycytidine, N6- [4-deoxy-4- [N2- [2 (E) , 4 (E) -tetradeca-dienoyl] glycylamino] -L-glycero-B-L-manno-heptopyranosyl] adenine, aplidine, ecteinascidin,
troxacitabine, aminopterin, 5-flurouracil, floxuridine, methotrexate, leucovarin, hydroxyurea, thioguanine (6-TG) , mercaptopurine (6-MP) , cytarabine, pentostatin, fludarabine phosphate, cladribine (2-CDA) , asparaginase, gemcitabine, alanosine, swainsonine, lometrexol, dexrazoxane, methioninase, and 3-aminopyridine-2-carboxaldehyde thiosemicarbazone; (6) prenyl-protein transferase inhibitor including farnesyl-protein transferase (FPTase) , geranylgeranyl-protein transferase type I (GGPTase-I) , and geranylgeranyl-protein transferase type-II (GGPTase-II, also called Rab GGPTase) ; (7) HMG-CoA reductase inhibitor such as lovastatin, simvastatin, pravastatin, atorvastatin, fluvastatin and rosuvastatin; (8) angiogenesis inhibitor such as inhibitors of the tyrosine kinase receptors Flt-1 (VEGFR1) and Flk-1/KDR (VEGFR2) , inhibitors of epidermal-derived, fibroblast-derived, or platelet derived growth factors, MMP (matrix metalloprotease) inhibitors, integrin blockers, interferon-, interleukin-12, erythropoietin (epoietin-α) , granulocyte-CSF (filgrastin) , granulocyte, macrophage-CSF (sargramostim) , pentosan polysulfate, cyclooxygenase inhibitors, steroidal anti-inflammatories, carboxyamidotriazole, combretastatin A-4, squalamine, 6-O-chloroacetyl-carbonyl) -fumagillol, thalidomide, angiostatin, troponin-1, angiotensin II antagonists, heparin, carboxypeptidase U inhibitors, and antibodies to VEGF, endostatin, ukrain, ranpirnase, IM862, acetyldinanaline, 5-amino-1- [ [3, 5-dichloro-4- (4-chlorobenzoyl) phenyl] methyl] -1H-1, 2, 3-triazole-4-carboxamide, CM101, squalamine, combretastatin, RPI4610, NX31838, sulfated mannopentaose phosphate, and 3- [ (2, 4-dimethylpyrrol-5-yl) methylene] -2-indolinone (SU5416) ; (9) PPAR-γagonists, PPAR-δ agonists, thiazolidinediones (such as DRF2725, CS-011, troglitazone, rosiglitazone, and pioglitazone) , fenofibrate, gemfibrozil, clofibrate, GW2570, SB219994, AR-H039242, JTT-501, MCC-555, GW2331, GW409544, NN2344, KRP297, NP0110, DRF4158, NN622, GI262570, PNU182716, DRF552926, 2- [ (5, 7-dipropyl-3-trifluoromethyl-1, 2-benzisoxazol-6-yl) oxy] -2-methylpropionic acid (disclosed in USSN 09/782,856) , and (2R) -7- (3-(2-chloro-4- (4-fluorophenoxy) phenoxy) propoxy) -2-ethylchromane-2-carboxylic acid (disclosed in USSN 60/235,708 and 60/244,697) ; (9) inhibitor of inherent multidrug resistance including inhibitors of p-glycoprotein (P-gp) , such as LY335979, XR9576, OC144-093, R101922, VX853 and PSC833 (valspodar) ; (10) inhibitor of cell proliferation and survival signaling such as inhibitors of EGFR (for example gefitinib and erlotinib) , inhibitors of ERB-2 (for example trastuzumab) , inhibitors of IGF1R such as MK-0646 (dalotuzumab) , inhibitors of CD20 (rituximab) , inhibitors of cytokine receptors, inhibitors of MET, inhibitors of PI3K family kinase (for example LY294002) , serine/threonine kinases (including but not limited to inhibitors of Akt such as described in (WO 03/086404, WO 03/086403, WO 03/086394, WO 03/086279, WO
02/083675, WO 02/083139, WO 02/083140 and WO 02/083138) , inhibitors of Raf kinase (for example BAY-43-9006 ) , inhibitors of MEK (for example CI-1040 and PD-098059) and inhibitors of mTOR (for example Wyeth CCI-779 and Ariad AP23573) ; (11) a bisphosphonate such as etidronate, pamidronate, alendronate, risedronate, zoledronate, ibandronate, incadronate or cimadronate, clodronate, EB-1053, minodronate, neridronate, piridronate and tiludronate; (12) γ-secretase inhibitors, (13) agents that interfere with receptor tyrosine kinases (RTKs) including inhibitors of c-Kit, Eph, PDGF, Flt3 and c-Met; (14) agent that interferes with a cell cycle checkpoint including inhibitors of ATR, ATM, the Chk1 and Chk2 kinasesand cdk and cdc kinase inhibitors and are specifically exemplified by 7-hydroxystaurosporin, flavopiridol, CYC202 (Cyclacel) and BMS-387032; (15) BTK inhibitors such as PCI32765, AVL-292 and AVL-101; (16) PARP inhibitors including iniparib, olaparib, AGO14699, ABT888 and MK4827; (16) ERK inhibitors; (17) mTOR inhibitors such as sirolimus, ridaforolimus, temsirolimus, everolimus; (18) cytotoxic/cytostatic agents.
“Cytotoxic/cytostatic agents” refer to compounds which cause cell death or inhibit cell proliferation primarily by interfering directly with the cell’s functioning or inhibit or interfere with cell mytosis, including alkylating agents, tumor necrosis factors, intercalators, hypoxia activatable compounds, microtubule inhibitors/microtubule-stabilizing agents, inhibitors of mitotic kinesins, inhibitors of histone deacetylase, inhibitors of kinases involved in mitotic progression, antimetabolites; biological response modifiers; hormonal/anti-hormonal therapeutic agents, haematopoietic growth factors, monoclonal antibody targeted therapeutic agents, topoisomerase inhibitors, proteasome inhibitors and ubiquitin ligase inhibitors.
Examples of cytotoxic agents include, but are not limited to, sertenef, cachectin, chlorambucil, cyclophosphamide, ifosfamide, mechlorethamine, melphalan, uracil mustard, thiotepa, busulfan, carmustine, lomustine, streptozocin, tasonermin, lonidamine, carboplatin, altretamine, dacarbazine, procarbazine, prednimustine, dibromodulcitol, ranimustine, fotemustine, nedaplatin, oxaliplatin, temozolomide, heptaplatin, estramustine, improsulfan tosilate, trofosfamide, nimustine, dibrospidium chloride, pumitepa, lobaplatin, satraplatin, profiromycin, cisplatin, irofulven, dexifosfamide, cis-aminedichloro (2-methyl-pyridine) platinum, benzylguanine, glufosfamide, GPX100, (trans, trans, trans) -bis-mu- (hexane-1, 6-diamine) -mu-[diamine-platinum (II) ] bis [diamine (chloro) platinum (II) ] tetrachloride, diarizidinylspermine, arsenic trioxide, 1- (11-dodecylamino-10-hydroxyundecyl) -3, 7-dimethylxanthine, zorubicin, doxorubicin, daunorubicin, idarubicin, anthracenedione, bleomycin, mitomycin C, dactinomycin, plicatomycin, bisantrene, mitoxantrone, pirarubicin, pinafide, valrubicin, amrubicin, antineoplaston, 3’ -deamino-3’ -morpholino-13-deoxo-10-hydroxycarminomycin, annamycin,
galarubicin, elinafide, MEN10755, and 4-demethoxy-3-deamino-3-aziridinyl-4-methylsulphonyl-daunorubicin.
An example of a hypoxia activatable compound is tirapazamine.
Examples of proteasome inhibitors include but are not limited to lactacystin and bortezomib.
Examples of microtubule inhibitors/microtubule-stabilising agents include vincristine, vinblastine, vindesine, vinzolidine, vinorelbine, vindesine sulfate, 3’ , 4’ -didehydro-4’ -deoxy-8’ -norvincaleukoblastine, podophyllotoxins (e.g., etoposide (VP-16) and teniposide (VM-26) ) , paclitaxel, docetaxol, rhizoxin, dolastatin, mivobulin isethionate, auristatin, cemadotin, RPR109881, BMS184476, vinflunine, cryptophycin, anhydrovinblastine, N, N-dimethyl-L-valyl-L-valyl-N-methyl-L-valyl-L-prolyl-L-proline-t-butylamide, TDX258, the epothilones (see for example U.S. Pat. Nos. 6,284,781 and 6,288,237) and BMS188797.
Some examples of topoisomerase inhibitors are topotecan, hycaptamine, irinotecan, rubitecan, 6-ethoxypropionyl-3’ , 4’ -O-exo-benzylidene-chartreusin, lurtotecan, 7- [2-(N-isopropylamino) ethyl] - (20S) camptothecin, BNP1350, BNPI1100, BN80915, BN80942, etoposide phosphate, teniposide, sobuzoxane, 2’ -dimethylamino-2’ -deoxy-etoposide, GL331, N-[2- (dimethylamino) ethyl] -9-hydroxy-5, 6-dimethyl-6H-pyrido [4, 3-b] carbazole-1-carboxamide, asulacrine, 2, 3- (methylenedioxy) -5-methyl-7-hydroxy-8-methoxybenzo [c] -phenanthridinium, 5-(3-aminopropylamino) -7, 10-dihydroxy-2- (2-hydroxyethylaminomethyl) -6H-pyrazolo [4, 5, 1-de] acridin-6-one, N- [1- [2- (diethylamino) ethylamino] -7-methoxy-9-oxo-9H-thioxanthen-4-ylmethyl] formamide, N- (2- (dimethylamino) ethyl) acridine-4-carboxamide, 6- [ [2-(dimethylamino) ethyl] amino] -3-hydroxy-7H-indeno [2, 1-c] quinolin-7-one, and dimesna.
Examples of inhibitors of mitotic kinesins include, but are not limited to inhibitors of KSP, inhibitors of MKLP1, inhibitors of CENP-E, inhibitors of MCAK, inhibitors of Kif14, inhibitors of Mphosph1 and inhibitors of Rab6-KIFL.
Examples of “histone deacetylase inhibitors” include, but are not limited to, vorinostat, trichostatin A, oxamflatin, PXD101, MG98, valproic acid and scriptaid.
“Inhibitors of kinases involved in mitotic progression” include, but are not limited to, inhibitors of aurora kinase, inhibitors of Polo-like kinases (PLK; in particular inhibitors of PLK-1) , inhibitors of bub-1 and inhibitors of bub-R1. An example of an “aurora kinase inhibitor” is VX-680.
“Antiproliferative agents” includes antisense RNA and DNA oligonucleotides such as G3139, ODN698, RVASKRAS, GEM231, and INX3001, and antimetabolites such as enocitabine, carmofur, tegafur, pentostatin, doxifluridine, trimetrexate, fludarabine, capecitabine,
galocitabine, cytarabine ocfosfate, fosteabine sodium hydrate, raltitrexed, paltitrexid, emitefur, tiazofurin, decitabine, nolatrexed, pemetrexed, nelzarabine, 2’ -deoxy-2’ -methylidenecytidine, 2’ -fluoromethylene-2’ -deoxycytidine, N6- [4-deoxy-4- [N2- [2, 4-tetradecadienoyl] glycylamino] -L-glycero-B-L-manno-heptopyranosyl] adenine, aplidine, ecteinascidin, troxacitabine, aminopterin, 5-flurouracil, floxuridine, methotrexate, leucovarin, hydroxyurea, thioguanine (6-TG) , mercaptopurine (6-MP) , cytarabine, pentostatin, fludarabine phosphate, cladribine (2-CDA) , asparaginase, gemcitabine, alanosine, swainsonine, lometrexol, dexrazoxane, methioninase, and 3-aminopyridine-2-carboxaldehyde thiosemicarbazone.
Non-limiting examples of suitable agents used in cancer therapy that may be combined with compounds of Formula I include, but are not limited to, abarelix; aldesleukin; alemtuzumab; alitretinoin; allopurinol; altretamine; amifostine; anastrozole; arsenic trioxide; asparaginase; azacitidine; bendamustine; bevacuzimab; bexarotene; bleomycin; bortezomib; busulfan; calusterone; capecitabine; carboplatin; carmustine; cetuximab; chlorambucil; cisplatin; cladribine; clofarabine; cyclophosphamide; cytarabine; dacarbazine; dactinomycin, actinomycin D; dalteparin; darbepoetin alfa; dasatinib; daunorubicin; degarelix; denileukin diftitox; dexrazoxane; docetaxel; doxorubicin; dromostanolone propionate; eculizumab; Elliott's B Solution; eltrombopag; epirubicin; epoetin alfa; erlotinib; estramustine; etoposide phosphate; etoposide; everolimus; exemestane; filgrastim; floxuridine; fludarabine; fluorouracil; fulvestrant; gefitinib; gemcitabine; gemtuzumab ozogamicin; goserelin acetate; histrelin acetate; hydroxyurea; ibritumomab tiuxetan; idarubicin; ifosfamide; imatinib mesylate; interferon alfa 2a; interferon alfa-2b; irinotecan; ixabepilone; lapatinib; lenalidomide; letrozole; leucovorin; leuprolide acetate; levamisole; lomustine; meclorethamine, nitrogen mustard; megestrol acetate; melphalan, L-PAM; mercaptopurine; mesna; methotrexate; methoxsalen; mitomycin C; mitotane; mitoxantrone; nandrolone phenpropionate; nelarabine; nilotinib; Nofetumomab; ofatumumab; oprelvekin; oxaliplatin; paclitaxel; palifermin; pamidronat; panitumumab; pazopanib; pegademase; pegaspargase; Pegfilgrastim; pemetrexed disodium; pentostatin; pipobroman; plerixafor; plicamycin, mithramycin) ; porfimer sodium; pralatrexate; procarbazine; quinacrine; Rasburicase; raloxifene hydrochloride; Rituximab; romidepsin; romiplostim; sargramostim; sargramostim; satraplatin; sorafenib; streptozocin; sunitinib maleate; tamoxifen; temozolomide; temsirolimus; teniposide; testolactone; thioguanine; thiotepa; topotecan; toremifene; tositumomab; trastuzumab; tretinoin; uracil mustard; valrubicin; vinblastine; vincristine; vinorelbine; vorinostat; and zoledronate.
It will be clear to a person skilled in the art that, where appropriate, the other therapeutic ingredient (s) may be used in the form of salts, for example as alkali metal or amine
salts or as acid addition salts, or prodrugs, or as esters, for example lower alkyl esters, or as solvates, for example hydrates, to optimise the activity and/or stability and/or physical characteristics, such as solubility, of the therapeutic ingredient. It will be clear also that, where appropriate, the therapeutic ingredients may be used in optically pure form.
The combinations referred to above may conveniently be presented for use in the form of a pharmaceutical composition and thus pharmaceutical compositions comprising a combination as defined above together with a pharmaceutically acceptable diluent, carrier or excipient represent a further aspect of the invention. These combinations are of particular interest in respiratory diseases and are conveniently adapted for inhaled or intranasal delivery.
The individual compounds of such combinations may be administered either sequentially or simultaneously in separate or combined pharmaceutical compositions. Preferably, the individual compounds will be administered simultaneously in a combined pharmaceutical composition. Appropriate doses of known therapeutic agents will be readily appreciated by those skilled in the art.
Pharmaceutical Compositions
While it is possible that, for use in therapy, a compound of Formula I, as well as salts, solvates and physiological functional derivatives thereof, may be administered as the raw chemical, it is possible to present the active ingredient as a pharmaceutical composition. Accordingly, the invention further provides a pharmaceutical composition which comprises a compound of Formula I and salts, solvates and physiological functional derivatives thereof, and one or more pharmaceutically acceptable carriers, diluents, or excipients. The compounds of the Formula I and salts, solvates and physiological functional derivatives thereof, are as described above. The carrier (s) , diluent (s) or excipient (s) must be acceptable in the sense of being compatible with the other ingredients of the formulation and not deleterious to the recipient thereof. In accordance with another aspect of the invention there is also provided a process for the preparation of a pharmaceutical composition including admixing a compound of the Formula I, or salts, solvates and physiological functional derivatives thereof, with one or more pharmaceutically acceptable carriers, diluents or excipients.
Routes of Administration
Pharmaceutical compositions of the present invention may be presented in unit dose forms containing a predetermined amount of active ingredient per unit dose. Such a unit may contain, for example, 5μg to 1 g, preferably 1 mg to 700 mg, more preferably 5 mg to 100 mg of a compound of the Formula I, depending on the condition being treated, the route of administration and the age, weight and condition of the patient. Such unit doses may therefore be
administered more than once a day. Preferred unit dosage compositions are those containing a daily dose or sub-dose (for administration more than once a day) , as herein above recited, or an appropriate fraction thereof, of an active ingredient. Furthermore, such pharmaceutical compositions may be prepared by any of the methods well known in the pharmacy art.
Pharmaceutical compositions of the present invention may be adapted for administration by any appropriate route, for example by the oral (including buccal or sublingual) , rectal, topical, inhaled, nasal, ocular, sublingual, subcutaneous, local or parenteral (including intravenous and intramuscular) route, and the like, all in unit dosage forms for administration. Such compositions may be prepared by any method known in the art of pharmacy, for example by bringing into association the active ingredient with the carrier (s) or excipient (s) . Dosage forms include tablets, troches, dispersions, suspensions, solutions, capsules, creams, ointments, aerosols, and the like.
In a further embodiment, the present invention provides a pharmaceutical composition adapted for administration by the oral route, for treating, for example, rheumatoid arthritis.
In a further embodiment, the present invention provides a pharmaceutical composition adapted for administration by the nasal route, for treating, for example, allergic rhinitis.
In a further embodiment, the present invention provides a pharmaceutical composition adapted for administration by the inhaled route, for treating, for example, asthma, Chronic Obstructive Pulmonary disease (COPD) or Acute Respiratory Distress Syndrome (ARDS) .
In a further embodiment, the present invention provides a pharmaceutical composition adapted for administration by the ocular route, for treating, diseases of the eye, for example, conjunctivitis.
In a further embodiment, the present invention provides a pharmaceutical composition adapted for administration by the parenteral (including intravenous) route, for treating, for example, cancer.
For parenteral administration, the pharmaceutical composition of the invention may be presented in unit-dose or multi-dose containers, e.g. injection liquids in predetermined amounts, for example in sealed vials and ampoules, and may also be stored in a freeze dried (lyophilized) condition requiring only the addition of sterile liquid carrier, e.g. water, prior to use.
Mixed with such pharmaceutically acceptable auxiliaries, e.g. as described in the standard reference, Gennaro, A.R. et al., Remington: The Science and Practice of Pharmacy
(20th Edition., Lippincott Williams &Wilkins, 2000, see especially Part 5: Pharmaceutical Manufacturing) , the active agent may be compressed into solid dosage units, such as pills, tablets, or be processed into capsules or suppositories. By means of pharmaceutically acceptable liquids the active agent can be applied as a fluid composition, e.g. as an injection preparation, in the form of a solution, suspension, emulsion, or as a spray, e.g. a nasal spray.
For making solid dosage units, the use of conventional additives such as fillers, colorants, polymeric binders and the like is contemplated. In general any pharmaceutically acceptable additive which does not interfere with the function of the active compounds can be used. Suitable carriers with which the active agent of the invention can be administered as solid compositions include lactose, starch, cellulose derivatives and the like, or mixtures thereof, used in suitable amounts. For parenteral administration, aqueous suspensions, isotonic saline solutions and sterile injectable solutions may be used, containing pharmaceutically acceptable dispersing agents and/or wetting agents, such as propylene glycol or butylene glycol.
Pharmaceutical compositions of the present invention which are adapted for oral administration may be presented as discrete units such as capsules or tablets; powders or granules; solutions or suspensions in aqueous or non-aqueous liquids; edible foams or whips; or oil-in-water liquid emulsions or water-in-oil liquid emulsions.
For instance, for oral administration in the form of a tablet or capsule, the active drug component can be combined with an oral, non-toxic pharmaceutically acceptable inert carrier such as ethanol, glycerol, water and the like. Powders are prepared by comminuting the compound to a suitable fine size and mixing with a similarly comminuted pharmaceutical carrier such as an edible carbohydrate, as, for example, starch or mannitol. Flavoring, preservative, dispersing and coloring agent can also be present.
Capsules are made by preparing a powder mixture, as described above, and filling formed gelatin sheaths. Glidants and lubricants such as colloidal silica, talc, magnesium stearate, calcium stearate or solid polyethylene glycol can be added to the powder mixture before the filling operation. A disintegrating or solubilizing agent such as agar-agar, calcium carbonate or sodium carbonate can also be added to improve the availability of the medicament when the capsule is ingested.
Moreover, when desired or necessary, suitable binders, lubricants, disintegrating agents and coloring agents can also be incorporated into the mixture. Suitable binders include starch, gelatin, natural sugars such as glucose or beta-lactose, corn sweeteners, natural and synthetic gums such as acacia, tragacanth or sodium alginate, carboxymethylcellulose, polyethylene glycol, waxes and the like. Lubricants used in these dosage forms include sodium
oleate, sodium stearate, magnesium stearate, sodium benzoate, sodium acetate, sodium chloride and the like. Disintegrators include, without limitation, starch, methyl cellulose, agar, bentonite, xanthan gum and the like. Tablets are formulated, for example, by preparing a powder mixture, granulating or slugging, adding a lubricant and disintegrant and pressing into tablets. A powder mixture is prepared by mixing the compound, suitably comminuted, with a diluent or base as described above, and optionally, with a binder such as carboxymethylcellulose, an aliginate, gelatin, or polyvinyl pyrrolidone, a solution retardant such as paraffin, a resorption accelerator such as a quaternary salt and/or an absorption agent such as bentonite, kaolin or dicalcium phosphate. The powder mixture can be granulated by wetting with a binder such as syrup, starch paste, acadia mucilage or solutions of cellulosic or polymeric materials and forcing through a screen. As an alternative to granulating, the powder mixture can be run through the tablet machine and the result is imperfectly formed slugs broken into granules. The granules can be lubricated to prevent sticking to the tablet forming dies by means of the addition of stearic acid, a stearate salt, talc or mineral oil. The lubricated mixture is then compressed into tablets. The compounds of the present invention can also be combined with a free flowing inert carrier and compressed into tablets directly without going through the granulating or slugging steps. A clear or opaque protective coating consisting of a sealing coat of shellac, a coating of sugar or polymeric material and a polish coating of wax can be provided. Dyestuffs can be added to these coatings to distinguish different unit dosages.
Oral fluids such as solution, syrups and elixirs can be prepared in dosage unit form so that a given quantity contains a predetermined amount of the compound. Syrups can be prepared by dissolving the compound in a suitably flavored aqueous solution, while elixirs are prepared through the use of a non-toxic alcoholic vehicle. Suspensions can be formulated by dispersing the compound in a non-toxic vehicle. Solubilizers and emulsifiers such as ethoxylated isostearyl alcohols and polyoxy ethylene sorbitol ethers, preservatives, flavor additive such as peppermint oil or natural sweeteners or saccharin or other artificial sweeteners, and the like can also be added.
Where appropriate, dosage unit compositions for oral administration can be microencapsulated. The formulation can also be prepared to prolong or sustain the release, for example, by coating or embedding particulate material in polymers, wax or the like.
The compounds of Formula I, and salts, solvates and physiological functional derivatives thereof, can also be administered in the form of liposome delivery systems, such as small unilamellar vesicles, large unilamellar vesicles and multilamellar vesicles. Liposomes can
be formed from a variety of phospholipids, such as cholesterol, stearylamine or phosphatidylcholines.
The compounds of Formula I and salts, solvates and physiological functional derivatives thereof may also be delivered by the use of monoclonal antibodies as individual carriers to which the compound molecules are coupled. The compounds may also be coupled with soluble polymers as targetable drug carriers. Such polymers can include polyvinylpyrrolidone, pyran copolymer, polyhydroxypropylmethacrylamide-phenol, polyhydroxyethylaspartamidephenol, or polyethyleneoxidepolylysine substituted with palmitoyl residues. Furthermore, the compounds may be coupled to a class of biodegradable polymers useful in achieving controlled release of a drug, for example, polylactic acid, polyepsilon caprolactone, polyhydroxy butyric acid, polyorthoesters, polyacetals, polydihydropyrans, polycyanoacrylates and cross-linked or amphipathic block copolymers of hydrogels.
Dosage forms for inhaled administration may conveniently be formulated as aerosols or dry powders.
For compositions suitable and/or adapted for inhaled administration, it is preferred that the compound or salt of Formula I is in a particle-size-reduced form, and more preferably the size-reduced form is obtained or obtainable by micronisation. The preferable particle size of the size-reduced (e.g. micronised) compound or salt or solvate is defined by a D50 value of about 0.5 to about 10 microns (for example as measured using laser diffraction) .
Aerosol formulations, e.g. for inhaled administration, can comprise a solution or fine suspension of the active substance in a pharmaceutically acceptable aqueous or non-aqueous solvent. Aerosol formulations can be presented in single or multidose quantities in sterile form in a sealed container, which can take the form of a cartridge or refill for use with an atomising device or inhaler. Alternatively the sealed container may be a unitary dispensing device such as a single dose nasal inhaler or an aerosol dispenser fitted with a metering valve (metered dose inhaler) which is intended for disposal once the contents of the container have been exhausted.
Where the dosage form comprises an aerosol dispenser, it preferably contains a suitable propellant under pressure such as compressed air, carbon dioxide or an organic propellant such as a hydrofluorocarbon (HFC) . Suitable HFC propellants include 1, 1, 1, 2, 3, 3, 3-heptafluoropropane and 1, 1, 1, 2-tetrafluoroethane. The aerosol dosage forms can also take the form of a pump-atomiser. The pressurised aerosol may contain a solution or a suspension of the active compound. This may require the incorporation of additional excipients e.g. co-solvents and/or surfactants to improve the dispersion characteristics and homogeneity of suspension formulations. Solution formulations may also require the addition of co-solvents such as ethanol.
Other excipient modifiers may also be incorporated to improve, for example, the stability and/or taste and/or fine particle mass characteristics (amount and/or profile) of the formulation.
For pharmaceutical compositions suitable and/or adapted for inhaled administration, it is preferred that the pharmaceutical composition is a dry powder inhalable composition. Such a composition can comprise a powder base such as lactose, glucose, trehalose, mannitol or starch, the compound of Formula I or salt or solvate thereof (preferably in particle-size-reduced form, e.g. in micronised form) , and optionally a performance modifier such as L-leucine or another amino acid, and/or metals salts of stearic acid such as magnesium or calcium stearate. Preferably, the dry powder inhalable composition comprises a dry powder blend of lactose and the compound of Formula I or salt thereof. The lactose is preferably lactose hydrate e.g. lactose monohydrate and/or is preferably inhalation-grade and/or fine-grade lactose. Preferably, the particle size of the lactose is defined by 90% or more (by weight or by volume) of the lactose particles being less than 1000 microns (micrometres) (e.g. 10-1000 microns e.g. 30-1000 microns) in diameter, and/or 50% or more of the lactose particles being less than 500 microns (e.g. 10-500 microns) in diameter. More preferably, the particle size of the lactose is defined by 90% or more of the lactose particles being less than 300 microns (e.g. 10-300 microns e.g. 50-300 microns) in diameter, and/or 50% or more of the lactose particles being less than 100 microns in diameter. Optionally, the particle size of the lactose is defined by 90% or more of the lactose particles being less than 100-200 microns in diameter, and/or 50% or more of the lactose particles being less than 40-70 microns in diameter. It is preferable that about 3 to about 30% (e.g. about 10%) (by weight or by volume) of the particles are less than 50 microns or less than 20 microns in diameter. For example, without limitation, a suitable inhalation-grade lactose is E9334 lactose (10% fines) (Borculo Domo Ingredients, Hanzeplein 25, 8017 J D Zwolle, Netherlands) .
Optionally, in particular for dry powder inhalable compositions, a pharmaceutical composition for inhaled administration can be incorporated into a plurality of sealed dose containers (e.g. containing the dry powder composition) mounted longitudinally in a strip or ribbon inside a suitable inhalation device. The container is rupturable or peel-openable on demand and the dose of e.g. the dry powder composition can be administered by inhalation via the device such as the device (GlaxoSmithKline) . Other dry powder inhalers are well known to those of ordinary skill in the art, and many such devices are commercially available, with representative devices including
device (GlaxoSmithKline) . Other dry powder inhalers are well known to those of ordinary skill in the art, and many such devices are commercially available, with representative devices including (Novartis) , AirmaxTM (IVAX) ,
(Novartis) , AirmaxTM (IVAX) ,  (Innovata Biomed) ,
(Innovata Biomed) ,  (GlaxoSmithKline) , Accuhaler (GlaxoSmithKline) ,
(GlaxoSmithKline) , Accuhaler (GlaxoSmithKline) ,  (Orion Pharma) , EclipseTM (Aventis) ,
(Orion Pharma) , EclipseTM (Aventis) ,  (Hovione) ,
(Hovione) ,  (Boehringer
Ingelheim) ,
(Boehringer
Ingelheim) ,  (Chiesi) ,
(Chiesi) ,  (GlaxoSmithKline) , SkyeHalerTM or CertihalerTM(SkyePharma) , Twisthaler (Schering-Plough) ,
(GlaxoSmithKline) , SkyeHalerTM or CertihalerTM(SkyePharma) , Twisthaler (Schering-Plough) ,  (AstraZeneca) ,
(AstraZeneca) ,  (Aventis) , and the like.
(Aventis) , and the like.
Dosage forms for ocular administration may be formulated as solutions or suspensions with excipients suitable for ophthalmic use.
Dosage forms for nasal administration may conveniently be formulated as aerosols, solutions, drops, gels or dry powders.
Pharmaceutical compositions adapted for administration by inhalation include fine particle dusts or mists, which may be generated by means of various types of metered, dose pressurized aerosols, nebulizers or insufflators.
For pharmaceutical compositions suitable and/or adapted for intranasal administration, the compound of Formula I or a pharmaceutically acceptable salt or solvate thereof may be formulated as a fluid formulation for delivery from a fluid dispenser. Such fluid dispensers may have, for example, a dispensing nozzle or dispensing orifice through which a metered dose of the fluid formulation is dispensed upon the application of a user-applied force to a pump mechanism of the fluid dispenser. Such fluid dispensers are generally provided with a reservoir of multiple metered doses of the fluid formulation, the doses being dispensable upon sequential pump actuations. The dispensing nozzle or orifice may be configured for insertion into the nostrils of the user for spray dispensing of the fluid formulation into the nasal cavity. A fluid dispenser of the aforementioned type is described and illustrated in WO-A-2005/044354, the entire content of which is hereby incorporated herein by reference. The dispenser has a housing which houses a fluid discharge device having a compression pump mounted on a container for containing a fluid formulation. The housing has at least one finger-operable side lever which is movable inwardly with respect to the housing to cam the container upwardly in the housing to cause the pump to compress and pump a metered dose of the formulation out of a pump stem through a nasal nozzle of the housing. A particularly preferred fluid dispenser is of the general type illustrated in FIGS. 30-40 of WO-A-2005/044354.
The invention further includes a pharmaceutical composition of a compound of Formula I or pharmaceutically acceptable salts thereof, as hereinbefore described, in combination with packaging material suitable for said composition, said packaging material including instructions for the use of the composition for the use as hereinbefore described.
The following are examples of representative pharmaceutical dosage forms for the compounds of this invention:

Water for injection to a total volume of 1 ml



It will be appreciated that when the compound of the present invention is administered in combination with other therapeutic agents normally administered by the inhaled, intravenous, oral or intranasal route, that the resultant pharmaceutical composition may be administered by the same routes.
It should be understood that in addition to the ingredients particularly mentioned above, the compositions may include other agents conventional in the art having regard to the
type of formulation in question, for example those suitable for oral administration may include flavoring agents.
A therapeutically effective amount of a compound of the present invention will depend upon a number of factors including, for example, the age and weight of the animal, the precise condition requiring treatment and its severity, the particular compound having Formula I, the nature of the formulation, and the route of administration, and will ultimately be at the discretion of the attendant physician or veterinarian. However, an effective amount of a compound of Formula I for the treatment of diseases or conditions associated with inappropriate Btk activity, will generally be in the range of 5 μg to 100 mg/kg body weight of recipient (mammal) per day and more usually in the range of 5 μg to 10 mg/kg body weight per day. This amount may be given in a single dose per day or more usually in a number (such as two, three, four, five or six) of sub-doses per day such that the total daily dose is the same. An effective amount of a salt or solvate, thereof, may be determined as a proportion of the effective amount of the compound of Formula I per se.
In general parenteral administration requires lower dosages than other methods of administration which are more dependent upon absorption. However, a dosage for humans preferably contains 0.0001-25 mg of a compound of Formula I or pharmaceutically acceptable salts thereof per kg body weight. The desired dose may be presented as one dose or as multiple subdoses administered at appropriate intervals throughout the day, or, in case of female recipients, as doses to be administered at appropriate daily intervals throughout the menstrual cycle. The dosage as well as the regimen of administration may differ between a female and a male recipient.
General Synthesis
The compounds of the present invention can be prepared by methods well known in the art of organic chemistry. See, for example, J. March, ‘Advanced Organic Chemistry’ 4th Edition, John Wiley and Sons. During synthetic sequences it may be necessary and/or desirable to protect sensitive or reactive groups on any of the molecules concerned. This is achieved by means of conventional protecting groups, such as those described in T. W. Greene and P.G.M. Wutts ‘Protective Groups in Organic Synthesis’ 3rd Edition, John Wiley and Sons, 1999. The protective groups are optionally removed at a convenient subsequent stage using methods well known in the art.
The products of the reactions are optionally isolated and purified, if desired, using conventional techniques, but not limited to, filtration, distillation, crystallization,
chromatography and the like. Such materials are optionally characterized using conventional means, including physical constants and spectral data.
The compounds of Formula I can be prepared by the general synthetic routes shown in the schemes below.
Scheme I

Reduction of 3-chloropyrazine-2-carbonitrile (II) can be accomplished by hydrogenation in the presence of a suitable catalyst system and solvent, for example Raney-Nickel ethanol to provide (3-chloropyrazin-2-yl) methanamine (III) . This amine can then be reacted with the acid (IV) . The reaction of IV can be carried out in a solvent such as DMF, THF or DCM in the presence of a base such as DIPEA, N-methylmorpholine, 4-DMAP or triethylamine and in the presence of a coupling reagent such as PyBOP, TBTU, EDCI or HATU to form N- ( (3-chloropyrazin-2-yl) methyl) amide (V) . Cyclization chloropyrazine (V) can be performed using condensation reagents like phosphorousoxychloride under heating conditions to provide the 8-chloroimidazo [1, 5-a] pyrazine derivatives VI. Subsequent bromination can be accomplished using bromine or N-bromosuccinimide in a suitable solvent like DCM or DMF at
appropriate temperature to obtain compounds of formula VII. 8-Aminoimidazo [1, 5-a] pyrazine derivatives (VIII) can be prepared from compounds VII using ammonia (gas) in isopropanol at elevated temperature in a pressure vessel (>4 atm) or with primary amine (such as dimethoxybenzylamine) under heating. Compounds of formula I can be prepared from compounds of formula VIII using an appropriate boronic acid or pinacol ester (IX) , in the presence of a suitable palladium catalyst system, for example bis (diphenylphosphino) ferrocene palladium (II) chloride complex or tetrakis (triphenylphosphine) palladium (0) in the presence of an inorganic base like potassium carbonate, cesium carbonate or potassium phosphate in a suitable solvent system like combinations of dioxane and water. Palladium catalysts and conditions to form either the pinacol esters or to couple the boronic acids or pinacol esters with the 1-bromoimidazo [1, 5-a] pyrazin-8-amine are well known to the skilled organic chemist –see, for example, Ei-ichi Negishi (Editor) , Armin de Meijere (Associate Editor) , Handbook of Organopalladium Chemistry for Organic Synthesis, John Wiley and Sons, 2002. The acid intermediates IV are commercially available or can be readily prepared using methods well known to the skilled organic chemist.
Scheme II

An alternative route was shown in Scheme II for the preparation of compound with formula I. The aldehyde VIII was converted to carboxylic acid XI using an appropriate boronic acid or pinacol ester (X) , in the presence of a suitable palladium catalyst system, similar to the condition decribed in Scheme 1. The reaction of I can be carried out in a solvent such as DMF, THF or DCM in the presence of a base such as DIPEA, N-methylmorpholine, 4-DMAP or triethylamine and in the presence of a coupling reagent such as PyBOP, TBTU, EDCI or HATU to form the final targets.
The invention is illustrated by the following examples.
EXAMPLES
The following examples are illustrative embodiments of the invention, not limiting the scope of the invention in any way. Reagents are commercially available or are prepared according to procedures in the literature.
Mass Spectrometry: Electron Spray spectra were recorded on the Applied Biosystems API-165 single quad mass spectrometer in alternating positive and negative ion mode using Flow Injection. The mass range was 120-2000 Da. and scanned with a step rate of 0.2 Da. and the capillary voltage was set to 5000 V. N2gas was used for nebulisation.
LC-MS spectrometer (Waters) Detector: PDA (200-320 nm) , Mass detector: ZQ and Eluent : A: acetonitrile with 0.05% trifluoroacetic acid , B: acetronitrile/water = 1/9 (v/v) with 0.05% trifluoroacetic acid.
Method A: LC-MS

Method B: LC-MS


Method C:
Sample Info : Easy-Access Method: '1-Short_TFA_Pos'
Method Info : B222 Column Agilent SBC (3.0x50 mm, 1.8 μm) ; Flow 1.0 mL/min; solvent A: H2O-0.1% TFA;
solvent B: MeCN-0.1% TFA;
GRADIENT TABLE: 0 min: 10% B, 0.3 min: 10%B, 1.5min: 95% B, 2.70min: 95% B, 2.76 min: 10% B
stop time 3.60 min, PostTime 0.70 min.
Method D:
Sample Info : Easy-Access Method: '1_Fast'
Method Info : A330 Column Agilent Zorbax SB-C18 (2.1x30 mm, 3.5 μm) ; Flow 2.0 mL/min;
solvent A: H2O-0.1% TFA;
solvent B: MeCN-0.1% TFA;
GRADIENT TABLE: 0.01 min: 10% B, 1.01 min: 95% B, 1.37 min: 95% B, 1.38 min: 10% B,
stop time 1.7min, PostTime=OFF
The following abbreviations are used throughout the application with respect to chemical terminology:
HATU O- (7-Azabenzotriazol-1-yl) -1, 1, 3, 3-tetramethyluroniumhexafluoro phosphate
Cbz Benzyloxycarbonyl
DMF N, N-Dimethylformamide
DCM Dichloromethane
EtOAc Ethyl acetate
DIPEA N, N-Diisopropylethylamine
THF Tetrahydrofuran
EtOH Ethanol
EDCI. HCl 1- (3-Dimethylaminopropyl) -3-ethylcarbodiimide. hydrochloride
4-DMAP 4-Dimethylamino pyridine
PyBOP O-Benzotriazole-1-yl-oxy-trispyrrolidinophosphonium
hexafluorophosphate
TBTU O-Benzotriazol-1-yl-N, N, N’ , N’ -tetramethyluronium tetrafluoroborate
HBr Hydrogen bromide
HCl Hydrogen chloride
HOAc Acetic acid
POCl3 Phosphorous oxychloride
HPLC High Pressure Liquid Chromatography
UPLC Ultra Performance Liquid Chromatography
LiHMDS Lithium hexamethyldisilazide
MeOH Methanol
DCM Dichloromethane
n-BuLi n-Butyllithium
CO2 Carbondioxide
NaHCO3 Sodiumbicarbonate
K3PO4 Potassium phosphate
P(Cy) 3 Tricyclohexylphosphine
Pd(OAc) 2 Palladium (II) acetate
Na2SO4 Sodium sulfate
Na2CO3 Sodium carbonate
DAST Diethylaminosulfur trifluoride
Cs2CO3 Cesium carbonate
Et2O Diethylether
Na2S2O3 Sodium thiosulfate
Na2S2O4 Sodium hydrosulfite
NaCNBH3 Sodium cyanoborohydride
NH4Cl Ammonium chloride
MgSO4 Magnesium sulfate
LiOH Lithium hydroxide
IPA Isopropylamine
TFA Trifluoro acetic acid
Cbz-Cl Benzylchloroformate
PE Petroleum ether
EA Ethyl acetate
NaHMDS Sodium hexamethyldisilazide
10% Pd/C 10% Palladium on carbon
TEA Triethyl amine
CDI 1, 1'-Carbonyl diimidazole
DMI 1, 3-Dimethyl-2-imidazolidinone
NBS N-Bromosuccinimide
i-PrOH 2-Propanol
K2CO3 Potassium carbonate
Pd (dppf) Cl2 1, 1'-Bis (diphenylphosphino) ferrocene palladium (II) chloride,
complex withdichloromethane
Et3N Triethylamine
2-BuOH 2-Butanol
LCMS Liquid Chromatography /Mass Spectrometry
MeCN Acetonitril
NH3 Ammonia
CD3I Trideuteromethyl iodide
CD3OD Tetradeuteromethanol
CH3I Iodomethane
CBr4 Carbon tetrabromide
Tris-HCl Tris (hydroxymethyl) aminomethane. hydrochloride
MgCl2 Magnesium chloride
NaN3 Sodium azide
DTT Dithiothreitol
DMSO Dimethyl sulfoxide
IMAP Immobilized Metal Ion Affinity-Based Fluorescence Polarization
ATP Adenosine triphosphate
MnCl2 Manganese (II) chloride
DMA Dimethylacetamide
IPA Isopropyl alcohol
TPP triphenylphosphine
DIAD Diisopropyl azodicarboxylate
DMB 2, 4-dimethoxybenzyl
DCE Dichloroethane
DEAD Diethyl azodicarboxylate
ACN Acetonitrile
RT (rt) Room Temperature
Aq Aqueous
EtOH Ethanol
MPLC Medium Pressure Liquid Chromoatography
Xantphos 4, 5-Bis (diphenylphosphino) -9, 9-dimethylxanthene
X-phos 2-Dicyclohexylphosphino-2', 4', 6'-triisopropylbiphenyl
TFA trifluoroacetic acid
Intermediates
Intermediate 1 and 2

(7R, 9aS) -7- (8-Amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -2-isopropylhexahydro-1H-
pyrido [1, 2-a] pyrazin-4 (6H) -one
(7R, 9aR) -7- (8-Amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -2-isopropylhexahydro-1H-
pyrido [1, 2-a] pyrazin-4 (6H) -one
Step 1: (3R, 6S) -1-benzyl 3-methyl 6- ( ( (tert-butyldiphenylsilyl) oxy) methyl) piperidine-1, 3-
dicarboxylate
To a solution of (3R, 6S) -1-benzyl 3-methyl 6- (hydroxymethyl) piperidine-1, 3-dicarboxylate (2.00 g, 6.51 mmol) , imidazole (0.532 g, 7.81 mmol) in DMF (10 ml) was added TBDPS-Cl (2.0 ml, 7.81 mmol) . It was stirred at room temperature for 2 h. The reaction mixture was diluted with ethyl acetate, washed with water three times, then brine once. The organic layer was dried over sodium sulfate, filtered and concentrated. The residue was purified by ISCO (Gold 120g, 0-50% ethyl acetate in hexane ) to give the title compound. LC-MS: C32H39NO5Si, calc. = 546.27; found = 546.29 (M+H) +.
Step 2: (3R, 6S) -1- ( (benzyloxy) carbonyl) -6- ( ( (tert-butyldiphenylsilyl) oxy) methyl) piperidine-3-
carboxylic acid
To a solution of (3R, 6S) -1-benzyl 3-methyl 6- ( ( (tert-butyldiphenylsilyl) oxy) methyl) piperidine-1, 3-dicarboxylate (3.40 g, 6.23 mmol) in THF (40 ml) , MeOH (40.0 ml) and water (40.0 ml) was added LiOH (5 M, 8 ml, 40.0 mmol) slowly. The mixture was stirred at room temperature for 3 h and acidified by 1 M HCl (about 40 mL) to adjust pH to 5. The mixture was extracted with
ethyl acetate (3x100 mL) . The combined organic layers were washed with brine (100 mL) , dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was used without purification. LC-MS: C31H37NO5Si, calc. = 532.25; found = 532.34 (M+H) +.
Step 3: (2S, 5R) -benzyl 2- ( ( (tert-butyldiphenylsilyl) oxy) methyl) -5- ( ( (3-chloropyrazin-2-
yl) methyl) carbamoyl) piperidine-1-carboxylate
To a mixture of (3R, 6S) -1- ( (benzyloxy) carbonyl) -6- ( ( (tert-butyldiphenylsilyl) oxy) methyl) piperidine-3-carboxylic acid (3.3 g, 6.21 mmol) , (3-chloropyrazin-2-yl) methanamine bis-hydrocloride salt (1.478 g, 6.83 mmol) and HATU (2.83 g, 7.45 mmol) in DMF (20 ml) was added DIEA (3.25 ml, 18.62 mmol) . The mixture was stirred at room temperature for 1 h. Most solvent was removed under reduced pressure and the residue was diluted with ethyl acetate (100 mL) , washed with water (3x50 mL) , then brine (100 mL) . The organic layer was seperated and dried over sodium sulfate, filtered and concentrated under redueced pressure. The residue was purified by ISCO (Gold 80g, 0-100% EtOAc/EtOH (3: 1) in hexane) to give the title compound. LC-MS: C36H41ClN4O4Si, calc. = 656.27; found = 656.41 (M+H) +.
Step 4: (2S, 5R) -benzyl 2- ( ( (tert-butyldiphenylsilyl) oxy) methyl) -5- (8-chloroimidazo [1, 5-
a] pyrazin-3-yl) piperidine-1-carboxylate
To a mixture of (2S, 5R) -benzyl 2- ( ( (tert-butyldiphenylsilyl) oxy) methyl) -5- ( ( (3-chloropyrazin-2-yl) methyl) carbamoyl) piperidine-1-carboxylate (2.2 g, 3.35 mmol) and sodium carbonate (2.84 g, 26.8 mmol) in acetonitrile (12 mL) and DMF (12.00 mL) at 0 ℃ was added POCl3 (1 mL, 10.73 mmol) dropwise. It was warmed to 45 ℃ and stirred for 1 h. The mixture was diluted with100 mL of ethyl acetate and washed with water (3x50 mL) . The combined aqueous layers were extracted with ethyl acetate (100 mL) . The combined organic layers were washed with brine, dried over sodium sulfate, filtered and concentrated. The residue was purified by ISCO (Gold 40g, 0-100% EtOAc/EtOH (3: 1) in hexane) to give the title compound. LC-MS: C36H39ClN4O3Si, calc. = 639.26; found = 639.45 (M+H) +. (Step 5: (2S, 5R) -benzyl 5- (1-bromo-8-
chloroimidazo [1, 5-a] pyrazin-3-yl) -2- ( ( (tert-butyldiphenylsilyl) oxy) methyl) piperidine-1-
carboxylate
To a solution of (2S, 5R) -benzyl 2- ( ( (tert-butyldiphenylsilyl) oxy) methyl) -5- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) piperidine-1-carboxylate (1.33 g, 2.081 mmol) in acetonitrile (15 ml) was added NBS (0.444 g, 2.497 mmol) . The mixture was stirred at room temperature for 15 min. It was concentrated and purified by ISCO (gold 40g, 0-50% ethyl acetate in hexane) to give the title compound. LC-MS: C36H38BrClN4O3Si, calc. = 717.17, 719.17; found = 717.35, 719.30 (M+H) +.
Step 6: (2S, 5R) -benzyl 5- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-
yl) -2- ( ( (tert-butyldiphenylsilyl) oxy) methyl) piperidine-1-carboxylate
To a solution of (2S, 5R) -benzyl 5- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -2- ( ( (tert-butyldiphenylsilyl) oxy) methyl) piperidine-1-carboxylate (1.35 g, 1.880 mmol) in DMF (6 ml) was added 2, 4-dimethoxybenzylamine (0.40 g, 2.392 mmol) and triethylamine (0.42 ml, 3.01 mmol) . The mixture was stirred at 60 ℃ for 4 h. It was diluted with ethyl acetate, washed with water three times and brine once. The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by ISCO (Gold 40g, 0-50% ethyl acetate in hexane) to give the title compound. LC-MS: C45H50BrN5O5Si, calc. = 848.36, 850.31; found = 848.28, 850.29 (M+H) +.
Step 7: (2S, 5R) -benzyl 5- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-
yl) -2- (hydroxymethyl) piperidine-1-carboxylate
A solution of (2S, 5R) -benzyl 5- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -2- ( ( (tert-butyldiphenylsilyl) oxy) methyl) piperidine-1-carboxylate (880 mg, 1.037 mmol) in THF (5 ml) in a plastic vial was treated with HF (70 wt% in pyridine, 0.8 ml, 1.037 mmol) at room temperature for 3 h. The mixture was diluted with ethyl acetate, washed with sodium bicarbonate, brine. The organic layer was dried over sodium sulfate, filtered and concentrated. The residue was purified by ISCO (Gold 40g, 0-100% EtOAc/EtOH in hexane) to give the title compound. LC-MS: C29H32BrN5O5, calc. = 610.17, 612.17; found = 610.21, 612.16 (M+H) +.
Step 8: (2S, 5R) -benzyl 5- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-
yl) -2-formylpiperidine-1-carboxylate
To a solution of (2S, 5R) -benzyl 5- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -2- (hydroxymethyl) piperidine-1-carboxylate (550 mg, 0.901 mmol) in DCM (9 mL) was added Dess-Martin periodinane (535 mg, 1.261 mmol) . It was stirred at rt for 30 min. The reaction was quenched with aqueous sodium bicarbonate and sodium thiosulfate. The mixture was extracted with ethyl acetate. The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure to give the title compound, which was used without further purification.
Step 9: (2S, 5R) -benzyl 5- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-
yl) -2- ( (isopropyl (2-methoxy-2-oxoethyl) amino) methyl) piperidine-1-carboxylate
A solution of (2S, 5R) -benzyl 5- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -2-formylpiperidine-1-carboxylate (548 mg, 0.901 mmol) and methyl 2-(isopropylamino) acetate (354 mg, 1.351 mmol) in DCM (6 ml) was stirred for 2 h. To the
solution was added sodium triacetoxyborohydride (382 mg, 1.801 mmol) and the mixture was stirred for 30 min. It was directly purified by ISCO (gold 40g, 0-10% methanol in DCM) to give the title compound. LC-MS: C35H43BrN6O6, calc. = 723.25, 725.25; found = 723.34, 725.21 (M+H) +. 1H NMR (CHCl3-d, 500 MHz) : δ= 0.99 (3 H, d, J = 6.51 Hz) , 1.04 (3 H, d, J = 6.58 Hz) , 1.80-1.76 (1 H, m) , 2.19-2.04 (3 H, m) , 2.45-2.35 (1 H, m) , 2.81 (2 H, d, J = 7.75 Hz) , 3.06 (1 H, t, J = 6.60 Hz) , 3.19 (1 H, s) , 3.33 (2 H, s) , 3.48 (1 H, dd, J = 13.89, 4.17 Hz) , 3.69-3.65 (3 H, m) , 3.83 (3 H, s) , 3.91 (3 H, s) , 4.17 (1 H, d, J = 13.94 Hz) , 4.29-4.24 (1 H, m) , 4.70 (2 H, d, J = 5.54 Hz) , 5.04-4.98 (2 H, m) , 6.48 (1 H, dd, J = 8.24, 2.42 Hz) , 6.53 (1 H, d, J = 2.37 Hz) , 6.75 (1 H, t, J = 5.57 Hz) , 6.98 (1 H, d, J = 5.05 Hz) , 7.08 (1 H, d, J = 5.02 Hz) , 7.19 (2 H, d, J = 7.34 Hz) , 7.28-7.35 (3H, m) ppm.
Diastereomer (2R, 5R) -benzyl 5- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -2- ( (isopropyl (2-methoxy-2-oxoethyl) amino) methyl) piperidine-1-carboxylate was isolated as a side product (115 mg, 18% yield) . LC-MS: C35H43BrN6O6, calc. = 723.25, 725.25; found = 723.32, 725.22 (M+H) +. 1H NMR (CHCl3-d, 500 MHz) : = 1.06-0.93 (6 H, m) , 1.72 (1 H, br s) , 2.19-1.98 (3 H, m) , 2.65 (1 H, br s) , 2.88 (1 H, dd, J = 13.49, 9.29 Hz) , 3.15-2.98 (2 H, m) , 3.27 (1 H, d, J = 7.47 Hz) , 3.40 (1 H, s) , 3.65 (1 H, s) , 3.71 (1 H, s) , 3.83 (3 H, s) , 3.91 (3 H, d, J = 2.75 Hz) , 4.35-4.19 (2 H, m) , 4.70 (2 H, t, J = 6.16 Hz) , 5.24-5.10 (2 H, m) , 6.47 (1 H, d, J = 8.34 Hz) , 6.52 (1 H, s) , 6.75 (1 H, s) , 6.98 (1 H, dd, J = 31.21, 5.03 Hz) , 7.18-7.14 (1 H, m) , 7.39-7.34 (5 H, m) ppm.
Step 9: (7R, 9aS) -7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -2-isopropylhexahydro-1H-
pyrido [1, 2-a] pyrazin-4 (6H) -one
(2S, 5R) -Benzyl 5- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -2-( (isopropyl (2-methoxy-2-oxoethyl) amino) methyl) piperidine-1-carboxylate (320 mg, 0.442 mmol) was treated with TFA (6 mL, 78 mmol) at 100 ℃ for 1 h. Most TFA was removed under reduced pressure and the residue was dissolved in DCM. It was concentrated again to give a crude product, which was used without purification. LC-MS: C17H23BrN6O, calc. = 407.12, 409.12; found = 407.08, 409.01 (M+H) +. 1H NMR (CHCl3-d, 500 MHz) : = 1.08 (3 H, d, J = 4.17 Hz) , 1.10 (3 H, d, J = 4.19 Hz) , 1.70 (1 H, t, J = 11.40 Hz) , 1.89 (1 H, dd, J = 13.45, 3.75 Hz) , 2.20-2.18 (2 H, m) , 2.46 (1 H, dd, J = 11.87, 6.68 Hz) , 2.78-2.73 (2 H, m) , 3.00-2.97 (2 H, m) , 3.19 (1 H, d, J = 5.39 Hz) , 3.34 (1 H, d, J = 15.92 Hz) , 3.48 (1 H, br s) , 4.92 (1 H, dd, J = 13.17, 4.04 Hz) , 7.01 (1 H, d, J = 5.26 Hz) , 7.30 (1 H, d, J = 5.29 Hz) ppm.
(7R, 9aR) -7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -2-isopropylhexahydro-1H-pyrido [1, 2-
a] pyrazin-4 (6H) -one
(2R, 5R) -Benzyl 5- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -2-( (isopropyl (2-methoxy-2-oxoethyl) amino) methyl) piperidine-1-carboxylate (76 mg, 0.105 mmol) was treated with TFA (2 mL, 26.0 mmol) at 100 ℃ for 2 h. It was concentrated and the residue was dissolved in 2 mL DCM and 1 mL of TEA. It was loaded on a silica gel sampler and purified by ISCO (Gold 12g, 0-100% ethyl acetate/ethanol (3: 1) in hexane with 2% TEA) to give the title compound. LC-MS: C17H23BrN6O, calc. = 407.12, 409.12; found = 406.99, 408.95 (M+H) +.
Intermediate 3

(7R, 9aS) -7- (8-Amino-1-bromo-5-chloroimidazo [1, 5-a] pyrazin-3-yl) -2-isopropylhexahydro-1H-
pyrido [1, 2-a] pyrazin-4 (6H) -one
To a solution of (7R, 9aS) -7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -2-isopropylhexahydro-1H-pyrido [1, 2-a] pyrazin-4 (6H) -one (12 mg, 0.029 mmol) in acetic acid (0.3 ml) was added NCS (4.5 mg, 0.034 mmol) and the reaction was heated at 80 ℃ for 1 h. It was concentrated under reduced pressure and the residue was purified by ISCO (4g, 0-100% ethyl acetate/ethanol (3: 1) in hexane with 2% TEA) to give the title compound. LC-MS: C17H22BrClN6O, calc. = 441.08, 443.08; found = 441.00, 443.01 (M+H) +.
Intermediate 4

(6R, 8aS) -6- (8-Amino-1-bromo-5-fluoroimidazo [1, 5-a] pyrazin-3-yl) tetrahydro-1H-oxazolo [3, 4-
a] pyridin-3 (5H) -one
Step 1: (6R, 8aS) -6- (1-Bromo-8- ( (2, 4-dimethoxybenzyl) amino) -5-fluoroimidazo [1, 5-a] pyrazin-3-
yl) tetrahydro-1H-oxazolo [3, 4-a] pyridin-3 (5H) -one
A solution of (6R, 8aS) -6- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) tetrahydro-1H-oxazolo [3, 4-a] pyridin-3 (5H) -one (396 mg, 0.788 mmol, preparation described
in patent application US20140206681) and (307 mg, 0.867 mmol) in methanol (4 mL) was heated at 60 ℃ for 1 h. The mixture was concentrated under reduced pressure. To the residue was added 10 mL of acetonitrile and the mixture was concentrated under reduced pressure. The residue was dissolved in acetonitrile (4 ml) along with Cs2CO3 (2568 mg, 7.88 mmol) . The mixture was heated at 90 ℃ for 30 min. It was cooled to room temperature, filtered. The filtrate was concentrated and the residue was purified by ISCO (Gold 40g, 0-10% Methanol in DCM) to give the title compound.
(307 mg, 0.867 mmol) in methanol (4 mL) was heated at 60 ℃ for 1 h. The mixture was concentrated under reduced pressure. To the residue was added 10 mL of acetonitrile and the mixture was concentrated under reduced pressure. The residue was dissolved in acetonitrile (4 ml) along with Cs2CO3 (2568 mg, 7.88 mmol) . The mixture was heated at 90 ℃ for 30 min. It was cooled to room temperature, filtered. The filtrate was concentrated and the residue was purified by ISCO (Gold 40g, 0-10% Methanol in DCM) to give the title compound.
Step 2: (6R, 8aS) -6- (8-Amino-1-bromo-5-fluoroimidazo [1, 5-a] pyrazin-3-yl) tetrahydro-1H-
oxazolo [3, 4-a] pyridin-3 (5H) -one
(6R, 8aS) -6- (1-Bromo-8- ( (2, 4-dimethoxybenzyl) amino) -5-fluoroimidazo [1, 5-a] pyrazin-3-yl) tetrahydro-1H-oxazolo [3, 4-a] pyridin-3 (5H) -one (200 mg, 0.384 mmol) was treated with TFA (2 mL, 26.0 mmol) at 100 ℃ for 2 h. The mixture was concentrated under reduced pressure. The residue was dissolved in 2 mL of DCM and 0.5 mL of TEA. It was purified by ISCO (Gold 24g, 0-100% ethyl acetate-EtOH (3: 1) in hexane) to give the title compound. LC-MS: C17H22BrClN6O, calc. = 370.03, 372.03; found = 369.95, 371.94 (M+H) +.
Intermediate 5

(3R, 8aR) -3- (8-Amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) tetrahydro-1H-pyrrolo [2, 1-
c] [1, 4] oxazin-6 (7H) -one
Step 1: (2R, 5R) -tert-butyl 2- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -5-
(hydroxymethyl) morpholine-4-carboxylate
A solution of (2R, 5S) -tert-butyl 5- ( ( (tert-butyldiphenylsilyl) oxy) methyl) -2- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) morpholine-4-carboxylate (1 g, 1.647 mmol, preparation described in patent application US20140206681) in THF (15 ml) was treated with TBAF (3.3 ml, 3.30 mmol) at 23 ℃ for 1 h. It was loaded on a silica cartridge, dried and purified ISCO (gold 24 g, 0-5% Methanol in DCM) to give the title compound. LC-MS: C16H21ClN4O4, calc. = 369.13; found = 369.13 (M+H) +. 1H NMR (CHCl3-d, 500 MHz) : δ= 1.53 (9 H, s) , 3.45 (1 H, d, J = 11.98 Hz) , 3.83-3.76 (2 H, m) , 3.94-3.93 (3 H, m) , 4.63 (1 H, d, J = 13.75 Hz) , 5.19 (1 H, t, J = 3.67 Hz) , 5.33 (1 H, s) , 7.39 (1 H, d, J = 5.02 Hz) , 7.85 (1 H, s) , 8.06 (1 H, d, J = 5.03 Hz) ppm.
Step 2: (2R, 5S) -tert-butyl 2- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -5-formylmorpholine-4-
carboxylate
To a solution of (2R, 5R) -tert-butyl 2- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -5-(hydroxymethyl) morpholine-4-carboxylate (200 mg, 0.542 mmol) in DCM (5 ml) was added Dess-Martin periodinane (276 mg, 0.651 mmol) . The mixture was stirred at room temperature for 30 min. It was quenched with saturated aqueous sodium bicarbonate and sodium thiosulfate solution. The mixture was extracted with ethyl acetate (20 mL x3) . The combined organic layers were washed with brine, dried over sodium sulfate, filtered and concentrated under reduced pressure. The product was used without purification.
Step3: (2R, 5R) -tert-butyl 2- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -5- (3-methoxy-3-oxoprop-1-en-
1-yl) morpholine-4-carboxylate
A solution of (carbomethoxymethyl) triphenylphosphonium bromide (300 mg, 0.722 mmol) in THF (3 mL) was added 1 M solution of LHMDS in THF (0.720 ml, 0.720 mmol) at 0 ℃, It was stirred for 15 min, then a solution of (2R, 5S) -tert-butyl 2- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -5-formylmorpholine-4-carboxylate (220 mg, 0.600 mmol) in THF (2 mL) was added. The mixture was warmed to room temperature and stirred for 1 h. It was quenched with pH 5 phosphate buffer, extracted with ethyl acetate (2x20 mL) . The combined organic layers were washed with brine, dried over sodium sulfate, filtered and concentrated. The residue was purified by ISCO (Gold 40g, 0-50% ethyl acetate in hexane) to give the title compound as a mixture of E/Z isomers (4: 1) . LC-MS: C19H23ClN4O5, calc. = 423.15; found = 423.05 (M+H) +.
Step4: (2R, 5R) -tert-butyl 2- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -5- (3-methoxy-3-
oxopropyl) morpholine-4-carboxylate
To a solution of (2R, 5R) -tert-butyl 2- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -5- (3-methoxy-3-oxoprop-1-en-1-yl) morpholine-4-carboxylate (88 mg, 0.208 mmol) and nickel (II) chloride hexahydrate (25 mg, 0.105 mmol) in MeOH (4 ml) cooled in an ice-acetone bath was added sodium borohydride (7.87 mg, 0.208 mmol) . It was stirred for 20 min, then quenched with saturated aqueous ammonium chloride. The mixture was extracted with ethyl acetate twice. The combined organic layers were washed with brine, dried over sodium sulfate, filtered and concentrated. The residue was purified by ISCO (Gold 24g, 0-50% ethyl acetate in hexane) to give the title compound. LC-MS: C19H25ClN4O5, calc. = 425.16; found = 425.10 (M+H) +. 1H NMR (CHCl3-d, 500 MHz) : = 1.51 (9 H, s) , 1.93-1.88 (1 H, m) , 2.42-2.38 (3 H, m) , 3.30 (1 H, dd, J = 11.80, 3.39 Hz) , 3.61-3.52 (2 H, m) , 3.71 (3 H, s) , 4.04 (1 H, s) , 4.90 (1 H, d, J = 13.96
Hz) , 5.21 (1 H, d, J = 4.48 Hz) , 7.37 (1 H, d, J = 5.02 Hz) , 7.84 (1 H, s) , 8.06 (1 H, d, J = 5.03 Hz) ppm.
Step 5: (2R, 5R) -2- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -5- (3-methoxy-3-oxopropyl) morpholin-
4-ium chloride
(2R, 5R) -tert-Butyl 2- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -5- (3-methoxy-3-oxopropyl) morpholine-4-carboxylate (57 mg, 0.134 mmol) was treated with 4 M HCl in dioxane (3 ml, 12.00 mmol) . The mixture was stirred at room temperature for 1 h. It was concentrated under reduced pressure and used directly for the next step. LC-MS: C14H17ClN4O3, calc. = 325.11; found = 325.01 (M+H) +.
Step 6: (3R, 8aR) -3- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) tetrahydro-1H-pyrrolo [2, 1-
c] [1, 4] oxazin-6 (7H) -one
A solution of (2R, 5R) -2- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -5- (3-methoxy-3-oxopropyl) morpholin-4-ium chloride (48.5 mg, 0.134 mmol) in dioxane (2 ml) was added TEA (50 μl, 0.359 mmol) . The mixture was heated at 110 ℃ for 3 h. It was purified directly by ISCO (gold 12g, 0-10% methanol in DCM) to give the title compound (37 mg, 0.126 mmol, 94 % yield) as a powder. LC-MS: C13H13ClN4O2, calc. = 293.08; found = 293.09 (M+H) +. 1H NMR (CHCl3-d, 500 MHz) : = 2.27 (1 H, dtd, J = 12.85, 8.69, 4.68 Hz) , 2.58-2.51 (2 H, m) , 3.45 (1 H, t, J = 10.87 Hz) , 3.54 (1 H, t, J = 12.22 Hz) , 3.86 (1 H, dtd, J = 10.67, 7.30, 3.83 Hz) , 4.23 (1 H, dd, J = 11.09, 3.87 Hz) , 4.58 (1 H, dd, J = 13.62, 2.98 Hz) , 4.80 (1 H, dd, J = 10.83, 3.15 Hz) , 7.39 (1 H, d, J = 4.96 Hz) , 7.84 (1 H, s) , 7.98 (1 H, d, J = 4.99 Hz) ppm.
Step 7: (3R, 8aR) -3- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) tetrahydro-1H-pyrrolo [2, 1-
c] [1, 4] oxazin-6 (7H) -one
A solution of (3R, 8aR) -3- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) tetrahydro-1H-pyrrolo [2, 1-c] [1, 4] oxazin-6 (7H) -one (37 mg, 0.126 mmol) in acetonitrile (1 ml) was treated with NBS (27.0 mg, 0.152 mmol) at room temperature for 30 min. LC-MS showed the completion of the reaction. It was concentrated and purified by ISCO (gold 12 g, 0-10% methanol in DCM) to give the title compound. LC-MS: C13H12BrClN4O2, calc. = 370.99, 372.99; found = 370.94, 372.94 (M+H) +.
Step 8: (3R, 8aR) -3- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) tetrahydro-1H-pyrrolo [2, 1-
c] [1, 4] oxazin-6 (7H) -one
A mixture of (3R, 8aR) -3- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) tetrahydro-1H-pyrrolo [2, 1-c] [1, 4] oxazin-6 (7H) -one (37 mg, 0.100 mmol) and 2 M ammonia in propanol (3 ml, 6.00 mmol) was heated in a sealed microwave tube at 110 ℃ for 16 h. It was concentrated and used without purification. LC-MS: C13H14BrN5O2, calc. = 352.04, 354.04; found = 351.98, 353.96 (M+H) +.
Intermediate 6

(3R, 9aR) -3- (8-Amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -8-isopropylhexahydropyrazino [2, 1-
c] [1, 4] oxazin-6 (1H) -one
Step 1: Methyl 2- (isopropylamino) acetate
To a solution of methyl bromoacetate (1 g, 6.54 mmol) in THF (10 ml) was added TEA (1.1 ml, 7.89 mmol) and isopropylamine (0.56 ml, 6.57 mmol) at 0 ℃. The mixture was stirred at room temperature for 20 h. It was diluted with ethyl acetate and the solution was washed with water then brine. The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by ISCO (Gold 40g, 0-100% ethyl acetate in hexane) to give the title compound as an oil. 1H NMR (CHCl3-d, 500 MHz) : = 1.06 (6 H, d, J = 6.24 Hz) , 2.82-2.77 (1 H, m) , 3.42 (2 H, s) , 3.73 (3 H, s) ppm.
Step 2: (2R, 5S) -tert-butyl 2- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -5- ( ( (tert-
butyldiphenylsilyl) oxy) methyl) morpholine-4-carboxylate
To a solution of (2R, 5S) -tert-butyl 5- ( ( (tert-butyldiphenylsilyl) oxy) methyl) -2- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) morpholine-4-carboxylate (3 g, 4.94 mmol) in acetonitrile (25 ml) was added NBS (1.055 g, 5.93 mmol) . The mixture was stirred at room temperature for 15 min. It was concentrated and purified by ISCO (gold 80g, 0-50% ethyl acetate in hexane) to give the title compound. LC-MS: C32H38BrClN4O4Si, calc. = 685.23; found = 687.23 (M+H) +.
Step 3: (2R, 5S) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-
yl) -5- ( ( (tert-butyldiphenylsilyl) oxy) methyl) morpholine-4-carboxylate
To a solution of (2R, 5S) -tert-butyl 2- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -5- ( ( (tert-butyldiphenylsilyl) oxy) methyl) morpholine-4-carboxylate (3.39 g, 4.94 mmol) in DMF (10 ml) was added 2, 4-dimethoxybenzylamine (0.991 g, 5.93 mmol) and TEA (1 mL, 7.17 mmol) . The mixture was stirred at 60 ℃ overnight. It was diluted with ethyl acetate, washed with water three times, brine once. The organic layer was dried over sodium sulfate, filtered and concentrated. The residue was purified by ISCO (Gold 4g, 0-50% ethyl acetate in hexane) to give the title compound. LC-MS: C41H50BrN5O6Si, calc. = 816.25, 818.25; found = 816.36, 818.36 (M+H) +.
Step 4: (2R, 5R) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-
yl) -5- (hydroxymethyl) morpholine-4-carboxylate
To a solution of (2R, 5S) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -5- ( ( (tert-butyldiphenylsilyl) oxy) methyl) morpholine-4-carboxylate (2 g, 2.448 mmol) in THF (20 ml) was treated with TBAF (4 ml, 4.00 mmol) at room temperature for 30 min. It was purified by ISCO (gold 24g, 0-10% methanol in DCM) to give the title compound.
LC-MS: C25H32BrN5O6, calc. = 578.16, 580.16; found = 578.11, 580.03 (M+H) +.
Step 5: (2R, 5S) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-
yl) -5-formylmorpholine-4-carboxylate
To a solution of (2R, 5R) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -5- (hydroxymethyl) morpholine-4-carboxylate (1.35 g, 2.334 mmol) in DCM (20 mL) was added Dess-Martin periodinane (1.386 g, 3.27 mmol) . The mixture was stirred at room temperature for 30 min. The reaction was quenched with aqueous sodium bicarbonate, sodium thiosulfate, extracted with ethyl acetate. The organic layer was dried over sodium sulfate, filtered and concentrated to give the title compound (1.34 g, 2.325 mmol, 100 % yield) . which was used without further purification.
Step 6: (2R, 5R) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-
yl) -5- ( (isopropyl (2-methoxy-2-oxoethyl) amino) methyl) morpholine-4-carboxylate
A mixture of (2R, 5S) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -5-formylmorpholine-4-carboxylate (255 mg, 0.442 mmol) and methyl 2-(isopropylamino) acetate (87 mg, 0.664 mmol) in DCM (2 ml) were stirred for 1 h. To the mixture was added sodium triacetoxyborohydride (188 mg, 0.885 mmol) . It was stirred for 30 min. The reaction mixture was directly purified by ISCO (gold 40g, 0-10% methanol in DCM) to give the title compound. LC-MS: C31H43BrN6O7, calc. = 691.25, 693.25; found = 691.21, 693.21 (M+H) +.
Step 7: (3R, 9aR) -3- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -8-
isopropylhexahydropyrazino [2, 1-c] [1, 4] oxazin-6 (1H) -one
(2R, 5R) -tert-Butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -5-( (isopropyl (2-methoxy-2-oxoethyl) amino) methyl) morpholine-4-carboxylate (230 mg, 0.333 mmol) was treated with TFA (4 mL, 51.9 mmol) at 100 ℃ for 0.5 h. It was concentrated to give a red residue, which was dissolved into 5 ml of DCM and added a few drops of TEA to make a slight yellow solution. It was purified by ISCO (0-10% MeOH in DCM with 2% TEA) to give (3R, 9aR) -3- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -8-isopropylhexahydropyrazino [2, 1-
c] [1, 4] oxazin-6 (1H) -one. LC-MS: C16H21BrN6O2, calc. = 409.10, 411.10; found = 409.12, 411.12 (M+H) +.
Intermediate 7

(3R, 9aR) -3- (8-Amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -8-
cyclopropylhexahydropyrazino [2, 1-c] [1, 4] oxazin-6 (1H) -one
Step 1: Methyl 2- (cyclopropylamino) acetate
To a solution of methyl bromoacetate (1 g, 6.54 mmol) in THF (10 ml) was added TEA (1.1 ml, 7.89 mmol) and cyclopropanamine (0.56 ml, 6.54 mmol) at 0 ℃. The mixture was stirred at room temperature for 5 h. It was diluted with ethyl acetate and washed with water then brine. The organic layer was separated and dried over sodium sulfate. It was filtered and concentrated under reduced pressure. The residue was purified by ISCO (Gold 40g, 0-100% ethyl acetate in hexane) to give the title compound. 1H NMR (CHCl3-d, 500 MHz) : = 0.47-0.37 (4 H, m) , 2.25-2.22 (2 H, m) , 3.48 (2 H, s) , 3.75 (3 H, s) ppm.
Step 2: (2R, 5R) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-
yl) -5- ( (cyclopropyl (2-methoxy-2-oxoethyl) amino) methyl) morpholine-4-carboxylate
A mixture of (2R, 5S) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -5-formylmorpholine-4-carboxylate (300 mg, 0.520 mmol) and methyl 2-(cyclopropylamino) acetate (101 mg, 0.781 mmol) in DCE (2 ml) were stirred for 2 h. To the mixture was added sodium triacetoxyborohydride (221 mg, 1.041 mmol) . The mixture was stirred for 30 min. It was directly purified by ISCO (gold 40g, 0-10% methanol in DCM) to give the title compound. LC-MS: C31H41BrN6O7, calc. = 689.23, 691.23; found = 689.29, 691.29 (M+H) +.
Step 3: (3R, 9aR) -3- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -8-
cyclopropylhexahydropyrazino [2, 1-c] [1, 4] oxazin-6 (1H) -one
(2R, 5R) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -5-( (cyclopropyl (2-methoxy-2-oxoethyl) amino) methyl) morpholine-4-carboxylate (359 mg, 0.521 mmol) was treated with TFA (4 mL, 51.9 mmol) at 100 ℃ for 0.5 h. The mixture was concentrated. The residue was dissolved in 2 mL DCM and added 0.5 mL TEA. It was purified
by ISCO (gold 40g, 0-10% methanol in DCM) to give the title compound. LC-MS: C16H19BrN6O2, calc. = 407.09, 409.08; found = 407.02, 409.02 (M+H) +. 1H NMR (CHCl3-d, 500 MHz) : = 0.51-0.47 (2 H, m) , 0.56-0.53 (2 H, m) , 1.74-1.71 (1 H, m) , 2.52 (1 H, dd, J = 11.97, 7.72 Hz) , 3.11 (1 H, dd, J = 12.13, 5.31 Hz) , 3.33 (1 H, dd, J = 13.82, 10.89 Hz) , 3.52 (3 H, s) , 3.75-3.70 (1 H, m) , 4.00 (1 H, dd, J = 11.14, 3.23 Hz) , 4.75 (1 H, dd, J = 10.90, 2.83 Hz) , 4.99 (1 H, dd, J = 13.79, 2.82 Hz) , 5.79 (2 H, br s) , 7.09 (1 H, d, J = 5.04 Hz) , 7.44 (1 H, d, J = 5.06 Hz) ppm.
Intermediate 8

(3R, 9aR) -3- (8-Amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -8- (tert-butyl) hexahydropyrazino [2, 1-
c] [1, 4] oxazin-6 (1H) -one
Step 1: Methyl 2- (tert-butylamino) acetate
To a solution of methyl bromoacetate (1 g, 6.54 mmol) in THF (10 ml) was added TEA (1.1 ml, 7.89 mmol) and 2-methylpropan-2-amine (0.56 ml, 6.54 mmol) at 0 ℃. The mixture was stirred at room temperature for 5 h. It was diluted with ethyl acetate and washed with water, then brine. The organic layer was separated and dried over sodium sulfate. It was filtered and concentrated. The residue was purified by ISCO (Gold 40g, 0-100% ethyl acetate in hexane) to give the title compound. 1H NMR (CHCl3-d, 500 MHz) : = 1.12 (9 H, s) , 3.43 (2 H, s) , 3.75 (3 H, s) ppm.
Step 2: (2R, 5R) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-
yl) -5- ( (tert-butyl (2-methoxy-2-oxoethyl) amino) methyl) morpholine-4-carboxylate
A mixture of (2R, 5S) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -5-formylmorpholine-4-carboxylate (300 mg, 0.520 mmol) and methyl 2- (tert-butylamino) acetate (113 mg, 0.781 mmol) were stirred in DCE (2 ml) for 16 h. To the mixture was added sodium triacetoxyborohydride (221 mg, 1.041 mmol) . It was stirred for 30 min. The reaction mixture was directly purified by ISCO (gold 40g, 0-10% methanol in DCM) and then ISCO (Gold 24g, 0-100% Ethyl Acetate-EtOH (3: 1) in Hexane) to give the title compound. LC-MS:C16H19BrN6O2, calc. = 705.26, 707.26; found = 705.22, 707.22 (M+H) +.
Step 3: (3R, 9aR) -3- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -8- (tert-
butyl) hexahydropyrazino [2, 1-c] [1, 4] oxazin-6 (1H) -one
(2R, 5R) -tert-Butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -5-( (tert-butyl (2-methoxy-2-oxoethyl) amino) methyl) morpholine-4-carboxylate (173 mg, 0.245 mmol) was treated with TFA (4 mL, 51.9 mmol) at 100 ℃. The mixture was cooled to room temperature and concentrated to give a red residue. It was dissolved into 2 mL of DCM and added TEA until it became yellow. The mixture was purified by ISCO (Gold 40g, 0-100% EtOAC/EtOH in Hexane with 2% TEA) to give the title compound. LC-MS: C17H23BrN6O2, calc. = 423.12, 425.12; found = 423.07, 425.07 (M+H) +. 1H NMR (CHCl3-d, 500 MHz) : = 1.06 (9 H, s) , 2.05 (1 H, dd, J = 11.82, 9.26 Hz) , 2.94 (1 H, ddd, J = 11.65, 4.46, 2.28 Hz) , 3.15-3.06 (2 H, m) , 3.29-3.23 (1 H, m) , 3.55 (1 H, dd, J = 15.83, 2.20 Hz) , 3.63 (1 H, dd, J = 11.30, 3.44 Hz) , 3.69-3.66 (1 H, m) , 5.28-5.23 (2 H, m) , 5.82 (2 H, s) , 7.07 (1 H, d, J = 5.04 Hz) , 7.54 (1 H, d, J = 5.04 Hz) ppm.
Intermediate 9

(3R, 9aR) -3- (8-Amino-1-bromo-5-fluoroimidazo [1, 5-a] pyrazin-3-yl) -8-
isopropylhexahydropyrazino [2, 1-c] [1, 4] oxazin-6 (1H) -one
Step 1: (2R, 5S) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) -5-fluoroimidazo [1, 5-
a] pyrazin-3-yl) -5- ( ( (tert-butyldiphenylsilyl) oxy) methyl) morpholine-4-carboxylate
To a solution of (2R, 5S) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -5- ( ( (tert-butyldiphenylsilyl) oxy) methyl) morpholine-4-carboxylate (1450 mg, 1.775 mmol) in MeOH (20 ml) was added (692 mg, 1.953 mmol) and the reaction was heated at 60 ℃ for 2 h. It was filtered through a syringe filter and concentrated. The residue was added acetonitrile (20 ml) and Cs2CO3 (5784 mg, 17.75 mmol) . The mixture was heated at 90 ℃ for 75 min. It was cooled to room temperature and directly purified by ISCO (Gold 40g, 0-50% ethyl acetate in hexane) to give the title compound. LC-MS: C41H49BrFN5O6Si, calc. = 834.27, 836.27; found = 834.48, 836.38 (M+H) +.
(692 mg, 1.953 mmol) and the reaction was heated at 60 ℃ for 2 h. It was filtered through a syringe filter and concentrated. The residue was added acetonitrile (20 ml) and Cs2CO3 (5784 mg, 17.75 mmol) . The mixture was heated at 90 ℃ for 75 min. It was cooled to room temperature and directly purified by ISCO (Gold 40g, 0-50% ethyl acetate in hexane) to give the title compound. LC-MS: C41H49BrFN5O6Si, calc. = 834.27, 836.27; found = 834.48, 836.38 (M+H) +.
Step 2: (2R, 5R) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) -5-fluoroimidazo [1, 5-
a] pyrazin-3-yl) -5- (hydroxymethyl) morpholine-4-carboxylate
To a solution of (2R, 5S) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) -5-fluoroimidazo [1, 5-a] pyrazin-3-yl) -5- ( ( (tert-butyldiphenylsilyl) oxy) methyl) morpholine-4-
carboxylate (840 mg, 1.006 mmol) in THF (5 mL) was treated with TBAF (1.5 ml, 1.500 mmol) at room temperature for 1 h. The mixture was concentrated and purified by ISCO (Gold 40g, 0-100% EtOAc/EtOH to Hexane) to give the title compound. LC-MS: C25H31BrFN5O6, calc. = 596.15, 598.15; found = 596.18, 598.18 (M+H) +.
Step 3: (2R, 5S) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) -5-fluoroimidazo [1, 5-
a] pyrazin-3-yl) -5-formylmorpholine-4-carboxylate
To a solution of (2R, 5R) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) -5-fluoroimidazo [1, 5-a] pyrazin-3-yl) -5- (hydroxymethyl) morpholine-4-carboxylate (520 mg, 0.872 mmol) in DCM (6 ml) was added Dess-Martin periodinane (481 mg, 1.133 mmol) and stirred for 1 h. The mixture was diluted with ethyl acetate and washed with sodium bicarbonate and sodium thiosulfate. It was dried over sodium sulfate, filtered and concentrated. The residue was used for the next step without purification.
Step 4: (2R, 5R) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) -5-fluoroimidazo [1, 5-
a] pyrazin-3-yl) -5- ( (isopropyl (2-methoxy-2-oxoethyl) amino) methyl) morpholine-4-carboxylate
To a solution of (2R, 5S) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) -5-fluoroimidazo [1, 5-a] pyrazin-3-yl) -5-formylmorpholine-4-carboxylate (518 mg, 0.871 mmol) in ClCH2CH2Cl (6 ml) was added methyl 2- (isopropylamino) acetate (171 mg, 1.307 mmol) . The reaction mixture was stirred for 2 h at room temperature and added sodium triacetoxyborohydride (369 mg, 1.743 mmol) . The mixture was stirred for 30 min. It was purified by ISCO to give the title compound.
Step 5: (3R, 9aR) -3- (8-amino-1-bromo-5-fluoroimidazo [1, 5-a] pyrazin-3-yl) -8-isopropylhexahydropyrazino [2, 1-c] [1, 4] oxazin-6 (1H) -one
(2R, 5R) -tert-Butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) -5-fluoroimidazo [1, 5-a] pyrazin-3-yl) -5- ( (isopropyl (2-methoxy-2-oxoethyl) amino) methyl) morpholine-4-carboxylate (200 mg, 0.282 mmol) was treated with TFA (5 mL, 64.9 mmol) at room temperature overnight. It was heated at 100 ℃ for 15 min and cooled to room temperature. It was concentrated and the residue was added 5 mL DCM and 1 mL of TEA. It was purified by ISCO (Gold 24g, 0-100% EtOAc/EtOH (3: 1) in Hexane with 2% TEA) to give the title compound. LC-MS: C16H20BrFN6O2, calc. = 427.09, 429.09; found = 427.04, 429.04 (M+H) +. 1H NMR (CHCl3-d, 500 MHz) : = 1.09-1.08 (6 H, m) , 2.34 (1 H, dd, J = 11.80, 7.85 Hz) , 2.61 (1 H, q, J = 7.19 Hz) , 2.80-2.75 (1 H, m) , 2.96 (1 H, dd, J = 11.94, 4.93 Hz) , 3.19-3.16 (1 H, m) , 3.48-3.39 (2 H, m) , 3.59 (1 H, t, J = 11.00 Hz) , 3.98 (1 H, dd, J = 11.16, 3.30 Hz) , 4.93-4.90 (2 H, m) , 5.63 (2 H, s) , 6.93 (1 H, s) ppm.
Intermediate 10

(6R, 8aS) -6- (8-amino-1-bromo-5-fluoroimidazo [1, 5-a] pyrazin-3-yl) hexahydroindolizin-3 (2H) -
one
Step1: ( (6R, 8aS) -6- (8-amino-1-bromo-5-fluoro-6-methoxy-5, 6-dihydroimidazo [1, 5-a] pyrazin-3-
yl) hexahydroindolizin-3 (2H) -one
1-chloromethyl-4-fluoro-1, 4-diazoniabicyclic [2.2.2] octane bis (tetrafluoroborate) 1- (1.214 g, 3.43 mmol) was added to a stirred mixture of (6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) hexahydroindolizin-3 (2H) -one (1g, 2.86 mmol, preparation described in patent application US20140206681) in MeOH (10 ml) and acetonitrile (10 ml) and the mixture was stirred at room temperature for overnight and concentrated. The residue was purified by column chromatography on silica gel (ISCO 80g) , eluting with CH2Cl2/MeOH (10/1) to give ( (6R, 8aS) -6- (8-amino-1-bromo-5-fluoro-6-methoxy-5, 6-dihydroimidazo [1, 5-a] pyrazin-3-yl) hexahydroindolizin-3 (2H) -one. LC-MS: Rt 0.92 min (method A) ; m/z 402.06 (M+H) +.
Step 2: (6R, 8aS) -6- (8-amino-1-bromo-5-fluoroimidazo [1, 5-a] pyrazin-3-yl) hexahydroindolizin-
3 (2H) -one
A stirred mixture of (6R, 8aS) -6- (8-amino-1-bromo-5-fluoro-6-methoxy-5, 6-dihydroimidazo [1, 5-a] pyrazin-3-yl) hexahydroindolizin-3 (2H) -one (300 mg, 0.750 mmol) in Pyridine (8 ml) was stirred under microwave irradiation at 180℃ for 15 min. and concentrated. The residue was purified by column chromatography on silica gel (ISCO 80 g) , eluting with CH2Cl2/MeOH (20/1) to give (6R, 8aS) -6- (8-amino-1-bromo-5-fluoroimidazo [1, 5-a] pyrazin-3-yl) hexahydroindolizin-3 (2H) -one. LC-MS: Rt 0.90 min; m/z 369.96 (M+H) +. 1H NMR (CHCl3-d, 500 MHz) : δ= 6.80 (s, 1H) , 4.41 (dd, 1H, J = 13.5 and 3.0 Hz) , 3.53-3.59 (m, 1H) , 3.26-3.31 (m, 1H) , 3.08 (s, 1H) , 2.45 (t, 2H, J = 10 Hz) , 2.28-2.33 (m, 1H) , 2.18-2.21 (m, 1H) , 1.96-2.08 (m, 2H) , 1.65-1.72 (m, 1H) , 1.34-1.42 (m, 1H) ppm.
Intermediate 11-14

(7R, 9aR) -7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-
quinolizin-4 (6H) -one
(7S, 9aS) -7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-quinolizin-
4 (6H) -one
(7R, 9aS) -7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-
quinolizin-4 (6H) -one
(7S, 9aR) -7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-
quinolizin-4 (6H) -one
Step 1 : methyl 6-formylnicotinate
To a solution of methyl 6- (hydroxymethyl) nicotinate (30 g, 179.6 mmol) in DCM (500 mL) was added MnO2 (93.8 g, 1077.8 mmol) . The mixture was stirred at room temperture overnight. The reaction mixture was filtered and concentrated. The residue was purified by column chromatography on silica gel eluted with PE/EA = 10/1 to give methyl 6-formylnicotinate. 1H NMR (400 MHz, CDCl3) δ ppm 3.98 (d, J = 0.78 Hz, 3 H) , 8.01 (d, J = 8.0 Hz, 1 H) , 8.40 -8.49 (m, 1 H) , 9.29 -9.38 (m, 1 H) , 10.11 (s, 1 H) .
Step 2 : methyl 6- (4-methoxy-4-oxobutanoyl) nicotinate
To a suspension of 3-benzyl-5- (2-hydroxyethyl) -4-methylthlazolium chloride (CAS: 4568-71-2, 5.9 g, 21.8 mmol) in [bmim] [PF6] (CAS: 174501-64-5, 15 mL) was added TEA (11 g, 109 mmol) , methyl acrylate (18.8 g, 218 mmol) and methyl 6-formylnicotinate (18 g, 109 mmol) at room temperture. The mixture was stirred at 80 ℃ for 3 hrs. The reaction mixture was treated with EA and water. The organic layer was dried and concentrated. The residue was purified by column chromatography on silica gel eluted with PE/EA = 15/1 to give methyl 6- (4-methoxy-4-oxobutanoyl) nicotinate. 1H NMR (400 MHz, CDCl3) δ ppm 2.74 (t, J = 8.0 Hz, 2 H) , 3.54 (t, J = 8.0 Hz, 2 H) , 3.66 (s, 3 H) , 3.95 (s, 3 H) , 8.06 (d, J = 8.0 Hz, 1 H) , 8.35 -8.41 (m, 1 H) , 9.20 -9.25 (m, 1 H) .
Step 3 : methyl 6- (1, 1-difluoro-4-methoxy-4-oxobutyl) nicotinate
To a solution of methyl 6- (4-methoxy-4-oxobutanoyl) nicotinate (10 g, 39.8 mmol) in DCM (100 mL) was added DAST (25.6 g, 159.4 mmol) . The mixture was stirred at room temperture
overnight. Then another batch of DAST (25.6 g, 159.4 mmol) was added and the mixture was stirred overnight again. The reaction mixture was treated with NaHCO3 aqueous solution and DCM. The organic layer was dried and concentrated, and the residue was purified by column chromatography on silica gel eluted with PE/EA = 25/1 to give methyl 6- (1, 1-difluoro-4-methoxy-4-oxobutyl) nicotinate. 1H NMR (400 MHz, CDCl3) δ ppm 2.52 -2.58 (m, 2 H) , 2.61 -2.75 (m, 2 H) , 3.66 (s, 3 H) , 3.96 (s, 3 H) , 7.70 (d, J = 8.0 Hz, 1 H) , 8.36 -8.43 (m, 1 H) , 9.20 (s, 1 H) .
Step 4 : methyl 6- (1, 1-difluoro-4-methoxy-4-oxobutyl) piperidine-3-carboxylate
A mixture of methyl 6- (1, 1-difluoro-4-methoxy-4-oxobutyl) nicotinate (5.5 g, 20.1 mmol) and PtO2 (1 g) in MeOH/HCl (100 mL/10 mL) was hydrogenated under 50 PSi at 50 ℃ overnight. The suspension was filtered through a pad of Celite and the filter cake was washed with MeOH. The combined filtrates were concentrated to give methyl 6- (1, 1-difluoro-4-methoxy-4-oxobutyl) piperidine-3-carboxylate which was used in next step directly. MS-ESI (m/z) : 280 (M+1) + (Acq Method D; Rt: 0.769 min) .
Step 5 : methyl 9, 9-difluoro-6-oxooctahydro-1H-quinolizine-3-carboxylate
A solution of methyl 6- (1, 1-difluoro-4-methoxy-4-oxobutyl) piperidine-3-carboxylate (5.61 g, 20.1 mmol) in 1, 4-Dioxane (100 ml) was stirred at 100 ℃ overnight. The solvent was evaporated. The residue was purified by column chromatography on silica gel, eluting with PE/EA = 1/1 to give methyl 9, 9-difluoro-6-oxooctahydro-1H-quinolizine-3-carboxylate. 1H NMR (400 MHz, CDCl3) δ ppm 1.63 -1.76 (m, 1 H) , 1.83 -1.92 (m, 2 H) , 2.09 -2.36 (m, 3 H) , 2.53 -2.60 (m, 2 H) , 2.66 -2.73 (m, 2 H) , 3.47 -3.58 (m, 1 H) , 3.68 (s, 3 H) , 5.07 -5.17 (m, 1 H) .
Step 6 : 9, 9-difluoro-6-oxooctahydro-1H-quinolizine-3-carboxylic acid
To a solution of methyl 9, 9-difluoro-6-oxooctahydro-1H-quinolizine-3-carboxylate (2.8 g, 11.33 mmol) in THF/MeOH/water (30 mL/30 mL/9 mL) was added LiOH·H2O (1.426 g, 34.0 mmol) . The mixture was stirred at room temperture for 2 hrs. The reaction mixture was acidified by 1 M HCl, and extracted with EA. The organic layer was dried and concentrated to give 9, 9-difluoro-6-oxooctahydro-1H-quinolizine-3-carboxylic acid, which was used in next step directly. MS-ESI (m/z) : 234 (M+1) + (Acq Method D; Rt: 0.296/0.381 min) .
Step 7 : N- ( (3-chloropyrazin-2-yl) methyl) -9, 9-difluoro-6-oxooctahydro-1H-quinolizine-3-
carboxamide
To a solution of 9, 9-difluoro-6-oxooctahydro-1H-quinolizine-3-carboxylic acid (1.9 g, 8.15 mmol) in THF (40 ml) was added (3-chloropyrazin-2-yl) methanamine hydrochloride (2.2 g, 12.22 mmol) , HATU (4.65 g, 12.22 mmol) and TEA (6.81 ml, 48.9 mmol) . The mixture was
stirred at room temperature overnight. The reaction mixture was treated with EA (200 ml) and water (200 ml) , the organic layer was dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel eluted with PE/DCM/THF = 1/1/1 to give N- ( (3-chloropyrazin-2-yl) methyl) -9, 9-difluoro-6-oxooctahydro-1H-quinolizine-3-carboxamide. 1H NMR (400 MHz, CDCl3) δ ppm 1.63 -1.85 (m, 2 H) , 1.99 -2.29 (m, 3 H) , 2.32 -2.81 (m, 5 H) , 3.46 -3.62 (m, 1 H) , 4.33 -4.98 (m, 3 H) , 8.15 -8.24 (m, 1 H) , 8.30 -8.40 (m, 1 H) .
Step 8 : 7- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-quinolizin-4 (6H) -one
To a solution of N- ( (3-chloropyrazin-2-yl) methyl) -9, 9-difluoro-6-oxooctahydro-1H-quinolizine-3-carboxamide (2.4 g, 6.69 mmol) in acetonitrile (20 ml) was added PCl5 (2.79 g, 13.38 mmol) . The mixture was stirred at room temperature for 2 hrs. The reaction mixture was poured into NaHCO3aqueous solution (300 ml) and extracted with DCM (200 ml) . The organic layer was dried over Na2SO4, filtered and the filtrate was evaporated under reduced pressure. The residue was purified by column chromatography on silica gel eluting with DCM/THF = 10/1 to give 7-(8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-quinolizin-4 (6H) -one. MS-ESI (m/z) : 341 (M+1) + (Acq Method D; Rt: 1.119 min) .
Step 9: 7- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-quinolizin-
4 (6H) -one
To a solution of 7- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-quinolizin-4 (6H) -one (1.07 g, 3.14 mmol) in acetonitrile (40 ml) was added a solution of NBS (0.615 g, 3.45 mmol) in acetonitrile (10 ml) . The mixture was stirred at room temperture for 1 hr. The reaction mixture was treated with EA (100 ml) and water (100 ml) , the organic layer was dried over Na2SO4 and concentrated. The residue was purified by column chromatography on silica gel, eluting with DCM/THF = 10/1 to give 7- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-quinolizin-4 (6H) -one. MS-ESI (m/z) : 421 (M+1) + (Acq Method D; Rt: 1.027 min) .
Step 10: 7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-quinolizin-
4 (6H) -one
To a solution of 7- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-quinolizin-4 (6H) -one (0.6 g, 1.430 mmol) in 2-propanol (5 ml) was added ammonia and i-PrOH (3.57 ml, 14.30 mmol) in 100 mL seal tube, and the resulting mixture was stirred at 95 ℃overnight. The valotiles were evaporated, and the residue was purified by column chromatography on silica gel eluting with DCM/MeOH = 30/1 to give 7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-quinolizin-4 (6H) -one.
Step 11:
(7R, 9aR) -7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-
quinolizin-4 (6H) -one
(7S, 9aS) -7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-quinolizin- 4 (6H) -one
(7R, 9aS) -7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-
quinolizin-4 (6H) -one
(7S, 9aR) -7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-
quinolizin-4 (6H) -one
7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-quinolizin-4 (6H) -one was separated by SFC condition: ["Column: Chiralpak AS-H 250×4.6mm I.D., 5um Mobile phase: methanol (0.05% DEA) in CO2 from 5% to 40% Flow rate: 2.5mL/min Wavelength: 220nm"] to give P1 ( (7R, 9aR) -7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-quinolizin-4 (6H) -one) , P2 (trans: (7R, 9aS) -7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-quinolizin-4 (6H) -one) , P3 ( (7S, 9aR) -7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-quinolizin-4 (6H) -one) , P4 ( (7S, 9aS) -7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-quinolizin-4 (6H) -one) .
For cis-products:
1H NMR (400 MHz, CDCl3) δ ppm 1.89 -2.26 (m, 3 H) , 2.32 -2.52 (m, 4 H) , 2.60 -2.75 (m, 1 H) , 2.83 -2.93 (m, 1 H) , 3.23 -3.35 (m, 1 H) , 3.63 -3.78 (m, 1 H) , 4.87 (d, J = 12.0 Hz, 1 H) , 5.71 (br. s., 2 H) , 7.03 (d, J = 5.09 Hz, 1 H) , 7.11 (d, J = 5.09 Hz, 1 H) .
For trans-products:
1H NMR (400 MHz, CDCl3) δ ppm 1.54 -1.64 (m, 1 H) , 2.08 -2.41 (m, 5 H) , 2.58 -2.78 (m, 3 H) , 2.93 -3.07 (m, 1 H) , 3.59 -3.75 (m, 1 H) , 4.89 (d, J = 12.0 Hz, 1 H) , 5.69 (br. s., 2 H) , 7.05 (d, J = 5.09 Hz, 1 H) , 7.26 (d, J = 5.09 Hz, 1 H) .
Intermediate 15

(1S, 6R, 8aR) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1-
(trifluoromethyl) hexahydroindolizin-3 (2H) -one
Step 1: (3R, 6R) -1-benzyl 3-methyl 6-formylpiperidine-1, 3-dicarboxylate
To a mixture of (3R, 6R) -1-benzyl 3-methyl 6- (hydroxymethyl) piperidine-1, 3-dicarboxylate (10 g, 32 mmol) in DCM (100 mL) was added DMP (16 g, 38 mmol) in portions with stirring at 0 ℃and then stirred at room temperature for 3 h, the mixture was quenched with sat. Na2SO3, extracted with DCM. After concentrated in vacuo to give (3R, 6R) -1-benzyl 3-methyl 6-formylpiperidine-1, 3-dicarboxylate.
Step 2: (3R, 6R) -1-benzyl 3-methyl 6- ( (S) -2, 2, 2-trifluoro-1- ( (trimethylsilyl) oxy) ethyl) piperidine- 1, 3-dicarboxylate
To a mixture of (3R, 6R) -1-benzyl 3-methyl 6-formylpiperidine-1, 3-dicarboxylate (8.9 g, 29 mmol) in DMF (50 mL) was added TMSCF3 (8.3 g, 29 mmol) and LiOAc (970 mg, 14 mmol) . The mixture was stirring at room temperature for 4 h, The organic phase was washed two times with water, extracted with EtOAc. The combined organic phases were dried over Na2SO4, and the solvent was removed under reduced pressure. The resulting mixture was purified by column chromatography on silica gel eluted with Pet. ether : EtOAc =10: 1 to afford (3R, 6R) -1-benzyl 3-methyl 6- ( (S) -2, 2, 2-trifluoro-1- ( (trimethylsilyl) oxy) ethyl) piperidine-1, 3-dicarboxylate.
MS:448 (M+1) .
Step 3: (3R, 6R) -1-benzyl 3-methyl 6- ( (S) -2, 2, 2-trifluoro-1-hydroxyethyl) piperidine-1, 3-
dicarboxylate
To a mixture of (3R, 6R) -1-benzyl 3-methyl 6- ( (S) -2, 2, 2-trifluoro-1-( (trimethylsilyl) oxy) ethyl) piperidine-1, 3-dicarboxylate (8.9 g, 29 mmol) in THF (50 mL) was added TBAF (4.4 g, 31 mmol) . The mixture was stirred at room temperature overnight. The organic phase was washed twice with water, extracted with EtOAc. The combined organic phases were dried over Na2SO4, and concentrated in vacuo to afford (3R, 6R) -1-benzyl 3-methyl 6- ( (S) -2, 2, 2-trifluoro-1-hydroxyethyl) piperidine-1, 3-dicarboxylate. MS: 376 (M+1) .
Step 4: (3R, 6R) -1-benzyl 3-methyl 6- (2, 2, 2-trifluoroacetyl) piperidine-1, 3-dicarboxylate
To a mixture of (3R, 6R) -1-benzyl 3-methyl 6- ( (S) -2, 2, 2-trifluoro-1-hydroxyethyl) piperidine-1, 3-dicarboxylate (6.2 g, 16.5 mmol) in DCM (100 mL) was added DMP (8.4 g, 19.8 mmol) in portions with stirring at 0 ℃ and then stirred at room temperature for 3 h. The mixture was quenched with sat Na2SO3, extracted with DCM. After concentrated in vacuo, the residual was purified by column chromatography on silica gel eluted with Pet. ether : EtOAc = 5 : 1 to afford (3R, 6R) -1-benzyl 3-methyl 6- (2, 2, 2-trifluoroacetyl) piperidine-1, 3-dicarboxylate.
Step 5: (3R, 6R) -1-benzyl 3-methyl 6- ( (E) -4-ethoxy-1, 1, 1-trifluoro-4-oxobut-2-en-2-
yl) piperidine-1, 3-dicarboxylate
To the solution of t-BuOK (20 mL, 20 mmol) in THF (20 mL) was added ethyl 2-(diethoxyphosphoryl) acetate (4.7 g, 20 mmol) . The reaction was stirred at room temperature for 0.5 hour. Then to the reaction mixture was added (3R, 6R) -1-benzyl 3-methyl 6- (2, 2, 2-trifluoroacetyl) piperidine-1, 3-dicarboxylate (3.9 g, 10 mmol) in 10 mL THF dropwise. The reaction mixture was stirred at room temperature for overnight. The reaction mixture was quenched with water and extracted with EtOAc, the combined organic phases were dried over Na2SO4, and the solvent was removed under reduced pressure. The resulting mixture was purified by column chromatography on silica gel eluted with Pet. ether : THF =5: 1 to afford (3R, 6R) -1-benzyl 3-methyl 6- ( (E) -4-ethoxy-1, 1, 1-trifluoro-4-oxobut-2-en-2-yl) piperidine-1, 3-dicarboxylate. MS: 444.3 (M+1) .
Step 6: (1S, 6R, 8aR) -methyl 3-oxo-1- (trifluoromethyl) octahydroindolizine-6-carboxylate
To the solution of (3R, 6R) -1-benzyl 3-methyl 6- ( (E) -4-ethoxy-1, 1, 1-trifluoro-4-oxobut-2-en-2-yl) piperidine-1, 3-dicarboxylate (2.4 g, 5.4 mmol) in MeOH (50 mL) was added Pd (OH) 2 (200 mg) . The reaction was purged with nitrogen and then was stirred under H2 balloon overnight at room temperature. The reaction mixture was filtered though celite, and the filtrate was concentrated to give (1S, 6R, 8aR) -methyl 3-oxo-1- (trifluoromethyl) octahydroindolizine-6-carboxylate. MS: 444.3 (M+1) .
Step 7: (1S, 6R, 8aR) -3-oxo-1- (trifluoromethyl) octahydroindolizine-6-carboxylic acid
To a solution of (1S, 6R, 8aR) -methyl 3-oxo-1- (trifluoromethyl) octahydroindolizine-6-carboxylate (1.1 g, 4.1 mmol) in MeOH/H2O (30 mL) was added LiOH (504 mg, 12 mmol) . The resulting mixture was stirred at room temperature overnight. The mixture was adjusted to pH= 2 with 1N HCl and extracted with DCM, and the combined organic layers were dried over anhydrous sodium sulfate. Then it was filtered and concentrated in vacuum to give (1S, 6R, 8aR) -3-oxo-1- (trifluoromethyl) octahydroindolizine-6-carboxylic acid, which was used directly to next step without further purification.
Step 8: (1S, 6R, 8aR) -N- ( (3-chloropyrazin-2-yl) methyl) -3-oxo-1-
(trifluoromethyl) octahydroindolizine-6-carboxamide
To a solution of (1S, 6R, 8aR) -3-oxo-1- (trifluoromethyl) octahydroindolizine-6-carboxylic acid (0.7 g, 2.8 mmol) in THF (100 mL) was added (3-chloropyrazin-2-yl) methanamine (0.5 g, 2.8 mmol) , DIEA (1.1 g, 8.4 mmol) and HATU (1 g, 2.8 mmol) . The mixture was stirred at room temperature for 2 hrs. The reaction mixture was extracted with DCM and water, the organic layer was dried and concentrated to give the crude product which was purified by column chromatography on silica gel eluted with Pet. ether : EtOAc =1: 1 to afford (1S, 6R, 8aR) -
N- ( (3-chloropyrazin-2-yl) methyl) -3-oxo-1- (trifluoromethyl) octahydroindolizine-6-carboxamide as light green oil. MS: 377 (M+1) .
Step 9: (1S, 6R, 8aR) -6- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1-
(trifluoromethyl) hexahydroindolizin-3 (2H) -one
To a solution of (1S, 6R, 8aR) -N- ( (3-chloropyrazin-2-yl) methyl) -3-oxo-1-(trifluoromethyl) octahydroindolizine-6-carboxamide (730 mg, 1.9 mmol) in MeCN (30 mL) was added PCl5 (1.2 g, 5.8 mmol) . The mixture was stirred at room temperature overnight. Then it was quenched by pouring into sat. NaHCO3 aqueous, and extracted with DCM. The organic layer was dried and concentrated to afford (1S, 6R, 8aR) -6- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1-(trifluoromethyl) hexahydroindolizin-3 (2H) -one. MS: 359 (M+1) .
Step 10: (1S, 6R, 8aR) -6- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1-
(trifluoromethyl) hexahydroindolizin-3 (2H) -one
To a solution of (1S, 6R, 8aR) -6- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1-(trifluoromethyl) hexahydroindolizin-3 (2H) -one (375 g, 1 mmol) in MeCN (10 mL) was added NBS (203 mg, 1.1 mmol) . The mixture was stirred at room temperature for 1 h. The reaction solution was poured into Na2SO3 solution and extracted with DCM. The organic layer was washed with water and brine, dried over Na2SO4, filtered and concentrated to afford (1S, 6R, 8aR) -6- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1-(trifluoromethyl) hexahydroindolizin-3 (2H) -one, which was used in next step without further purification. MS: 439 (M+1) .
Step 11: (1S, 6R, 8aR) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1-
(trifluoromethyl) hexahydroindolizin-3 (2H) -one
To a 30 mL seal tube, was added a solution of (1S, 6R, 8aR) -6- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1- (trifluoromethyl) hexahydroindolizin-3 (2H) -one (320 mg, 0.7 mmol) in NH3·H2O/i-PrOH (10 mL/10 mL) . The mixture was stirred at 110 ℃ overnight. After cooled to room temperature, the mixture was diluted with EtOAc, washed with water and brine, dried over Na2SO4. After concentrated in vacuum, the resulting mixture was purified by pre-HPLC to afford (1S, 6R, 8aR) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1-(trifluoromethyl) hexahydroindolizin-3 (2H) -one with minor isomer impurity. MS: 418 (M+1) .
MS-ESI (m/z) : 418 (M+1) + (Acq Method D; Rt: 1.067min) .
Step 12: (single) (1S, 6R, 8aR) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1-(trifluoromethyl) hexahydroindolizin-3 (2H) -one
Pure (1S, 6R, 8aR) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1-(trifluoromethyl) hexahydroindolizin-3 (2H) -one was obtained by SFC by the following
condition: Column: Chiralpak AS-H 250×4.6mm I.D., 5um;
Mobile phase: methanol (0.05% DEA) in CO2 from5% to 40%;
Flow rate: 2.35mL/min; Wavelength: 220nm.
Intermediate 16

(1R, 6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1-
(trifluoromethyl) hexahydroindolizin-3 (2H) -one
Step 1: (3R, 6S) -1-benzyl 3-methyl 6-formylpiperidine-1, 3-dicarboxylate
Dess-MartinPeriodinane (6.07 g, 14.32 mmol) was added to a stirred (3R, 6S) -1-benzyl 3-methyl 6- (hydroxymethyl) piperidine-1, 3-dicarboxylate (4 g, 13.01 mmol) in CH2Cl2 (40 mL) and the mixture was stirred at 25 ℃ overnight. The mixture was concentrated and extracted by DCM and washed with Na2SO3 aqueous and NaHCO3 aqueous. The organic layers were dried with Na2SO4, filtered and concentrated to give (3R, 6S) -1-benzyl 3-methyl 6-(hydroxymethyl) piperidine-1, 3-dicarboxylate.
Step 2: (3R, 6S) -1-benzyl 3-methyl 6- ( (R) -2, 2, 2-trifluoro-1- ( (trimethylsilyl) oxy) ethyl) piperidine- 1, 3-dicarboxylate
To a mixture of (3R, 6S) -1-benzyl 3-methyl 6- (hydroxymethyl) piperidine-1, 3-dicarboxylate (4 g, 13.10 mmol) in DMF (30 mL) was added trimethyl (trifluoromethyl) silane (3.73 g, 26.2 mmol) and lithium acetate (0.432 g, 6.55 mmol) at 0 ℃. The mixture was stirred at 25 ℃ under N2 protection overnight. The mixture was concentrated to remove solvent and water was added. The mixture was extracted by EtOAc (80 mL×3) . The organic layers were dried and purified with silica gel chromatography (40 g, Pet. ether: THF = 10 : 1) to give (3R, 6S) -1-benzyl 3-methyl 6-( (R) -2, 2, 2-trifluoro-1- ( (trimethylsilyl) oxy) ethyl) piperidine-1, 3-dicarboxylate. MS: 448 (M+1) .
Step 3: (3R, 6S) -1-benzyl 3-methyl 6- ( (R) -2, 2, 2-trifluoro-1-hydroxyethyl) piperidine-1, 3-
dicarboxylate
To a mixture of (3R, 6S) -1-benzyl 3-methyl 6- ( (R) -2, 2, 2-trifluoro-1-( (trimethylsilyl) oxy) ethyl) piperidine-1, 3-dicarboxylate (2 g, 4.47 mmol) in THF (30 mL) was
added TBAF (8.94 mL, 8.94 mmol) . The mixture was stirred at room temperature overnight. The organic phase was washed twice with water, extracted with EtOAc (200 mL) . The combined organic phases were dried over Na2SO4, and the solvent was removed under reduced pressure. The resulting mixture was purified by column chromatography on silica gel eluted with (Pet. Ether : THF =5: 1) to afford (3R, 6S) -1-benzyl 3-methyl 6- ( (R) -2, 2, 2-trifluoro-1-hydroxyethyl) piperidine-1, 3-dicarboxylate. MS: 376 (M+1) .
Step 4: (3R, 6S) -1-benzyl 3-methyl 6- (2, 2, 2-trifluoroacetyl) piperidine-1, 3-dicarboxylate
To a mixture of (3R, 6S) -1-benzyl 3-methyl 6- ( (R) -2, 2, 2-trifluoro-1-hydroxyethyl) piperidine-1, 3-dicarboxylate (1.4 g, 3.73 mmol) in DCM (30 mL) was added DMP (1.898 g, 4.48 mmol) in portions with stirring at 0 ℃ and then the reaction was stirred at room temperature for 3 h. The mixture was quenched with sat. Na2SO3 aqueous, extracted with DCM (120 mL) . The organic layer was concentrated in vacuo, the residual was purified by column chromatography on silica gel eluted with (Pet. Ether/EtOAc = 5/1) to afford (3R, 6S) -1-benzyl 3-methyl 6- (2, 2, 2-trifluoroacetyl) piperidine-1, 3-dicarboxylate.
Step 5: (3R, 6S) -1-benzyl 3-methyl 6- ( (Z) -4-ethoxy-1, 1, 1-trifluoro-4-oxobut-2-en-2-
yl) piperidine-1, 3-dicarboxylate
To the solution of potassium 2-methylpropan-2-olate (0.601 g, 5.36 mmol) in THF (15 mL) was added ethyl 2- (diethoxyphosphoryl) acetate (1.201 g, 5.36 mmol) . The reaction was stirred at room temperature for 0.5 hour. Then to the reaction mixture was added (3R, 6S) -1-benzyl 3-methyl 6- (2, 2, 2-trifluoroacetyl) piperidine-1, 3-dicarboxylate (1 g, 2.68 mmol) in 10 mL THF dropwise. The reaction mixture was stirred at room temperature overnight. Then it was quenched with water and extracted with EtOAc (100 mL ×2 ) , the combined organic phases were dried over Na2SO4, and the solvent was removed under reduced pressure. The resulting mixture was purified by column chromatography on silica gel eluted with (Pet. Ether : THF = 5 : 1) to afford (3R, 6S) -1-benzyl 3-methyl 6- ( (Z) -4-ethoxy-1, 1, 1-trifluoro-4-oxobut-2-en-2-yl) piperidine-1, 3-dicarboxylate. MS: 444.3 (M+1) .
Step 6: (1R, 6R, 8aS) -methyl 3-oxo-1- (trifluoromethyl) octahydroindolizine-6-carboxylate
To the solution of (3R, 6S) -1-benzyl 3-methyl 6- ( (Z) -4-ethoxy-1, 1, 1-trifluoro-4-oxobut-2-en-2-yl) piperidine-1, 3-dicarboxylate (780 mg, 1.759 mmol) in MeOH (30 mL) was added Pd (OH) 2 (124 mg, 0.176 mmol) . The reaction was purged with nitrogen then was stirred under a H2 balloon (45 psi) for overnight at 30 ℃. The reaction was filtered though celite and the filtrate was concentrated to give (1R, 6R, 8aS) -methyl 3-oxo-1- (trifluoromethyl) octahydroindolizine-6-carboxylate. MS: 266 (M+1) .
Step 7: (1R, 6R, 8aS) -3-oxo-1- (trifluoromethyl) octahydroindolizine-6-carboxylic acid
To a solution of (1R, 6R, 8aS) -methyl 3-oxo-1- (trifluoromethyl) octahydroindolizine-6-carboxylate (420 mg, 1.584 mmol) in MeOH (15 mL) and water (15 mL) was added LiOH (114 mg, 4.75 mmol) . The resulting mixture was stirred at room temperature overnight. Then it was adjusted to pH = 2 with 1N HCl. The mixture was extracted with DCM (50 mL×3) , and the combined organic layers were dried over anhydrous sodium sulfate, concentrated in vacuum to give (1R, 6R, 8aS) -3-oxo-1- (trifluoromethyl) octahydroindolizine-6-carboxylic acid.
Step 8: (1R, 6R, 8aS) -N- ( (3-chloropyrazin-2-yl) methyl) -3-oxo-1-
(trifluoromethyl) octahydroindolizine-6-carboxamide
To a solution of (1R, 6S, 8aS) -3-oxo-1- (trifluoromethyl) octahydroindolizine-6-carboxylic acid (285 mg, 1.135 mmol) in DMF (10 mL) was added HATU (475 mg, 1.248 mmol) , DIEA (0.594 ml, 3.40 mmol) and (3-chloropyrazin-2-yl) methanamine (179 mg, 1.248 mmol) . The mixture was stirred at room temperature for 2 hrs. The reaction mixture was extracted with EtOAc (100 mL) and water, the organic layer was dried and concentrated. The residue was purified by column chromatography on silica gel eluted with (Pet. Ether /EtOAc = 1/1) to afford (1R, 6S, 8aS) -N- ( (3-chloropyrazin-2-yl) methyl) -3-oxo-1- (trifluoromethyl) octahydroindolizine-6-carboxamide. MS: 377 (M+1) .
Step 9: (1R, 6R, 8aS) -6- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1-
(trifluoromethyl) hexahydroindolizin-3 (2H) -one
To a solution of (1R, 6S, 8aS) -N- ( (3-chloropyrazin-2-yl) methyl) -3-oxo-1-(trifluoromethyl) octahydroindolizine-6-carboxamide (415 mg, 1.101 mmol) in acetonitrile (20 mL) was added pentachlorophosphorane (917 mg, 4.41 mmol) . The mixture was stirred at room temperature overnight. Then it was quenched by pouring into sat. NaHCO3 aqueous, and extracted with EtOAc (100 mL) . The organic layer was dried and concentrated to afford (1R, 6R, 8aS) -6- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1- (trifluoromethyl) hexahydroindolizin-3 (2H) -one. MS: 359 (M+1) .
Step 10: (1R, 6R, 8aS) -6- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1-
(trifluoromethyl) hexahydroindolizin-3 (2H) -one
To a solution of (1R, 6R, 8aS) -6- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1-(trifluoromethyl) hexahydroindolizin-3 (2H) -one (380 mg, 1.059 mmol) in acetonitrile (20 mL) was added NBS (207 mg, 1.165 mmol) . The mixture was stirred at room temperature for 1 h. The reaction solution was poured into Na2SO3 solution and extracted with DCM (100 mL) . The organic layer was washed with water and brine, dried over Na2SO4, concentrated to afford yellow solid (1R, 6R, 8aS) -6- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1-
(trifluoromethyl) hexahydroindolizin-3 (2H) -one, which was used in next step without further purification. MS: 439 (M+1) .
Step 11: (1R, 6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1-
(trifluoromethyl) hexahydroindolizin-3 (2H) -one
To a 100 mL seal tube, was added a solution of (1R, 6R, 8aS) -6- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1- (trifluoromethyl) hexahydroindolizin-3 (2H) -one (420 mg, 0.960 mmol) in 2-propanol (15 mL) , ammonia hydrate (15mL) , and stirred at 110 ℃ overnight. After cooled to room temperature, the mixture was extracted with EtOAc (50 mL × 2) , washed with water and brine, dried over Na2SO4. After concentrated in vacuum, the residual was purified by column chromatography on silica gel eluted with (DCM : MeOH = 10: 1) to afford solid (1R, 6R, 8aS) -6-(8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1- (trifluoromethyl) hexahydroindolizin-3 (2H) -one.
MS:420 (M+1) .
Step 12: (two iosmers) (1R, 6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1-
(trifluoromethyl) hexahydroindolizin-3 (2H) -one
The compound (1R, 6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1-(trifluoromethyl) hexahydroindolizin-3 (2H) -one (intermediate X3, Rt = 9.630 min) and its epimer (Rt = 8.651 min, peak 2) were separated by SFC from diastereomeric (1R, 6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1- (trifluoromethyl) hexahydroindolizin-3 (2H) -one by SFC using the following condition: Column: Chiralcel OD-H 250×4.6mm I.D., 5um Mobile phase: ethanol (0.05% DEA) in CO2 from5% to 40% Flow rate: 2.35mL/min Wavelength: 220nm.
Intermediate 17 &18

(1S, 6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1-methyltetrahydro-1H-
oxazolo [3, 4-a] pyridin-3 (5H) -one
(1R, 6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1-methyltetrahydro-1H-
oxazolo [3, 4-a] pyridin-3 (5H) -one
Step 1: (3R, 6S) -1-benzyl 3-methyl 6- (1-hydroxyethyl) piperidine-1, 3-dicarboxylate
To a solution of (3R, 6S) -1-benzyl 3-methyl 6-formylpiperidine-1, 3-dicarboxylate (12.4 g, 40. mmol) in dry THF (190 mL) at -78 ℃, MeMgBr (19 ml, 3M /L) was added dropwise. The
reaction mixture was stirred at -78 ℃ for 2 h. The reaction was quenched with NH4Cl aq. (100 mL) , then dulited with DCM (100 mL) and H2O (100 mL) , the organic layer was washed with brine (100 mL) , dried over Na2SO4. The crude product was purified by column chromatography on silica gel eluted with PE/EA (100 %-40 %) to give (3R, 6S) -1-benzyl 3-methyl 6- (1-hydroxyethyl) piperidine-1, 3-dicarboxylate. 1H NMR (400MHz, CD3OD) δ = 7.41 -7.26 (m, 5H) , 5.19 -5.04 (m, 2H) , 4.51 -4.42 (m, 1H) , 4.05 -3.88 (m, 1H) , 3.55 (br. s., 3H) , 3.10 (d, J=9.8 Hz, 1H) , 2.66 (br. s., 1H) , 1.99 -1.82 (m, 3H) , 1.77 -1.56 (m, 1H) , 1.19 (d, J=6.0 Hz, 1H) , 1.08 (d, J=6.0 Hz, 2H) ppm.
Step 2: (6R, 8aS) -1-methyl-3-oxohexahydro-1H-oxazolo [3, 4-a] pyridine-6-carboxylic acid
To a solution of (3R, 6S) -1-benzyl 3-methyl 6- (1-hydroxyethyl) piperidine-1, 3-dicarboxylate (6.5 g, 20.2 mmol) in THF/H2O (1: 1, 150 ml) was added LiOH. H2O (1.7 g, 40.4 mmol) portionwise. The resulting solution was stirred at 25 ℃ for 12 h under N2. The reaction was acidified to pH 5-6 with 2M HCl, dried by lyophilization to give solid. The solid was extracted by DCM, then the organic layer was concentrated under reduced pressure to get (6R, 8aS) -1-methyl-3-oxohexahydro-1H-oxazolo [3, 4-a] pyridine-6-carboxylic acid. 1H NMR (400MHz, CD3OD) δ = 4.76 (quin, J=6.9 Hz, 1H) , 4.29 (quin, J=6.3 Hz, 1H) , 4.00 (dtd, J=1.5, 4.9, 13.2 Hz, 1H) , 3.77 -3.64 (m, 1H) , 3.31 -3.27 (m, 1H) , 3.06 -2.87 (m, 1H) , 2.53 -2.36 (m, 1H) , 2.32 -2.17 (m, 1H) , 2.04 -1.93 (m, 1H) , 1.73 (qd, J=3.3, 12.5 Hz, 1H) , 1.68 -1.45 (m, 2H) , 1.44 -1.32 (m, 3H) ppm.
Step 3: (6R, 8aS) -N- ( (3-chloropyrazin-2-yl) methyl) -1-methyl-3-oxohexahydro-1H-oxazolo [3, 4-
a] pyridine-6-carboxamide
To a solution of (6R, 8aS) -1-methyl-3-oxohexahydro-1H-oxazolo [3, 4-a] pyridine-6-carboxylic acid (600 mg, 3.02 mmol) in dry DCM (12 mL) at 5 ℃, Oxalyl dichloride (1.15 g, 9.04 mmol) was added dropwise and DMF (3 drops) was added. The reaction mixture was stirred at 30 ℃for 2 h. The reaction mixture was concentrated in vacuo to give crude acyl chloride. The solution of crude acyl chloride in DCM (3 ml) was added to a solution of (3-chloropyrazin-2-yl) methanamine hydrochloride (594 mg, 3.32 mmol) , TEA (640 mg, 6.04 mmol) in DCM (10 ml) , and then the reaction mixture was stirred at 30 ℃ for 12h. The reaction was dulited with DCM (20 mL) and H2O (20 mL) , the organic layer was washed with brine (20 mL) , dried over Na2SO4, The crude product was purified by column chromatography on silica gel eluted with PE/THF (100 %-40 %) to give (6R, 8aS) -N- ( (3-chloropyrazin-2-yl) methyl) -1-methyl-3-oxohexahydro-1H-oxazolo [3, 4-a] pyridine-6-carboxamide. 1H NMR (400MHz, CD3OD) δ = 8.55 (d, J=2.5 Hz, 1H) , 8.36 (d, J=2.5 Hz, 1H) , 4.77 (quin, J=6.8 Hz, 1H) , 4.65 (s, 2H) , 4.30 (quin, J=6.2 Hz, 1H) , 3.98 -3.90 (m, 1H) , 3.78 -3.70 (m, 1H) , 3.12 -3.00 (m, 1H) , 2.51 (ttd, J=4.1,
12.1, 16.0 Hz, 1H) , 2.19 -2.07 (m, 1H) , 2.00 (dd, J=3.5, 13.1 Hz, 1H) , 1.81 -1.66 (m, 2H) , 1.55 -1.39 (m, 3H) , 1.35 (d, J=6.5 Hz, 2H) ppm.
Step 4: (6R, 8aS) -6- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1-methyltetrahydro-1H-oxazolo [3, 4-
a] pyridin-3 (5H) -one
To a solution of (6R, 8aS) -N- ( (3-chloropyrazin-2-yl) methyl) -1-methyl-3-oxohexahydro-1H-oxazolo [3, 4-a] pyridine-6-carboxamide (700 mg, 2.16 mmol) in anhydrous acetonitrile (10 mL) was added POCl3 (1.6 g, 10.77 mmol) at an ice-water bath, and then dimethylformamide (160 mg, 2.16 mmol) was added to the mixture. The resulting mixture was stirred at 25 ℃ for 12 h. The reaction was poured to an ice-water mixture, neutralized with powdered sodium bicarbonate, extracted with DCM (20 mL x 3) . The organic layer was washed with brine (30 mL) , dried over Na2SO4, concentrated to give (6R, 8aS) -6- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1-methyltetrahydro-1H-oxazolo [3, 4-a] pyridin-3 (5H) -one. 1H NMR (400MHz, CD3OD) δ = 8.26 -8.20 (m, 1H) , 7.85 (s, 1H) , 7.39 (d, J=5.0 Hz, 1H) , 4.84 -4.76 (m, 1H) , 4.36 (quin, J=6.2 Hz, 1H) , 4.07 -3.97 (m, 1H) , 3.87 (ddd, J=3.6, 7.7, 11.7 Hz, 1H) , 3.54 -3.33 (m, 2H) , 2.30 -2.14 (m, 1H) , 2.10 -1.78 (m, 3H) , 1.77 -1.54 (m, 1H) , 1.47 -1.38 (m, 3H) ppm.
Step 5: (6R, 8aS) -6- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1-methyltetrahydro-1H-
oxazolo [3, 4-a] pyridin-3 (5H) -one
To a solution of compound 6 (6R, 8aS) -6- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1-methyltetrahydro-1H-oxazolo [3, 4-a] pyridin-3 (5H) -one (50 mg, 0.163 mmol) in anhydrous DMF (1 mL) was added 1-bromopyrrolidine-2, 5-dione (30.5 mg, 0.17 mmol) , and stirred for 2 hrs. Then the reaction mixture was poured into an ice-water (10 mL) and sta. NaHCO3 (aq. ) was added to the stirred mixture, filtered and the cake was dissolved with ethyl acetate (50 mL) , washed with H2O (30 mL x 10) . Then the organic layer was washed with brine (30 mL) , dried over Na2SO4, and evaporated to get (6R, 8aS) -6- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1-methyltetrahydro-1H-oxazolo [3, 4-a] pyridin-3 (5H) -one. 1H NMR (400MHz, CD3OD) δ = 8.21 (d, J=5.0 Hz, 1H) , 7.36 (d, J=5.3 Hz, 1H) , 4.82 -4.79 (m, 1H) , 4.60 (s, 1H) , 4.01 (d, J=10.5 Hz, 1H) , 3.86 (ddd, J=3.6, 7.9, 11.8 Hz, 1H) , 3.33 (br. s., 1H) , 2.22 (d, J=14.1 Hz, 1H) , 1.96 -1.85 (m, 1H) , 1.83 -1.77 (m, 1H) , 1.70 (dt, J=3.4, 12.5 Hz, 1H) , 1.39 (d, J=6.5 Hz, 3H) ppm.
Step 6: (6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1-methyltetrahydro-1H-
oxazolo [3, 4-a] pyridin-3 (5H) -one
To a solution of (6R, 8aS) -6- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1-methyltetrahydro-1H-oxazolo [3, 4-a] pyridin-3 (5H) -one (400 mg, 1.04 mmol) in propan-2-ol (4 mL) was added ammonia hydrate (4 mL) , and heated at 100 ℃ for 12 hrs. The reaction mixture was evaporated
to get (6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1-methyltetrahydro-1H-oxazolo [3, 4-a] pyridin-3 (5H) -one.
Step 7: (1S, 6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1-methyltetrahydro-1H-
oxazolo [3, 4-a] pyridin-3 (5H) -one
(1R, 6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1-methyltetrahydro-1H-
oxazolo [3, 4-a] pyridin-3 (5H) -one
(6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1-methyltetrahydro-1H-oxazolo [3, 4-a] pyridin-3 (5H) -one was separated bySFC separation (Instrument: Berger MultiGramTM SFC, Mettler Toledo Co, Ltd. Column: AS 250mm*30mm, 5um. Mobile phase: A:Supercritical CO2 , B: MeOH(0.05% NH3 H2O) , A: B =73: 27 at 60ml/min. Column Temp: 38℃. Nozzle Pressure: 100Bar. Nozzle Temp: 60℃. Evaporator Temp: 20℃. Trimmer Temp: 25℃. Wavelength: 220nm) to get the (1S, 6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1-methyltetrahydro-1H-oxazolo [3, 4-a] pyridin-3 (5H) -one.
1H NMR (400MHz, CD3OD) = 7.59 (d, J=5.0 Hz, 1H) , 6.98 (d, J=5.0 Hz, 1H) , 4.60 (br. s., 1H) , 4.35 (t, J=6.1 Hz, 1H) , 3.97 (dd, J=2.9, 12.7 Hz, 1H) , 3.49 -3.41 (m, 1H) , 3.27 -3.16 (m, 1H) , 2.15 (d, J=12.5 Hz, 1H) , 2.09 -2.01 (m, 1H) , 1.91 -1.79 (m, 1H) , 1.65 -1.53 (m, 1H) , 1.45 (d, J=6.3 Hz, 3H) ppm. and
(1R, 6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1-methyltetrahydro-1H-oxazolo [3, 4-a] pyridin-3 (5H) -one.
1H NMR (400MHz, CD3OD) = 7.58 (d, J=5.0 Hz, 1H) , 6.98 (d, J=5.3 Hz, 1H) , 4.60 (br. s., 1H) , 3.97 (d, J=8.5 Hz, 1H) , 3.89 -3.78 (m, 1H) , 3.28 -3.17 (m, 2H) , 2.20 (d, J=13.6 Hz, 1H) , 1.88 (d, J=13.8 Hz, 1H) , 1.82 -1.75 (m, 1H) , 1.69 (dt, J=3.4, 12.5 Hz, 1H) , 1.38 (d, J=6.5 Hz, 3H) ppm.
Intermediate 19

(7R, 9aR) -7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) tetrahydro-1H- [1, 4] oxazino [3, 4-
c] [1, 4] oxazin-4 (3H) -one
Step 1: (2R, 5R) -tert-butyl 2- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -5-
(hydroxymethyl) morpholine-4-carboxylate
To a solution of (2R, 5S) -tert-butyl 5- ( ( (tert-butyldiphenylsilyl) oxy) methyl) -2- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) morpholine-4-carboxylate (0.5 g, 0.823 mmol) in THF (15 mL)
was added TBAF (2.47 mL, 2.47 mmol, 1.0M in THF) at 0 ℃. The reaction was warmed to 18 ℃and stirred for 18 h. The reaction mixture was diluted with H2O (40 mL) , extracted with ethyl acetate (10 mL×2) . The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated and purified by combi flash (DCM: THF= 10-30%) to give (2R, 5R) -tert-butyl 2- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -5- (hydroxymethyl) morpholine-4-carboxylate. MS (ESI) m/z (M+H) +: 369.0. Acq Method D Rt =1.065 min) 1H NMR (400MHz, CDCl3) δ= 8.02 (d, J=5.1 Hz, 1H) , 7.80 (s, 1H) , 7.34 (d, J=5.1 Hz, 1H) , 5.14 (br. s., 1H) , 4.59 (d, J=13.3 Hz, 1H) , 3.89 (br. s., 3H) , 3.76 (d, J=11.7 Hz, 2H) , 3.39 (d, J=10.6 Hz, 1H) , 2.55 (br. s., 2H) , 1.48 (s, 9H) ppm.
Step 2: (2R, 5R) -tert-butyl 5- ( (2- (tert-butoxy) -2-oxoethoxy) methyl) -2- (8-chloroimidazo [1, 5-
a] pyrazin-3-yl) morpholine-4-carboxylate
To a vigorously stirred mixture of (2R, 5R) -tert-butyl 2- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -5-(hydroxymethyl) morpholine-4-carboxylate (1.4 g, 3.80 mmol) and TBAI (0.070 g, 0.190 mmol) in toluene (12 mL) at 0 ℃ was added dropwise 25 % w/v NaOH (12 mL) . Following complete addition, to the vigorously stirred reaction mixture at 0 ℃was added dropwise a 1 : 1 (v/v) mixture of tert-butyl 2-bromoacetate (2.221 g, 11.39 mmol) and toluene (1 mL) . The reaction mixture was allowed to warm to ambient temperature overnight. The organic layer was separated, washed with sequential portions of water until the washings were neutral. The organic layers were dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (PE: THF = 0-30%) to give (2R, 5R) -tert-butyl 5- ( (2- (tert-butoxy) -2-oxoethoxy) methyl) -2- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) morpholine-4-carboxylate. 1H NMR (400MHz, CDCl3) δ= 8.07 (d, J=5.1 Hz, 1H) , 7.82 (s, 1H) , 7.34 (d, J=5.1 Hz, 1H) , 5.23 (d, J=3.5 Hz, 1H) , 4.83 (d, J=13.7 Hz, 1H) , 4.08 (br. s., 1H) , 4.01 (s, 2H) , 3.94 -3.84 (m, 2H) , 3.66 (dd, J=5.7, 8.8 Hz, 1H) , 3.57 (dd, J=4.5, 13.9 Hz, 1H) , 3.25 (dd, J=3.1, 11.7 Hz, 1H) , 1.49 (d, J=4.7 Hz, 18H) ppm.
Step 3: (2R, 5R) -tert-butyl 2- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -5- ( (2- (tert-butoxy) -
2-oxoethoxy) methyl) morpholine-4-carboxylate
To the solution of (2R, 5R) -tert-butyl 5- ( (2- (tert-butoxy) -2-oxoethoxy) methyl) -2- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) morpholine-4-carboxylate (0.2 g, 0.414 mmol) in DMF (5 mL) was added NBS (0.088 g, 0.497 mmol) . The resulting mixture was stirred at 30 ℃ for 1.5 h. LCMS showed the reaction was complete. The mixture was diluted with H2O (40 mL) , extracted with ethyl acetate (10 mL×2) . The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuo to give crude (2R, 5R) -tert-butyl 2- (1-bromo-8-
chloroimidazo [1, 5-a] pyrazin-3-yl) -5- ( (2- (tert-butoxy) -2-oxoethoxy) methyl) morpholine-4-carboxylate. MS (ESI) m/z (M+H) +: 563.2. Acq Method D Rt = 1.358 min) 1H NMR (400MHz, CDCl3) δ= 8.07 (d, J=5.1 Hz, 1H) , 7.34 (d, J=4.7 Hz, 1H) , 5.15 (d, J=3.5 Hz, 1H) , 4.79 (d, J=14.1 Hz, 1H) , 4.14 (br. s., 1H) , 4.01 (s, 2H) , 3.93 -3.84 (m, 2H) , 3.66 (dd, J=5.9, 9.0 Hz, 1H) , 3.58 (dd, J=4.3, 13.7 Hz, 1H) , 3.27 (dd, J=3.5, 12.1 Hz, 1H) , 1.57 -1.44 (m, 18H) ppm.
Step4: (2R, 5R) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-
yl) -5- ( (2- (tert-butoxy) -2-oxoethoxy) methyl) morpholine-4-carboxylate
To a solution of (2R, 5R) -tert-butyl 2- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -5- ( (2- (tert-butoxy) -2-oxoethoxy) methyl) morpholine-4-carboxylate (0.22 g, 0.392 mmol) in DMF (5 mL) was added the (2, 4-dimethoxyphenyl) methanamine (0.079 g, 0.470 mmol) , K2CO3 (0.162 g, 1.175 mmol) . The resulting mixture was stirred at 85 ℃ for 2.5 h. The mixture was washed with H2O (50 mL) , extracted with EtOAc (10 mL× 2) . The EA layer wasdried over sodium sulfate, filtered concentrated, purified by combi flash (Pet. ether : THF = 0-50%) to give (2R, 5R) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -5- ( (2- (tert-butoxy) -2-oxoethoxy) methyl) morpholine-4-carboxylate. MS (ESI) m/z (M+H) +: 694.1. Acq Method D Rt=1.142 min) . 1H NMR (400MHz, CDCl3) δ = 7.45 (d, J=5.1 Hz, 1H) , 7.10 (d, J=5.1 Hz, 1H) , 6.82 -6.71 (m, 1H) , 6.54 -6.40 (m, 2H) , 5.04 (d, J=3.1 Hz, 1H) , 4.76 -4.62 (m, 3H) , 4.12 (br. s., 1H) , 4.00 (s, 2H) , 3.90 -3.84 (m, 4H) , 3.84 -3.77 (m, 4H) , 3.67 (dd, J=6.1, 8.8 Hz, 1H) , 3.51 (dd, J=4.3, 13.7 Hz, 1H) , 3.32 (dd, J=3.1, 12.1 Hz, 1H) , 1.52 (s, 9H) , 1.48 (s, 9H) ppm.
Step5: 2- ( ( (3R, 6R) -6- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-
yl) morpholin-3-yl) methoxy) acetic acid
The solution of (2R, 5R) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -5- ( (2- (tert-butoxy) -2-oxoethoxy) methyl) morpholine-4-carboxylate (0.25 g, 0.361 mmol) in TFA (3.5 mL) was stirred at 18 ℃ for 2.5 h. The reaction solution was concentrated in vacuo to give 2- ( ( (3R, 6R) -6- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) morpholin-3-yl) methoxy) acetic acid which was used in next step without further purification.
MS (ESI) m/z (M+H) +: 536.2. Acq Method D Rt=0.854 min) .
Step6: (7R, 9aR) -7- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-
yl) tetrahydro-1H- [1, 4] oxazino [3, 4-c] [1, 4] oxazin-4 (3H) -one
To the solution of 2- ( ( (3R, 6R) -6- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) morpholin-3-yl) methoxy) acetic acid (1.5 g, 2.80 mmol) in DCM (20 mL) was added EDCI (0.804 g, 4.19 mmol) , followed by DMAP (0.512 g, 4.19 mmol) . The residue was
stirred at 30 ℃ for 18 h. The mixture was diluted with H2O (100 mL) , extracted with DCM (30 mL×2) . The combined organic phases weredried over sodium sulfate, filtered, concentrated, and purified by combi flash (PE: THF = 0-50%) to give (7R, 9aR) -7- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) tetrahydro-1H- [1, 4] oxazino [3, 4-c] [1, 4] oxazin-4 (3H) -one. MS (ESI) m/z (M+H) +: 520 . 1. Acq Method D Rt = 1.065 min) . 1H NMR (400MHz, CDCl3) δ = 7.30 -7.27 (m, 1H) , 7.15 (d, J=5.1 Hz, 1H) , 6.79 (br. s., 1H) , 6.54 -6.39 (m, 2H) , 4.94 (dd, J=2.5, 13.9 Hz, 1H) , 4.73 (dd, J=2.3, 11.0 Hz, 1H) , 4.68 (d, J=5.5 Hz, 2H) , 4.28 -4.14 (m, 2H) , 4.07 -3.96 (m, 2H) , 3.88 (s, 3H) , 3.83 -3.75 (m, 4H) , 3.64 -3.52 (m, 2H) , 3.38 (dd, J=11.3, 13.7 Hz, 1H) ppm.
Step7: (7R, 9aR) -7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) tetrahydro-1H-
[1, 4] oxazino [3, 4-c] [1, 4] oxazin-4 (3H) -one
The solution of (7R, 9aR) -7- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl)tetrahydro-1H- [1, 4] oxazino [3, 4-c] [1, 4] oxazin-4 (3H) -one (134 mg, 0.259 mmol) in TFA (2 mL) was heated to reflux for 3 hrs. The reaction solution was cooled and concentrated in vacuo to give
(7R, 9aR) -7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) tetrahydro-1H- [1, 4] oxazino [3, 4-c] [1, 4] oxazin-4 (3H) -one, which was used in next step without further purification. MS (ESI) m/z (M+H) +: 370.1. Acq Method D Rt = 0.922 min) .
Intermediate 20

(6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -2, 2-dimethylhexahydroindolizin- 3 (2H) -one
Step 1: (6R, 8aS) -N-methoxy-N-methyl-3-oxooctahydroindolizine-6-carboxamide
To a solution of (6R, 8aS) -3-oxooctahydroindolizine-6-carboxylic acid (9.5g, 51.89 mmol) in anhydrous DCM (250mL) was added isopropyl carbonochloridate (7.63g, 62.26 mmol) and Et3N (6.3g, 62.26 mmol) at 0 ℃. The reaction mixture was stirred for 1 hour at room temperature. Then Et3N (15.75g, 155.66 mmol) and N, O-dimethylhydroxylamine hydrochloride (10.07g, 103.77 mmol) was added , the reaction was stirred for 5 hours at room temperature. After the solvent was removed in vacuo and the residue was extracted with EA and the combined organic layer was washed with water, brine and dried over anhydrous Na2SO4. The organic layer was
concentrated in vacuo and the residue was purified by silica gel column chromatography (PE/THF = 50/50) to afford (6R, 8aS) -N-methoxy-N-methyl-3-oxooctahydroindolizine-6-carboxamide. 1H NMR (400 MHz, CDCl3) δ ppm 1.14 -1.28 (m, 1 H) , 1.49 -1.76 (m, 2 H) , 1.86 -1.98 (m, 2 H) , 2.12 -2.27 (m, 1 H) , 2.36 (t, J = 8.8 Hz, 2 H) , 2.65 -2.89 (m, 2 H) , 3.14 (s, 3 H) , 3.37 -3.51 (m, 1 H) , 3.67 (s, 3 H) , 4.02 -4.22 (m, 1 H) ppm.
Step 2: (6R, 8aS) -6- (hydroxymethyl) hexahydroindolizin-3 (2H) -one
To a solution (6R, 8aS) -N-methoxy-N-methyl-3-oxooctahydroindolizine-6-carboxamide (8.5 g, 37.59 mmol) in EtOH (185 mL) was added NaBH4 (4.17 g, 112.77 mmol) at 0 ℃. The reaction mixture was warmed to room temperature and stirred for overnight. The reaction was quenched with aq. HCl to pH = 8, then extracted with DCM and the combined organic layer was washed with water, brine and dried over anhydrous Na2SO4. The organic layer was concentrated in vacuo to afford (6R, 8aS) -6- (hydroxymethyl) hexahydroindolizin-3 (2H) -one. 1H NMR (400 MHz, CDCl3) δ ppm 1.09 -1.28 (m, 2 H) , 1.48 -1.68 (m, 2 H) , 1.78 -2.00 (m, 2 H) , 2.10 -2.24 (m, 1 H) , 2.25 -2.45 (m, 3 H) , 2.83 (m, 1 H) , 3.30 -3.59 (m, 3 H) , 4.20 (dd, J1= 12.8 Hz, J2= 2.4 Hz, 1 H) .
Step 3: (6R, 8aS) -6- ( ( (tert-butyldimethylsilyl) oxy) methyl) hexahydroindolizin-3 (2H) -one
Imidazole (2.84 g, 41.69 mmol) and 4-dimethylaminopyridine (678.58 mg, 5.56 mmol) followed (6R, 8aS) -6- (hydroxymethyl) hexahydroindolizin-3 (2H) -one (4.7 g, 527.79 mmol) were dissolved in anhydrous dichloromethane (140 ml) under a nitrogen atmosphere. To this solution, tert-butyldimethylsilylchloride (6.26 g, 841.69 mmol) was added and the reaction was stirred at room temperature for 20 h. After 20 h, the resulting mixture was filtered to remove the solid phase and extracted with DCM, and the combined organic layer was washed with water, brine and dried over anhydrous Na2SO4. The organic layer was concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (PE/THF = 5/1) to afford (6R, 8aS) -6-( ( (tert-butyldimethylsilyl) oxy) methyl) hexahydroindolizin-3 (2H) -one. 1H NMR (400 MHz, CDCl3) δ ppm 0.01 (s, 6 H) , 0.86 (s, 9 H) , 1.12 -1.29 (m, 2 H) , 1.56 (m, 2 H) , 1.76 -1.93 (m, 2 H) , 2.10 -2.25 (m, 1 H) , 2.36 (m, 3 H) , 3.34 (m, 1 H) , 3.47 (s, 2 H) , 4.13 (d, J = 11.6 Hz, 1 H) .
Step 4: (6R, 8aS) -6- ( ( (tert-butyldimethylsilyl) oxy) methyl) -2, 2-dimethylhexahydroindolizin-
3 (2H) -one
A solution of (6R, 8aS) -6- ( ( (tert-butyldimethylsilyl) oxy) methyl) hexahydroindolizin-3 (2H) -one (7.1 g, 25.07 mmol) in toluene (125 mL) was cooled down to -78 ℃. LDA (50.1 mL, 100.28 mmol) was slowly added dropwise to the reaction mixture and stirring was continued for 1.5 hours at -78 ℃. After this time, CH3I (10.7 g, 75.21 mmol) were slowly added. The reaction was warmed up to room temperature and stirred for overnight. The reaction was quenched with
saturated ammonium chloride solution, then extracted with EA and the combined organic layer was washed with water, brine and dried over anhydrous Na2SO4. The organic layer was concentrated in vacuo and purified by silica gel column chromatography (PE/THF = 10/1) to affording the desired (6R, 8aS) -6- ( ( (tert-butyldimethylsilyl) oxy) methyl) -2, 2-dimethylhexahydroindolizin-3 (2H) -one. 1H NMR (400 MHz, CDCl3) δ ppm 0.01 (s, 6 H) , 0.86 (s, 9 H) , 1.02 -1.28 (m, 8 H) , 1.42 (m, 1 H) , 1.56 (m, 1 H) , 1.81 (m, 1 H) , 1.86 -1.97 (m, 1 H) , 2.03 (t, J = 12.4Hz, 1 H) , 2.36 (t, J = 12.8 Hz, 1 H) , 3.19 -3.32 (m, 1 H) , 3.40 -3.54 (m, 2 H) , 4.06 -4.18 (m, 1 H) .
Step 5: (6R, 8aS) -6- (hydroxymethyl) -2, 2-dimethylhexahydroindolizin-3 (2H) -one
To a solution (6R, 8aS) -6- ( ( (tert-butyldimethylsilyl) oxy) methyl) -2, 2-dimethylhexahydroindolizin-3 (2H) -one (5.8g, 18.64 mmol) in MeOH (93 mL) was added TsOH·H2O (3.54g, 18.64 mmol) . The reaction was stirred for 2 hours at room temperature. Then the mixture was neutralized with aq. NaHCO3 to adjust to pH = 8. The resulting mixture was extracted with DCM and the combined organic layer was washed with water, brine and dried over anhydrous Na2SO4. The organic layer was concentrated in vacuo and the residue was purified by silica gel column chromatography (PE/THF = 100/30) to afford (6R, 8aS) -6-(hydroxymethyl) -2, 2-dimethylhexahydroindolizin-3 (2H) -one. 1H NMR (400 MHz, CDCl3) δppm 0.98 -1.21 (m, 7 H) , 1.32 -1.43 (m, 1 H) , 1.55 (s, 1 H) , 1.74 -1.93 (m, 2 H) , 1.99 (d, J = 12.4 Hz, 1 H) , 2.24 -2.38 (m, 1 H) , 3.19 -3.52 (m, 4 H) , 4.12 (dd, J1= 10.0, J2= 2.8 Hz, 1 H) .
Step 6: (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizine-6-carboxylic acid
A solution of (6R, 8aS) -6- (hydroxymethyl) -2, 2-dimethylhexahydroindolizin-3 (2H) -one (3.5 g, 17.75 mmol) in acetone (88 mL) and water (17 mL) was added sodium hydrogen carbonate (4.47 g, 53.26 mmol) . To the mixture, cooled at 0 ℃, sodium bromide (361.86 mg, 3.55 mmol) , 2, 2, 6, 6-tetramethylpiperidine-N-oxyl radical (TEMPO, 55.48 mg, 0.36 mmol) , and trichloroisocyanuric acid (TCCA, 4.13 g, 17.75 mmol) were successively added. The mixture was stirred at room temperature for 2 hours and filtered through a celite layer. The filtrate was treated with saturated aqueous sodium carbonate and washed with DCM. The aqueous layer was acidified with 2 M HCl, then extracted with DCM/i-PrOH = 10/1. The combined organic extracts were dried over Na2SO4, and concentrated under reduced pressure to give (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizine-6-carboxylic acid. 1H NMR (400 MHz, CDCl3) δ ppm 1.05 -1.38 (m, 7 H) , 1.50 (dd, J = 12.33, 7.63 Hz, 1 H) , 1.56 -1.72 (m, 1 H) , 2.02 (d, J = 11.74 Hz, 1 H) , 2.12 (dd, J = 12.33, 6.85 Hz, 1 H) , 2.24 (d, J = 13.30 Hz, 1 H) , 2.44 (t, J = 11.54 Hz, 1 H) , 2.78 (t, J = 12.33 Hz, 1 H) , 3.37 (d, J = 7.04 Hz, 1 H) , 4.38 (d, J = 12.52 Hz, 1 H) .
Step 7: (6R, 8aS) -N- ( (3-chloropyrazin-2-yl) methyl) -2, 2-dimethyl-3-oxooctahydroindolizine-6-
carboxamide
To a solution of (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizine-6-carboxylic acid (2.9 g, 13.74 mmol) in anhydrous DCM (137 mL) was added isopropyl carbonochloridate (2.02 g, 16.48 mmol) and Et3N (1.67 g, 16.48 mmol) at 0 ℃. The reaction mixture was stirred for 1 hour at room temperature. Then Et3N (2.78 g, 27.47 mmol) and (3-chloropyrazin-2-yl) methanamine hydrochloride (3.2 g, 17.86 mmol) was added , the reaction was stirred for 5 hours at room temperature. The solution was quenched with water, extracted with DCM and the combined organic layer was washed with water, brine and dried over anhydrous Na2SO4. The organic layer was concentrated in vacuo. The crude was purified by silica gel column chromatography (PE/THF = 3/1) to afford (6R, 8aS) -N- ( (3-chloropyrazin-2-yl) methyl) -2, 2-dimethyl-3-oxooctahydroindolizine-6-carboxamide. 1H NMR (400 MHz, CDCl3) δ ppm 1.08 -1.28 (m, 6 H) , 1.40 -1.55 (m, 1 H) , 1.70 -1.91 (m, 2 H) , 1.95 -2.21 (m, 3 H) , 2.36 (t, J = 12.4 Hz, 1 H) , 2.87 (t, J = 12.4 Hz, 1 H) , 3.40 (d, J = 7.6 Hz, 1 H) , 4.32 (dd, J1= 13.2, J2= 3.2 Hz, 1 H) , 4.70 (m, 2 H) , 7.05 (s, 1 H) , 8.32 (s, 1 H) , 8.44 (s, 1 H) .
Step 8: (6R, 8aS) -N- ( (3-chloropyrazin-2-yl) methyl) -2, 2-dimethyl-3-oxooctahydroindolizine-6-
carboxamide
To a solution of (6R, 8aS) -N- ( (3-chloropyrazin-2-yl) methyl) -2, 2-dimethyl-3-oxooctahydroindolizine-6-carboxamide (1.8 g, 5.34 mmol) in acetonitrile (50 ml) was added PCl5 (3.34 g, 16.03 mmol) in portions in an ice bath. After addition, the mixture was allowed to warme to room temperture (20 ℃) and stirred for 12 hrs under N2 atmosphere. TLC (PE: EA=1: 1) showed that the reaction was complete, then the mixture was poured into ice aq. NaHCO3 (100 mL) slowly, and stirred for 20 min. The mixture was then extracted with DCM (80mLx3) . The organic layer was washed with brine (60 mL) , dried over Na2SO4, and concentrated in vacuo to give the residue, which was then purified with flash chromatography (EA/PE=10 %~ 80 %) to give (6R, 8aS) -6- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -2, 2-dimethylhexahydroindolizin-3 (2H) -one. C16H19ClN4O [M+H] + 319, found 319.1H NMR (400MHz, CDCl3) δ 7.81 (s, 1H) , 7.69 (d, J=4.7 Hz, 1H) , 7.35 (d, J=5.1 Hz, 1H) , 4.34 (d, J=9.8 Hz, 1H) , 3.56 -3.46 (m, 1H) , 3.10 -2.95 (m, 2H) , 2.20 -2.12 (m, 4H) , 1.56 (dd, J=7.4, 12.9 Hz, 1H) , 1.40 -1.29 (m, 1H) , 1.24 (s, 3H) , 1.15 (s, 3H) ppm.
Step 9: (6R, 8aS) -6- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -2, 2-
dimethylhexahydroindolizin-3 (2H) -one
1-bromopyrrolidine-2, 5-dione (0.893 g, 5.02 mmol) was added to a stirred mixture of (6R, 8aS) -6- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -2, 2-dimethylhexahydroindolizin-3 (2H) -one (1.6 g, 5.02
mmol) in DMF (30 ml) and the mixture was stirred at room temperture for 2h. The reaction was quenched by into water. The solid was filtered and was washed with water (30 mL) and EA (10 mL) and dried to give the crude product (6R, 8aS) -6- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -2, 2-dimethylhexahydroindolizin-3 (2H) -one. C16H18BrClN4O [M/M+2] + 397/399, found 397/399.1H NMR (400MHz, DMSO-d6) δ8.44 (d, J=5.1 Hz, 1H) , 7.38 (d, J=5.1 Hz, 1H) , 4.09 -3.99 (m, 1H) , 3.51 -3.42 (m, 1H) , 3.33 -3.25 (m, 1H) , 2.07 (dd, J=6.7, 12.5 Hz, 1H) , 2.03 -1.90 (m, 2H) , 1.77 -1.65 (m, 1H) , 1.42 (dd, J=7.8, 12.1 Hz, 1H) , 1.35 -1.22 (m, 1H) , 1.07 (s, 3H) , 1.00 (s, 3H) ppm.
Step 10: (6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -2, 2-
dimethylhexahydroindolizin-3 (2H) -one
To a solution of (6R, 8aS) -6- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -2, 2-dimethylhexahydroindolizin-3 (2H) -one (1.6 g, 4.02 mmol) in 2-propanol (10 ml) was added ammonia hydrate (13.43 ml, 121 mmol) in 100 mL of stealed tube and the mixture was stirred at 110℃ overnight. Then the reaction mixture was filtered and the filtrate was concentrated in vacuo. The residue was purified with flash cloumn (PE/THF=0%~80%) to afford (6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -2, 2-dimethylhexahydroindolizin-3 (2H) -one.C16H18BrClN4O [M/M+2] + 397/399, found 397/399.1H NMR (400MHz, DMSO-d6) δ7.65 (d, J=4.7 Hz, 1H) , 6.97 (d, J=4.3 Hz, 1H) , 6.63 (br. s., 2H) , 4.07 (d, J=4.3 Hz, 1H) , 3.98 (d, J=12.5 Hz, 1H) , 3.44 (d, J=7.4 Hz, 1H) , 2.90 (t, J=12.1 Hz, 1H) , 2.12 -2.03 (m, 1H) , 1.95 (t, J=16.0 Hz, 2H) , 1.74 -1.63 (m, 1H) , 1.48 -1.38 (m, 1H) , 1.35 -1.22 (m, 1H) , 1.07 (br. s., 3H) , 0.99 (br. s., 3H) ppm.
Intermediate 21

(6R, 8aS) -6- (8-amino-1-bromo-5-fluoroimidazo [1, 5-a] pyrazin-3-yl) -2, 2-
dimethylhexahydroindolizin-3 (2H) -one
Step 1: (6R, 8aS) -6- (8-amino-1-bromo-5-fluoro-6-methoxy-5, 6-dihydroimidazo [1, 5-a] pyrazin-3-
yl) -2, 2-dimethylhexahydroindolizin-3 (2H) -one
SELECTFLUOR (412 mg, 1.163 mmol) was added to a stirred, cooled 0 ℃ (6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -2, 2-dimethylhexahydroindolizin-3 (2H) -one (400 mg,
1.057 mmol) inMeCN (12 ml) and MeOH (12.00 ml) and the mixture was stirred at room temperature for Overnight. The mixture was then concentrated and purified by silica gel chromatography (40g, DCM: MeOH=100%~90%) to give a (6R, 8aS) -6- (8-amino-1-bromo-5-fluoro-6-methoxy-5, 6-dihydroimidazo [1, 5-a] pyrazin-3-yl) -2, 2-dimethylhexahydroindolizin-3 (2H) -one. C17H23BrFN5O2 [M/M+2] + 428/430, found 428/430.1H NMR (400MHz, CD3OD) δ6.82 -6.57 (m, 1H) , 5.14 (br. s., 1H) , 4.16 (d, J=9.4 Hz, 1H) , 3.63 -3.52 (m, 1H) , 3.49 -3.42 (m, 3H) , 3.19 -3.11 (m, 1H) , 3.02 -2.88 (m, 1H) , 2.20 (dd, J=7.2, 12.7 Hz, 1H) , 2.09 (d, J=11.0 Hz, 2H) , 1.93 -1.81 (m, 1H) , 1.57 (dd, J=8.0, 12.7 Hz, 1H) , 1.45 -1.33 (m, 1H) , 1.19 (s, 3H) , 1.16 -1.07 (m, 3H) ppm.
Step 2: (6R, 8aS) -6- (8-amino-1-bromo-5-fluoroimidazo [1, 5-a] pyrazin-3-yl) -2, 2-
dimethylhexahydroindolizin-3 (2H) -one
To a solution of (6R, 8aS) -6- (8-amino-1-bromo-5-fluoro-6-methoxy-5, 6-dihydroimidazo [1, 5-a] pyrazin-3-yl) -2, 2-dimethylhexahydroindolizin-3 (2H) -one (200 mg, 0.467 mmol) in acetonitrile (8 ml) was added Cs2CO3 (1521 mg, 4.67 mmol) and the mixture was heated to 90℃ for 0.5 h under Micwave for cooling condition. The reaction mixture (brown suspension) was cooled to room temperature, filtered, washed with acetonitrile and the solution was concentrated under reduced pressure. The residue was purified by column chromatography on 20g silica gel [Column] , eluting with CH2Cl2/MeOH (gradient 0% to 15%) to give the product (6R, 8aS) -6- (8-amino-1-bromo-5-fluoroimidazo [1, 5-a] pyrazin-3-yl) -2, 2-dimethylhexahydroindolizin-3 (2H) -one. C16H19BrFN5O [M/M+2] + 396/398, found 396/398.1H NMR (400MHz, CDCl3) δ 7.75 -7.41 (m, 1H) , 6.14 (br. s., 2H) , 4.82 (dd, J=3.1, 12.9 Hz, 1H) , 3.89 (tdd, J=3.7, 7.2, 10.9 Hz, 1H) , 3.76 (t, J=11.5 Hz, 1H) , 3.53 (t, J=12.1 Hz, 1H) , 2.67 -2.52 (m, 3H) , 2.47 -2.36 (m, 1H) , 1.98 (dd, J=7.8, 12.5 Hz, 1H) , 1.88 (s, 3H) , 1.82 -1.72 (m, 1H) , 1.68 (s, 3H) ppm.
Intermediate 22

(6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-dimethylhexahydroindolizin- 3 (2H) -one
Step 1: methyl 2- (5-bromopyridin-2-yl) -2-methylpropanoate
A solution of diisoproylamine (18.05 mL, 127 mmol) in THF (360 mL) was cooled to -10 ℃under nitrogen and treated with n-BuLi (50.7 mL, 127 mmol) over five minutes. After 30 min, the solution was cooled to -78 ℃ and treated with methyl isobutyrate (12.93 g, 127 mmol) . After 45 min at -78 ℃, a solution of 2, 5-dibromopyridine (20 g, 84 mmol) in THF (40 mL) was added via syringe, resulting in a bright yellow solution. After the addition was completed, the reaction was removed from the cooling bath and warmed to room temperature. After overnight at room temperature, the reaction was quenched by the addition of aqueous NH4Cl (100 mL) and the reaction mixture was extracted with EtOAc. The organic layer was dried over Na2SO4, filtered and concentrated. The crude product was purified by silica gel chromatography (Pet. ether/EtOAc = 90: 10) to give methyl 2- (5-bromopyridin-2-yl) -2-methylpropanoate.
Step 2: 2- (5-bromopyridin-2-yl) -2-methylpropanoic acid
To a solution of methyl 2- (5-bromopyridin-2-yl) -2-methylpropanoate (18.7 g, 72.4 mmol) and NaOH (39.8 mL, 80 mmol) in MeOH (180 mL) was stirred at 50℃ for overnight. The solvent was removed under reduced pressure. The residue was washed with EtOAc, the water layer was added HCl to adjust to pH = 5, extracted with EtOAc. The organic layers were dried over Na2SO4, concentrated to give 2- (5-bromopyridin-2-yl) -2-methylpropanoic acid.
Step 3: 2- (5-bromopyridin-2-yl) -2-methylpropanoyl chloride
To a solution of 2- (5-bromopyridin-2-yl) -2-methylpropanoic acid (1.1 g, 4.51 mmol) in DCM (30 mL) at 0 ℃ was added oxalyl chloride (1.183 mL, 13.52 mmol) and DMF (3.49 μL, 0.045 mmol) . The mixture was stirred at 15℃ overnight. The mixture was concentrated in vacuuo to give the title product, which was used directly in next step.
Step 4: 3- (5-bromopyridin-2-yl) -1-diazo-3-methylbutan-2-one
2- (5-bromopyridin-2-yl) -2-methylpropanoyl chloride (11 g, 41.9 mmol) was dissolved in a 1: 1 solution of MeCN and THF (30 mL) , and the solution was added dropwise to an ice-water cooled solution of 2M TMSCHN2 (41.9 mL, 84 mmol) and Et3N (9.34 mL, 67.0 mmol) in a 1: 1 solution of MeCN and THF (70 mL) . The resulting yellow reaction mixture was allowed to warm to ambient temperature overnight. The solvent was removed under reduced pressure and the residue was dissolved in EtOAc (50 mL) and washed with water (50 mL) , NaHCO3 (50 mL) , and brine (50 mL) . The organic phase was dried over MgSO4 and concentrated to give a red-brown gum, which was purified by chromatography (Pet. ether/EtOAc = 10: 1) to give 3- (5-bromopyridin-2-yl) -1-diazo-3-methylbutan-2-one. 1H NMR (400 MHz, CDCl3) δ 8.64 (d, J=1.57 Hz, 1H) , 7.78 (dd, J=1.96, 8.61 Hz, 1H) , 7.24 (d, J=8.61 Hz, 1H) , 5.14 (s, 1H) , 1.57 (s, 6H) ppm.
Step 5: methyl 3- (5-bromopyridin-2-yl) -3-methylbutanoate
To a solution of (benzoyloxy) silver (1.025 g, 4.48 mmol) and triethylamine (9.06 g, 90 mmol) in MeOH (50 mL) was added a solution of 3- (5-bromopyridin-2-yl) -1-diazo-3-methylbutan-2-one (6 g, 22.38 mmol) in MeOH (150 mL) . The mixture was stirred at 15℃ (r.t.) overnight. The mixture was concentrated and EtOAc was added. The organic layers were washed with a saturated NaHCO3 solution and purified with silica gel chromatography (120 g, Pet. Ether/THF=5/1) to give methyl 3- (5-bromopyridin-2-yl) -3-methylbutanoate. 1H NMR (400 MHz, CDCl3) δ 8.58 (d, J=1.56 Hz, 1H) , 7.75 (dd, J=1.96, 8.61 Hz, 1H) , 7.27 (d, J=6.26 Hz, 1H) , 3.55 (s, 3H) , 2.83 (s, 2H) , 1.44 (s, 6H) . MS: 274 (M+1) ppm.
Step 6: methyl 6- (4-methoxy-2-methyl-4-oxobutan-2-yl) nicotinate
A mixture of methyl 3- (5-bromopyridin-2-yl) -3-methylbutanoate (4 g, 14.70 mmol) , Et3N (6.15 mL, 44.1 mmol) , Pd (OAc) 2 (0.330 g, 1.470 mmol) and DPPF (1.630 g, 2.94 mmol) in MeOH (20 mL) and DMF (60 mL) was stirred at 90 ℃ under CO (50 psi) atmosphere overnight. The reaction mixture was concentrated and water was added, extracted by EtOAc. The organic layers were washed with brine, purified with silica gel chromatography (40 g, Pet. Ether/EtOAc =10: 1) to give methyl 6- (4-methoxy-2-methyl-4-oxobutan-2-yl) nicotinate. 1H NMR (400 MHz, CDCl3) δ 9.13 (s, 1H) , 8.16-8.29 (m, 1H) , 7.36-7.49 (m, 1H) , 3.94 (s, 3H) , 3.53 (s, 3H) , 2.88 (s, 2H) , 1.40-1.56 (m, 6H) ppm.
Step 7: (trans) methyl 1, 1-dimethyl-3-oxooctahydroindolizine-6-carboxylate
To a mixture of methyl 6- (4-methoxy-2-methyl-4-oxobutan-2-yl) nicotinate (500 mg, 1.990 mmol) in AcOH (15 mL) was added NaCNBH3 (375 mg, 5.97 mmol) at 0 ℃. The mixture was stirred at 25 ℃ overnight. The reaction mixture was concentrated to remove the solvent and quenched with NaHCO3 solution to pH=8. The mixture was extracted by EtOAc, the organic layers were dried and concentrated to give a residual, which was dissolved in MeOH and stirred at 70 ℃ overnight. The reaction mixture was concentrated and purified with silica gel chromatography (20 g, P Pet. Ether/THF = 1/1) to give an oil (trans) methyl 1, 1-dimethyl-3-oxooctahydroindolizine-6-carboxylate. 1H NMR (400 MHz, CDCl3) δ 4.36 (dd, J=3.72, 13.11 Hz, 1H) , 3.65-3.78 (m, 3H) , 2.99 (dd, J=2.93, 11.93 Hz, 1H) , 2.75 (t, J=12.33 Hz, 1H) , 2.36 (tt, J=3.96, 11.88 Hz, 1H) , 2.15-2.28 (m, 3H) , 1.75 (dd, J=3.13, 12.91 Hz, 1H) , 1.57 (dq, J=2.74, 12.91 Hz, 1H) , 1.22-1.33 (m, 1H) , 1.16 (s, 3H) , 1.02 (s, 3H) ppm.
Step 8: (trans) 1, 1-dimethyl-3-oxooctahydroindolizine-6-carboxylic acid
A mixture of (trans) methyl 1, 1-dimethyl-3-oxooctahydroindolizine-6-carboxylate (1.7 g, 7.55 mmol) and LiOH·H2O (0.633 g, 15.09 mmol) in MeOH (20 mL) and water (7 mL) was stirred at 25 ℃ overnight. The mixture was concentrated and adjusted pH=3 with 1N HCl. The mixture
was extracted by DCM, dried and concentrated to give a white solid (trans) 1, 1-dimethyl-3-oxooctahydroindolizine-6-carboxylic acid.
Step 9: (trans) N- ( (3-chloropyrazin-2-yl) methyl) -1, 1-dimethyl-3-oxooctahydroindolizine-6-
carboxamide
To a mixture of (trans) 1, 1-dimethyl-3-oxooctahydroindolizine-6-carboxylic acid (1.2 g, 5.68 mmol) in DMF (30 mL) was added (3-chloropyrazin-2-yl) methanamine (1.023 g, 5.68 mmol) and N-ethyl-N-isopropylpropan-2-amine (2.202 g, 17.04 mmol) and HATU (2.160 g, 5.68 mmol) at 25 ℃. The reaction was stirred at 25 ℃ overnight. Water was added and extracted by DCM (3 times) . The organic layer was dried and concentrated to give a residual, which was purified with silica gel chromatography (40 g, DCM: MeOH = 20: 1) to give (trans) N- ( (3-chloropyrazin-2-yl) methyl) -1, 1-dimethyl-3-oxooctahydroindolizine-6-carboxamide. MS: 337 (M+1) .
Step 10: (trans) 6- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1, 1-dimethylhexahydroindolizin-3 (2H) -
one
PCl5 (2.040 g, 9.80 mmol) was added to a stirred solution of (trans) N- ( (3-chloropyrazin-2-yl) methyl) -1, 1-dimethyl-3-oxooctahydroindolizine-6-carboxamide (1.1 g, 3.27 mmol) in acetonitrile (20 mL) . The mixture was stirred at 25 ℃ overnight. The reaction was quenched with NaHCO3 and extracted by DCM and dried to give (trans) 6- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1, 1-dimethylhexahydroindolizin-3 (2H) -one. MS: 319 (M+1) .
Step 11: (trans) 6- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1, 1-
dimethylhexahydroindolizin-3 (2H) -one
NBS (0.860 g, 4.83 mmol) was added to a stirred solution of (trans) 6- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1, 1-dimethylhexahydroindolizin-3 (2H) -one (1.4 g, 4.39 mmol) in acetonitrile (30 mL) and the mixture was stirred at 25 ℃ overnight. The reaction was quenched with NaHCO3 and extracted by EA and dried to give (trans) 6- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1, 1-dimethylhexahydroindolizin-3 (2H) -one. MS: 399 (M+1) .
Step 12: (trans) 6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-
dimethylhexahydroindolizin-3 (2H) -one
(trans) 6- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1, 1-dimethylhexahydroindolizin-3 (2H) -one (1.7 g, 4.27 mmol) was added to a stirred ammonia·H2O (15 mL, 94 mmol) in 2-propanol (15 mL) and the mixture was stirred 110 ℃ overnight. The reaction mixture was concentrated and extracted by DCM and dried to give (trans) 6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-dimethylhexahydroindolizin-3 (2H) -one. MS: 380 (M+1) .
Step 13: (6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-
dimethylhexahydroindolizin-3 (2H) -one
(trans) 6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-dimethylhexahydroindolizin-3 (2H) -one(1.5 g, 3.97 mmol) was transfered to SFC seperation group and separated under the following condition (Column: Chiralpak AS-H 250×4.6mm I. D., 5um; Mobile phase: ethanol (0.05% DEA) in CO2 from5% to 40%; Flow rate: 2.35mL/min; Wavelength: 220nm. ) to give (6R, 8aS) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-dimethylhexahydroindolizin-3 (2H) -one (Rt = 8.268 min) and its enantiomer (6S, 8aR) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-dimethylhexahydroindolizin-3 (2H) -one (Rt = 6.625 min) .
Intermediate 23

(6R, 8aS) -6- (8-amino-1-bromo-5-fluoroimidazo [1, 5-a] pyrazin-3-yl) -1, 1-
dimethylhexahydroindolizin-3 (2H) -one
Step 1: (6R, 8aS) -6- (8-amino-1-bromo-5-fluoro-6-methoxy-5, 6-dihydroimidazo [1, 5-a] pyrazin-3-
yl) -1, 1-dimethylhexahydroindolizin-3 (2H) -one
SelectFluor (562 mg, 1.586 mmol) was added to a stirred solution of (6S, 8aR) -6- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-dimethylhexahydroindolizin-3 (2H) -one (500 mg, 1.322 mmol) in MeOH (10 mL) and acetonitrile (10 mL) at 0 ℃. The mixture was stirred at room temperature overnight. The mixture was concentrated and water was added, extracted with EtOAc (20 mL × 1) . The aqueous layer was adjusted pH = 8~9 with NaHCO3 and extracted by DCM (20 mL × 3) . The combined organic layers were dried and concentrated to give (6R, 8aS) -
6- (8-amino-1-bromo-5-fluoro-6-methoxy-5, 6-dihydroimidazo [1, 5-a] pyrazin-3-yl) -1, 1-
dimethylhexahydroindolizin-3 (2H) -one. MS: 428/430 (M+1) .
Step 2: (6R, 8aS) -6- (8-amino-1-bromo-5-fluoroimidazo [1, 5-a] pyrazin-3-yl) -1, 1-
dimethylhexahydroindolizin-3 (2H) -one
A mixture of (6R, 8aS) -6- (8-amino-1-bromo-5-fluoro-6-methoxy-5, 6-dihydroimidazo [1, 5-a] pyrazin-3-yl) -1, 1-dimethylhexahydroindolizin-3 (2H) -one (500 mg, 1.167 mmol) and cesium carbonate (3804 mg, 11.67 mmol) in acetonitrile (15 mL) was stirred under microwave at 90 ℃for 0.5 h.. Water was added and extracted by DCM (20 mL × 2) , washed with brine and dried to give (6R, 8aS) -6- (8-amino-1-bromo-5-fluoroimidazo [1, 5-a] pyrazin-3-yl) -1, 1-dimethylhexahydroindolizin-3 (2H) -one. MS: 396/398 (M+1) .
Intermediate 24

(6R, 8aS) -6- (8-amino-1-bromo-5-methylimidazo [1, 5-a] pyrazin-3-yl) -2, 2-
dimethylhexahydroindolizin-3 (2H) -one
Step 1: (6R, 8aS) -6- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -2, 2-dimethylhexahydroindolizin-
3 (2H) -one
PCl5 (2.78 g, 13.36 mmol) was added to a stirred (6S, 8aR) -N- ( (3-chloropyrazin-2-yl) methyl) -2, 2-dimethyl-3-oxooctahydroindolizine-6-carboxamide (1.5g, 4.45 mmol) in acetonitrile (30 mL) and the mixture was stirred at 25 ℃ overnight. The reaction was quenched with NaHCO3 and extracted by DCM and dried to give a (6S, 8aR) -6- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -2, 2-dimethylhexahydro-indolizin-3 (2H) -one. 1H NMR (400 MHz, CDCl3) δ7.80 (s, 1H) , 7.70 (d, J=4.70 Hz, 1H) , 7.35 (d, J=5.09 Hz, 1H) , 4.34 (d, J=9.78 Hz, 1H) , 3.47-3.57 (m, 1H) , 2.94-3.11 (m, 2H) , 2.09-2.22 (m, 4H) , 1.56 (dd, J=7.43, 12.52 Hz, 1H) , 1.29-1.39 (m, 1H) , 1.24 (s, 3H) , 1.15 (s, 3H) ppm.
Step 2: (6R, 8aS) -6- (8-chloro-5-methylimidazo [1, 5-a] pyrazin-3-yl) -2, 2-
dimethylhexahydroindolizin-3 (2H) -one
To a solution of (6R, 8aS) -6- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -2, 2-dimethylhexa-hydroindolizin-3 (2H) -one (200 mg, 0.627 mmol) in THF (10 mL) was added BuLi (0.376 ml, 0.941 mmol, 2.5 M) at -70 ℃ in 10 min. Then iodomethane (178 mg, 1.255 mmol) was dropwise added in 30 min. The mixture was stirred at this temperature and room temperature overnight. The reaction was quenched with a saturated NH4Cl solution and extracted by EtOAc (2 × 10 mL) , dried and concentrated to give (6R, 8aS) -6- (8-chloro-5-methylimidazo [1, 5-a] pyrazin-3-yl) -2, 2-dimethylhexahydroindolizine. MS: 233 (M+1) . 1H NMR (400 MHz, CDCl3) δ7.69-7.81 (m, 1H) , 6.98 (s, 1H) , 4.19-4.36 (m, 1H) , 3.21-3.56 (m, 2H) , 2.90-3.14 (m, 1H) , 2.61-2.71 (m, 2H) , 1.99-2.18 (m, 4H) , 1.42-1.65 (m, 2H) , 1.18-1.25 (m, 3H) , 1.03-1.11 (m, 3H) ppm.
Step 3: (6R, 8aS) -6- (1-bromo-8-chloro-5-methylimidazo [1, 5-a] pyrazin-3-yl) -2, 2-
dimethylhexahydroindolizin-3 (2H) -one
NBS (118 mg, 0.661 mmol) was added to a stirred (6S, 8aR) -6- (8-chloro-5-methyl-imidazo [1, 5-a] pyrazin-3-yl) -1, 1-dimethylhexahydroindolizin-3 (2H) -one (200 mg, 0.601 mmol) in acetonitrile (10 mL) and the mixture was stirred at 25 ℃ overnight. The reaction was quenched with
NaHCO3 solution and extracted by EtOAc (50 mL x 2) , dried and concentrated to give (6S, 8aR) -6- (1-bromo-8-chloro-5-methyl-imidazo [1, 5-a] pyrazin-3-yl) -1, 1-dimethylhexahydroindolizin-3 (2H) -one. MS: 413 (M+1) .
Step 4: (6R, 8aS) -6- (8-amino-1-bromo-5-methylimidazo [1, 5-a] pyrazin-3-yl) -2, 2-
dimethylhexahydroindolizin-3 (2H) -one
To a solution (6S, 8aR) -6- (1-bromo-8-chloro-5-methylimidazo [1, 5-a] pyrazin-3-yl) -2, 2-dimethylhexahydroindolizin-3 (2H) -one (170 mg, 0.413 mmol) was added to a stirred ammonia·H2O (5 mL, 0.413 mmol) in 2-propanol (7 mL) and the mixture was stirred 110 ℃ in a seal tube for overnight. The reaction mixture concentrated and extracted by DCM (30 mL x 2) and washed with water, dried and concentrated to give (6S, 8aR) -6- (8-amino-1-bromo-5-methylimidazo [1, 5-a] pyrazin-3-yl) -2, 2-dimethylhexahydroindolizin-3 (2H) -one. MS:392/394 (M+1) .
Intermediate 25

(trans, single, isomer 1 &isomer 2) 7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -2-
ethyloctahydro-1H-pyrido [1, 2-c] pyrimidin-1-one
Step 1: methyl 6-vinylnicotinate
To a solution of methyl 6-chloronicotinate (50 g, 230 mmol) in propan-2-ol (500 mL) was added potassium vinyltrifluopobopate (62.3 g, 460 mmol) , Et3N (70.4 g, 700 mmol) , Pd (dppf) Cl2·DCM (5.7 g) . The mixture was stirred at 100 ℃ for 2 hours under N2. The mixture was concentrated and the residue was purified by column chromatography on silica gel eluted with Pet. ether : EtOAc =15: 1 to give methyl 6-vinylnicotinate. 1HNMR (400 MHz, CDCl3) δ: 9.14 (s, 1H) , 8.28-8.17 (m, 1H) , 7.38 (d, J=7.8 Hz, 1H) , 6.84 (dd, J=10.8, 17.4 Hz, 1H) , 6.32 (d, J=17.6 Hz, 1H) , 5.60 (d, J=11.0 Hz, 1H) , 3.93 (s, 3H) ppm.
Step 2: methyl 6- (2- ( (2, 4-dimethoxybenzyl) amino) ethyl) nicotinate
To a solution of methyl 6-vinylnicotinate (50 g, 306 mmol) in THF (400 mL) and MeOH (80 mL) was added (2, 4-dimethoxyphenyl) methanamine (102 g, 613 mmol) and K2CO3 (42.3 g, 306 mmol) . The mixture was stirred at 70℃ for 16 h. The solvent was evaporated and the residue was diluted with EtOAc (600 mL) , and then was washed with water (200 mL) . The organic layer
was dried over Na2SO4, filtered and concentrated to give the residue, which was purified by column chromatography on silica gel (EtOAc: THF=1: 1) to give methyl 6- (2- ( (2, 4-dimethoxybenzyl) amino) ethyl) nicotinate. 1H NMR (400 MHz, CDCl3) δ 9.09 (s, 1H) , 8.15 (d, J= 9.6 Hz, 1H) , 7.23 (d, J= 8.0 Hz, 1H) , 7.07 (d, J= 8.4 Hz, 1H) , 6.39-6.36 (m, 2H) , 3.91 (s, 3H) , 3.76 (s, 6H) , 3.73-3.71 (m, 2H) , 3.05-2.96 (m, 4H) ppm.
Step 3: methyl 6- (2- ( (2, 4-dimethoxybenzyl) (phenoxycarbonyl) amino) ethyl) nicotinate
To a solution of methyl 6- (2- ( (2, 4-dimethoxybenzyl) amino) ethyl) nicotinate (45 g, 136 mmol) in THF (400 mL) was added triethylamine (55.1 g, 545 mmol) . The mixture was cooled to 0℃. Then the phenyl carbonochloridate (42.7 g, 272 mmol) was added by dropwise. After the addition, the mixture was warmmed to room temperature, and stirred for 1.5h. The reaction was quenched with water (300 mL) , extracted with EtOAc (300 mL × 3) . The combined organic layers were dried over Na2SO4, filtered and concentrated in vacuum to give the crude, which was purified by column chromatography (EtOAc in petroleum ether=10%~50%) to give methyl 6- (2-( (2, 4-dimethoxybenzyl) (phenoxycarbonyl) amino) ethyl) nicotinate. 1H NMR (400 MHz, CDCl3) δ 9.10 (s, 1H) , 8.16 (d, J= 8.0 Hz, 1H) , 7.32-7.24 (m, 3H) , 7.17-7.14 (m, 2H) , 7.06-7.02 (m, 2H) , 6.44-6.42 (m, 2H) , 4.50-4.46 (m, 2H) , 3.92 (s, 3H) , 3.76 (s, 6H) , 3.73-3.71 (m, 2H) , 3.15 (t, J= 7.2 Hz, 2H) ppm.
Step 4: Methyl 6- (2- ( (2, 4-dimethoxybenzyl) (phenoxycarbonyl) amino) ethyl) piperidine-3-
carboxylate
To a solution of methyl 6- (2- ( (2, 4-dimethoxybenzyl) (phenoxycarbonyl) amino) ethyl) nicotinate (6.9 g, 15.32 mmol) in acetic acid (80 mL) was added NaCNBH3 (3.85 g, 61.3 mmol) . The mixture was stirred at 20 ℃ for 16h. The solvent was evaporated and basified with sat. NaHCO3 aqueous. Then the mixture was extracted with EtOAc (100 mL×3) . The organic layer was dried over Na2SO4, filtered and concentrated to give the crude of methyl 6- (2- ( (2, 4-dimethoxybenzyl) (phenoxycarbonyl) amino) ethyl) piperidine-3-carboxylate, which was used in next step whithout further purification. MS: 456 (M+1) .
Step 5: methyl 2- (2, 4-dimethoxybenzyl) -1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7-
carboxylate
A solution of methyl 6- (2- ( (2, 4-dimethoxybenzyl) (phenoxycarbonyl) amino) ethyl) piperidine-3-carboxylate (7 g, 15.33 mmol) in MeCN (100 mL) was heated to 80 ℃ for 16 h. The mixture was concentrated in vacuum to give the crude product, which was purified by column chromatography (EtOAc in petroleum ether = 10%~50%) to give the trans-methyl 2- (2, 4-dimethoxybenzyl) -1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7-carboxylate as a solid. 1H
NMR (400MHz, CDCl3) δ: 7.20 (d, J=7.8 Hz, 1H) , 6.48 -6.40 (m, 2H) , 4.81 (dd, J=1.6, 12. Hz, 1H) , 4.58 (dd, J=3.2, 3.2 Hz, 2H) , 3.79 (s, 6H) , 3.67 (s, 3H) , 3.24-3.09 (m, 3H) , 2.65-2.55 (m, 1H) , 2.51-2.40 (m, 1H) , 2.10 (d, J=12.9 Hz, 1H) , 2.05-1.96 (m, 1H) , 1.79-1.64 (m, 2H) , 1.56 (dd, J=3.3, 12.7 Hz, 1H) , 1.37-1.28 (m, 1H) ppm.
Step 6: (trans) methyl 1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7-carboxylate
A solution of (trans) methyl 2- (2, 4-dimethoxybenzyl) -1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7-carboxylate (9 g, 24.83 mmol) in TFA (50 mL) was stirred at 80℃ for 30 mins. The mixture was concentrated in vacuo, the residue was diluted with DCM (200 mL) , and basified with aq. NaHCO3 solution. The organic layer was separated. The water layer was extracted with DCM (60 mL×4) . The combined organic layers were concentrated in vacuum to give a solid, which was washed with a solution of EtOAc and Petroleum ether (50 mL) (v/v=1: 2) to give the (trans) methyl 1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7-carboxylate as solid. MS: 213 (M+1) . 1H NMR (400 MHz, DMSO-d6) δ: 6.32 (brs, 1H) , 4.44 (d, J= 11.6 Hz, 1H) , 3.57 (s, 3H) , 3.08-3.01 (m, 2H) , 2.39-2.36 (m, 1H) , 2.32-2.28 (m, 1H) , 1.96-1.88 (m, 2H) , 1.65 (d, J= 11.2 Hz, 1H) , 1.62-1.54 (m, 1H) , 1.45-1.41 (m, 1H) , 1.28-1.22 (m, 1H) ppm.
Step 7: (trans) methyl 2-ethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7-carboxylate
To a solution of (trans) methyl 1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7-carboxylate (1 g, 4.71 mmol) in DMF (15 mL) was added NaH (0.283 g, 7.07 mmol) . 10 min later, the iodoethane (0.772 g, 4.95 mmol) was added. The mixture was stirred at 25 ℃ for 2 h. The mixture was quenched with water (2 mL) . The mixture was concentrated in vacuum to give the crude of a mixture of methyl 2-ethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7-carboxylate and 2-ethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7-carboxylic acid, which was used for next step without any purification. MS: 227/241 (M+1) .
Step 8: (trans) 2-ethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7-carboxylic acid
To a solution of (trans) methyl 2-ethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7-carboxylate (800 mg, 3.33 mmol) in a mixture solution of THF (10 mL) and MeOH (10 mL) was added LiOH aqueous (1M) (6.66 mL, 6.66 mmol) . The mixture was stirred at 20℃ for 2 h.. The mixture was concentrated, and the residue was diluted with water (20 mL) , washed with EtOAc (10 mL) . The water layer was acidified to pH=2 with 1N HCl solution. The residue solution was extracted with DCM (15 mL×4) . The organic layers were dried over Na2SO4, filtered and concentrated in vacuum to give the crude of 2-ethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7-carboxylic acid, which was used for next step without further purification. MS: 227 (M+1) .
Step 9: (trans) N- ( (3-chloropyrazin-2-yl) methyl) -2-ethyl-1-oxooctahydro-1H-pyrido [1, 2-
c] pyrimidine-7-carboxamide
To a solution of (trans) 2-ethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7-carboxylic acid (700 mg, 3.09 mmol) in DMF (15 mL) was added HATU (1764 mg, 4.64 mmol) and Et3N (1.294 mL, 9.28 mmol) followed by (3-chloropyrazin-2-yl) methanamine hydrochloride (835 mg, 4.64 mmol) . The mixture was stirred at 20 ℃ for 3 h.. The mixture was quenched with water (10 mL) , and extracted with DCM (20 mL×3) . The combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated in vacuum to give the crude, which was purified over combi-flash (20 g, DCM: THF=10: 1) to give (trans) N- ( (3-chloropyrazin-2-yl) methyl) -2-ethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7-carboxamide. 1H NMR (400 MHz, CDCl3) δ 8.43 (d, J= 2.4 Hz, 1H) , 8.31 (d, J= 2.4 Hz, 1H) , 7.03 (s, 1H) , 4.73-4.63 (m, 3H) , 3.42-3.31 (m, 2H) , 3.27-3.16 (m, 3H) , 2.69 (t, J= 12.4 Hz, 1H) , 2.43-2.36 (m, 1H) , 2.10-1.98 (m, 2H) , 1.83-1.69 (m, 3H) , 1.38-1.32 (m, 1H) , 1.09 (t, J= 7.2 Hz, 3H) ppm.
Step 10: (trans) 7- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -2-ethyloctahydro-1H-pyrido [1, 2-
c] pyrimidin-1-one
To a solution of (trans) N- ( (3-chloropyrazin-2-yl) methyl) -2-ethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7-carboxamide (600 mg, 1.705 mmol) in MeCN (15 mL) was added PCl5 (1065 mg, 5.12 mmol) at 0℃. After 10 minutes, then mixture was warmed to 15 ℃ and stirred for 2h. The mixture was poured into sat. NaHCO3 aqueous (100 mL) , then was extracted with EtOAc (30 mL×3) , the combined organic layers were washed with brine (30 mL) , dried over Na2SO4, filtered and concentrated in vacuum to give the crude of (trans) 7- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -2-ethyloctahydro-1H-pyrido [1, 2-c] pyrimidin-1-one, which was used for next step without further purification. 1H NMR (400 MHz, CDCl3) δ: 7.85 (d, J= 4.4 Hz, 1H) , 7.35 (d, J= 6.4 Hz, 1H) , 4.77-4.73 (m, 2H) , 3.46-3.40 (m, 1H) , 3.38-3.36 (m, 2H) , 3.30-3.27 (m, 2H) , 2.26-2.24 (m, 1H) , 2.71 (t, J= 5.2Hz, 1H) , 2.22-2.17 (m, 3H) , 1.93-1.92 (m, 1H) , 1.81-1.75 (m, 1H) , 1.49-1.52 (m, 1H) , 1.13 (t, J= 7.2 Hz, 3H) ppm.
Step 11: (trans) 7- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -2-ethyloctahydro-1H-
pyrido [1, 2-c] pyrimidin-1-one
To a solution of (trans) 7- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -2-ethyloctahydro-1H-pyrido [1, 2-c] pyrimidin-1-one (450 mg, 1.348 mmol) in MeCN (10 mL) was added NBS (264 mg, 1.483 mmol) . The mixture was stirred at 15℃ for 1h. The mixture was poured into sat. NaHCO3 aqueous (50 mL) , then was extracted with EtOAc (30 mL×3) . The combined organic layers were washed with brine (30 mL) , filtered and concentrated in vacuum to give the crude, which was purified by combi-flash (20 g, DCM: THF=10: 1) to give the (trans) 7- (1-bromo-8-
chloroimidazo [1, 5-a] pyrazin-3-yl) -2-ethyloctahydro-1H-pyrido [1, 2-c] pyrimidin-1-one. 1H NMR (400 MHz, CDCl3) δ7.85 (d, J= 5.2 Hz, 1H) , 7.32 (d, J= 4.8 Hz, 1H) , 4.70 (d, J = 13.2 Hz, 1H) , 3.57-3.48 (m, 1H) , 3.48-3.38 (m, 2H) , 3.35-3.30 (m, 2H) , 3.14-3.07 (m, 1H) , 2.72 (t, J= 12.0 Hz, 1H) , 2.35-2.25 (m, 3H) , 2.00-1.96 (m, 1H) , 1.88-1.81 (m, 1H) , 1.61-1.54 (m, 1H) , 1.13 (t, J= 7.2 Hz, 3H) ppm.
Step 12: (trans) 7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -2-ethyloctahydro-1H-
pyrido [1, 2-c] pyrimidin-1-one
To a solution of (trans) 7- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -2-ethyloctahydro-1H-pyrido [1, 2-c] pyrimidin-1-one (340 mg, 0.824 mmol) in 2-propanol (8 mL) was added ammonium hydroxide (8 mL, 57.5 mmol) . The mixture was sealed and heated to 100℃, and stirred for 16 hours. The mixture was cooled to room temperature, diluted with DCM (50 mL) , and then was washed with water (10 mL×3) . The organic layer was dried over Na2SO4, concentrated in vacuum to give the (trans) 7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -2-ethyloctahydro-1H-pyrido [1, 2-c] pyrimidin-1-one. 1H NMR (400 MHz, CDCl3) δ: 7.39 (d, J=5.1 Hz, 1H) , 7.04 (d, J=5.1 Hz, 1H) , 5.63 (br. s., 2H) , 4.74 (dd, J=2.9, 13.1 Hz, 1H) , 3.45 (tt, J=6.9, 13.8 Hz, 1H) , 3.39-3.31 (m, 2H) , 3.30-3.22 (m, 2H) , 3.06-2.95 (m, 1H) , 2.72-2.62 (m, 1H) , 2.24-2.11 (m, 3H) , 1.90 (dd, J=2.7, 12.9 Hz, 1H) , 1.79 (dt, J=7.6, 13.2 Hz, 1H) , 1.56-1.43 (m, 1H) , 1.13 (t, J=7.0 Hz, 3H) ppm.
Step 13: (trans, single, isomer 1 &isomer 2) 7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -2-
ethyloctahydro-1H-pyrido [1, 2-c] pyrimidin-1-one
The (trans) racemic 7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -2-ethyloctahydro-1H-pyrido [1, 2-c] pyrimidin-1-one (300 mg, 0.763 mmol) was separated by SFC to give 7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -2-ethyloctahydro-1H-pyrido [1, 2-c] pyrimidin-1-one (trans, single, isomer 1, Ret. time : 8.786 min) , and 7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -2-ethyloctahydro-1H-pyrido [1, 2-c] pyrimidin-1-one (trans, single, isomer 2. Ret time: 9.960 min) . SFC condition: Column: Chiralcel OD-H 250×4.6mm I. D., 5um; Mobile phase: methanol (0.05% DEA) in CO2 from 5% to 40%; Flow rate: 2.35mL/min; Wavelength: 220nm.
Intermediate 26

(3R, 8aR) -3- (8-amino-1-bromo-5-chloroimidazo [1, 5-a] pyrazin-3-yl) tetrahydro-1H-pyrrolo [2, 1-
c] [1, 4] oxazin-6 (7H) -one
Step 1: (2R, 5S) -tert-butyl 5- ( ( (tert-butyldiphenylsilyl) oxy) methyl) -2- (5, 8-dichloroimidazo [1, 5-
a] pyrazin-3-yl) morpholine-4-carboxylate
To (2R, 5S) -tert-butyl 5- ( ( (tert-butyldiphenylsilyl) oxy) methyl) -2- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) morpholine-4-carboxylate (1 g, 1.647 mmol) in THF (100 ml) at -78 ℃, was added n-butyllithium (2.5 M in hexane) (0.790 ml, 1.976 mmol) slowly and the reaction mixture was stirred for 30 min. Perchloroethane (0.437 g, 1.844 mmol) dissolved in 8 mL of THF was added slowly while maintaining the temp at -78℃ and the reaction mixture was stirred for another 30 min. The reaction mixture was quenched with sat. NH4Cl (aq) (50 mL) and extracted with EtOAc (3x80 mL) . The combined organic phase was dried over anhydrous Na2SO4 and concentrated to dryness to give crude residue which was column purified (using 20-30% EtOAc in hex ) to give (2R, 5S) -tert-butyl 5- ( ( (tert-butyldiphenylsilyl) oxy) methyl) -2- (5, 8-dichloroimidazo [1, 5-a] pyrazin-3-yl) morpholine-4-carboxylate. LCMS: C32H38Cl2N4O4Si ; [M+H] +: 641.23, Rt= 1.28 min, 1.73 min) .
Step 2: (2R, 5S) -tert-butyl 2- (1-bromo-8-chloro-5- (2-hydroxypropan-2-yl) imidazo [1, 5-a] pyrazin-
3-yl) -5-methylmorpholine-4-carboxylate
(2R, 5S) -tert-butyl 5- ( ( (tert-butyldiphenylsilyl) oxy) methyl) -2- (5, 8-dichloroimidazo [1, 5-a] pyrazin-3-yl) morpholine-4-carboxylate (1 g, 1.558 mmol) was dissolved in DMF and cooled to 0 ℃ in an ice bath. N-bromosuccimide (0.333 g, 1.870 mmol) was added and the reaction mixture was stirred at room temperature for 1 hour under a stream of nitrogen. The reaction mixture was quenched with sat. NaHCO3 (20 mL) and extracted with EtOAC (3x30 mL) . The combined organic phases were washed with sat. NaCl (3x30 mL) , dried over anhydrous Na2SO4 and filtered. The organic phase was concentrated under vacuum to afford the crude residue (2R, 5S) -tert-butyl 2- (1-bromo-8-chloro-5- (2-hydroxypropan-2-yl) imidazo [1, 5-a] pyrazin-3-yl) -5-methylmorpholine-4-carboxylate (1.43g, 2.92 mmol, 100% yield) , which was used for next step without further purification. LCMS: C32H37BrCl2N4O4Si [M+H] +: 621.06, Rt= 1.91 min) .
Step 3: ( (2R, 5S) -tert-butyl 2- (1-bromo-5-chloro-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-
a] pyrazin-3-yl) -5- ( ( (tert-butyldiphenylsilyl) oxy) methyl) morpholine-4-carboxylate
The crude of (2R, 5S) -tert-butyl 2- (1-bromo-5, 8-dichloroimidazo [1, 5-a] pyrazin-3-yl) -5- ( ( (tert-butyldiphenylsilyl) oxy) methyl) morpholine-4-carboxylate (1.0g, 1.388 mmol) was disolved in 1, 4-dioxane, along with N-ethyl-N-isopropylpropan-2-amine (0.846 ml, 4.86 mmol) followed sequentially by (2, 4-dimethoxyphenyl) methanamine (0.730 ml, 4.86 mmol) . The reaction
mixture was stirred at room temperature for overnight under a stream of nitrogen. The mixture was then concentrated to dryness under reduced pressure to give a crude residue, which was column purified on silica gel eluting with (30% EtOAc: Hexane) to give ( (2R, 5S) -tert-butyl 2- (1-bromo-5-chloro-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -5- ( ( (tert-butyldiphenylsilyl) oxy) methyl) morpholine-4-carboxylate. LCMS: C41H49BrClN5O6Si; [M+H] +: 852.22, Method A Rt= 4.86 min) .
Step 4: (2R, 5R) -tert-butyl 2- (1-bromo-5-chloro-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-
a] pyrazin-3-yl) -5- (hydroxymethyl) morpholine-4-carboxylate
(2R, 5S) -tert-butyl 2- (1-bromo-5-chloro-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -5- ( ( (tert-butyldiphenylsilyl) oxy) methyl) morpholine-4-carboxylate (1 g, 1.175 mmol) was dissolved in THF (20 ml) and then was treated with TBAF (1.175 ml, 1.175 mmol) at 23 ℃ for 1 h. The solvent was evaporated and the residue was loaded on a silica cartridge, dried and purified by ISCO (gold 120g, 0-5% Methanol in DCM) to give (2R, 5R) -tert-butyl 2- (1-bromo-5-chloro-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -5-(hydroxymethyl) morpholine-4-carboxylate. LCMS: C25H31BrClN5O6 ; [M+H] +: 614.12, Method B Rt= 1.40 min) .
Step 5: (2R, 5S) -tert-butyl 2- (1-bromo-5-chloro-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-
a] pyrazin-3-yl) -5-formylmorpholine-4-carboxylate
To (2R, 5R) -tert-butyl 2- (1-bromo-5-chloro-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -5- (hydroxymethyl) morpholine-4-carboxylate (600 mg, 0.979 mmol) in DCM (10 ml) was added Dess-Martin periodinane (498 mg, 1.175 mmol) . The mixture was stirred at room temperature for 30 min. It was quenched with saturated aqueous sodium bicarbonate and sodium thiosulfate solution. The mixure was extracted with ethyl acetate (20 mL x3) . The combined organic layers were washed with brine, dried over sodium sulfate, filtered and concentrated to give (2R, 5S) -tert-butyl 2- (1-bromo-5-chloro-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -5-formylmorpholine-4-carboxylate. The product was used without purification.
LCMS: C25H29BrClN5O6; [M+H] +: 612.03, Method B Rt= 1.48 min) .
Step 6: (2R, 5R) -tert-butyl 2- (1-bromo-5-chloro-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-
a] pyrazin-3-yl) -5- ( (E) -3-methoxy-3-oxoprop-1-en-1-yl) morpholine-4-carboxylate
To (Carbomethoxymethyl) triphenylphosphonium bromide (408 mg, 0.982 mmol) in THF (3 mL) was added 1 M solution of LHMDS in THF (0.982 ml, 0.982 mmol) at 0 ℃. It was stirred for 15 min, then a solution of (2R, 5S) -tert-butyl 2- (1-bromo-5-chloro-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -5-formylmorpholine-4-carboxylate (500 mg, 0.818 mmol) in THF (2 mL) was added. The mixture was warmed to room temperature and
stirred for 1 h. The reaction mixture was quenched with pH 5 phosphate buffer, extracted with ethyl acetate (2x20 mL) . The combined organic layers were washed with brine, dried over sodium sulfate, filtered and concentrated. The residue was purified by ISCO (gold 40g, 0-50% ethyl acetate in hexane) to give (2R, 5R) -tert-butyl 2- (1-bromo-5-chloro-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -5- ( (E) -3-methoxy-3-oxoprop-1-en-1-yl) morpholine-4-carboxylate. as a mixture. LCMS: C28H33BrClN5O7; [M+H] +: 667.99, Method B Rt= 1.54 min) .
Step 7: (2R, 5R) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-
yl) -5- (3-methoxy-3-oxopropyl) morpholine-4-carboxylate
To a solution of (2R, 5R) -tert-butyl 2- (1-bromo-5-chloro-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -5- ( (E) -3-methoxy-3-oxoprop-1-en-1-yl) morpholine-4-carboxylate (100 mg, 0.150 mmol) and nickel (II) chloride hexahydrate (17.82 mg, 0.075 mmol) in MeOH (10 ml) was added sodiumborohydride (5.67 mg, 0.150 mmol) at -20 ℃. The reaction was stirred for 30 min. It was quenched with saturated aqueous ammonium chloride (10 mL) , extracted with ethyl acetate (3X10 mL) . The combined organic layers were washed with brine, dried over sodium sulfate, fitlered and concentrated. The residue was purified by ISCO (gold 24g, 0-40% ethyl acetate in hexane) to give (2R, 5R) -tert-butyl 2- (1-bromo-8-( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -5- (3-methoxy-3-oxopropyl) morpholine-4-carboxylate. (as a mixture)
LCMS: C28H35BrClN5O7; [M+H] +: 668.02, Method B Rt= 1.50 min) .
Step 8: (3R, 8aR) -3- (8-amino-1-bromo-5-chloroimidazo [1, 5-a] pyrazin-3-yl) tetrahydro-1H-
pyrrolo [2, 1-c] [1, 4] oxazin-6 (7H) -one
(2R, 5R) -tert-butyl 2- (1-bromo-5-chloro-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -5- (3-methoxy-3-oxopropyl) morpholine-4-carboxylate (90 mg, 0.135 mmol) was treated with TFA (2 ml, 26.0 mmol) at 100 ℃ for 2h. The crude was dissolved in 10 mL of DMF and filtered, and subjected to purification using the mass directed reverse phase purification system (using CH3CN: H2O, TFA system) to isolate products, which was lyophilized to afford (3R, 8aR) -3- (8-amino-1-bromo-5-chloroimidazo [1, 5-a] pyrazin-3-yl) tetrahydro-1H-pyrrolo [2, 1-c] [1, 4] oxazin-6 (7H) -one. LCMS: C13H13BrClN5O2; [M+H] +: 385.81, Method B Rt= 0.32 min) .
Intermediate 27

(3R, 8aR) -3- (8-amino-1-bromo-5-fluoroimidazo [1, 5-a] pyrazin-3-yl) tetrahydro-1H-pyrrolo [2, 1-
c] [1, 4] oxazin-6 (7H) -one
Step 1: (2R, 5S) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) -5-fluoro-6-methoxy-5, 6-
dihydroimidazo [1, 5-a] pyrazin-3-yl) -5- ( ( (tert-butyldiphenylsilyl) oxy) methyl) morpholine-4-
carboxylate
(2R, 5S) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -5-( ( (tert-butyldiphenylsilyl) oxy) methyl) morpholine-4-carboxylate (1.6 g, 1.959 mmol) dissolved in MeOH (25 ml) was treated with 1-chloromethyl-4-fluoro-1, 4-diazoniabicyclo [2.2.2] octane bis (tetrafluoroborate (0.763 g, 2.155 mmol) and was heated at 60℃ for 2 h. The mixture was filtered, and the solvent was evaporated to dryness. The residue was dissioved in acetonitrile (25.00 ml) and treated with Cs2CO3 (6.38 g, 19.59 mmol) and was heated at 90℃ for 2 h. The reaction mixture was filtered, washed with acteonitrile. Filtrate was concentrated and the residue was loaded on a silica cartridge, dried and purified ISCO (gold 120g, 0-25% EtOAc in Hexane) to give (2R, 5S) -tert-butyl 2- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) -5-fluoro-6-methoxy-5, 6-dihydroimidazo [1, 5-a] pyrazin-3-yl) -5- ( ( (tert-butyldiphenylsilyl) oxy) methyl) morpholine-4-carboxylate. LCMS: C42H53BrFN5O7Si; [M+H] +: 836.33, method B Rt= 1.85 min) .
Step 2 to 6: (3R, 8aR) -3- (8-amino-1-bromo-5-fluoroimidazo [1, 5-a] pyrazin-3-yl) tetrahydro-1H-
pyrrolo [2, 1-c] [1, 4] oxazin-6 (7H) -one
Following the prcedures that were used to prepare intermediate 26 (step 4 to 8) , (3R, 8aR) -3- (8-amino-1-bromo-5-fluoroimidazo [1, 5-a] pyrazin-3-yl) tetrahydro-1H-pyrrolo [2, 1-c] [1, 4] oxazin-6(7H) -one was prepared. LCMS: C13H13BrFN5O2; [M+H] +: 371.92, Rt= 1.21 min) .
Intermediate 28

(3R, 7R, 9aS) -7- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -3-
methyloctahydro-4H-pyrido [1, 2-a] pyrazin-4-one
Step 1: (2S, 5R) -tert-butyl 5- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-
yl) -2-formylpiperidine-1-carboxylate
To a 50 ml one necked round bottom flask was charged with (2S, 5R) -tert-butyl 5- (1-bromo-8-( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -2- (hydroxymethyl) piperidine-1-carboxylate (800 mg, 1.388 mmol) along with sodium bicarbonate (233 mg, 2.78 mmol) , molecularsieves (4A) and CH2Cl2 (8 mL) . The mixture was stirred and Dess-Martin periodinane (706 mg, 1.665 mmol) was added in one portion. The resulting reaction mixture was then stirred at room temperature for 2 hrs. The yellow mixture was loaded directly to Combi flash column (24 g silica gel) through a filtered cartridge. The product was eluted with ethyl acetate in Pet. ether (0% to 80%) to provide product (2S, 5R) -tert-butyl 5- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -2-formylpiperidine-1-carboxylate. MS: 574/576 (M+1) . 1H NMR (400MHz, CDCl3) = 9.61 (s, 1H) , 7.13 (d, J=4.7 Hz, 1H) , 7.08 (br. s., 1H) , 6.78 (br. s., 1H) , 6.49 (s, 1H) , 6.44 (d, J=8.2 Hz, 1H) , 4.67 (d, J=5.5 Hz, 2H) , 4.01 (br. s., 1H) , 3.88 (s, 3H) , 3.80 (s, 3H) , 3.53 (br. s., 1H) , 3.06 (br. s., 1H) , 2.32 (dd, J=5.9, 12.9 Hz, 1H) , 2.10 (br. s., 2H) , 1.82 (s, 2H) , 1.36 (s, 9H) ppm.
Step 2: (2S, 5R) -tert-butyl 5- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-
yl) -2- ( ( ( (R) -1-methoxy-1-oxopropan-2-yl) amino) methyl) piperidine-1-carboxylate
To a solution of (2S, 5R) -tert-butyl 5- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -2-formylpiperidine-1-carboxylate (150 mg, 0.261 mmol) in MeOH (2 mL) was added a solution of (R) -methyl 2-aminopropanoate hydrochloride (146 mg, 1.044 mmol) and potassium acetate (256 mg, 2.61 mmol) in MeOH (1 mL) , then followed by NaCNBH4 (49.2 mg, 0.783 mmol) . The mixture was stirred at 20 ℃ for 2 h. Then the reaction mixture was poured into aq. NaHCO3 (10 mL) , extracted with DCM (15 mL×3) . The organic layer was washed with brine, dried over Na2SO4, concentrated in vacuo and purified by silica gel chromatography (4g, DCM: THF=100%~30%) to give (2S, 5R) -tert-butyl 5- (1-bromo-8- ( (2, 4-
dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -2- ( ( ( (R) -1-methoxy-1-oxopropan-2-yl) amino) methyl) piperidine-1-carboxylate. MS: 661/663 (M+1) . 1H NMR (400MHz, CD3OD) = 7.40 (d, J=5.3 Hz, 1H) , 7.22 (d, J=8.3 Hz, 1H) , 7.03 (d, J=5.3 Hz, 1H) , 6.58 (d, J=2.3 Hz, 1H) , 6.45 (dd, J=2.3, 8.3 Hz, 1H) , 4.70 -4.63 (m, 1H) , 4.58 -4.53 (m, 1H) , 4.38 (br. s., 1H) , 4.08 (d, J=13.3 Hz, 1H) , 3.89 (s, 3H) , 3.78 (s, 3H) , 3.75 (s, 3H) , 3.51 (q, J=6.9 Hz, 1H) , 3.36 (d, J=11.0 Hz, 2H) , 3.13 -3.05 (m, 1H) , 2.64 (dd, J=5.1, 12.2 Hz, 2H) , 2.11 (d, J=16.3 Hz, 2H) , 1.50 (dd, J=3.4, 13.7 Hz, 1H) , 1.30 (d, J=7.0 Hz, 3H) , 1.02 (br. s., 9H) ppm.
Step 3: (3R, 7R, 9aS) -7- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -3-
methylhexahydro-1H-pyrido [1, 2-a] pyrazin-4 (6H) -one
A solution of (2S, 5R) -tert-butyl 5- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -2- ( ( ( (R) -1-methoxy-1-oxopropan-2-yl) amino) methyl) piperidine-1-carboxylate (120 mg, 0.181 mmol) in HCl/Dioxane (4M) (0.907 ml, 3.63 mmol) was stirred at room temperture (15 ℃) for 2 h, then concentrated in vacuo. The residue was dissolved in MeOH (2 mL) and K2CO3 (75 mg, 0.544 mmol) was then added. The mixture was stirred at 90 ℃ for 2h and then concentrated in vacuo. The residue was purified with 12 g of silica gel column (THF/dichloromethane 0%~ 80%) to give the product (3R, 7R, 9aS) -7- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -3-methylhexahydro-1H-pyrido [1, 2-a] pyrazin-4 (6H) -one. MS: 529/531 (M+1) . Acq Method D Rt =1.063 min) . 1H NMR (400MHz, CDCl3) δ = 9.61 (s, 1H) , 7.13 (d, J=4.7 Hz, 1H) , 7.08 (br. s., 1H) , 6.78 (br. s., 1H) , 6.49 (s, 1H) , 6.44 (d, J=8.2 Hz, 1H) , 4.67 (d, J=5.5 Hz, 2H) , 4.01 (br. s., 1H) , 3.88 (s, 3H) , 3.80 (s, 3H) , 3.53 (br. s., 1H) , 3.06 (br. s., 1H) , 2.32 (dd, J=5.9, 12.9 Hz, 1H) , 2.10 (br. s., 2H) , 1.82 (s, 2H) , 1.36 (s, 9H) ppm.
Intermediate 29

(3S, 7R, 9aS) -7- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -3-
methylhexahydro-1H-pyrido [1, 2-a] pyrazin-4 (6H) -one
This intermediate (3S, 7R, 9aS) -7- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -3-methylhexahydro-1H-pyrido [1, 2-a] pyrazin-4 (6H) -one was prepared, in an analogues manner as described for intermediate 28 (step 2 to step 4) , from (2S, 5R) -tert-butyl 5-(1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -2-formylpiperidine-1-carboxylate (2S, 5R) -tert-butyl (200 mg, 0.348 mmol) and (S) -methyl 2-aminopropanoate hydrochloride (194 mg, 1.393 mmol) . MS (ESI) : 529/531 (M+1) . Acq Method D Rt =0.993 min) . 1H NMR (400MHz, CDCl3) δ = 7.20 (d, J=5.1 Hz, 1H) , 7.11 (d, J=4.7 Hz, 1H) , 6.73 (br. s., 1H) , 6.49 (s, 1H) , 6.44 (d, J=8.2 Hz, 1H) , 4.92 -4.85 (m, 1H) , 4.67 (d, J=5.5 Hz, 2H) , 3.88 (s, 3H) , 3.80 (s, 3H) , 3.52 (d, J=7.0 Hz, 1H) , 3.39 (br. s., 1H) , 3.28 (dd, J=4.7, 12.9 Hz, 1H) , 2.96 (d, J=9.8 Hz, 2H) , 2.67 (t, J=12.3 Hz, 1H) , 2.17 (d, J=4.3 Hz, 1H) , 1.81 (br. s., 1H) , 1.41 (d, J=7.0 Hz, 3H) , 1.33 -1.27 (m, 1H) , 0.87 (br. s., 1H) ppm.
Intermediate 30
(7R, 9aS) -7- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -3, 3-
dimethylhexahydro-1H-pyrido [1, 2-a] pyrazin-4 (6H) -one

Step 1: (2S, 5R) -tert-butyl 5- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-
yl) -2- ( ( (1-methoxy-2-methyl-1-oxopropan-2-yl) amino) methyl) piperidine-1-carboxylate
To a solution of (2S, 5R) -tert-butyl 5- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -2-formylpiperidine-1-carboxylate (200 mg, 0.348 mmol) in MeOH (4 mL) was added methyl 2-amino-2-methylpropanoate hydrochloride (214 mg, 1.393 mmol) and potassium acetate (342 mg, 3.48 mmol) , then followed by NaCNBH3 (65.6 mg, 1.044 mmol) . The mixture was stirred at 18 ℃ for 2 h. Then to the reaction mixture was added aq. NaHCO3 (20 mL) , the mixture extracted with DCM (25 mL×3) . The organic layer was washed with brine (20 mL) , dried over Na2SO4, concentrated in vacuo and residue was purified by silica gel chromatography (12g, DCM: THF=100%~300%) to give (2S, 5R) -tert-butyl 5- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -2- ( ( (1-methoxy-2-methyl-1-oxopropan-2-yl) amino) methyl) piperidine-1-carboxylate. MS (ESI) : 675/673 (M+1) . (Acq Method DRt =1.115 min) . 1H NMR (400MHz, CDCl3) δ = 7.28 (d, J=2.3 Hz, 1H) , 7.11 (d, J=5.0 Hz, 1H) , 7.02 (d,
J=5.0 Hz, 1H) , 6.77 (t, J=5.5 Hz, 1H) , 6.50 (d, J=2.3 Hz, 1H) , 6.45 (dd, J=2.4, 8.2 Hz, 1H) , 4.69 (d, J=5.8 Hz, 2H) , 4.30 -4.22 (m, 1H) , 4.08 (d, J=13.6 Hz, 1H) , 3.88 (s, 3H) , 3.81 (s, 3H) , 3.73 (s, 3H) , 3.35 (dd, J=4.0, 14.1 Hz, 1H) , 3.16 (br. s., 1H) , 2.85 (dd, J=9.0, 10.8 Hz, 1H) , 2.60 (dd, J=5.9, 11.2 Hz, 1H) , 2.54 -2.45 (m, 1H) , 2.24 -2.18 (m, 1H) , 2.07 -2.01 (m, 1H) , 1.58 -1.51 (m, 1H) , 1.33 (d, J=4.8 Hz, 6H) , 1.21 (s, 9H) ppm.
Step 2: (7R, 9aS) -7- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -3, 3-
dimethylhexahydro-1H-pyrido [1, 2-a] pyrazin-4 (6H) -one
A solution of (2S, 5R) -tert-butyl 5- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -2- ( ( (1-methoxy-2-methyl-1-oxopropan-2-yl) amino) methyl) piperidine-1-carboxylate (70 mg, 0.104 mmol) in 4M hydrogen chloride/Dioxane (0.518 mL, 2.072 mmol) was stirred at room temperture (15 ℃) for 2 h. TLC showed the starting materials was comused and the mixture concentracted in vacuo. The residue was dissloved into MeOH (2 mL) and K2CO3 (43.0 mg, 0.311 mmol) was then added. The mixture was stirred at 90 ℃ for 2h then concentracted in vacuo. The residue was purified with silica gel column chromatography (THF/dichloromethane 0%~ 80%) to give the product (7R, 9aS) -7- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -3, 3-dimethylhexahydro-1H-pyrido [1, 2-a] pyrazin-4 (6H) -one. MS (ESI) : 543/545 (M+1) . Acq Method D Rt =1.073 min) . 1H NMR (400MHz, CDCl3) δ = 7.24 -7.17 (m, 1H) , 7.14 (d, J=5.1 Hz, 1H) , 7.04 (d, J=5.1 Hz, 1H) , 6.67 (t, J=5.1 Hz, 1H) , 6.42 (s, 1H) , 6.37 (dd, J=2.0, 8.2 Hz, 1H) , 4.82 (dd, J=2.9, 13.1 Hz, 1H) , 4.60 (d, J=5.5 Hz, 2H) , 3.81 (s, 3H) , 3.76 -3.68 (m, 3H) , 3.39 -3.30 (m, 1H) , 3.23 (dd, J=5.3, 13.9 Hz, 1H) , 2.94 -2.85 (m, 1H) , 2.80 (dd, J=6.8, 13.9 Hz, 1H) , 2.55 (t, J=12.3 Hz, 1H) , 2.07 (d, J=6.3 Hz, 1H) , 1.80 (dd, J=2.3, 13.3 Hz, 1H) , 1.53 -1.45 (m, 1H) , 1.40 -1.26 (m, 6H) , 1.22 -1.14 (m, 1H) ppm.
Intermediate 31
7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -2, 3, 3-trimethyloctahydro-1H-pyrido [1, 2-
c] pyrimidin-1-one

Step 1: (R) -2-methyl-N- (propan-2-ylidene) propane-2-sulfinamide
To a solution of (R) -2-methylpropane-2-sulfinamide (25 g, 206 mmol) in THF (650 mL) was added acetone (59.9 g, 1031 mmol) and tetraethoxytitanium (235 g, 1031 mmol) . The solution was stirred at 80 ℃ for 30 h. The reaction mixture was poured to saturated aqueous NaHCO3 (1000 mL) . The mixture was filtered through a Celite pad, and the filtrate was extracted with EtOAc (1000 mL×2) . The organic layer was washed with brine (2000 mL) , dried over Na2SO4, concentrated in vacuo, the residue was purified by silica gel column chromatography (120 g, Pet. ether/EtOAc = 2/1, 30min) to give (R) -2-methyl-N- (propan-2-ylidene) propane-2-sulfinamide. 1H NMR (400MHz, CDCl3) δ 2.34 (s, 3H) , 2.19 (s, 3H) , 1.23 (s, 9H) ppm.
Step 2: (R) -N- (1- (5-bromopyridin-2-yl) -2-methylpropan-2-yl) -2-methylpropane-2-sulfinamide
To a solution of 5-bromo-2-methylpyridine (13 g, 76 mmol) in THF (600 mL) was added LDA (45.3 mL, 91 mmol) dropwise at -78 ℃. The solution was stirred at -78 ℃ for 15 min. Then a solution of (R) -2-methyl-N- (propan-2-ylidene) propane-2-sulfinamide (14.62 g, 91 mmol) in THF (50 mL) was added to above mixture with stirring at -78 ℃ for 4 h. The reaction mixture was poured to saturated aqueous NH4Cl (800 mL) , extracted with EtOAc (500 mL×2) . The organic layer was washed with brine (800 mL) , dried over Na2SO4, concentrated in vacuo, and the residue was purified by silica gel column chromatography (120 g, Pet. ether/EtOAc = 1/1, 40 min) to give (R) -N- (1- (5-bromopyridin-2-yl) -2-methylpropan-2-yl) -2-methylpropane-2-sulfinamide. 1H NMR (400MHz, CDCl3) δ 8.57 (d, J = 1.6 Hz, 1H) , 7.72 (dd, J = 2.3, 8.2 Hz, 1H) , 7.12 (d, J = 8.2 Hz, 1H) , 2.96 -2.87 (m, 2H) , 1.34 (s, 3H) , 1.26 (s, 3H) , 1.19 (s, 9H) ppm.
Step 3: (R) -N- (1- (5-bromopyridin-2-yl) -2-methylpropan-2-yl) -N, 2-dimethylpropane-2-
sulfinamide
To a solution of (R) -N- (1- (5-bromopyridin-2-yl) -2-methylpropan-2-yl) -2-methylpropane-2-sulfinamide (12 g, 36.0 mmol) in DMF (200 mL) was added NaH (2.88 g, 72.0 mmol) at 0 ℃. The solution was stirred at 0 ℃ for 15min. Then iodomethane (20.44 g, 144 mmol) was added, the mixture was stirred at 20 ℃ for 18 h. The reaction mixture was poured to saturated aqueous NH4Cl (500 mL) , extracted with EtOAc (300 mL×2) . The organic layer was washed with brine (600 mL) , dried over Na2SO4, concentrated in vacuo, and the residue was purified by silica gel column chromatography (120 g, Pet. ether/EtOAc = 1/1, 40 min) to give (R) -N- (1- (5-bromopyridin-2-yl) -2-methylpropan-2-yl) -N, 2-dimethylpropane-2-sulfinamide. 1H NMR (400MHz, CDCl3) δ 8.59 (s, 1H) , 7.72 (dd, J = 2.0, 8.2 Hz, 1H) , 7.15 (d, J = 8.2 Hz, 1H) , 3.14 (d, J = 12.9 Hz, 1H) , 2.91 (d, J = 12.9 Hz, 1H) , 2.62 (s, 3H) , 1.30 (s, 3H) , 1.26 (s, 3H) , 1.14 (s, 9H) ppm.
Step 4: (R) -methyl 6- (2- (N, 2-dimethylpropan-2-ylsulfinamido) -2-methylpropyl) nicotinate
To a solution of (R) -N- (1- (5-bromopyridin-2-yl) -2-methylpropan-2-yl) -N, 2-dimethylpropane-2-sulfinamide (11 g, 31.7 mmol) in MeOH (100 mL) and DMF (100 mL) was added Et3N (13.24 mL, 95 mmol) , DPPF (1.756 g, 3.17 mmol) and Pd (OAc) 2 (1.422 g, 6.33 mmol) . The mixture was stirred at 80 ℃ for 24 hours under CO (50psi) . Then the reaction mixture was filtered, and the filtrate was concentrated in vacuo. Then the residue was taken into water (500 mL) and extracted with EtOAc (300 mL×3) . The combined organic layers were washed with water (800 mL) , brine (800 mL) , dried over Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (120 g, Pet. ether/EtOAc = 1/1, 30 min) to afford (R) -methyl 6- (2- (N, 2-dimethylpropan-2-ylsulfinamido) -2-methylpropyl) nicotinate. 1H NMR (400MHz, CDCl3) δ 9.13 (s, 1H) , 8.25 -8.14 (m, 1H) , 7.32 (d, J = 8.2 Hz, 1H) , 3.94 (s, 3H) , 3.26 (d, J = 12.9 Hz, 1H) , 3.02 (d, J = 12.9 Hz, 1H) , 2.64 (s, 3H) , 1.32 (s, 3H) , 1.28 (s, 3H) , 1.13 (s, 9H) ppm.
Step 5: methyl 6- (2-methyl-2- (methylamino) propyl) nicotinate
To a mixture of (R) -methyl 6- (2- (N, 2-dimethylpropan-2-ylsulfinamido) -2-methylpropyl) nicotinate (3 g, 9.19 mmol) in Dioxane (10 mL) was added hydrogen chloride/dioxane (4M) (4.59 mL, 18.38 mmol) . Then the mixture was stirred at 20 ℃ for 2 h and concentrated in vacuo to give methyl 6- (2-methyl-2- (methylamino) propyl) nicotinate. MS: 222 (M+1) (Acq Method D Rt: 0.199 min) .
Step 6: methyl 6- (2- ( ( (benzyloxy) carbonyl) (methyl) amino) -2-methylpropyl) nicotinate
To a solution of methyl 6- (2-methyl-2- (methylamino) propyl) nicotinate (2 g, 9.00 mmol) in THF (20 mL) and water (20 mL) was added Na2CO3 (2.86 g, 27.0 mmol) . Then CbzCl (2.57 mL, 18.00 mmol) was added at 0 ℃, the mixture was stirred at 20 ℃ for 18 h. Then the reaction mixture was extracted with EtOAc (50 mL×2) . The organic layer was washed with brine (100 mL) , dried over Na2SO4, concentrated in vacuo to give methyl 6- (2-( ( (benzyloxy) carbonyl) (methyl) amino) -2-methylpropyl) nicotinate. 1H NMR (400MHz, CDCl3) δ 9.09 (s, 1H) , 8.04 -7.96 (m, 1H) , 7.43 -7.34 (m, 5H) , 6.97 (d, J = 8.2 Hz, 1H) , 5.16 (s, 2H) , 3.94 (s, 3H) , 3.38 (s, 2H) , 2.60 (s, 3H) , 1.48 (s, 6H) ppm.
Step 7: methyl 6- (2- ( ( (benzyloxy) carbonyl) (methyl) amino) -2-methylpropyl) piperidine-3-
carboxylate
To a solution of methyl 6- (2- ( ( (benzyloxy) carbonyl) (methyl) amino) -2-methylpropyl) nicotinate (3 g, 8.42 mmol) in AcOH (30 mL) was added NaCNBH3 (1.587 g, 25.3 mmol) at 0 ℃. Then the mixture was stirred at 0 ℃ to 20 ℃ for 18 h. The reaction mixture was poured to saturated of NaHCO3 (200 mL) , extracted with EtOAc (150 mL×2) . The organic layer was washed with brine (300 mL) , dried over Na2SO4, concentrated in vacuo to give crude methyl 6- (2-
( ( (benzyloxy) carbonyl) (methyl) amino) -2-methylpropyl) piperidine-3-carboxylate. MS: 363.2 (M+1) (Acq Method D Rt: 0.882 min) .
Step 8: 1-benzyl 3-methyl 6- (2- ( ( (benzyloxy) carbonyl) (methyl) amino) -2-
methylpropyl) piperidine-1, 3-dicarboxylate
To a solution of methyl 6- (2- ( ( (benzyloxy) carbonyl) (methyl) amino) -2-methylpropyl) piperidine-3-carboxylate (3 g, 8.28 mmol) in THF (30 mL) and water (30 mL) was added Na2CO3 (2.63 g, 24.83 mmol) . Then CBZ-Cl (1.772 mL, 12.42 mmol) was added at 0 ℃ the mixture was stirred at 0 ℃ to 20 ℃ for 18 h. The reaction mixture was poured to water (50 mL) , extracted with EtOAc (50 mL×2) . The organic layer was washed with brine (100 mL) , dried over Na2SO4, concentrated in vacuo to give crude1-benzyl 3-methyl 6- (2-( ( (benzyloxy) carbonyl) (methyl) amino) -2-methylpropyl) piperidine-1, 3-dicarboxylate. MS: 497.3 (M+1) (Acq Method D Rt: 1.383 min) .
Step 9: methyl 6- (2-methyl-2- (methylamino) propyl) piperidine-3-carboxylate
To a mixture of 1-benzyl 3-methyl 6- (2- ( ( (benzyloxy) carbonyl) (methyl) amino) -2-methylpropyl) piperidine-1, 3-dicarboxylate (3.5 g, 7.05 mmol) in MeOH (50 mL) was added Pd-C (0.150 g, 1.410 mmol) . Then the mixture was stirred under H2 balloon at 20 ℃ for 2 hrs, then the mixture was filtered and the filtrate was concentrated in vacuo to give methyl 6- (2-methyl-2-(methylamino) propyl) piperidine-3-carboxylate as yellow liquid. MS: 229.2 (M+1) (Acq Method D Rt: 0.122 min) .
Step 10: methyl 2, 3, 3-trimethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7- carboxylate (trans)
To a solution of methyl 6- (2-methyl-2- (methylamino) propyl) piperidine-3-carboxylate (1.6 g, 7.01 mmol) in THF (50 mL) was added CDI (4.54 g, 28.0 mmol) . Then the mixture was stirred at 20 ℃ for 18 h. The reaction mixture was poured into water (20 mL) , extracted with EtOAc (20 mL×2) . The organic layer was washed with brine (50 mL) , dried over Na2SO4, concentrated in vacuo. The residue was purified by prep-HPLC (ACN/water with 0.1% TFA modifier) to give methyl 2, 3, 3-trimethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7-carboxylate (cis) . 1H NMR (400MHz, CD3OD) δ 4.83 (d, J = 12.9 Hz, 1H) , 3.67 (s, 3H) , 3.27 -3.20 (m, 1H) , 2.84 (s, 3H) , 2.79 -2.71 (m, 2H) , 2.19 (d, J = 12.5 Hz, 1H) , 1.87 (dd, J = 4.1, 13.5 Hz, 1H) , 1.68 -1.60 (m, 3H) , 1.47 -1.38 (m, 1H) , 1.28 (s, 3H) , 1.20 (s, 3H) ppm and methyl 2, 3, 3-trimethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7-carboxylate (trans) . 1H NMR (400MHz, CD3OD) δ 4.63 (d, J = 11.0 Hz, 1H) , 3.68 (s, 3H) , 3.26 -3.16 (m, 1H) , 2.86 (s, 3H) , 2.63 -2.52 (m, 1H) , 2.46 -2.36 (m, 1H) , 2.09 (d, J = 13.3 Hz, 1H) , 1.93 (dd, J = 4.3, 13.3 Hz, 1H) , 1.85 (dd, J = 3.1,
13.3 Hz, 1H) , 1.72 -1.61 (m, 1H) , 1.58 -1.47 (m, 1H) , 1.30 (s, 3H) , 1.28 -1.23 (m, 1H) , 1.21 (s, 3H) ppm.
Step 11: 2, 3, 3-trimethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7-carboxylic acid (trans)
To a solution of methyl 2, 3, 3-trimethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7-carboxylate (trans) (600 mg, 2.359 mmol) in THF (4 mL) and water (4.00 mL) was added lithium hydroxide, H2O (198 mg, 4.72 mmol) . Then the mixture was stirred at 20 ℃ for 2 h. The reaction mixture was poured into water (20 mL) , pH was adjusted to 2 with HCl (1M) . The mixture was extracted with DCM (20 mL×2) . The organic layer was washed with brine (40 mL) , dried over Na2SO4, concentrated in vacuo to give 2, 3, 3-trimethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7-carboxylic acid (trans) . 1H NMR (400MHz, CD3OD) δ 4.64 (d, J = 12.9 Hz, 1H) , 3.22 (t, J = 11.3 Hz, 1H) , 2.86 (s, 3H) , 2.57 (t, J = 12.3 Hz, 1H) , 2.35 (t, J = 11.9 Hz, 1H) , 2.15 -2.07 (m, 1H) , 1.97 -1.83 (m, 2H) , 1.72 -1.62 (m, 1H) , 1.58 -1.47 (m, 1H) , 1.31 (s, 3H) , 1.24 (br. s., 1H) , 1.21 (s, 3H) ppm.
Step 12: N- ( (3-chloropyrazin-2-yl) methyl) -2, 3, 3-trimethyl-1-oxooctahydro-1H-pyrido [1, 2-
c] pyrimidine-7-carboxamide (trans)
To a solution of 2, 3, 3-trimethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7-carboxylic acid (560 mg, 2.330 mmol) in DMF (25 mL) was added HATU (1329 mg, 3.50 mmol) , the mixture was stirred at 20 ℃ for 0.5 h. Then (3-chloropyrazin-2-yl) methanamine, HCl (503 mg, 2.80 mmol) was added, followed by Et3N (1.299 mL, 9.32 mmol) . The mixture was stirred at 20 ℃ for 18 h. TLC showed the material was consumed completely, then the reaction mixture was poured into water (20 mL) , extracted with DCM (20 mL×2) . The organic layer was washed with brine (40 mL) , dried over Na2SO4, concentrated in vacuo to give N- ( (3-chloropyrazin-2-yl) methyl) -2, 3, 3-trimethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7-carboxamide (trans) . 1H NMR (400MHz, CD3OD) δ 8.52 (d, J = 2.3 Hz, 1H) , 8.33 (s, 1H) , 4.62 (s, 2H) , 4.56 (d, J = 12.9 Hz, 1H) , 3.26 -3.21 (m, 1H) , 2.86 (s, 3H) , 2.65 (t, J = 12.3 Hz, 1H) , 2.43 (t, J = 11.7 Hz, 1H) , 2.03 -1.82 (m, 4H) , 1.67 (br. s., 1H) , 1.31 (s, 3H) , 1.29 -1.24 (m, 1H) , 1.21 (s, 3H) ppm.
Step 13: 7- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -2, 3, 3-trimethyloctahydro-1H-pyrido [1, 2-
c] pyrimidin-1-one (trans)
To a solution of N- ( (3-chloropyrazin-2-yl) methyl) -2, 3, 3-trimethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidine-7-carboxamide (800 mg, 2.187 mmol) in acetonitrile (15 mL) was added PCl5 (1366 mg, 6.56 mmol) at 0 ℃. Then the mixture was stirred at 20 ℃ for 5 h. The reaction mixture was poured into saturated of NaHCO3 (50 mL) , extracted with DCM (50 mL×2) . The organic layer was washed with brine (100 mL) , dried over Na2SO4, concentrated in vacuo to give 7- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -2, 3, 3-trimethyloctahydro-1H-pyrido [1, 2-
c] pyrimidin-1-one (trans) . 1H NMR (400MHz, CD3OD) δ 8.25 (d, J = 4.7 Hz, 1H) , 7.84 (s, 1H) , 7.39 (d, J = 5.1 Hz, 1H) , 4.61 (d, J = 11.7 Hz, 1H) , 3.46 -3.37 (m, 1H) , 3.33 (br. s., 1H) , 2.88 (s, 3H) , 2.86 -2.80 (m, 1H) , 2.13 (d, J = 12.1 Hz, 1H) , 2.05 -1.90 (m, 3H) , 1.75 (t, J = 12.5 Hz, 1H) , 1.49 -1.40 (m, 1H) , 1.34 (s, 3H) , 1.25 (s, 3H) ppm.
Step 14: 7- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -2, 3, 3-trimethyloctahydro-1H-
pyrido [1, 2-c] pyrimidin-1-one (trans)
To a solution of 7- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -2, 3, 3-trimethyloctahydro-1H-pyrido [1, 2-c] pyrimidin-1-one (600 mg, 1.725 mmol) in DMF (10 mL) was added NBS (461 mg, 2.59 mmol) at 0 ℃. Then the mixture was stirred at 20 ℃ for 5 h. The reaction mixture was poured into saturated of NaHCO3 (50 mL) , extracted with DCM (50 mL×2) . The organic layer was washed with brine (100 mL) , dried over Na2SO4, concentrated in vacuo to give 7- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -2, 3, 3-trimethyloctahydro-1H-pyrido [1, 2-c] pyrimidin-1-one (trans) as white solid. MS: 428.1, 430.1 (M+1) (Acq Method D Rt: 1.212 min) .
Step 15: 7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -2, 3, 3-trimethyloctahydro-1H-
pyrido [1, 2-c] pyrimidin-1-one (trans)
The mixture of 7- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -2, 3, 3-trimethyloctahydro-1H-pyrido [1, 2-c] pyrimidin-1-one (600 mg, 1.406 mmol) in 2-propanol (15 mL) and ammonium hydroxide (15 mL, 108 mmol) was stirred at 100 ℃ for 18 h in a 100 mL of sealed tube. The reaction mixture was poured into water (50 mL) , extracted with DCM (50 mL×2) . The organic layer was washed with brine (100 mL) , dried over Na2SO4, concentrated in vacuo to give 7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -2, 3, 3-trimethyloctahydro-1H-pyrido [1, 2-c] pyrimidin-1-one (trans) as yellow solid. MS: 407.1, 409.1 (M+1) (Acq Method D Rt: 0.911 min) .
Intermediate 32
7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -3, 3-dimethylhexahydropyrido [1, 2-
c] [1, 3] oxazin-1 (3H) -one

Step 1: 1- (5-bromopyridin-2-yl) -2-methylpropan-2-ol
To a solution of 5-bromo-2-methylpyridine (20 g, 116 mmol) in THF (250 mL) was added LDA
(69.8 mL, 140 mmol) dropwise at -78 ℃ under N2 protection. The solution was stirred at -78 ℃ for 30 minutes. To the solution was added propan-2-one (20 g, 344 mmol) and the mixture was stirred at 13℃ for 16 hours. To the solution was added saturated NH4Cl (100 mL) and extracted with EtOAc (200 mL) . The combined organic was washed with 100 mL water and brine (50 mL) , dried over Na2SO4, concentrated in vacuo and purified by chromatography on silica gel (40 g) (pet. Ether : EtOAc = 5 : 95) to give the compound 1- (5-bromopyridin-2-yl) -2-methylpropan-2-ol.1H NMR (400MHz, CDCl3) δ 8.59 (d, J = 1.6 Hz, 1H) , 7.77 (dd, J = 2.3, 8.2 Hz, 1H) , 7.06 (d, J = 8.2 Hz, 1H) , 5.00 (s, 1H) , 2.88 (s, 2H) , 1.22 (s, 6H) ppm. MS-ESI (m/z) : 230.1, 232.1 (M+1) +(Acq Method D 0.309 min)
Step 2: Methyl 6- (2-hydroxy-2-methylpropyl) nicotinate
A solution of 1- (5-bromopyridin-2-yl) -2-methylpropan-2-ol (1 g, 4.35 mmol) , TEA (0.606 mL, 4.35 mmol) , PdOAc2 (0.976 g, 4.35 mmol) and DPPF (2.409 g, 4.35 mmol) in MeOH (20 mL) and DMF (20 mL) was stirred at 80 ℃ for 72 hours under CO (55 psi) . The solution was concentrated in vacuo and dissolved in EtOAc (1000 mL) . The mixture was washed with water (1000 mL) and brine (500 mL) . The combined organic was dried over Na2SO4 and purified by chromatography over silica gel (120 g) (Pet. Ehter: EtOAc = 20 : 80 to 30 : 70) to give the compound methyl 6- (2-hydroxy-2-methylpropyl) nicotinate. 1H NMR (400MHz, CDCl3) δ 9.13 (s, 1H) , 8.25 (dd, J = 1.8, 8.0 Hz, 1H) , 7.24 (d, J = 7.8 Hz, 1H) , 5.33 (s, 1H) , 3.96 (s, 3H) , 3.00 (s, 2H) , 1.23 (s, 6H) ppm. MS-ESI (m/z) : 210 (M+1) + (Acq Method DRt: 0.834 min)
Step 3: methyl 6- (2-hydroxy-2-methylpropyl) piperidine-3-carboxylate
To a solution of methyl 6- (2-hydroxy-2-methylpropyl) nicotinate (10 g, 47.8 mmol) in acetic acid (100 mL) was added sodium cyanoborohydride (10 g, 159 mmol) and the mixture was stirred at 13 ℃ for 16 hours. The solution was then poured into saturated Na2CO3 (20 mL) and extracted with EtOAc (50 mL) . The combined organic layers were dried over Na2SO4 and concentrated in vacuo to give the compound methyl 6- (2-hydroxy-2-methylpropyl) piperidine-3-carboxylate (racemic) without purification. 1H NMR (400MHz, CDCl3) δ 3.80 -3.69 (m, 3H) , 3.45 -3.30 (m, 1H) , 3.08 -2.73 (m, 2H) , 2.60 (d, J = 3.8 Hz, 1H) , 2.43 (tt, J = 4.0, 11.5 Hz, 1H) , 1.77 -1.69 (m, 1H) , 1.59 -1.50 (m, 1H) , 1.33 -1.20 (m, 6H) ppm.
Step 4: methyl 3, 3-dimethyl-1-oxooctahydropyrido [1, 2-c] [1, 3] oxazine-7-carboxylate
To a solution of methyl 6- (2-hydroxy-2-methylpropyl) piperidine-3-carboxylate (racemic) (7.3 g, 33.9 mmol) in MeCN (150 mL) was added di (1H-imidazol-1-yl) methanone (16.49 g, 102 mmol) and mixture was stirred at 15 ℃ for 72 hours. The mixture was purified by chromatography on silica (80 g) (DCM : MeOH = 5 : 95) to give compound methyl 3, 3-dimethyl-1-oxooctahydropyrido [1, 2-c] [1, 3] oxazine-7-carboxylate (racemic, trans) and compound methyl
3, 3-dimethyl-1-oxooctahydropyrido [1, 2-c] [1, 3] oxazine-7-carboxylate (racemic, maybe cis) . NOE showed compound methyl 3, 3-dimethyl-1-oxooctahydropyrido [1, 2-c] [1, 3] oxazine-7-carboxylate (racemic) as trans conformation. 1H NMR (400MHz, CDCl3) δ 4.76 (dd, J = 1.8, 13.1 Hz, 1H) , 3.69 (s, 3H) , 3.36 -3.20 (m, 1H) , 2.75 (t, J = 12.5 Hz, 1H) , 2.50 (tt, J = 3.6, 12.0 Hz, 1H) , 2.15 (d, J = 13.3 Hz, 1H) , 1.97 (dd, J = 5.5, 14.1 Hz, 1H) , 1.89 (dd, J = 3.1, 13.3 Hz, 1H) , 1.75 -1.65 (m, 1H) , 1.58 (dq, J = 3.5, 13.0 Hz, 1H) , 1.37 (d, J = 16.0 Hz, 6H) , 1.32 -1.20 (m, 1H) ppm.
Step 5: 3, 3-dimethyl-1-oxooctahydropyrido [1, 2-c] [1, 3] oxazine-7-carboxylic acid
A solution of lithium hydroxide, H2O (104 mg, 2.487 mmol) and methyl 3, 3-dimethyl-1-oxooctahydropyrido [1, 2-c] [1, 3] oxazine-6-carboxylate (TRANS) (500 mg, 2.072 mmol) in water (15 mL) and THF (15 mL) was stirred at 15 ℃ for 16 hours. The solution was concentrated in vacuo and acidified with 1 N HCl to pH = 1, then extracted with EtOAc (20 mLx3) . The combined organic layers were dried over Na2SO4 and concentrated in vacuo to give the compound 3, 3-dimethyl-1-oxooctahydropyrido [1, 2-c] [1, 3] oxazine-6-carboxylic acid (trans) without purification. 1H NMR (400MHz, CDCl3) δ 8.88 -7.53 (m, 1H) , 4.76 (dd, J = 1.6, 13.3 Hz, 1H) , 3.36 -3.21 (m, 1H) , 2.76 (t, J = 12.7 Hz, 1H) , 2.51 (ddd, J = 3.7, 8.4, 11.7 Hz, 1H) , 2.20 (d, J = 12.9 Hz, 1H) , 1.97 (dd, J = 5.5, 13.7 Hz, 1H) , 1.90 (dd, J = 2.7, 13.3 Hz, 1H) , 1.76 -1.66 (m, 1H) , 1.59 (dq, J = 3.5, 13.0 Hz, 1H) , 1.38 (s, 3H) , 1.34 (s, 3H) , 1.32 -1.17 (m, 1H) ppm.
Step 6: N- ( (3-chloropyrazin-2-yl) methyl) -3, 3-dimethyl-1-oxooctahydropyrido [1, 2-
c] [1, 3] oxazine-7-carboxamide
To a solution of DIEA (1.084 mL, 6.20 mmol) , 3, 3-dimethyl-1-oxooctahydropyrido [1, 2-c] [1, 3] oxazine-6-carboxylic acid (trans) (470 mg, 2.068 mmol) and (3-chloropyrazin-2-yl) methanamine, 2HCl (537 mg, 2.482 mmol) in MeCN (30 mL) was added HATU (1180 mg, 3.10 mmol) and the mixture was stirred at 15 ℃ for 16 hours. The mixture was then diluted with water (30 mL) and extracted with DCM (80 mLx3) . The combined organic layers were dried over Na2SO4 and purified by chromatography on silica gel (12 g) (Pet. Ether : EtOAc = 30 : 70 to DCM : MeOH = 80 : 20) to give the compound N- ( (3-chloropyrazin-2-yl) methyl) -3, 3-dimethyl-1-oxooctahydropyrido [1, 2-c] [1, 3] oxazine-6-carboxamide (trans) . 1H NMR (400MHz, CDCl3) δ 8.46 (d, J = 2.0 Hz, 1H) , 8.33 (br. s., 1H) , 7.00 (br. s., 1H) , 4.82 -4.56 (m, 3H) , 3.49 (s, 1H) , 3.35 (d, J = 2.7 Hz, 1H) , 2.94 -2.85 (m, 1H) , 2.53 -2.41 (m, 1H) , 2.10 -1.66 (m, 6H) , 1.42 -1.32 (m, 6H) , 1.30 -1.24 (m, 1H) MS-ESI (m/z) : 353 (M+1) + (Acq Method DRt: 0.908 min)
Step 7: 7- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -3, 3-dimethylhexahydropyrido [1, 2-c] [1, 3] oxazin-
1 (3H) -one
To a solution of N- ( (3-chloropyrazin-2-yl) methyl) -3, 3-dimethyl-1-oxooctahydropyrido [1, 2-c] [1, 3] oxazine-7-carboxamide (450 mg, 1.275 mmol) in MeCN (20 mL) was added PCl5 (1 g, 4.80 mmol) and the mixture was stirred at 18 ℃ for 16 hours. The red solution was poured into Na2CO3 (30 g) in 30 mL water slowly with stirring. The mixture was extracted with EtOAc (100 mL x 2) . The combined organic layers were dried over Na2CO3 (10 g) and Na2SO4 (10 g) then filtered. The filtrate was concentrated in vacuo to give the compound 7- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -3, 3-dimethylhexahydropyrido [1, 2-c] [1, 3] oxazin-1 (3H) -one. 1H NMR (400MHz CDCl3) δ 7.81 (s, 1H) , 7.76 (d, J = 5.1 Hz, 1H) , 7.39 (d, J = 4.7 Hz, 1H) , 4.73 (dd, J = 2.2, 13.1 Hz, 1H) , 3.54 -3.42 (m, 1H) , 3.23 -3.08 (m, 1H) , 3.03 -2.92 (m, 1H) , 2.27 -2.13 (m, 2H) , 2.08 (dd, J = 5.7, 13.9 Hz, 2H) , 1.83 -1.72 (m, 1H) , 1.41 (d, J = 16.4 Hz, 6H) , 1.32 -1.20 (m, 1H) ppm. MS-ESI (m/z) : 335 (M+1) + (Acq Method DRt: 0.986 min)
Step 8: 7- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -3, 3-dimethylhexahydropyrido [1, 2-
c] [1, 3] oxazin-1 (3H) -one
To a solution of 7- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -3, 3-dimethylhexahydropyrido [1, 2-c] [1, 3] oxazin-1 (3H) -one (trans, racemic) (400 mg, 1.195 mmol) in MeCN (25 mL) was added NBS (400 mg, 2.247 mmol) and the mixture was stirred at 18 ℃ for 2 hours. The solution was poured into saturated Na2SO3 (20 mL) and extracted with EtOAc (50 mL) . The combined organic layers were washed with 20 mL saturated aq. Na2CO3, dried over Na2SO4 (20 g) and Na2CO3 (20 g) . The solution was filtered and the filtrate was concentrated in vacuo to give the compound 7- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -3, 3-dimethylhexahydropyrido [1, 2-c] [1, 3] oxazin-1 (3H) -one (trans, racemic) without purification. 1H NMR (400MHz, CDCl3) δ 8.46 (br. s., 1H) , 7.75 (d, J = 5.1 Hz, 1H) , 7.36 (d, J = 4.7 Hz, 1H) , 4.69 (dd, J = 2.7, 13.3 Hz, 1H) , 3.52 -3.40 (m, 1H) , 3.20 -3.07 (m, 1H) , 2.99 -2.91 (m, 1H) , 2.80 (s, 2H) , 2.76 (s, 4H) , 2.21 -2.12 (m, 2H) , 2.07 (dd, J = 6.1, 13.9 Hz, 2H) , 1.79 -1.72 (m, 1H) , 1.40 (d, J = 18.0 Hz, 7H) ppm. MS-ESI (m/z) : 413, 415 (M+1) + (Acq Method DRt: 1.133 min)
Step 9: 7- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -3, 3-
dimethylhexahydropyrido [1, 2-c] [1, 3] oxazin-1 (3H) -one
A mixture of K2CO3 (200 mg, 1.450 mmol) , (2, 4-dimethoxyphenyl) methanamine (97 mg, 0.580 mmol) and 7- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -3, 3-dimethylhexahydropyrido [1, 2-c] [1, 3] oxazin-1 (3H) -one (trans) (200 mg, 0.483 mmol) in DMF (10 mL) was stirred at 80 ℃ for 2 hours. The mixture was concentrate in vacuo and purified by chromatography on silica gel (12 g) (DCM : MeOH = 95 : 5) to give the compound 7- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -3, 3-dimethylhexahydropyrido [1, 2-c] [1, 3] oxazin-1 (3H) -one (trans) . 1H NMR (400MHz, CDCl3) δ 8.01 (s, 1H) , 7.13 (q, J = 5.1 Hz,
2H) , 6.73 (br. s.,1H) , 6.49 (s, 1H) , 6.46 -6.38 (m, 1H) , 4.67 (d, J = 5.5 Hz, 1H) , 3.88 (s, 2H) , 3.83 -3.77 (m, 4H) , 3.39 (br. s., 1H) , 2.95 (s, 4H) , 2.88 (s, 3H) , 3.12 -2.85 (m, 1H) , 2.14 -1.96 (m, 4H) , 1.80 -1.71 (m, 1H) , 1.47 -1.33 (m, 6H) , 1.26 (t, J = 7.0 Hz, 1H) ppm. MS-ESI (m/z) : 544, 546 (M+1) + (Acq Method DRt: 0.972 min)
Step 10: 7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -3, 3-dimethylhexahydropyrido [1, 2-
c] [1, 3] oxazin-1 (3H) -one
A solution of 7- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -3, 3-dimethylhexahydropyrido [1, 2-c] [1, 3] oxazin-1 (3H) -one (trans, racemic) (450 mg, 0.827 mmol) in TFA (5 mL, 64.9 mmol) was stirred at 80 ℃ for 2 hours under N2 protection. The mixture was concentrated in vacuo and to the residue was added sat. aq. Na2CO3 (10 mL) and extracted with DCM (30 mL) . The organic layer was dried over Na2SO4 and purified by chromatography on silica gel (4 g) to give the compound 7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -3, 3-dimethylhexahydropyrido [1, 2-c] [1, 3] oxazin-1 (3H) -one (trans, racemic) . 1H NMR (400MHz, CDCl3) δ 7.27 (br. s., 1H) , 7.04 (d, J = 4.7 Hz, 1H) , 5.73 (br. s., 2H) , 4.71 (d, J = 11.7 Hz, 1H) , 3.91 -3.69 (m, 2H) , 3.42 (d, J = 3.1 Hz, 1H) , 3.11 -3.01 (m, 1H) , 2.98 -2.92 (m, 1H) , 2.17 -2.08 (m, 2H) , 2.07 -1.98 (m, 3H) , 1.88 -1.68 (m, 4H) , 1.39 (d, J = 19.6 Hz, 7H) ppm. MS-ESI (m/z) : 394, 396 (M+1) + (Acq Method DRt: 0.757 min)
Intermediate 33

2-chloro-5-ethoxy-N- (1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl) -4- (4, 4, 5, 5-tetramethyl-
1, 3, 2-dioxaborolan-2-yl) benzamide
Step 1: 2-chloro-5-hydroxy-4-iodobenzoic acid
A solution of odine (0.75g, 2.95mmol) and potassium iodide (0.55g, 3.31mmol) in water (5ml) was added dropwise to a solution of 2-Chloro-5-hydroxybenzoic acid (0.5g, 2.90mmol) in ammonium hydroxide (conc, 4ml, 48mmol) and then stirred 2 hours at room temperature. The reaction mixture was diluted with water (5ml) and acidified to pH 3 with conc. HCl. The mixture was then extracted with EtOAc (50ml) and H2O (20ml) . Organic layer was separated , dried over Na2SO4, then filtered and the filtrate was concentrated to yield 2-Chloro-5-hydroxy-4-iodobenzoic acid as a solid. LCMS : C7H4ClIO3 MH Found [M+H] +: 298.84 RT = 1.26 min, Method B
Step 2: Methyl 2-chloro-5-hydroxy-4-iodobenzoate
Thionyl chloride (0.5ml, 6.85mmol) was added dropwise to a solution of 2-chloro-5-hydroxy-4-iodobenzoic acid (0.6g, 2.01mmol) in MeOH (10ml) at 0℃ , then stirred overnight at room temperature. The solvent was evaporated and residue was extracted with EtOAc (100ml) . The organic layer was washed with saturated NaHCO3 (40ml) , dried over Na2SO4 then filtered and the filtrate was concentrated to afford the title compound Methyl -2-Chloro-5-hydroxy-4-iodobenzoate.
Step 3: methyl 2-chloro-5-ethoxy-4-iodobenzoate
Iodoethane (0.3ml, 3.75mmol) was added to a suspension of Methyl -2-Chloro-5-hydroxy-4-iodobenzoate (0.9g, 2.87mmol) and potassium carbonate (0.4g, 2.89mmol) in DMF (4ml) , and then stirred for 2 hours at room temperature. The mixture was extracted with EtOAc (100ml) and H2O (40ml) . The organic layer was dried over Na2SO4, then filtered and the filtrate was concentrated. The crude was purified by chromatography (40g silica gel, 20% ethylacetate in Hexanes) yielding title compound methyl-2-chloro-5-ethoxy-4-iodobenzoate. LCMS : C10H10ClIO3, Found [M+H] + 340.89, RT = 1.56 min, method B..
Step 4: 2-Chloro-5-ethoxy-4-iodobenzoic acid
Lithium hydroxide monohydrate (0.2g , 4.77mmol) was added to a solution of methyl-2-chloro-5-ethoxy-4-iodobenzoate (0.3g , 0.846mmol) in Methanol : THF: H2O (7ml, 3: 3: 1) then stirred at 40℃ overnight. The reaction mixture was cooled to room temperature , the solvent was evaporated , and residue was diluted with H2O (20ml) , and acidified with conc HCl to pH 3. The precipitated white solid was filtered and washed with H2O (10ml) . Solid was dissolved in THF (20ml) and solvent was evaporated yielding title compound 2-chloro-5-ethoxy-4-iodobenzoic acid.
Step 5: 2-chloro-5-ethoxy-4-iodobenzoyl chloride
Oxalyl chloride (0.3ml, 3.16mmol) was added to solution of 2-Chloro-5-Ethoxy-4-Iodobenzoic acid (0.3g , 1.101mmol) and DMF (0.05ml , 3.16mmol) in CH2Cl2 (10ml) then stirred at room temperature for 1 hour. Solvent was evaporated, yielding the title compound 2-Chloro-5-Ethoxy-4-Iodobenzoyl chloride.
Step 6: 2-Chloro-5-ethoxy-4-iodo-N- (1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl) benzamide DMAP (3mg , 0.025mmol) and N-Ethyl-N-isopropylpropan-2-amine (0.2ml, 1.148mmol) were added to mixture of 2-Chloro-5-Ethoxy-4-Iodobenzoyl chloride (0.27g, 0.783mmol) and 1-Methyl-5-trifluoromethyl-1H-Pyrazol-3-amine (150mg, 0.908mmol) in THF (3ml) . The reaction mixture was stirred at room temperature for 2 hours. The reaction was quencehed with EtOAc (50ml) and H2O (20ml) , then organic layer was separated, dried over Na2SO4, then filtered, and concentrated. The residue was purified by chromatography (24g silica gel, 15% ethylacetate in
Hexanes) yielding title compound 2-Chloro-5-Ethoxy-4-Iodo-N- (1-Methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl) benzamide.
LCMS : C14H12ClF3IN3O2, Found [M+H] + 473.96, RT = 1.53 min, method B.
Step 7: 2-Chloro-5-ethoxy-N- (1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl) -4- (4, 4, 5, 5-
tetramethyl-1, 3, 2-dioxaborolan-2-yl) benzamide
DMSO (1ml) was added to a mixture of Bis (Pinacolato) Diboron (160mg, 0.630mmol) , potassium acetate (80mg , 0.815mmol) ; [1, 1’ -
Bis (Diphenylphosphino) ferrocene] Dichloropalladium (11) (20mg, 0.027mmol) and 2-Chloro-5-Ethoxy-4-Iodo-N- (1-Methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl) benzamide (200mg, 0.422mmol) . The reaction mixture wasdegassed and then stirred at 80℃ for 4 hours. After cooled to room temperature, the mixture was then extracted with EtOAc (50ml) and H2O (20ml) . The organic layer was separated and dried over Na2SO4, then filtered and concentrated. The residue was purified by chromatography (24g silica gel, 45% ethylacetate in Hexanes) yielding title compound 2-chloro-5-ethoxy-N- (1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl) -4- (4, 4, 5-trimethyl-1, 3, 2-dioxaborolan-2-yl) benzamide ) . (160mg, 80%)
LCMS : C14H12ClF3N3O2, Found [M+H] + 474.12, RT = 1.55 min, method B.
Intermediate 34
2-chloro-5-ethoxy-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -N- (4-
(trifluoromethyl) pyridin-2-yl) benzamide

Following the procedure of intermediate 33; but in Step 6 substituting an equivalent quantity of 4- (trifluoromethyl) pyridin-2-amine for 1-Methyl-5-trifluoromethyl-1H-Pyrazol-3-amine , then following the procedure of intermediate 33 step 7 the title compound 2-chloro-5-ethoxy-4-(4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -N- (4- (trifluoromethyl) pyridin-2-yl) benzamide was obtained.
LCMS : C21H23BClF3N2O4, Found [M+H] + 470.99, 472.97; RT = 1.57 min, method B.
Intermediate 35
2-Chloro-5-ethoxy-N- (pyridin-2-yl) -4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) benzamide

Following the procedure of intermediate 33; in Step 6 substituting an equivalent quantity of 2-amino pyridine for 1-Methyl-5-trifluoromethyl-1H-Pyrazol-3-amine, then following procedure of intermediate 33 step 7 , the title compound 2-chloro-5-ethoxy-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -N- (4- (trifluoromethyl) pyridin-2-yl) benzamide was obtained.
LCMS : C20H24BClN2O4, Found [M+H] + 402.88, 404.87; RT = 3.42 min, method A.
Intermediate 36
5-ethoxy-2, 3-difluoro-N- (1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl) -4- (4, 4, 5, 5-tetramethyl-
1, 3, 2-dioxaborolan-2-yl) benzamide

Step 1: 2, 3-Difluoro-5-hydroxy-4-iodobenzoic acid
A solution of Iodine (0.88g, 3.47mmol) and potassium iodide (0.65g, 3.92mmol) in water (6ml) was added dropwise to a solution of 2, 3-difluoro-5-hydroxybenzoic acid (0.6g, 3.45 mmol) in ammonium hydroxide (conc, 5ml, 60 mmol) then stirred 2 hours at room temperature. The reaction mixture was diluted with water (5ml) and acidified to pH 3 with conc. HCl. The precipitated yellow solid was filtered , washed with water (10ml) . The solid was dissolved in 10% MeOH/CH2Cl2 and solvent evaporated yielding title compound 2, 3-difluoro-5-hydroxy-4-iodobenzoic acid.
LCMS : C7H3F2IO3 MH Found [M+H] +: 301.02 RT = 3.01 min, method A.
Step 2: Ethyl 5-ethoxy-2, 3-difluoro-4-iodobenzoate
A solution of Iodoethane (2.3g, 14.75mmol) in DMF (3ml) was added to a suspension of 2, 3-Difluoro-5-hydroxy-4-iodobenzoic acid (1g, 3.33 mmol) and potassium carbonate (2g, 14.47mmol) in DMF (8ml) . The mixture was then stirred overnight. The reaction mixture was concentrated and the residue was purified by chromatography (40g silica gel, 15% ethylacetate in Hexanes) yielding title compound ethyl 5-ethoxy-2, 3-difluoro-4-iodobenzoate.
LCMS : C11H11F2IO3, Found [M+H] + 356.89, RT = 1.56 min, method B.
Step 3: 5-Ethoxy-2, 3-difluoro-4-iodobenzoic acid
Lithium Hydroxide monohydrate (8ml, 8 mmol) was added to a solution of 5-Ethoxy-2, 3 difluoro-4-Iodobenzoate (1.2g , 3.37 mmol) in THF: MeOH (20ml, v/v 1: 1) then stirred at 40℃ overnight. The reaction mixture was cooled to room temperature , the solvent was evaporated , and residue was diluted with H2O (20ml) , and acidified with conc HCl to pH 3. The mixture was extracted with EtOAc (100ml) , washed with H2O (30ml) . The organic layer was seperated and dried over Na2SO4, then filtered and concentrated yielding title compound 5-Ethoxy-2, 3 difluoro-4-Iodobenzoic acid.
LCMS : C9H7F2IO3, Found [M+H] + 329.06, RT = 1.37 min, method B.
Step 4: 5-Ethoxy-2, 3-difluoro-4-iodo-N- (1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-
yl) benzamide
1- (3-dimethylaminopropyl) -3-ethylcarbodiimide hydrochloride (300 mg, 1.565 mmol) and HOBT (200 mg, 1.306 mmol) were added to solution of 5-ethoxy-2, 3-difluoro-4-iodobenzoic acid (400 mg, 1.219 mmol) and 1-methyl-5- (trifluoromethyl) -1H-pyrazole-3-amine (220 mg, 1.332 mmol) in DMF (5 ml) and then stirred 2 hours at room temp. The reaction mixture was extracted with EtOAc (60ml) and H2O (30ml) . The organic layer was separated and washed with brine (20ml) , dried organics over Na2SO4, filtered and concentrated. The residue was purified by chromatography (40g silica gel) eluting with 15 % EtOAc-Hexanes yielding title compound 5-ethoxy-2, 3-difluoro-4-iodo-N- (1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl) benzamide.
LCMS : C14H11F5IN3O2, Found [M+H] + 475.93, RT = 1.51 min method B.
Step 5: 5-Ethoxy-2, 3-difluoro-N- (1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl) -4- (4, 4, 5, 5-
tetramethyl-1, 3, 2-dioxaborolan-2-yl) benzamide
DMSO (1 ml) was added to mixture of 5-ethoxy-2, 3-difluoro-4-iodo-N- (1-methyl-5-(trifluoromethyl) -1H-pyrazol-3-yl) benzamide (110 mg, 0.232 mmol) ; bis (pinacolato) diboron (80 mg, 0.315 mmol) ; [1, 1'-Pd (dppf) Cl2 (20 mg, 0.027 mmol) and potassium acetate (40mg, 0.408 mmol) The mixture was degassed 3 times, then stirred at 80℃ for 4 hours. After cooled to room temperature, the mixture was. extracted with EtOAc (50ml) and H2O (20ml) , The organic layer was seperated and washed with H2O (20ml) , dried over Na2SO4, filtered and concentrated. The residue was purified by chromatography (24g silica gel) eluting with 1/2 EtOAc-Hexanes yielding 5-ethoxy-2, 3-difluoro-N- (1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl) -4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) benzamide.
LCMS : C20H23BF5N3O4, Found [M+H] + 476.23, RT = 1.59 min, method B.
Intermediate 37

5-ethoxy-2-fluoro-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -N- (4-
(trifluoromethyl) pyridin-2-yl) benzamide
Step 1: 5-ethoxy-2-fluoro-4-iodo-N- (4- (trifluoromethyl) pyridin-2-yl) benzamide
To a solution of 5-ethoxy-2-fluoro-4-iodobenzoic acid (5 g, 16.13 mmol) in anhydrous DCM (70 mL) was added oxalyl chloride (10.23 g, 81 mmol) at 0 ℃, then DMF (one drop) was added and the mixture was stirred at 20 ℃ for 1.5 hrs. The mixture was concentrated in vacuo and diluted with THF (80 mL) . 4- (trifluoromethyl) pyridin-2-amine (5.23 g, 32.3 mmol) was addedto the mixtureat 0 ℃. The reaction was stirred at 80 ℃ for 16 hrs. After cooling to room temperature, the mixture was concentrated to give the crude product, which was purified by column chromatography on silica gel eluted with (EtOAc: Pet ether = 1%~ 50%) to give 5-ethoxy-2-fluoro-4-iodo-N- (4- (trifluoromethyl) pyridin-2-yl) benzamide as a foam. 1H NMR (400MHz, CDCl3) δ = 9.23 (d, J = 15.3 Hz, 1H) , 8.65 (s, 1H) , 8.49 (d, J = 4.7 Hz, 1H) , 7.67 (d, J = 10.6 Hz, 1H) , 7.50 (d, J = 6.7 Hz, 1H) , 7.30 (d, J = 4.3 Hz, 1H) , 4.15 (q, J = 6.8 Hz, 2H) , 1.49 (t, J = 6.8 Hz, 3H) ppm.
Step 2: 5-ethoxy-2-fluoro-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -N- (4-
(trifluoromethyl) pyridin-2-yl) benzamide
To a mixture of 5-ethoxy-2-fluoro-4-iodo-N- (4- (trifluoromethyl) pyridin-2-yl) benzamide (3.15 g, 6.94 mmol) , KOAc (2.042 g, 20.81 mmol) , 4, 4, 4', 4', 5, 5, 5', 5'-octamethyl-2, 2'-bi (1, 3, 2-dioxaborolane) (2.64 g, 10.40 mmol) and PdCl2 (dppf) (0.508 g, 0.694 mmol) under N2 atmosphere was added DMSO (15 mL) and the mixture was immediately evacuated and backfilled with N2 three times. The reaction was then stirred at 60 ℃ overnight. The mixture was diluted with EtOAc (50 mL) and water (10 mL) . The organic layer was separated and the aqueous layer was extracted by EtOAc (3 ×20 mL) . The combined organic layers were washed with water (10 mL) , brine (10 mL) , dried over Na2SO4 and concentrated to afford the crude product, which was pruified by column chromatography on silica gel eluted with (EtOAc: Pet ether = 1% -50%) to give 5-ethoxy-2-fluoro-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -N- (4-(trifluoromethyl) pyridin-2-yl) benzamide as solid. 1H NMR (400MHz, CDCl3) δ = 9.37 (d, J =
16.4 Hz, 1H) , 8.76-8.68 (m, 1H) , 8.55-8.48 (m, 1H) , 7.58 (d, J = 5.9 Hz, 1H) , 7.44 (d, J = 11.7 Hz, 1H) , 7.36-7.29 (m, 1H) , 4.11 (q, J = 6.8 Hz, 2H) , 1.45 (t, J = 6.8 Hz, 3H) , 1.38 (s, 12H) ppm.
Intermediate 38

3-ethoxy-5-fluoro-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -N- (4-
(trifluoromethyl) pyridin-2-yl) benzamide
Step1: 3-Ethoxy-5-fluorobenzoic acid
Sodium (6.54 g, 285 mmol) was dissolved in EtOH (150 ml) and concentrated to give a white solid. The solid was dissolved in DMSO (100 ml) and then added 3, 5-difluorobenzoic acid (18g, 114 mmol) . The mixture was stirred at 80 ℃ for 12 hours. The mixture was cooled to room temperature and then the mixture was acided to ph=5 with 2M HCl , extracted with ethyl acetate (50 mL×3) . The combined organic layers were washed with brine (20 mL) , dried over anhydrous sodium sulfate, concentrated to afford the product 3-ethoxy-5-fluorobenzoic acid.
1H NMR (400MHz, CDCl3) δ = 7.44 -7.33 (m, 2H) , 6.83 (d, J=10.2 Hz, 1H) , 4.06 (q, J=7.0 Hz, 2H) , 1.43 (t, J=7.0 Hz, 3H) ppm.
Step 2: 4-borono-3-ethoxy-5-fluorobenzoic acid
To a solution of 3-ethoxy-5-fluorobenzoic acid (4 g, 21.72 mmol) in THF (30 ml) was added LDA (32.6 ml, 65.2 mmol) dropwise at -78 ℃ under N2 atmosphere. The resultant solution was stirred for 15 min followed by slow addition of triisopropyl borate (4.90 g, 26.1 mmol) . The mixture was stirred for 30 min and then hydrolyzed with 1M HCl. The mixture was extracted with EA (20 mL × 3) . The organic layer was washed with water (10 mL) , brine (10 mL) , dried over Na2SO4, concentrated to afford the crude product, then the crude product was pruified by column chromatography on silica gel eluted with (THF: PE = 10%-100%) to give 4-borono-3-ethoxy-5-fluorobenzoic acid.
1H NMR (400MHz, DMSO-d6) δ = 8.40 (s, 1H) , 7.21 (s, 1H) , 7.14 (d, J=7.8 Hz, 1H) , 4.03 (q, J=7.0 Hz, 2H) , 1.28 (t, J=6.8 Hz, 3H) ppm.
Step 3: 3-Ethoxy-5-fluoro-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) benzoic acid
To a solution of 4-borono-3-ethoxy-5-fluorobenzoic acid (2.45g, 10.75 mmol) in toluene (50 ml) was added 2, 3-dimethylbutane-2, 3-diol (1.397 g, 11.82 mmol) in one portion at room temperature under N2 atmosphere. The resultant solution was heated to 120 ℃ and stirred at this temperature for 14h. The mixture was cooled to room temperature and concentrated to afford the crude product, then the crude product was pruified by column chromatography on silica gel eluted with (THF: PE = 10%-50%) to give 3-ethoxy-5-fluoro-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) benzoic acid.
1H NMR (400MHz, CDCl3) = 7.34 (d, J=8.2 Hz, 1H) , 7.29 (s, 1H) , 4.07 (q, J=6.7 Hz, 2H) , 1.46 -1.31 (m, 15H) ppm.
Step 4: 3-Ethoxy-5-fluoro-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -N- (4-
(trifluoromethyl) pyridin-2-yl) benzamide
To a solution of 3-ethoxy-5-fluoro-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) benzoic acid (300 mg, 0.967 mmol) in anhydrous DCM (10 ml) was added oxalyl chloride (614 mg, 4.84 mmol) at 0 ℃, then DMF (one drop) was added and the mixture was stirred at 20 ℃ for 1.5 hrs. The mixture was concentrated in vacuo, which then was diluted with THF (6 ml) , to the mixture was added 4- (trifluoromethyl) pyridin-2-amine (314 mg, 1.935 mmol) at 0 ℃. The mixture was stirred at 80 ℃ for 16 hrs. After cooling to room temperature, the mixture was concentrated to give the crude product. The crude product was purified by column chromatography on silica gel eluted with (EA: PE = 1%~ 50%) to give 3-ethoxy-5-fluoro-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -N- (4- (trifluoromethyl) pyridin-2-yl) benzamide. The compound structure was confirmed by NOE.
1H NMR (400MHz, CDCl3) = 8.71 -8.66 (m, 2H) , 8.47 (d, J=5.0 Hz, 1H) , 7.31 (d, J=5.0 Hz, 1H) , 7.18 (s, 1H) , 7.12 (dd, J=0.9, 8.2 Hz, 1H) , 4.10 (q, J=6.9 Hz, 2H) , 1.46 -1.38 (m, 15H) ppm.
Intermediate 39

N- (4-cyclopropylpyridin-2-yl) -3-ethoxy-5-fluoro-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-
yl) benzamide
Step 1: 3-ethoxy-5-fluoro-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) benzoic acid
To a solution of 4-borono-3-ethoxy-5-fluorobenzoic acid (2.45g, 10.75 mmol) in toluene (50 mL) was added 2, 3-dimethylbutane-2, 3-diol (1.397 g, 11.82 mmol) in one portion at room temperature under N2 atmosphere. The resultant solution was heated to120 ℃ and stirred at this temperature for 14h. The mixture was cooled to room temperature and concentrated to afford the crude product, then the crude product was pruified by column chromatography on silica gel eluted with (THF: Pet. Ether = 10%-50%) to give 3-ethoxy-5-fluoro-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) benzoic acid.
1H NMR (400MHz, CDCl3) δ= 7.34 (d, J=8.2 Hz, 1H) , 7.29 (s, 1H) , 4.07 (q, J=6.7 Hz, 2H) , 1.46 -1.31 (m, 15H) ppm.
Step 2: N- (4-cyclopropylpyridin-2-yl) -5-ethoxy-2-fluoro-4-iodobenzamide
To a solution of 5-ethoxy-2-fluoro-4-iodobenzoic acid (1g, 3.23 mmol) in anhydrous CH2Cl2 (10 mL) was added oxalyl chloride (2.047 g, 16.13 mmol) at 0 ℃, then DMF (one drop) was added and the mixture was stirred at 20 ℃ for 1.5 hrs. The mixture was concentrated in vacuo, which then was diluted with THF (10 mL) , to the mixture was added 4-cyclopropylpyridin-2-amine (0.865 g, 6.45 mmol) at 0 ℃. The mixture was stirred at 80 ℃ for 16 hrs. After cooling to room temperature, the mixture was concentrated to give the crude product. The crude product was purified by column chromatography on silica gel eluted with (EtOAc: Pet ether = 1%~ 50%) to give N- (4-cyclopropylpyridin-2-yl) -5-ethoxy-2-fluoro-4-iodobenzamide. 1H NMR (400MHz, CDCl3) δ = 9.01 (d, J = 15.3 Hz, 1H) , 8.14 (d, J = 5.1 Hz, 1H) , 8.07 (s, 1H) , 7.65 (d, J = 10.6 Hz, 1H) , 7.50 (d, J = 6.7 Hz, 1H) , 6.78 (d, J = 4.3 Hz, 1H) , 4.15 (q, J = 7.0 Hz, 2H) , 1.99-1.87 (m, 1H) , 1.50 (t, J = 6.8 Hz, 3H) , 1.11 (q, J = 6.7 Hz, 2H) , 0.92-0.83 (m, 2H) ppm.
Intermediate 40

N- (4-cyclopropylpyridin-2-yl) -5-ethoxy-2-fluoro-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-
yl) benzamide
Step 1: N- (4-cyclopropylpyridin-2-yl) -5-ethoxy-2-fluoro-4-iodobenzamide
To a solution of 5-ethoxy-2-fluoro-4-iodobenzoic acid (2g, 6.45 mmol) in anhydrous CH2Cl2 (20 mL) was added oxalyl chloride (4.09 g, 32.3 mmol) at 0 ℃, then DMF (one drop) was added and the mixture was stirred at 20 ℃ for 1.5 hrs. The mixture was concentrated in vacuo, which then diluted with THF (20 mL) , to the mixture was added 4-cyclopropylpyridin-2-amine (1.731 g, 12.90 mmol) at 0 ℃. The mixture was stirred at 80 ℃ for 16 hrs. After cooling to 20 ℃, the mixture was concentrated to give the crude product. The crude product was purified by column chromatography on silica gel eluted with (40 g, DCM: Pet ether = 1: 1) to give N- (4-cyclopropylpyridin-2-yl) -5-ethoxy-2-fluoro-4-iodobenzamide. 1H NMR (400MHz, CDCl3) δ = 9.03 (d, J = 14.9 Hz, 1H) , 8.14 (d, J = 5.1 Hz, 1H) , 8.07 (s, 1H) , 7.65 (d, J = 10.6 Hz, 1H) , 7.50 (d, J = 6.3 Hz, 1H) , 6.78 (d, J = 4.7 Hz, 1H) , 4.14 (quin, J = 6.7 Hz, 2H) , 1.92 (dd, J = 3.9, 8.6 Hz, 1H) , 1.50 (t, J = 7.0 Hz, 3H) , 1.11 (d, J = 6.7 Hz, 2H) , 0.87 (d, J = 3.1 Hz, 2H) ppm.
Step 2: N- (4-cyclopropylpyridin-2-yl) -5-ethoxy-2-fluoro-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-
dioxaborolan-2-yl) benzamide
To a mixture of N- (4-cyclopropylpyridin-2-yl) -5-ethoxy-2-fluoro-4-iodobenzamide (500 mg, 1.173 mmol) , KOAc (345 mg, 3.52 mmol) , 4, 4, 4', 4', 5, 5, 5', 5'-octamethyl-2, 2'-bi (1, 3, 2-dioxaborolane) (447 mg, 1.760 mmol) and PdCl2 (dppf) (86 mg, 0.117 mmol) under N2 atmosphere was added 1, 4-Dioxane (5 mL) and immediately evacuated and backfilled with N2 three times. The mixture was then stirred at 70 ℃ for 16h. The mixture was diluted with EtOAc (10 mL) and water (4 mL) . The organic layer was separated and the aqueous layer was extracted by EtOAc (3 ×10 mL) . The organic layer was washed with water (10 mL) , brine (10 mL) , dried over Na2SO4, concentrated to afford the crude product, which was pruified by column chromatography on silica gel eluted with (4g, EtOAc: Pet ether =1: 10 ) to give N- (4-cyclopropylpyridin-2-yl) -5-ethoxy-2-fluoro-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) benzamide. 1H NMR (400MHz, CDCl3) δ = 9.11 (d, J = 15.7 Hz, 1H) , 8.14 (d, J = 5.1 Hz, 1H) , 8.09 (s, 1H) , 7.56 (d, J = 5.9 Hz, 1H) , 7.41 (d, J = 11.7 Hz, 1H) , 6.77 (d, J = 5.1 Hz, 1H) , 4.09 (q, J = 7.0 Hz, 2H) , 1.92 (br. s., 1H) , 1.43 (t, J = 6.8 Hz, 3H) , 1.36 (s, 12H) , 1.15-1.06 (m, 2H) , 0.87 (d, J = 6.3 Hz, 2H) ppm.
Intermediate 41

3-ethoxy-5-fluoro-N- (1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl) -4- (4, 4, 5, 5-tetramethyl-
1, 3, 2-dioxaborolan-2-yl) benzamide
To a solution of 3-ethoxy-5-fluoro-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) benzoic acid (1.5g, 4.84 mmol) in anhydrous CH2Cl2 (20 mL) was added oxalyl chloride (3.07 g, 24.18 mmol) at 0 ℃, then DMF (one drop) was added and the mixture was stirred at 20 ℃ for 1.5 hrs. The mixture was concentrated in vacuo, which was then diluted with THF (20 mL) . To the mixture was added a solution of 1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-amine (1.597 g, 9.67 mmol) at 0 ℃. The mixture was stirred at 80 ℃ for 16 hrs. After cooling to room temperature, the mixture was concentrated to give the crude product, which was purified by column chromatography on silica gel eluted with (EtOAc: Pet ether = 1%~ 50%) to give 3-ethoxy-5-fluoro-N- (1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl) -4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) benzamideas. 1H NMR (400MHz, CDCl3) δ = 8.43 (br. s., 1H) , 7.21 (s, 1H) , 7.12 (s, 1H) , 7.03 (d, J = 8.2 Hz, 1H) , 4.06 (q, J = 6.8 Hz, 2H) , 3.88 (s, 3H) , 1.47-1.35 (m, 15H) ppm.
Intermediate 42

4-ethoxy-5- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -N- (4- (trifluoromethyl) pyridin-2-
yl) picolinamide
Step 1: 2, 5-dichloro-4-ethoxypyridine
To a mixture of 2, 5-dichloropyridin-4-ol (1 g, 6.10 mmol) and K2CO3 (1.686 g, 12.20 mmol) in DMF (10 mL) was added iodoethane (0.951 g, 6.10 mmol) , the reaction mixture was stirred at 25 ℃ for 18h. The reaction mixture was added to ice-water (100 mL) , extracted with EtOAc (2 × 60 mL) . The combined organic phase was washed with brine (120 mL) , dried over Na2SO4, concentrated in vacuo, and the residue was purified by silica gel column chromatography (Pet. Ether : EtOAc = 5: 1) to give 2, 5-dichloro-4-ethoxypyridine. 1H NMR (400MHz, CDCl3) δ8.20 (s, 1H) , 6.82 (s, 1H) , 4.15 (q, J = 6.7 Hz, 2H) , 1.55 -1.44 (m, 3H) ppm.
Step 2: 5-chloro-4-ethoxy-2-vinylpyridine
To a mixture of 2, 5-dichloro-4-ethoxypyridine (800 mg, 4.17 mmol) and potassium vinyltrifluoroborate (1674 mg, 12.50 mmol) in 2-propanol (10 mL) was added Et3N (4.06 mL, 29.2 mmol) , then to the reaction mixture was added PdCl2 (dppf) (305 mg, 0.417 mmol) under N2, the mixture was stirred at 100 ℃ for 18h. The reaction mixture was added to water (50 mL) , extracted with EtOAc (2 × 50 mL) . The combined organic phase was washed with brine (100 mL) , dried over Na2SO4, concentrated in vacuo, and the crude was purified by silica gel column chromatography (Pet. Ether : EtOAc = 5: 1) to give 5-chloro-4-ethoxy-2-vinylpyridine. 1H NMR (400MHz, CDCl3) δ8.39 (s, 1H) , 6.84 (s, 1H) , 6.73 (dd, J = 10.8, 17.4 Hz, 1H) , 6.17 (d, J = 17.6 Hz, 1H) , 5.49 (d, J = 10.6 Hz, 1H) , 4.19 (q, J = 7.0 Hz, 2H) , 1.51 (t, J = 6.8 Hz, 3H) ppm.
Step 3: 5-chloro-4-ethoxypicolinic acid
To a mixture of 5-chloro-4-ethoxy-2-vinylpyridine (400 mg, 2.178 mmol) in acetone (20 mL) was added a solution of KMnO4 (1721 mg, 10.89 mmol) in water (20.00 mL) , then the reaction mixture was stirred at 25 ℃ for 18h. The reaction mixture was added to a solution of Na2SO3 (100 mL) , filtered, the filtrate was extracted with EtOAc (2 × 100 mL) . The combined organic phase was washed with brine (200 mL) , dried over Na2SO4, concentrated in vacuo to give 5-chloro-4-ethoxypicolinic acid. 1H NMR (400MHz, DMSO-d6) δ13.40 (s, 1H) , 8.56 (s, 1H) , 7.68 (s, 1H) , 4.29 (d, J = 7.0 Hz, 2H) , 1.36 (t, J = 6.7 Hz, 3H) ppm.
Step 4: 5-chloro-4-ethoxy-N- (4- (trifluoromethyl) pyridin-2-yl) picolinamide
To a mixture of 5-chloro-4-ethoxypicolinic acid (200 mg, 0.992 mmol) in DMF (5 mL) was added HATU (566 mg, 1.488 mmol) , the mixture was stirred at 25 ℃ for 1h. Then to the mixture was added 4- (trifluoromethyl) pyridin-2-amine (241 mg, 1.488 mmol) and Et3N (0.277 mL, 1.984 mmol) , the reaction mixture was stirred at 25 ℃ for 18h. The reaction mixture was added to ice-water (15 mL) , extracted with EtOAc (2 × 10 mL) . The combined organic phase was washed with brine (20 mL) , dried over Na2SO4, concentrated in vacuo to give 5-chloro-4-ethoxy-N- (4-(trifluoromethyl) pyridin-2-yl) picolinamide. 1H NMR (400MHz, CDCl3) δ10.53 (br. s., 1H) , 8.63
(s, 1H) , 8.47 (d, J = 5.1 Hz, 1H) , 8.40 (s, 1H) , 7.78 (s, 1H) , 7.24 (d, J = 4.7 Hz, 1H) , 4.25 (q, J = 6.8 Hz, 2H) , 1.50 -1.45 (m, 3H) ppm.
Step 5: 4-ethoxy-5- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -N- (4- (trifluoromethyl) pyridin-
2-yl) picolinamide
To a mixture of 5-chloro-4-ethoxy-N- (4- (trifluoromethyl) pyridin-2-yl) picolinamide (150 mg, 0.434 mmol) and 4, 4, 4', 4', 5, 5, 5', 5'-octamethyl-2, 2'-bi (1, 3, 2-dioxaborolane) (132 mg, 0.521 mmol) in Dioxane (4 mL) was added potassium acetate (128 mg, 1.302 mmol) , then to the reaction mixture was added TRICYCLOHEXYLPHOSPHINE (6.08 mg, 0.022 mmol) and Pd2 (dba) 3 (19.87 mg, 0.022 mmol) under N2, the mixture was stirred at 100 ℃ for 15 h. The reaction mixture was added to water (30 mL) , extracted with EtOAc (2 × 30 mL) . The combined organic phase was washed with brine (60 mL) , dried over Na2SO4, concentrated in vacuo, and the crude was purified by silica gel column chromatography (4 g, Pet. Ether: EtOAc = 5/1) to give 4-ethoxy-5- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -N- (4- (trifluoromethyl) pyridin-2-yl) picolinamide. MS 438 [M+1] .
Intermediate 43

4-methyl-5- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -N- (4- (trifluoromethyl) pyridin-2-
yl) picolinamide
Step 1: 5-bromo-4-methylpicolinic acid
To a solution of 2, 5-dibromo-4-methylpyridine (3 g, 11.96 mmol) in anhydrous toluene (40 ml) ) was added BuLi (5.74 ml, 14.35 mmol) at -78 ℃ dropwise. The reaction mixture was stirred at -78 ℃ for 1 h. Then the reaction mixture was poured into a 100 mL beaker with dry carbon dioxide (4 g, 91 mmol) . The reaction mixture was stirred at 20 ℃ for another 10 minutes. Then the mixture was poured into saturated citric acid and a yellow precipitate was formed. The suspension was filtered and the solid was collected, dried in vacuo to give 5-bromo-4-methylpicolinic acid. 1HNMR (400 MHz, DMSO-d6) : δ = 8.71 (s, 1 H) , 7.99 (s, 1 H) , 2.40 (s, 3 H) . MS: 215.9 (M+1) ppm.
Step 2: 5-bromo-4-methyl-N- (4- (trifluoromethyl) pyridin-2-yl) picolinamide
5-bromo-4-methylpicolinic acid (600 mg, 2.78 mmol) and SOCl2 (10 mL, 137 mmol) were heated to reflux at 90 ℃. Excess SOCl2 was removed by distillation after 1.5 hours. Anhydrous toluene (5 mL) was added to the residue, and then evaporated to remove remaining SOCl2 to give the corresponding acid chloride. To a solution of 4- (trifluoromethyl) pyridin-2-amine (540 mg, 3.33 mmol) and Et3N (1.161 mL, 8.33 mmol) in CH2Cl2 (6 mL) , the solution of acid chloride in anhydrous CH2Cl2 (10 mL) was added over a period of 10 min at 0 ℃. The reaction mixture was then stirred at room temperature for 2 hours. The solvent was removed under reduced pressure and the residue was purified by silica gel chromatography using EtOAc to give 5-bromo-4-methyl-N- (4- (trifluoromethyl) pyridin-2-yl) picolinamide. 1HNMR (400 MHz, CDCl3) : δ = 10.52 (s, 1 H) , 8.66 (d, J = 14.8 Hz, 2 H) , 8.51 (d, J = 4.4 Hz, 1 H) , 8.13 (s, 1 H) , 7.28 (d, J = 4.8 Hz, 1 H) , 2.51 (s, 3 H) . MS: 359.9 (M+1) ppm.
Step 3: 4-methyl-5- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -N- (4- (trifluoromethyl) pyridin-
2-yl) picolinamide
A solution of 5-bromo-4-methyl-N- (4- (trifluoromethyl) pyridin-2-yl) picolinamide (30 mg, 0.083 mmol) , PdCl2 (dppf) (12.19 mg, 0.017 mmol) , potassium acetate (16.35 mg, 0.167 mmol) and 4,4, 4', 4', 5, 5, 5', 5'-octamethyl-2, 2'-bi (1, 3, 2-dioxaborolane) (31.7 mg, 0.125 mmol) in 1, 4-dioxane (1.5 mL) and water (0.2 mL) . The reaction mixture was refilled with nitrogen three times and stirred at 100 ℃ for 2 hours. After cooled to room temperature, the solution was filtered and the filtrate was concentrated under reduced pressure to get the crude product, which was purified by pre-TLC (petroleum ether : ethyl acetate=1: 1) to afford the compound 4-methyl-5- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -N- (4- (trifluoromethyl) pyridin-2-yl) picolinamide. 1H NMR (400 MHz, CDCl3) : δ = 10.77 (s, 1 H) , 8.83 (s, 1 H) , 8.72 (s, 1 H) , 8.51 (d, J = 5.2 Hz, 1 H) , 8.04 (s, 1 H) , 7.27 (d, J = 4.8 Hz, 1 H) , 2.62 (s, 3 H) , 1.24 (s, 12 H) . MS: 408.1 (M+1) ppm.
Intermediate 44

3-fluoro-5-methoxy-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -N- (4-
(trifluoromethyl) pyridin-2-yl) benzamide
Step 1: 2-fluoro-4-formyl-6-methoxyphenyl trifluoromethanesulfonate
A solution of commercially available 3-fluoro-4-hydroxy-5-methoxybenzaldehyde (2.0 g, 11.76 mmol) and pyridine (2.88 ml, 35.3 mmol) in DCM (58.8 ml) at 0 ℃ was treated with TriflicAnhydride (2.185 ml, 12.93 mmol) dissolved in DCM (5 ml) dropwise via an additional funnel. The mixture was then stirred at 0 ℃ for 4 h. The mixture was poured into water and
extracted with DCM (x2) . The organic layer was dried (MgSO4) and concentrated to afford an oil. Purification by chromatography on silica 0-15 % EtOAc /Hexane afforded the title compound. MS: calc. = 303.2, found = 303.0 (M+1) .
Step 2: 3-fluoro-5-methoxy-4- ( ( (trifluoromethyl) sulfonyl) oxy) benzoic acid
A solution of 2-fluoro-4-formyl-6-methoxyphenyl trifluoromethanesulfonate, (1000 mg, 3.31 mmol) and 2-methylbut-2-ene (1050 μl, 9.93 mmol) in t-BuOH (1.10E+04 μl) was treated slowly via an additional funnel a solution of sodiumchlorite (688 mg, 7.61 mmol) and sodiumdihydrogenphosphate (794 mg, 6.62 mmol) dissolved in Water (1.10E+04 μl) and the mixture was stirred at room temperature for 3 h. The solvent was evaporated and the resulting solid was dissolved with water. The aqueous mixture was acidified with 1 N HCl (pH=4) and extracted with EtOAc (x2) . The organic layer was dried (MgSO4) and concentrated to afford the title compound. MS: calc. = 318.9, found = 318.9 (M+1) . 1H NMR (DMSO-d, 500 MHz) : 3.99 (3 H, s) , 7.57 (1 H, d, J = 9.8 Hz) , 7.60 (1 H, s) ppm.
Step 3: 2-fluoro-6-methoxy-4- ( (4- (trifluoromethyl) pyridin-2-yl) carbamoyl) phenyl trifluoromethanesulfonate
A solution of 3-fluoro-5-methoxy-4- ( ( (trifluoromethyl) sulfonyl) oxy) benzoic acid, (600 mg, 1.886 mmol) and DMF (51.1 μl, 0.660 mmol) in DCM (4714 μl) was treated with thionylchloride (1376 μl, 18.86 mmol) and the mixture was stirred at 35 ℃ for 18 h. The solvent was evaporated and the residue co-evaporated with with toluene (x2) . The resulting oil was then diluted with MeCN (5 ml) and treated with DMAP (299 mg, 2.451 mmol) . The mixture was stirred at rt for 5 min and 4- (trifluoromethyl) pyridin-2-amine (374 mg, 2.263 mmol) was added and the mixture was stirred at 35 ℃ for 6 h to afford a suspension. The suspension was diluted with DCM and washed with water (x2) and brine. The organic layer was dried (MgSO4) and concentrated to afford an oil. The oily residue was purified by chromatography on silica eluting with 0-10 % EtOAc /Hexane) to afford the title compound. MS: 463.2 Found = 463.1 (M+1) .
Step 4: 3-fluoro-5-methoxy-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -N- (4-
(trifluoromethyl) pyridin-2-yl) benzamide
A sealed vial containing 2-fluoro-6-methoxy-4- ( (4- (trifluoromethyl) pyridin-2-yl) carbamoyl) phenyl trifluoromethanesulfonate, (1000 mg, 2.163 mmol) , 4, 4, 4', 4', 5, 5, 5', 5'-octamethyl-2, 2'-bi (1, 3, 2-dioxaborolane) (824 mg, 3.24 mmol) , potassiumacetate (637 mg, 6.49 mmol) and PdCl2 (dppf) -CH2Cl2Adduct (177 mg, 0.216 mmol) was treated with Dioxane (2.16E+04 μl) and the system degassed and backfilled with N2 (x3) . The mixture was then stirred at 100 ℃ for 24 h. The mixture was diluted with EtOAc and filtered. The filtrate was washed with water and brine, dried (MgSO4) and concentrated to afford a black oil. Purification
by chromatography on silica (0-20 % EtOAc /Hexane) afforded 3-fluoro-5-methoxy-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -N- (4- (trifluoromethyl) pyridin-2-yl) benzamide. MS: calc. = 441.2, found = 441.2 (M+1) .
Please refer to patent application US20140206681 (A1) for preparation of certain boronic esters.
Example 1

4- {8-amino-5-fluoro-3- [ (3R, 9aR) -8- (1-methylethyl) -6-oxooctahydropyrazino [2, 1-c] [1, 4] oxazin-
3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [4- (trifluoromethyl) pyridin-2-
yl] benzamide
A mixture of Pd (dppf) Cl2 (4, 6.14 μmol) , 3-ethoxy-5-fluoro-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -N- (4- (trifluoromethyl) pyridin-2-yl) benzamide (41.5 mg, 0.091 mmol) and (3R, 9aR) -3- (8-amino-1-bromo-5-fluoroimidazo [1, 5-a] pyrazin-3-yl) -8-isopropylhexahydropyrazino [2, 1-c] [1, 4] oxazin-6 (1H) -one (30 mg, 0.070 mmol) in THF (0.7 ml) was degassed by bubbling nitrogen for 15 min. To the mixture was added K2CO3 (0.211 ml, 0.211 mmol) . The reaction mixture was heated at 100 ℃ for 5 h. It was concentrated and directly purified by ISCO (gold 12g, 0-100%EtOAc/EtOH (3: 1) in hexane) to give 4- (8-amino-5-fluoro-3- ( (3R, 9aR) -8-isopropyl-6-oxooctahydropyrazino [2, 1-c] [1, 4] oxazin-3-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-ethoxy-5-fluoro-N- (4- (trifluoromethyl) pyridin-2-yl) benzamide. LC-MS: (M+1) + 675.4, Rt = 1.21 min (Method B) . 1HNMR (500 MHz, CDCl3) , δ = 8.88 (1H, d, J = 4.7 Hz) , 8.71 (1H, s) , 8.53 (1H, d, J = 5.0 Hz) , 7.45 (1H, d, J = 3.5 Hz) , 7.29-7.39 (2H, m) , 6.97 (1H, s) , 5.05 (2H, ddd) , 4.82 (2H, d, J = 20.4 Hz) , 4.14-4.20 (2H, m) , 4.03 (1H, m) , 3.72-3.79 (2H, m) , 3.65 (H, td, J = 11.0 Hz, 2.9 Hz) , 3.52 (1H, m) , 3.41 (1H, dd, J = 16.1, 6.3 Hz) , 3.20 (1H, d, J = 16.o Hz) , 2.97 (1H, m) , 2.78 (1H, m) , 2.36 (1H, m) , 1.31 (3H, t) , 1.09 (6H, d) ppm.
Example 2

4- (8-amino-5-chloro-3- ( (6R, 8aS) -3-oxooctahydroindolizin-6-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-
fluoro-5-methoxy-N- (1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl) benzamide
Step 1: Methyl 3-fluoro-5-methoxy-4- ( ( (trifluoromethyl) sulfonyl) oxy) benzoate
A solution of commercially available methyl 3-fluoro-4-hydroxy-5-methoxybenzoate (1.0 g, 5.00 mmol) and pyridine (1.212 ml, 14.99 mmol) in DCM (24.98 ml) at 0 ℃ was treated with TriflicAnhydride (0.928 ml, 5.50 mmol) slowly via a syringe and needle and the mixture stirred at 0 ℃ for 2 h then at room temperature for 1h. The mixture was added to water, diluted with DCM and the layers separated. The aqueous layer was extracted with DCM and the combined organics was dried (MgSO4) and concentrated to afford a clear oil. Purification by chromatography on silica 0 to 20 % EtOAc /Hexane afforded 1.43 g of thetitle compound.
MS: calc. = 333.2, found 333.1.1H NMR (DMSO-d, 500 MHz) : 3.97 (3 H, s) , 4.02 (3 H, s) 7.54-7.56 (2 H, m) .
Step 2: Methyl 3-fluoro-5-methoxy-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) benzoate
In a sealed flask, a suspension of methyl 3-fluoro-5-methoxy-4- ( ( (trifluoromethyl) sulfonyl) oxy) benzoate, (800 mg, 2.408 mmol) , 4, 4, 4', 4', 5, 5, 5', 5'-octamethyl-2, 2'-bi (1, 3, 2-dioxaborolane) (917 mg, 3.61 mmol) , PdCl2 (dppf) -CH2Cl2Adduct (197 mg, 0.241 mmol) and potassium acetate (709 mg, 7.22 mmol) in Dioxane (2.41E+04 μl) was evacuated with house vacuum and backfilled with N2 (x3) . The resulting mixture was then stirred at 100 ℃ overnight. The mixture was diluted with EtOAc and filtered. The filtrate was washed with brine, dried (MgSO4) , filtered and concentrated under reduced pressure. The residue was purified by chromatography on silica (0-10 % EtOAc /Hexane) to afford 477 mg of the title compound. MS: calc. = 311.1, found 311.0.
Step 3: 4- (8-amino-5-chloro-3- ( (6R, 8aS) -3-oxooctahydroindolizin-6-yl) imidazo [1, 5-a] pyrazin-
1-yl) -3-fluoro-5-methoxybenzoic acid
A sealed tube containing (6R, 8aS) -6- (8-amino-1-bromo-5-chloroimidazo [1, 5-a] pyrazin-3-yl) hexahydroindolizin-3 (2H) -one (300 mg, 0.780 mmol) , methyl 3-fluoro-5-methoxy-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) benzoate, (484 mg, 1.560 mmol) and Pd (dppf) Cl2 (50.8 mg,
0.078 mmol) was treated with degassed THF (3900 μl) via a syringe and needle. With house vacuum the mixture was degassed and backfilled with N2 (x2) and then treated with degassed K2CO3 (2340 μl, 2.340 mmol) 1M aq solution. The mixture was degassed again and backfilled with N2 (x2) then stirred at 100 ℃ for 4 h. The mixture was diluted with EtOAc and filtered. The filtrate was concentrated to afford an oil. The oil was purified by reverse phase HPLC (ACN /water with 0.05 % TFA) to afford the title compound. MS: calc. = 474.1, found 474.1 (M+1) .
Step 4: 4- (8-amino-5-chloro-3- ( (6R, 8aS) -3-oxooctahydroindolizin-6-yl) imidazo [1, 5-a] pyrazin-1-
yl) -3-fluoro-5-methoxy-N- (1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl) benzamide
A solution of 4- (8-amino-5-chloro-3- ( (6R, 8aS) -3-oxooctahydroindolizin-6-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-fluoro-5-methoxybenzoic acid, (18 mg, 0.038 mmol) in DMF (190 μl) at 0 ℃ was treated with HATU (14.44 mg, 0.038 mmol) and the mixture stirred at 0 ℃ for 10 min. 1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-amine (6.27 mg, 0.038 mmol) dissolved in DMF (190 μl) was then added via a syringe slowly followed by DIEA (6.63 μl, 0.038 mmol) and the mixture stirred at 0 ℃ for 30 min, then at room temperature for 1 h. The mixture was purified by reverse phase HPLC (ACN /water with 0.05 % TFA) to afford the title compound as the TFA salt. MS: calc. 621.2 found 621.2 (M+1) .
Example 3
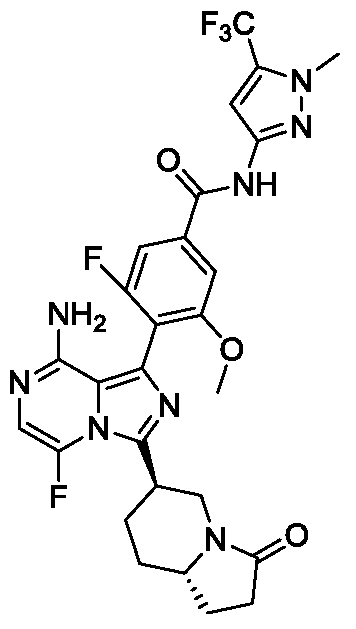
4- (8-amino-5-fluoro-3- ( (6R, 8aS) -3-oxooctahydroindolizin-6-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-
fluoro-5-methoxy-N- (1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl) benzamide
Following the procedure described fro Example 2 but with (6R, 8aS) -6- (8-amino-1-bromo-5-fluoroimidazo [1, 5-a] pyrazin-3-yl) hexahydroindolizin-3 (2H) -one in Step 3, 4- (8-amino-5-fluoro-3- ( (6R, 8aS) -3-oxooctahydroindolizin-6-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-fluoro-5-methoxy-N-(1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl) benzamide was prepared. MS: calc. 605.5 found 605.2 (M+1) .
Examples 4, 5, 6

4- (8-amino-3- (9, 9-difluorooctahydro-1H-quinolizin-3-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-ethoxy-
N- (4- (trifluoromethyl) pyridin-2-yl) benzamide
Step 1: 9, 9-difluoro-6-oxooctahydro-1H-quinolizine-3-carboxylic acid
To a solution of methyl 9, 9-difluoro-6-oxooctahydro-1H-quinolizine-3-carboxylate (cis) (1.4 g, 5.66 mmol) in MeOH (10 mL) was added sodium methoxide (6.47 mL, 28.3 mmol) . The resulting mixture was heated to 50 ℃ and stirred at 50 ℃ for 24 h. To the resulting mixture was added aq. 1 M HCl to pH= 3~4 slowly. The mixture was evaporated in vacuo to give crude, which was added MeOH (5 mL) , THF (10 mL) and DCM (10 mL) and stirred for 1 h. The resulting mixture was filtered and evaporated in vacuo to give 9, 9-difluoro-6-oxooctahydro-1H-quinolizine-3-carboxylic acid, which was used in the next step directly. MS-ESI (m/z) : 234.2 (M+1) + (Acq Method D Rt = 0.30 min)
Step 2: N- ( (3-chloropyrazin-2-yl) methyl) -9, 9-difluoro-6-oxooctahydro-1H-quinolizine-3-
carboxamide
To a solution of 9, 9-difluoro-6-oxooctahydro-1H-quinolizine-3-carboxylic acid (2.1 g, 6.30 mmol) and (3-chloropyrazin-2-yl) methanamine, 2HCl (1.638 g, 7.56 mmol) in THF (20 mL) was added HATU (2.88 g, 7.56 mmol) and TEA (2.196 mL, 15.76 mmol) . The mixture was stirred at 30 ℃ for 4 h. The mixture was poured into water (50 mL) and extracted with EtOAc (3x30 mL) . The organic layers were washed with brine (3x30 mL) , dried over Na2SO4, filtered and the filtrate was concentrated in vacuo to give crude N- ( (3-chloropyrazin-2-yl) methyl) -9, 9-difluoro-6-oxooctahydro-1H-quinolizine-3-carboxamide, which was used in the next step directly. MS-ESI (m/z) : 359.0 (M+1) + (Acq Method D Rt: 0.939 min)
Step 3: 7- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-quinolizin-4 (6H) -one
To a solution of N- ( (3-chloropyrazin-2-yl) methyl) -9, 9-difluoro-6-oxooctahydro-1H-quinolizine-3-carboxamide (3.1 g, 6.20 mmol) in acetonitrile (30 mL) was added pentachlorophosphorane (4.52 g, 21.69 mmol) . The resulting mixture was heated to 85 ℃ and
stirred at 85 ℃ for 1 h. The resulting mixture was poured into aqueous NaHCO3 (100 mL) and extracted with EtOAc (3x50 mL) . The combined organic layers were washed with aqueous NaHCO3 (3x50 mL) , dried over Na2SO4, filtered and evaporated in vacuo to give crude, which was purified by silica gel chromatography (EtOAc in DCM: 0-25%) to give 7- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-quinolizin-4 (6H) -one. MS-ESI (m/z) : 341.0 (M+1) + (Acq Method D Rt: 1.040 min)
Step 4: 7- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-quinolizin-
4 (6H) -one
To a solution of 7- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-quinolizin-4 (6H) -one (450 mg, 1.189 mmol) in DMF (10 mL) was added NBS (254 mg, 1.426 mmol) . The resulting mixture was stirred at 14 ℃ (room temperature) for 1 h. The resulting mixture was poured into aqueous NaCl (50 mL) and extracted with EtOAc (3x50 mL) . The combined organic layers were washed with aqueous NaCl (6x50 mL) , dried over Na2SO4, filtered and evaporated in vacuo to give 7- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-quinolizin-4 (6H) -one, which was used in the next step directly. MS-ESI (m/z) : 421.0 (M+1) + (Acq Method D Rt: 1.178 min)
Step 5: 7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-quinolizin-
4 (6H) -one
In a 100 mL seal tube, to a solution of 7- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -1,1-difluorohexahydro-1H-quinolizin-4 (6H) -one (500 mg, 1.132 mmol) in 2-propanol (5 mL) was added ammonia, H2O (10 mL, 1.132 mmol) , and stirred at 95 ℃ for 20 h. The solvent was evaporated to give crude. To a solution of crude in 2-propanol (5 mL) was added ammonia, H2O (10 mL, 1.132 mmol) . The resulting mixture was heated to 95 ℃ and stirred at 95 ℃ for 20 h. The brown resulting mixture was poured into water (20 mL) and extracted with EtOAc (3x20 mL) . The combined organic layers were washed with brine (3x20 mL) , dried over anhydrous Na2SO4, filtered and evaporated in vacuo to give 7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-quinolizin-4 (6H) -one, which was used in the next step directly.
MS-ESI (m/z) : 402.0 (M+1) + (Acq Method D Rt: 0.849 min)
Step 6: 1-bromo-3- (9, 9-difluorooctahydro-1H-quinolizin-3-yl) imidazo [1, 5-a] pyrazin-8-amine
To a solution of 7- (8-amino-1-bromoimidazo [1, 5-a] pyrazin-3-yl) -1, 1-difluorohexahydro-1H-quinolizin-4 (6H) -one (250 mg, 0.500 mmol) in THF (5 mL) was added BH3. DMS (0.237 mL, 2.499 mmol) at 0 ℃. After the condition was completed, the yellow resulting mixture was warmed to 9-11 ℃ (room temperature) and stirred at 9-11 ℃ (room temperature) for 4 h. The resulting mixture was quenched with MeOH (10 mL) slowly at 0 ℃. The resulting mixture was
evaporated in vacuo to give crude, which was purified by silica gel chromatography (MeOH in DCM: 0-10%) to give 1-bromo-3- (9, 9-difluorooctahydro-1H-quinolizin-3-yl) imidazo [1, 5-a] pyrazin-8-amine. MS-ESI (m/z) : 386.0 (M+1) + (Acq Method D Rt: 0.912 min)
Step 7: 4- (8-amino-3- (9, 9-difluorooctahydro-1H-quinolizin-3-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-
ethoxy-N- (4- (trifluoromethyl) pyridin-2-yl) benzamide
To a solution of 1-bromo-3- (9, 9-difluorooctahydro-1H-quinolizin-3-yl) imidazo [1, 5-a] pyrazin-8-amine (50 mg, 0.129 mmol) and 3-ethoxy-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -N- (4- (trifluoromethyl) pyridin-2-yl) benzamide (67.8 mg, 0.155 mmol) in THF (2 mL) and water (0.500 mL) was added PdCl2DtBPF (8.46 mg, 0.013 mmol) and Na2CO3 (41.2 mg, 0.388 mmol) under nitrogen protection. Then the mixture was heated to 90 ℃ by microwave and at 90 ℃ for 1 hour. The resulring mixture was diluted with EtOAc (5 mL) and water (10 mL) , extracted with EtOAc (3x5 mL) . The combined organic layers were washed with brine (3x10 mL) , dried over anhydrous Na2SO4, filtered and evaporated in vacuo to give crude. It was purified by HPLC (Column: YMC-Actus Triart C18 150*30mm*5um; Condition: CH3CN/water with 0.1% TFA modifier) to give 4- (8-amino-3- (9, 9-difluorooctahydro-1H-quinolizin-3-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-ethoxy-N- (4- (trifluoromethyl) pyridin-2-yl) benzamide, TFA (trans, racemic) and 4- (8-amino-3- (9, 9-difluorooctahydro-1H-quinolizin-3-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-ethoxy-N- (4- (trifluoromethyl) pyridin-2-yl) benzamide, TFA (cis, racemic) . The 4- (8-amino-3- (9, 9-difluorooctahydro-1H-quinolizin-3-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-ethoxy-N- (4- (trifluoromethyl) pyridin-2-yl) benzamide, TFA (trans, racemic) was chiral separated by SFC (Column : AS 250mmx30mmx10um; Mobile phase: A: Supercritical CO2 , B: MeOH (0.1% NH3. H2O) , A: B =85: 15 at 50 mL/min; Column Temp: 38 ℃;Nozzle Pressure: 100 Bar; Wavelength: 220nm) to give 4- (8-amino-3- (9, 9-difluorooctahydro-1H-quinolizin-3-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-ethoxy-N- (4-(trifluoromethyl) pyridin-2-yl) benzamide (trans, isomer 1) as a solid and 4- (8-amino-3- (9, 9-difluorooctahydro-1H-quinolizin-3-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-ethoxy-N- (4-(trifluoromethyl) pyridin-2-yl) benzamide (trans, enantiomer 2) as a solid. The 4- (8-amino-3-(9, 9-difluorooctahydro-1H-quinolizin-3-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-ethoxy-N- (4-(trifluoromethyl) pyridin-2-yl) benzamide (trans, enantiomer 2) which contained minor impurities was purified by again HPLC (Column: Phenomenex Synergi C18 250x21.2mmx4um; Condition: CH3CN/water with 0.1% TFA modifier) to give 4- (8-amino-3- (9, 9-difluorooctahydro-1H-quinolizin-3-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-ethoxy-N- (4- (trifluoromethyl) pyridin-2-yl) benzamide, TFA (trans, enantiomer 2) .
Cis, racemic: cis 4- (8-amino-3- (9, 9-difluorooctahydro-2H-quinolizin-3-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-ethoxy-N- (4- (trifluoromethyl) pyridin-2-yl) benzamide
MS-ESI (m/z) : 616.0 (M+1) + (Acq Method D Rt: 0.994 min)
1H NMR (400MHz, CD3OD) δ 8.63 (br. s., 2H) , 7.91-7.71 (m, 4H) , 7.46 (d, J =3.9 Hz, 1H) , 7.09 (br. s., 1H) , 4.37-3.92 (m, 6H) , 3.83-3.66 (m, 2H) , 2.42 (br. s., 1H) , 2.35-2.08 (m, 6H) , 2.02 (br. s., 1H) , 1.36 (d, J =4.3 Hz, 3H) .
SFC check (Column: Chiralpak AS-3 150×4.6mm I.D., 3um Mobile phase: methanol (0.05% DEA) in CO2 from 5% to 40% Flow rate: 2.5mL/min Wavelength: 220nm)
Trans, isomer 1: 4- (8-amino-3- ( (3S, 9aR) -9, 9-difluorooctahydro-2H-quinolizin-3-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-ethoxy-N- (4- (trifluoromethyl) pyridin-2-yl) benzamide
Rt = 3.568 min, Method C
1H NMR (400MHz, CD3OD) δ 8.67-8.59 (m, 2H) , 7.78-7.71 (m, 2H) , 7.61-7.55 (m, 2H) , 7.44 (d, J =4.7 Hz, 1H) , 7.03 (d, J =5.1 Hz, 1H) , 4.21 (q, J =6.7 Hz, 2H) , 3.45-3.39 (m, 1H) , 3.13 (d, J =11.0 Hz, 1H) , 2.90 (d, J =11.3 Hz, 1H) , 2.57-2.50 (m, 1H) , 2.40-2.08 (m, 5H) , 1.95-1.71 (m, 5H) , 1.33-1.28 (m, 3H)
Trans, isomer 2: 4- (8-amino-3- ( (3R, 9aR) -9, 9-difluorooctahydro-2H-quinolizin-3-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-ethoxy-N- (4- (trifluoromethyl) pyridin-2-yl) benzamide
Rt = 3.914 min, Method C
1H NMR (400MHz, CD3OD) δ 8.66-8.60 (m, 2H) , 7.85 (d, J =5.9 Hz, 1H) , 7.80-7.74 (m, 2H) , 7.69 (d, J =7.8 Hz, 1H) , 7.46 (d, J =4.7 Hz, 1H) , 7.05 (d, J =5.5 Hz, 1H) , 4.26 (q, J =6.7 Hz, 2H) , 3.75-3.64 (m, 2H) , 3.39 (d, J =11.7 Hz, 3H) , 3.00 (br. s., 1H) , 2.35 (t, J =15.3 Hz, 3H) , 2.11-1.91 (m, 5H) , 1.34 (t, J =7.0 Hz, 3H) ppm.
Following the procedure in Example 1, using appropriate boronic ester and imidazopyrazine bromo intermediates for the Suzuki coupling catylzed by different palladium catalyst, the examples in Table 1 were prepared.
Table 1









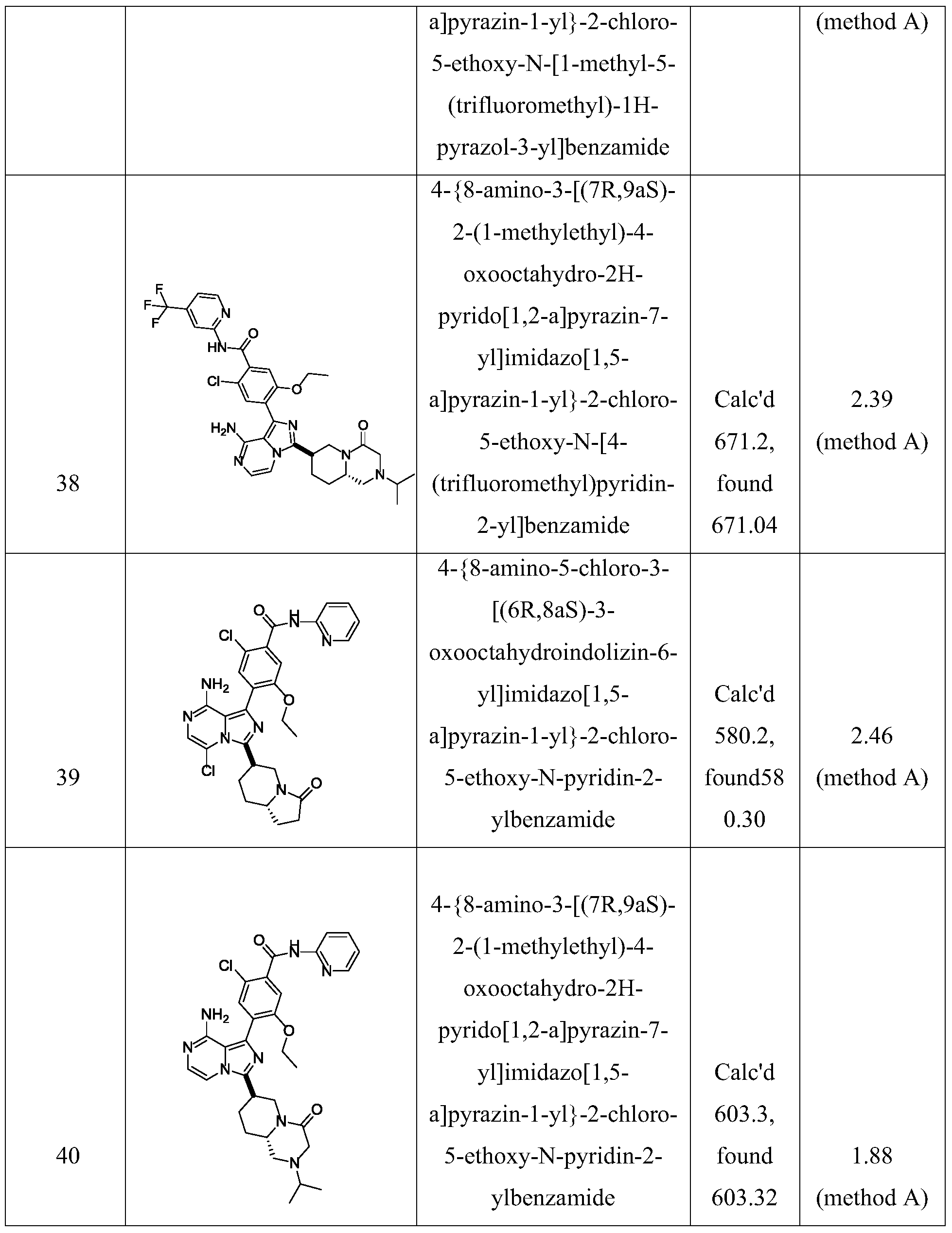































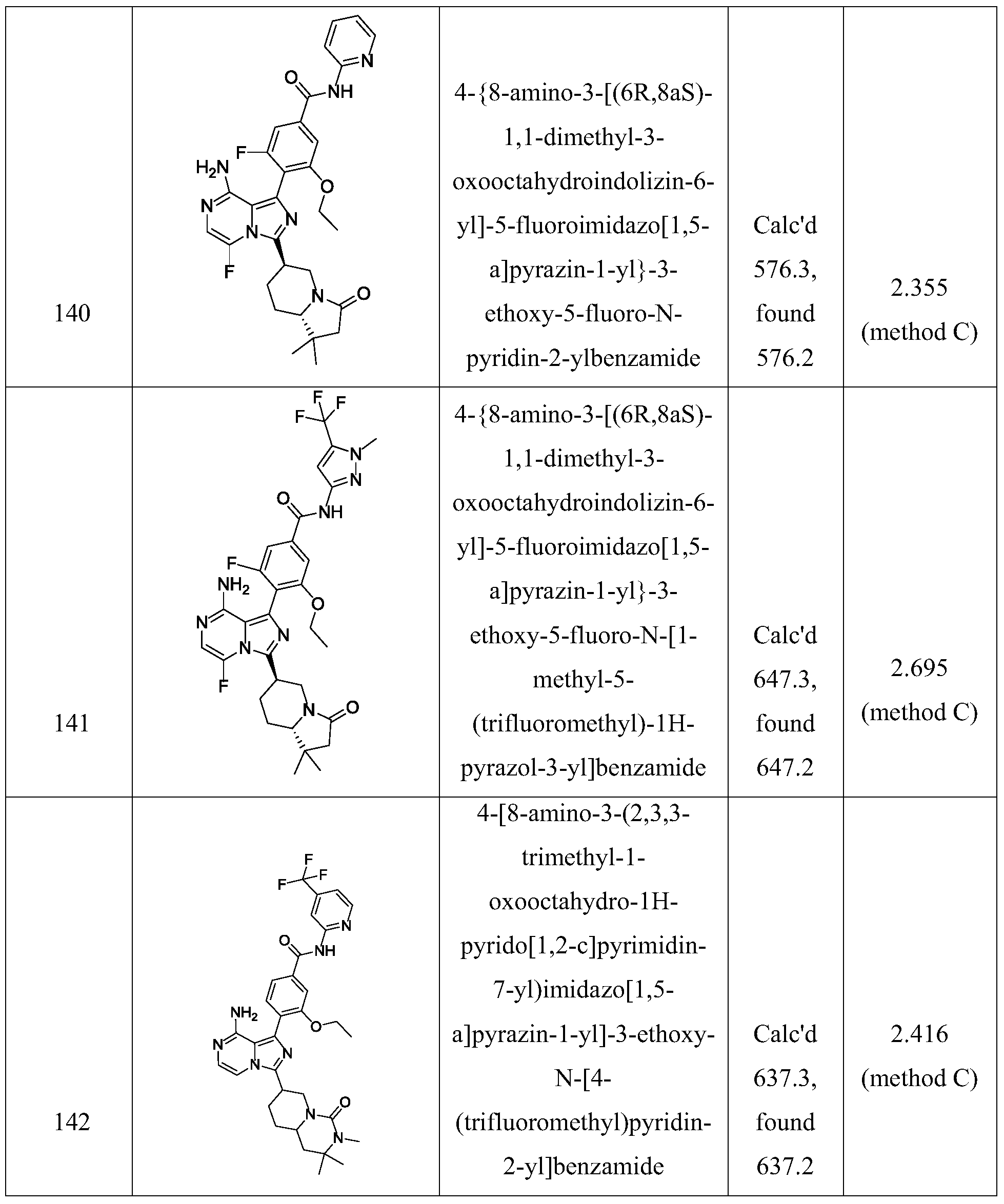






Example 159.

4- (8-amino-3- (2- (2-methoxyethyl) -4-oxooctahydro-1H-pyrido [1, 2-a] pyrazin-7-yl) imidazo [1, 5-
a] pyrazin-1-yl) -3-ethoxy-N- (4- (trifluoromethyl) pyridin-2-yl) benzamide
Step 1: (7R, 9aS) -2-benzyl 7-methyl 4-oxohexahydro-1H-pyrido [1, 2-a] pyrazine-2, 7 (6H) -
dicarboxylate
A mixture of (7R, 9aS) -2-tert-butyl 7-methyl 4-oxohexahydro-1H-pyrido [1, 2-a] pyrazine-2,7 (6H) -dicarboxylate (660 mg, 2.113 mmol) in HCl/dioxane (4M) (6 mL, 24.00 mmol) was stirred at 10 ℃ for 30 min. The mixture was evaporated in vacuo to give the product (7R, 9aS) -methyl 4-oxooctahydro-1H-pyrido [1, 2-a] pyrazine-7-carboxylate. The above residue was then dissolved in DCM (10 mL) and sat. aq. NaHCO3 (6 mL) was added, followed by benzyl carbonochloridate (541 mg, 3.17 mmol) . The mixture was stirred at 25 ℃ for 12 hrs. The mixture was then diluted with H2O (20 mL) , and extracted with EtOAc (10 mL × 3) . The organic layer was washed with brine (10 mL) , dried over Na2SO4, filtered, and evaporated to
give the crude product, which was then purified by flash chromatography (Pet. ether/THF= 70 ~60%, 220 nm) to afford the title compound. 1H NMR (400MHz, CDCl3) d = 7.35 (s, 5H) , 5.20 -5.10 (m, 2H) , 4.94 (d, J=12.1 Hz, 1H) , 4.28 (d, J=18.0 Hz, 1H) , 4.10 -3.86 (m, 2H) , 3.69 (s, 3H) , 3.35 (br. s., 2H) , 2.63 -2.53 (m, 1H) , 2.49 -2.37 (m, 1H) , 2.18 (d, J=12.1 Hz, 1H) , 1.84 (br. s., 1H) , 1.71 -1.62 (m, 1H) , 1.41 -1.29 (m, 1H) ppm.
Step 2: (7R, 9aS) -2- ( (benzyloxy) carbonyl) -4-oxooctahydro-1H-pyrido [1, 2-a] pyrazine-7-
carboxylic acid
A mixture of (7R, 9aS) -2-benzyl 7-methyl 4-oxohexahydro-1H-pyrido [1, 2-a] pyrazine-2, 7 (6H) -dicarboxylate (600 mg, 1.732 mmol) in THF (4 mL) was treated with lithium hydroxide, H2O (2 mL, 4.00 mmol) at 10 ℃ for 12 hrs, then diluted with H2O (5 mL) and acidified to pH=2 ~ 3 with 2M HCl (aq. ) . The mixture was extracted with DCM/i-PrOH (10 mL ×10, 3: 1) . The organic layer was evaporated to afford the title compound.
1H NMR (400MHz, DMSO-d6) δ = 7.42 -7.20 (m, 4H) , 5.14 -4.96 (m, 2H) , 4.02 -3.90 (m, 2H) , 3.58 -3.36 (m, 3H) , 3.23 (br. s., 1H) , 2.83 (t, J=12.3 Hz, 1H) , 2.65 (d, J=7.4 Hz, 1H) , 2.02 (br. s., 1H) , 1.84 (d, J=11.0 Hz, 1H) , 1.54 -1.36 (m, 2H) ppm.
Step 3: (7S, 9aR) -benzyl 7- ( ( (3-chloropyrazin-2-yl) methyl) carbamoyl) -4-oxohexahydro-1H-
pyrido [1, 2-a] pyrazine-2 (6H) -carboxylate
To a solution of (7R, 9aS) -2- ( (benzyloxy) carbonyl) -4-oxooctahydro-1H-pyrido [1, 2-a] pyrazine-7-carboxylic acid (480 mg, 1.444 mmol) in anhydrous DMF (10 mL) was added EDC (415 mg, 2.166 mmol) , DMAP (265 mg, 2.166 mmol) , (3-chloropyrazin-2-yl) methanamine, HCl (312 mg, 1.733 mmol) , and TEA (0.302 mL, 2.166 mmol) under N2, and the mixture was stirred at 25 ℃ for 12 hrs. The mixture was poured into H2O (150 mL) , and extracted with EtOAc (20 mL × 5) . The organic layer was washed with H2O (10 mL × 6) , brine (30 mL) , dried over Na2SO4, filtered, and evaporated in vacuo to give the crude product, which was then purified by flash chromatography (DCM/THF=85 ~ 70 %) to afford the title compound.
1H NMR (400MHz CDCl3) δ = 8.44 (d, J=2.0 Hz, 1H) , 8.33 (s, 1H) , 6.98 (br. s., 1H) , 5.20 -5.10 (m, 2H) , 4.88 (d, J=12.9 Hz, 1H) , 4.68 (d, J=3.9 Hz, 2H) , 4.29 -4.18 (m, 1H) , 4.16 -4.05 (m, 1H) , 3.94 (br. s., 1H) , 3.41 (br. s., 2H) , 2.73 (t, J=12.3 Hz, 1H) , 2.39 (t, J=11.7 Hz, 1H) , 2.08 (d, J=11.3 Hz, 1H) , 1.95 -1.86 (m, 2H) , 1.41 -1.32 (m, 1H) ppm.
Step 4: (7R, 9aS) -benzyl 7- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -4-oxohexahydro-1H-
pyrido [1, 2-a] pyrazine-2 (6H) -carboxylate
To a solution of (7R, 9aS) -benzyl 7- ( ( (3-chloropyrazin-2-yl) methyl) carbamoyl) -4-oxohexahydro-1H-pyrido [1, 2-a] pyrazine-2 (6H) -carboxylate (440 mg, 0.673 mmol) in acetonitrile (12 mL) was added PCl5 (560 mg, 2.69 mmol) in portions at 0 ℃ under N2. After addition, the mixture was
stirred for 3 hours and then poured into 40 mL of sat. aq. NaHCO3, and extracted with DCM (10 mL × 4) . The organic layer was washed with brine (10 mL) , dried over Na2SO4, filtered, and evaporated to give the crude product, which was then purified by flash chromatography (DCM/THF=90~70%) to afford the title compound.
1H NMR (400MHz, CDCl3) δ = 7.87 (s, 1H) , 7.77 (d, J=4.8 Hz, 1H) , 7.45 -7.40 (m, 6H) , 5.23 (s, 2H) , 4.98 (d, J=13.3 Hz, 1H) , 4.27 (br. s., 2H) , 4.01 (s, 1H) , 3.61 (br. s., 2H) , 3.15 (s, 1H) , 2.88 (br. s., 1H) , 2.35 -2.26 (m, 2H) , 2.07 (s, 1H) , 1.63 (br. s., 1H) ppm.
Step 5: (7R, 9aS) -benzyl 7- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -4-oxohexahydro-1H-
pyrido [1, 2-a] pyrazine-2 (6H) -carboxylate
To a solution of (7R, 9aS) -benzyl 7- (8-chloroimidazo [1, 5-a] pyrazin-3-yl) -4-oxohexahydro-1H-pyrido [1, 2-a] pyrazine-2 (6H) -carboxylate (250 mg, 0.568 mmol) in anhydrous DMF (3 mL) was added 1-bromopyrrolidine-2, 5-dione (111 mg, 0.625 mmol) under N2. The mixture was stirred at 15 ℃ for 2 hours, then poured into an ice-water (20 mL) and 2 mL of sat. aq. NaHCO3 was added and the mixture was stirred for 2min. The mixture was then filtered, and the filter cake was washed with H2O (5 mL) , and then taken into EtOAc (60 mL) . The organic layer was washed with H2O (5 mL × 6) , brine (10 mL) , dried over Na2SO4, filtered and evaporated to afford the title compound, which was used in the next step directly.
1H NMR (400MHz, CDCl3) δ = 7.69 (d, J=5.1 Hz, 1H) , 7.37 (s, 6H) , 5.17 (s, 2H) , 4.89 (d, J=13.3 Hz, 1H) , 4.20 (br. s., 2H) , 3.91 (br. s., 1H) , 3.54 (br. s., 2H) , 3.07 -2.98 (m, 1H) , 2.78 (br. s., 1H) , 2.28 -2.17 (m, 2H) ppm.
Step 6: (7R, 9aS) -benzyl 7- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-
yl) -4-oxohexahydro-1H-pyrido [1, 2-a] pyrazine-2 (6H) -carboxylate
To a solution of (7R, 9aS) -benzyl 7- (1-bromo-8-chloroimidazo [1, 5-a] pyrazin-3-yl) -4-oxohexahydro-1H-pyrido [1, 2-a] pyrazine-2 (6H) -carboxylate (210 mg, 0.405 mmol) in anhydrous DMF (3 mL) was added K2CO3 (112 mg, 0.810 mmol) and followed by (2, 4-dimethoxyphenyl) methanamine (88 mg, 0.526 mmol) under N2. The mixture was stirred at 80 ℃ for 2 h, then poured into H2O (30 mL) , and extracted with EtOAc (10 mL × 3) . The organic layer was washed with H2O (5 mL × 5) , brine (10 mL) , dried over Na2SO4, filtered and evaporated. The residue was then purified by flash chromatography (DCM/THF=90 ~ 80%) to afford the title compound.
1H NMR (400MHz, CDCl3) δ = 7.36 (s, 5H) , 7.28 -7.26 (m, 1H) , 7.12 (s, 1H) , 6.74 (br. s., 1H) , 6.49 (s, 1H) , 6.44 (d, J=8.2 Hz, 1H) , 5.16 (s, 2H) , 4.88 (d, J=12.9 Hz, 1H) , 4.67 (d, J=5.5 Hz, 2H) , 4.19 (d, J=9.8 Hz, 2H) , 3.99 -3.91 (m, 1H) , 3.88 (s, 3H) , 3.80 (s, 3H) , 3.75 (br. s., 1H) ,
3.49 (br. s., 2H) , 2.95 (br. s., 1H) , 2.77 (d, J=11.7 Hz, 1H) , 2.14 (br. s., 2H) , 1.85 (br. s., 1H) ppm.
Step 7: (7R, 9aS) -benzyl 7- (8- ( (2, 4-dimethoxybenzyl) amino) -1- (2-ethoxy-4- ( (4-
(trifluoromethyl) pyridin-2-yl) carbamoyl) phenyl) imidazo [1, 5-a] pyrazin-3-yl) -4-oxohexahydro-
1H-pyrido [1, 2-a] pyrazine-2 (6H) -carboxylate
To a solution of (7R, 9aS) -benzyl 7- (1-bromo-8- ( (2, 4-dimethoxybenzyl) amino) imidazo [1, 5-a] pyrazin-3-yl) -4-oxohexahydro-1H-pyrido [1, 2-a] pyrazine-2 (6H) -carboxylate (150 mg, 0.231 mmol) , 3-ethoxy-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -N- (4- (trifluoromethyl) pyridin-2-yl) benzamide (111 mg, 0.254 mmol) and K2CO3 (96 mg, 0.693 mmol) in 1, 4-Dioxane (4.5 mL) and Water (1.5 mL) was added Pd (dppf) Cl2 (16.90 mg, 0.023 mmol) under N2. The mixture was stirred at 80 ℃ for 1.5 hrs then diluted with 5 mL of H2O, extracted with EtOAc (5 mL × 4) . The organic layer was washed with brine (10 mL) , dried over Na2SO4, filtered and evaporated to get the crude product, which was then purified by flash chromatography (DCM/THF=80 ~ 70%) to afford the title compound.
1H NMR (400MHz, CDCl3) δ = 8.73 (d, J=4.3 Hz, 1H) , 8.52 (d, J=5.0 Hz, 1H) , 7.59 -7.55 (m, 1H) , 7.52 -7.48 (m, 2H) , 7.41 -7.33 (m, 6H) , 7.19 (s, 2H) , 7.16 -7.12 (m, 1H) , 6.99 (s, 1H) , 6.41 -6.38 (m, 2H) , 5.53 (t, J=5.1 Hz, 1H) , 5.18 (s, 2H) , 5.01 -4.95 (m, 1H) , 4.56 (br. s., 2H) , 4.23 (d, J=8.5 Hz, 2H) , 3.95 (br. s., 1H) , 3.77 (s, 4H) , 3.53 (s, 5H) , 3.10 -3.01 (m, 1H) , 2.93 -2.84 (m, 1H) , 2.27 -2.19 (m, 2H) , 1.94 (s, 1H) , 1.56 (br. s., 1H) , 1.13 (t, J=6.9 Hz, 3H) ppm.
Step 8: 4- (8-amino-3- ( (7R, 9aS) -4-oxooctahydro-1H-pyrido [1, 2-a] pyrazin-7-yl) imidazo [1, 5-
a] pyrazin-1-yl) -3-ethoxy-N- (4- (trifluoromethyl) pyridin-2-yl) benzamide
A mixture of (7R, 9aS) -benzyl 7- (8- ( (2, 4-dimethoxybenzyl) amino) -1- (2-ethoxy-4- ( (4-(trifluoromethyl) pyridin-2-yl) carbamoyl) phenyl) imidazo [1, 5-a] pyrazin-3-yl) -4-oxohexahydro-1H-pyrido [1, 2-a] pyrazine-2 (6H) -carboxylate (170 mg, 0.193 mmol) in TFA (4 mL) was stirred at 80 ℃ for 4 hours. The mixture was cooled to room temperature and evaporated to give a residue, which was then dissolve with H2O (5 mL) , and basified to pH=8~9 with sat. aq. N NaHCO3. The mixture was extracted with DCM (10 mL x 3) . The organic layer was washed with brine (10 mL) , dried over Na2SO4evaporated in vacuo to afford the title compound, MS: 595.2 (M+1) , which was used in the next step directly without further purification.
Step 9: 4- (8-amino-3- (2- (2-methoxyethyl) -4-oxooctahydro-1H-pyrido [1, 2-a] pyrazin-7-
yl) imidazo [1, 5-a] pyrazin-1-yl) -3-ethoxy-N- (4- (trifluoromethyl) pyridin-2-yl) benzamide
To a solution of 4- (8-amino-3- (4-oxooctahydro-1H-pyrido [1, 2-a] pyrazin-7-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-ethoxy-N- (4- (trifluoromethyl) pyridin-2-yl) benzamide (50 mg, 0.00 mmol) in DMF (1 mL) was added sodium hydrogen carbonate (14.13 mg, 0.168 mmol) , followed by 1-
bromo-2-methoxyethane (11.69 mg, 0.084 mmol) , K2CO3 (35 mg, 0.25 mmol) and NaI (3 mg) . The mixture was stirred for 3 hrs at 30 ~ 40 ℃, poured into H2O (10 mL) , and extracted with DCM (5 mL×3) . The organic layer was evaporated to give a crude product, which was then purified by prep-HPLC (column: YMC-Actus Triart C18 150x30mmx5um; Condition: CH3CN/water with 0.1% TFA modifier) to afford the title compound. LCMS Method C, Retention time: 1.912 min, (M+H) + m/z : 653.3.
1H NMR (400MHz, CD3OD) δ = 8.66 -8.58 (m, 2H) , 7.84 (d, J=5.9 Hz, 1H) , 7.80 -7.74 (m, 2H) , 7.72 -7.67 (m, 1H) , 7.45 (d, J=4.7 Hz, 1H) , 7.02 (d, J=5.9 Hz, 1H) , 4.26 (q, J=6.7 Hz, 2H) , 4.05 -3.79 (m, 5H) , 3.75 (d, J=4.3 Hz, 2H) , 3.43 (s, 6H) , 3.28 -3.23 (m, 1H) , 3.08 (t, J=12.5 Hz, 1H) , 2.29 (d, J=13.7 Hz, 1H) , 2.13 -1.96 (m, 2H) , 1.72 -1.57 (m, 1H) , 1.34 (t, J=6.8 Hz, 3H) ppm.
Biological Activity
The Btk inhibitor compounds of the invention having Formula I inhibit the Btk kinase activity. All compounds of the invention have an IC50 of 10 μM or lower. In another aspect the invention relates to compounds of Formula I which have an IC50 of less than 100 nM. In yet another aspect the invention relates to compounds of Formula I which have an IC50 of less than 10 nM.
The term IC50 means the concentration of the test compound that is required for 50% inhibition of its maximum effect in vitro.
Btk enzyme activity Assay Methods
BTK enzymatic activity was determined with the LANCE (Lanthanide Chelate Excite) TR-FRET (Time-resolved fluorescence resonance energy transfer) assay. In this assay, the potency (IC50) of each compound was determined from an eleven point (1: 3 serial dilution; final compound concentration range in assay from 1 μM to 0.017 nM) titration curve using the following outlined procedure. To each well of a black non-binding surface Corning 384-well microplate (Corning Catalog #3820) , 5 nL of compound (2000 fold dilution in final assay volume of 10 μL) was dispensed, followed by the addition of 7.5 μL of 1x kinase buffer (50 mM Hepes 7.5, 10 mM MgCl2, 0.01% Brij-35, 1 mM EGTA, 0.05% BSA & 1 mM DTT) containing 5.09 pg/L (66.67 pM) of BTK enzyme (recombinant protein from baculovirus-transfected Sf9 cells: full-length BTK, 6HIS-tag cleaved) . Following a 60 minute compound & enzyme incubation, each reaction was initiated by the addition of 2.5 μL 1x kinase buffer containing 8 μM biotinylated “A5” peptide (Biotin-EQEDEPEGDYFEWLE-NH2) (SEQ. ID. NO. : 1) , and 100 μM ATP. The final reaction in each well of 10 μL consists of 50 pM hBTK, 2 μM biotin-A5-peptide, and 25 μM ATP. Phosphorylation reactions were allowed to proceed for 120 minutes. Reactions were immediately quenched by the addition of 20 uL of 1x quench buffer (15 mM EDTA, 25 mM Hepes 7.3, and 0.1%
Triton X-100) containing detection reagents (0.626 nM of LANCE-Eu-W1024-anti-phosphoTyrosine antibody, PerkinElmer and 86.8 nM of Streptavidin-conjugated Dylight 650, Dyomics/ThermoFisher Scientific) . After 60 minutes incubation with detection reagents, reaction plates were read on a PerkinElmer EnVision plate reader using standard TR-FRET protocol. Briefly, excitation of donor molecules (Eu-chelate: anti-phospho-antibody) with a laser light source at 337 nm produces energy that can be transferred to Dylight-650 acceptor molecules if this donor: acceptor pair is within close proximity. Fluorescence intensity at both 665 nm (acceptor) and 615 nm (donor) are measured and a TR-FRET ratio calculated for each well (acceptor intensity/donor intensity) . IC50 values were determined by 4 parameter robust fit of TR-FRET ratio values vs. (Log10) compound concentrations.
The following Table 2 provides specific IC50 values for all the examples. The IC50 values set forth below were determined according to Assay method described above.
Table 2. Compounds BTK binding potency




Claims (14)
- A compound according to Formula I, or a pharmaceutically acceptable salt, thereof
 wherein:R11 is independently selected from the group consisting of:a) H,b) halogen,c) cyano,d) (1-6C) alkyl,wherein in aromatic ring KB1 is N or C (R7) ;B2 is N or C (R8) ;R7 is H, halogen, (1-3C) alkyl, (1-6C) alkoxy, (3-6C) cycloalkoxy, amino, or (1-3C) alkylamino;R8 is H, halogen, (1-3C) alkyl, (1-6C) alkoxy, (3-6C) cycloalkoxy, amino, or (1-3C) alkylamino;R9 is H, halogen, (1-3C) alkyl, (1-6C) alkoxy, (3-6C) cycloalkoxy, amino, or (1-3C) alkylamino;R10 is H, halogen, (1-3C) alkyl, (1-6C) alkoxy, (3-6C) cycloalkoxy, amino, or (1-3C) alkylamino;wherein heteroaromatic ring L is selected from the group consisting of:
wherein:R11 is independently selected from the group consisting of:a) H,b) halogen,c) cyano,d) (1-6C) alkyl,wherein in aromatic ring KB1 is N or C (R7) ;B2 is N or C (R8) ;R7 is H, halogen, (1-3C) alkyl, (1-6C) alkoxy, (3-6C) cycloalkoxy, amino, or (1-3C) alkylamino;R8 is H, halogen, (1-3C) alkyl, (1-6C) alkoxy, (3-6C) cycloalkoxy, amino, or (1-3C) alkylamino;R9 is H, halogen, (1-3C) alkyl, (1-6C) alkoxy, (3-6C) cycloalkoxy, amino, or (1-3C) alkylamino;R10 is H, halogen, (1-3C) alkyl, (1-6C) alkoxy, (3-6C) cycloalkoxy, amino, or (1-3C) alkylamino;wherein heteroaromatic ring L is selected from the group consisting of: R5 is H, cyano, (1-4C) alkyl, (3-6C) cycloalkyl, or (3-6C) cycloalkoxy,wherein R5 may optionally be substituted with one, two or three halogens;wherein ring M is selected from the group consisting of:
R5 is H, cyano, (1-4C) alkyl, (3-6C) cycloalkyl, or (3-6C) cycloalkoxy,wherein R5 may optionally be substituted with one, two or three halogens;wherein ring M is selected from the group consisting of: Q is C=O or CH2;T is C (Re) 2, O, NRe, or a bond;U is C (Rd) 2, O, or NRd;V is CH2 or O;each Rc is independently selected from H, fluoro, methyl or trifluoromethyl;each Rd is independently selected from H, (1-3C) alkyl, (1-3C) alkoxy, cyclopropyl or cyclopropylmethylene;each Re is independently selected from H or (1-6C) alkyl; andwith the proviso that:when Q is CH2, then T is C (Re) 2.
Q is C=O or CH2;T is C (Re) 2, O, NRe, or a bond;U is C (Rd) 2, O, or NRd;V is CH2 or O;each Rc is independently selected from H, fluoro, methyl or trifluoromethyl;each Rd is independently selected from H, (1-3C) alkyl, (1-3C) alkoxy, cyclopropyl or cyclopropylmethylene;each Re is independently selected from H or (1-6C) alkyl; andwith the proviso that:when Q is CH2, then T is C (Re) 2. - The compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein the ring M is selected from the group consisting of

- The compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein the heteroaromatic ring L isand
 B1 is CH; andB2 is CH.
B1 is CH; andB2 is CH. - The compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein R5 is selected from the group consisting of hydrogen, CN, cyclopropyl, (1-4C) alkyl, and (3-6C) cycloalkoxy; wherein the alkyl may optionally be substituted with one, two or three halogen.
- The compound of claim 4, or a pharmaceutically acceptable salt thereof, wherein R5 is selected from the group consisting of hydrogen, methyl, cyclopropyl, cyclopropoxyl, and trifluoromethyl.
- The compound of claim 1 selected from the group consisting of:4- (8-amino-5-fluoro-3- ( (3R, 9aR) -8-isopropyl-6-oxooctahydropyrazino [2, 1-c] [1, 4] oxazin-3-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-ethoxy-5-fluoro-N- (4- (trifluoromethyl) pyridin-2-yl) benzamide4- (8-amino-5-chloro-3- ( (6R, 8aS) -3-oxooctahydroindolizin-6-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-fluoro-5-methoxy-N- (1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl) benzamide4- (8-amino-5-fluoro-3- ( (6R, 8aS) -3-oxooctahydroindolizin-6-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-fluoro-5-methoxy-N- (1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl) benzamide4- (8-amino-3- ( (3S, 9aR) -9, 9-difluorooctahydro-2H-quinolizin-3-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-ethoxy-N- (4- (trifluoromethyl) pyridin-2-yl) benzamide4- (8-amino-3- ( (3R, 9aR) -9, 9-difluorooctahydro-2H-quinolizin-3-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-ethoxy-N- (4- (trifluoromethyl) pyridin-2-yl) benzamidecis 4- (8-amino-3- (9, 9-difluorooctahydro-2H-quinolizin-3-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-ethoxy-N- (4- (trifluoromethyl) pyridin-2-yl) benzamide4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- (1-tert-butyl-1H-1, 2, 3-triazol-4-yl) -5-ethoxy-2-fluorobenzamide4- {8-amino-5-chloro-3- [ (3R, 8aR) -6-oxohexahydro-1H-pyrrolo [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-fluoro-5-methoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (3R, 8aR) -6-oxohexahydro-1H-pyrrolo [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-fluoro-5-methoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-5-fluoro-3- [ (3R, 8aR) -6-oxohexahydro-1H-pyrrolo [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-fluoro-5-methoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (difluoromethyl) pyridin-2-yl] -3-ethoxybenzamide4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (difluoromethyl) pyridin-2-yl] -3-methoxybenzamide4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (difluoromethyl) pyridin-2-yl] -3-methoxybenzamide4- {8-amino-5-chloro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (difluoromethyl) pyridin-2-yl] -3-ethoxybenzamide4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (1, 1-difluoroethyl) pyridin-2-yl] -3-ethoxybenzamide4- {8-amino-5-chloro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (1, 1-difluoroethyl) pyridin-2-yl] -3-ethoxybenzamide4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-propoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-propoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-fluoro-5-methoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-fluoro-5-methoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-5-cyano-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-5-fluoro-3- [ (6R) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3- (cyclopropyloxy) -N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-5-chloro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-5-chloro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N-pyridin-2-ylbenzamide4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N-pyridin-2-ylbenzamide4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- (4-cyanopyridin-2-yl) -3-ethoxy-5-fluorobenzamide4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [6- (trifluoromethyl) pyrimidin-4-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -2-chloro-5-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -2-chloro-5-ethoxy-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -5-ethoxy-2, 3-difluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -2-chloro-5-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -2-chloro-5-ethoxy-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -2-chloro-5-ethoxy-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide4- {8-amino-5-chloro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -2-chloro-5-ethoxy-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide4- {8-amino-5-fluoro-3- [ (3R, 9aR) -8- (1-methylethyl) -6-oxooctahydropyrazino [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -2-chloro-5-ethoxy-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -2-chloro-5-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-5-chloro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -2-chloro-5-ethoxy-N-pyridin-2-ylbenzamide4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -2-chloro-5-ethoxy-N-pyridin-2-ylbenzamide4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N-pyridin-2-ylbenzamide4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -5-ethoxy-2-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide5- {8-amino-5-chloro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -4-methyl-N- [4- (trifluoromethyl) pyridin-2-yl] pyridine-2-carboxamide4- {8-amino-3- [ (6S, 8aR) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (6S, 8aR) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (6S, 8aR) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (7S, 9aR) -octahydropyrido [2, 1-c] [1, 4] oxazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (3R, 9aR) -9, 9-difluoro-6-oxooctahydro-2H-quinolizin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (3R, 9aR) -9, 9-difluoro-6-oxooctahydro-2H-quinolizin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (7R, 9aS) -octahydropyrido [2, 1-c] [1, 4] oxazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (6S, 8aR) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (3S, 9aS) -9, 9-difluoro-6-oxooctahydro-2H-quinolizin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (3S, 9aS) -9, 9-difluoro-6-oxooctahydro-2H-quinolizin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (3R, 8aR) -6-oxohexahydro-1H-pyrrolo [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (1S, 6R, 8aR) -3-oxo-1- (trifluoromethyl) octahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (3R, 9aS) -9, 9-difluoro-6-oxooctahydro-2H-quinolizin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (3R, 9aS) -9, 9-difluoro-6-oxooctahydro-2H-quinolizin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (3S, 9aR) -9, 9-difluoro-6-oxooctahydro-2H-quinolizin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (3S, 9aR) -9, 9-difluoro-6-oxooctahydro-2H-quinolizin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-fluoro-5-methoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (1S, 6R, 8aS) -1-methyl-3-oxotetrahydro-1H- [1, 3] oxazolo [4, 3-c] [1, 4] oxazin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (1R, 6R, 8aS) -1-methyl-3-oxotetrahydro-1H- [1, 3] oxazolo [4, 3-c] [1, 4] oxazin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (3R, 9aR) -6-oxohexahydro-1H- [1, 4] oxazino [3, 4-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] -5-fluoroimidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] -5-fluoroimidazo [1, 5-a] pyrazin-1-yl} -3-fluoro-5-methoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizin-6-yl] -5-fluoroimidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (1R, 6R, 8aS) -3-oxo-1- (trifluoromethyl) octahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (1R, 6R, 8aS) -3-oxo-1- (trifluoromethyl) octahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -5-ethoxy-2-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizin-6-yl] -5-methylimidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxotetrahydro-1H- [1, 3] oxazolo [4, 3-c] [1, 4] oxazin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizin-6-yl] -5-methylimidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -5-ethoxy-2-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide5- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -4-ethoxy-N-[4- (trifluoromethyl) pyridin-2-yl] pyridine-2-carboxamide4- {8-amino-3- [ (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizin-6-yl] -5-fluoroimidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizin-6-yl] -5-fluoroimidazo [1, 5-a] pyrazin-1-yl} -3-fluoro-5-methoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- (4-cyclopropylpyridin-2-yl) -3-ethoxy-5-fluorobenzamide4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- (4-cyclopropylpyridin-2-yl) -3-ethoxy-5-fluorobenzamide4- {8-amino-3- [ (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -5-ethoxy-2-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -5-ethoxy-2-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizin-6-yl] -5-fluoroimidazo [1, 5-a] pyrazin-1-yl} -5-ethoxy-2-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizin-6-yl] -5-fluoroimidazo [1, 5-a] pyrazin-1-yl} -5-ethoxy-2-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -5-ethoxy-2-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- (4-cyclopropylpyridin-2-yl) -3-ethoxy-5-fluorobenzamide4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (difluoromethyl) pyridin-2-yl] -3-ethoxy-5-fluorobenzamide4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (1, 1-difluoroethyl) pyridin-2-yl] -3-ethoxy-5-fluorobenzamide4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- (4-cyclopropylpyridin-2-yl) -3-ethoxy-5-fluorobenzamide4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -2, 2-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- (4-cyclopropylpyridin-2-yl) -5-ethoxy-2-fluorobenzamide4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- (4-cyclopropylpyridin-2-yl) -5-ethoxy-2-fluorobenzamide4- [8-amino-3- (2-ethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidin-7-yl) imidazo [1, 5-a] pyrazin-1-yl] -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- [8-amino-3- (2-ethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidin-7-yl) imidazo [1, 5-a] pyrazin-1-yl] -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- [8-amino-3- (1, 1-dimethyl-3-oxooctahydroindolizin-6-yl) imidazo [1, 5-a] pyrazin-1-yl] -3-ethoxy-5-fluoro-N-pyridin-2-ylbenzamide4- [8-amino-3- (1-oxohexahydro-3H-pyrido [1, 2-c] [1, 3] oxazin-7-yl) imidazo [1, 5-a] pyrazin-1-yl] -3-ethoxy-5-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- [8-amino-3- (1-oxohexahydro-3H-pyrido [1, 2-c] [1, 3] oxazin-7-yl) imidazo [1, 5-a] pyrazin-1-yl] -3-ethoxy-5-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4-amino-5- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (trifluoromethyl) pyridin-2-yl] pyridine-2-carboxamide5- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -6-methoxy-N- [4- (trifluoromethyl) pyridin-2-yl] pyridine-2-carboxamide5- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (trifluoromethyl) pyridin-2-yl] pyrimidine-2-carboxamide5- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -6- (ethylamino) -N- [4- (trifluoromethyl) pyridin-2-yl] pyridine-2-carboxamide4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-chloro-5-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-chloro-5-ethoxy-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-chloro-5-ethoxy-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-chloro-N- [4- (difluoromethyl) pyridin-2-yl] -5-ethoxybenzamide4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-chloro-5-ethoxy-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-chloro-5-methoxy-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide4- {8-amino-3- [ (3R, 8aR) -6-oxohexahydro-1H-pyrrolo [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (3R, 9aR) -8- (1-methylethyl) -6-oxooctahydropyrazino [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (3R, 9aR) -8- (1-methylethyl) -6-oxooctahydropyrazino [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide4- {8-amino-3- [ (3R, 9aR) -8- (1-methylethyl) -6-oxooctahydropyrazino [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (3R, 9aR) -8-cyclopropyl-6-oxooctahydropyrazino [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (3R, 9aR) -8-cyclopropyl-6-oxooctahydropyrazino [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (3R, 9aR) -8-tert-butyl-6-oxooctahydropyrazino [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (3R, 9aR) -8-tert-butyl-6-oxooctahydropyrazino [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-5-chloro-3- [ (3R, 9aR) -8- (1-methylethyl) -6-oxooctahydropyrazino [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-5-fluoro-3- [ (3R, 9aR) -8- (1-methylethyl) -6-oxooctahydropyrazino [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-5-fluoro-3- [ (3R, 9aR) -8- (1-methylethyl) -6-oxooctahydropyrazino [2, 1-c] [1, 4] oxazin-3-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxohexahydro [1, 3] oxazolo [3, 4-a] pyridin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-5-chloro-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (7R, 9aR) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (7R, 9aR) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N-pyridin-2-ylbenzamide4- {8-amino-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N-pyridin-2-ylbenzamide4- {8-amino-5-chloro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N-pyridin-2-ylbenzamide4- {8-amino-5-chloro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-chloro-5-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N-pyridin-2-ylbenzamide4- [8-amino-3- (octahydropyrido [2, 1-c] [1, 4] oxazin-7-yl) imidazo [1, 5-a] pyrazin-1-yl] -3-ethoxy-5-fluoro-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- [8-amino-3- (octahydropyrido [2, 1-c] [1, 4] oxazin-7-yl) imidazo [1, 5-a] pyrazin-1-yl] -3-ethoxy-5-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N-pyridin-2-ylbenzamide4- {8-amino-5-chloro-3- [ (2R, 6R, 8aS) -2- (cyclopropylmethyl) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-5-chloro-3- [ (2R, 6R, 8aS) -2- (cyclopropylmethyl) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (3R, 7R, 9aS) -3-methyl-4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-methoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] -5-fluoroimidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N-pyridin-2-ylbenzamide4- {8-amino-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] -5-fluoroimidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide4- [8-amino-3- (2, 3, 3-trimethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidin-7-yl) imidazo [1, 5-a] pyrazin-1-yl] -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (1, 1-difluoroethyl) pyridin-2-yl] -3-ethoxybenzamide4- [8-amino-3- (2, 3, 3-trimethyl-1-oxooctahydro-1H-pyrido [1, 2-c] pyrimidin-7-yl) imidazo [1, 5-a] pyrazin-1-yl] -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -5-ethoxy-2-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (difluoromethyl) pyridin-2-yl] -3-ethoxybenzamide4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- (4-cyclopropylpyridin-2-yl) -3-ethoxybenzamide4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (cyclopropyloxy) pyridin-2-yl] -3-ethoxy-5-fluorobenzamide4- {8-amino-5-fluoro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- [4- (cyclopropyloxy) pyridin-2-yl] -3-ethoxybenzamide4- {8-amino-3- [ (7R, 9aS) -2- (1-methylethyl) -4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-fluoro-5-methoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (7R, 9aS) -3, 3-dimethyl-4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-3- [ (3S, 7R, 9aS) -3-methyl-4-oxooctahydro-2H-pyrido [1, 2-a] pyrazin-7-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-N- [4- (trifluoromethyl) pyridin-2-yl] benzamide4- {8-amino-5-chloro-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N- [1-methyl-5- (trifluoromethyl) -1H-pyrazol-3-yl] benzamide4- {8-amino-5-chloro-3- [ (6R, 8aS) -1, 1-dimethyl-3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -3-ethoxy-5-fluoro-N-pyridin-2-ylbenzamide4- [8-amino-3- (3, 3-dimethyl-1-oxohexahydro-3H-pyrido [1, 2-c] [1, 3] oxazin-7-yl) imidazo [1, 5-a] pyrazin-1-yl] -N- [4- (difluoromethyl) pyridin-2-yl] -3-ethoxy-5-fluorobenzamide4- {8-amino-5-chloro-3- [ (6R, 8aS) -3-oxooctahydroindolizin-6-yl] imidazo [1, 5-a] pyrazin-1-yl} -N- (4-cyanopyridin-2-yl) -3-ethoxy-5-fluorobenzamide4- [8-amino-3- (3, 3-dimethyl-1-oxohexahydro-3H-pyrido [1, 2-c] [1, 3] oxazin-7-yl) imidazo [1, 5-a] pyrazin-1-yl] -N- [4- (difluoromethyl) pyridin-2-yl] -3-ethoxy-5-fluorobenzamide4- (8-amino-3- (2- (2-methoxyethyl) -4-oxooctahydro-1H-pyrido [1, 2-a] pyrazin-7-yl) imidazo [1, 5-a] pyrazin-1-yl) -3-ethoxy-N- (4- (trifluoromethyl) pyridin-2-yl) benzamideor a pharmaceutically acceptable salt thereof
- A pharmaceutical composition which comprises the compound of claim 1 or a pharmaceutically acceptable salt thereof and one or more pharmaceutically acceptable carriers.
- The pharmaceutical composition of claim 7, which further comprises at least one additional therapeutically active agent.
- The compound of claim 1 or a pharmaceutically acceptable salt thereof for use in therapy.
- The compound of claim 1 or a pharmaceutically acceptable salt thereof for use in the treatment of Bruton’s Tyrosine Kinase (Btk) mediated disorders.
- Use of the compound of Formula I according to claim 1 or a pharmaceutically acceptable salt thereof for the manufacture of a medicament for the treatment of Bruton’s Tyrosine Kinase (Btk) mediated disorders.
- A method for treating a subject suffering with a Bruton's Tyrosine Kinase (Btk) mediated disorder comprising administering to the subject the compound of claim 1 in an amount effective to treat the Btk mediated disorder, thereby treating the subject.
- The method of claim 12, wherein the Btk mediated disorder is selected from the group consisting of rheumatoid arthritis, psoriatic arthritis, infectious arthritis, progressive chronic arthritis, deforming arthritis, osteoarthritis, traumatic arthritis, gouty arthritis, Reiter’s syndrome, polychondritis, acute synovitis and spondylitis, glomerulonephritis (with or without nephrotic syndrome) , autoimmune hematologic disorders, hemolytic anemia, aplasic anemia, idiopathic thrombocytopenia, and neutropenia, autoimmune gastritis, and autoimmune inflammatory bowel diseases, ulcerative colitis, Crohn’s disease, host versus graft disease, allograft rejection, chronic thyroiditis, Graves’ disease, schleroderma, diabetes (type I and type II) , active hepatitis (acute and chronic) , pancreatitis, primary billiary cirrhosis, myasthenia gravis, multiple sclerosis, systemic lupus erythematosis, psoriasis, atopic dermatitis, contact dermatitis, eczema, skin sunburns, vasculitis (e.g. Behcet’s disease) chronic renal insufficiency, Stevens-Johnson syndrome, inflammatory pain, idiopathic sprue, cachexia, sarcoidosis, Guillain-Barré syndrome, uveitis, conjunctivitis, kerato conjunctivitis, otitis media, periodontal disease, pulmonary interstitial fibrosis, asthma, bronchitis, rhinitis, sinusitis, pneumoconiosis, pulmonary insufficiency syndrome, pulmonary emphysema, pulmonary fibrosis, silicosis, chronic inflammatory pulmonary disease, and chronic obstructive pulmonary disease.
- The method of claim 13, wherein the Btk mediated disorder is rheumatoid arthritis, psoriatic arthritis, or osteoarthritis.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/CN2014/095766 WO2016106625A1 (en) | 2014-12-31 | 2014-12-31 | Btk inhibitors |
| PCT/US2015/066229 WO2016109221A1 (en) | 2014-12-31 | 2015-12-17 | Btk inhibitors |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/CN2014/095766 WO2016106625A1 (en) | 2014-12-31 | 2014-12-31 | Btk inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016106625A1 true WO2016106625A1 (en) | 2016-07-07 |
Family
ID=56283899
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2014/095766 Ceased WO2016106625A1 (en) | 2014-12-31 | 2014-12-31 | Btk inhibitors |
| PCT/US2015/066229 Ceased WO2016109221A1 (en) | 2014-12-31 | 2015-12-17 | Btk inhibitors |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2015/066229 Ceased WO2016109221A1 (en) | 2014-12-31 | 2015-12-17 | Btk inhibitors |
Country Status (1)
| Country | Link |
|---|---|
| WO (2) | WO2016106625A1 (en) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020015735A1 (en) * | 2018-07-20 | 2020-01-23 | 正大天晴药业集团股份有限公司 | Bruton tyrosine kinase inhibitors |
| US11071730B2 (en) | 2018-10-31 | 2021-07-27 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds |
| US11203591B2 (en) | 2018-10-31 | 2021-12-21 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds |
| US11453681B2 (en) | 2019-05-23 | 2022-09-27 | Gilead Sciences, Inc. | Substituted eneoxindoles and uses thereof |
| WO2023110970A1 (en) | 2021-12-14 | 2023-06-22 | Netherlands Translational Research Center Holding B.V | Macrocyclic btk inhibitors |
| WO2024246287A1 (en) | 2023-06-02 | 2024-12-05 | Crossfire Oncology Holding B.V. | Medical use of a macrocyclic reversible btk inhibitor |
| WO2024245578A1 (en) | 2023-06-02 | 2024-12-05 | Netherlands Translational Research Center Holding B.V. | Therapeutic combinations of an irreversible btk inhibitor and a macrocyclic reversible btk inhibitor |
| WO2024245577A1 (en) | 2023-06-02 | 2024-12-05 | Netherlands Translational Research Center Holding B.V. | Therapeutic combinations of an irreversible btk inhibitor and a reversible btk inhibitor |
| WO2024256568A1 (en) | 2023-06-13 | 2024-12-19 | Crossfire Oncology Holding B.V. | Salt and crystal forms of a macrocyclic btk inhibitor |
| WO2024256574A1 (en) | 2023-06-13 | 2024-12-19 | Crossfire Oncology Holding B.V. | Process for preparing macrocyclic btk inhibitors |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN115443136A (en) * | 2020-04-22 | 2022-12-06 | 普林斯匹亚生物制药公司 | Treatment of acute respiratory distress syndrome and other disorders involving cytokine storm with BTK inhibitors |
| US20240100172A1 (en) | 2020-12-21 | 2024-03-28 | Hangzhou Jijing Pharmaceutical Technology Limited | Methods and compounds for targeted autophagy |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013010380A1 (en) * | 2011-07-19 | 2013-01-24 | Merck Sharp & Dohme Corp. | Btk inhibitors |
| WO2013010868A1 (en) * | 2011-07-19 | 2013-01-24 | Msd Oss B.V. | 4 - imidazopyridazin- 1 -yl-benzamides and 4 - imidazotriazin- 1 - yl - benzamides as btk- inhibitors |
| WO2014113942A1 (en) * | 2013-01-23 | 2014-07-31 | Merck Sharp & Dohme Corp. | Btk inhibitors |
| WO2014114185A1 (en) * | 2013-01-23 | 2014-07-31 | Merck Sharp & Dohme Corp. | Btk inhibitors |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8957080B2 (en) * | 2013-04-09 | 2015-02-17 | Principia Biopharma Inc. | Tyrosine kinase inhibitors |
-
2014
- 2014-12-31 WO PCT/CN2014/095766 patent/WO2016106625A1/en not_active Ceased
-
2015
- 2015-12-17 WO PCT/US2015/066229 patent/WO2016109221A1/en not_active Ceased
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013010380A1 (en) * | 2011-07-19 | 2013-01-24 | Merck Sharp & Dohme Corp. | Btk inhibitors |
| WO2013010868A1 (en) * | 2011-07-19 | 2013-01-24 | Msd Oss B.V. | 4 - imidazopyridazin- 1 -yl-benzamides and 4 - imidazotriazin- 1 - yl - benzamides as btk- inhibitors |
| WO2014113942A1 (en) * | 2013-01-23 | 2014-07-31 | Merck Sharp & Dohme Corp. | Btk inhibitors |
| WO2014114185A1 (en) * | 2013-01-23 | 2014-07-31 | Merck Sharp & Dohme Corp. | Btk inhibitors |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020015735A1 (en) * | 2018-07-20 | 2020-01-23 | 正大天晴药业集团股份有限公司 | Bruton tyrosine kinase inhibitors |
| CN112424203A (en) * | 2018-07-20 | 2021-02-26 | 正大天晴药业集团股份有限公司 | Bruton's tyrosine kinase inhibitors |
| US11897878B2 (en) | 2018-10-31 | 2024-02-13 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds |
| US11203591B2 (en) | 2018-10-31 | 2021-12-21 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds |
| US11071730B2 (en) | 2018-10-31 | 2021-07-27 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds |
| US11925631B2 (en) | 2018-10-31 | 2024-03-12 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds |
| US12258346B2 (en) | 2018-10-31 | 2025-03-25 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds |
| US11453681B2 (en) | 2019-05-23 | 2022-09-27 | Gilead Sciences, Inc. | Substituted eneoxindoles and uses thereof |
| US12037342B2 (en) | 2019-05-23 | 2024-07-16 | Gilead Sciences, Inc. | Substituted eneoxindoles and uses thereof |
| WO2023110970A1 (en) | 2021-12-14 | 2023-06-22 | Netherlands Translational Research Center Holding B.V | Macrocyclic btk inhibitors |
| WO2024246287A1 (en) | 2023-06-02 | 2024-12-05 | Crossfire Oncology Holding B.V. | Medical use of a macrocyclic reversible btk inhibitor |
| WO2024245578A1 (en) | 2023-06-02 | 2024-12-05 | Netherlands Translational Research Center Holding B.V. | Therapeutic combinations of an irreversible btk inhibitor and a macrocyclic reversible btk inhibitor |
| WO2024245577A1 (en) | 2023-06-02 | 2024-12-05 | Netherlands Translational Research Center Holding B.V. | Therapeutic combinations of an irreversible btk inhibitor and a reversible btk inhibitor |
| WO2024256568A1 (en) | 2023-06-13 | 2024-12-19 | Crossfire Oncology Holding B.V. | Salt and crystal forms of a macrocyclic btk inhibitor |
| WO2024256574A1 (en) | 2023-06-13 | 2024-12-19 | Crossfire Oncology Holding B.V. | Process for preparing macrocyclic btk inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2016109221A1 (en) | 2016-07-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9481682B2 (en) | Substituted benzamides and substituted pyridinecarboxamides as Btk inhibitors | |
| US9446130B2 (en) | BTK inhibitors | |
| US10040805B2 (en) | Substituted imidazo[1,5-a]pyrazines as Btk inhibitors | |
| EP3240543B1 (en) | Btk inhibitors | |
| WO2016106625A1 (en) | Btk inhibitors | |
| EP3082811B1 (en) | Btk inhibitors | |
| WO2016106624A1 (en) | Tertiary alcohol imidazopyrazine btk inhibitors | |
| WO2016161570A1 (en) | Azacarbazole btk inhibitors | |
| EP3082809A1 (en) | Btk inhibitors | |
| WO2014113932A1 (en) | Btk inhibitors | |
| WO2016109219A1 (en) | Biarylether imidazopyrazine btk inhibitors | |
| WO2016106627A1 (en) | Btk inhibitors | |
| WO2016192074A1 (en) | Btk inhibitors | |
| EP3240542B1 (en) | Btk inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14909434 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 14909434 Country of ref document: EP Kind code of ref document: A1 |