WO2011018894A1 - Pyrrolopyrimidine derivatives as potassium channel modulators - Google Patents
Pyrrolopyrimidine derivatives as potassium channel modulators Download PDFInfo
- Publication number
- WO2011018894A1 WO2011018894A1 PCT/JP2010/005024 JP2010005024W WO2011018894A1 WO 2011018894 A1 WO2011018894 A1 WO 2011018894A1 JP 2010005024 W JP2010005024 W JP 2010005024W WO 2011018894 A1 WO2011018894 A1 WO 2011018894A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- substituted
- pyrrolo
- pyrimidin
- unsubstituted
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 Cc1c(c(C(O)=O)c[n]2*)c2ncn1 Chemical compound Cc1c(c(C(O)=O)c[n]2*)c2ncn1 0.000 description 2
- GVDQXTOWBFFOGV-UHFFFAOYSA-N C[n]1c2ncnc(Nc3ccccc3)c2c(C(O)=O)c1 Chemical compound C[n]1c2ncnc(Nc3ccccc3)c2c(C(O)=O)c1 GVDQXTOWBFFOGV-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
Definitions
- the present invention relates to pyrrolopyrimidine derivatives which have activation activities of potassium channels as the SK channels, and which are useful in the treatment or prevention of disorders and diseases in which potassium channels are involved.
- the invention also relates to pharmaceutical compositions comprising these compounds and the use of these compounds and compositions in the prevention or treatment of such diseases in which potassium channels are involved.
- the present invention also relates to the prevention or treatment of such diseases, which comprises administering the above-mentioned pyrrolopyrimidine derivatives to a subject in need of such a prevention or treatment a therapeutically effective amount of said derivative.
- K + channels play a fundamental role in the homeostasis of cell function through the regulation of the transmembrane movement of ions, including K + , Na + , Ca 2+ and Cl - .
- Cellular activity can be affected by modifications of the activities of the ion channels.
- K + channels are a diverse and ubiquitous group of ion channels and have several critical roles in cell function. These channels principally regulate the resting membrane potential of the cell and attenuate the level of excitation of cells. For example, the outward flow of K + upon opening of K + channels makes the interior of the cell more negative, counteracting depolarizing voltages applied to the cells.
- K + channels are regulated by voltage, cell metabolism, second messengers, and receptor mediated processes.
- SK channels small conductance Ca 2+ -activated K + channels
- Ca 2+ -activated K + channels consist of three subtypes, namely SK1, SK2 and SK3. All have a topology that is typical for K + channels with six transmembrane helices and a P-loop region.
- SK channels are voltage-insensitive and can be activated by increases in the levels of intracellular Ca 2+ , such as what occurs during an action potential. Their activation is important for regulating afterhyperpolarization (AHP) and limits the firing frequency of action potentials in neurons and other cell types.
- AHP afterhyperpolarization
- SK channels are widely expressed in the brain.
- SK1 and SK2 channels are particular enriched in the cortex and hippocampus, while SK3 is also prominent in subcortical areas, notably the striatum, thalamus and monoaminergic nuclei.
- SK channels are involved in neurophysiological processes like spike frequency adaptation, pacemaking, as well as synaptic integration and plasticity.
- SK2 and SK3, in contrast to SK1, are also expressed in many other peripheral tissues, particular those rich in smooth muscle. Activation of SK channels leads to membrane potential hyperpolarization and relaxation of a variety of smooth muscles.
- SK3 channels are the only known SK subtype expressed in denervated skeletal muscle and breast tumor tissue.
- SK3 channel contains two polymorphic polyglutamine repeats in its N-terminus. Genetic studies find over-representation of longer alleles of the second SK3 polyglutamine repeat in patients with schizophrenia, anorexia nervosa, and spinocerebellar ataxia. In one schizophrenia patient a truncation mutant of SK3 channel, acting as a dominant-negative suppressor of SK channels, is also identified. Mutations in SK3 channel are thus suspected to be a possible underlying cause for several psychiatric disorders.
- SK channel activity i.e., opener or positive gating modulator
- SK channel activity i.e., opener or positive gating modulator
- diseases including, schizophrenia, ataxia, epilepsy, pain, hypertension, urinary incontinence, erectile dysfunction, irritable bowel syndrome (Curr Med Chem. 2007;14) and Parkinson's disease (Exp Neurol. 2008; 213).
- WO 2005/047289 Structurally close compounds are disclosed in WO 2005/047289. However, the compounds there don't have any substituents at amino group on the pyrrolopyrimidine ring, which is quite different from the compounds of this invention.
- the compounds in WO 2005/047289 are useful for the treatment of abnormal cell growth such as hyperproliferative disease represented by cancers, whereas the compounds of this invention are useful for the treatment of a condition or disorder mediated by potassium channel such as schizophrenia, ataxia, epilepsy, pain, hypertension, urinary incontinence, erectile dysfunction, irritable bowel syndrome and Parkinson's disease.
- the present invention is directed to pyrrolopyrimidine derivatives compounds which are SK channel modulators, and which are useful in the treatment or prevention of neurological and psychiatric disorders and diseases in which SK channels are involved.
- the invention is also directed to pharmaceutical compositions comprising these compounds and the use of these compounds and compositions in the prevention or treatment of such diseases in which SK channels are involved.
- the invention also relates to a method for prevention or treatment of such diseases comprising administering to mammals in need for the prevention or treatment of the disorders or diseases.
- the present invention provides compounds of formula (I) and salts thereof:
- R 1 is independently selected from the group consisting of: (1) hydrogen, (2) C 1-6 alkyl, which is unsubstituted or substituted with one or more substituents selected from R 5 , and (3) cycloalkyl which is unsubstituted or substituted with one or more substituents selected from R 5 :
- R 2 is selected from the group consisting of: (1) hydrogen, (2) C 1-6 alkyl, which is unsubstituted or substituted with one or more substituents selected from R 5 , (3) C 3-6 cycloalkyl which is unsubstituted or substituted with one or more substituents selected from R 5 , (4) phenyl, which is unsubstituted or substituted with one or more substituents selected from R 5 , (5) heterocycle, which is unsubstituted or substituted with one or more substituents selected from R 5 , and (6) heterocycle substituted C 1-6 alkyl, said heterocycle may be unsubstituted or substituted with one or more substituents selected from R 5 , i.e., a C 1-6 alkyl substituted by heterocycle, the heterocycle being unsubstituted or substituted with one or more substituents selected from R 5 ;
- R 3 is selected from the group consisting of: (1) C 1-6 alkyl, which is unsubstituted or substituted with one or more substituents selected from R 5 , (2) C 3-6 cycloalkyl which is unsubstituted or substituted with one or more substituents selected from R 5 , (3) phenyl, which is unsubstituted or substituted with one or more substituents selected from R 5 , (4) heterocycle, which is unsubstituted or substituted with one or more substituents selected from R 5 , and (5) heterocycle substituted C 1-6 alkyl, said heterocycle may be unsubstituted or substituted with one or more substituents selected from R 5 ;
- n is 1, 2, 3 or 4; when n is two or more than two, R 4 may be same or different;
- R 6 and R 7 are independently selected from the group consisting of: (1) hydrogen, (2) C 1-6 alkyl, which is unsubstituted or substituted with R 9 , (3) C 3-6 alkenyl, which is unsubstituted or substituted with R 9 , (4) cycloalkyl which is unsubstituted or substituted with R 9 , (5) phenyl, which is unsubstituted or substituted with R 5 , and (6) heterocycle, which is unsubstituted or substituted with R 9 , or R 6 and R 7 taken together with the nitrogen atom to which they are attached form a 3 to 8 membered ring, where the ring may contain one to four heteroatoms independently selected from nitrogen, oxygen, and sulfur; where the ring may be saturated or partially saturated or unsaturated; which is unsubstituted or substituted one or more substituents selected from R 9 ;
- R 8 is selected from the definitions of R 6 and R 7 ;
- R 2 is selected from the group consisting of: (1) hydrogen, (2) C 1-6 alkyl, which is unsubstituted or substituted with one or more substituents selected from R 5 , and (3) C 3-6 cycloalkyl which is unsubstituted or substituted with one or more substituents selected from R 5 ;
- R 3 is selected from the group consisting of: (1) C 1-6 alkyl, which is unsubstituted or substituted with one or more substituents selected from R 5 , (2) C 3-6 cycloalkyl which is unsubstituted or substituted with one or more substituents selected from R 5 , and (3) phenyl, which is unsubstituted or substituted with one or more substituents selected from R 5 ;
- R 4 is independently selected from the group consisting of: (1) hydrogen, (2) halogen, (3) hydroxy, (4) -O p -C 1-6 alkyl, where p is 0
- R 9 is selected from the group consisting of: (1) hydroxy, (2) halogen, (3) C 1-6 alkyl, (4) -C 3-6 cycloalkyl, (5) -O-C 1-6 alkyl, (6) -CN, and (7) -O p -(C 1-3 )perfluoroalkyl, where p is 0 or 1 (wherein if p is 0, a chemical bond is present in the place of O p or a pharmaceutically acceptable salt thereof.
- the more preferable compounds are selected from: 5-(1H-1,3-benzodiazol-2-yl)-7-methyl-N-(2-phenoxyethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine, 5-(1H-1,3-benzodiazol-2-yl)-N-(4-fluorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine, 5-(1H-1,3-benzodiazol-2-yl)-N-[2-(4-fluorophenyl)ethyl]-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine, 5-(1H-1,3-benzodiazol-2-yl)-N-(cyclohexylmethyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine, 5-(1H-1,3-benzodiazol-2-yl)-7-methyl-N-(oxolan-2-ylmethyl)-7H-
- the present invention provides the use of a compound of formula (I) or a pharmaceutically acceptable salt thereof, each as described herein, for the manufacture of a medicament for the treatment of a condition or disorder mediated by potassium channel; in particular, SK channels activation activity.
- a pharmaceutical composition which comprises a compound of formula (I) or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier or excipient.
- the present invention also provides the use of a compound of formula (I) or a pharmaceutically acceptable salt thereof, each as described herein, for the manufacture of a medicament for the treatment of diseases selected from potassium channels related diseases.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof, each as described herein, together with a pharmaceutically acceptable carrier for said compound.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof, each as described herein, together with a pharmaceutically acceptable carrier for said compound and another pharmacologically active agent.
- the present invention provides a process for preparing a pharmaceutical composition, the process comprising mixing a compound of formula (I) or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable carrier or excipient.
- the present invention provides an intermediate in a process for preparing a compound of formula (I) or a pharmaceutically acceptable salt thereof.
- the present invention provides a method of treatment of a condition or disorder mediated by potassium channels activation activity, in a mammalian subject, which comprises administering to a mammal in need of such treatment a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof, each as described herein.
- the present invention provides a process for preparing a pharmaceutical composition, the process comprising mixing a compound of formula (I) or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable carrier or excipient.
- Examples of conditions or disorders mediated by potassium channels activation activity include, but are not limited to, potassium channels related diseases.
- the compounds of the present invention show the potassium channels activation activity.
- the compounds of the present invention may show less toxicity, good absorption, distribution, good solubility, less protein binding affinity other than potassium channels, less drug-drug interaction, and good metabolic stability.
- halogen or “halo” as used herein are intended to include fluoro, chloro, bromo and iodo.
- C 1-6 as in C 1-6 alkyl is defined to identify the group as having 1, 2, 3, 4, 5 or 6 carbons in a linear or branched arrangement, such that C 1-6 alkyl specifically includes methyl, ethyl, n-propyl, iso-propyl, n-butyl, iso-butyl, tert-butyl, pentyl, and hexyl.
- C 2-6 alkenyl is defined to identify the group as having 2, 3, 4, 5 or 6 carbons which incorporates at least one double bond, which may be in a E- or a Z- arrangement.
- a group which is designated as being independently substituted with substituents may be independently substituted with multiple numbers of such substituents.
- alkenyl means a hydrocarbon radical having at least one double bond including, but not limited to, ethenyl, propenyl, 1-butenyl, 2-butenyl and the like.
- cycloalkyl means a mono- or bicyclic ring, but not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, norboranyl, and adamantyl groups and the like.
- heterocycle includes both unsaturated and saturated heterocyclic moieties, wherein the unsaturated heterocyclic moieties include benzoimidazolyl, benzimidazolonyl, benzofuranyl, benzofurazanyl, benzopyrazolyl, benzotriazolyl, benzothiophenyl, benzoxazolyl,carbazolyl, carbolinyl, cinnolinyl, furanyl, imidazolyl, indolinyl, indolyl, indolazinyl, indazolyl, isobenzofuranyl, isoindolyl, isoquinolyl, isothiazolyl, isoxazolyl, naphthpyridinyl, oxadiazolyl, oxazolyl, oxazolinyl, isoxazolinyl, oxetanyl, pyrazinyl, pyrazolyl,
- Any of the reactive groups such as amino, carboxyl and hydroxyl may be protected by a protecting group.
- protecting group means a hydroxy or amino protecting group which is selected from typical hydroxy or amino protecting groups described in Protective Groups in Organic Synthesis edited by T. W. Greene et al. (John Wiley & Sons, 1991);
- treating refers to curative, palliative and prophylactic treatment, including reversing, alleviating, inhibiting the progress of, or preventing the disorder or condition to which such term applies, or one or more symptoms of such disorder or condition.
- Ca 2+ ionophore means a lipid-soluble molecule usually synthesized by microorganisms to transport ions across the lipid bilayer of the cell membrane, but not limited to, ionomycin, A23187, and the like.
- the compounds of formula (I) can form acid addition salts thereof. It will be appreciated that for use in medicine the salts of the compounds of formula (I) should be pharmaceutically acceptable. Suitable pharmaceutically acceptable salts will be apparent to those skilled in the art and include those described in J. Pharm. Sci, 1977, 66, 1-19, such as acid addition salts formed with inorganic acids e.g. hydrochloric, hydrobromic, sulfuric, nitric or phosphoric acid; and organic acids e.g.
- succinic maleic, formic, acetic, trifluoroacetic, propionic, fumaric, citric, tartaric, benzoic, p-toluenesulfonic, methanesulfonic or naphthalenesulfonic acid.
- Certain of the compounds of formula (I) may form acid addition salts with one or more equivalents of the acid.
- the present invention includes within its scope all possible stoichiometric and non-stoichiometric forms.
- certain compounds containing an acidic function such as a carboxy can be isolated in the form of their inorganic salt in which the counter ion can be selected from sodium, potassium, lithium, calcium, magnesium and the like, as well as from organic bases.
- the compounds of formula (I) and salts thereof may be prepared in crystalline or non-crystalline form, and, if crystalline, may optionally be hydrated or solvated.
- This invention includes within its scope stoichiometric hydrates or solvates as well as compounds containing variable amounts of water and/or solvent.
- Salts and solvates having non-pharmaceutically acceptable counter-ions or associated solvents are within the scope of the present invention, for example, for use as intermediates in the preparation of other compounds of formula (I) and their pharmaceutically acceptable salts.
- the compounds of formula (I) may have polymorphs in crystalline form, which are within the scope of the present invention.
- a prodrug of a compound of formula (I) is a functional derivative of the compound which, upon administration to a patient, eventually liberates the compound of formula (I) in vivo.
- Administration of a compound of formula (I) as a prodrug may enable the skilled artisan to do one or more of the following: (a) modify the onset of action of the compound in vivo; (b) modify the duration of action of the compound in vivo; (c) modify the transportation or distribution of the compound in vivo; (d) modify the solubility of the compound in vivo; and (e) overcome a side effect or other difficulty encountered with the compound.
- Typical functional derivatives used to prepare prodrugs include modifications of the compound that are chemically or enzymatically cleaved in vivo. Such modifications, which include the preparation of phosphates, amides, esters, thioesters, carbonates, and carbamates, are well known to those skilled in the art.
- compounds of formula (I) there may be some chiral carbon atoms.
- compounds of formula (I) exist as stereoisomers.
- the invention extends to all optical isomers such as stereoisomeric forms of the compounds of formula (I) including enantiomers, diastereoisomers and mixtures thereof, such as racemates.
- the different stereoisomeric forms may be separated or resolved one from the other by conventional methods or any given isomer may be obtained by conventional stereoselective or asymmetric syntheses.
- the invention also includes isotopically-labeled compounds, which are identical to those described herein, but for the fact that one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number usually found in nature.
- isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine, iodine, and chlorine, such as 3 H, 11 C, 14 C, 18 F, 123 I and 125 I.
- Compounds of the invention that contain the aforementioned isotopes and/or other isotopes of other atoms are within the scope of the present invention.
- Isotopically-labeled compounds of the present invention for example those into which radioactive isotopes such as 3 H, 14 C are incorporated, are useful in drug and/or substrate tissue distribution assays. Tritiated, i.e., 3 H, and carbon-14, i.e., 14 C, isotopes are particularly preferred for their ease of preparation and detectability. 11 C and 18 F isotopes are particularly useful in PET (positron emission tomography), and 125 I isotopes are particularly useful in SPECT (single photon emission computerized tomography), all useful in brain imaging.
- lsotopically labeled compounds of the invention can generally be prepared by carrying out the procedures disclosed in the Schemes and/or in the Examples below, then substituting a readily available isotopically labeled reagent for a non-isotopically labeled reagent.
- the present compounds exhibit unexpected properties, such as with respect to duration of action and/or metabolism, such as increased metabolic stability, enhanced oral bioavailability or absorption, and/or decreased drug-drug interactions.
- the compounds of formula (I), being potassium channel modulators, are potentially useful in the treatment of a number of diseases, including, schizophrenia, ataxia, epilepsy, pain, hypertension, urinary incontinence, erectile dysfunction, irritable bowel syndrome (Curr Med Chem. 2007;14) and Parkinson's disease (Exp Neurol. 2008; 213). Particularly chronic, inflammatory, neuropathic, nociceptive and visceral pain, is a preferred use.
- Potassium channels have been implicated in a wide range of biological functions. This has suggested a potential role for these receptors in a variety of disease processes in humans or other species.
- the compounds of the present invention have utility in treating, preventing, ameliorating, controlling or reducing the risk of a variety of neurological and psychiatric disorders associated with potassium channels, including one or more of the following conditions or diseases: a respiratory disease, epilepsy, convulsions, seizures, absence seizures, vascular spasms, coronary artery spasms, renal disorders, polycystic kidney disease, bladder spasms, urinary incontinence, bladder outflow obstruction, erectile dysfunction, gastrointestinal dysfunction, secretory diarrhoea, ischaemia, cerebral ischaemia, ischaemic heart disease, angina pectoris, coronary heart disease, autism, ataxia, traumatic brain injury, Parkinson's disease, bipolar disorder, psychosis, schizophrenia, anxiety, depression, mania, mood disorders, dementia, memory and attention deficits, Alzheimer's
- the dosage of active ingredient in the compositions of this invention may be varied, however, it is necessary that the amount of the active ingredient be such that a suitable dosage form is obtained.
- the active ingredient may be administered to patients (animals and human) in need of such treatment in dosages that will provide optimal pharmaceutical efficacy.
- the selected dosage depends upon the desired therapeutic effect, on the route of administration, and on the duration of the treatment.
- the dose will vary from patient to patient depending upon the nature and severity of disease, the patient's weight, special diets then being followed by a patient, concurrent medication, and other factors which those skilled in the art will recognize.
- the total daily dose of the compounds of the invention is typically in the range 0.1 mg to 1000 mg depending, of course, on the mode of administration.
- oral administration may require a total daily dose of from 1 mg to 1000 mg, while an intravenous dose may only require from 0.1 mg to 100 mg.
- the total daily dose may be administered in single or divided doses and may, at the physician's discretion, fall outside of the typical range given herein.
- These dosages are based on an average human subject having a weight of about 60kg to 70kg. The physician will readily be able to determine doses for subjects whose weight falls outside this range, such as infants and the elderly.
- references herein to "treatment” include references to curative, palliative and prophylactic treatment.
- the dosage range will be about 0.5 mg to 500 mg per patient per day; in another embodiment about 0.5 mg to 200 mg per patient per day; in another embodiment about 1 mg to 100 mg per patient per day; and in another embodiment about 5 mg to 50 mg per patient per day; in yet another embodiment about 1 mg to 30 mg per patient per day.
- Pharmaceutical compositions of the present invention may be provided in a solid dosage formulation such as comprising about 0.5 mg to 500 mg active ingredient, or comprising about 1 mg to 250 mg active ingredient.
- the pharmaceutical composition may be provided in a solid dosage formulation comprising about 1 mg, 5 mg, 10mg, 25 mg, 50 mg, 100 mg, 200 mg or 250 mg active ingredient.
- compositions may be provided in the form of tablets containing 1.0 to 1000 milligrams of the active ingredient, such as 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400, 500, 600, 750, 800, 900, and 1000 milligrams of the active ingredient for the symptomatic adjustment of the dosage to the patient to be treated.
- the compounds may be administered on a regimen of 1 to 4 times per day, such as once or twice per day.
- the compounds of the present invention may be used in combination with one or more other drugs in the treatment, prevention, control, amelioration, or reduction of risk of diseases or conditions for which compounds of the present invention or the other drugs may have utility, where the combination of the drugs together are safer or more effective than either drug alone.
- Such other drug(s) may be administered, by a route and in an amount commonly used therefore, contemporaneously or sequentially with a compound of the present invention.
- a pharmaceutical composition in unit dosage form containing such other drugs and the compound of the present invention is envisioned.
- the combination therapy may also include therapies in which the compound of the present invention and one or more other drugs are administered on different overlapping schedules. It is also contemplated that when used in combination with one or more other active ingredients, the compounds of the present invention and the other active ingredients may be used in lower doses than when each is used singly.
- compositions of the present invention include those that contain one or more other active ingredients, in addition to a compound of the present invention.
- the above combinations include combinations of a compound of the present invention not only with one other active compound, but also with two or more other active compounds.
- compounds of the present invention may be used in combination with other drugs that are used in the prevention, treatment, control, amelioration, or reduction of risk of the diseases or conditions for which compounds of the present invention are useful.
- Such other drugs may be administered, by a route and in an amount commonly used therefore, contemporaneously or sequentially with a compound of the present invention.
- a pharmaceutical composition containing such other drugs in addition to the compound of the present invention is envisioned.
- the pharmaceutical compositions of the present invention include those that also contain one or more other active ingredients, in addition to a compound of the present invention.
- the weight ratio of the compound of the compound of the present invention to the second active ingredient may be varied and will depend upon the effective dose of each ingredient. Generally, an effective dose of each will be used. Thus, for example, when a compound of the present invention is combined with another agent, the weight ratio of the compound of the present invention to the other agent will generally range from about 1000:1 to about 1:1000, including about 200: 1 to about 1:200. Combinations of a compound of the present invention and other active ingredients will generally also be within the aforementioned range, but in each case, an effective dose of each active ingredient should be used. In such combinations the compound of the present invention and other active agents may be administered separately or in conjunction. In addition, the administration of one element may be prior to, concurrent to, or subsequent to the administration of other agent(s).
- a potassium channels modulator may be usefully combined with another pharmacologically active compound, or with two or more other pharmacologically active compounds, particularly in the treatment of inflammatory, pain and urological diseases or disorders.
- a potassium channels modulator particularly a compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof, as defined above, may be administered simultaneously, sequentially or separately in combination with one or more agents selected from:
- an opioid analgesic e.g. morphine, heroin, hydromorphone, oxymorphone, levorphanol, levallorphan, methadone, meperidine, fentanyl, cocaine, codeine, dihydrocodeine, oxycodone, hydrocodone, propoxyphene, nalmefene, nalorphine, naloxone, naltrexone, buprenorphine, butorphanol, nalbuphine or pentazocine;
- opioid analgesic e.g. morphine, heroin, hydromorphone, oxymorphone, levorphanol, levallorphan, methadone, meperidine, fentanyl, cocaine, codeine, dihydrocodeine, oxycodone, hydrocodone, propoxyphene, nalmefene, nalorphine, naloxone, naltrexone, buprenorphine, butorphanol, nal
- NSAID nonsteroidal antiinflammatory drug
- diclofenac diflusinal, etodolac
- fenbufen fenoprofen
- flufenisal flurbiprofen
- ibuprofen indomethacin
- ketoprofen ketorolac
- meclofenamic acid mefenamic acid, meloxicam
- nabumetone naproxen
- nimesulide nitroflurbiprofen
- olsalazine oxaprozin
- phenylbutazone piroxicam
- sulfasalazine sulindac
- tolmetin or zomepirac a nonsteroidal antiinflammatory drug
- a barbiturate sedative e.g. amobarbital, aprobarbital, butabarbital, butabital, mephobarbital, metharbital, methohexital, pentobarbital, phenobartital, secobarbital, talbutal, theamylal or thiopental;
- a benzodiazepine having a sedative action e.g. chlordiazepoxide, clorazepate, diazepam, flurazepam, lorazepam, oxazepam, temazepam or triazolam;
- an H1 antagonist having a sedative action e.g. diphenhydramine, pyrilamine, promethazine, chlorpheniramine or chlorcyclizine;
- a sedative such as glutethimide, meprobamate, methaqualone or dichloralphenazone;
- a skeletal muscle relaxant e.g. baclofen, carisoprodol, chlorzoxazone, cyclobenzaprine, methocarbamol or orphrenadine;
- an NMDA receptor antagonist e.g. dextromethorphan ((+)-3-hydroxy-N-methylmorphinan) or its metabolite dextrorphan ((+)-3-hydroxy-N-methylmorphinan), ketamine, memantine, pyrroloquinoline quinine, cis-4-(phosphonomethyl)-2-piperidinecarboxylic acid, budipine, EN-3231 (MorphiDex(registered trademark), a combination formulation of morphine and dextromethorphan), topiramate, neramexane or perzinfotel including an NR2B antagonist, e.g.
- an NMDA receptor antagonist e.g. dextromethorphan ((+)-3-hydroxy-N-methylmorphinan) or its metabolite dextrorphan ((+)-3-hydroxy-N-methylmorphinan), ketamine, memantine, pyrroloquinoline quinine, cis-4-(phosphon
- an alpha-adrenergic e.g. doxazosin, tamsulosin, clonidine, guanfacine, dexmetatomidine, modafinil, or 4-amino-6,7-dimethoxy-2-(5-methane-sulfonamido-1,2,3,4-tetrahydroisoquinol-2-yl)-5-(2-pyridyl) quinazoline;
- a tricyclic antidepressant e.g. desipramine, imipramine, amitriptyline or nortriptyline;
- an anticonvulsant e.g. carbamazepine, lamotrigine, topiratmate or valproate;
- a tachykinin (NK) antagonist particularly an NK-3, NK-2 or NK-1 antagonist, e.g. (alphaR,9R)-7-[3,5-bis(trifluoromethyl)benzyl]-8,9,10,11-tetrahydro-9-methyl-5-(4-methylphenyl)-7H-[1,4]diazocino[2,1-g][1,7]-naphthyridine-6-13-dione (TAK-637), 5-[[(2R,3S)-2-[(1R)-1-[3,5-bis(trifluoromethyl)phenyl]ethoxy-3-(4-fluorophenyl)-4-morpholinyl]-methyl]-1,2-dihydro-3H-1,2,4-triazol-3-one (MK-869), aprepitant, lanepitant, dapitant or 3-[[2-methoxy-5-(trifluoromethoxy)phenyl]-methylamin
- a muscarinic antagonist e.g oxybutynin, tolterodine, propiverine, tropsium chloride, darifenacin, solifenacin, temiverine and ipratropium;
- COX-2 selective inhibitor e.g. celecoxib, rofecoxib, parecoxib, valdecoxib, deracoxib, etoricoxib, or lumiracoxib;
- coal-tar analgesic in particular paracetamol
- a neuroleptic such as droperidol, chlorpromazine, haloperidol, perphenazine, thioridazine, mesoridazine, trifluoperazine, fluphenazine, clozapine, olanzapine, risperidone, ziprasidone, quetiapine, sertindole, aripiprazole, sonepiprazole, blonanserin, iloperidone, perospirone, raclopride, zotepine, bifeprunox, asenapine, lurasidone, amisulpride, balaperidone, palindore, eplivanserin, osanetant, rimonabant, meclinertant, Miraxion(registered trademark) or sarizotan;
- vanilloid receptor agonist e.g. resinferatoxin
- antagonist e.g. capsazepine
- beta-adrenergic such as propranolol
- corticosteroid such as dexamethasone
- a 5-HT receptor agonist or antagonist particularly a 5-HT1B/1D agonist such as eletriptan, sumatriptan, naratriptan, zolmitriptan or rizatriptan;
- a 5-HT2A receptor antagonist such as R(+)-alpha-(2,3-dimethoxy-phenyl)-1-[2-(4-fluorophenylethyl)]-4-piperidinemethanol (MDL-100907);
- a cholinergic (nicotinic) analgesic such as ispronicline (TC-1734), (E)-N-methyl-4-(3-pyridinyl)-3-buten-1-amine (RJR-2403), (R)-5-(2-azetidinylmethoxy)-2-chloropyridine (ABT-594) or nicotine;
- a PDEV inhibitor such as 5-[2-ethoxy-5-(4-methyl-1-piperazinyl-sulphonyl)phenyl]-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (sildenafil), (6R,12aR)-2,3,6,7,12,12a-hexahydro-2-methyl-6-(3,4-methylenedioxyphenyl)-pyrazino[2',1':6,1]-pyrido[3,4-b]indole-1,4-dione (IC-351 or tadalafil), 2-[2-ethoxy-5-(4-ethyl-piperazin-1-yl-1-sulphonyl)-phenyl]-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one (vardenafil), 5-(5-acetyl-
- an alpha-2-delta ligand such as gabapentin, pregabalin, 3-methylgabapentin, (1alpha,3alpha,5alpha)(3-amino-methyl-bicyclo[3.2.0]hept-3-yl)-acetic acid, (3S,5R)-3 aminomethyl-5 methyl-heptanoic acid, (3S,5R)-3 amino-5 methyl-heptanoic acid, (3S,5R)-3 amino-5 methyl-octanoic acid, (2S,4S)-4-(3-chlorophenoxy)proline, (2S,4S)-4-(3-fluorobenzyl)-proline, [(1R,5R,6S)-6-(aminomethyl)bicyclo[3.2.0]hept-6-yl]acetic acid, 3-(1-aminomethyl-cyclohexylmethyl)-4H-[1,2,4]oxadiazol-5-one, C-[1-(1H-te
- mGluR1 metabotropic glutamate subtype 1 receptor
- a serotonin reuptake inhibitor such as sertraline, sertraline metabolite demethylsertraline, fluoxetine, norfluoxetine (fluoxetine desmethyl metabolite), fluvoxamine, paroxetine, citalopram, citalopram metabolite desmethylcitalopram, escitalopram, d,l-fenfluramine, femoxetine, ifoxetine, cyanodothiepin, litoxetine, dapoxetine, nefazodone, cericlamine and trazodone;
- noradrenaline (norepinephrine) reuptake inhibitor such as maprotiline, lofepramine, mirtazepine, oxaprotiline, fezolamine, tomoxetine, mianserin, buproprion, buproprion metabolite hydroxybuproprion, nomifensine and viloxazine (VivalanR), especially a selective noradrenaline reuptake inhibitor such as reboxetine, in particular (S,S)-reboxetine;
- a dual serotonin-noradrenaline reuptake inhibitor such as venlafaxine, venlafaxine metabolite O-desmethylvenlafaxine, clomipramine, clomipramine metabolite desmethylclomipramine, duloxetine, milnacipran and imipramine;
- an inducible nitric oxide synthase (iNOS) inhibitor such as S-[2-[(1-iminoethyl)amino]ethyl]-L-homocysteine, S-[2-[(1-iminoethyl)-amino]ethyl]-4,4-dioxo-L-cysteine, S-[2-[(1-iminoethyl)amino]ethyl]-2-methyl-L-cysteine, (2S,5Z)-2-amino-2-methyl-7-[(1-iminoethyl)amino]-5-heptenoic acid, 2-[[(1R,3S)-3-amino-4- hydroxy-1-(5-thiazolyl)-butyl]thio]-5-chloro-3-pyridinecarbonitrile; 2-[[[(1R,3S)-3-amino-4-hydroxy-1-(5-thiazolyl
- an acetylcholinesterase inhibitor such as donepezil
- a prostaglandin E2 subtype 4 (EP4) antagonist such as N-[( ⁇ 2-[4-(2-ethyl-4,6-dimethyl-1H-imidazo[4,5-c]pyridin-1-yl)phenyl]ethyl ⁇ amino)-carbonyl]-4-methylbenzenesulfonamide or 4-[(1S)-1-( ⁇ [5-chloro-2-(3-fluorophenoxy)pyridin-3-yl]carbonyl ⁇ amino)ethyl]benzoic acid;
- leukotriene B4 antagonist such as 1-(3-biphenyl-4-ylmethyl-4-hydroxy-chroman-7-yl)-cyclopentanecarboxylic acid (CP-105696), 5-[2-(2-Carboxyethyl)-3-[6-(4-methoxyphenyl)-5E- hexenyl]oxyphenoxy]-valeric acid (ONO-4057) or DPC-11870,
- a 5-lipoxygenase inhibitor such as zileuton, 6-[(3-fluoro-5-[4-methoxy-3,4,5,6-tetrahydro-2H-pyran-4-yl])phenoxy-methyl]-1-methyl-2-quinolone (ZD-2138), or 2,3,5-trimethyl-6-(3-pyridylmethyl),1,4-benzoquinone (CV-6504);
- a sodium channel blocker such as lidocaine
- 5-HT3 antagonist such as ondansetron
- pharmaceutically acceptable salts and solvates thereof
- a pharmaceutical composition of the invention which may be prepared by admixture, suitably at ambient temperature and atmospheric pressure, is usually adapted for oral, parenteral or rectal administration and, as such, may be in the form of tablets, capsules, oral liquid preparations, powders, granules, lozenges, reconstitutable powders, injectable or infusible solutions or suspensions or suppositories. Orally administrate compositions are generally preferred. Tablets and capsules for oral administration may be in unit dose form, and may contain conventional excipients, such as binding agents (e.g. pregelatinised maize starch, polyvinylpyrrolidone or hydroxypropyl methylcellulose); fillers (e.g.
- binding agents e.g. pregelatinised maize starch, polyvinylpyrrolidone or hydroxypropyl methylcellulose
- fillers e.g.
- lactose microcrystalline cellulose or calcium hydrogen phosphate
- tabletting lubricants e.g. magnesium stearate, talc or silica
- disintegrants e.g. potato starch or sodium starch glycollate
- acceptable wetting agents e.g. sodium lauryl sulphate.
- the tablets may be coated according to methods well known in normal pharmaceutical practice.
- Oral liquid preparations may be in the form of, for example, aqueous or oily suspension, solutions, emulsions, syrups or elixirs, or may be in the form of a dry product for reconstitution with water or other suitable vehicle before use.
- Such liquid preparations may contain conventional additives such as suspending agents (e.g. sorbitol syrup, cellulose derivatives or hydrogenated edible fats), emulsifying agents (e.g. lecithin or acacia), non-aqueous vehicles (which may include edible oils e.g. almond oil, oily esters, ethyl alcohol or fractionated vegetable oils), preservatives (e.g.
- Preparations for oral administration may be suitably formulated to give controlled release of the active compound or pharmaceutically acceptable salt thereof.
- fluid unit dosage forms are prepared utilising a compound of formula (I) or pharmaceutically acceptable salt thereof and a sterile vehicle.
- Formulations for injection may be presented in unit dosage form e.g. in ampoules or in multi-dose, utilising a compound of formula (I) or pharmaceutically acceptable salt thereof and a sterile vehicle, optionally with an added preservative.
- the compositions may take such forms as suspensions, solutions or emulsions in oily or aqueous vehicles, and may contain formulatory agents such as suspending, stabilising and/or dispersing agents.
- the active ingredient may be in powder form for constitution with a suitable vehicle, e.g. sterile pyrogen-free water, before use.
- the compound depending on the vehicle and concentration used, can be either suspended or dissolved in the vehicle.
- the compound can be dissolved for injection and filter sterilised before filling into a suitable vial or ampoule and sealing.
- adjuvants such as a local anaesthetic, preservatives and buffering agents are dissolved in the vehicle.
- the composition can be frozen after filling into the vial and the water removed under vacuum.
- Parenteral suspensions are prepared in substantially the same manner, except that the compound is suspended in the vehicle instead of being dissolved, and sterilisation cannot be accomplished by filtration.
- the compound can be sterilised by exposure to ethylene oxide before suspension in a sterile vehicle.
- a surfactant or wetting agent is included in the composition to facilitate uniform distribution of the compound.
- Lotions may be formulated with an aqueous or oily base and will in general also contain one or more emulsifying agents, stabilising agents, dispersing agents, suspending agents, thickening agents, or colouring agents. Drops may be formulated with an aqueous or non-aqueous base also comprising one or more dispersing agents, stabilising agents, solubilising agents or suspending agents. They may also contain a preservative.
- the compounds of formula (I) or pharmaceutically acceptable salts thereof may also be formulated in rectal compositions such as suppositories or retention enemas, e.g. containing conventional suppository bases such as cocoa butter or other glycerides.
- the compounds of formula (I) or pharmaceutically acceptable salts may also be formulated as depot preparations. Such long acting formulations may be administered by implantation (for example subcutaneously or intramuscularly) or by intramuscular injection.
- the compounds of formula (I) or pharmaceutically acceptable salts may be formulated with suitable polymeric or hydrophobic materials (for example as an emulsion in an acceptable oil) or ion exchange resins, or as sparingly soluble derivatives, for example, as a sparingly soluble salt.
- the compounds formula (I) or pharmaceutically acceptable salts thereof may be formulated as solutions for administration via a suitable metered or unitary dose device or alternatively as a powder mix with a suitable carrier for administration using a suitable delivery device.
- compounds of formula (I) or pharmaceutically acceptable salts thereof may be formulated for oral, buccal, parenteral, topical (including ophthalmic and nasal), depot or rectal administration or in a form suitable for administration by inhalation or insufflation (either through the mouth or nose).
- the compounds of formula (I) and pharmaceutically acceptable salts thereof may be formulated for topical administration in the form of ointments, creams, gels, lotions, pessaries, aerosols or drops (e.g.
- Ointments and creams may, for example, be formulated with an aqueous or oily base with the addition of suitable thickening and/or gelling agents.
- Ointments for administration to the eye may be manufactured in a sterile manner using sterilized components.
- bases are likewise no particular restriction on the nature of the bases used, and any base commonly used in reactions of this type may equally be used here.
- bases include: alkali metal hydroxides, such as lithium hydroxide, sodium hydroxide, potassium hydroxide, and barium hydroxide; alkali metal hydrides, such as lithium hydride, sodium hydride, and potassium hydride; alkali metal alkoxides, such as sodium methoxide, sodium ethoxide, and potassium t-butoxide; alkali metal carbonates, such as lithium carbonate, sodium carbonate, potassium carbonate, and cesium carbonate; alkali metal hydrogencarbonates, such as lithium hydrogencarbonate, sodium hydrogencarbonate, and potassium hydrogencarbonate; amines, such as N-methylmorpholine, triethylamine, tripropylamine, tributylamine, diisopropylethylamine, N-methylpiperidine, pyridine, 4-pyrrolidinopyridine, picoline,
- the reactions are normally and preferably effected in the presence of inert solvent.
- solvent there is no particular restriction on the nature of the solvent to be employed, provided that it has no adverse effect on the reaction or the reagents involved and that it can dissolve reagents, at least to some extent.
- Suitable solvents include, but not limited to: halogenated hydrocarbons, such as dichloromethane, chloroform, carbon tetrachloride, and dichloroethane; ethers, such as diethyl ether, diisopropyl ether, THF, and dioxane; aromatic hydrocarbons, such as benzene, toluene and nitrobenzene; amides, such as, DMF, N,N-dimethylacetamide, and hexamethylphosphoric triamide; amines, such as N-methylmorpholine, triethylamine, tripropylamine, tributylamine, diisopropylethylamine, N-methylpiperidine, pyridine, 4-pyrrolidinopyridine, N,N-dimethylaniline, and N,N-diethylaniline; alcohols, such as methanol, ethanol, propanol, isopropanol, and butanol; n
- solvents including but not limited to DMF, DMSO, THF, diethylether, diisopropylether, dimethoxyethane, acetonitrile, dichloromethane, dichloroethane and chloroform are preferred.
- Flash column chromatography was carried out using Merck silica gel 60 (230-400 mesh ASTM) or Fuji Silysia Chromatorex(registered trademark) DU3050 (Amino Type, 30-50 micrometer) or Biotage silica (32-63 mm, KP-Sil) or Biotage amino bounded silica (35-75 mm, KP-NH).
- Process A The purification of compounds using HPLC was performed by the following apparatus and conditions ("process A”); Apparatus; Waters MS-trigger AutoPurification TM system Column; Waters XTerra C18, 19X50mm, 5mm particle, Method A; Methanol or acetonitrile / 0.05%(v/v) formic acid aqueous solution, Method B; Methanol or acetonitrile / 0.01%(v/v) ammonia aqueous solution.
- HPLC("Process B") The purification using HPLC("Process B") was performed by the following apparatus and conditions: Apparatus; UV-trigger preparative HPLC system, Waters (Column; XTerra MS C18, 5 micrometer, 19 x 50 mm or 30 x 50 mm), Detector; UV 254 nm, Conditions; acetonitrile:0.05% formic acid aqueous solution or acetonitrile:0.01%(v/v) aqueous ammonia solution; 20 mL/min (19 x 50 mm) or 40 mL/min (30 x 50 mm) at room temperature.
- ESI mass spectral data
- Apparatus Waters Alliance HPLC system on ZQ or ZMD mass spectrometer and UV detector.
- pyrrolopyrimidine derivatives of the formula (I) can be prepared by the procedures described in the general methods presented below or by the specific methods described in the Examples section and the Preparations section, or by routine modifications thereof.
- the present invention also encompasses any one or more of these processes for preparing the pyrrolopyrimidine derivatives of formula (I), in addition to any novel intermediates used therein.
- R 1 , R 2 , R 3 , R 4 , and n are as previously defined for pyrrolopyrimidine derivatives of the formula (I) unless otherwise stated. All starting materials in the following general syntheses may be commercially available or obtained by conventional methods known to those skilled in the art.
- an intermediate of formula (III) can be prepared from a compound of formula (II) by S N Ar reaction with amines (R 2 R 3 NH) in a suitable solvent such as ethanol and DMF at a temperature of from about 20 to about 150 o C for about 1-24 hours.
- an intermediate of formula (IV) can be prepared from a compound of formula (III) by amidation with (R 4 )n-benzene-1,2-diamines with using a suitable coupling reagent such as EDC preferably under the presence of a base such a combination of N,N-Diisopropylethylamine and HOBT in a suitable solvent such as DMF at a temperature of from about 20 to about 100 o C for about 1-24 hours.
- a suitable coupling reagent such as EDC preferably under the presence of a base such a combination of N,N-Diisopropylethylamine and HOBT in a suitable solvent such as DMF at a temperature of from about 20 to about 100 o C for about 1-24 hours.
- a compound of formula (I) can be prepared from a compound of formula (IV) by cyclization in an acidic media such as acetic acid at a temperature of from about 20 to about 120 o C for about 1-24 hours.
- STEP 2 4-Chloro-7-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carboxylic acid
- STEP A 7-Methyl-4-(phenylamino)-7H-pyrrolo[2,3-d]pyrimidine-5-carboxylic acid
- STEP B N-(2-Aminophenyl)-7-methyl-4-(phenylamino)-7H-pyrrolo[2,3-d]pyrimidine-5-carboxamide
- STEP C 5-(1H-1,3-Benzodiazol-2-yl)-7-methyl-N-phenyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine
- Examples 2-64 End products of Examples 2-64 were prepared from 4-chloro-7-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carboxylic acid (STEP 2 of Example 1) or 4-chloro-7H-pyrrolo[2,3-d]pyrimidine-5-carboxylic acid (WO2008/075007) according to STEP A, B and C of Example 1 (3 steps).
- STEP B The intermediates IV (2B-64B) listed in Table 4 were prepared from the intermediates III (2A-64A) and (R 4 )n-benzene-1,2-diamines according to the procedure described in STEP B of Example 1, respectively.
- LC-MS Method A was used for STEP B of Examples 2-64 (2B-64B).
- 2B stands for an intermediate of STEP B of Example 2.
- the ability of the pyrrolopyrimidine derivatives of the formula (I) to activate the potassium channel may be measured using the assay described below.
- SK3 channel assay Human SK3 channel (hSK3) activity was detected by channel assay. Approximately 25,000 cells/well of CHO-K1 cells stably expressing hSK3 channels were plated into black walled, clear-bottom 96-well plates. Following overnight incubation in a 5% CO 2 incubator at 37 o C. The cell was treated with the FluxOR TM detection kits (Invitrogen; Catalog nos. F10016, F10017, Revised: 27-March-2008 MP 10016) according to the literatures (J Gen Physiol. 61, 669 (1973); Journal of Biomolecular Screening 9, 8 (2004)) in the presence of a Ca 2+ ionophore (e.g. ionomycin, A23187).
- a Ca 2+ ionophore e.g. ionomycin, A23187
- EC 50 values were calculated by fitting the resulting fluorescence intensities to a four-parametric logistic equation. EC 50 was defined as the concentration of test compound required to yield 50% of the maximal response.
- the compounds of the examples were tested in the above-described assay.
- Electrophysiology assay Membrane current though SK3 channel was recorded using the whole-cell configuration of the patch-clamp technique.
- CHO-K1 cells stably expressing human SK3 channel were grown in Ham's F-12 medium supplemented with 10% FBS, 100 units/mL of penicillin, 100 microgram/mL of streptomycin and 500 microgram/mL of geneticin at 37 degree C in 5% CO 2 .
- Cells were dissociated by brief exposure to trypsin-EDTA, suspended in supplemented F-12 medium and plated on 3.5-mm glass cover slips.
- Grass pipettes were pulled from borosilicate capillary glass with an outside diameter of 1.5 mm using a Sutter P-97 puller (Sutter Instrument Co.).
- the pipettes were fire polished and had a resistance of 1-3 Mohm when filled with the pipette solution as follows (in mM); 155 KCl, 10 EGTA, 8.23 CaCl 2 , 1.11 MgCl 2 , 10 HEPES, pH7.2 with KOH.
- the extracellular solution contained (in mM); 155 KCl, 2 CaCl 2 , 1 MgCl 2 , 10 HEPES, pH7.4 with NaOH.
- the test compound was dissolved in DMSO and serially diluted in the extracellular solution to the appropriate concentrations. Cells plated on cover slips were placed in a perfusion chamber superfused with the extracellular solution. Voltage clamp command pulses were generated by PULSE software (HEKA).
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Neurology (AREA)
- Heart & Thoracic Surgery (AREA)
- Biomedical Technology (AREA)
- Cardiology (AREA)
- Neurosurgery (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Urology & Nephrology (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present invention relates to pyrrolopyrimidine derivatives of formula(I). They have activation activities of potassium channels as the SK channels, and are useful in the treatment or prevention of disorders and diseases in which potassium channels are involved. The invention also relates to pharmaceutical compositions comprising these compounds and the use of these compounds and compositions in the prevention or treatment of such diseases in which potassium channels are involved. The present invention also relates to the prevention or treatment of such diseases, which comprises administering the above-mentioned pyrrolopyrimidine derivatives to a subject in need of such a prevention or treatment a therapeutically effective amount of said derivative.(I) (R1=Hydrogen etc, R2=Hydrogen etc, R3=C1-6alkyl etc, R4= Hydrogen etc, n=1-4)
Description
The present invention relates to pyrrolopyrimidine derivatives which have activation activities of potassium channels as the SK channels, and which are useful in the treatment or prevention of disorders and diseases in which potassium channels are involved. The invention also relates to pharmaceutical compositions comprising these compounds and the use of these compounds and compositions in the prevention or treatment of such diseases in which potassium channels are involved. The present invention also relates to the prevention or treatment of such diseases, which comprises administering the above-mentioned pyrrolopyrimidine derivatives to a subject in need of such a prevention or treatment a therapeutically effective amount of said derivative.
Ion channels play a fundamental role in the homeostasis of cell function through the regulation of the transmembrane movement of ions, including K+, Na+, Ca2+ and Cl-. Cellular activity can be affected by modifications of the activities of the ion channels. K+ channels are a diverse and ubiquitous group of ion channels and have several critical roles in cell function. These channels principally regulate the resting membrane potential of the cell and attenuate the level of excitation of cells. For example, the outward flow of K+ upon opening of K+ channels makes the interior of the cell more negative, counteracting depolarizing voltages applied to the cells. K+ channels are regulated by voltage, cell metabolism, second messengers, and receptor mediated processes. Because of the crucial role in cell physiology, their malfunction can lead to many diseases.
SK channels (small conductance Ca2+-activated K+ channels) are subfamily of Ca2+-activated K+ channels and consist of three subtypes, namely SK1, SK2 and SK3. All have a topology that is typical for K+ channels with six transmembrane helices and a P-loop region. SK channels are voltage-insensitive and can be activated by increases in the levels of intracellular Ca2+, such as what occurs during an action potential. Their activation is important for regulating afterhyperpolarization (AHP) and limits the firing frequency of action potentials in neurons and other cell types.
SK channels (small conductance Ca2+-activated K+ channels) are subfamily of Ca2+-activated K+ channels and consist of three subtypes, namely SK1, SK2 and SK3. All have a topology that is typical for K+ channels with six transmembrane helices and a P-loop region. SK channels are voltage-insensitive and can be activated by increases in the levels of intracellular Ca2+, such as what occurs during an action potential. Their activation is important for regulating afterhyperpolarization (AHP) and limits the firing frequency of action potentials in neurons and other cell types.
From molecular distribution, electrophysiology and pharmacological studies, an array of functions for SK channels have been proposed. All subtypes of SK channels are widely expressed in the brain. SK1 and SK2 channels are particular enriched in the cortex and hippocampus, while SK3 is also prominent in subcortical areas, notably the striatum, thalamus and monoaminergic nuclei. SK channels are involved in neurophysiological processes like spike frequency adaptation, pacemaking, as well as synaptic integration and plasticity. SK2 and SK3, in contrast to SK1, are also expressed in many other peripheral tissues, particular those rich in smooth muscle. Activation of SK channels leads to membrane potential hyperpolarization and relaxation of a variety of smooth muscles. SK3 channels are the only known SK subtype expressed in denervated skeletal muscle and breast tumor tissue.
Mutations in ion channel genes are responsible for neuromuscular and other neurological disorders. SK3 channel contains two polymorphic polyglutamine repeats in its N-terminus. Genetic studies find over-representation of longer alleles of the second SK3 polyglutamine repeat in patients with schizophrenia, anorexia nervosa, and spinocerebellar ataxia. In one schizophrenia patient a truncation mutant of SK3 channel, acting as a dominant-negative suppressor of SK channels, is also identified. Mutations in SK3 channel are thus suspected to be a possible underlying cause for several psychiatric disorders.
Compounds that enhance SK channel activity (i.e., opener or positive gating modulator) have potential utility for the treatment of a number of diseases, including, schizophrenia, ataxia, epilepsy, pain, hypertension, urinary incontinence, erectile dysfunction, irritable bowel syndrome (Curr Med Chem. 2007;14) and Parkinson's disease (Exp Neurol. 2008; 213).
Structurally close compounds are disclosed in WO 2005/047289. However, the compounds there don't have any substituents at amino group on the pyrrolopyrimidine ring, which is quite different from the compounds of this invention. In addition the compounds in WO 2005/047289 are useful for the treatment of abnormal cell growth such as hyperproliferative disease represented by cancers, whereas the compounds of this invention are useful for the treatment of a condition or disorder mediated by potassium channel such as schizophrenia, ataxia, epilepsy, pain, hypertension, urinary incontinence, erectile dysfunction, irritable bowel syndrome and Parkinson's disease.
The present invention is directed to pyrrolopyrimidine derivatives compounds which are SK channel modulators, and which are useful in the treatment or prevention of neurological and psychiatric disorders and diseases in which SK channels are involved. The invention is also directed to pharmaceutical compositions comprising these compounds and the use of these compounds and compositions in the prevention or treatment of such diseases in which SK channels are involved. The invention also relates to a method for prevention or treatment of such diseases comprising administering to mammals in need for the prevention or treatment of the disorders or diseases.
The present invention provides compounds of formula (I) and salts thereof:

wherein:
R1 is independently selected from the group consisting of:
(1) hydrogen, (2) C1-6 alkyl, which is unsubstituted or substituted with one or more substituents selected from R5, and (3) cycloalkyl which is unsubstituted or substituted with one or more substituents selected from R5:
R1 is independently selected from the group consisting of:
(1) hydrogen, (2) C1-6 alkyl, which is unsubstituted or substituted with one or more substituents selected from R5, and (3) cycloalkyl which is unsubstituted or substituted with one or more substituents selected from R5:
R2 is selected from the group consisting of:
(1) hydrogen, (2) C1-6 alkyl, which is unsubstituted or substituted with one or more substituents selected from R5, (3) C3-6 cycloalkyl which is unsubstituted or substituted with one or more substituents selected from R5, (4) phenyl, which is unsubstituted or substituted with one or more substituents selected from R5, (5) heterocycle, which is unsubstituted or substituted with one or more substituents selected from R5, and (6) heterocycle substituted C1-6 alkyl, said heterocycle may be unsubstituted or substituted with one or more substituents selected from R5, i.e., a C1-6 alkyl substituted by heterocycle, the heterocycle being unsubstituted or substituted with one or more substituents selected from R5;
(1) hydrogen, (2) C1-6 alkyl, which is unsubstituted or substituted with one or more substituents selected from R5, (3) C3-6 cycloalkyl which is unsubstituted or substituted with one or more substituents selected from R5, (4) phenyl, which is unsubstituted or substituted with one or more substituents selected from R5, (5) heterocycle, which is unsubstituted or substituted with one or more substituents selected from R5, and (6) heterocycle substituted C1-6 alkyl, said heterocycle may be unsubstituted or substituted with one or more substituents selected from R5, i.e., a C1-6 alkyl substituted by heterocycle, the heterocycle being unsubstituted or substituted with one or more substituents selected from R5;
R3 is selected from the group consisting of:
(1) C1-6 alkyl, which is unsubstituted or substituted with one or more substituents selected from R5, (2) C3-6 cycloalkyl which is unsubstituted or substituted with one or more substituents selected from R5, (3) phenyl, which is unsubstituted or substituted with one or more substituents selected from R5, (4) heterocycle, which is unsubstituted or substituted with one or more substituents selected from R5, and (5) heterocycle substituted C1-6 alkyl, said heterocycle may be unsubstituted or substituted with one or more substituents selected from R5;
(1) C1-6 alkyl, which is unsubstituted or substituted with one or more substituents selected from R5, (2) C3-6 cycloalkyl which is unsubstituted or substituted with one or more substituents selected from R5, (3) phenyl, which is unsubstituted or substituted with one or more substituents selected from R5, (4) heterocycle, which is unsubstituted or substituted with one or more substituents selected from R5, and (5) heterocycle substituted C1-6 alkyl, said heterocycle may be unsubstituted or substituted with one or more substituents selected from R5;
R4 is independently selected from the group consisting of:
(1) hydrogen, (2) halogen, (3) hydroxy, (4) -Op-C1-6 alkyl, where p is 0 or 1 (wherein if p is 0, a chemical bond is present in the place of Op) and where the alkyl is unsubstituted or substituted with one or more substituents selected from R5, (5) -Op-C3-6 cycloalkyl, where p is 0 or 1 (wherein if p is 0, a bond is present in the place of Op) and where the cycloalkyl is unsubstituted or substituted with one or more substituents selected from R5, (6) C2-4 alkenyl, where the alkenyl is unsubstituted or substituted with one or more substituents selected from R5, (7) -(C=O)-NR6R7, (8) -NR6R7, (9) -S(O)2-NR6R7, (10) -NR6-S(O)2R7, (11) -S(O)r-R8, where r is 0, 1 or 2 and where R8 is selected from the definitions of R6 and R7 , (12) -CO2H, and (13) -CN;
(1) hydrogen, (2) halogen, (3) hydroxy, (4) -Op-C1-6 alkyl, where p is 0 or 1 (wherein if p is 0, a chemical bond is present in the place of Op) and where the alkyl is unsubstituted or substituted with one or more substituents selected from R5, (5) -Op-C3-6 cycloalkyl, where p is 0 or 1 (wherein if p is 0, a bond is present in the place of Op) and where the cycloalkyl is unsubstituted or substituted with one or more substituents selected from R5, (6) C2-4 alkenyl, where the alkenyl is unsubstituted or substituted with one or more substituents selected from R5, (7) -(C=O)-NR6R7, (8) -NR6R7, (9) -S(O)2-NR6R7, (10) -NR6-S(O)2R7, (11) -S(O)r-R8, where r is 0, 1 or 2 and where R8 is selected from the definitions of R6 and R7 , (12) -CO2H, and (13) -CN;
n is 1, 2, 3 or 4; when n is two or more than two, R4 may be same or different;
R5 is selected from the group consisting of:
(1) halogen, (2) hydroxy, (3) -(C=O)q-Or-C1-6 alkyl, where q is 0 or 1 and r is 0 or 1 (wherein if q is 0 or r is 0, a bond is present in the place of (C=O)q or Or, and wherein if q is 0 and r is 0, a single bond is present in the place of (C=O)q-Or) where the alkyl is unsubstituted or substituted with one or more substituents selected from R9, (4) -Op-(C1-3)perfluoroalkyl, where p is 0 or 1 (wherein if p is 0, a chemical bond is present in the place of Op) , (5) -(C=O)q-Or-C3-6 cycloalkyl, where q is 0 or 1 and r is 0 or 1 (wherein if q is 0 or r is 0, a bond is present in the place of (C=O)q or Or, and wherein if q is 0 and r is 0, a single bond is present in the place of (C=O)q-Or) and where the cycloalkyl is unsubstituted or substituted with one or more substituents selected from R9, (6) -(C=O)q-C2-4 alkenyl, where q is 0 or 1 (wherein if q is 0, a single bond is present in the place of (C=O)q) and the alkenyl is unsubstituted or substituted with one or more substituents selected from R9, (7) -(C=O)q-Or-phenyl or -(C=O)q-Or-napthyl, where q is 0 or 1 and r is 0 or 1 (wherein if q is 0 or r is 0, a bond is present in the place of (C=O)q or Or, and wherein if q is 0 and r is 0, a single bond is present in the place of (C=O)q-Or) and where the phenyl or napthyl is unsubstituted or substituted with one or more substituents selected from R9, (8) -(C=O)q-Or-heterocycle, where the heterocycle is unsubstituted or substituted with one or more substituents selected from R9, (9) -(C=O)-NR6R7, (10) -NR6R7, (11) -S(O)2-NR6R7, (12) -S(O)t-R8, where t is 0, 1 or 2, (13) -CO2H, (14) -CN, and (15) -NO2;
(1) halogen, (2) hydroxy, (3) -(C=O)q-Or-C1-6 alkyl, where q is 0 or 1 and r is 0 or 1 (wherein if q is 0 or r is 0, a bond is present in the place of (C=O)q or Or, and wherein if q is 0 and r is 0, a single bond is present in the place of (C=O)q-Or) where the alkyl is unsubstituted or substituted with one or more substituents selected from R9, (4) -Op-(C1-3)perfluoroalkyl, where p is 0 or 1 (wherein if p is 0, a chemical bond is present in the place of Op) , (5) -(C=O)q-Or-C3-6 cycloalkyl, where q is 0 or 1 and r is 0 or 1 (wherein if q is 0 or r is 0, a bond is present in the place of (C=O)q or Or, and wherein if q is 0 and r is 0, a single bond is present in the place of (C=O)q-Or) and where the cycloalkyl is unsubstituted or substituted with one or more substituents selected from R9, (6) -(C=O)q-C2-4 alkenyl, where q is 0 or 1 (wherein if q is 0, a single bond is present in the place of (C=O)q) and the alkenyl is unsubstituted or substituted with one or more substituents selected from R9, (7) -(C=O)q-Or-phenyl or -(C=O)q-Or-napthyl, where q is 0 or 1 and r is 0 or 1 (wherein if q is 0 or r is 0, a bond is present in the place of (C=O)q or Or, and wherein if q is 0 and r is 0, a single bond is present in the place of (C=O)q-Or) and where the phenyl or napthyl is unsubstituted or substituted with one or more substituents selected from R9, (8) -(C=O)q-Or-heterocycle, where the heterocycle is unsubstituted or substituted with one or more substituents selected from R9, (9) -(C=O)-NR6R7, (10) -NR6R7, (11) -S(O)2-NR6R7, (12) -S(O)t-R8, where t is 0, 1 or 2, (13) -CO2H, (14) -CN, and (15) -NO2;
R6 and R7 are independently selected from the group consisting of:
(1) hydrogen, (2) C1-6 alkyl, which is unsubstituted or substituted with R9, (3) C3-6 alkenyl, which is unsubstituted or substituted with R9, (4) cycloalkyl which is unsubstituted or substituted with R9, (5) phenyl, which is unsubstituted or substituted with R5, and (6) heterocycle, which is unsubstituted or substituted with R9, or R6 and R7 taken together with the nitrogen atom to which they are attached form a 3 to 8 membered ring, where the ring may contain one to four heteroatoms independently selected from nitrogen, oxygen, and sulfur; where the ring may be saturated or partially saturated or unsaturated; which is unsubstituted or substituted one or more substituents selected from R9;
(1) hydrogen, (2) C1-6 alkyl, which is unsubstituted or substituted with R9, (3) C3-6 alkenyl, which is unsubstituted or substituted with R9, (4) cycloalkyl which is unsubstituted or substituted with R9, (5) phenyl, which is unsubstituted or substituted with R5, and (6) heterocycle, which is unsubstituted or substituted with R9, or R6 and R7 taken together with the nitrogen atom to which they are attached form a 3 to 8 membered ring, where the ring may contain one to four heteroatoms independently selected from nitrogen, oxygen, and sulfur; where the ring may be saturated or partially saturated or unsaturated; which is unsubstituted or substituted one or more substituents selected from R9;
R8 is selected from the definitions of R6 and R7;
R9 is selected from the group consisting of:
(1) hydroxy, (2) halogen, (3) C1-6 alkyl, (4) -C3-6 cycloalkyl, (5) -O-C1-6 alkyl, (6) -O(C=O)-C1-6 alkyl, (7) -NH-C1-6 alkyl, (8) phenyl, (9) heterocycle, (10) -CO2H, (11) -CN, and (12) -Op-(C1-3)perfluoroalkyl, where p is 0 or 1 (wherein if p is 0, a chemical bond is present in the place of Op);
or a pharmaceutically acceptable salt thereof.
(1) hydroxy, (2) halogen, (3) C1-6 alkyl, (4) -C3-6 cycloalkyl, (5) -O-C1-6 alkyl, (6) -O(C=O)-C1-6 alkyl, (7) -NH-C1-6 alkyl, (8) phenyl, (9) heterocycle, (10) -CO2H, (11) -CN, and (12) -Op-(C1-3)perfluoroalkyl, where p is 0 or 1 (wherein if p is 0, a chemical bond is present in the place of Op);
or a pharmaceutically acceptable salt thereof.
Preferable compounds of this invention are in formula (I) wherein the definition described above:
R2 is selected from the group consisting of:
(1) hydrogen, (2) C1-6 alkyl, which is unsubstituted or substituted with one or more substituents selected from R5, and (3) C3-6 cycloalkyl which is unsubstituted or substituted with one or more substituents selected from R5;
R3 is selected from the group consisting of:
(1) C1-6 alkyl, which is unsubstituted or substituted with one or more substituents selected from R5, (2) C3-6 cycloalkyl which is unsubstituted or substituted with one or more substituents selected from R5, and (3) phenyl, which is unsubstituted or substituted with one or more substituents selected from R5;
R4 is independently selected from the group consisting of:
(1) hydrogen, (2) halogen, (3) hydroxy, (4) -Op-C1-6 alkyl, where p is 0 or 1 (wherein if p is 0, a chemical bond is present in the place of Op) and where the alkyl is unsubstituted or substituted with one or more substituents selected from R5, (5) -Op-C3-6 cycloalkyl, where p is 0 or 1 (wherein if p is 0, a bond is present in the place of Op) and where the cycloalkyl is unsubstituted or substituted with one or more substituents selected from R5, (6) C2-4 alkenyl, where the alkenyl is unsubstituted or substituted with one or more substituents selected from R5, and (7) -CN;
R2 is selected from the group consisting of:
(1) hydrogen, (2) C1-6 alkyl, which is unsubstituted or substituted with one or more substituents selected from R5, and (3) C3-6 cycloalkyl which is unsubstituted or substituted with one or more substituents selected from R5;
R3 is selected from the group consisting of:
(1) C1-6 alkyl, which is unsubstituted or substituted with one or more substituents selected from R5, (2) C3-6 cycloalkyl which is unsubstituted or substituted with one or more substituents selected from R5, and (3) phenyl, which is unsubstituted or substituted with one or more substituents selected from R5;
R4 is independently selected from the group consisting of:
(1) hydrogen, (2) halogen, (3) hydroxy, (4) -Op-C1-6 alkyl, where p is 0 or 1 (wherein if p is 0, a chemical bond is present in the place of Op) and where the alkyl is unsubstituted or substituted with one or more substituents selected from R5, (5) -Op-C3-6 cycloalkyl, where p is 0 or 1 (wherein if p is 0, a bond is present in the place of Op) and where the cycloalkyl is unsubstituted or substituted with one or more substituents selected from R5, (6) C2-4 alkenyl, where the alkenyl is unsubstituted or substituted with one or more substituents selected from R5, and (7) -CN;
R5 is selected from the group consisting of:
(1) halogen, (2) hydroxy, (3) -(C=O)q-Or-C1-6 alkyl, where q is 0 or 1 and r is 0 or 1 (wherein if q is 0 or r is 0, a bond is present in the place of (C=O)q or Or, and wherein if q is 0 and r is 0, a single bond is present in the place of (C=O)q-Or) where the alkyl is unsubstituted or substituted with one or more substituents selected from R9, (4) -Op-(C1-3)perfluoroalkyl, where p is 0 or 1 (wherein if p is 0, a chemical bond is present in the place of Op) , (5) -(C=O)q-Or-C3-6 cycloalkyl, where q is 0 or 1 and r is 0 or 1 (wherein if q is 0 or r is 0, a bond is present in the place of (C=O)q or Or, and wherein if q is 0 and r is 0, a single bond is present in the place of (C=O)q-Or) and where the cycloalkyl is unsubstituted or substituted with one or more substituents selected from R9, (6) -(C=O)q-C2-4 alkenyl, where q is 0 or 1 (whrein if q is 0, a single bond is present in the place of (C=O)q) and where the alkenyl is unsubstituted or substituted with one or more substituents selected from R9, (7) -(C=O)q-Or-phenyl or -(C=O)p-Or-napthyl, where q is 0 or 1 and r is 0 or 1 (wherein if q is 0 or r is 0, a bond is present in the place of (C=O)q or Or, and wherein if q is 0 and r is 0, a single bond is present in the place of (C=O)q-Or) and where the phenyl or napthyl is unsubstituted or substituted with one or more substituents selected from R9, (8) -(C=O)q-Or-heterocycle, where the heterocycle is unsubstituted or substituted with one or more substituents selected from R9, and (9) -CN;
(1) halogen, (2) hydroxy, (3) -(C=O)q-Or-C1-6 alkyl, where q is 0 or 1 and r is 0 or 1 (wherein if q is 0 or r is 0, a bond is present in the place of (C=O)q or Or, and wherein if q is 0 and r is 0, a single bond is present in the place of (C=O)q-Or) where the alkyl is unsubstituted or substituted with one or more substituents selected from R9, (4) -Op-(C1-3)perfluoroalkyl, where p is 0 or 1 (wherein if p is 0, a chemical bond is present in the place of Op) , (5) -(C=O)q-Or-C3-6 cycloalkyl, where q is 0 or 1 and r is 0 or 1 (wherein if q is 0 or r is 0, a bond is present in the place of (C=O)q or Or, and wherein if q is 0 and r is 0, a single bond is present in the place of (C=O)q-Or) and where the cycloalkyl is unsubstituted or substituted with one or more substituents selected from R9, (6) -(C=O)q-C2-4 alkenyl, where q is 0 or 1 (whrein if q is 0, a single bond is present in the place of (C=O)q) and where the alkenyl is unsubstituted or substituted with one or more substituents selected from R9, (7) -(C=O)q-Or-phenyl or -(C=O)p-Or-napthyl, where q is 0 or 1 and r is 0 or 1 (wherein if q is 0 or r is 0, a bond is present in the place of (C=O)q or Or, and wherein if q is 0 and r is 0, a single bond is present in the place of (C=O)q-Or) and where the phenyl or napthyl is unsubstituted or substituted with one or more substituents selected from R9, (8) -(C=O)q-Or-heterocycle, where the heterocycle is unsubstituted or substituted with one or more substituents selected from R9, and (9) -CN;
R9 is selected from the group consisting of:
(1) hydroxy, (2) halogen, (3) C1-6 alkyl, (4) -C3-6 cycloalkyl, (5) -O-C1-6 alkyl, (6) -CN, and (7) -Op-(C1-3)perfluoroalkyl, where p is 0 or 1 (wherein if p is 0, a chemical bond is present in the place of Op
or a pharmaceutically acceptable salt thereof.
(1) hydroxy, (2) halogen, (3) C1-6 alkyl, (4) -C3-6 cycloalkyl, (5) -O-C1-6 alkyl, (6) -CN, and (7) -Op-(C1-3)perfluoroalkyl, where p is 0 or 1 (wherein if p is 0, a chemical bond is present in the place of Op
or a pharmaceutically acceptable salt thereof.
The more preferable compounds are selected from:
5-(1H-1,3-benzodiazol-2-yl)-7-methyl-N-(2-phenoxyethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(4-fluorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-[2-(4-fluorophenyl)ethyl]-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(cyclohexylmethyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-7-methyl-N-(oxolan-2-ylmethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(2-ethoxyethyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(3-fluorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-7-methyl-N-(2-methylpropyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(2-fluorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-7-methyl-N-[3-(propan-2-yloxy)propyl]-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-7-methyl-N-[3-(trifluoromethoxy)phenyl]-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(2-chlorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(3-chlorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(2,5-difluorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(2,4-difluorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-7-methyl-N-(2-methylphenyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(2,3-difluorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-7-methyl-N-[3-(propan-2-yl)phenyl]-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(3,5-dichlorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(2,3-dichlorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
N-(3-chlorophenyl)-7-methyl-5-(6-methyl-1H-1,3-benzodiazol-2-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
N-(3-chlorophenyl)-7-methyl-5-(7-methyl-1H-1,3-benzodiazol-2-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
N-(3-chlorophenyl)-5-(6-fluoro-1H-1,3-benzodiazol-2-yl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(6-chloro-1H-1,3-benzodiazol-2-yl)-N-(3-chlorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
N-(3-chlorophenyl)-5-(6-methoxy-1H-1,3-benzodiazol-2-yl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-7-methyl-N-[3-(propan-2-yloxy)phenyl]-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(2,5-dichlorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(3-chloro-4-fluorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(6-chloro-1H-1,3-benzodiazol-2-yl)-N-(4-fluorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(4-bromophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-benzodiazol-2-yl)-N-(4-fluorophenyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-benzodiazol-2-yl)-N-(3-chlorophenyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-benzodiazol-2-yl)-N-(2-phenoxyethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(6-fluoro-1H-benzodiazol-2-yl)-N-(3-isopropoxypropyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
N-(3-isopropoxypropyl)-7-methyl-5-(6-methyl-1H-benzodiazol-2-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(6-fluoro-1H-benzodiazol-2-yl)-N-(2-isopropoxyethyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
N-(2-isopropoxyethyl)-7-methyl-5-(6-methyl-1H-benzodiazol-2-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(6-chloro-1H-benzodiazol-2-yl)-N-(3-ethoxypropyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine, and
N-(3-ethoxypropyl)-7-methyl-5-(6-methyl-1H-benzodiazol-2-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
or a pharmaceutically acceptable salt thereof.
5-(1H-1,3-benzodiazol-2-yl)-7-methyl-N-(2-phenoxyethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(4-fluorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-[2-(4-fluorophenyl)ethyl]-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(cyclohexylmethyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-7-methyl-N-(oxolan-2-ylmethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(2-ethoxyethyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(3-fluorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-7-methyl-N-(2-methylpropyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(2-fluorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-7-methyl-N-[3-(propan-2-yloxy)propyl]-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-7-methyl-N-[3-(trifluoromethoxy)phenyl]-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(2-chlorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(3-chlorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(2,5-difluorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(2,4-difluorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-7-methyl-N-(2-methylphenyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(2,3-difluorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-7-methyl-N-[3-(propan-2-yl)phenyl]-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(3,5-dichlorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(2,3-dichlorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
N-(3-chlorophenyl)-7-methyl-5-(6-methyl-1H-1,3-benzodiazol-2-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
N-(3-chlorophenyl)-7-methyl-5-(7-methyl-1H-1,3-benzodiazol-2-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
N-(3-chlorophenyl)-5-(6-fluoro-1H-1,3-benzodiazol-2-yl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(6-chloro-1H-1,3-benzodiazol-2-yl)-N-(3-chlorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
N-(3-chlorophenyl)-5-(6-methoxy-1H-1,3-benzodiazol-2-yl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-7-methyl-N-[3-(propan-2-yloxy)phenyl]-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(2,5-dichlorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(3-chloro-4-fluorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(6-chloro-1H-1,3-benzodiazol-2-yl)-N-(4-fluorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(4-bromophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-benzodiazol-2-yl)-N-(4-fluorophenyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-benzodiazol-2-yl)-N-(3-chlorophenyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-benzodiazol-2-yl)-N-(2-phenoxyethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(6-fluoro-1H-benzodiazol-2-yl)-N-(3-isopropoxypropyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
N-(3-isopropoxypropyl)-7-methyl-5-(6-methyl-1H-benzodiazol-2-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(6-fluoro-1H-benzodiazol-2-yl)-N-(2-isopropoxyethyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
N-(2-isopropoxyethyl)-7-methyl-5-(6-methyl-1H-benzodiazol-2-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(6-chloro-1H-benzodiazol-2-yl)-N-(3-ethoxypropyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine, and
N-(3-ethoxypropyl)-7-methyl-5-(6-methyl-1H-benzodiazol-2-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
or a pharmaceutically acceptable salt thereof.
Also, the present invention provides the use of a compound of formula (I) or a pharmaceutically acceptable salt thereof, each as described herein, for the manufacture of a medicament for the treatment of a condition or disorder mediated by potassium channel; in particular, SK channels activation activity. In order to use the compounds of formula (I) and pharmaceutically acceptable salts thereof in therapy, they will normally be formulated into a pharmaceutical composition in accordance with standard pharmaceutical practice. The present invention also provides a pharmaceutical composition, which comprises a compound of formula (I) or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier or excipient.
Preferably, the present invention also provides the use of a compound of formula (I) or a pharmaceutically acceptable salt thereof, each as described herein, for the manufacture of a medicament for the treatment of diseases selected from potassium channels related diseases.
Also, the present invention provides a pharmaceutical composition comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof, each as described herein, together with a pharmaceutically acceptable carrier for said compound.
Also, the present invention provides a pharmaceutical composition comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof, each as described herein, together with a pharmaceutically acceptable carrier for said compound and another pharmacologically active agent.
Also, the present invention provides a process for preparing a pharmaceutical composition, the process comprising mixing a compound of formula (I) or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable carrier or excipient.
Also, the present invention provides an intermediate in a process for preparing a compound of formula (I) or a pharmaceutically acceptable salt thereof.
Further, the present invention provides a method of treatment of a condition or disorder mediated by potassium channels activation activity, in a mammalian subject, which comprises administering to a mammal in need of such treatment a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof, each as described herein.
In a further aspect, the present invention provides a process for preparing a pharmaceutical composition, the process comprising mixing a compound of formula (I) or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable carrier or excipient.
Examples of conditions or disorders mediated by potassium channels activation activity include, but are not limited to, potassium channels related diseases. The compounds of the present invention show the potassium channels activation activity. The compounds of the present invention may show less toxicity, good absorption, distribution, good solubility, less protein binding affinity other than potassium channels, less drug-drug interaction, and good metabolic stability.
As appreciated by those of skill in the art, "halogen" or "halo" as used herein are intended to include fluoro, chloro, bromo and iodo. Similarly, C1-6, as in C1-6 alkyl is defined to identify the group as having 1, 2, 3, 4, 5 or 6 carbons in a linear or branched arrangement, such that C1-6 alkyl specifically includes methyl, ethyl, n-propyl, iso-propyl, n-butyl, iso-butyl, tert-butyl, pentyl, and hexyl. Similarly, C2-6 alkenyl is defined to identify the group as having 2, 3, 4, 5 or 6 carbons which incorporates at least one double bond, which may be in a E- or a Z- arrangement. A group which is designated as being independently substituted with substituents may be independently substituted with multiple numbers of such substituents.
The term "alkenyl", as used herein, means a hydrocarbon radical having at least one double bond including, but not limited to, ethenyl, propenyl, 1-butenyl, 2-butenyl and the like.
The term "cycloalkyl", as used herein, means a mono- or bicyclic ring, but not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, norboranyl, and adamantyl groups and the like.
The term "heterocycle" as used herein includes both unsaturated and saturated heterocyclic moieties, wherein the unsaturated heterocyclic moieties include benzoimidazolyl, benzimidazolonyl, benzofuranyl, benzofurazanyl, benzopyrazolyl, benzotriazolyl, benzothiophenyl, benzoxazolyl,carbazolyl, carbolinyl, cinnolinyl, furanyl, imidazolyl, indolinyl, indolyl, indolazinyl, indazolyl, isobenzofuranyl, isoindolyl, isoquinolyl, isothiazolyl, isoxazolyl, naphthpyridinyl, oxadiazolyl, oxazolyl, oxazolinyl, isoxazolinyl, oxetanyl, pyrazinyl, pyrazolyl, pyridazinyl, pyridopyridinyl, pyridazinyl, pyridyl, pyrimidinyl, pyrrolyl, quinazolinyl, quinolyl, quinoxalinyl, tetrazolyl, tetrazolopyridyl, thiadiazolyl, thiazolyl, thienyl, triazolyl, and N-oxides thereof, and wherein the saturated heterocyclic moieties include azetidinyl, 1,4-dioxanyl, hexahydroazepinyl, piperazinyl, piperidinyl, pyridin-2-onyl, pyrrolidinyl, morpholinyl, tetrahydrofuranyl, thiomorpholinyl, and tetrahydrothienyl, and N-oxides thereof and S-oxides thereof.
Any of the reactive groups such as amino, carboxyl and hydroxyl may be protected by a protecting group.
The term "protecting group", as used herein, means a hydroxy or amino protecting group which is selected from typical hydroxy or amino protecting groups described in Protective Groups in Organic Synthesis edited by T. W. Greene et al. (John Wiley & Sons, 1991);
The term "treating" and "treatment", as used herein, refers to curative, palliative and prophylactic treatment, including reversing, alleviating, inhibiting the progress of, or preventing the disorder or condition to which such term applies, or one or more symptoms of such disorder or condition.
The term "Ca2+ ionophore", as used herein, means a lipid-soluble molecule usually synthesized by microorganisms to transport ions across the lipid bilayer of the cell membrane, but not limited to, ionomycin, A23187, and the like.
As used herein, the article "a" or "an" refers to both the singular and plural form of the object to which it refers unless indicated otherwise.
Included within the scope of the "compounds of the invention" are all salts, solvates, hydrates, complexes, polymorphs, prodrugs, radiolabeled derivatives, stereoisomers and optical isomers of the compounds of formula (I).
The compounds of formula (I) can form acid addition salts thereof. It will be appreciated that for use in medicine the salts of the compounds of formula (I) should be pharmaceutically acceptable. Suitable pharmaceutically acceptable salts will be apparent to those skilled in the art and include those described in J. Pharm. Sci, 1977, 66, 1-19, such as acid addition salts formed with inorganic acids e.g. hydrochloric, hydrobromic, sulfuric, nitric or phosphoric acid; and organic acids e.g. succinic, maleic, formic, acetic, trifluoroacetic, propionic, fumaric, citric, tartaric, benzoic, p-toluenesulfonic, methanesulfonic or naphthalenesulfonic acid. Certain of the compounds of formula (I) may form acid addition salts with one or more equivalents of the acid. The present invention includes within its scope all possible stoichiometric and non-stoichiometric forms. In addition, certain compounds containing an acidic function such as a carboxy can be isolated in the form of their inorganic salt in which the counter ion can be selected from sodium, potassium, lithium, calcium, magnesium and the like, as well as from organic bases.
The compounds of formula (I) and salts thereof may be prepared in crystalline or non-crystalline form, and, if crystalline, may optionally be hydrated or solvated. This invention includes within its scope stoichiometric hydrates or solvates as well as compounds containing variable amounts of water and/or solvent.
Salts and solvates having non-pharmaceutically acceptable counter-ions or associated solvents are within the scope of the present invention, for example, for use as intermediates in the preparation of other compounds of formula (I) and their pharmaceutically acceptable salts.
The compounds of formula (I) may have polymorphs in crystalline form, which are within the scope of the present invention.
Additionally, the compounds of formula (I) may be administered as prodrugs. As used herein, a "prodrug" of a compound of formula (I) is a functional derivative of the compound which, upon administration to a patient, eventually liberates the compound of formula (I) in vivo. Administration of a compound of formula (I) as a prodrug may enable the skilled artisan to do one or more of the following: (a) modify the onset of action of the compound in vivo; (b) modify the duration of action of the compound in vivo; (c) modify the transportation or distribution of the compound in vivo; (d) modify the solubility of the compound in vivo; and (e) overcome a side effect or other difficulty encountered with the compound. Typical functional derivatives used to prepare prodrugs include modifications of the compound that are chemically or enzymatically cleaved in vivo. Such modifications, which include the preparation of phosphates, amides, esters, thioesters, carbonates, and carbamates, are well known to those skilled in the art.
In certain of the compounds of formula (I), there may be some chiral carbon atoms. In such cases, compounds of formula (I) exist as stereoisomers. The invention extends to all optical isomers such as stereoisomeric forms of the compounds of formula (I) including enantiomers, diastereoisomers and mixtures thereof, such as racemates. The different stereoisomeric forms may be separated or resolved one from the other by conventional methods or any given isomer may be obtained by conventional stereoselective or asymmetric syntheses.
Certain of the compounds herein can exist in various tautomeric forms and it is to be understood that the invention encompasses all such tautomeric forms.
The invention also includes isotopically-labeled compounds, which are identical to those described herein, but for the fact that one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number usually found in nature. Examples of isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine, iodine, and chlorine, such as 3H, 11C, 14C, 18F, 123I and 125I. Compounds of the invention that contain the aforementioned isotopes and/or other isotopes of other atoms are within the scope of the present invention. Isotopically-labeled compounds of the present invention, for example those into which radioactive isotopes such as 3H, 14C are incorporated, are useful in drug and/or substrate tissue distribution assays. Tritiated, i.e., 3H, and carbon-14, i.e., 14C, isotopes are particularly preferred for their ease of preparation and detectability. 11C and 18F isotopes are particularly useful in PET (positron emission tomography), and 125I isotopes are particularly useful in SPECT (single photon emission computerized tomography), all useful in brain imaging. Further, substitution with heavier isotopes such as deuterium, i.e., 2H, can afford certain therapeutic advantages resulting from greater metabolic stability, for example increased in vivo half-life or reduced dosage requirements and, hence, may be preferred in some circumstances. lsotopically labeled compounds of the invention can generally be prepared by carrying out the procedures disclosed in the Schemes and/or in the Examples below, then substituting a readily available isotopically labeled reagent for a non-isotopically labeled reagent.
With respect to other compounds disclosed in the art, the present compounds exhibit unexpected properties, such as with respect to duration of action and/or metabolism, such as increased metabolic stability, enhanced oral bioavailability or absorption, and/or decreased drug-drug interactions.
The compounds of formula (I), being potassium channel modulators, are potentially useful in the treatment of a number of diseases, including, schizophrenia, ataxia, epilepsy, pain, hypertension, urinary incontinence, erectile dysfunction, irritable bowel syndrome (Curr Med Chem. 2007;14) and Parkinson's disease (Exp Neurol. 2008; 213). Particularly chronic, inflammatory, neuropathic, nociceptive and visceral pain, is a preferred use.
Potassium channels have been implicated in a wide range of biological functions. This has suggested a potential role for these receptors in a variety of disease processes in humans or other species. The compounds of the present invention have utility in treating, preventing, ameliorating, controlling or reducing the risk of a variety of neurological and psychiatric disorders associated with potassium channels, including one or more of the following conditions or diseases:
a respiratory disease, epilepsy, convulsions, seizures, absence seizures, vascular spasms, coronary artery spasms, renal disorders, polycystic kidney disease, bladder spasms, urinary incontinence, bladder outflow obstruction, erectile dysfunction, gastrointestinal dysfunction, secretory diarrhoea, ischaemia, cerebral ischaemia, ischaemic heart disease, angina pectoris, coronary heart disease, autism, ataxia, traumatic brain injury, Parkinson's disease, bipolar disorder, psychosis, schizophrenia, anxiety, depression, mania, mood disorders, dementia, memory and attention deficits, Alzheimer's disease, amyotrophic lateral sclerosis (ALS), dysmenorrhea, narcolepsy, Reynaud's disease, intermittent claudication, Sjogren's syndrome, arrhythmia, hypertension, myotonic muscle dystrophia, spasticity, xerostomi, diabetes type II, hyperinsulinemia, premature labour, baldness, cancer, irritable bowel syndrome, immune suppression, migraine or pain, or withdrawal symptoms caused by the termination of abuse of chemical substances, in particular opioids, heroin, cocaine and morphine, benzodiazepines and benzodiazepine-like drugs, and alcohol.
a respiratory disease, epilepsy, convulsions, seizures, absence seizures, vascular spasms, coronary artery spasms, renal disorders, polycystic kidney disease, bladder spasms, urinary incontinence, bladder outflow obstruction, erectile dysfunction, gastrointestinal dysfunction, secretory diarrhoea, ischaemia, cerebral ischaemia, ischaemic heart disease, angina pectoris, coronary heart disease, autism, ataxia, traumatic brain injury, Parkinson's disease, bipolar disorder, psychosis, schizophrenia, anxiety, depression, mania, mood disorders, dementia, memory and attention deficits, Alzheimer's disease, amyotrophic lateral sclerosis (ALS), dysmenorrhea, narcolepsy, Reynaud's disease, intermittent claudication, Sjogren's syndrome, arrhythmia, hypertension, myotonic muscle dystrophia, spasticity, xerostomi, diabetes type II, hyperinsulinemia, premature labour, baldness, cancer, irritable bowel syndrome, immune suppression, migraine or pain, or withdrawal symptoms caused by the termination of abuse of chemical substances, in particular opioids, heroin, cocaine and morphine, benzodiazepines and benzodiazepine-like drugs, and alcohol.
The dosage of active ingredient in the compositions of this invention may be varied, however, it is necessary that the amount of the active ingredient be such that a suitable dosage form is obtained. The active ingredient may be administered to patients (animals and human) in need of such treatment in dosages that will provide optimal pharmaceutical efficacy.
The selected dosage depends upon the desired therapeutic effect, on the route of administration, and on the duration of the treatment. The dose will vary from patient to patient depending upon the nature and severity of disease, the patient's weight, special diets then being followed by a patient, concurrent medication, and other factors which those skilled in the art will recognize.
For administration to human patients, the total daily dose of the compounds of the invention is typically in the range 0.1 mg to 1000 mg depending, of course, on the mode of administration. For example, oral administration may require a total daily dose of from 1 mg to 1000 mg, while an intravenous dose may only require from 0.1 mg to 100 mg. The total daily dose may be administered in single or divided doses and may, at the physician's discretion, fall outside of the typical range given herein.
These dosages are based on an average human subject having a weight of about 60kg to 70kg. The physician will readily be able to determine doses for subjects whose weight falls outside this range, such as infants and the elderly.
For the avoidance of doubt, references herein to "treatment" include references to curative, palliative and prophylactic treatment.
In one embodiment, the dosage range will be about 0.5 mg to 500 mg per patient per day; in another embodiment about 0.5 mg to 200 mg per patient per day; in another embodiment about 1 mg to 100 mg per patient per day; and in another embodiment about 5 mg to 50 mg per patient per day; in yet another embodiment about 1 mg to 30 mg per patient per day. Pharmaceutical compositions of the present invention may be provided in a solid dosage formulation such as comprising about 0.5 mg to 500 mg active ingredient, or comprising about 1 mg to 250 mg active ingredient. The pharmaceutical composition may be provided in a solid dosage formulation comprising about 1 mg, 5 mg, 10mg, 25 mg, 50 mg, 100 mg, 200 mg or 250 mg active ingredient. For oral administration, the compositions may be provided in the form of tablets containing 1.0 to 1000 milligrams of the active ingredient, such as 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400, 500, 600, 750, 800, 900, and 1000 milligrams of the active ingredient for the symptomatic adjustment of the dosage to the patient to be treated. The compounds may be administered on a regimen of 1 to 4 times per day, such as once or twice per day.
The compounds of the present invention may be used in combination with one or more other drugs in the treatment, prevention, control, amelioration, or reduction of risk of diseases or conditions for which compounds of the present invention or the other drugs may have utility, where the combination of the drugs together are safer or more effective than either drug alone. Such other drug(s) may be administered, by a route and in an amount commonly used therefore, contemporaneously or sequentially with a compound of the present invention. When a compound of the present invention is used contemporaneously with one or more other drugs, a pharmaceutical composition in unit dosage form containing such other drugs and the compound of the present invention is envisioned. However, the combination therapy may also include therapies in which the compound of the present invention and one or more other drugs are administered on different overlapping schedules. It is also contemplated that when used in combination with one or more other active ingredients, the compounds of the present invention and the other active ingredients may be used in lower doses than when each is used singly.
Accordingly, the pharmaceutical compositions of the present invention include those that contain one or more other active ingredients, in addition to a compound of the present invention. The above combinations include combinations of a compound of the present invention not only with one other active compound, but also with two or more other active compounds.
Likewise, compounds of the present invention may be used in combination with other drugs that are used in the prevention, treatment, control, amelioration, or reduction of risk of the diseases or conditions for which compounds of the present invention are useful. Such other drugs may be administered, by a route and in an amount commonly used therefore, contemporaneously or sequentially with a compound of the present invention. When a compound of the present invention is used contemporaneously with one or more other drugs, a pharmaceutical composition containing such other drugs in addition to the compound of the present invention is envisioned. Accordingly, the pharmaceutical compositions of the present invention include those that also contain one or more other active ingredients, in addition to a compound of the present invention.
The weight ratio of the compound of the compound of the present invention to the second active ingredient may be varied and will depend upon the effective dose of each ingredient. Generally, an effective dose of each will be used. Thus, for example, when a compound of the present invention is combined with another agent, the weight ratio of the compound of the present invention to the other agent will generally range from about 1000:1 to about 1:1000, including about 200: 1 to about 1:200. Combinations of a compound of the present invention and other active ingredients will generally also be within the aforementioned range, but in each case, an effective dose of each active ingredient should be used. In such combinations the compound of the present invention and other active agents may be administered separately or in conjunction. In addition, the administration of one element may be prior to, concurrent to, or subsequent to the administration of other agent(s).
A potassium channels modulator may be usefully combined with another pharmacologically active compound, or with two or more other pharmacologically active compounds, particularly in the treatment of inflammatory, pain and urological diseases or disorders. For example, a potassium channels modulator, particularly a compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof, as defined above, may be administered simultaneously, sequentially or separately in combination with one or more agents selected from:
- an opioid analgesic, e.g. morphine, heroin, hydromorphone, oxymorphone, levorphanol, levallorphan, methadone, meperidine, fentanyl, cocaine, codeine, dihydrocodeine, oxycodone, hydrocodone, propoxyphene, nalmefene, nalorphine, naloxone, naltrexone, buprenorphine, butorphanol, nalbuphine or pentazocine;
- a nonsteroidal antiinflammatory drug (NSAID), e.g. aspirin, diclofenac, diflusinal, etodolac, fenbufen, fenoprofen, flufenisal, flurbiprofen, ibuprofen, indomethacin, ketoprofen, ketorolac, meclofenamic acid, mefenamic acid, meloxicam, nabumetone, naproxen, nimesulide, nitroflurbiprofen, olsalazine, oxaprozin, phenylbutazone, piroxicam, sulfasalazine, sulindac, tolmetin or zomepirac;
- a barbiturate sedative, e.g. amobarbital, aprobarbital, butabarbital, butabital, mephobarbital, metharbital, methohexital, pentobarbital, phenobartital, secobarbital, talbutal, theamylal or thiopental;
- a benzodiazepine having a sedative action, e.g. chlordiazepoxide, clorazepate, diazepam, flurazepam, lorazepam, oxazepam, temazepam or triazolam;
- an H1 antagonist having a sedative action, e.g. diphenhydramine, pyrilamine, promethazine, chlorpheniramine or chlorcyclizine;
- a sedative such as glutethimide, meprobamate, methaqualone or dichloralphenazone;
- a skeletal muscle relaxant, e.g. baclofen, carisoprodol, chlorzoxazone, cyclobenzaprine, methocarbamol or orphrenadine;
- an NMDA receptor antagonist, e.g. dextromethorphan ((+)-3-hydroxy-N-methylmorphinan) or its metabolite dextrorphan ((+)-3-hydroxy-N-methylmorphinan), ketamine, memantine, pyrroloquinoline quinine, cis-4-(phosphonomethyl)-2-piperidinecarboxylic acid, budipine, EN-3231 (MorphiDex(registered trademark), a combination formulation of morphine and dextromethorphan), topiramate, neramexane or perzinfotel including an NR2B antagonist, e.g. ifenprodil, traxoprodil or (-)-(R)-6-{2-[4-(3-fluorophenyl)-4-hydroxy-1-piperidinyl]-1-hydroxyethyl-3,4-dihydro-2(1H)-quinolinone;
- an alpha-adrenergic, e.g. doxazosin, tamsulosin, clonidine, guanfacine, dexmetatomidine, modafinil, or 4-amino-6,7-dimethoxy-2-(5-methane-sulfonamido-1,2,3,4-tetrahydroisoquinol-2-yl)-5-(2-pyridyl) quinazoline;
- a tricyclic antidepressant, e.g. desipramine, imipramine, amitriptyline or nortriptyline;
- an anticonvulsant, e.g. carbamazepine, lamotrigine, topiratmate or valproate;
- a tachykinin (NK) antagonist, particularly an NK-3, NK-2 or NK-1 antagonist, e.g. (alphaR,9R)-7-[3,5-bis(trifluoromethyl)benzyl]-8,9,10,11-tetrahydro-9-methyl-5-(4-methylphenyl)-7H-[1,4]diazocino[2,1-g][1,7]-naphthyridine-6-13-dione (TAK-637), 5-[[(2R,3S)-2-[(1R)-1-[3,5-bis(trifluoromethyl)phenyl]ethoxy-3-(4-fluorophenyl)-4-morpholinyl]-methyl]-1,2-dihydro-3H-1,2,4-triazol-3-one (MK-869), aprepitant, lanepitant, dapitant or 3-[[2-methoxy-5-(trifluoromethoxy)phenyl]-methylamino]-2-phenylpiperidine (2S,3S);
- a muscarinic antagonist, e.g oxybutynin, tolterodine, propiverine, tropsium chloride, darifenacin, solifenacin, temiverine and ipratropium;
- a COX-2 selective inhibitor, e.g. celecoxib, rofecoxib, parecoxib, valdecoxib, deracoxib, etoricoxib, or lumiracoxib;
- a coal-tar analgesic, in particular paracetamol;
- a neuroleptic such as droperidol, chlorpromazine, haloperidol, perphenazine, thioridazine, mesoridazine, trifluoperazine, fluphenazine, clozapine, olanzapine, risperidone, ziprasidone, quetiapine, sertindole, aripiprazole, sonepiprazole, blonanserin, iloperidone, perospirone, raclopride, zotepine, bifeprunox, asenapine, lurasidone, amisulpride, balaperidone, palindore, eplivanserin, osanetant, rimonabant, meclinertant, Miraxion(registered trademark) or sarizotan;
- a vanilloid receptor agonist (e.g. resinferatoxin) or antagonist (e.g. capsazepine);
- a beta-adrenergic such as propranolol;
- a local anaesthetic such as mexiletine;
- a corticosteroid such as dexamethasone;
- a 5-HT receptor agonist or antagonist, particularly a 5-HT1B/1D agonist such as eletriptan, sumatriptan, naratriptan, zolmitriptan or rizatriptan;
- a 5-HT2A receptor antagonist such as R(+)-alpha-(2,3-dimethoxy-phenyl)-1-[2-(4-fluorophenylethyl)]-4-piperidinemethanol (MDL-100907);
- a cholinergic (nicotinic) analgesic, such as ispronicline (TC-1734), (E)-N-methyl-4-(3-pyridinyl)-3-buten-1-amine (RJR-2403), (R)-5-(2-azetidinylmethoxy)-2-chloropyridine (ABT-594) or nicotine;
- Tramadol(registered trademark);
- a PDEV inhibitor, such as 5-[2-ethoxy-5-(4-methyl-1-piperazinyl-sulphonyl)phenyl]-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (sildenafil), (6R,12aR)-2,3,6,7,12,12a-hexahydro-2-methyl-6-(3,4-methylenedioxyphenyl)-pyrazino[2',1':6,1]-pyrido[3,4-b]indole-1,4-dione (IC-351 or tadalafil), 2-[2-ethoxy-5-(4-ethyl-piperazin-1-yl-1-sulphonyl)-phenyl]-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one (vardenafil), 5-(5-acetyl-2-butoxy-3-pyridinyl)-3-ethyl-2-(1-ethyl-3-azetidinyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one, 5-(5-acetyl-2-propoxy-3-pyridinyl)-3-ethyl-2-(1-isopropyl-3-azetidinyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one, 5-[2-ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-[2-methoxyethyl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one, 4-[(3-chloro-4-methoxybenzyl)amino]-2-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-N-(pyrimidin-2-ylmethyl)pyrimidine-5-carboxamide, 3-(1-methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-N-[2-(1-methylpyrrolidin-2-yl)ethyl]-4-propoxybenzenesulfonamide;
- an alpha-2-delta ligand such as gabapentin, pregabalin, 3-methylgabapentin, (1alpha,3alpha,5alpha)(3-amino-methyl-bicyclo[3.2.0]hept-3-yl)-acetic acid, (3S,5R)-3 aminomethyl-5 methyl-heptanoic acid, (3S,5R)-3 amino-5 methyl-heptanoic acid, (3S,5R)-3 amino-5 methyl-octanoic acid, (2S,4S)-4-(3-chlorophenoxy)proline, (2S,4S)-4-(3-fluorobenzyl)-proline, [(1R,5R,6S)-6-(aminomethyl)bicyclo[3.2.0]hept-6-yl]acetic acid, 3-(1-aminomethyl-cyclohexylmethyl)-4H-[1,2,4]oxadiazol-5-one, C-[1-(1H-tetrazol-5-ylmethyl)-cycloheptyl]-methylamine, (3S,4S)-(1-aminomethyl-3,4-dimethyl-cyclopentyl)-acetic acid, (3S,5R)-3 aminomethyl-5 methyl-octanoic acid, (3S,5R)-3 amino-5 methyl-nonanoic acid, (3S,5R)-3 amino-5 methyl-octanoic acid, (3R,4R,5R)-3-amino-4,5-dimethyl-heptanoic acid and (3R,4R,5R)-3-amino-4,5-dimethyl-octanoic acid;
- a cannabinoid;
- metabotropic glutamate subtype 1 receptor (mGluR1) antagonist;
- a serotonin reuptake inhibitor such as sertraline, sertraline metabolite demethylsertraline, fluoxetine, norfluoxetine (fluoxetine desmethyl metabolite), fluvoxamine, paroxetine, citalopram, citalopram metabolite desmethylcitalopram, escitalopram, d,l-fenfluramine, femoxetine, ifoxetine, cyanodothiepin, litoxetine, dapoxetine, nefazodone, cericlamine and trazodone;
- a noradrenaline (norepinephrine) reuptake inhibitor, such as maprotiline, lofepramine, mirtazepine, oxaprotiline, fezolamine, tomoxetine, mianserin, buproprion, buproprion metabolite hydroxybuproprion, nomifensine and viloxazine (VivalanR), especially a selective noradrenaline reuptake inhibitor such as reboxetine, in particular (S,S)-reboxetine;
- a dual serotonin-noradrenaline reuptake inhibitor, such as venlafaxine, venlafaxine metabolite O-desmethylvenlafaxine, clomipramine, clomipramine metabolite desmethylclomipramine, duloxetine, milnacipran and imipramine;
- an inducible nitric oxide synthase (iNOS) inhibitor such as S-[2-[(1-iminoethyl)amino]ethyl]-L-homocysteine, S-[2-[(1-iminoethyl)-amino]ethyl]-4,4-dioxo-L-cysteine, S-[2-[(1-iminoethyl)amino]ethyl]-2-methyl-L-cysteine, (2S,5Z)-2-amino-2-methyl-7-[(1-iminoethyl)amino]-5-heptenoic acid, 2-[[(1R,3S)-3-amino-4- hydroxy-1-(5-thiazolyl)-butyl]thio]-5-chloro-3-pyridinecarbonitrile; 2-[[(1R,3S)-3-amino-4-hydroxy-1-(5-thiazolyl)butyl]thio]-4-chlorobenzonitrile, (2S,4R)-2-amino-4-[[2-chloro-5-(trifluoromethyl)phenyl]thio]-5-thiazolebutanol, 2-[[(1R,3S)-3-amino-4-hydroxy-1-(5-thiazolyl) butyl]thio]-6-(trifluoromethyl)-3 pyridinecarbonitrile, 2-[[(1R,3S)-3- amino-4-hydroxy- 1 -(5-thiazolyl)butyl]thio]-5-chlorobenzonitrile, N-[4-[2-(3-chlorobenzylamino)ethyl]phenyl]thiophene-2-carboxamidine, or guanidinoethyldisulfide;
- an acetylcholinesterase inhibitor such as donepezil;
- a prostaglandin E2 subtype 4 (EP4) antagonist such as N-[({2-[4-(2-ethyl-4,6-dimethyl-1H-imidazo[4,5-c]pyridin-1-yl)phenyl]ethyl}amino)-carbonyl]-4-methylbenzenesulfonamide or 4-[(1S)-1-({[5-chloro-2-(3-fluorophenoxy)pyridin-3-yl]carbonyl}amino)ethyl]benzoic acid;
- a leukotriene B4 antagonist; such as 1-(3-biphenyl-4-ylmethyl-4-hydroxy-chroman-7-yl)-cyclopentanecarboxylic acid (CP-105696), 5-[2-(2-Carboxyethyl)-3-[6-(4-methoxyphenyl)-5E- hexenyl]oxyphenoxy]-valeric acid (ONO-4057) or DPC-11870,
- a 5-lipoxygenase inhibitor, such as zileuton, 6-[(3-fluoro-5-[4-methoxy-3,4,5,6-tetrahydro-2H-pyran-4-yl])phenoxy-methyl]-1-methyl-2-quinolone (ZD-2138), or 2,3,5-trimethyl-6-(3-pyridylmethyl),1,4-benzoquinone (CV-6504);
- a sodium channel blocker, such as lidocaine;
- a 5-HT3 antagonist, such as ondansetron;
and the pharmaceutically acceptable salts and solvates thereof.
and the pharmaceutically acceptable salts and solvates thereof.
Such combinations offer significant advantages, including synergistic activity, in therapy.
A pharmaceutical composition of the invention, which may be prepared by admixture, suitably at ambient temperature and atmospheric pressure, is usually adapted for oral, parenteral or rectal administration and, as such, may be in the form of tablets, capsules, oral liquid preparations, powders, granules, lozenges, reconstitutable powders, injectable or infusible solutions or suspensions or suppositories. Orally administrate compositions are generally preferred. Tablets and capsules for oral administration may be in unit dose form, and may contain conventional excipients, such as binding agents (e.g. pregelatinised maize starch, polyvinylpyrrolidone or hydroxypropyl methylcellulose); fillers (e.g. lactose, microcrystalline cellulose or calcium hydrogen phosphate); tabletting lubricants (e.g. magnesium stearate, talc or silica); disintegrants (e.g. potato starch or sodium starch glycollate); and acceptable wetting agents (e.g. sodium lauryl sulphate). The tablets may be coated according to methods well known in normal pharmaceutical practice.
Oral liquid preparations may be in the form of, for example, aqueous or oily suspension, solutions, emulsions, syrups or elixirs, or may be in the form of a dry product for reconstitution with water or other suitable vehicle before use. Such liquid preparations may contain conventional additives such as suspending agents (e.g. sorbitol syrup, cellulose derivatives or hydrogenated edible fats), emulsifying agents (e.g. lecithin or acacia), non-aqueous vehicles (which may include edible oils e.g. almond oil, oily esters, ethyl alcohol or fractionated vegetable oils), preservatives (e.g. methyl or propyl-p-hydroxybenzoates or sorbic acid), and, if desired, conventional flavourings or colorants, buffer salts and sweetening agents as appropriate. Preparations for oral administration may be suitably formulated to give controlled release of the active compound or pharmaceutically acceptable salt thereof.
For parenteral administration, fluid unit dosage forms are prepared utilising a compound of formula (I) or pharmaceutically acceptable salt thereof and a sterile vehicle. Formulations for injection may be presented in unit dosage form e.g. in ampoules or in multi-dose, utilising a compound of formula (I) or pharmaceutically acceptable salt thereof and a sterile vehicle, optionally with an added preservative. The compositions may take such forms as suspensions, solutions or emulsions in oily or aqueous vehicles, and may contain formulatory agents such as suspending, stabilising and/or dispersing agents. Alternatively, the active ingredient may be in powder form for constitution with a suitable vehicle, e.g. sterile pyrogen-free water, before use. The compound, depending on the vehicle and concentration used, can be either suspended or dissolved in the vehicle. In preparing solutions, the compound can be dissolved for injection and filter sterilised before filling into a suitable vial or ampoule and sealing. Advantageously, adjuvants such as a local anaesthetic, preservatives and buffering agents are dissolved in the vehicle. To enhance the stability, the composition can be frozen after filling into the vial and the water removed under vacuum. Parenteral suspensions are prepared in substantially the same manner, except that the compound is suspended in the vehicle instead of being dissolved, and sterilisation cannot be accomplished by filtration. The compound can be sterilised by exposure to ethylene oxide before suspension in a sterile vehicle. Advantageously, a surfactant or wetting agent is included in the composition to facilitate uniform distribution of the compound.
Lotions may be formulated with an aqueous or oily base and will in general also contain one or more emulsifying agents, stabilising agents, dispersing agents, suspending agents, thickening agents, or colouring agents. Drops may be formulated with an aqueous or non-aqueous base also comprising one or more dispersing agents, stabilising agents, solubilising agents or suspending agents. They may also contain a preservative.
The compounds of formula (I) or pharmaceutically acceptable salts thereof may also be formulated in rectal compositions such as suppositories or retention enemas, e.g. containing conventional suppository bases such as cocoa butter or other glycerides.
The compounds of formula (I) or pharmaceutically acceptable salts may also be formulated as depot preparations. Such long acting formulations may be administered by implantation (for example subcutaneously or intramuscularly) or by intramuscular injection. Thus, for example, the compounds of formula (I) or pharmaceutically acceptable salts may be formulated with suitable polymeric or hydrophobic materials (for example as an emulsion in an acceptable oil) or ion exchange resins, or as sparingly soluble derivatives, for example, as a sparingly soluble salt.
For intranasal administration, the compounds formula (I) or pharmaceutically acceptable salts thereof may be formulated as solutions for administration via a suitable metered or unitary dose device or alternatively as a powder mix with a suitable carrier for administration using a suitable delivery device. Thus compounds of formula (I) or pharmaceutically acceptable salts thereof may be formulated for oral, buccal, parenteral, topical (including ophthalmic and nasal), depot or rectal administration or in a form suitable for administration by inhalation or insufflation (either through the mouth or nose). The compounds of formula (I) and pharmaceutically acceptable salts thereof may be formulated for topical administration in the form of ointments, creams, gels, lotions, pessaries, aerosols or drops (e.g. eye, ear or nose drops). Ointments and creams may, for example, be formulated with an aqueous or oily base with the addition of suitable thickening and/or gelling agents. Ointments for administration to the eye may be manufactured in a sterile manner using sterilized components.
General Synthesis
Throughout the instant application, the following abbreviations are used with the following meanings:
BOP: 1H-Benzotriazol-1-yloxytris(dimethylamino)phosphonium Hexafluorophosphate
DMF: N,N-dimethylformamide
EDC: 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Hydrochloride
FMOC: 9-fluorenylmethoxycarbonyl
HOBT: 1-Hydroxybenztriazole
HBTU: O-(Benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium Hexafluorophosphate
HPLC: High pressure liquid chromatography
tR: Retention time
MHz: Megahertz
NMR: Nuclear Magnetic Resonance
TFA: Trifluoroacetic acid
THF: Tetrahydrofuran
TLC: Thin layer chromatography
Throughout the instant application, the following abbreviations are used with the following meanings:
BOP: 1H-Benzotriazol-1-yloxytris(dimethylamino)phosphonium Hexafluorophosphate
DMF: N,N-dimethylformamide
EDC: 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Hydrochloride
FMOC: 9-fluorenylmethoxycarbonyl
HOBT: 1-Hydroxybenztriazole
HBTU: O-(Benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium Hexafluorophosphate
HPLC: High pressure liquid chromatography
tR: Retention time
MHz: Megahertz
NMR: Nuclear Magnetic Resonance
TFA: Trifluoroacetic acid
THF: Tetrahydrofuran
TLC: Thin layer chromatography
The term of "base" is likewise no particular restriction on the nature of the bases used, and any base commonly used in reactions of this type may equally be used here. Examples of such bases include: alkali metal hydroxides, such as lithium hydroxide, sodium hydroxide, potassium hydroxide, and barium hydroxide; alkali metal hydrides, such as lithium hydride, sodium hydride, and potassium hydride; alkali metal alkoxides, such as sodium methoxide, sodium ethoxide, and potassium t-butoxide; alkali metal carbonates, such as lithium carbonate, sodium carbonate, potassium carbonate, and cesium carbonate; alkali metal hydrogencarbonates, such as lithium hydrogencarbonate, sodium hydrogencarbonate, and potassium hydrogencarbonate; amines, such as N-methylmorpholine, triethylamine, tripropylamine, tributylamine, diisopropylethylamine, N-methylpiperidine, pyridine, 4-pyrrolidinopyridine, picoline, 2,6-di(t-butyl)-4-methylpyridine, quinoline, N,N-dimethylaniline, N,N-diethylaniline, 1,5-diazabicyclo[4.3.0]non-5-ene (DBN), 1,4-diazabicyclo[2.2.2]octane (DABCO), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), lutidine, and colidine; alkali metal amides, such as lithium amide, sodium amide, potassium amide, lithium diisopropyl amide, potassium diisopropyl amide, sodium diisopropyl amide, lithium bis(trimethylsilyl)amide and potassium bis(trimethylsilyl)amide. Of these, triethylamine, diisopropylethylamine, DBU, DBN, DABCO, pyridine, lutidine, colidine, sodium carbonate, sodium hydrogencarbonate, sodium hydroxide, potassium carbonate, potassium hydrogencarbonate, potassium hydroxide, barium hydroxide, and cesium carbonate are preferred.
The reactions are normally and preferably effected in the presence of inert solvent. There is no particular restriction on the nature of the solvent to be employed, provided that it has no adverse effect on the reaction or the reagents involved and that it can dissolve reagents, at least to some extent. Examples of suitable solvents include, but not limited to: halogenated hydrocarbons, such as dichloromethane, chloroform, carbon tetrachloride, and dichloroethane; ethers, such as diethyl ether, diisopropyl ether, THF, and dioxane; aromatic hydrocarbons, such as benzene, toluene and nitrobenzene; amides, such as, DMF, N,N-dimethylacetamide, and hexamethylphosphoric triamide; amines, such as N-methylmorpholine, triethylamine, tripropylamine, tributylamine, diisopropylethylamine, N-methylpiperidine, pyridine, 4-pyrrolidinopyridine, N,N-dimethylaniline, and N,N-diethylaniline; alcohols, such as methanol, ethanol, propanol, isopropanol, and butanol; nitriles, such as acetonitrile and benzonitrile; sulfoxides, such as dimethyl sulfoxide (DMSO) and sulfolane; ketones, such as acetone and diethylketone. Of these solvents, including but not limited to DMF, DMSO, THF, diethylether, diisopropylether, dimethoxyethane, acetonitrile, dichloromethane, dichloroethane and chloroform are preferred.
The invention is illustrated in the following non-limiting examples in which, unless stated otherwise: all reagents are commercially available, all operations were carried out at room or ambient temperature, that is, in the range of about 18-25 oC; evaporation of solvent was carried out using a rotary evaporator under reduced pressure with a bath temperature of up to about 60 oC; reactions were monitored by thin layer chromatography (tlc) and reaction times are given for illustration only; the structure and purity of all isolated compounds were assured by at least one of the following techniques: tlc (Merck silica gel 60 F254 precoated TLC plates or Merck NH2 F254 precoated HPTLC plates), mass spectrometry or nuclear magnetic resonance (NMR). Yields are given for illustrative purposes only. Flash column chromatography was carried out using Merck silica gel 60 (230-400 mesh ASTM) or Fuji Silysia Chromatorex(registered trademark) DU3050 (Amino Type, 30-50 micrometer) or Biotage silica (32-63 mm, KP-Sil) or Biotage amino bounded silica (35-75 mm, KP-NH). The purification of compounds using HPLC was performed by the following apparatus and conditions ("process A"); Apparatus; Waters MS-trigger AutoPurificationTM system Column; Waters XTerra C18, 19X50mm, 5mm particle, Method A; Methanol or acetonitrile / 0.05%(v/v) formic acid aqueous solution, Method B; Methanol or acetonitrile / 0.01%(v/v) ammonia aqueous solution. The purification using HPLC("Process B") was performed by the following apparatus and conditions: Apparatus; UV-trigger preparative HPLC system, Waters (Column; XTerra MS C18, 5 micrometer, 19 x 50 mm or 30 x 50 mm), Detector; UV 254 nm, Conditions; acetonitrile:0.05% formic acid aqueous solution or acetonitrile:0.01%(v/v) aqueous ammonia solution; 20 mL/min (19 x 50 mm) or 40 mL/min (30 x 50 mm) at room temperature. Low-resolution mass spectral data (ESI) were obtained by the following apparatus and conditions: Apparatus; Waters Alliance HPLC system on ZQ or ZMD mass spectrometer and UV detector. NMR data was determined at 270 MHz (JEOL JNM-LA 270 spectrometer) or 300 MHz (JEOL JNM-LA300) using deuterated chloroform (99.8%(v/v) D) or dimethylsulfoxide (99.9%(v/v) D) as solvent unless indicated otherwise, relative to tetramethylsilane (TMS) as internal standard in parts per million (ppm); conventional abbreviations used are: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad, etc. Chemical symbols have their usual meanings;

Conditions for determining HPLC retention time:




All of the pyrrolopyrimidine derivatives of the formula (I) can be prepared by the procedures described in the general methods presented below or by the specific methods described in the Examples section and the Preparations section, or by routine modifications thereof. The present invention also encompasses any one or more of these processes for preparing the pyrrolopyrimidine derivatives of formula (I), in addition to any novel intermediates used therein.
In the following general methods, R1, R2, R3, R4, and n are as previously defined for pyrrolopyrimidine derivatives of the formula (I) unless otherwise stated. All starting materials in the following general syntheses may be commercially available or obtained by conventional methods known to those skilled in the art.

In Step A, an intermediate of formula (III) can be prepared from a compound of formula (II) by SNAr reaction with amines (R2R3NH) in a suitable solvent such as ethanol and DMF at a temperature of from about 20 to about 150 oC for about 1-24 hours.
In Step B, an intermediate of formula (IV) can be prepared from a compound of formula (III) by amidation with (R4)n-benzene-1,2-diamines with using a suitable coupling reagent such as EDC preferably under the presence of a base such a combination of N,N-Diisopropylethylamine and HOBT in a suitable solvent such as DMF at a temperature of from about 20 to about 100 oC for about 1-24 hours.
In Step C, a compound of formula (I) can be prepared from a compound of formula (IV) by cyclization in an acidic media such as acetic acid at a temperature of from about 20 to about 120 oC for about 1-24 hours.
Example 1
5-(1H-1,3-Benzodiazol-2-yl)-7-methyl-N-phenyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine
5-(1H-1,3-Benzodiazol-2-yl)-7-methyl-N-phenyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine
STEP 1: 5-Bromo-4-chloro-7-methyl-7H-pyrrolo[2,3-d]pyrimidine

To a stirred solution of 4-chloro-7-methyl-7H-pyrrolo[2,3-d]pyrimidine (WO2005/095357, 6.02 g, 35.9 mmol) in dichloromethane (120 mL) was added N-bromosuccinimide (7.67 g, 43.1 mmol) at 0 oC. The resulting mixture was stirred at room temperature for 2 hrs. The reaction mixture was quenched with saturated sodium bicarbonate aqueous solution (200 mL), and extracted with dichloromethane (200 mL). The organic layer was dried over magnesium sulfate, and concentrated under reduced pressure. The residue was purified by silica-gel column chromatography (dichloromethane/ethyl acetate=10/1) to give the title compound (8.27 g, 93%) as a white solid.;
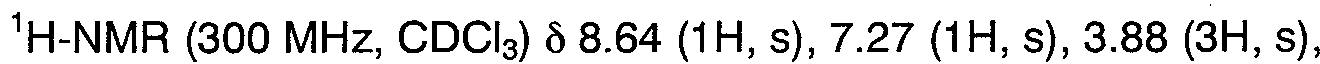 LCMS (Method A) m/z: M+1 247.9; tR = 2.70 min.
LCMS (Method A) m/z: M+1 247.9; tR = 2.70 min.
STEP 2: 4-Chloro-7-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carboxylic acid

To a stirred solution of 5-bromo-4-chloro-7-methyl-7H-pyrrolo[2,3-d]pyrimidine (STEP 1, 8.20 g, 33.3 mmol) in tetrahydrofuran (120 mL) was added 1.59 M n-butyllithium in hexane solution (25.1 mL, 39.9 mmol) slowly at -78 oC. After stirring at -78 oC for 1 hr, solid CO2 was added to the mixture. The resulting was warmed to room temperature gradually, and quenched with 1M HCl aqueous solution (200 mL). The mixture was extracted with dichloromethane/methanol (10/1, 200 mLx4), and the combined organic fraction was dried over magnesium sulfate. After removal of the solvent by evaporation, the residue was triturated with ethyl acetate (100 mL) and collected by filtration to give the title compound (6.11 g, 87%) as an off-white solid.;
 LCMS (Method A) m/z: M+1 212.0; tR = 1.30 min.
LCMS (Method A) m/z: M+1 212.0; tR = 1.30 min.
STEP A: 7-Methyl-4-(phenylamino)-7H-pyrrolo[2,3-d]pyrimidine-5-carboxylic acid

A mixture of 4-chloro-7-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carboxylic acid (STEP 2, 50 mg, 0.24 mmol) and aniline (26 mg, 0.28 mmol) in ethanol (2 mL) was refluxed for 1 hr. After removal of the solvent, the residue was triturated with ethylacetate, and collected by filtration to give the title compound (49 mg, 77%) as a white solid.; LCMS (Method A) m/z: M+1 269.0; tR = 2.44 min.
STEP B:
N-(2-Aminophenyl)-7-methyl-4-(phenylamino)-7H-pyrrolo[2,3-d]pyrimidine-5-carboxamide

A mixture of 7-methyl-4-(phenylamino)-7H-pyrrolo[2,3-d]pyrimidine-5-carboxylic acid (STEP A, 49 mg, 0.183 mmol), o-phenylenediamine (40 mg, 0.365 mmol), HOBT (42 mg, 0.274 mmol), EDC (53 mg, 0.274 mmol), and N,N-Diisopropylethylamine (0.13 mL, 0.731 mmol) in N,N-dimethylformamide (2 mL) was stirred at 60 oC for 2 hrs. After cooing to room temperature, the mixture was diluted with ethylacetate (30 mL) and washed with water (30 mLx2). The organic layer was dried over magnesium sulfate. After removal of the solvent by evaporation, the residue was diluted with methanol and applied onto a strong cation exchange cartridge (BondElute(registered trademark) SCX, 1 g/6 mL, Varian Inc.), and the solid phase matrix was rinsed with methanol (8 mL). The crude mixture was eluted with 1 mol/L ammonia in methanol (8 mL) and concentrated under reduced pressure. The residue was crystallized from diethyl ether/diisopropyl ether to give the title compound (12 mg, 14%) as a white solid.; LCMS (Method A) m/z: M+1 359.0; tR = 2.90 min.
N-(2-Aminophenyl)-7-methyl-4-(phenylamino)-7H-pyrrolo[2,3-d]pyrimidine-5-carboxamide
STEP C: 5-(1H-1,3-Benzodiazol-2-yl)-7-methyl-N-phenyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine

A mixture of N-(2-aminophenyl)-7-methyl-4-(phenylamino)-7H-pyrrolo[2,3-d]pyrimidine-5-carboxamide (STEP B, 12 mg, 0.033 mmol) and acetic acid (2 mL) was stirred at 95 oC for 2 hrs. After cooling to room temperature, the reaction mixture was concentrated under reduced pressure. The residue was diluted with methanol and applied onto a strong cation exchange cartridge (BondElute(registered trademark) SCX, 1 g/6 mL, Varian Inc.), and the solid phase matrix was rinsed with methanol (8 mL). The crude mixture was eluted with 1 mol/L ammonia in methanol (8 mL) and concentrated under reduced pressure. The residue was purified by preparative HPLC to give 7.2 mg (63%) of the title compound.; LCMS (Method B) m/z: M+1 341; tR = 1.91 min.
Examples 2-64
End products of Examples 2-64 were prepared from 4-chloro-7-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carboxylic acid (STEP 2 of Example 1) or 4-chloro-7H-pyrrolo[2,3-d]pyrimidine-5-carboxylic acid (WO2008/075007) according to STEP A, B and C of Example 1 (3 steps).
End products of Examples 2-64 were prepared from 4-chloro-7-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carboxylic acid (STEP 2 of Example 1) or 4-chloro-7H-pyrrolo[2,3-d]pyrimidine-5-carboxylic acid (WO2008/075007) according to STEP A, B and C of Example 1 (3 steps).
STEP A
The intermediates III (2A-64A) listed in Table 3 were prepared from 4-chloro-7-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carboxylic acid (STEP 2 of Example 1) or 4-chloro-7H-pyrrolo[2,3-d]pyrimidine-5-carboxylic acid (WO2008/075007) and amines (R2R3NH) according to the procedure described in STEP A of Example 1. LC-MS (Method A) was used for STEP A of Examples 2-64 (2A-64A). For example, 2A stands for an intermediate of STEP A of Example 2.
The intermediates III (2A-64A) listed in Table 3 were prepared from 4-chloro-7-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carboxylic acid (STEP 2 of Example 1) or 4-chloro-7H-pyrrolo[2,3-d]pyrimidine-5-carboxylic acid (WO2008/075007) and amines (R2R3NH) according to the procedure described in STEP A of Example 1. LC-MS (Method A) was used for STEP A of Examples 2-64 (2A-64A). For example, 2A stands for an intermediate of STEP A of Example 2.
Table 3.







STEP B
The intermediates IV (2B-64B) listed in Table 4 were prepared from the intermediates III (2A-64A) and (R4)n-benzene-1,2-diamines according to the procedure described in STEP B of Example 1, respectively. LC-MS (Method A) was used for STEP B of Examples 2-64 (2B-64B). For example, 2B stands for an intermediate of STEP B of Example 2.
The intermediates IV (2B-64B) listed in Table 4 were prepared from the intermediates III (2A-64A) and (R4)n-benzene-1,2-diamines according to the procedure described in STEP B of Example 1, respectively. LC-MS (Method A) was used for STEP B of Examples 2-64 (2B-64B). For example, 2B stands for an intermediate of STEP B of Example 2.
Table 4














STEP C
End products of the Examples 2-64 listed in Table 5 were prepared from the intermediates IV (2B-64B) according to the procedure described in STEP C of Example 1, respectively. LC-MS (Method A) for Example 2 and LC-MS (Method B) for Example 3-64 were described in Table 5.
End products of the Examples 2-64 listed in Table 5 were prepared from the intermediates IV (2B-64B) according to the procedure described in STEP C of Example 1, respectively. LC-MS (Method A) for Example 2 and LC-MS (Method B) for Example 3-64 were described in Table 5.
Table 5













The ability of the pyrrolopyrimidine derivatives of the formula (I) to activate the potassium channel may be measured using the assay described below.
SK3 channel assay
Human SK3 channel (hSK3) activity was detected by channel assay. Approximately 25,000 cells/well of CHO-K1 cells stably expressing hSK3 channels were plated into black walled, clear-bottom 96-well plates. Following overnight incubation in a 5% CO2 incubator at 37oC. The cell was treated with the FluxORTM detection kits (Invitrogen; Catalog nos. F10016, F10017, Revised: 27-March-2008 MP 10016) according to the literatures (J Gen Physiol. 61, 669 (1973); Journal of Biomolecular Screening 9, 8 (2004)) in the presence of a Ca2+ ionophore (e.g. ionomycin, A23187). Resulting cell plates were loaded onto FDSS6000 (Hamamatsu Photonics). Images were collected at 0.3 Hz. Thirty seconds of baseline were collected and then 25 microL of compound solutions were added to the cells. Images were collected for an additional 4 minutes. EC50 values were calculated by fitting the resulting fluorescence intensities to a four-parametric logistic equation. EC50 was defined as the concentration of test compound required to yield 50% of the maximal response.
The compounds of the examples were tested in the above-described assay. The results (SK3 activation) are as follows:
All examples of the invention have an EC50 = <10 microM in the above assay. Example 2, 3, 4, 6, 7, 8, 11, 14, 16, 17, 18, 19, 21, 29, 30, 33, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 47, 50, 53, 54, 55, 56, 57, 58, 59, 61, 63, and 64 of this invention have an EC50 = <3 microM in the assay.
Human SK3 channel (hSK3) activity was detected by channel assay. Approximately 25,000 cells/well of CHO-K1 cells stably expressing hSK3 channels were plated into black walled, clear-bottom 96-well plates. Following overnight incubation in a 5% CO2 incubator at 37oC. The cell was treated with the FluxORTM detection kits (Invitrogen; Catalog nos. F10016, F10017, Revised: 27-March-2008 MP 10016) according to the literatures (J Gen Physiol. 61, 669 (1973); Journal of Biomolecular Screening 9, 8 (2004)) in the presence of a Ca2+ ionophore (e.g. ionomycin, A23187). Resulting cell plates were loaded onto FDSS6000 (Hamamatsu Photonics). Images were collected at 0.3 Hz. Thirty seconds of baseline were collected and then 25 microL of compound solutions were added to the cells. Images were collected for an additional 4 minutes. EC50 values were calculated by fitting the resulting fluorescence intensities to a four-parametric logistic equation. EC50 was defined as the concentration of test compound required to yield 50% of the maximal response.
The compounds of the examples were tested in the above-described assay. The results (SK3 activation) are as follows:
All examples of the invention have an EC50 = <10 microM in the above assay. Example 2, 3, 4, 6, 7, 8, 11, 14, 16, 17, 18, 19, 21, 29, 30, 33, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 47, 50, 53, 54, 55, 56, 57, 58, 59, 61, 63, and 64 of this invention have an EC50 = <3 microM in the assay.
Electrophysiology assay
Membrane current though SK3 channel was recorded using the whole-cell configuration of the patch-clamp technique.
CHO-K1 cells stably expressing human SK3 channel were grown in Ham's F-12 medium supplemented with 10% FBS, 100 units/mL of penicillin, 100 microgram/mL of streptomycin and 500 microgram/mL of geneticin at 37 degree C in 5% CO2. Cells were dissociated by brief exposure to trypsin-EDTA, suspended in supplemented F-12 medium and plated on 3.5-mm glass cover slips.
Grass pipettes were pulled from borosilicate capillary glass with an outside diameter of 1.5 mm using a Sutter P-97 puller (Sutter Instrument Co.). The pipettes were fire polished and had a resistance of 1-3 Mohm when filled with the pipette solution as follows (in mM); 155 KCl, 10 EGTA, 8.23 CaCl2, 1.11 MgCl2, 10 HEPES, pH7.2 with KOH. The extracellular solution contained (in mM); 155 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, pH7.4 with NaOH. The test compound was dissolved in DMSO and serially diluted in the extracellular solution to the appropriate concentrations. Cells plated on cover slips were placed in a perfusion chamber superfused with the extracellular solution. Voltage clamp command pulses were generated by PULSE software (HEKA). Following establishment of the whole-cell configuration, 1 seconds of linear voltage-ramps (-110 mV to +60 mV) were applied to the cell every 10 seconds from a holding potential of 0 mV. Leak subtraction was not performed. Cell capacitance and series resistance were electronically compensated and updated during the experiments. All experiments were conducted at room temperature.
Current recordings were performed with an EPC9 amplifier (HEKA) in the voltage-clamp mode. Data were filtered at 1-3 kHz and acquired on a PC computer with the PULSE software. For analyses, currents at -80 mV were measured and analyzed. The current in the absence of the test compound was set to 100%.
The compounds of this invention showed good results in electrophysiology assay. For example, the compound in example 3 increased the current over 200% at 10 microM.
Membrane current though SK3 channel was recorded using the whole-cell configuration of the patch-clamp technique.
CHO-K1 cells stably expressing human SK3 channel were grown in Ham's F-12 medium supplemented with 10% FBS, 100 units/mL of penicillin, 100 microgram/mL of streptomycin and 500 microgram/mL of geneticin at 37 degree C in 5% CO2. Cells were dissociated by brief exposure to trypsin-EDTA, suspended in supplemented F-12 medium and plated on 3.5-mm glass cover slips.
Grass pipettes were pulled from borosilicate capillary glass with an outside diameter of 1.5 mm using a Sutter P-97 puller (Sutter Instrument Co.). The pipettes were fire polished and had a resistance of 1-3 Mohm when filled with the pipette solution as follows (in mM); 155 KCl, 10 EGTA, 8.23 CaCl2, 1.11 MgCl2, 10 HEPES, pH7.2 with KOH. The extracellular solution contained (in mM); 155 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, pH7.4 with NaOH. The test compound was dissolved in DMSO and serially diluted in the extracellular solution to the appropriate concentrations. Cells plated on cover slips were placed in a perfusion chamber superfused with the extracellular solution. Voltage clamp command pulses were generated by PULSE software (HEKA). Following establishment of the whole-cell configuration, 1 seconds of linear voltage-ramps (-110 mV to +60 mV) were applied to the cell every 10 seconds from a holding potential of 0 mV. Leak subtraction was not performed. Cell capacitance and series resistance were electronically compensated and updated during the experiments. All experiments were conducted at room temperature.
Current recordings were performed with an EPC9 amplifier (HEKA) in the voltage-clamp mode. Data were filtered at 1-3 kHz and acquired on a PC computer with the PULSE software. For analyses, currents at -80 mV were measured and analyzed. The current in the absence of the test compound was set to 100%.
The compounds of this invention showed good results in electrophysiology assay. For example, the compound in example 3 increased the current over 200% at 10 microM.
Claims (10)
- A compound of formula (I):

R1 is selected from the group consisting of:
(1) hydrogen, (2) C1-6 alkyl, which is unsubstituted or substituted with one or more substituents selected from R5, and (3) C3-6 cycloalkyl which is unsubstituted or substituted with one or more substituents selected from R5;
R2 is selected from the group consisting of:
(1) hydrogen, (2) C1-6 alkyl, which is unsubstituted or substituted with one or more substituents selected from R5, (3) C3-6 cycloalkyl which is unsubstituted or substituted with one or more substituents selected from R5, (4) phenyl, which is unsubstituted or substituted with one or more substituents selected from R5, (5) heterocycle, which is unsubstituted or substituted with one or more substituents selected from R5, and (6) heterocycle substituted C1-6 alkyl, said heterocycle may be unsubstituted or substituted with one or more substituents selected from R5, i.e., a C1-6 alkyl substituted by heterocycle, the heterocycle being unsubstituted or substituted with one or more substituents selected from R5;
R3 is selected from the group consisting of:
(1) C1-6 alkyl, which is unsubstituted or substituted with one or more substituents selected from R5, (2) C3-6 cycloalkyl which is unsubstituted or substituted with one or more substituents selected from R5, (3) phenyl, which is unsubstituted or substituted with one or more substituents selected from R5, (4) heterocycle, which is unsubstituted or substituted with one or more substituents selected from R5, and (5) heterocycle substituted C1-6 alkyl, said heterocycle may be unsubstituted or substituted with one or more substituents selected from R5;
R4 is independently selected from the group consisting of:
(1) hydrogen, (2) halogen, (3) hydroxy, (4) -Op-C1-6 alkyl, where p is 0 or 1 (wherein if p is 0, a chemical bond is present in the place of Op) and where the alkyl is unsubstituted or substituted with one or more substituents selected from R5, (5) -Op-C3-6 cycloalkyl, where p is 0 or 1 (wherein if p is 0, a bond is present in the place of Op) and where the cycloalkyl is unsubstituted or substituted with one or more substituents selected from R5, (6) C2-4 alkenyl, where the alkenyl is unsubstituted or substituted with one or more substituents selected from R5, (7) -(C=O)-NR6R7, (8) -NR6R7, (9) -S(O)2-NR6R7, (10) -NR6-S(O)2R7, (11) -S(O)r-R8, where r is 0, 1 or 2 and where R8 is selected from the definitions of R6 and R7 , (12) -CO2H, and (13) -CN;
n is 1, 2, 3 or 4; when n is two or more than two, R4 may be same or different;
R5 is selected from the group consisting of:
(1) halogen, (2) hydroxy, (3) -(C=O)q-Or-C1-6 alkyl, where q is 0 or 1 and r is 0 or 1 (wherein if q is 0 or r is 0, a bond is present in the place of (C=O)q or Or, and wherein if q is 0 and r is 0, a single bond is present in the place of (C=O)q-Or) where the alkyl is unsubstituted or substituted with one or more substituents selected from R9, (4) -Op-(C1-3)perfluoroalkyl, where p is 0 or 1 (wherein if p is 0, a chemical bond is present in the place of Op) , (5) -(C=O)q-Or-C3-6 cycloalkyl, where q is 0 or 1 and r is 0 or 1 (wherein if q is 0 or r is 0, a bond is present in the place of (C=O)q or Or, and wherein if q is 0 and r is 0, a single bond is present in the place of (C=O)q-Or) and where the cycloalkyl is unsubstituted or substituted with one or more substituents selected from R9, (6) -(C=O)q-C2-4 alkenyl, where q is 0 or 1 (wherein if q is 0, a single bond is present in the place of (C=O)q ) and the alkenyl is unsubstituted or substituted with one or more substituents selected from R9, (7) -(C=O)q-Or-phenyl or -(C=O)q-Or-napthyl, where q is 0 or 1 and r is 0 or 1 (wherein if q is 0 or r is 0, a bond is present in the place of (C=O)q or Or, and wherein if q is 0 and r is 0, a single bond is present in the place of (C=O)q-Or) and where the phenyl or napthyl is unsubstituted or substituted with one or more substituents selected from R9, (8) -(C=O)q-Or-heterocycle, where the heterocycle is unsubstituted or substituted with one or more substituents selected from R9, (9) -(C=O)-NR6R7, (10) -NR6R7, (11) -S(O)2-NR6R7, (12) -S(O)t-R8, where t is 0, 1 or 2, (13) -CO2H, (14) -CN, and (15) -NO2;
R6 and R7 are independently selected from the group consisting of:
(1) hydrogen, (2) C1-6 alkyl, which is unsubstituted or substituted with R9, (3) C3-6 alkenyl, which is unsubstituted or substituted with R9, (4) cycloalkyl which is unsubstituted or substituted with R9, (5) phenyl, which is unsubstituted or substituted with R9, and (6) heterocycle, which is unsubstituted or substituted with R9, or R6 and R7 taken together with the nitrogen atom to which they are attached form a 3 to 8 membered ring, where the ring may contain one to four heteroatom independently selected from nitrogen, oxygen, and sulfur; where the ring may be saturated or partially saturated or unsaturated; which is unsubstituted or substituted one or more substituents selected from R9;
R8 is selected from the definitions of R6 and R7;
R9 is selected from the group consisting of:
(1) hydroxy, (2) halogen, (3) C1-6 alkyl, (4) -C3-6 cycloalkyl, (5) -O-C1-6 alkyl, (6) -O(C=O)-C1-6 alkyl, (7) -NH-C1-6 alkyl, (8) phenyl, (9) heterocycle, (10) -CO2H, (11) -CN, and (12) -Op-(C1-3)perfluoroalkyl, where p is 0 or 1 (wherein if p is 0, a chemical bond is present in the place of Op);
or a pharmaceutically acceptable salt thereof. - The compound as claimed in claim 1 wherein
R2 is selected from the group consisting of:
(1) hydrogen, (2) C1-6 alkyl, which is unsubstituted or substituted with one or more substituents selected from R5, and (3) C3-6 cycloalkyl which is unsubstituted or substituted with one or more substituents selected from R5;
R3 is selected from the group consisting of:
(1) C1-6 alkyl, which is unsubstituted or substituted with one or more substituents selected from R5, (2) C3-6 cycloalkyl which is unsubstituted or substituted with one or more substituents selected from R5, and (3) phenyl, which is unsubstituted or substituted with one or more substituents selected from R5;
R4 is independently selected from the group consisting of:
(1) hydrogen, (2) halogen, (3) hydroxy, (4) -Op-C1-6 alkyl, where p is 0 or 1 (wherein if p is 0, a chemical bond is present in the place of Op) and where the alkyl is unsubstituted or substituted with one or more substituents selected from R5, (5) -Op-C3-6 cycloalkyl, where p is 0 or 1 (wherein if p is 0, a bond is present in the place of Op) and where the cycloalkyl is unsubstituted or substituted with one or more substituents selected from R5, (6) C2-4 alkenyl, where the alkenyl is unsubstituted or substituted with one or more substituents selected from R5, and (7) -CN;
R5 is selected from the group consisting of:
(1) halogen, (2) hydroxy, (3) -(C=O)q-Or-C1-6 alkyl, where q is 0 or 1 and r is 0 or 1 (wherein if q is 0 or r is 0, a bond is present in the place of (C=O)q or Or, and wherein if q is 0 and r is 0, a single bond is present in the place of (C=O)q-Or) where the alkyl is unsubstituted or substituted with one or more substituents selected from R9, (4) -Op-(C1-3)perfluoroalkyl, where p is 0 or 1 (wherein if p is 0, a chemical bond is present in the place of Op) , (5) -(C=O)q-Or-C3-6 cycloalkyl, where q is 0 or 1 and r is 0 or 1 (wherein if q is 0 or r is 0, a bond is present in the place of (C=O)q or Or, and wherein if q is 0 and r is 0, a single bond is present in the place of (C=O)q-Or) and where the cycloalkyl is unsubstituted or substituted with one or more substituents selected from R9, (6) -(C=O)q-C2-4 alkenyl, where q is 0 or 1 (whrein if q is 0, a single bond is present in the place of (C=O)q) and where the alkenyl is unsubstituted or substituted with one or more substituents selected from R9, (7) -(C=O)q-Or-phenyl or -(C=O)p-Or-napthyl, where q is 0 or 1 and r is 0 or 1 (wherein if q is 0 or r is 0, a bond is present in the place of (C=O)q or Or, and wherein if q is 0 and r is 0, a single bond is present in the place of (C=O)q-Or) and where the phenyl or napthyl is unsubstituted or substituted with one or more substituents selected from R9, (8) -(C=O)q-Or-heterocycle, where the heterocycle is unsubstituted or substituted with one or more substituents selected from R9, and (9) -CN;
R9 is selected from the group consisting of:
(1) hydroxy, (2) halogen, (3) C1-6 alkyl, (4) -C3-6 cycloalkyl, (5) -O-C1-6 alkyl, (6) -CN, and (7) -Op-(C1-3)perfluoroalkyl, where p is 0 or 1 (wherein if p is 0, a chemical bond is present in the place of Op);
or a pharmaceutically acceptable salt thereof. - The compound as claimed in claim 1 or 2, which is selected from:
5-(1H-1,3-benzodiazol-2-yl)-7-methyl-N-(2-phenoxyethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(4-fluorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-[2-(4-fluorophenyl)ethyl]-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(cyclohexylmethyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-7-methyl-N-(oxolan-2-ylmethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(2-ethoxyethyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(3-fluorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-7-methyl-N-(2-methylpropyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(2-fluorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-7-methyl-N-[3-(propan-2-yloxy)propyl]-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-7-methyl-N-[3-(trifluoromethoxy)phenyl]-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(2-chlorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(3-chlorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(2,5-difluorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(2,4-difluorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-7-methyl-N-(2-methylphenyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(2,3-difluorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-7-methyl-N-[3-(propan-2-yl)phenyl]-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(3,5-dichlorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(2,3-dichlorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
N-(3-chlorophenyl)-7-methyl-5-(6-methyl-1H-1,3-benzodiazol-2-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
N-(3-chlorophenyl)-7-methyl-5-(7-methyl-1H-1,3-benzodiazol-2-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
N-(3-chlorophenyl)-5-(6-fluoro-1H-1,3-benzodiazol-2-yl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(6-chloro-1H-1,3-benzodiazol-2-yl)-N-(3-chlorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
N-(3-chlorophenyl)-5-(6-methoxy-1H-1,3-benzodiazol-2-yl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-7-methyl-N-[3-(propan-2-yloxy)phenyl]-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(2,5-dichlorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(3-chloro-4-fluorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(6-chloro-1H-1,3-benzodiazol-2-yl)-N-(4-fluorophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-1,3-benzodiazol-2-yl)-N-(4-bromophenyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-benzodiazol-2-yl)-N-(4-fluorophenyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-benzodiazol-2-yl)-N-(3-chlorophenyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(1H-benzodiazol-2-yl)-N-(2-phenoxyethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(6-fluoro-1H-benzodiazol-2-yl)-N-(3-isopropoxypropyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
N-(3-isopropoxypropyl)-7-methyl-5-(6-methyl-1H-benzodiazol-2-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(6-fluoro-1H-benzodiazol-2-yl)-N-(2-isopropoxyethyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
N-(2-isopropoxyethyl)-7-methyl-5-(6-methyl-1H-benzodiazol-2-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
5-(6-chloro-1H-benzodiazol-2-yl)-N-(3-ethoxypropyl)-7-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine, and
N-(3-ethoxypropyl)-7-methyl-5-(6-methyl-1H-benzodiazol-2-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine,
or a pharmaceutically acceptable salt thereof. - A pharmaceutical composition comprising the compound or the pharmaceutically acceptable salt thereof, as claimed in any one of claims 1 to 3, and a pharmaceutically acceptable carrier.
- A pharmaceutical composition as claimed in claim 4, further comprising another pharmacologically active agent.
- A method for the treatment of a condition or disorder in which potassium channel modulators are involved, in a mammalian subject, including a human, which comprises administering to a mammal in need of such treatment a therapeutically effective amount of the compound or the pharmaceutically acceptable salt thereof, as claimed in any one of claims 1 to 3.
- The method as claimed in claim 6, wherein said condition or disorder is selected from the group consisting of: a respiratory disease, epilepsy, convulsions, seizures, absence seizures, vascular spasms, coronary artery spasms, renal disorders, polycystic kidney disease, bladder spasms, urinary incontinence, bladder outflow obstruction, erectile dysfunction, gastrointestinal dysfunction, secretory diarrhoea, ischaemia, cerebral ischaemia, ischaemic heart disease, angina pectoris, coronary heart disease, autism, ataxia, traumatic brain injury, Parkinson's disease, bipolar disorder, psychosis, schizophrenia, anxiety, depression, mania, mood disorders, dementia, memory and attention deficits, Alzheimer's disease, amyotrophic lateral sclerosis (ALS), dysmenorrhea, narcolepsy, Reynaud's disease, intermittent claudication, Sjogren's syndrome, arrhythmia, hypertension, myotonic muscle dystrophia, spasticity, xerostomi, diabetes type II, hyperinsulinemia, premature labour, baldness, cancer, irritable bowel syndrome, immune suppression, migraine or pain, or withdrawal symptoms caused by the termination of abuse of chemical substances, in particular opioids, heroin, cocaine and morphine, benzodiazepines and benzodiazepine-like drugs, and alcohol;
and combinations thereof.
- A compound of formula (I) or a pharmaceutically acceptable salt, solvate or composition thereof, as claimed in any one of claims 1 to 3, for use in the treatment of a condition or disorder in which potassium modulators are involved.
- The use of a compound of the formula (I), or a pharmaceutically acceptable salt, solvate or composition thereof, as claimed in any one of claims 1 to 3, for the manufacture of a medicament for the treatment of a condition or disorder in which potassium channel modulators are involved.
- The use as claimed in claim 9, wherein the condition or disorder is selected from the group consisting of: a respiratory disease, epilepsy, convulsions, seizures, absence seizures, vascular spasms, coronary artery spasms, renal disorders, polycystic kidney disease, bladder spasms, urinary incontinence, bladder outflow obstruction, erectile dysfunction, gastrointestinal dysfunction, secretory diarrhoea, ischaemia, cerebral ischaemia, ischaemic heart disease, angina pectoris, coronary heart disease, autism, ataxia, traumatic brain injury, Parkinson's disease, bipolar disorder, psychosis, schizophrenia, anxiety, depression, mania, mood disorders, dementia, memory and attention deficits, Alzheimer's disease, amyotrophic lateral sclerosis (ALS), dysmenorrhea, narcolepsy, Reynaud's disease, intermittent claudication, Sjogren's syndrome, arrhythmia, hypertension, myotonic muscle dystrophia, spasticity, xerostomi, diabetes type II, hyperinsulinemia, premature labour, baldness, cancer, irritable bowel syndrome, immune suppression, migraine or pain, or withdrawal symptoms caused by the termination of abuse of chemical substances, in particular opioids, heroin, cocaine and morphine, benzodiazepines and benzodiazepine-like drugs, and alcohol;
and combinations thereof.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US27203509P | 2009-08-10 | 2009-08-10 | |
| US61/272,035 | 2009-08-10 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011018894A1 true WO2011018894A1 (en) | 2011-02-17 |
Family
ID=43586077
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/005024 Ceased WO2011018894A1 (en) | 2009-08-10 | 2010-08-10 | Pyrrolopyrimidine derivatives as potassium channel modulators |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2011018894A1 (en) |
Cited By (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9266892B2 (en) | 2012-12-19 | 2016-02-23 | Incyte Holdings Corporation | Fused pyrazoles as FGFR inhibitors |
| JP2016512503A (en) * | 2013-03-11 | 2016-04-28 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | Pyrrolotriazines as potassium ion channel inhibitors |
| JP2016512504A (en) * | 2013-03-11 | 2016-04-28 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | Pyrrolopyridazine as a potassium ion channel inhibitor |
| JP2016512505A (en) * | 2013-03-11 | 2016-04-28 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | Pyrrolotriazines as potassium ion channel inhibitors |
| US9388185B2 (en) | 2012-08-10 | 2016-07-12 | Incyte Holdings Corporation | Substituted pyrrolo[2,3-b]pyrazines as FGFR inhibitors |
| US9533954B2 (en) | 2010-12-22 | 2017-01-03 | Incyte Corporation | Substituted imidazopyridazines and benzimidazoles as inhibitors of FGFR3 |
| US9533984B2 (en) | 2013-04-19 | 2017-01-03 | Incyte Holdings Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US9580423B2 (en) | 2015-02-20 | 2017-02-28 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| WO2017043550A1 (en) * | 2015-09-08 | 2017-03-16 | 大鵬薬品工業株式会社 | Condensed pyrimidine compound or salt thereof |
| US9611267B2 (en) | 2012-06-13 | 2017-04-04 | Incyte Holdings Corporation | Substituted tricyclic compounds as FGFR inhibitors |
| US9708318B2 (en) | 2015-02-20 | 2017-07-18 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9890156B2 (en) | 2015-02-20 | 2018-02-13 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| KR20180112044A (en) | 2016-02-23 | 2018-10-11 | 다이호야쿠힌고교 가부시키가이샤 | A novel condensed pyrimidine compound or salt thereof |
| US10611762B2 (en) | 2017-05-26 | 2020-04-07 | Incyte Corporation | Crystalline forms of a FGFR inhibitor and processes for preparing the same |
| WO2020140054A1 (en) | 2018-12-28 | 2020-07-02 | Spv Therapeutics Inc. | Cyclin-dependent kinase inhibitors |
| US10851105B2 (en) | 2014-10-22 | 2020-12-01 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| WO2021095805A1 (en) * | 2019-11-13 | 2021-05-20 | 日本新薬株式会社 | Therapeutic agent and prophylactic agent for functional gastrointestinal disorders and xerostomia |
| US11174257B2 (en) | 2018-05-04 | 2021-11-16 | Incyte Corporation | Salts of an FGFR inhibitor |
| US11407750B2 (en) | 2019-12-04 | 2022-08-09 | Incyte Corporation | Derivatives of an FGFR inhibitor |
| US11466004B2 (en) | 2018-05-04 | 2022-10-11 | Incyte Corporation | Solid forms of an FGFR inhibitor and processes for preparing the same |
| US11566028B2 (en) | 2019-10-16 | 2023-01-31 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11591329B2 (en) | 2019-07-09 | 2023-02-28 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11607416B2 (en) | 2019-10-14 | 2023-03-21 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11628162B2 (en) | 2019-03-08 | 2023-04-18 | Incyte Corporation | Methods of treating cancer with an FGFR inhibitor |
| WO2023064361A1 (en) * | 2021-10-12 | 2023-04-20 | Biosplice Therapeutics, Inc. | 7h-pyrrolo[2,3-d]pyrimidines and preparation as dyrk1a inhibitors |
| US11897891B2 (en) | 2019-12-04 | 2024-02-13 | Incyte Corporation | Tricyclic heterocycles as FGFR inhibitors |
| US11939331B2 (en) | 2021-06-09 | 2024-03-26 | Incyte Corporation | Tricyclic heterocycles as FGFR inhibitors |
| US11993586B2 (en) | 2018-10-22 | 2024-05-28 | Novartis Ag | Crystalline forms of potassium channel modulators |
| US12012409B2 (en) | 2020-01-15 | 2024-06-18 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US12065494B2 (en) | 2021-04-12 | 2024-08-20 | Incyte Corporation | Combination therapy comprising an FGFR inhibitor and a Nectin-4 targeting agent |
| US12122767B2 (en) | 2019-10-01 | 2024-10-22 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US12134625B2 (en) | 2021-10-12 | 2024-11-05 | Biosplice Therapeutics, Inc. | Pyrrolo[2,1-f][1,2,4]triazines and preparation and uses thereof |
| US12258376B2 (en) | 2017-08-21 | 2025-03-25 | Taiho Pharmaceutical Co., Ltd. | Fusion protein of DCTN1 protein with RET protein |
| US12319683B2 (en) | 2018-05-08 | 2025-06-03 | Nippon Shinyaku Co., Ltd. | Azabenzimidazole compounds and pharmaceutical |
| US12428420B2 (en) | 2021-06-09 | 2025-09-30 | Incyte Corporation | Tricyclic heterocycles as FGFR inhibitors |
| US12528813B2 (en) | 2019-11-13 | 2026-01-20 | Nippon Shinyaku Co., Ltd. | Substituted imidazo[4,5-b]pyridines as M3 receptor PAMs |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000505109A (en) * | 1996-11-27 | 2000-04-25 | ファイザー・インク | Fused bicyclic pyrimidine derivatives |
| JP2002519412A (en) * | 1998-07-02 | 2002-07-02 | ニューロサーチ、アクティーゼルスカブ | Potassium channel blocker |
| JP2003503501A (en) * | 1999-06-29 | 2003-01-28 | ニューロサーチ、アクティーゼルスカブ | Potassium channel blocker |
| JP2005508300A (en) * | 2001-06-23 | 2005-03-31 | アベンティス・ファーマスーティカルズ・インコーポレイテツド | Pyrrolopyrimidines as protein kinase inhibitors |
| JP2005530713A (en) * | 2002-03-21 | 2005-10-13 | アボット・ラボラトリーズ | Kinase inhibitor |
| JP2005538118A (en) * | 2002-08-06 | 2005-12-15 | アストラゼネカ アクチボラグ | Condensed pyridine and pyrimidine with TIE2 (TEK) activity |
| WO2006009425A1 (en) * | 2004-07-20 | 2006-01-26 | Quest International Services B.V. | Taste improving substances |
| JP2007511596A (en) * | 2003-11-17 | 2007-05-10 | ファイザー・プロダクツ・インク | Pyrrolopyrimidine compounds useful in the treatment of cancer |
-
2010
- 2010-08-10 WO PCT/JP2010/005024 patent/WO2011018894A1/en not_active Ceased
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000505109A (en) * | 1996-11-27 | 2000-04-25 | ファイザー・インク | Fused bicyclic pyrimidine derivatives |
| JP2002519412A (en) * | 1998-07-02 | 2002-07-02 | ニューロサーチ、アクティーゼルスカブ | Potassium channel blocker |
| JP2003503501A (en) * | 1999-06-29 | 2003-01-28 | ニューロサーチ、アクティーゼルスカブ | Potassium channel blocker |
| JP2005508300A (en) * | 2001-06-23 | 2005-03-31 | アベンティス・ファーマスーティカルズ・インコーポレイテツド | Pyrrolopyrimidines as protein kinase inhibitors |
| JP2005530713A (en) * | 2002-03-21 | 2005-10-13 | アボット・ラボラトリーズ | Kinase inhibitor |
| JP2005538118A (en) * | 2002-08-06 | 2005-12-15 | アストラゼネカ アクチボラグ | Condensed pyridine and pyrimidine with TIE2 (TEK) activity |
| JP2007511596A (en) * | 2003-11-17 | 2007-05-10 | ファイザー・プロダクツ・インク | Pyrrolopyrimidine compounds useful in the treatment of cancer |
| WO2006009425A1 (en) * | 2004-07-20 | 2006-01-26 | Quest International Services B.V. | Taste improving substances |
Cited By (74)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10213427B2 (en) | 2010-12-22 | 2019-02-26 | Incyte Corporation | Substituted imidazopyridazines and benzimidazoles as inhibitors of FGFR3 |
| US10813930B2 (en) | 2010-12-22 | 2020-10-27 | Incyte Corporation | Substituted imidazopyridazines and benzimidazoles as inhibitors of FGFR3 |
| US9533954B2 (en) | 2010-12-22 | 2017-01-03 | Incyte Corporation | Substituted imidazopyridazines and benzimidazoles as inhibitors of FGFR3 |
| US11840534B2 (en) | 2012-06-13 | 2023-12-12 | Incyte Corporation | Substituted tricyclic compounds as FGFR inhibitors |
| US11053246B2 (en) | 2012-06-13 | 2021-07-06 | Incyte Corporation | Substituted tricyclic compounds as FGFR inhibitors |
| US10131667B2 (en) | 2012-06-13 | 2018-11-20 | Incyte Corporation | Substituted tricyclic compounds as FGFR inhibitors |
| US9611267B2 (en) | 2012-06-13 | 2017-04-04 | Incyte Holdings Corporation | Substituted tricyclic compounds as FGFR inhibitors |
| US12534463B2 (en) | 2012-06-13 | 2026-01-27 | Incyte Corporation | Substituted tricyclic compounds as FGFR inhibitors |
| US9388185B2 (en) | 2012-08-10 | 2016-07-12 | Incyte Holdings Corporation | Substituted pyrrolo[2,3-b]pyrazines as FGFR inhibitors |
| US9745311B2 (en) | 2012-08-10 | 2017-08-29 | Incyte Corporation | Substituted pyrrolo[2,3-b]pyrazines as FGFR inhibitors |
| US9266892B2 (en) | 2012-12-19 | 2016-02-23 | Incyte Holdings Corporation | Fused pyrazoles as FGFR inhibitors |
| JP2016512505A (en) * | 2013-03-11 | 2016-04-28 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | Pyrrolotriazines as potassium ion channel inhibitors |
| JP2016512504A (en) * | 2013-03-11 | 2016-04-28 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | Pyrrolopyridazine as a potassium ion channel inhibitor |
| JP2016512503A (en) * | 2013-03-11 | 2016-04-28 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | Pyrrolotriazines as potassium ion channel inhibitors |
| US9533984B2 (en) | 2013-04-19 | 2017-01-03 | Incyte Holdings Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11530214B2 (en) | 2013-04-19 | 2022-12-20 | Incyte Holdings Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US10947230B2 (en) | 2013-04-19 | 2021-03-16 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US10450313B2 (en) | 2013-04-19 | 2019-10-22 | Incyte Holdings Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US10040790B2 (en) | 2013-04-19 | 2018-08-07 | Incyte Holdings Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US10851105B2 (en) | 2014-10-22 | 2020-12-01 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9890156B2 (en) | 2015-02-20 | 2018-02-13 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10214528B2 (en) | 2015-02-20 | 2019-02-26 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US11667635B2 (en) | 2015-02-20 | 2023-06-06 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US11173162B2 (en) | 2015-02-20 | 2021-11-16 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10251892B2 (en) | 2015-02-20 | 2019-04-09 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10016438B2 (en) | 2015-02-20 | 2018-07-10 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9708318B2 (en) | 2015-02-20 | 2017-07-18 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US11014923B2 (en) | 2015-02-20 | 2021-05-25 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10632126B2 (en) | 2015-02-20 | 2020-04-28 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9580423B2 (en) | 2015-02-20 | 2017-02-28 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10738048B2 (en) | 2015-02-20 | 2020-08-11 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9801889B2 (en) | 2015-02-20 | 2017-10-31 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| CN108026101A (en) * | 2015-09-08 | 2018-05-11 | 大鹏药品工业株式会社 | Condensed pyramidine compounds or its salt |
| US10233189B2 (en) | 2015-09-08 | 2019-03-19 | Taiho Pharmaceutical Co., Ltd. | Fused pyrimidine compound or salt thereof |
| JP6113379B1 (en) * | 2015-09-08 | 2017-04-12 | 大鵬薬品工業株式会社 | Fused pyrimidine compound or salt thereof |
| US10787457B2 (en) | 2015-09-08 | 2020-09-29 | Taiho Pharmaceutical Co., Ltd. | Fused pyrimidine compound or salt thereof |
| RU2729636C2 (en) * | 2015-09-08 | 2020-08-11 | Тайхо Фармасьютикал Ко., Лтд. | Condensed pyrimidine compound or salt thereof |
| US11236096B2 (en) | 2015-09-08 | 2022-02-01 | Taiho Pharmaceutical Co., Ltd. | Fused pyrimidine compound or salt thereof |
| US11014930B2 (en) | 2015-09-08 | 2021-05-25 | Taiho Pharmaceutical Co., Ltd. | Fused pyrimidine compound or salt thereof |
| WO2017043550A1 (en) * | 2015-09-08 | 2017-03-16 | 大鵬薬品工業株式会社 | Condensed pyrimidine compound or salt thereof |
| KR102058366B1 (en) | 2015-09-08 | 2019-12-23 | 다이호야쿠힌고교 가부시키가이샤 | Condensed pyrimidine compounds or salts thereof |
| US11046696B2 (en) | 2016-02-23 | 2021-06-29 | Taiho Pharmaceutical Co., Ltd. | Fused pyrimidine compound or salt thereof |
| US10807986B2 (en) | 2016-02-23 | 2020-10-20 | Taiho Pharmaceutical Co., Ltd. | Fused pyrimidine compound or salt thereof |
| KR20180112044A (en) | 2016-02-23 | 2018-10-11 | 다이호야쿠힌고교 가부시키가이샤 | A novel condensed pyrimidine compound or salt thereof |
| US10155768B2 (en) | 2016-02-23 | 2018-12-18 | Taiho Pharmaceutical Co., Ltd. | Fused pyrimidine compound or salt thereof |
| US10611762B2 (en) | 2017-05-26 | 2020-04-07 | Incyte Corporation | Crystalline forms of a FGFR inhibitor and processes for preparing the same |
| US11472801B2 (en) | 2017-05-26 | 2022-10-18 | Incyte Corporation | Crystalline forms of a FGFR inhibitor and processes for preparing the same |
| US12258376B2 (en) | 2017-08-21 | 2025-03-25 | Taiho Pharmaceutical Co., Ltd. | Fusion protein of DCTN1 protein with RET protein |
| US11466004B2 (en) | 2018-05-04 | 2022-10-11 | Incyte Corporation | Solid forms of an FGFR inhibitor and processes for preparing the same |
| US11174257B2 (en) | 2018-05-04 | 2021-11-16 | Incyte Corporation | Salts of an FGFR inhibitor |
| US12552792B2 (en) | 2018-05-04 | 2026-02-17 | Incyte Corporation | Solid forms of an FGFR inhibitor and processes for preparing the same |
| US12473286B2 (en) | 2018-05-04 | 2025-11-18 | Incyte Corporation | Salts of an FGFR inhibitor |
| US12024517B2 (en) | 2018-05-04 | 2024-07-02 | Incyte Corporation | Salts of an FGFR inhibitor |
| US12319683B2 (en) | 2018-05-08 | 2025-06-03 | Nippon Shinyaku Co., Ltd. | Azabenzimidazole compounds and pharmaceutical |
| US11993586B2 (en) | 2018-10-22 | 2024-05-28 | Novartis Ag | Crystalline forms of potassium channel modulators |
| WO2020140054A1 (en) | 2018-12-28 | 2020-07-02 | Spv Therapeutics Inc. | Cyclin-dependent kinase inhibitors |
| US11628162B2 (en) | 2019-03-08 | 2023-04-18 | Incyte Corporation | Methods of treating cancer with an FGFR inhibitor |
| US11591329B2 (en) | 2019-07-09 | 2023-02-28 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US12122767B2 (en) | 2019-10-01 | 2024-10-22 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11607416B2 (en) | 2019-10-14 | 2023-03-21 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US12083124B2 (en) | 2019-10-14 | 2024-09-10 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11566028B2 (en) | 2019-10-16 | 2023-01-31 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US12528813B2 (en) | 2019-11-13 | 2026-01-20 | Nippon Shinyaku Co., Ltd. | Substituted imidazo[4,5-b]pyridines as M3 receptor PAMs |
| WO2021095805A1 (en) * | 2019-11-13 | 2021-05-20 | 日本新薬株式会社 | Therapeutic agent and prophylactic agent for functional gastrointestinal disorders and xerostomia |
| US11407750B2 (en) | 2019-12-04 | 2022-08-09 | Incyte Corporation | Derivatives of an FGFR inhibitor |
| US11897891B2 (en) | 2019-12-04 | 2024-02-13 | Incyte Corporation | Tricyclic heterocycles as FGFR inhibitors |
| US12552804B2 (en) | 2019-12-04 | 2026-02-17 | Incyte Corporation | Tricyclic heterocycles as FGFR inhibitors |
| US12168660B2 (en) | 2019-12-04 | 2024-12-17 | Incyte Corporation | Derivatives of an FGFR inhibitor |
| US12012409B2 (en) | 2020-01-15 | 2024-06-18 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US12065494B2 (en) | 2021-04-12 | 2024-08-20 | Incyte Corporation | Combination therapy comprising an FGFR inhibitor and a Nectin-4 targeting agent |
| US12428420B2 (en) | 2021-06-09 | 2025-09-30 | Incyte Corporation | Tricyclic heterocycles as FGFR inhibitors |
| US11939331B2 (en) | 2021-06-09 | 2024-03-26 | Incyte Corporation | Tricyclic heterocycles as FGFR inhibitors |
| US12134625B2 (en) | 2021-10-12 | 2024-11-05 | Biosplice Therapeutics, Inc. | Pyrrolo[2,1-f][1,2,4]triazines and preparation and uses thereof |
| WO2023064361A1 (en) * | 2021-10-12 | 2023-04-20 | Biosplice Therapeutics, Inc. | 7h-pyrrolo[2,3-d]pyrimidines and preparation as dyrk1a inhibitors |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2011018894A1 (en) | Pyrrolopyrimidine derivatives as potassium channel modulators | |
| US20240400544A1 (en) | N-(hydroxyalkyl (hetero)aryl) tetrahydrofuran carboxamide analogs as modulators of sodium channels | |
| US20240294512A1 (en) | Hydroxy and (halo)alkoxy substituted tetrahydrofurans as modulators of sodium channels | |
| US12440481B2 (en) | Esters and carbamates as modulators of sodium channels | |
| EP2951155B1 (en) | Amides as modulators of sodium channels | |
| US8969383B2 (en) | Picolinamide derivatives as TTX-S blockers | |
| EP2951168A1 (en) | Quinoline and quinazoline amides as modulators of sodium channels | |
| EP2771335A2 (en) | (4-phenylimidazol-2-yl) ethylamine derivatives useful as sodium channel modulators | |
| EP2593431A2 (en) | Chemical compounds | |
| WO2023205463A1 (en) | Heteroaryl compounds for the treatment of pain | |
| WO2013061297A1 (en) | Pyridazine Derivatives Useful in Therapy | |
| EP2593433A2 (en) | N-sulfonylbenzamides as inhibitors of voltage-gated sodium channels | |
| EP2435407A1 (en) | Aryl substituted carboxamide derivatives as calcium or sodium channel blockers | |
| EP2791108A1 (en) | Sulfonamide derivatives | |
| WO2007129188A1 (en) | Cyclopropanecarboxamide compound | |
| EP3487839A1 (en) | Amide derivatives as nav1.7 and nav1.8 blockers | |
| WO2012020567A1 (en) | Acyl piperazine derivatives as ttx-s blockers | |
| WO2007125398A2 (en) | : sulfonamide compounds as antagonists of the n-type calcium channel | |
| WO2023008585A1 (en) | Crystalline forms | |
| US9901571B2 (en) | Tetrahydropyrazolopyridine derivatives as ghrelin receptor agonists | |
| EP3131916A1 (en) | Serine derivatives as ghrelin receptor agonists | |
| HK40056551A (en) | Process for preparing pyridone amides which are modulators of sodium channels and intermediate compounds used therein | |
| HK1244269A1 (en) | Pyridone amides as modulators of sodium channels | |
| HK1217691B (en) | Quinoline and quinazoline amides as modulators of sodium channels | |
| HK1217692B (en) | Amides as modulators of sodium channels |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10808079 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 10808079 Country of ref document: EP Kind code of ref document: A1 |