ISOTHIAZOLE DERIVATIVES AND THEIR USE AS PESTICIDES
The present invention relates to azole derivatives, to processes for preparing them, to fungicidal, insecticidal, acaricidal, molluscicidal and nematicidal compositions comprising them, to methods of using them to combat fungal diseases (especially fungal diseases of plants) and to methods of using them to combat and control insect, acarine, mollusc and nematode pests.
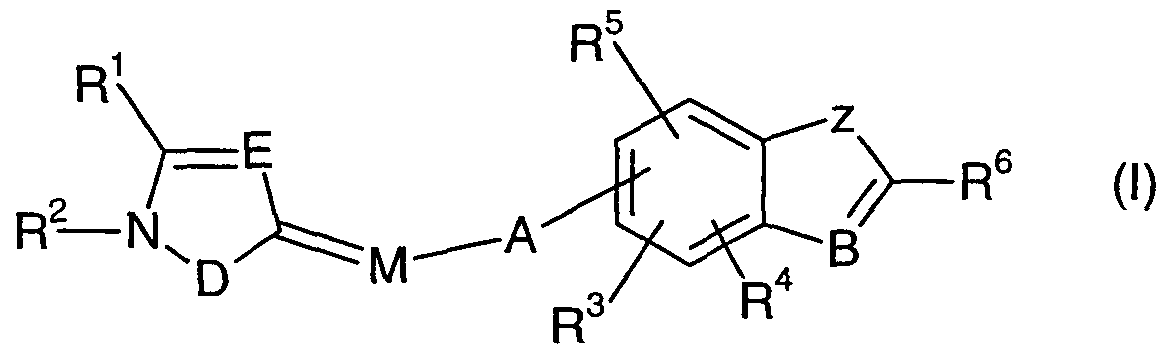
Azole and azine derivatives are disclosed in WO95/31448, WO97/18198, WO98/02424, WO98/05670 and WO98/17630. The present invention provides a compound of formula (I):
where A is optionally substituted Cι-
6 alkylene, optionally substituted C
2.
6 alkenylene, optionally substituted C
2.
6 alkynylene, optionally substituted cycloalkylene, optionally substituted Cι.
6 alkylenoxy, optionally substituted oxy(Cι.
6)alkylene, optionally substituted Cι-
6 alkylenethio, optionally substituted thio(Cι-
6)alkylene, optionally substituted Cι_
6 alkylenamino, optionally substituted amino(Cι.6)alkylene, optionally substituted [Cι.
6 alkyleneoxy(Cι_
6)alkylene], optionally substituted [Cι_
6 alkylenethio(Cι_
6)alkylene], optionally substituted [Cι.
6 alkylenesulfιnyl(C
1.
6)alkylene], optionally substituted [ -
6 alkylenesulfonyl(C
1-
6)alkylene] or optionally substituted [d.
6 alkyleneamino(C
1.
6)alkylene]; B is N, N-oxide or CR
18;
D is S or NR7; E is N or CR12;
M is N-C(=Y), where N is the atom of attachment to the ring containing D and E; Y is O, S or NR13; Z is O, S or NR14;
R1 is hydrogen, halogen, optionally substituted d.6 alkyl, optionally substituted C2.6 alkenyl, optionally substituted C2.6 alkynyl, optionally substituted d_6 alkoxy, optionally substituted Cι.6 alkylthio, optionally substituted C3.7 cycloalkyl, cyano, nitro or SF5;
R2 is optionally substituted C^o alkyl, optionally substituted [C2.6 alkenyl(d-6)- alkyl], optionally substituted [C2.6 alkynyl (C1.6)alkyl], optionally substituted C3.7 cycloalkyl, optionally substituted CMO alkylcarbonyl, optionally substituted d_10 alkoxycarbonyl, formyl, optionally substituted d-ϊoalkylaminocarbonyl, optionally substituted di(Cι-1o)alkylaminocarbonyl, optionally substituted phenoxycarbonyl, optionally substituted _6 alkylthio, optionally substituted d_6 alkylsulfinyl, optionally substituted Cι_6 alkylsulfonyl, optionally substituted d.6 arylthio, optionally substituted d-6 arylsulfinyl, optionally substituted d.6 arylsulfonyl or R20R21NS(O)P; p is 0,1 or 2;
R3, R4 and R5 are, independently, hydrogen, halogen, optionally substituted d.6 alkyl, optionally substituted d.6 alkoxy, optionally substituted .6 alkylthio, optionally substituted Ci.6 alkylsulfinyl, optionally substituted -6 alkylsulfonyl, cyano, nitro, optionally substituted d.6 alkylcarbonyl, optionally substituted d-6 alkoxycarbonyl or SF5;
R6 is hydrogen, halogen, cyano, optionally substituted d-20 alkyl, optionally substituted C2.2o alkenyl, optionally substituted C2.20 alkynyl, optionally substituted C3.7 cycloalkyl, optionally substituted C5..3 cycloalkenyl, formyl, optionally substituted d-20 alkoxycarbonyl, optionally substituted C1-20 alkylcarbonyl, aminocarbonyl, optionally substituted d.2oalkylaminocarbonyl, optionally substituted di(Cι.20)alkylaminocarbonyl, optionally substituted aryloxycarbonyl, optionally substituted arylcarbonyl, optionally substituted arylaminocarbonyl, optionally substituted N-(C1.6)alkyl-N-arylaminocarbonyl, optionally substituted diarylaminocarbonyl, optionally substituted heteroaryloxycarbonyl, optionally substituted heteroarylcarbonyl, optionally substituted heteroarylaminocarbonyl, optionally substituted N-(C1-6)alkyl-N-heteroarylaminocarbonyl, optionally substituted diheteroarylaminocarbonyl, optionally substituted phenyl, optionally substituted heteroaryl, optionally substituted heterocyclyl, SH, optionally substituted d-2o alkylthio, optionally substituted _2o alkylsulfinyl, optionally substituted d.2o alkylsulfonyl, optionally substituted arylthio, optionally substituted arylsulfinyl, optionally substituted arylsulfonyl, R26O, R28R29N or R31ON=C(R27);
R7 is hydrogen or . alkyl;
R is hydrogen, halogen, optionally substituted Cj.6 alkyl, optionally substituted C2.6 alkenyl, optionally substituted C .6 alkynyl, optionally substituted d-6 alkoxy, optionally substituted -6 alkylthio, optionally substituted d.6 alkylsulfinyl, optionally substituted d_6 alkylsulfonyl, cyano, nitro, formyl, optionally substituted Cj.6 alkylcarbonyl, optionally
substituted d_6 alkoxycarbonyl, SF5 or R3 ON=C(R30); or R1 and R12 together with the atoms to which they are attached may be joined to form a five, six or seven-membered saturated or unsaturated, carbocylic or heterocyclic ring which may contain one or two heteroatoms selected from O, N or S and which is optionally substituted by d„6 alkyl, d_6 haloalkyl or halogen;
R13 is hydrogen, cyano, nitro, optionally substituted d_6 alkyl, optionally substituted C3.7 cycloalkyl, optionally substituted (C2.6)alkenyl(d_6)alkyl, optionally substituted (C2-6)alkynyl(Cι-6)alkyl, optionally substituted phenyl, optionally substituted heteroaryl, optionally substituted d_6 alkylcarbonyl, optionally substituted d-6 alkoxycarbonyl, optionally substituted d.6 alkylamino, optionally substituted di(d-6)alkylamino, optionally substituted d_6 alkylcarbonylamino, optionally substituted d-6 alkoxycarbonylamino, optionally substituted d.6 alkoxy, optionally substituted d-6 alkylthio, optionally substituted -6 alkylsulfinyl, optionally substituted d_6 alkylsulfonyl, optionally substituted arylthio, optionally substituted arylsulfinyl, optionally substituted arylsulfonyl or optionally substituted C ι .6 alkylcarbonyloxy ;
R14 is hydrogen, cyano, optionally substituted d.8 alkyl, optionally substituted [C2_6 alkenyl (d_6)alkyl], optionally substituted [C2.6 alkynyl(d-6)alkyl], optionally substituted C3-7 cycloalkyl, optionally substituted [C3.7 cycloalkyl (C1.6)alkyl], d-6 alkoxy(d.6)alkyl, optionally substituted d.6 alkoxycarbonyl, optionally substituted d-6 alkylcarbonyl, optionally substituted d-6 alkylaminocarbonyl, optionally substituted di(d. ^alkylaminocarbonyl, optionally substituted phenyl, optionally substituted heteroaryl, optionally substituted alkylsulfonyl or optionally substituted arylsulfonyl;
R18 is hydrogen, halogen, nitro, cyano, optionally substituted d_8 alkyl, optionally substituted C2.6 alkenyl, optionally substituted C2.6 alkynyl, optionally substituted C3.7 cycloalkyl, optionally substituted d.6 alkoxycarbonyl, optionally substituted d_6 alkylcarbonyl, optionally substituted Cι_6 alkylaminocarbonyl, optionally substituted di(C1.6)alkylaminocarbonyl, optionally substituted phenyl or optionally substituted heteroaryl;
R20 and R21 are, independently, optionally substituted d_6 alkyl or R20and R21 together with the N atom to which they are attached form a five, six or seven-membered heterocyclic ring which may contain one or two further hetero atoms selected from O, N or S and which may be optionally substituted by one or two d. alkyl groups;
R32 and R31 are, independently, hydrogen, optionally substituted phenyl (Cι_2)alkyl or optionally substituted d.20 alkyl;
R and R are independently hydrogen, optionally substituted phenyl or optionally substituted d.6 alkyl; R26 is hydrogen, optionally substituted d_2o alkyl, optionally substituted [C2.20 alkenyl(Cι_6)alkyl], optionally substituted [C2.20 alkynyl(d-6) alkyl], optionally substituted C3. cycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted heterocyclyl, (C1-6)alkylCH=N, optionally substituted arylCH=N, optionally substituted [aryl(d.6)aTkyl]CH=N, optionally substituted heteroarylCH=N, optionally substituted [heterocyclyl(C1.6)alkyl]CH=N, optionally substituted arylC(CH3)=N, optionally substituted heteroarylC(CH3)=N or optionally substituted di(d-6)alkylC=N; and R28 and R29 are, independently, hydrogen, optionally substituted d-20 alkyl, optionally substituted C3.7 cycloalkyl, optionally substituted [C2.2o alkenyl(d-6)alkyl], optionally substituted [C2.20 alkynyl(Ci.6)alkyl], optionally substituted Cι.20 alkoxycarbonyl, optionally substituted phenoxycarbonyl, formyl, optionally substituted C1-20 alkylcarbonyl, optionally substituted d_2o alkylsulfonyl or optionally substituted phenylsulfonyl.
One group of preferred compounds of formula (I) is a group wherein A is optionally substituted d-6 alkylene, optionally substituted C2.6 alkenylene, optionally substituted C2_ alkynylene, optionally substituted d-6 alkylenoxy, optionally substituted oxy(Cι_6)alkylene, optionally substituted d-6 alkylenethio, optionally substituted thio(Cι_6)alkylene, optionally substituted d_6 alkylenamino, optionally substituted amino(Cι-6)alkylene, optionally substituted [d-6 alkyleneoxy(d-6)alkylene], optionally substituted [Cχ_6 alkylenethio(Cι.6)alkylene], optionally substituted [d.6 alkylenesulfinyl(Cι.6)alkylene], optionally substituted [d-6 alkylenesulfonyl(Cι-6)alkylene] or optionally substituted [d-6 alkyleneamino(C1.6)alkylene];
B is N, N-oxide or CR18;
D is S or NR7;
E is N or CR12;
M is N-C(=Y), where N is the atom of attachment to the ring containing D and E; Y is O, S or NR13;
Z is O, S or NR14;
R1 is hydrogen, halogen, optionally substituted d_6 alkyl, optionally substituted C2.6 alkenyl, optionally substituted C2_6 alkynyl, optionally substituted d_6 alkoxy, optionally substituted d-6 alkylthio, optionally substituted C3.7 cycloalkyl, cyano, nitro or SF5;
R3, R4 and R5 are, independently, hydrogen, halogen, optionally substituted Cι_6 alkyl, optionally substituted d.6 alkoxy, optionally substituted Cι-6 alkylthio, optionally substituted d-6 alkylsulfinyl, optionally substituted d_6 alkylsulfonyl, cyano, nitro, optionally substituted d-6 alkylcarbonyl, optionally substituted Cι-6 alkoxycarbonyl or SF5;
R6 is hydrogen, halogen, cyano, optionally substituted d_2o alkyl, optionally substituted C2.20 alkenyl, optionally substituted C2.2o alkynyl, optionally substituted C3.7 cycloalkyl, optionally substituted C5-6 cycloalkenyl, formyl, optionally substituted d-2o alkoxycarbonyl, optionally substituted C1-20 alkylcarbonyl, aminocarbonyl, optionally substituted Cj.2o alkylaminocarbonyl, optionally substituted di(C1.2o)alkylaminocarbonyl, optionally substituted aryloxycarbonyl, optionally substituted arylcarbonyl, optionally substituted arylaminocarbonyl, optionally substituted N-alkyl-N-arylaminocarbonyl, optionally substituted diarylaminocarbonyl, optionally substituted heteroaryloxycarbonyl, optionally substituted heteroarylcarbonyl, optionally substituted heteroarylaminocarbonyl, optionally substituted alkylheteroarylaminocarbonyl, optionally substituted diheteroarylaminocarbonyl, optionally substituted phenyl, optionally substituted heteroaryl, optionally substituted heterocyclyl, R O, HS, optionally substituted d-2o alkylthio, optionally substituted d.20 alkylsulfinyl, optionally substituted Cι. 0 alkylsulfonyl, optionally substituted arylthio, optionally substituted arylsulfinyl, optionally substituted arylsulfonyl, R28R29N or R31ON=C(R27);
R7 is hydrogen or Cχ.β alkyl;
R2 is optionally substituted CMO alkyl, optionally substituted [C2_6 alkenyl(d_6)- alkyl], optionally substituted [C2-6 alkynyl(Cι_6)alkyl], optionally substituted C3.7 cycloalkyl, optionally substituted CMO alkylcarbonyl, optionally substituted C^o alkoxycarbonyl, formyl, optionally substituted C O alkylaminocarbonyl, optionally substituted di(C1.1o)alkylaminocarbonyl, optionally substituted phenoxycarbonyl, optionally substituted d.6 alkylthio, optionally substituted d_6 alkylsulfinyl, optionally substituted Cι_6 alkylsulfonyl, optionally substituted d_6 arylthio, optionally substituted d_6 arylsulfinyl, optionally substituted d-6 arylsulfonyl or R20R21NS(O)P; p is 0,1 or 2;
R12 is hydrogen, halogen, optionally substituted Cι.6 alkyl, optionally substituted C2.6 alkenyl, optionally substituted C2-6 alkynyl, optionally substituted d_6 alkoxy, optionally substituted Cι_6 alkylthio, optionally substituted d.6 alkylsulfinyl, optionally substituted d.6 alkylsulfonyl, cyano, nitro, formyl, R32ON=C(R30), optionally substituted d.6 alkylcarbonyl, optionally substituted Cι-6 alkoxycarbonyl or SF5; or R1 and R12 together with the atoms to which they are attached may be joined to form a five, six or seven-membered saturated or unsaturated ring carbocylic or heterocyclic ring which may contain one or two heteroatoms selected from O, N or S and which may be optionally substituted by Cι_6 alkyl, d.6 haloalkyl or halogen; R13 is hydrogen, cyano, nitro, optionally substituted Cι-6 alkyl, optionally substituted
C
3.
7 cycloalkyl, optionally substituted (C
2-
6)alkenyl(C
1.
6)alkyl, optionally substituted (C
2-
6)alkynyl(C
1.
6)alkyl, optionally substituted phenyl, optionally substituted heteroaryl, optionally substituted d-
6 alkylcarbonyl, optionally substituted Cι_
6 alkoxycarbonyl, optionally substituted d-6 alkylamino, optionally substituted di(C).
6)alkylamino, optionally substituted d-
6 alkylcarbonylamino, optionally substituted Cι-
6 alkoxycarbonylamino, optionally substituted d-6 alkoxy, optionally substituted Cι.
6 alkylthio, optionally substituted d-
6 alkylsulfinyl, optionally substituted Cι-6 alkylsulfonyl, optionally substituted arylthio, optionally substituted arylsulfinyl, optionally substituted arylsulfonyl or
R14 is hydrogen, cyano, optionally substituted d.8 alkyl, optionally substituted [C2.6 alkenyl(C1.6)alkyl], optionally substituted [C2.6 alkynyl(Cι_6)alkyl], optionally substituted C3.7 cycloalkyl, optionally substituted [C3.7 cycloalkyl(C1.6)alkyl], Cj.6 alkoxy(d-6)alkyl, optionally substituted Cι_6 alkoxycarbonyl, optionally substituted Cι-6 alkylcarbonyl, optionally substituted d-6 alkylaminocarbonyl, optionally substituted di(d.6)alkylamino- carbonyl, optionally substituted phenyl, optionally substituted heteroaryl, optionally substituted alkylsulfonyl or optionally substituted arylsulfonyl;
R18 is hydrogen, halogen, nitro, cyano, optionally substituted d-8 alkyl, optionally substituted C2_6 alkenyl, optionally substituted C2.6 alkynyl, optionally substituted C3.7 cycloalkyl, optionally substituted d-6 alkoxycarbonyl, optionally substituted Cι-6 alkylcarbonyl, optionally substituted Cι-6 alkylaminocarbonyl, optionally substituted di(C1.6)alkylaminocarbonyl, optionally substituted phenyl or optionally substituted heteroaryl;
R20 and R21 are, independently, optionally substituted d_6 alkyl or R20 and R21 together with the N atom to which they are attached form a five, six or seven-membered heterocyclic ring which may contain one or two further hetero atoms selected from O, N or S and which may be optionally substituted by one or two d_6 alkyl groups; R26 is hydrogen, optionally substituted d.2o alkyl, optionally substituted [C2.2o alkenyl(C1.6)alkyl], optionally substituted [C2.2o alkynyl(d_6)alkyl], optionally substituted C3.7 cycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted heterocyclyl, (C1.6)alkylCH=N or di(C1.6)alkylC=N;
R28 and R29 are, independently, hydrogen, optionally substituted d-20 alkyl, optionally substituted C3-7 cycloalkyl, optionally substituted [C2. 0 alkenyl (d_6)alkyl], optionally substituted [C2.2o alkynyl(d-ό)alkyl], optionally substituted d_ o alkoxycarbonyl, optionally substituted phenoxycarbonyl, formyl, optionally substituted d.20 alkylcarbonyl, optionally substituted Cι- 0 alkylsulfonyl or optionally substituted phenylsulfonyl; or R28 and R29 together with the N atom to which they are attached form a five, six or seven-membered heterocyclic ring which may contain one or two further hetero atoms selected from O, N or S and which may be optionally substituted by one or two d-6 alkyl groups;
R27 and R30 are independently hydrogen, optionally substituted phenyl or optionally substituted d-6 alkyl; and
R31 and R32 are, independently, hydrogen, optionally substituted phenyl (C1.2)alkyl or optionally substituted d-20 alkyl.
The compounds of formula (I) may exist in different geometric or optical isomers or tautomeric forms. This invention covers all such isomers and tautomers and mixtures thereof in all proportions as well as isotopic forms such as deuterated compounds.
When present, optional substituents on alkylene, alkenylene or alkynylene moieties include (subject to valency constraints) one or more of hydroxy, halogen, Cι-6 alkyl, d_6 haloalkyl, d_6 cyanoalkyl, d-6 alkoxy(d_6) alkyl, d_6 alkoxy, cyano, =O, =NR33 and =CR34R35; and, especially, one or more of halogen, d.6 alkyl, Cι_6 haloalkyl, Cι_6 cyanoalkyl, d.6 alkoxy(d.6) alkyl, d-6 alkoxy, cyano, =O, =NR33 and =CR34R35; wherein R33 is Q.6 alkyl, C1-6 haloalkyl, OR36 or NR37R38; where R34 and R35 are, independently, hydrogen, C,-6 alkyl, d.6 alkoxy, d.6 haloalkyl, cyano, d-6 alkoxycarbonyl, C]_6 alkylcarbonyl or NR39R40; R36 is d-6 alkyl, Cι_6 haloalkyl or phenyl(d.2)alkyl; R37 and R38 are, independently, hydrogen, Cι_8 alkyl, C3.7 cycloalkyl, C2.6 alkenyl(Cι_6)alkyl, C2.6 alkynyl(d_6)alkyl, C2_6
haloalkyl, d_6 alkoxy(C1_6)alkyl, d-6 alkoxycarbonyl (d.6)alkyl, carboxy(d-6)alkyl or phenyl(Cι. )alkyl; or R and R together with the N atom to which they are attached form a five, six or seven-membered heterocyclic ring which may contain one or two further hetero atoms selected from O, N or S and which is optionally substituted by one or two d-6 alkyl groups; R39 and R40 are, independently, hydrogen, d.8 alkyl, C3.7 cycloalkyl, C2_6 alkenyl(d_6)alkyl, C2.6 alkynyl(Cι-6)alkyl, C2.6 haloalkyl, Cι_6 alkoxy(d.6)alkyl, Cι_6 alkoxycarbonyl(C1.6)alkyl, carboxy(d.6)alkyl or phenyl(C1. )alkyl; or R39 and R40 together with the N atom to which they are attached form a five, six or seven-membered heterocyclic ring which may contain one or two further heteroatoms selected from O, N or S and which is optionally substituted by one or two d_6 alkyl groups.
Each alkyl moiety is a straight or branched chain and is, for example, methyl, ethyl, n-propyl, rc-butyl, rc-pentyl, n-hexyl, wo-propyl, «-butyl, sec-butyl, wobutyl, tert-butyl or neσ-pentyl.
When present, the optional substituents on an alkyl moiety include one or more of halogen, nitro, cyano, NCS-, C3.7 cycloalkyl (itself optionally substituted with d-6 alkyl or halogen), C5.7 cycloalkenyl (itself optionally substituted with Cι-6 alkyl or halogen), hydroxy, d-io alkoxy, d-io alkoxy(d- ^alkoxy, tri(C1^)alkylsilyl(C1.6)alkoxy, d-6 alkoxycarbonyl(C1.1o)alkoxy, Cuo haloalkoxy, aryl(Cι_4)alkoxy (where the aryl group is optionally substituted), C .7 cycloalkyloxy (where the cycloalkyl group is optionally substituted with d-6 alkyl or halogen), d-io alkenyloxy, C^o alkynyloxy, SH, d-io alkylthio, CMO haloalkylthio, aryl(C1^)alkylthio (where the aryl group is optionally substituted), C3.7 cycloalkylthio (where the cycloalkyl group is optionally substituted with Cι- alkyl or halogen), tri(Cι^)alkylsilyl(Cι-6)alkylthio, arylthio (where the aryl group is optionally substituted), d-6 alkylsulfonyl, d_6 haloalkylsulfonyl, Cι_6 alkylsulfinyl, d.6 haloalkylsulfinyl, arylsulfonyl (where the aryl group may be further optionally substituted), tri(C1.4)alkylsilyl, aryldi(Cι^)alkylsilyl, (C1. )alkyldiarylsilyl, triarylsilyl, d_ιo alkylcarbonyl, HO2C, Cj-io alkoxycarbonyl, aminocarbonyl, C e alkylaminocarbonyl, di(Cι-6 alkylaminocarbonyl, N-(Cι_3 alkyl)-N-(d.3 alkoxy)aminocarbonyl, Cι.6 alkylcarbonyloxy, arylcarbonyloxy (where the aryl group is optionally substituted), di(C1-6)alkylaminocarbonyloxy, aryl (itself optionally substituted), heteroaryl (itself optionally substituted), heterocyclyl (itself optionally substituted with d_6 alkyl or halogen), aryloxy (where the aryl group is optionally substituted), heteroaryloxy, (where the heteroaryl
group is optionally substituted), heterocyclyloxy (where the heterocyclyl group is optionally substituted with d.6 alkyl or halogen), amino, d_6 alkylamino, di(d-6)alkylamino, d.6 alkylcarbonylamino and N-(C1.6)alkylcarbonyl-N-(C1.6)alkylamino.
Alkenyl and alkynyl moieties can be in the form of straight or branched chains, and the alkenyl moieties, where appropriate, can be of either the (E)- or ©-configuration. Examples are vinyl, allyl and propargyl.
When present, the optional substituents on alkenyl or alkynyl include those optional substituents given above for an alkyl moiety.
In the context of this specification acyl is optionally substituted d.6 alkylcarbonyl (for example acetyl), optionally substituted C2_6 alkenylcarbonyl, optionally substituted C2-6 alkynylcarbonyl, optionally substituted arylcarbonyl (for example benzoyl) or optionally substituted heteroarylcarbonyl.
Halogen is fluorine, chlorine, bromine or iodine.
Haloalkyl groups are alkyl groups which are substituted with one or more of the same or different halogen atoms and are, for example, CF3, CF2C1, CF3CH2 or CHF2CH2.
Aryl includes naphthyl, anthracyl, fluorenyl and indenyl but is preferably phenyl.
The term heteroaryl refers to an aromatic ring containing up to 10 atoms including one or more heteroatoms (preferably one or two heteroatoms) selected from O, S and N. Examples of such rings include pyridine, pyrimidine, furan, quinoline, quinazoline, pyrazole, thiophene, thiazole, oxazole and isoxazole.
The terms heterocycle and heterocyclyl refer to a non-aromatic ring containing up to 10 atoms including one or more (preferably one or two) heteroatoms selected from O, S and N. Examples of such rings include 1,3-dioxolane, tetrahydrofuran and morpholine.
When present, the optional substituents on heterocyclyl include C\.(, alkyl as well as those optional substituents given above for an alkyl moiety.
Cycloalkyl includes cyclopropyl, cyclopentyl and cyclohexyl.
Cycloalkenyl includes cyclopentenyl and cyclohexenyl.
When present, the optional substituents on cycloalkyl or cycloalkenyl include d_3 alkyl as well as those optional substituents given above for an alkyl moiety. Carbocyclic rings include aryl, cycloalkyl and cycloalkenyl groups.
When present, the optional substituents on aryl or heteroaryl are selected, independently, from halogen, nitro, cyano, NCS-, d_6 alkyl, d_6 haloalkyl, Cι-6
alkoxy(C!.6)alkyl, C2.6 alkenyl, C2_6 haloalkenyl, C2.6 alkynyl, C3.7 cycloalkyl (itself optionally substituted with Cι-6 alkyl or halogen), C5.7 cycloalkenyl (itself optionally substituted with d-6 alkyl or halogen), hydroxy, CMO alkoxy, CMO alkoxy(C1.1o)alkoxy, tri(C1. )alkylsilyl(C1.6)alkoxy, d.6 alkoxycarbonyl(C1.1o)alkoxy, Cno haloalkoxy, aryl(C1. )alkoxy (where the aryl group is optionally substituted), C3.7 cycloalkyloxy (where the cycloalkyl group is optionally substituted with d_6 alkyl or halogen), CMO alkenyloxy, Ci-io alkynyloxy, SH, CMO alkylthio, C O haloalkylthio, aryl(C1^)alkylthio (where the aryl group may be further optionally substituted), C3.7 cycloalkylthio (where the cycloalkyl group is optionally substituted with d-6 alkyl or halogen), tri(C1.4)alkylsilyl(C1.6)alkylthio, arylthio (where the aryl group is optionally substituted), d_6 alkylsulfonyl, C e haloalkylsulfonyl, C . alkylsulfinyl, d-6 haloalkylsulfinyl, arylsulfonyl (where the aryl group is optionally substituted), tri(d^)alkylsilyl, aryldi(d^)alkylsilyl, (Cι.4)alkyldiarylsilyl, triarylsilyl, Cuio alkylcarbonyl, HO2C, C O alkoxycarbonyl, aminocarbonyl, d_6 alkylaminocarbonyl, di(d.6 alkylaminocarbonyl, N-(d-3 alkyl)-N-(Cι.3 alkoxy)aminocarbonyl, Cι-6 alkylcarbonyloxy, arylcarbonyloxy (where the aryl group is optionally substituted), di(C1-6)alkylaminocarbonyloxy, aryl (itself optionally substituted), heteroaryl (which itself may be further optionally substituted), heterocyclyl (itself optionally substituted with d-6 alkyl or halogen), aryloxy (where the aryl group is optionally substituted), heteroaryloxy (where the heteroaryl group is optionally substituted), heterocyclyloxy (where the heterocyclyl group is optionally substituted with d_6 alkyl or halogen), amino, d.6 alkylamino, di(C1.6)alkylamino, d.6 alkylcarbonylamino and N-(C i _6)alkylcarbonyl-N-(C i _6)alkylamino.
For substituted phenyl moieties, heterocyclyl and heteroaryl groups it is preferred that one or more substituents are independently selected from halogen, d_6 alkyl, d_ haloalkyl, d.6 alkoxy(d.6)alkyl, d-6 alkoxy, d-6 haloalkoxy, d_6 alkylthio, d.6 haloalkylthio, d_6 alkylsulfinyl, d_6 haloalkylsulfinyl, d.6 alkylsulfonyl, Ci-6 haloalkylsulfonyl, C2_6 alkenyl, C2_6 haloalkenyl, C .6 alkynyl, C3. cycloalkyl, nitro, cyano, CO2H, d-6 alkylcarbonyl, Cι_6 alkoxycarbonyl, R41R42N or R43R44NC(O); wherein R41, R42, R43 and R44 are, independently, hydrogen or d-6 alkyl. Haloalkenyl groups are alkenyl groups which are substituted with one or more of the same or different halogen atoms.
O 01/55139
- li ¬
lt is to be understood that dialkylamino substituents include those where the dialkyl groups together with the N atom to which they are attached form a five, six or seven- membered heterocyclic ring which may contain one or two further heteroatoms selected from O, N or S and which is optionally substituted by one or two independently selected (d-^alkyl groups. When heterocyclic rings are formed by joining two groups on an N atom, the resulting rings are suitably pyrrolidine, piperidine, thiomorpholine and morpholine each of which may be substituted by one or two independently selected (d_6) alkyl groups.
Preferably the optional substituents on an alkyl moiety include one or more of halogen, nitro, cyano, HO2C, CMO alkoxy (itself optionally substituted by C O alkoxy), aryl(Cι_4)alkoxy, CMO alkylthio, CMO alkylcarbonyl, C O alkoxycarbonyl, Ci-6 alkylaminocarbonyl, di(Cι-6 alkyl)aminocarbonyl, (C1.6)alkylcarbonyloxy, optionally substituted phenyl, heteroaryl, aryloxy, arylcarbonyloxy, heteroaryloxy, heterocyclyl, heterocyclyloxy, C3.7 cycloalkyl (itself optionally substituted with (Cι-6)alkyl or halogen), C3.7 cycloalkyloxy, C5.7 cycloalkenyl, d_6 alkylsulfonyl, d.6 alkylsulfinyl, tri(Cι_4)alkylsilyl, tri(Ci_4)alkylsilyl(Ci.6)alkoxy, aryldi(d-4)alkylsilyl, (Cι. )alkyldiarylsilyl and triarylsilyl.
Preferably the optional substituents on alkenyl or alkynyl include one or more of halogen, aryl and C . cycloalkyl.
It is more preferred that heterocyclyl is optionally substituted by Cue alkyl.
Preferably the optional substituents for cycloalkyl include halogen, cyano and d-3 alkyl.
Preferably the optional substituents for cycloalkenyl include d.3 alkyl, halogen and cyano.
In a further aspect, the present invention provides a compound of formula (IA):
wherein A, B, D, E, M, Z, R1, R2, R3, R4, R5 and R6 are as defined above for a compound of formula (I).
One group of preferred compounds of formula (IA) is a group where R1 is hydrogen, halogen, Ci-6 alkyl, C2.6 alkenyl, C2.6 alkynyl, d_6 cyanoalkyl, d.6 haloalkyl, Ci-6 alkoxy, d_6 haloalkoxy, d.6 alkylthio, d-6 haloalkylthio, C3.6 cycloalkyl, C3.7 cycloalkyl(Cι_4)alkyl, Cue alkoxy(Cι_6)alkyl, cyano, nitro or SF5; A is Cue alkylene, Cue alkenylene, d-6 alkylenoxy, oxy(d-6)alkylene, d-6 alkylenamino or d_6 alkylenethio, each of which is optionally substituted by d_3 alkyl, d_3 haloalkyl, d_3 cyanoalkyl, halogen, d_3 alkoxy, C].6 alkoxycarbonyl, cyano, =O, =NR33 or =CR34R35;
B is N or CR18; D is S or NR7 where R7 is hydrogen or d_6 alkyl;
E is N or CR12;
M is NC(=Y) where N is the atom of attachment to the ring containing D and E;
Y is O, S or NR13;
Z is O, S or NR14; R3, R4 and R5 are independently selected from hydrogen, halogen, d-6 alkyl, d_6 alkoxy, d.6 haloalkoxy, d-6 alkylthio, Cι.6 haloalkylthio, d-6 alkylsulfinyl, Cι.6 haloalkylsulfinyl, Cu6 alkylsulfonyl, d.6 haloalkylsulfonyl, d-6 haloalkyl, cyano, nitro, d.6 alkylcarbonyl, d-6 alkoxycarbonyl or SF5;
R6 is cyano, C1-8 alkyl, d-6 haloalkyl, d.6 cyanoalkyl, C2.6 alkenyl, C2.6 alkynyl, C3.7 cycloalkyl, C3.7 halocycloalkyl, C3.7 cyanocycloalkyl, d.3 alkyl(C3.7)cycloalkyl, d_3 alkyl(C3.7)halocycloalkyl, C5.6 cycloalkenyl, C3.7 cycloalkyl(C1.6)alkyl, C5.6 cycloalkenyl(Cμ6)alkyl, C2_6 haloalkenyl, d_6cyanoalkenyl, Cue alkoxy(C1_6)alkyl, C3_6 alkenyloxy(C1.6)alkyl, C3_6 alkynyloxy(d-6)alkyl, aryloxy(C1.6)alkyl, formyl, d.6 carboxyalkyl, d.6 alkylcarbonyl(C1.6)alkyl, C2.6 alkenylcarbonyl(d_6)alkyl, C2.6 alkynylcarbonyl(C1-6)alkyl, Ci.6 alkoxycarbonyl(Cι.6)alkyl, C3.6 alkenyloxycarbonyl- (C!-6)alkyl, C3.6 alkynyloxycarbonyl(d-6)alkyl, aryloxycarbonyl(d.6)alkyl, Cue alkylthio(Cι.6)alkyl, d.6 alkylsulfinyl(d.6)alkyl, C1.6 alkylsulfonyl(d.6)alkyl, aminocarbonyl(C1.6)alkyl, aminocarbonyl(C2.6)alkenyl, aminocarbonyl(C2.6)alkynyl, d_6 alkylaininocarbonyl(C1-6)alkyl, di(C1.6)alkylaminocarbonyl(C1.6)alkyl, Ci.6 alkylaminocarbonyl(C1.6)alkenyl, di(Cι-6)alkylaminocarbonyl(C1.6)alkenyl, alkylaminocarbonyl(C \ ,6)alkynyl, di(C 1 _6)alkylaminocarbonyl(C 1.6)alkynyl, C 1 _6 alkoxycarbonyl, Cι_6 alkylcarbonyl, aminocarbonyl, Cι_6 alkylaminocarbonyl,
di(C1-6)alkylaminocarbonyl, phenyl (optionally substituted by halo, nitro, cyano, _6 alkyl, d-6 haloalkyl, Cue alkoxy or d_6 haloalkoxy), phenyl(Cj. )alkyl (wherein the phenyl group is optionally substituted by halo, nitro, cyano, d^ alkyl, d_6 haloalkyl, d.6 alkoxy or Cue haloalkoxy), phenyl(C2.4)alkenyl, (wherein the phenyl group is optionally substituted by halo, nitro, cyano, d_6 alkyl, Ci-6 haloalkyl, d_6 alkoxy or d.6 haloalkoxy), heteroaryl (optionally substituted by halo, nitro, cyano, Cue alkyl, d_6 haloalkyl, d_6 alkoxy or d_ haloalkoxy), heterocyclyl (optionall substituted by halo, nitro, cyano, d-6 alkyl, d-6 haloalkyl, Cu alkoxy or d-6 haloalkoxy),, heteroaryl(d.4)alkyl (where the heteroaryl may be substituted by halo, nitro, cyano, Cι_6 alkyl, Ci-6 haloalkyl, d-6 alkoxy or d-6 haloalkoxy), heterocyclyl (C1.4)alkyl (where the heterocyclyl may be substituted by halo, cyano, d_6 alkyl, Ci-6 haloalkyl, d.6 alkoxy or d-6 haloalkoxy), R26O, d.8 alkylthio, R28R29N or R31ON=C(R27);
R2 is CMO alkyl, benzyloxymethyl, benzoyloxymethyl, C1_6alkoxy(C1.6)alkyl, C2.6 alkenyl (C1.6)alkyl (especially allyl), C2.6 alkynyl(Cι-6)alkyl (especially propargyl), CMO alkylcarbonyl or CMO alkoxycarbonyl;
R12 is hydrogen, halogen, d_6 alkyl, C2.6 alkenyl, Ci-6 alkynyl, -6 haloalkyl, Ci-6 alkoxy, -6 alkoxy (C1_6)alkyl, Ci_6 haloalkoxy, Ci-6 alkylthio, Cue haloalkylthio, d-6 alkylsulfinyl, d-6 haloalkylsulfinyl, Cι-6 alkylsulfonyl, d_6 haloalkylsulfonyl, -6 haloalkyl, cyano, nitro, formyl, CH=NOR32, Cue alkylcarbonyl, d-6 alkoxycarbonyl or SF5; or together R1 and R12 together with the atoms to which they are attached may be joined to form a five, six or seven-membered saturated or unsaturated ring carbocylic or heterocyclic ring which may contain one or two hetero atoms selected from O, N or S and which may be optionally substituted by d_6 alkyl, Cι_6 haloalkyl or halogen;
R13 is cyano, nitro, d-6 alkyl, Cι.6 haloalkyl, C3.7 cycloalkyl, C3.7 cycloalkyl(C1-6)alkyl, CH2(C2.6)alkenyl, CH2(C2_6)alkynyl, phenyl (optionally substituted by halo, nitro, cyano, d_6 alkyl, _6 haloalkyl, d-6 alkoxy or d_6 haloalkoxy) heteroaryl (optionally substituted by halo, nitro, cyano, d_6 alkyl, Cue haloalkyl, d_6 alkoxy or Cμ6 haloalkoxy), d_6 alkylcarbonyl, Cι_6 alkoxycarbonyl, d-6 alkylamino, di(C ^alkylamino, d-6 alkylcarbonylamino, d.6 alkoxycarbonylamino, Cι_6 alkoxy, Cι_6 alkylthio, Cu alkylsulfinyl, d_6 alkylsulfonyl, Cj-6 haloalkylthio, Cι.6 haloalkylsulfinyl, Ci-6 haloalkylsulfonyl, arylthio, arylsulfinyl, arylsulfonyl or OCO(C]-6)alkyl;
R14 is hydrogen, d_8 alkyl, d_6 haloalkyl, d.6 cyanoalkyl, C2.6 alkenyl, C2_6 alkynyl, C3.7 cycloalkyl, C2.6 haloalkenyl, C3.7 cycloalkyl(d_6)alkyl, Cue alkoxy(d-6)alkyl, d.6 alkoxycarbonyl, Cj.6 alkylcarbonyl, d_6 alkylaminocarbonyl, di(d-6)alkylaminocarbonyl, phenyl (optionally substituted by halo, nitro, cyano, d_6 alkyl, d-6 haloalkyl, Cue alkoxy or Cue haloalkoxy) or heteroaryl (optionally substituted by halo, nitro, cyano, Cι_6 alkyl, d-6 haloalkyl, d.6 alkoxy or Cι.6 haloalkoxy); R33 is d.6 alkyl, OR36 or NR37R38; R34 is hydrogen, d-6 alkyl or d_6 haloalkyl;
R35 is hydrogen, Cue alkyl, d-6 haloalkyl, Ci-β alkoxy, cyano, d-6 alkoxycarbonyl, d.6 alkylcarbonyl or NR39R40;
R18 is hydrogen, halogen, nitro, cyano, Cι-8 alkyl, d-6 haloalkyl, d-6 cyanoalkyl, C2.6 alkenyl, C2.6 alkynyl, C3-7 cycloalkyl, C2_6 haloalkenyl, C3.7 cycloalkyl(d-6)alkyl, d.6 alkoxy(Cι.6)alkyl, Cue alkoxycarbonyl, Cι_6 alkylcarbonyl, Ci-6 alkylaminocarbonyl, di(C1-6)alkylaminocarbonyl, Cue alkoxycarbonyl(C1_6)alkyl, Cue alkylcarbonyl(C1_6)alkyl, d-6 alkylaminocarbonyl (C1_6)alkyl, di(d_6)alkylaminocarbonyl(d_6)alkyl, phenyl (optionally substituted by halo, nitro, cyano, d-6 alkyl, d-6 haloalkyl, d-6 alkoxy or d_6 haloalkoxy), phenyl(C!.6)alkyl (wherein the phenyl group is optionally substituted by halo, nitro, cyano, Cue alkyl, d-6 haloalkyl, Cι_6 alkoxy or d-6 haloalkoxy), heteroaryl (optionally substituted by halo, nitro, cyano, d_6 alkyl, d-6 haloalkyl, d_6 alkoxy or d.6 haloalkoxy) or heteroaryl(d_6)alkyl (wherein the heteroaryl group is optionally substituted by halo, nitro, cyano, d_6 alkyl, d.6 haloalkyl, d_6 alkoxy or d.6 haloalkoxy); R36 is Ct-6 alkyl or optionally substituted phenyl(d.2)alkyl;
R37 and R38 are, independently, hydrogen, d_8 alkyl or phenyl (optionally substituted by halo, nitro, cyano, d.6 alkyl, Ci-6 haloalkyl, d_6 alkoxy or d_ haloalkoxy); R26 is hydrogen, d_8 alkyl, d.6 haloalkyl, d_6 cyanoalkyl, C2.6 alkenyl, C2.6 alkynyl,
Cue alkoxy(d_6)alkyl, phenyl (C1.4)alkyl , (wherein the phenyl group is optionally substituted by halo, nitro, cyano, d- alkyl, Cι_6 haloalkyl, d-6 alkoxy or d-6 haloalkoxy), heteroaryl(d- )alkyl (wherein the heteroaryl group is optionally substituted by halo, nitro, cyano, Ci-6 alkyl, Ci-6 haloalkyl, d_ alkoxy or d_ haloalkoxy), heterocyclyl (optionally substituted by halo, nitro, cyano, Ci-6 alkyl, Cι- haloalkyl, Cι_6 alkoxy or d-6 haloalkoxy), heterocyclyl(Cι_ )alkyl (wherein the heterocyclyl group is optionally substituted by halo,
nitro, cyano, d_6 alkyl, d_6 haloalkyl, d_6 alkoxy or d_6 haloalkoxy), d_6 alkoxycarbonyl(C1.6)alkyl or N=C(CH3)2;
R27 is d-6 alkyl, d-e haloalkyl or phenyl (optionally substituted by halo, nitro, cyano, d-6 alkyl, Ci-6 haloalkyl, d_6 alkoxy or Cι_6 haloalkoxy); R28 and R29 are, independently, hydrogen, d.8 alkyl, C3.7 cycloalkyl, C3.6 alkenyl,
C3-6 alkynyl, C3.7 cycloalkyl(d. )alkyl, C2.6 haloalkyl, d-6 alkoxy(C1.6)alkyl, Ci-6 alkoxycarbonyl, or R28 and R29 together with the N atom to which they are attached form a five, six or seven-membered heterocyclic ring which may contain one or two further hetero atoms selected from O, N or S and which may be optionally substituted by one or two d-6 alkyl groups;
R30 is hydrogen or d_3 alkyl;
R31 and R32 are, independently, Cι_6 alkyl or phenyl(d_2)alkyl (wherein the phenyl group is optionally substituted by halo, nitro, cyano, d_6 alkyl, Cι_6 haloalkyl, d.6 alkoxy or d-6 haloalkoxy); and R39 and R40 are, independently, hydrogen, d.8 alkyl, C3.7 cycloalkyl, C3.6 alkenyl,
C3.6 alkynyl, C2.6 haloalkyl, d-6 alkoxy(C1_6)alkyl, d-6 alkoxycarbonyl (Cι-6)alkyl, carboxy(d_6)alkyl or phenyl(C1.2)alkyl; or R39 and R40 together with the N atom to which they are attached form a five, six or seven-membered heterocyclic ring which may contain one or two further hetero atoms selected from O, N or S and which may be optionally substituted by one or two d-6 alkyl groups;
It is preferred that A is d_6 alkylene, d_6 alkenylene, d-6 alkylenoxy, oxy(d-6)alkylene or d_6 alkylenamino, each of which is optionally substituted by Cι_3 alkyl, C]-3 haloalkyl, d.3 cyanoalkyl, halogen, d.3 alkoxy, d.6 alkoxycarbonyl, cyano, =O, =NR33 or =CR34R35; where R33 is d.6 alkyl, OR36 or NR37R38; R36 is d.6 alkyl or phenyl(C,_2)alkyl (where the phenyl group is optionally substituted by halo, nitro, cyano, Cu6 alkyl, d-6 haloalkyl, Ci-6 alkoxy or d_6 haloalkoxy); R37 and R38 are, independently, hydrogen, d.8 alkyl or phenyl (which may be optionally substituted by halo, nitro, cyano, d_6 alkyl, Cι_6 haloalkyl, Cι_6 alkoxy or d.6 haloalkoxy); R34 is hydrogen, Cι_6 alkyl or Cμ6 haloalkyl; R35 is hydrogen, Cι-6 alkyl, Cι-6 haloalkyl, Cι_6 alkoxy, cyano, Cι_6 alkoxycarbonyl, Cι_6 alkylcarbonyl or NR39R40; and R39 and R40 are, independently, hydrogen, Cι-8 alkyl, C3.7 cycloalkyl, C3.6 alkenyl, C3-6 alkynyl, C2.6 haloalkyl, Cι_6 alkoxy(d-6)alkyl, Cι_6 alkoxycarbonyl(Cι-6)alkyl, carboxy(C1.6)alkyl or phenyl(Cι_2)alkyl; or R39 and R40 together
with the N atom to which they are attached form a five, six or seven-membered heterocyclic ring which may contain one or two further heteroatoms selected from O, N or S and which may be optionally substituted by one or two d_6 alkyl groups.
A is more preferably d_4 alkylene (optionally substituted by halogen, d.3 alkyl or Cι-3 alkoxy, -C(O)- or d-4 alkyleneoxy (which may be optionally substituted by d.3 alkyl).
It is even more preferred that A is d- alkyl-substituted d_4 alkylene, fluoro- substituted d-4 alkylene, methoxy-substituted d-4 alkylene, -C(O)- or C2. alkyleneoxy; still more preferably A is d_2 alkyl-substituted C alkylene, fluoro-substituted Cj.4 alkylene or methoxy-substituted Cj.4 alkylene. It is further preferred that A is CH(CH3)CH2, CH2CH(CH3), CH(CH3), CHF,
CH(OCH3) or CH(CH3)O; even further preferred that A is CH(CH3)CH2, CH2CH(CH3), CH(CH3), CHF or CH(CH3)O; especially preferred that A is CHF, CH(OCH3) or CH(CH3); and most preferably A is CHF or CH(CH3).
B is preferably N, C-H, C-halogen or C-C1.3 alkyl. B is more preferably N.
D is preferably S.
E is preferably CR12.
A preferred value of M is NC(=O) where N is the atom of attachment to the ring containing E and D. Y is preferably O or S.
Y is more preferably O.
Preferably Z is O or S.
Z is more preferably O.
It is preferred that R1 is hydrogen, halogen, d.6 alkyl, d.6 cyanoalkyl, Cue haloalkyl, C3-7 cycloalkyl(d_4)alkyl, d.6 alkoxy(d.6)alkyl, C2.6 alkenyl, C2.6 alkynyl, Cι_6 alkoxy, d_6 haloalkoxy, Cue alkylthio, Cι-6 haloalkylthio, d-6 cycloalkyl, cyano, nitro or SF5.
R1 is more preferably hydrogen, halogen, Cι_6 alkyl, C2.6 alkenyl, Ci-6 haloalkyl, Cι_6 alkoxy, d-6 haloalkoxy, Cι.6 alkylthio, Ci-6 haloalkylthio, C3_6 cycloalkyl, cyano, nitro or SF5. It is even more preferred that R1 is hydrogen, halogen, Ci-6 alkyl, Cι.6 haloalkyl, Cι.6 alkoxy(Cι_6)alkyl, C .6 alkenyl, Cι_6 alkoxy, Ci-6 haloalkoxy, Ci-6 alkylthio, Cι_6 haloalkylthio, C3.6 cycloalkyl or cyano.
It is most preferred that R1 is halogen, d.6 alkyl, d.6 haloalkyl, d.6 alkoxy or d-6 haloalkoxy.
R2 is preferably CMO alkyl, benzyloxymethyl, benzoyloxymethyl, C1.6alkoxy(C1.6)alkyl, C2.6 alkenyl(d.6)alkyl (especially allyl), C2.6 alkynyl(C1.6)alkyl (especially propargyl), C O alkylcarbonyl or CMO alkoxycarbonyl.
It is preferred that R3, R4 and R5 are, independently, hydrogen, halogen, C|.6 alkyl, d-6 haloalkyl, d_6 alkoxy, d-6 haloalkoxy, d.6 alkylthio, d.6 haloalkylthio, d_6 alkylsulfinyl, d.6 haloalkylsulfinyl, Cue alkylsulfonyl, Cue haloalkylsulfonyl, cyano, nitro, Cue alkylcarbonyl or d.6 alkoxycarbonyl. It is more preferred that R3, R4 and R5 are, independently, hydrogen, halogen or d.3 alkyl.
It is even more preferred that R3, R4 and R5 are, independently, hydrogen or halogen (especially fluorine).
It is preferred that R6 is cyano, Cι.8 alkyl, Cι.8 haloalkyl, d.8 cyanoalkyl, C3.7 cycloalkyl(d.6)alkyl, C5-6 cycloalkenyl(d-6)alkyl, d-6 alkoxy(d-6)alkyl, C3.6 alkenyloxy(C1.6)alkyl, C3.6 alkynyloxy(C1.6)alkyl, aryloxy(C1.6)alkyl, Cue carboxyalkyl, d.6 alkylcarbonyl(d-6)alkyl, C2.6 alkenylcarbonyl (d.6)alkyl, C2.6 alkynylcarbonyl(C1.6)alkyl, d-6 alkoxycarbonyl(d-6)alkyl, C3_6 alkenyloxycarbonyl(Cι.6)alkyl, C3.6 alkynyloxycarbonyl(C1.6)alkyl, aryloxycarbonyl(d-6)alkyl, d_6 alkylthio(C].6)alkyl, Ci-6 alkylsulfinyl(Cι-6)alkyl, d-6 alkylsulfonyl(d-6)alkyl, aminocarbonyl(d-6)alkyl, d.6 alkylaminocarbonyl (Cι.6)alkyl, di(Cι.6)alkylaminocarbonyl(d.6)alkyl, phenyl(d.4)alkyl (wherein the phenyl group is optionally substituted by halo, nitro, cyano, Cι_6 alkyl, Cι_6 haloalkyl, d_ alkoxy or d-6 haloalkoxy), heteroaryl(d.4)alkyl (where the heteroaryl group is optionally substituted by halo, nitro, cyano, d-6 alkyl, d-6 haloalkyl, d_6 alkoxy or d_6 haloalkoxy), heterocyclyl(C1.4)alkyl (where the heterocyclyl group is optionally substituted by halo, cyano, d_6 alkyl, d-6 haloalkyl, d_6 alkoxy or d_6 haloalkoxy), C2_6 alkenyl, C2_6 haloalkenyl, d_6 cyanoalkenyl, C5-6 cycloalkenyl, aminocarbonyl(C2_6)alkenyl, d-6 alkylaminocarbonyl(d-6)alkenyl, di(C1-6)alkylaminocarbonyl(C1-6)alkenyl, phenyl(C2.4)alkenyl (wherein the phenyl group is optionally substituted by halo, nitro, cyano, d-6 alkyl, d_6 haloalkyl, d-6 alkoxy or Cι_6 haloalkoxy), C2-6 alkynyl, aminocarbonyl(C .6)alkynyl, alkylaminocarbonyl (C1_6)alkynyl, di(C1.6)alkylaminocarbonyl(C1-6)alkynyl, C3.7 cycloalkyl, C3-7 halocycloalkyl, C3.7
O 01/55139
- 18 -
cyanocycloalkyl, d.3 alkyl(C3-7)cycloalkyl, C1.3 alkyl (C3.7)halocycloalkyl, C5-6 cycloalkenyl, formyl, d_6 alkoxycarbonyl, Cue alkylcarbonyl, aminocarbonyl, Ci-6 alkylaminocarbonyl, di(C1.6)alkylaminocarbonyl, phenyl (optionally substituted by halo, nitro, cyano, d_6 alkyl, Ci-6 haloalkyl, d-6 alkoxy or Cue haloalkoxy), heteroaryl (optionally substituted by halo, nitro, cyano, Ci-6 alkyl, d_6 haloalkyl, Ci-6 alkoxy or Ci-6 haloalkoxy), heterocyclyl
(optionally substituted by halo, nitro, cyano, d-6 alkyl, d_6 haloalkyl, d_6 alkoxy or Cj_6 haloalkoxy), Cι.8 alkylthio, R26O, R28R29N or R3,ON=C(R27); where R26 is hydrogen, d-8 alkyl, Cι_6 haloalkyl, Cue cyanoalkyl, d.6 alkoxy(d.6)alkyl, phenyl(d.4)alkyl, (wherein the phenyl group is optionally substituted by halo, nitro, cyano, d-6 alkyl, d_6 haloalkyl, d.6 alkoxy or d-6 haloalkoxy), heteroaryl(d.4)alkyl (wherein the heteroaryl group is optionally substituted by halo, nitro, cyano, Cι-6 alkyl, d-6 haloalkyl, Ci-6 alkoxy or d-6 haloalkoxy), heterocyclyl (C1.4)alkyl (wherein the heterocyclyl group is optionally substituted by halo, nitro, cyano, Cι_6 alkyl, Cι_6 haloalkyl, d.6 alkoxy or Cι_6 haloalkoxy), d-6 alkoxycarbonyl(d-6)alkyl, C2.6 alkenyl, C2.6 alkynyl or N=C(CH3)2; R27 is phenyl (optionally substituted by halo, nitro, cyano, Cι-6 alkyl, Ci-6 haloalkyl, Cue alkoxy or Cue haloalkoxy), Ci-6 alkyl or Cι_6 haloalkyl; R28 and R29 are, independently, hydrogen, Cι_8 alkyl, C3.7cycloalkyl(C1-4)alkyl, C2.6 haloalkyl, d.6 alkoxy(Cι.6)alkyl, C3.7 cycloalkyl, C3.6 alkenyl, C3.6 alkynyl or d_6 alkoxycarbonyl; and R31 is phenyl(Cι.2)alkyl (wherein the phenyl group is optionally substituted by halo, nitro, cyano, Ci-6 alkyl, d_6 haloalkyl, Ci-6 alkoxy or d_6 haloalkoxy) or d-6 alkyl.
It is further preferred that R6 is d_8 alkyl, Cι-8 haloalkyl, d_8 cyanoalkyl, C3.7 cycloalkyl(C1.6)alkyl, C5.6 cycloalkenyl(Ci-6)alkyl, Ci-6 alkoxy(C1.6)alkyl, C3_6 alkenyloxy(d-6)alkyl, C3-6 alkynyloxy(C1-6)alkyl, aryloxy(C1.6)alkyl, Cι_6 carboxyalkyl, d-6 alkylcarbonyl(Cι-6)alkyl, C2.6 alkenylcarbonyl(Ci_6)alkyl, C .6 alkynylcarbonyl(Cι_6)alkyl, Cμ6 alkoxycarbonyl(C1.6)alkyl, C3.6 alkenyloxycarbonyl(C1-6)alkyl, C3-6 alkynyloxycarbonyl- (Ci.6)alkyl, aryloxycarbonyl(Cι.6)alkyl, Cue alkylthio(Cι-6)alkyl, d.6 alkylsulfinyl(Cι_6)alkyl, Ci-6 alkylsulfonyl(C]-6)alkyl, aminocarbonyl (d.6)alkyl, Ci-6 alkylaminocarbonyl(Cι_6)alkyl, di(Cι-6)alkylaminocarbonyl(Cι-6)alkyl, phenyl (Cι_4)alkyl (wherein the phenyl group is optionally substituted by halo, nitro, cyano, d_ alkyl, Cι.6 haloalkyl, d_6 alkoxy or Cι-6 haloalkoxy), heteroaryl(Cι-4)alkyl (wherein the heteroaryl group is optionally substituted by halo, nitro, cyano, Cj_6 alkyl, Cι-6 haloalkyl, Cι_6 alkoxy or Cι-6 haloalkoxy), heterocyclyl(Cι_ )alkyl (wherein the heterocyclyl group is optionally substituted by halo,
nitro, cyano, d.6 alkyl, d-6 haloalkyl, Cι.6 alkoxy or d.6 haloalkoxy), C2.6 alkenyl, C2.6 haloalkenyl, Cue cyanoalkenyl, C5.6 cycloalkenyl, aminocarbonyl(C2.6)alkenyl, Cue alkylaminocarbonyl(C1-6)alkenyl, di(C16)alkylaminocarbonyl(d_6)alkenyl, phenyl(C2 )alkenyl (wherein the phenyl group is optionally substituted by halo, nitro, cyano, d-6 alkyl, d.6 haloalkyl, d_6 alkoxy or d-6 haloalkoxy), C2.6 alkynyl, aminocarbonyl(C2-6)alkynyl, alkylaminocarbonyl (C ^alkynyl, di(C1.6)alkylaminocarbonyl(C].6)alkynyl, C3.7 cycloalkyl, C3.7 halocycloalkyl, C3.7 cyanocycloalkyl, C1-3 alkyl(C3.7)cycloalkyl, C1.3 alkyl(C3.7)halocycloalkyl, phenyl (optionally substituted by halo, nitro, cyano, d.6 alkyl, d_6 haloalkyl, d.6 alkoxy or Cι_6 haloalkoxy), heteroaryl (optionally substituted by halo, nitro, cyano, d.6 alkyl, d_ haloalkyl, d-6 alkoxy or d.6 haloalkoxy), heterocyclyl (optionally substituted by halo, nitro, cyano, Cι_6 alkyl, d.6 haloalkyl, d.6 alkoxy or d-6 haloalkoxy), d.8 alkylthio, R26O, R28R29N or R31ON=C(R27); where R26 is Cj.g alkyl or d-6 haloalkyl; R27 is phenyl (optionally substituted by halo, nitro, cyano, d_ alkyl, Cj-β haloalkyl, d.6 alkoxy or d.6 haloalkoxy), Ci-6 alkyl or d_6 haloalkyl; R28 and R29 are, independently, hydrogen, d_8 alkyl, C3.7 cycloalkyl(Cι^)alkyl, C2.6 haloalkyl, C1.6 alkoxy(C1.6)alkyl, C3.7 cycloalkyl, C3.6 alkenyl, C3_6 alkynyl or d-6 alkoxycarbonyl; and R is phenyl(C1.2)alkyl (wherein the phenyl group is optionally substituted by halo, nitro, cyano, d_6 alkyl, d-6 haloalkyl, Cue alkoxy or d.6 haloalkoxy) or d.6 alkyl. R6 is more preferably d-8 alkyl, d_8 haloalkyl, d_8 cyanoalkyl, C2.6 alkenyl, C2.6 alkynyl, C3.7 cycloalkyl, C3.7 halocycloalkyl, C3-7 cyanocycloalkyl, C1.3 alkyl(C3.7)cycloalkyl, Cι-3 alkyl(C3-7)halocycloalkyl, C5-6 cycloalkenyl, C3.7 cycloalkyl(d-6)alkyl, C5.6 cycloalkenyl(Cι.6)alkyl, C2.6 haloalkenyl, e cyanoalkenyl, d_6 alkoxy(Cι.6)alkyl, C3-6 alkenyloxy(d.6)alkyl, C3-6 alkynyloxy(d-6)alkyl, aryloxy(d-6)alkyl, Cι.6 carboxyalkyl, d.6 alkylcarbonyl(C1.6)alkyl, C2.6 alkenylcarbonyl(Ci_6)alkyl, C2_6 alkynylcarbonyl(Cι.6)alkyl, d-6 alkoxycarbonyl(C1.6)alkyl, C3.6 alkenyloxycarbonyl(d.6)alkyl, C3.6 alkynyloxycarbonyl(C1.6)alkyl, aryloxycarbonyl(Cι.6)alkyl, d.6 alkylthio(Cι.6)alkyl, Cι.6 alkylsulfιnyl(C].6)alkyl, Cue alkylsulfonyl(Cι.6)alkyl, aminocarbonyl(d.6)alkyl, aminocarbonyl(C .6)alkenyl, aminocarbonyl(C2-6)alkynyl, C)_6 alkylaminocarbonyl(Cι-6)alkyl, di(Cι-6)alkylaminocarbonyl(C1-6)alkyl, Cι.6 alkylaminocarbonyl(C1.6)alkenyl, di(Cι-6)alkylaminocarbonyl(Cι-6)alkenyl, alkylaminocarbonyl(Cι-6)alkynyl, di(Cι.6)alkylaminocarbonyl(Cι-6)alkynyl, phenyl (optionally substituted by halo, nitro, cyano,
d_6 alkyl, d.6 haloalkyl, d-6 alkoxy or d_6 haloalkoxy), phenyl(Ci-4)alkyl (wherein the phenyl group is optionally substituted by halo, nitro, cyano, Ci-6 alkyl, d_6 haloalkyl, Cι_6 alkoxy or d_6 haloalkoxy), phenyl(C2.4)alkenyl, (wherein the phenyl group is optionally substituted by halo, nitro, cyano, d_6 alkyl, d_6 haloalkyl, Ci-6 alkoxy or d_6 haloalkoxy), heteroaryl (optionally substituted by halo, nitro, cyano, d-6 alkyl, d.6 haloalkyl, d.6 alkoxy or Ci-6 haloalkoxy), heterocyclyl (wherein the heterocyclyl group is optionally substituted by halo, nitro, cyano, Cι-6 alkyl, d_6 haloalkyl, d_6 alkoxy or d_6 haloalkoxy), heteroaryl (Ci_4)alkyl (wherein the heteroaryl group is optionally substituted by halo, nitro, cyano, Cue alkyl, d_6 haloalkyl, Cue alkoxy or d_ haloalkoxy), heterocyclyl (C1.4)alkyl (wherein the heterocyclyl group is optionally substituted by halo, nitro, cyano, d_6 alkyl, Cι_6 haloalkyl, d.6 alkoxy or d-6 haloalkoxy), R26O, C,_8 alkylthio, R28R29N or R31ON=C(R27); where R26 is d.8 alkyl, d.6 haloalkyl; R27 is Cι.6 alkyl, Ci-6 haloalkyl or phenyl (optionally substituted by halo, nitro, cyano, d_ alkyl, d_6 haloalkyl, d-6 alkoxy or Cι-6 haloalkoxy); R28 and R29 are, independently, hydrogen, d_8 alkyl, C3.7 cycloalkyl, C .6 alkenyl, C3_6 alkynyl, C3_7 cycloalkyl(C1.4)alkyl, C2_6 haloalkyl, d_6 alkoxy(Cι.6)alkyl, Ci-6 alkoxycarbonyl, or R28 and R29 together with the N atom to which they are attached form a five, six or seven-membered heterocyclic ring which may contain one or two further heteroatoms selected from O, N or S and which may be optionally substituted by one or two d-6 alkyl groups; and R31 is d_6 alkyl or phenyl(Cι-2)alkyl (wherein the phenyl group is optionally substituted by halo, nitro, cyano, d_6 alkyl, d-6 haloalkyl, d_6 alkoxy or d.6 haloalkoxy); and R6 is still more preferably d.8 alkyl, Cι-8 haloalkyl, d-8 cyanoalkyl, C3.7 cycloalkyl, d.3 alkyl(C3.7)cycloalkyl, Cι_6 alkoxy(C1.6)alkyl, heterocyclyl (wherein the heterocyclyl group is optionally substituted by halo, nitro, cyano, Cι_6 alkyl, Ci-6 haloalkyl, d-6 alkoxy or d.6 haloalkoxy) or R28R29N; where R28 andR29 are, independently, Cι.8 alkyl or together with the N atom to which they are attached form a five, six or seven-membered heterocyclic ring which may contain one or two further heteroatoms selected from O, N or S and which may be optionally substituted by one or two Ci-6 alkyl groups.
It is even more preferred that R6 is Cι_8 alkyl, d-8 haloalkyl, d_8 cyanoalkyl, Cι_6 alkoxy (d_6) alkyl, C3.7 cycloalkyl, d_3 alkyl (C3_7) cycloalkyl, heterocyclyl (optionally substituted by halo, nitro, cyano, Cu alkyl, Cι.6 haloalkyl, d-6 alkoxy or d.6 haloalkoxy) or di(Cι_g)alkylamino.
It is yet more preferred that R6 is d.8 alkyl, d_8 haloalkyl, d.8 cyanoalkyl, d-6 alkoxy (d-β) alkyl, C3.7 cycloalkyl, d-3 alkyl (C3.7) cycloalkyl, heterocyclyl (optionally substituted by d_6 alkyl) or di(C1.8)alkylamino.
R6 is most preferably Cι.8 alkyl, d-g haloalkyl, Cι.8 cyanoalkyl, C3.7 cycloalkyl, d-3 alkyl(C3-7)cycloalkyl, d.6 alkoxy(C1.6)alkyl or R28R29N; where R28 and R29 are, independently, d.8 alkyl or together with the N atom to which they are attached form a five, six or seven-membered heterocyclic ring which may contain one further heteroatom selected from O, N or S and which may be optionally substituted by one or two d-6 alkyl groups. It is preferred that R12 is hydrogen, halogen, Cι-6 alkyl, Ci-6 haloalkyl, d_6 alkoxy(C1.6)alkyl, C2.6 alkenyl, d.6 alkynyl, d_6 alkoxy, d.6 haloalkoxy, d.6 alkylthio, Cι.6 haloalkylthio, d_6 alkylsulfinyl, d_6 haloalkylsulfinyl, d_6 alkylsulfonyl, d_6 haloalkylsulfonyl, cyano, nitro, formyl, d.6 alkylcarbonyl, d_6 alkoxycarbonyl, SF5 or CH=NOR32; or R1 and R12 together with the atoms to which they are attached may be joined to form a five, six or seven-membered saturated or unsaturated, carbocylic or heterocyclic ring which may contain one or two heteroatoms selected from O, N or S and which is optionally substituted by d_6 alkyl, d-6 haloalkyl or halogen; where R32 is phenyl(Cι_2)alkyl (wherein the phenyl group is optionally substituted by halo, nitro, cyano, d_6 alkyl, Ci-6 haloalkyl, d.6 alkoxy or d_6 haloalkoxy) or d-6 alkyl.
It is more preferred that R12 is hydrogen, halogen, d-6 alkyl, d_6 haloalkyl, d_6 alkoxy(C1-6)alkyl, Cue alkoxy, Cue haloalkoxy, d.6 alkylthio or SF5; or R1 and R12 together with the atoms to which they are attached form a cyclopentane or benzene ring optionally substituted by d.6 alkyl, d- haloalkyl or halogen.
R12 is even more preferably hydrogen, halogen, d.6 alkyl, d-6 haloalkyl, d_6 alkoxy, d-6 haloalkoxy, d_6 alkoxy(Cι_6)alkyl, d_6 alkylthio or SF5; or R1 and R12 together with the atoms to which they are attached form a benzene ring optionally substituted by d.6 alkyl, Cue haloalkyl or halogen; or alternatively the ring may be a cyclopentane ring.
It is further preferred that R12 is hydrogen, halogen, d.6 alkyl, Cι.6 haloalkyl, d-6 alkoxy(C1.6)alkyl, d-6 alkoxy, d_6 haloalkoxy, or R1 and R12 together with the atoms to which they are attached form a cyclopentane ring optionally substituted by d_6 alkyl, Cι_6 haloalkyl or halogen.
R12 is most preferably halogen, Cι.6 alkyl, Cι_6 haloalkyl, Ci-6 alkoxy, Cι-6 alkoxy(d-6)alkyl or d-6 haloalkoxy.
It is preferred that R13 is cyano, nitro, Cue alkyl, d_6 haloalkyl, C3.7 cycloalkyl(d-6)alkyl, C3.7 cycloalkyl, (C2.6)alkenylCH2, (C2.6)alkynylCH2, phenyl (optionally substituted by halo, nitro, cyano, d_6 alkyl, Ci- haloalkyl, d-6 alkoxy or d.6 haloalkoxy), heteroaryl (optionally substituted by halo, nitro, cyano, d.6 alkyl, d-6 haloalkyl, d.6 alkoxy or d-6 haloalkoxy), d_6 alkylcarbonyl, d_6 alkoxycarbonyl, Cu6 alkylamino, di(C1.6)alkylamino, Cue alkylcarbonylamino, d. alkoxycarbonylamino, Cue alkoxy, Cι_6 alkylthio, d-6 haloalkylthio, d.6 alkylsulfinyl, Ci-6 haloalkylsulfinyl, Ci-6 alkylsulfonyl, d_6 haloalkylsulfonyl, arylthio, arylsulfinyl, arylsulfonyl or (Cι_6)alkylcarbonyloxy.
It is preferred that R14 is hydrogen, d_8 alkyl, d-6 haloalkyl, d.6 cyanoalkyl, C2-6 alkenylCH2, C2.6 haloalkenylCH2, C2.6 alkynylCH2, C3.7 cycloalkyl, C3.7 cycloalkyl(C1.6)alkyl, Cue alkoxy(CI.6)alkyl, Cι.6 alkoxycarbonyl, Ci-6 alkylcarbonyl, Cι.6 alkylaminocarbonyl, di(Ci.6)alkylaminocarbonyl, phenyl (optionally substituted by halo, nitro, cyano, Ci-6 alkyl, Cι_6 haloalkyl, Cι_6 alkoxy or d_6 haloalkoxy) or heteroaryl (optionally substituted by halo, nitro, cyano, Cι_6 alkyl, Ci-6 haloalkyl, Ci-6 alkoxy or Cj-6 haloalkoxy).
It is more preferred that R14 is hydrogen, d-8 alkyl or d_6 haloalkyl. It is preferred that R18 is hydrogen, halogen, nitro, cyano, d_8 alkyl, d-6 haloalkyl, Ci-6 cyanoalkyl, C3.7 cycloalkyl(d-6)alkyl, Ci_6 alkoxy(Cι-6)alkyl, d-6 alkoxycarbonyl(Ci-6)alkyl, Cι.6 alkylcarbonyl(d.6)alkyl, Cι-6 alkylaminocarbonyl(Cι-6)alkyl, di(C1-6)alkylamino-carbonyl(Cι-6)alkyl, phenyl(Cι.6)alkyl (wherein the phenyl group is optionally substituted by halo, nitro, cyano, d_6 alkyl, Cι_6 haloalkyl, d- alkoxy or Cι_6 haloalkoxy), heteroaryl(Ci_6)alkyl (wherein the heteroaryl group is optionally substituted by halo, nitro, cyano, d.6 alkyl, d-6 haloalkyl, d.6 alkoxy or Cι.6 haloalkoxy), C2.6 alkenyl, C2.6 haloalkenyl, C2.6 alkynyl, C3.7 cycloalkyl, d-6 alkoxycarbonyl, Cι_6 alkylcarbonyl, d_6 alkylaminocarbonyl, di(C1.6)alkylaminocarbonyl, phenyl (optionally substituted by halo, nitro, cyano, d_6 alkyl, Ci-6 haloalkyl, d_6 alkoxy or d_6 haloalkoxy) or heteroaryl (optionally substituted by halo, nitro, cyano, Cι_6 alkyl, Ci- haloalkyl, Ci_6 alkoxy or Cι-6 haloalkoxy).
It is more preferred that R18 is hydrogen, halogen, Cι_8 alkyl or d_6 haloalkyl. The compounds in Tables 1 to 74 below illustrate compounds of the invention.
Table 1 provides 129 compounds of formula (1):
wherein R and R are as defined in Table 1.
-25
- 26
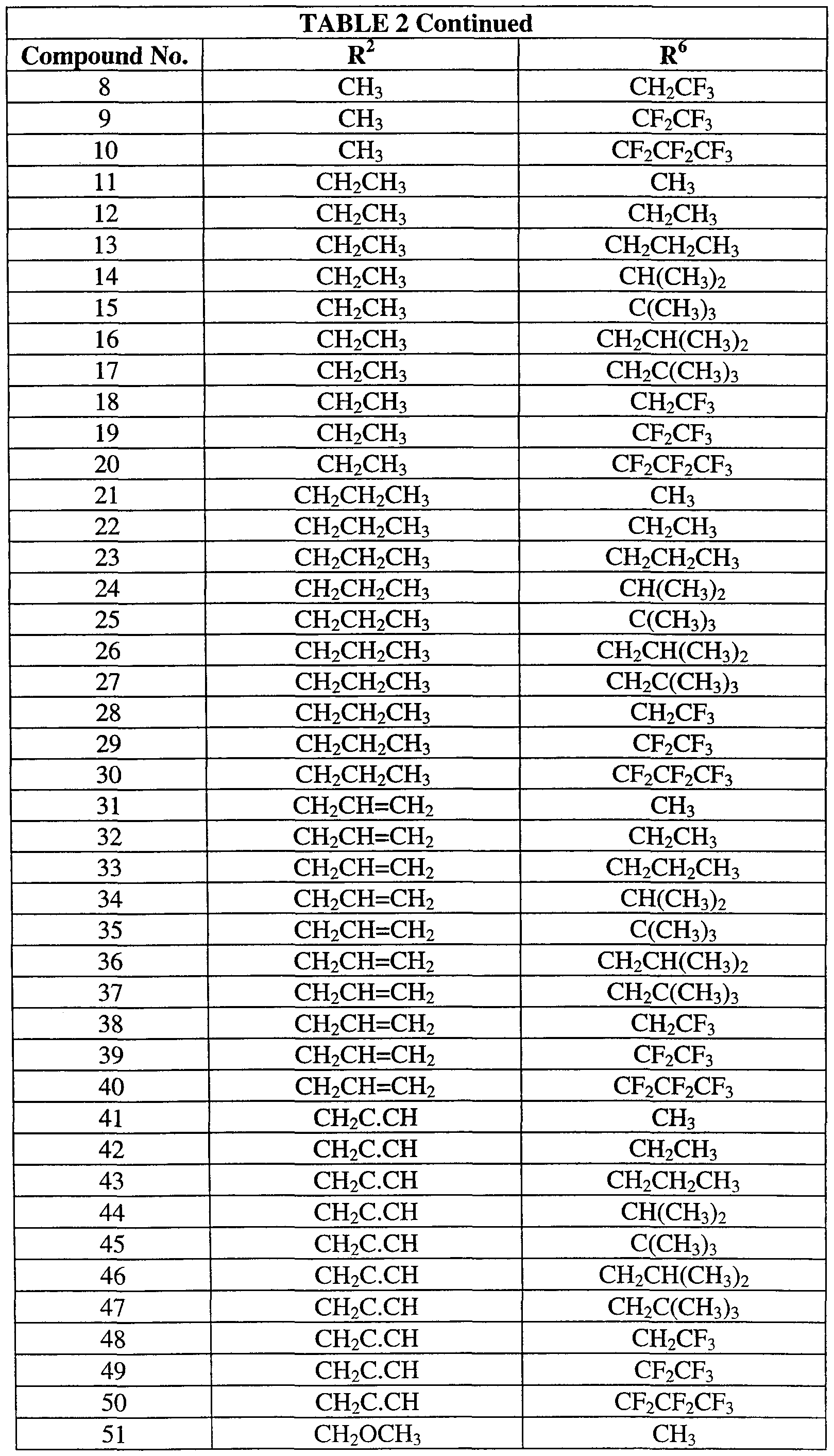
Table 2 provides 70 compounds of formula (2):
wherein R6 and R2 are as defined in Table 2.
Table 2
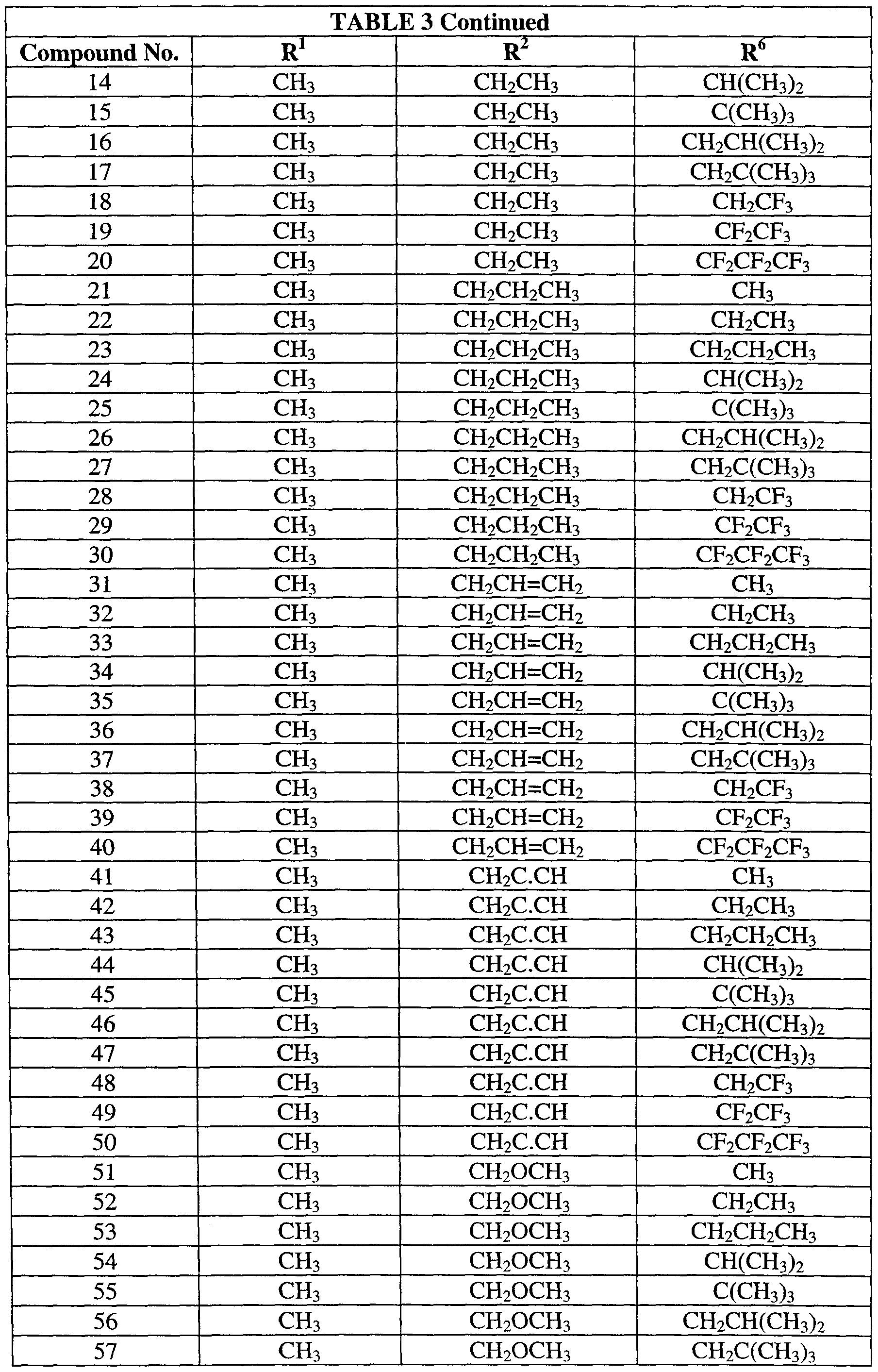
Table 3 provides 140 compounds of formula (3):
wherein R , 1 , r R>6 and R are as defined in Table 3.
Table 3
29
30
Table 4 provides 140 compounds of formula (4):
wherein R
and R are as defined in Table 3.
Table 5 provides 140 compounds of formula (5):
wherein R
1, R
6 and R
2 are as defined in Table 3.
Table 6 provides 140 compounds of formula (6):
wherein R1, R6 and R2 are as defined in Table 3.
Table 7 provides 140 compounds of formula (7):
wherein R1, R6 and R2 are as defined in Table 3.
Table 8 provides 140 compounds of formula (8):
wherein R , R and R are as defined in Table 3.
Table 9 provides 140 compounds of formula (9):
wherein R and R are as defined in Table 3.
Table 10 provides 140 compounds of formula (10):
wherein R1, R6 and R2 are as defined in Table 3 Table 11 provides 140 compounds of formula (11):
wherein R1, R6 and R2 are as defined in Table 3
Table 12 provides 140 compounds of formula (12):
1 f wherein R , R and R are as defined in Table 3
Table 13 provides 140 compounds of formula (13):
wherein R and R are as defined in Table 3
Table 14 provides 140 compounds of formula (14):
wherein R , 1 , R and R are as defined in Table 3
Table 15 provides 140 compounds of formula (15):
wherein R1, R6 and R2 are as defined in Table 3
Table 16 provides 140 compounds of formula (16):
wherein R1, R6 and R2 are as defined in Table 3
Table 17 provides 140 compounds of formula (17):
wherein R1, R6 and R2 are as defined in Table 3
Table 18 provides 140 compounds of formula (18):
wherein R
1, R
6 and R
2 are as defined in Table 3
Table 19 provides 140 compounds of formula (19):
wherein R1, R6 and R2 are as defined in Table 3.
Table 20 provides 140 compounds of formula (20):
wherein R1, R6 and R2 are as defined in Table 3. Table 21 provides 140 compounds of formula (21):
wherein R1, R6 and R2 are as defined in Table 3 Table 22 provides 140 compounds of formula (22):
wherein R and R are as defined in Table 3
Table 23 provides 140 compounds of formula (23):
wherein R , 1 , R and R are as defined in Table 3
Table 24 provides 140 compounds of formula (24):
wherein R1, R6 and R2 are as defined in Table 3
Table 25 provides 140 compounds of formula (25):
wherein R1, R6 and R2 are as defined in Table 3
Table 26 provides 140 compounds of formula (26):
wherein R ,ι , R and R are as defined in Table 3
Table 27 provides 140 compounds of formula (27):
wherein R , 1 , R and R are as defined in Table 3
Table 28 provides 140 compounds of formula (28):
wherein R , 1 , R and R are as defined in Table 3
Table 29 provides 140 compounds of formula (29):
wherein R ,ι , R and R are as defined in Table 3
Table 30 provides 140 compounds of formula (30):
wherein R1, R6 and R2 are as defined in Table 3
Table 31 provides 140 compounds of formula (31):
wherein R1, R6 and R2 are as defined in Table 3
Table 32 provides 140 compounds of formula (32):
wherein R
1, R
6 and R
2 are as defined in Table 3
Table 33 provides 140 compounds of formula (33):
wherein R1, R6 and R2 are as defined in Table 3
Table 34 provides 140 compounds of formula (34):
wherein R6 and R2 are as defined in Table 14 Table 35 provides 140 compounds of formula (35):
wherein R1, R6 and R2 are as defined in Table 3
Table 36 provides 140 compounds of formula (36):
wherein R , 1 , R and R are as defined in Table 3
Table 37 provides 140 compounds of formula (37):
wherein R
1, R
6 and R
2 are as defined in Table 3
Table 38 provides 140 compounds of formula (38):
wherein R and R are as defined in Table 3
Table 39 provides 140 compounds of formula (39):
wherein R1, R6 and R2 are as defined in Table 3
Table 40 provides 140 compounds of formula (40):
wherein R . 1 , r R>6 and R are as defined in Table 3
Table 41 provides 140 compounds of formula (41):
wherein R » 1 , r R>6 and R are as defined in Table 3
Table 42 provides 140 compounds of formula (42):
wherein R , 1 , R and R are as defined in Table 3
Table 43 provides 140 compounds of formula (43):
wherein R1, R6 and R2 are as defined in Table 3
Table 44 provides 140 compounds of formula (44):
wherein R1, R6 and R2 are as defined in Table 3
Table 45 provides 140 compounds of formula (45):
wherein R1, R6 and R2 are as defined in Table 3
Table 46 provides 140 compounds of formula (46):
wherein R6 and R2 are as defined in Table 3.
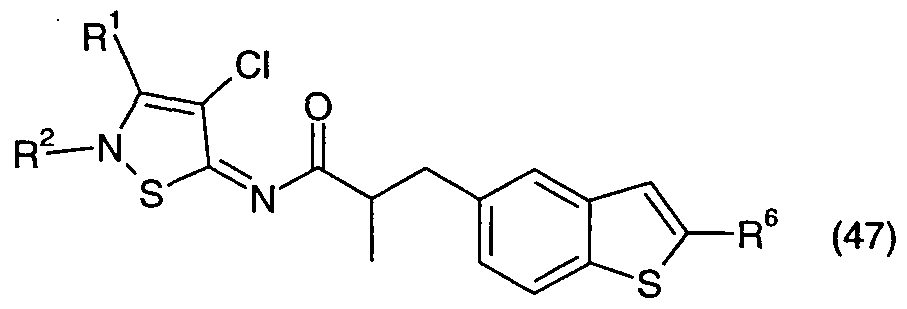
Table 47 provides 140 compounds of formula (47):
wherein R1, R6 and R2 are as defined in Table 3.
Table 48 provides 140 compounds of formula (48):
wherein R ,ι , R and R are as defined in Table 3
Table 49 provides 140 compounds of formula (49):
wherein R1, R6 and R2 are as defined in Table 3 Table 50 provides 140 compounds of formula (50):
wherein R1, R6 and R2 are as defined in Table 3
Table 51 provides 140 compounds of formula (51)
wherein R . 1 , r R>6 and R are as defined in Table 3
Table 52 provides 140 compounds of formula (52):
wherein R1, R6 and R2 are as defined in Table 3
Table 53 provides 140 compounds of formula (53):
wherein R1, R6 and R2 are as defined in Table 3
Table 54 provides 140 compounds of formula (54):
wherein R , ι , R and R are as defined in Table 3 Table 55 provides 140 compounds of formula (55):
wherein R , R and R are as defined in Table 3
Table 56 provides 140 compounds of formula (56):
wherein R , R and R are as defined in Table 3
Table 57 provides 140 compounds of formula (57):
wherein R1, R6 and R2 are as defined in Table 3
Table 58 provides 140 compounds of formula (58):
wherein R1, R6 and R2 are as defined in Table 3 Table 59 provides 140 compounds of formula (59):
wherein R .1 , r R>6 and R are as defined in Table 3
Table 60 provides 140 compounds of formula (60):
wherein R1, R6 and R2 are as defined in Table 3
Table 61 provides 140 compounds of formula (61):
wherein R6 and R2 are as defined in Table 3.
Table 62 provides 140 compounds of formula (62):
wherein R » 1 , r R>6 and R are as defined in Table 3
Table 63 provides 140 compounds of formula (63):
wherein R . 1 , r R>6 and R are as defined in Table 3
Table 64 provides 140 compounds of formula (64):
wherein R
are as defined in Table 3
Table 65 provides 140 compounds of formula (65):
wherein R , 1 , r R> 6 and R are as defined in Table 3
Table 66 provides 140 compounds of formula (66):
wherein R1, R6 and R2 are as defined in Table 3
Table 67 provides 140 compounds of formula (67):
wherein R1, R6 and R2 are as defined in Table 3
Table 68 provides 140 compounds of formula (68):
wherein R1, R6 and R2 are as defined in Table 3
Table 69 provides 140 compounds of formula (69):
wherein R1, R6 and R2 are as defined in Table 3
Table 70 provides 140 compounds of formula (70):
wherein R , 1 , r R>6 and R are as defined in Table 3
Table 71 provides 140 compounds of formula (71):
wherein R , R and R are as defined in Table 3
Table 72 provides 70 compounds of formula (72):
wherein R6 and R2 are as defined in Table 2
Table 73 provides 70 compounds of formula (73):
wherein R and R are as defined in Table 2
Table 74 provides 70 compounds of formula (74):
wherein R6 and R2 are as defined in Table 2.
Table 75 shows selected NMR data, all with CDC13, as the solvent (unless otherwise stated; if a mixture of solvents is present, this is indicated as, for example (CDC13 / dβ- DMSO)), (no attempt is made to list all characterising data in all cases) for compounds of Tables 1 to 74.
Table 75
54 -
The compounds of the invention may be made in a variety of ways. For example a compound of formula (I), which is a compound of formula (A) [wherein R1, R2, R3, R4, R5, R6, A, B, D, E and Z are as defined above in relation to a compound of formula (I)] may be made from a compound of formula (B) [wherein R1, R , R , R5, R , A, B, D, E and Z are as defined above in relation to a compound of formula (I)] by treatment with an alkylating agent (such as an alkyl halide, dialkyl sulfate or trialkyloxonium salt), an acylating agent (such as
an acid chloride) or similar reagent (such as a carbamoyl chloride, or sulfenyl chloride), optionally in the presence of a base. Frequently these reactions give rise to a mixture of a compound of formula (A), together with the isomeric product, a compound of formula (C). A compound of formula (A) may be separated from a compound of formula (C) and purified by routine techniques such as recrystallisation, chromatography or trituration with a suitable solvent.
Compound (B) Compound (A) Compound (C)
A compound of formula (A), (wherein R = alkoxyalkyl or acyloxyalkyl) may also be prepared from a compound of formula (B) by sequential reaction with formaldehyde and an alkylating or acylating agent. A compound of formula (B) [wherein B, D, E, Z, R1 R3, R4, R5, R6, are as defined above in relation to formula (I), and A is optionally substituted alkylene, alkenylene, alkynylene, alkylenoxy, alkylenamino or alkylenethio] may be prepared by reacting an amine of formula (II), [where D, E, and R1 are as defined above in relation to a compound of formula (I)] with an appropriate acid (ITJ) [where B, Z, R3, R4, R5 and R6 are as defined above in relation to formula (I), A is optionally substituted alkylene, alkenylene, alkynylene, alkylenoxy, alkylenamino or alkylenethio and X is hydroxy], preferably in the presence of a suitable coupling reagent such as 1,3-dicyclohexylcarbodiimide, 1,3- diisopropylcarbodiimide, l-(3-dimethylaminopropyl)-3-ethylcarbodiimide or 1,1'- carbonyldiimidazole), or with a suitable acid halide (lTf) [where X is halogen (especially chlorine) and where B, Z, R3, R4, R5 and R6 are as defined above in relation to formula (I), A is optionally substituted alkylene, alkenylene, alkynylene, alkylenoxy, alkylenamino or alkylenethio], acid anhydride (JIT) [where X = OC(O)alkyl and where B, Z, R3, R4, R5 and R6 are as defined above in relation to formula (I), and A is optionally substituted alkylene, alkenylene, alkynylene, alkylenoxy, alkylenamino or alkylenethio] or ester (IH) [where X = alkoxy, (especially methoxy) substituted alkoxy or aryloxy, and B, Z, R3, R4, R5 and R6 are as defined above in relation to formula (I), and A is optionally substituted alkylene, alkenylene, alkynylene, alkylenoxy, alkylenamino or alkylenethio] optionally in the presence
of a base such as triethylamine or sodium methoxide and in a suitable solvent (such as 1,1,2,2-tetrachloroethane, tetrahydrofuran, N,N-dimethylacetamide or mesitylene). Compounds (B) [wherein A is optionally substituted oxyalkylene and B, Z, R3, R4, R5 and R6 are as defined above in relation to formula (I)] may be prepared in an analogous manner starting from amine (II) and a suitable chloroformate (HI), [wherein X is chlorine, A is optionally substituted oxyalkylene and B, Z, R3, R4, R5 and R6 are as defined above in relation to formula (I)]
Compounds of formula (IT) are known compounds or may be made from known compounds by known methods.
Compounds (lTJ) may be prepared in a number of ways, the preferred method is dependent on the nature of the fused benzheterocyclic ring and on the nature of the moiety A- C(O)-X . For example, the moiety A-C(O)-X can be attached to a preformed fused heterocyclic ring:
Examples of such procedures include, but are not restricted to, (i) The coupling of a hydroxy-heterocycle of formula (IN) [wherein Z is OH and B, Z, R3, R4, R5 and R6 are as defined above in relation to formula (I)] to an alkyl halide of formula (V) (such as a haloalkyl ester, wherein X is alkoxy and A" is optionally substituted alkylene) under basic conditions to give a compound of formula (Hla):
(ii) The coupling of a suitably functionalised alkane (such as a malonate derivative), alkene (such as an acrylate) and alkyne with suitable fused heterocyclic halides (especially bromides or iodides) under transition-metal (especially Cu and Pd) mediated cross-coupling conditions. An example of this type of transformation is the reaction between an alkene (VI) (wherein X is alkoxy, A'" is optionally substituted alkylene and R
x and R
y are as defined for a substituent on alkenylene) and a halogenated heterocycle (TV) [wherein Z is chlorine or, especially, bromine or iodine and B, Z, R
3, R
4, R
5 and R are as defined above in relation to formula (I)] under Pd(0) catalysis to give a compound of formula (Hlb) :
(iii) By direct alkylation or acylation under, for example, Friedel-Craft conditions. Certain compounds of formula (HI) are amenable to modification to give further analogues. For example, a compound of formula (Hie), (wherein X is an alkoxy moiety and A is alkylene), undergo reactions typical of an aliphatic ester. Thus a compound of formula (Hie), [where J represents a single bond or suitable group (such as CH2)] may be treated with a suitable base, such as lithium diisopropylamide, sodium hydride or lithium hexamethyldisilazide in a suitable solvent such as tetrahydrofuran and then treated with an alkylating agent (such as an alkyl halide), a halogenating agent (such as an N- halosuccinimide, or N-fluorosulfonimide) or another electrophilic agent, Rf-LG, (LG designates a suitable leaving group such as a halide) to introduce a new substituent Rf . This procedure may be repeated to introduce a second substituent Rg, which may be the same or different to Rf:
Compound (Hie) As expected, a compound of formula (HI) bearing a fragment which is sufficiently chemically reactive undergoes reactions typical of that fragment. For example, a compound of formula (Hid) [wherein X is alkoxy and J represents a single bond or suitable group (such
as CH2)] will undergo certain reactions typical of an α-ketoester. Thus, a compound of formula (Hid) may be reduced by metal hydrides such as sodium borohydride in a suitable solvent such as ethanol to give the corresponding alcohol:
These compounds may be converted to a compound of formula (A) by similar procedures to those outlined previously.
The syntheses of substituted benzimidazoles, benzoxazoles and benzothiazoles from substituted benzenes are well known (see for example, Alan R. Katritzky and Charles W. Rees, Comprehensive Heterocyclic Chemistry, Vol. 6, Pergamon Press, 1984, Helmut M Hugel, Synth. Commun., 15 (12), 1075-1080, (1985), J. Scheigetz, R. Zamboni and B. Roy, Synth. Commun., 25 (18), 2791-2806, (1995), David W. Dunwell, Delme Evans, Terence A. Hicks, J. Med. Chem., 1975, 18, No. 1, 53; Abdou O. Abdelhamid, Cyril Parkanyi, S.M. Khaledur Rashid and Winston D. Lloyd, J. Heterocyclic Chem., 25, 403, (1988); Teruyuki Kondo, Sungbong Yang, Keun-Tae Huh, Masanobu Kobayashi, Shinju Kotachi and
Yoshihisa Watanabe, Chemistry Letters, 1275, 1991; Dale L. Boger, J. Org. Chem., 43, No 11, 2296, 1978) and similar processes may be utilised in the synthesis of (HI) from appropriate starting materials. Benzothiophenes may be made from appropriate thiophenols by processes similar to those described by Robert D Schuetz and Richard L Titus (J. Heterocycl. Chem., 4, No 4, 465 (1967); suitable thiophenols are known compounds or may be prepared by known methods. Benzofurans may be made from ort zo-halophenols as described by Henning Lutjens and Peter J Scammells, Tetrahedron Letters 39 (1998), 6581- 6584, Terence C Owen et al., Tetrahedron Letters 30, No 13, 1597 (1989) and Fred G Schreiber and Robert Stevenson J.C.S. Perkin 1, 90, 1977. Indoles may be made from ortho- haloanilines according to the methods of Cheng-yi Chen et al, J. Org. Chem. 1997, 62, 2676, Takao Sakamoto et al, J. Org. Chem. 1997, 62, 6507 and Alan D. Adams et al, WO9827974.
In an alternative approach to Compounds of formula (HI), the fused heterocyclic ring may be formed by ring synthesis from a suitably substituted benzene ring (VH) [wherein the
atoms or groups Q and G are suitable precursors for the formation of the desired heterocyclic rriinngg,, XX iiss aallkkooxxyy aanndd AA,, RR33,, RR44 aanndd RR55 aarree aass ddeefifinneedd i in relation to a compound of formula (I)] by processes analogous to those described above:
(NH) (HI)
This methodology may be extended to the following transformation [wherein the atoms or groups Q and G are suitable precursors for the formation of the desired heterocyclic ring, and A, D, E, R1, R3, R4 and R5 are as defined in relation to a compound of formula (I)]
ring synthesis


Compounds of formula (NH) are known compounds or may be made by known methods Heteroaryl Ν-oxides may be prepared by known methods. The compounds of formula (I) can be used to combat and control infestations of insect pests such as Lepidoptera, Diptera, Hemiptera, Thysanoptera, Orthoptera, Dictyoptera, Coleoptera, Siphonaptera, Hymenoptera and Isoptera and also other invertebrate pests, for example, acarine, nematode and mollusc pests. Insects, acarines, nematodes and molluscs are hereinafter collectively referred to as pests. The pests which may be combated and controlled by the use of the invention compounds include those pests associated with agriculture (which term includes the growing of crops for food and fibre products), horticulture and animal husbandry, companion animals, forestry and the storage of products of vegetable origin (such as fruit, grain and timber); those pests associated with the damage of man-made structures and the transmission of diseases of man and animals; and also nuisance pests (such as flies).
Examples of pest species which may be controlled by the compounds of formula (I) include: Myzus persicae (aphid), Aphis gossypii (aphid), Aphis fabae (aphid), Lygus spp. (capsids), Dysdercus spp. (capsids), Nilaparvata lugens (planthopper), Nephotettixc incticeps (leafhopper), Nezara spp. (stinkbugs), Euschistus spp. (stinkbugs), Leptocorisa spp. (stinkbugs), Frankliniella occidentalis (thrip), Thrips spp. (thrips), Leptinotarsa
decemlineata (Colorado potato beetle), Anthonomus grandis (boll weevil), Aonidiella spp. (scale insects), Trialeurodes spp. (white flies), Bemisia tabaci (white fly), Ostrinia nubilalis (European corn borer), Spodoptera littoralis (cotton leafworm), Heliothis virescens (tobacco budworm), Helicoverpa armigera (cotton bollworm), Helicoverpa zea (cotton bollworm), Sylepta derogata (cotton leaf roller), Pieris brassicae (white butterfly), Plutella xylostella (diamond back moth), Agrotis spp. (cutworms), Chilo suppressalis (rice stem borer), Locusta_ migratoria (locust), Chortiocetes terminifera (locust), Diabrotica spp. (rootworms), Panonychus ulmi (European red mite), Panonychus citri (citrus red mite), Tetranychus urticae (two-spotted spider mite), Tetranychus cinnabarinus (carmine spider mite), Phyllocoptruta oleivora (citrus rust mite), Polyphagotarsonemus lotus (broad mite),
Brevipalpus spp. (flat mites), Boophilus microplus (cattle tick), Dermacentor variabilis (American dog tick), Ctenocephalides felis (cat flea), Liriomyza spp. (leafminer), Musca domestica (housefly), Aedes aegypti (mosquito), Anopheles spp. (mosquitoes), Culex spp. (mosquitoes), Lucillia spp. (blowflies), Blattella germanica (cockroach), Periplaneta americana (cockroach), Blatta orientalis (cockroach), termites of the Mastotermitidae (for example Mastotermes spp.), the Kalotermitidae (for example Neotermes spp.), the Rhinotermitidae (for example Coptoterm.es formosanus, Reticulitermes flavipes, R. speratu, R. virginicus, R. hesperus, and R. santonensis) and the Termitidae (for example Globitermes sulphureus), Solenopsis geminata (fire ant), Monomorium pharaonis (pharaoh's ant), Damalinia spp. and Linognathus spp. (biting and sucking lice), Meloidogyne spp. (root knot nematodes), Globodera spp. and Heterodera spp. (cyst nematodes), Pratylenchus spp. (lesion nematodes), Rhodopholus spp. (banana burrowing nematodes), Tylenchulus spp. (citrus nematodes), Haemonchus contortus (barber pole worm), Caenorhabditis elegans_ (vinegar eelworm), Trichostrongylus spp. (gastro intestinal nematodes) and Deroceras reticulatum (slug).
The compounds of formula (I) are also active fungicides and may be used to control one or more of the following pathogens: Pyricularia oryz e (Magnaporthe grisea) on rice and wheat and other Pyricularia spp. on other hosts; Puccinia recondita, Puccinia striiformis and other rusts on wheat, Puccinia hordei, Puccinia striiformis and other rusts on barley, and rusts on other hosts (for example turf, rye, coffee, pears, apples, peanuts, sugar beet, vegetables and ornamental plants); Erysiphe cichoracearum on cucurbits (for example melon); Erysiphe graminis (powdery mildew) on barley, wheat, rye and turf and other
powdery mildews on various hosts, such as Sphaerotheca macularis on hops, Sphaerotheca fusca (Sphaerotheca fuliginea) on cucurbits (for example cucumber), Leveillula taurica on tomatoes, aubergine and green pepper, Podosphaera leucotricha on apples and Uncinula necator on vines; Cochliobolus spp., Helminthosporium spp., Drechslera spp. (Pyrenophora spp.), Rhynchosporium spp., Mycosphaerella graminicola (Septoria tritici) and Phaeosphaeria nodorum (Stagonospora nodorum or Septoria nodorum), Pseudocercosporella herpotrichoides and Gaeumannomyces graminis on cereals (for example wheat, barley, rye), turf and other hosts; Cercospora arachidicola and Cercosporidium personatum on peanuts and other Cercospora spp. on other hosts, for example sugar beet, bananas, soya beans and rice; Botrytis cinerea (grey mould) on tomatoes, strawberries, vegetables, vines and other hosts and other Botrytis spp. on other hosts; Alternaria spp. on vegetables (for example carrots), oil-seed rape, apples, tomatoes, potatoes, cereals (for example wheat) and other hosts; Venturia spp. (including Venturia inaequalis (scab)) on apples, pears, stone fruit, tree nuts and other hosts; Cladosporium spp. on a range of hosts including cereals (for example wheat) and tomatoes; Monilinia spp. on stone fruit, tree nuts and other hosts; Didymella spp. on tomatoes, turf, wheat, cucurbits and other hosts; Phoma spp. on oil-seed rape, turf, rice, potatoes, wheat and other hosts; Aspergillus spp. and Aureobasidium spp. on wheat, lumber and other hosts; Ascochyta spp. on peas, wheat, barley and other hosts; Stemphylium spp. (Pleospora spp.) on apples, pears, onions and other hosts; summer diseases (for example bitter rot (Glomerella cingulata), black rot or frogeye leaf spot (Botryosphaeria obtusa), Brooks fruit spot (Mycosphaerella pomϊ), Cedar apple rust (Gymnosporangiumjuniperi-virginianae), sooty blotch (Gloeodes pomigena), flyspeck (Schizothyrium pomϊ) and white rot (Botryosphaeria dothided)) on apples and pears; Plasmopara viticola on vines; other downy mildews, such as Bremia lactucae on lettuce, Peronospora spp. on soybeans, tobacco, onions and other hosts, Pseudoperonospora humuli on hops and Pseudoperonospora cubensis on cucurbits; Pythium spp. (including Pythium ultimum) on turf and other hosts; Phytophthora infestans on potatoes and tomatoes and other Phytophthora spp. on vegetables, strawberries, avocado, pepper, ornamentals, tobacco, cocoa and other hosts; Thanatephorus cucumeris on rice and turf and other Rhizoctonia spp. on various hosts such as wheat and barley, peanuts, vegetables, cotton and turf; Sclerotinia spp. on turf, peanuts, potatoes, oil-seed rape and other hosts; Sclerotium spp. on turf, peanuts and other hosts; Gibberella fujikuroi on rice; Colletotrichum spp. on a range of hosts
including turf, coffee and vegetables; Laetisaria fuciformis on turf; Mycosphaerella spp. on bananas, peanuts, citrus, pecans, papaya and other hosts; Diaporthe spp. on citrus, soybean, melon, pears, lupin and other hosts; Elsinoe spp. on citrus, vines, olives, pecans, roses and other hosts; Verticillium spp. on a range of hosts including hops, potatoes and tomatoes; Pyrenopeziza spp. on oil-seed rape and other hosts; Oncobasidium theobromae on cocoa causing vascular streak dieback; Fusarium spp., Typhula spp., Microdochium nivale, Ustilago spp., Urocystis spp., Tilletia spp. and Claviceps purpurea on a variety of hosts but particularly wheat, barley, turf and maize; Ramularia spp. on sugar beet, barley and other hosts; post-harvest diseases particularly of fruit (for example Penicillium digitatum, Penicillium italicum and Trichoderma viride on oranges, Colletotrichum musae and
Gloeosporium musarum on bananas and Botrytis cinerea on grapes); other pathogens on vines, notably Eutypa lota, Guignardia bidwellii, Phellinus igniarus, Phomopsis viticola, Pseudopeziza tracheiphila and Stereum hirsutum; other pathogens on trees (for example Lophodermium seditiosum) or lumber, notably Cephaloascus fragrans, Ceratocystis spp., Ophiostoma piceae, Penicillium spp., Trichoderma pseudokoningii, Trichoderma viride, Trichoderma harzianum, Aspergillus niger, Leptographium lindbergi and Aureobasidium pullulans; and fungal vectors of viral diseases (for example Polymyxa graminis on cereals as the vector of barley yellow mosaic virus (B YMN) and Polymyxa betae on sugar beet as the vector of rhizomania). A compound of formula (I) may move acropetally, basipetally or locally in plant tissue to be active against one or more fungi. Moreover, a compound of formula (I) may be volatile enough to be active in the vapour phase against one or more fungi on the plant. The invention therefore provides a method of combating and controlling insects, acarines, nematodes or molluscs which comprises applying an insecticidally, acaricidally, nematicidally or molluscicidally effective amount of a compound of formula (I), or a composition containing a compound of formula (I), to a pest, a locus of pest, or to a plant susceptible to attack by a pest, and a method of combating and controlling fungi which comprises applying a fungicidally effective amount of a compound of formula (I), or a composition containing a compound of formula (I), to a plant, to a seed of a plant, to the locus of the plant or seed, to soil or to any other growth medium (for example a nutrient solution). The compounds of formula (I) are preferably used against insects, acarines, nematodes or fungi.
The term "plant" as used herein includes seedlings, bushes and trees. Furthermore, the fungicidal method of the invention includes protectant, curative, systemic, eradicant and antisporulant treatments.
As fungicides, the compounds of formula (I) are preferably used for agricultural, horticultural and turfgrass purposes in the form of a composition.
In order to apply a compound of formula (I) as an insecticide, acaricide, nematicide or molluscicide to a pest, a locus of pest, or to a plant susceptible to attack by a pest, or, as a fungicide to a plant, to a seed of a plant, to the locus of the plant or seed, to soil or to any other growth medium, a compound of formula (I) is usually formulated into a composition which includes, in addition to the compound of formula (I), a suitable inert diluent or carrier and, optionally, a surface active agent (SFA). SFAs are chemicals which are able to modify the properties of an interface (for example, liquid/solid, liquid/air or liquid/liquid interfaces) by lowering the interfacial tension and thereby leading to changes in other properties (for example dispersion, emulsification and wetting). It is preferred that all compositions (both solid and liquid formulations) comprise, by weight, 0.0001 to 95%, more preferably 1 to 85%, for example 5 to 60%, of a compound of formula (I). The composition is generally used for the control of pests or fungi such that a compound of formula (I) is applied at a rate of from O.lg tolOkg per hectare, preferably from lg to 6kg per hectare, more preferably from lg to lkg per hectare. When used in a seed dressing, a compound of formula (I) is used at a rate of 0.000 lg to lOg (for example O.OOlg or 0.05g), preferably 0.005g to lOg, more preferably 0.005g to 4g, per kilogram of seed.
In another aspect the present invention provides an insecticidal, acaricidal, nematicidal, molluscicidal or fungicidal composition comprising an insecticidally, acaricidally, nematicidally, moUuscicidally or fungicidally effective amount of a compound of formula (I) and a suitable carrier or diluent therefor. The composition is preferably an insecticidal, acaricidal, nematicidal or fungicidal composition.
In a still further aspect the invention provides a method of combating and controlling pests or fungi at a locus which comprises treating the pests or fungi or the locus of the pests or fungi with an insecticidally, acaricidally, nematicidally, moUuscicidally or fungicidally effective amount of a composition comprising a compound of formula (I). The compounds of formula (I) are preferably used against insects, acarines, nematodes or fungi.
O 01/55139 „ „ PCT/GBOl/00301
- 64 -
The compositions can be chosen from a number of formulation types, including dustable powders (DP), soluble powders (SP), water soluble granules (SG), water dispersible granules (WG), wettable powders (WP), granules (GR) (slow or fast release), soluble concentrates (SL), oil miscible liquids (OL), ultra low volume liquids (UL), emulsifiable concentrates (EC), dispersible concentrates (DC), emulsions (both oil in water (EW) and water in oil (EO)), micro-emulsions (ME), suspension concentrates (SC), aerosols, fogging/smoke formulations, capsule suspensions (CS) and seed treatment formulations. The formulation type chosen in any instance will depend upon the particular purpose envisaged and the physical, chemical and biological properties of the compound of formula (I). Dustable powders (DP) may be prepared by mixing a compound of formula (I) with one or more solid diluents (for example natural clays, kaolin, pyrophyllite, bentonite, alumina, montmorillonite, kieselguhr, chalk, diatomaceous earths, calcium phosphates, calcium and magnesium carbonates, sulphur, lime, flours, talc and other organic and inorganic solid carriers) and mechanically grinding the mixture to a fine powder. Soluble powders (SP) may be prepared by mixing a compound of formula (I) with one or more water-soluble inorganic salts (such as sodium bicarbonate, sodium carbonate or magnesium sulphate) or one or more water-soluble organic solids (such as a polysaccharide) and, optionally, one or more wetting agents, one or more dispersing agents or a mixture of said agents to improve water dispersibility/solubility. The mixture is then ground to a fine powder. Similar compositions may also be granulated to form water soluble granules (SG). Wettable powders (WP) may be prepared by mixing a compound of formula (I) with one or more solid diluents or carriers, one or more wetting agents and, preferably, one or more dispersing agents and, optionally, one or more suspending agents to facilitate the dispersion in liquids. The mixture is then ground to a fine powder. Similar compositions may also be granulated to form water dispersible granules (WG).
Granules (GR) may be formed either by granulating a mixture of a compound of formula (I) and one or more powdered solid diluents or carriers, or from pre-formed blank granules by absorbing a compound of formula (I) (or a solution thereof, in a suitable agent) in a porous granular material (such as pumice, attapulgite clays, fuller's earth, kieselguhr, diatomaceous earths or ground corn cobs) or by adsorbing a compound of formula (I) (or a solution thereof, in a suitable agent) on to a hard core material (such as sands, silicates, mineral carbonates, sulphates or phosphates) and drying if necessary. Agents which are
- ΌJ -
commonly used to aid absorption or adsorption include solvents (such as aliphatic and aromatic petroleum solvents, alcohols, ethers, ketones and esters) and sticking agents (such as polyvinyl acetates, polyvinyl alcohols, dextrins, sugars and vegetable oils). One or more other additives may also be included in granules (for example an emulsifying agent, wetting agent or dispersing agent).
Dispersible Concentrates (DC) may be prepared by dissolving a compound of formula (I) in water or an organic solvent, such as a ketone, alcohol or glycol ether. These solutions may contain a surface active agent (for example to improve water dilution or prevent crystallisation in a spray tank). Emulsifiable concentrates (EC) or oil-in- water emulsions (EW) may be prepared by dissolving a compound of formula (I) in an organic solvent (optionally containing one or more wetting agents, one or more emulsifying agents or a mixture of said agents). Suitable organic solvents for use in ECs include aromatic hydrocarbons (such as alkylbenzenes or alkylnaphthalenes, exemplified by SOLVESSO 100, SOLVESSO 150 and SOLVESSO 200; SOLVESSO is a Registered Trade Mark), ketones (such as cyclohexanone or methylcyclohexanone) and alcohols (such as benzyl alcohol, furfuryl alcohol or butanol), N-alkylpyrrolidones (such as N-methylpyrrolidone or N-octylpyrrolidone), dimethyl amides of fatty acids (such as C8-Cio fatty acid dimethylamide) and chlorinated hydrocarbons. An EC product may spontaneously emulsify on addition to water, to produce an emulsion with sufficient stability to allow spray application through appropriate equipment. Preparation of an EW involves obtaining a compound of formula (I) either as a liquid (if it is not a liquid at room temperature, it may be melted at a reasonable temperature, typically below 70°C) or in solution (by dissolving it in an appropriate solvent) and then emulsifiying the resultant liquid or solution into water containing one or more SFAs, under high shear, to produce an emulsion. Suitable solvents for use in EWs include vegetable oils, chlorinated hydrocarbons (such as chlorobenzenes), aromatic solvents (such as alkylbenzenes or alkylnaphthalenes) and other appropriate organic solvents which have a low solubility in water.
Microemulsions (ME) may be prepared by mixing water with a blend of one or more solvents with one or more SFAs, to produce spontaneously a thermodynamically stable isotropic liquid formulation. A compound of formula (I) is present initially in either the water or the solvent/SFA blend. Suitable solvents for use in MEs include those hereinbefore described for use in in ECs or in EWs. An ME may be either an oil-in- water or a water-in-oil
O 01/55139 . , PCT/GBOl/00301
- 66 -
system (which system is present may be determined by conductivity measurements) and may be suitable for mixing water-soluble and oil-soluble pesticides in the same formulation. An ME is suitable for dilution into water, either remaining as a microemulsion or forming a conventional oil-in-water emulsion. Suspension concentrates (SC) may comprise aqueous or non-aqueous suspensions of finely divided insoluble solid particles of a compound of formula (I). SCs may be prepared by ball or bead milling the solid compound of formula (I) in a suitable medium, optionally with one or more dispersing agents, to produce a fine particle suspension of the compound. One or more wetting agents may be included in the composition and a suspending agent may be included to reduce the rate at which the particles settle. Alternatively, a compound of formula (I) may be dry milled and added to water, containing agents hereinbefore described, to produce the desired end product. — **" —
Aerosol formulations comprise a compound of formula (I) and a suitable propellant (for example n-butane). A compound of formula (I) may also be dissolved or dispersed in a suitable medium (for example water or a water miscible liquid, such as n-propanol) to provide compositions for use in non-pressurised, hand-actuated spray pumps.
A compound of formula (I) may be mixed in the dry state with a pyrotechnic mixture to form a composition suitable for generating, in an enclosed space, a smoke containing the compound. Capsule suspensions (CS) may be prepared in a manner similar to the preparation of
EW formulations but with an additional polymerisation stage such that an aqueous dispersion of oil droplets is obtained, in which each oil droplet is encapsulated by a polymeric shell and contains a compound of formula (I) and, optionally, a carrier or diluent therefor. The polymeric shell may be produced by either an interfacial polycondensation reaction or by a coacervation procedure. The compositions may provide for controlled release of the compound of formula (I) and they may be used for seed treatment. A compound of formula (I) may also be formulated in a biodegradable polymeric matrix to provide a slow, controlled release of the compound.
A composition may include one or more additives to improve the biological performance of the composition (for example by improving wetting, retention or distribution on surfaces; resistance to rain on treated surfaces; or uptake or mobility of a compound of formula (I)). Such additives include surface active agents, spray additives based on oils, for
example certain mineral oils or natural plant oils (such as soy bean and rape seed oil), and blends of these with other bio-enhancing adjuvants (ingredients which may aid or modify the action of a compound of formula (I)).
A compound of formula (I) may also be formulated for use as a seed treatment, for example as a powder composition, including a powder for dry seed treatment (DS), a water soluble powder (SS) or a water dispersible powder for slurry treatment (WS), or as a liquid composition, including a flowable concentrate (FS), a solution (LS) or a capsule suspension (CS). The preparations of DS, SS, WS, FS and LS compositions are very similar to those of, respectively, DP, SP, WP, SC and DC compositions described above. Compositions for treating seed may include an agent for assisting the adhesion of the composition to the seed (for example a mineral oil or a film-forming barrier).
Wetting agents, dispersing agents and emulsifying agents may be surface SFAs of the cationic, anionic, amphoteric or non-ionic type.
Suitable SFAs of the cationic type include quaternary ammonium compounds (for example cetyltrimethyl ammonium bromide), imidazolines and amine salts.
Suitable anionic SFAs include alkali metals salts of fatty acids, salts of aliphatic monoesters of sulphuric acid (for example sodium lauryl sulphate), salts of sulphonated aromatic compounds (for example sodium dodecylbenzenesulphonate, calcium dodecylbenzenesulphonate, butylnaphthalene sulphonate and mixtures of sodium di- isσpropyl- and tri-wopropyl-naphthalene sulphonates), ether sulphates, alcohol ether sulphates (for example sodium laureth-3-sulphate), ether carboxylates (for example sodium laureth-3-carboxylate), phosphate esters (products from the reaction between one or more fatty alcohols and phosphoric acid (predominately mono-esters) or phosphorus pentoxide (predominately di-esters), for example the reaction between lauryl alcohol and tetraphosphoric acid; additionally these products may be ethoxylated), sulphosuccinamates, paraffin or olefine sulphonates, taurates and lignosulphonates.
Suitable SFAs of the amphoteric type include betaines, propionates and glycinates. Suitable SFAs of the non-ionic type include condensation products of alkylene oxides, such as ethylene oxide, propylene oxide, butylene oxide or mixtures thereof, with fatty alcohols (such as oleyl alcohol or cetyl alcohol) or with alkylphenols (such as octylphenol, nonylphenol or octylcresol); partial esters derived from long chain fatty acids or hexitol anhydrides; condensation products of said partial esters with ethylene oxide; block
WO 01/55139 _ 68 . PCT/GBOl/00301
polymers (comprising ethylene oxide and propylene oxide); alkanolamides; simple esters (for example fatty acid polyethylene glycol esters); amine oxides (for example lauryl dimethyl amine oxide); and lecithins.
Suitable suspending agents include hydrophilic colloids (such as polysaccharides, polyvinylpyrrolidone or sodium carboxymethylcellulose) and swelling clays (such as bentonite or attapulgite).
A compound of formula (I) may be applied by any of the known means of applying pesticidal or fungicidal compounds. For example, it may be applied, formulated or unformulated, to the pests or to a locus of the pests (such as a habitat of the pests, or a growing plant liable to infestation by the pests) or to any part of the plant, including the foliage, stems, branches or roots, to the seed before it is planted or to other media in which plants are growing or are to be planted (such as soil surrounding the roots, the soil generally, paddy water or hydroponic culture systems), directly or it may be sprayed on, dusted on, applied by dipping, applied as a cream or paste formulation, applied as a vapour or applied through distribution or incorporation of a composition (such as a granular composition or a composition packed in a water-soluble bag) in soil or an aqueous environment.
A compound of formula (I) may also be injected into plants or sprayed onto vegetation using electrodynamic spraying techniques or other low volume methods, or applied by land or aerial irrigation systems. Compositions for use as aqueous preparations (aqueous solutions or dispersions) are generally supplied in the form of a concentrate containing a high proportion of the active ingredient, the concentrate being added to water before use. These concentrates, which may include DCs, SCs, ECs, EWs, MEs SGs, SPs, WPs, WGs and CSs, are often required to withstand storage for prolonged periods and, after such storage, to be capable of addition to water to form aqueous preparations which remain homogeneous for a sufficient time to enable them to be applied by conventional spray equipment. Such aqueous preparations may contain varying amounts of a compound of formula (I) (for example 0.0001 to 10%, by weight) depending upon the purpose for which they are to be used.
A compound of formula (I) may be used in mixtures with fertilisers (for example nitrogen-, potassium- or phosphorus-containing fertilisers). Suitable formulation types include granules of fertiliser. The mixtures suitably contain up to 25% by weight of the compound of formula (I).
WO 01/55139 ,_ PCT/GBOl/00301
- 69 -
The invention therefore also provides a fertiliser composition comprising a fertiliser and a compound of formula (I).
The compositions of this invention may contain other compounds having biological activity, for example micronutrients or compounds having similar or complementary fungicidal activity or which possess plant growth regulating, herbicidal, insecticidal, nematicidal or acaricidal activity.
By including another fungicide, the resulting composition may have a broader spectrum of activity or a greater level of intrinsic activity than the compound of formula (I) alone. Further the other fungicide may have a synergistic effect on the fungicidal activity of the compound of formula (I).
The compound of formula (I) may be the sole active ingredient of the composition or it may be admixed with one or more additional active ingredients such as a pesticide, fungicide, synergist, herbicide or plant growth regulator where appropriate. An additional active ingredient may: provide a composition having a broader spectrum of activity or increased persistence at a locus; synergise the activity or complement the activity (for example by increasing the speed of effect or overcoming repellency) of the compound of formula (I); or help to overcome or prevent the development of resistance to individual components. The particular additional active ingredient will depend upon the intended utility of the composition. Examples of suitable pesticides include the following: a) Pyrethroids, such as permethrin, cypermethrin, fenvalerate, esfenvalerate, deltamethrin, cyhalothrin (in particular lambda-cyhalothrin), bifenthrin, fenpropathrin, cyfluthrin, tefluthrin, fish safe pyrethroids (for example ethofenprox), natural pyrethrin, tetramethrin, s-bioallethrin, fenfluthrin, prallethrin or 5-benzyl-3-furylmethyl-(E)-(lR,3S)-2,2-dimethyl- 3-(2-oxothiolan-3-ylidenemethyl)cyclopropane carboxylate; b) Organophosphates, such as, profenofos, sulprofos, acephate, methyl parathion, azinphos-methyl, demeton-s-methyl, heptenophos, thiometon, fenamiphos, monocrotophos, profenofos, triazophos, methamidophos, dimethoate, phosphamidon, malathion, chlorpyrifos, phosalone, terbufos, fensulfothion, fonofos, phorate, phoxim, pirimiphos-methyl, pirimiphos-ethyl, fenitrothion, fosthiazate or diazinon; c) Carbamates (including aryl carbamates), such as pirimicarb, triazamate, cloethocarb, carbofuran, furathiocarb, ethiofencarb, aldicarb, thiofurox, carbosulfan, bendiocarb, fenobucarb, propoxur, methomyl or oxamyl;
d) Benzoyl ureas, such as diflubenzuron, triflumuron, hexaflumuron, flufenoxuron or chlorfluazuron; e) Organic tin compounds, such as cyhexatin, fenbutatin oxide or azocyclotin; f) Pyrazoles, such as tebufenpyrad and fenpyroximate; g) Macrolides, such as avermectins or milbemycins, for example abamectin, emamectin benzoate, ivermectin, milbemycin, spinosad or azadirachtin; h) Hormones or pheromones; i) Organochlorine compounds such as endosulfan, benzene hexachloride, DDT, chlordane or dieldrin; j) Amidines, such as chlordimeform or amitraz; k) Fumigant agents, such as chloropicrin, dichloropropane, methyl bromide or metam;
1) Chloronicotinyl compounds such as imidacloprid, thiacloprid, acetamiprid, nitenpyram or thiamethoxam; m) Diacylhydrazines, such as tebufenozide, chromafenozide or methoxyfenozide; n) Diphenyl ethers, such as diofenolan or pyriproxifen; o) Indoxacarb; p) Chlorfenapyr; or q) Pymetrozine.
In addition to the major chemical classes of pesticide listed above, other pesticides having particular targets may be employed in the composition, if appropriate for the intended utility of the composition. For instance, selective insecticides for particular crops, for example stemborer specific insecticides (such as cartap) or hopper specific insecticides (such as buprofezin) for use in rice may be employed. Alternatively insecticides or acaricides specific for particular insect species/stages may also be included in the compositions (for example acaricidal ovo-larvicides, such as clofentezine, flubenzimine, hexythiazox or tetradifon; acaricidal motilicides, such as dicofol or propargite; acaricides, such as bromopropylate or chlorobenzilate; or growth regulators, such as hydramethylnon, cyromazine, methoprene, chlorfluazuron or diflubenzuron).
Examples of fungicidal compounds which may be included in the composition of the invention are (E)-N-methyl-2-[2-(2,5-dimethylphenoxymethyl)phenyl]-2-methoxy- iminoacetamide (SSF-129), 4-bromo-2-cyano-N,N-dimethyl-6-trifluoromethylbenzimidazole-
1-sulphonamide, α-[N-(3-chloro-2,6-xylyl)-2-methoxyacetamido]-γ-butyrolactone, 4-chloro- 2-cyano-N,N-dimethyl-5-p-tolylimidazole-l-sulfonamide (IKF-916, cyamidazosulfamid), 3-5-dichloro-N-(3-chloro- 1 -ethyl- 1 -methyl-2-oxopropyl)-4-methylbenzamide (RH-7281 , zoxamide), N-allyl-4,5,-dimethyl-2-trimethylsilylthiophene-3-carboxamide (MOΝ65500), N- (1 -cyano- 1 ,2-dimethylpropyl)-2-(2,4-dichlorophenoxy)propionamide (AC382042),
N-(2-methoxy-5-pyridyl)-cyclopropane carboxamide, acibenzolar (CGA245704), alanycarb, aldimorph, anilazine, azaconazole, azoxystrobin, benalaxyl, benomyl, biloxazol, bitertanol, blasticidin S, bromuconazole, bupirimate, captafol, captan, carbendazim, carbendazim chlorhydrate, carboxin, carpropamid, carvone, CGA41396, CGA41397, chinomethionate, chlorothalonil, chlorozolinate, clozylacon, copper containing compounds such as copper oxychloride, copper oxyquinolate, copper sulphate, copper tallate and Bordeaux mixture, cymoxanil, cyproconazole, cyprodinil, debacarb, di-2-pyridyl disulphide 1,1 '-dioxide, dichlofluanid, diclomezine, dicloran, diethofencarb, difenoconazole, difenzoquat, diflumetorim, O,O-di-wo-propyl-S-benzyl thiophosphate, dimefluazole, dimetconazole, dimethomorph, dimethirimol, diniconazole, dinocap, dithianon, dodecyl dimethyl ammonium chloride, dodemorph, dodine, doguadine, edifenphos, epoxiconazole, ethirimol, ethyl(Z)-N-benzyl-N([methyl(methyl-thioethylideneaminooxycarbonyl)amino]thio)- -β-alaninate, etridiazole, famoxadone, fenamidone (RPA407213), fenarimol, fenbuconazole, fenfuram, fenhexamid (KBR2738), fenpiclonil, fenpropidin, fenpropimorph, fentin acetate, fentin hydroxide, ferbam, ferimzone, fluazinam, fludioxonil, flumetover, fluoroimide, fluquinconazole, flusilazole, flutolanil, flutriafol, folpet, fuberidazole, furalaxyl, furametpyr, guazatine, hexaconazole, hydroxyisoxazole, hymexazole, imazalil, imibenconazole, iminoctadine, iminoctadine triacetate, ipconazole, iprobenfos, iprodione, iprovalicarb (SZX0722), isopropanyl butyl carbamate, isoprothiolane, kasugamycin, kresoxim-methyl, LY186054, LY211795, LY248908, mancozeb, maneb, mefenoxam, mepanipyrim, mepronil, metalaxyl, metconazole, metiram, metiram-zinc, metominostrobin, myclobutanil, neoasozin, nickel dimethyldithiocarbamate, nitrothal-ώopropyl, nuarimol, ofurace, organomercury compounds, oxadixyl, oxasulfuron, oxolinic acid, oxpoconazole, oxycarboxin, pefurazoate, penconazole, pencycuron, phenazin oxide, phosetyl-Al, phosphorus acids, phthalide, picoxystrobin (ZA1963), polyoxin D, polyram, probenazole, prochloraz, procymidone, propamocarb, propiconazole, propineb, propionic acid, pyrazophos, pyrifenox, pyrimethanil, pyroquilon, pyroxyfur, pyrrolnitrin, quaternary ammonium compounds, quinomethionate,
quinoxyfen, quintozene, sipconazole (F-155), sodium pentachlorophenate, spiroxamine, streptomycin, sulphur, tebuconazole, tecloftalam, tecnazene, tetraconazole, thiabendazole, thifluzamid, 2-(thiocyanomethylthio)benzothiazole, thiophanate-methyl, thiram, timibenconazole, tolclofos-methyl, tolylfluanid, triadimefon, triadimenol, triazbutil, triazoxide, tricyclazole, tridemorph, trifloxystrobin (CGA279202), triforine, triflumizole, triticonazole, validamycin A, vapam, vinclozolin, zineb and ziram.
The compounds of formula (I) may be mixed with soil, peat or other rooting media for the protection of plants against seed-borne, soil-borne or foliar fungal diseases.
Examples of suitable synergists for use in the compositions include piperonyl butoxide, sesamex, safroxan and dodecyl imidazole.
Suitable herbicides and plant-growth regulators for inclusion in the compositions will depend upon the intended target and the effect required.
An example of a rice selective herbicide which may be included is propanil. An example of a plant growth regulator for use in cotton is PIX™. Some mixtures may comprise active ingredients which have significantly different physical, chemical or biological properties such that they do not easily lend themselves to the same conventional formulation type. In these circumstances other formulation types may be prepared. For example, where one active ingredient is a water insoluble solid and the other a water insoluble liquid, it may nevertheless be possible to disperse each active ingredient in the same continuous aqueous phase by dispersing the solid active ingredient as a suspension (using a preparation analogous to that of an SC) but dispersing the liquid active ingredient as an emulsion (using a preparation analogous to that of an EW). The resultant composition is a suspoemulsion (SE) formulation.
The invention is illustrated by the following Examples.
EXAMPLE 1
This Example illustrates the preparation of Compound No. 68 of Table No. 1. Step 1 - Preparation of N-(4-chloro-3-methylisothiazol-5-yl)-4- methoxyphenylacetamide. 4-Methoxyphenylacetic acid (33.2g, 0.2mol) and N,N-dimethylformamide (1ml) were dissolved in dichloromethane (300ml) and the mixture cooled in an ice-bath. Oxalyl chloride (27.94g, 0.22mol) was added dropwise, and once the addition was complete the cooling bath
O 01/55139 73
was removed and the mixture stirred for 3hours. The solvent was removed in vacuo, the residue taken up in xylene (300 ml) and 5-amino-4-chloro-3-methylisothiazole added. The mixture was heated under reflux for 2hours. The mixture was cooled to room temperature, and the solvent evaporated in vacuo. The residue was taken up in ethyl acetate and washed with aqueous sodium hydroxide solution. On standing, N-(4-chloro-3-methylisothiazol-5-yl)- 4-methoxyphenylacetamide, (27.3 g) precipitated from the aqueous solution.
1H NMR (CDC13) δ ppm: 2.4 (s, 3H); 3.8 (s, 2H); 3.85 (s, 3H); 6.95 (m, 2H); 7.25 (m, 2H); 8.1 (br, IH) Step 2 - Preparation of N-(4-chloro-3-methylisothiazol-5-yI)-4- hydroxyphenylacetamide
N-(4-Chloro-3-methylisothiazol-5-yl)-4-methoxyphenylacetamide (32.8g, 0.1 lmol) was dissolved in dichloromethane (300ml) and cooled to below -70°C. A solution of boron tribromide in dichloromethane (IM, 332ml, 0.332mol) was added dropwise, maintaining the internal temperature below -65°C. When the addition was complete, the mixture was allowed to warm to room temperature, and stirring continued for 3 hours. With ice-bath cooling, methanol (100 ml) cautiously was added, and the mixture stirred for several minutes. The solvent was removed in vacuo, methanol added, the mixture stirred, and the solvent evaporated once more. The residue was partitioned between ethyl acetate and saturated aqueous sodium bicarbonate solution. The organic extract was washed with brine, dried over magnesium sulfate, filtered and the filtrate evaporated in vacuo. The residue was triturated with hexane to afford N-(4-chloro-3-methylisothiazol-5-yl)-4-hydroxyphenylacetamide (28 g) as a solid.
1H NMR (Mixture ύ?6-DMSO / CDC13) δ ppm: 2.35 (s, 3H); 3.8 (s, 2H); 6.85 (m, 2H); 7.15 (m, 2H); 8.65 (br, 1); 9.1 (br, IH). Step 3 - Preparation of N-(4-chloro-3-methylisothiazol-5-yl)-4-hydroxy-3- nitrophenylacetamide
The phenol (28 g, 0.099 mol) obtained in Step 2 above was suspended in ethanol (200 ml) and ferric nitrate nonahydrate (40.0 g, 0.99 mol) added. The mixture was stirred at 50 °C for 90 minutes, then cooled to room temperature and the solvent removed in vacuo. The residue was partitioned between 2M aqueous hydrochloric acid and ethyl acetate. The organic extract was washed sequentially with 2M aqueous hydrochloric acid and brine, dried over magnesium sulfate, filtered and the filtrate evaporated in vacuo. Trituration with hexane
WO 01/55139 _ 7 PCT/GBOl/00301
afforded N-(4-chloro-3-methylisothiazol-5-yl)-4-hydroxy-3-nitrophenylacetamide (30.12 g) as a solid.
1H NMR (Mixture d6-DMSO / CDC13) δ ppm: 2.4 (s, 3H); 3.9 (s, 2H); 7.1 (d, IH);
7.65 (dd, IH); 8.15 (d, IH); 11.0 (s, IH). Step 4 - Preparation of N-(4-chloro-3-methyl-5-isothiazolyl)-3-amino-4-hydroxyphenyl- acetamide
N-(4-Chloro-3-methylisothiazol-5-yl)-4-hydroxy-3-nitrophenylacetamide (37.4 g,
0.114 mol) was hydrogenated at 15 bar over a 3% platinum on carbon catalyst (15 g) in N,N- dimethylformamide (180 ml). Once the reduction was complete, the catalyst was removed by filtration and the filtrate evaporated in vacuo to give N-(4-chloro-3-methylisothiazol-5-yl)-3- amino-4-hydroxyphenylacetamide (26.8 g) as a pale brown solid, used without further purification in the next step.
1H NMR (d6-DMSO) δ ppm: 2.25 (s, 3H); 3.6 (s, 2H); 6.3 (dd, IH); 6.5 (m, 2H); 8.9
(br, lH); 11.5 (s, IH) Step 5 - Preparation of N-(4-chloro-3-methylisothiazol-5-yl)-3-(2,2,2- trifluoropropionamido)-4-hydroxyphenylacetamide
To a stirred solution of N-(4-chloro-3-methylisothiazol-5-yl)-[3-amino-4- hydroxyphenylj-acetamide (11.2g) in dry N,N-dimethylacetamide (lOOmL) containing l-(3- dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (8.6g) at ambient temperature was added dropwise 3,3,3-trifluoropropionic acid (4.8g) over 0.3h. The mixture was stirred for a further 2h, poured onto ice then stirred for lh. The solid which had precipitated was filtered from solution, washed with cold water then dried to give N-(4-chloro-3-methylisothiazol-5- yl)-3-(2,2,2-trifluoropropionamido)-4-hydroxyphenylacetamide as a colourless solid, 14.6g, m.p. 184-189°C. 1H NMR -DMSO) δ: 2.25 (s, 3H); 3.60 (q, 2H); 3.75 (s, 2H); 6.78 (m, IH); 6.87
(m, IH); 7.80 (br, IH); 9.48 (br, IH); 9.82 (br, IH); 11.60 (s, IH).
Step 6 - Preparation of N-(4-chloro-3-methylisothiazol-5-yl)-[2-(2,2,2-trifluoroethyl)- benzoxazol-5-yl]acetamide.
N-(4-chloro-3-methylisothiazol-5-yl)-3-(2,2,2-trifluoropropionamido)-4-hydroxy- phenylacetamide (14.4g) was suspended in 1,1,2,2-tetrachloroethane (140 mL) containing jβr -toluenesulfonic acid (1.68g) and the mixture was heated to reflux with stirring for 2h.
The reaction was cooled and the solvent evaporated under reduced pressure. The residue
was treated with an aqueous solution of sodium chloride then extracted with ethyl acetate. The organic extracts were combined, dried (magnesium sulfate) and the solvent evaporated under reduced pressure. The residue was fractionated by chromatography (silica, hexane/ethyl acetate 2: 1 by volume) to give N-(4-chloro-3-methylisothiazol-5-yl)-[2-(2,2,2- trifluoroethyl)benzoxazol-5-yl]acetamide as a pale yellow solid, 7.75g, m.p. 154-155°C.
1H NMR (CDC13) δ: 2.40 (s, 3H); 3.85 (q, 2H); 4.00 (s, 2H); 7.40 (dd, IH); 7.61 (dd, IH); 7.71 (d, IH); 8.15 (br, IH).
Step 7 - Preparation of N-(4-chloro-2-ethoxymethyl-3-methylisothiazolin-5-ylidene)-[2- (2,2,2-trifluoroethyI)benzoxazol-5-yl]acetamide N-(4-chloro-3-methylisothiazol-5-yl)-[2-(2,2,2-trifluoroethyl)benzoxazol-5- yl]acetamide (1.50 g) in dry dichloromethane (15 mL) was stirred at ambient temperature under an atmosphere of nitrogen and N,O-bis(trimethylsilyl)acetamide (0.995 mL) was added. The solution was stirred for 15 minutes then chloromethyl ethyl ether (0.620 mL) was added. The reaction mixture was stirred for an additional 48 h, diluted with further dichloromethane then washed with saturated aqueous sodium bicarbonate solution. The organic phase was separated, dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated under reduced pressure to give a red oil which was fractionated by chromatography to give the desired product as a colourless solid, 0.34g, m.p. 148.5-149.5°C.
EXAMPLE 2 This Example illustrates the preparation of Compound No. 18 of Table No. 1.
Potassium carbonate (0.414 g) and N-(4-chloro-3-methylisothiazol-5-yl)-[2-(2,2,2- trifluoroethyl)benzoxazol-5-yl]acetamide (0.770 g) were heated together in refluxing methyl isobutyl ketone (10 ml) for 30 minutes. Ethyl iodide (2. 5 g) was added and the mixture heated at reflux overnight. The mixture was cooled to room temperature, poured into water, neutralised and extracted with ethyl acetate. The organic extract was dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. The residue was further purified by flash column chromatography, eluting with ethyl acetate : hexane 2:1, and then with ethyl acetate to give the desired product (0.054 g) as a yellow-orange solid, m.p. 133-135°C.
EXAMPLE 3 This Example illustrates the preparation of Compound No. 37 of Table No. 1. Step 1 - Preparation of 5-amino-4-chloro-3-methylisothiazole
5-Amino-3-methylisothiazole hydrochloride (250g, 1.66 moles) was suspended in dichloromethane(1.25 L) and stirred at 8 °C. Sulfuryl chloride (146.8ml, 1.83 moles) was added dropwise over 1 hour, and during this addition the temperature of the reaction mixture was maintained between 10 and 15 °C. As the sulfuryl chloride was added the suspension dissolved and a dark oil began to fall out of solution. The resultant two-phase mixture was stirred at 10°C for 15 minutes. The mixture was cooled to below 10 °C and quenched by careful addition of aqueous potassium carbonate solution (367.3g, 2.66 moles, of potassium carbonate in 1 L of water). The two phases were separated and the aqueous layer extracted with dichloromethane (600ml+400ml). The combined organic layers were dried over anhydrous magnesium sulfate, filtered and the filtrate concentrated in vacuo. The residue was slurried in hexane (~500ml) for 1 hour, filtered and dried to give 5-amino-4-chloro-3-methyl- isothiazole as a red-brown-solid (228.7g, 93%), m.p. 69-71°C. 1H NMR (CDC13) δ: 2.30 (s, 3H); 4.6 (br, 2H). Step 2 - Preparation of methyl (4-hydroxyphenyl)acetate
Hydrogen chloride was bubbled through a solution of (4-hydroxyphenyl)acetic acid (25 g , 0.16 mol) in methanol (100 ml) at room temperature. An exotherm resulted in the solution refluxing for about ten minutes. The mixture was allowed to cool to room temperature and the solvent evaporated in vacuo to afford methyl (4-hydroxyphenyl)acetate as a yellow oil (27.5 g) which crystallised on seeding, m.p. 46-52°C.
1H NMR (CDCI3) δ: 3.57(s,2H); 3.71(s,3H); 6.0(b,lH); 6.76(m,2H); 7.10(m,2H). Step 3 - Preparation of methyl (4-hydroxy-3-nitrophenyI)acetate Nitric acid (69% by weight, 16M, 20 ml) was added dropwise to a solution of methyl
(4-hydroxyphenyl)acetate (50.0 g, 0.3 mol) in acetic acid (500 ml), maintaining the temperature of the reaction below 15 °C by external cooling (an induction period was observed for this reaction). Once GC analysis confirmed that the reaction was complete, the mixture was carefully quenched into water (2 L.) with vigorous stirring. An emulsion was formed which subsequently crystallised. After filtration, the solid was washed with water and dried to give methyl (4-hydroxy-3-nitrophenyl)acetate (49.4 g) as a yellow powder.
WO 01/55139 _ 7? _ PCT/GB l
1H NMR (CDCI3) δ: 3.63 (s, 2H); 3.72 (s, 3H); 7.14 (d,lH); 7.52 (dd, IH); 8.02 (d, IH); 10.5 (s, IH). Step 4 Preparation of methyl (3-amino-4-hydroxyphenyl)acetate Methyl (4-hydroxy-3-nitrophenyl)acetate (48.9 g, 0.23 mol) and 5% palladium on carbon were suspended in methanol and the resulting mixture hydrogenated until all the starting material was consumed. The reaction mixture was filtered to remove the catalyst and the filter-cake was washed with methanol. The combined filtrate and washings were concentrated in vacuo, affording methyl (3-amino-4-hydroxyphenyl)acetate as a solid (41.0g).
1H NMR (d6-OMSO) δ: 3.51ppm (s, 2H), 4.45 (b, 2H); 6.20 (dd, IH); 6.40 (d, IH); 6.49 (d, IH); 8.87 (br, IH). Step 5 Preparation of methyl [3-(2,2-dimethylpropionamido)-4-hydroxyphenyl]acetate_ Sodium bicarbonate (19 g, 0.23 mol) was suspended in 1,2-dimethoxyethane (180ml), and methyl (3-amino-4-hydroxyphenyl)acetate (26.3 g, 0.145 mol) added. To this mixture was added, dropwise, a solution of tert-butylacetyl chloride in 1,2-dimethoxyethane (45ml), over 2 hours. Once the addition was complete the mixture was stirred at room temperature for 1 hour. The mixture was filtered, the inorganic solid washed with ethyl acetate (3x 50ml) and the filtrate and washings combined and concentrated. Trituration with hexane gave methyl [3-(2,2-dimethylpropionamido)-4-hydroxyphenyl]acetate (40.1 g) as an off-white solid, m.p. 112 - 113°C.
1H NMR (CDC13):1.1 (s, 9H,); 2.30 (s, 2H); 3.51 (s, 2H); 3.70 (s, 3H); 6.9-7.0 (m, 3H); 7.55 (br, IH); 8.85 (br, IH). Step 6
Preparation of methyl [2-(2,2-dimethylpropyl)benzoxazoI-5-yl]acetate
Pαr -toluenesulphonic acid (1.5g) in toluene (120ml) was stirred and heated to reflux with a Dean & Stark assembly fitted to remove water. After 1 hour at reflux, the solution cooled to ~80°C and methyl [3-(2,2-dimethylpropionamido)-4-hydroxyphenyl]acetate (20.0g, 0.07mol) was added portionwise. The reaction mixture was then heated at reflux for 6 hours, cooled, diluted with hexane (200ml) and filtered through a plug of silica gel, eluting with
ethyl acetate. The filtrate was evaporated in vacuo to give methyl [2-(2,2- dimethylpropyl)benz-oxazol-5-yl]acetate (17.5g) as an oil. 1H NMR (CDC13): 1.1 (s, 9H); 2.8 (s, 2H); 3.7 (s, 3H); 3.74 (s, 2H); 7.4 (m, 3H). Step 7 Preparation of [2-(2,2-dimethylpropyl)benzoxazoI-5-yl]acetic acid
Methyl [2-(2,2-dimethylpropyl)-5-benzoxazolyl]acetate (5.00 g, 0.0185 mol) was dissolved in methanol (5 ml), and a solution of sodium hydroxide (0.8 lg, 0.0204 mol) in water (5 ml) was added slowly over 20 minutes, maintaining the temperature below 25 °C by external cooling. Once the addition was complete, the mixture allowed to stir at room temperature for 1 hour. The reaction mixture was poured slowly into water (50 ml), and concentrated hydrochloride acid was added until the pH of the mixture was below pH 6. The mixture was stirred for 1 hour, filtered, the solid washed thoroughly with water and dried. Trituration with hexane gave [2-(2,2-dimethylpropyl)benzoxazol-5-yl]acetic acid (4.51 g) as a white solid, m.p. 108-109°C. 1H NMR (CDC13): 1.05 (s, 9H); 2.80 (s, 2H); 3.77 (s, 2H); 7.42 (m, 3H)
Step 8
Preparation of N-(4-chIoro-3-methylisothiazoI-5-yl)-[2-(2,2-dimethylpropyl)- benzoxazol-5-yl]acetamide
[2-(2,2-dimethylpropyl)benzoxazol-5-yl]acetic acid (0.800 g, 0.003 mol) was suspended in dichloromethane (10 ml) and one drop of N,N-dimethylformamide and oxalyl chloride (0.451 g, 0.004 mol) added sequentially. The mixture was stirred for 2 hours, and then the solvent removed in vacuo. The residue was taken up in xylene (10 ml), 5-amino-4- chloro-3-methylisothiazole (0.829 g, 0.006 mol) added and the mixture heated under reflux for 2 hours. The mixture was cooled to room temperature, diluted with ethyl acetate and washed with brine. The organic solution was dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. The residue was further purified by flash column chromatography on silica gel, eluting with a 2: 1 mixture of hexane : ethyl acetate, to give N- (4-chloro-3-methylisothiazol-5-yl)-[2-(2,2-dimethylpropyl)benzoxazol-5-yl]acetamide (0.325 g) as a pale orange solid, m.p. 144-145 °C. 1H NMR (CDCI3) δ ppm: 1.1 (s, 9H); 2.35 (s, 3H); 2.85 (s, 2H); 3.95 (s, 2H); 7.25
(dd, IH); 7.55 (d, IH); 7.65 (d, IH); 8.1 (br, IH)
Step 9
Preparation of N-(2-allyl-4-chloro-3-methylisothiazolin-5-yIidene)-(2-[2,2-dimethyl- propyI]benzoxazol-5-yl)acetamide
N-(4-chloro-3-methylisothiazol-5-yl)-(2-[2,2-dimethylpropyl]benzoxazol-5- yl)acetamide (0.755g) in methyl isobutyl ketone (lOmL) containing anhydrous potassium carbonate (0.414g) was stirred and heated to reflux for 0.5h then cooled to ambient temperature. Allyl bromide (1.94g) and potassium iodide (0.664g) were added and the mixture heated to reflux for 17h then cooled to ambient temperature. The reaction was poured into water and extracted with ethyl acetate (three times), washed with water, dried (magnesium sulfate) then evaporated under reduced pressure. The dark brown residue was fractionated by chromatography (silica; hexane /ethyl acetate 2:1 to neat ethyl acetate) to give the desired product, 0.176g, m.p.l43-145°C.
EXAMPLE 4 This Example illustrates the preparation of Compound No. 61 of Table No. 1. N-(4-chloro-3-methylisothiazol-5-yl)-(2-methylbenzoxazol-5-yl)acetamide (0.64 g) in dry tetrahydrofuran (10 mL) was stirred at ambient temperature under an atmosphere of nitrogen and treated with lithium bis(trimethylsilyl)amide (2.2mL of IM solution in tetrahydrofuran ). The mixture was stirred for 30 minutes at 50°C then cooled to ambient temperature. Ethyl chloromethyl ether ( 0.62 g) was added then the reaction stirred for 3 h. The reaction mixture was poured into water and extracted with ethyl acetate (twice). The extracts were combined, dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated under reduced pressure to give a brown oil. The oil was fractionated by chromatography (silica; hexane/ethyl acetate 2: 1 to ethyl acetate) to the desired product, 0.079g, m.p. 96-97°C. EXAMPLE 5
This Example illustrates the preparation of Compound No. 68 of Table No. 3. Step l
Preparation of methyl 4-methoxyphenylacetate A solution of methyl 4-hydroxyphenylacetate (25. Og, 0.147mol) in tetrahydrofuran (50ml) was added dropwise to a stirred suspension of sodium hydride (4.45g of an 80% dispersion in oil, 0.147 mol) in tetrahydrofuran (150 ml) and the mixture stirred for 90 minutes. A solution
of methyl iodide (20.9 g, 0.47 mol) in tetrahydrofuran (50 ml) was added dropwise and the mixture stirred overnight at room temperature.
The solvent was removed in vacuo, and the residue partitioned between water and ethyl acetate. The organic extract was washed with brine, dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. The residue was purified by chromatography on silica gel, eluting with ethyl acetate : hexane 1:9 to give methyl 4-methoxyphenylacetate (22.0 g) as a colourless oil.
1H NMR (CDC13) δ: 3.57(s,2H); 3.68(s,3H); 3.8(s,3H); 6.87(m,2H); 7.20(m,2H). Step 2 Preparation of methyl 2-(4-methoxyphenyl)propionate
A solution of lithium diisopropylamide (2.0M solution in tetrahydrofuran /ethyl benzene / heptane, 34.0ml, 0.0667mol) was added dropwise to a solution of methyl 4- methoxyphenyl-acetate (12.0g, 0.0667 mol) in tetrahydrofuran (150ml) at -70°C under a nitrogen atmosphere and the mixture stirred at -70 °C for lhour. A solution of methyl iodide (9.5 g, 0.0667mol) in tetrahydrofuran (20ml) was added dropwise and the mixture was stirred at -70°C for a further lhour. The cooling bath was removed and the mixture allowed to warm to room temperature overnight.
The reaction was quenched with water, acidified with dilute aqueous hydrochloric acid and extracted with ethyl acetate. The organic extract was washed with brine, dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. The residue was purified by flash column chromatography on silica gel, eluting with ethyl acetate : hexane 1 :9 to give methyl 2-(4-methoxyphenyl)propionate (8.8g) as a colourless oil.
1H NMR (CDC13) δ: 1.48(d,3H); 3.67(s,3H); 3.69(q,lH); 3.79(s,3H); 6.86(m,2H); 7.22 (m, 2H). Step 3
Preparation of N-(4-chloro-3-methyIisothiazol-5-yl)-2-(4-methoxyphenyl)propionamide 5-Amino-4-chloro-3-methylisothiazole (7.4 g, 0.050 mol) was added to a suspension of sodium methoxide (6.1 g, 0.112 mol) in tetrahydrofuran (160 ml) and the mixture stirred at room temperature for 20 minutes. A solution of methyl 2-(4-methoxyphenyl)propionate (8.8 g, 0.045 mol) in tetrahydrofuran (40 ml) was added dropwise, and the mixture stirred for 3hours at room temperature.
The reaction was quenched with water, acidified with dilute aqueous hydrochloric acid and extracted with ethyl acetate. The organic extract was washed with brine, dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. The residue was purified by flash column chromatography on silica gel, eluting with ethyl acetate : dichloromethane 2.5 : 97.5 to give N-(4-chloro-3-methylisothiazol-5-yl)-2-(4- methoxyphenyl)propionamide (12.5 g) as a colourless solid.
1H NMR (CDC13) δ: 1.63 (d, 3H); 2.35 (s, 3H); 3.83 (s, 3H); 3.83 (q, IH); 6.95 (m, 2H); 7.28 (m, 2H); 7.98 (b, IH). Step 4 Preparation of N-(4-chloro-3-methyIisothiazol-5-yI)-2-(4-hydroxyphenyl)propionamide Boron tribromide (1.0 M solution in dichloromethane, 110 ml, 0.11 mol) was added dropwise to a stirred solution of N-(4-chloro-3-methylisothiazol-5-yl)-2-(4-methoxyphenyl)- propionamide (12.5 g, 0.040 mol) in dichloromethane (200 ml) at -70 °C. Once the addition was complete the cooling bath was removed and the mixture allowed to warm to room temperature overnight. The mixture was cooled to 0 °C and excess methanol added cautiously. The solvent was evaporated in vacuo, and the residue partitioned between ethyl acetate and brine. The organic layer was dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo to give N-(4-chloro-3-methylisothiazol-5-yl)-2-(4- hydroxyphenyl)-propionamide (11.8 g) used without further purification in the next step. 1H NMR (CDCI3) δ: 1.62 (d, 3H); 2.38 (s, 3H); 3.83 (q, IH); 5.51 (b, IH); 6.90 (m, 2H); 7.23 (m, 2H); 7.98 (b, IH). Step 5
Preparation of N-(4-chloro-3-methylisothiazol-5-yl)-2-(4-hydroxy-3-nitrophenyl)- propionamide Ferric nitrate nonahydrate (16.16 g, 0.04 mol) was added to a solution of N-(4-chloro-
3-methylisothiazol-5-yl)-2-(4-hydroxyphenyl)proρionamide (11.8 g, 0.04 mol) in ethanol (100 ml) and the mixture stirred and warmed at 50 °C for 2 Vz hours. The mixture was cooled to room temperature and the solvent removed in vacuo. The residue was partitioned between 2M aqueous hydrochloric acid and ethyl acetate and the organic phase dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. Purification by flash column chromatography on silica gel, eluting with ethyl acetate : dichloromethane 5 : 95 gave N-(4-
chloro-3-methylisothiazol-5-yl)-2-(4-hydroxy-3-nitrophenyl)propionamide (11.1 g) as a yellow solid.
1H NMR (CDC13) δ: 1.66 (d, 3H); 2.38 (s, 3H); 3.93 (q, IH); 7.21 (d, IH); 7.65 (dd, IH); 8.10 (d, IH); 8.41 (b, IH); 10.55 (s, IH) Step 6
Preparation of N-(4-chloro-3-methylisothiazol-5-yl)-2-(3-amino-4-hydroxyphenyl)- propionamide
A mixture of N-(4-chloro-3-methylisothiazol-5-yl)-2-(4-hydroxy-3- nitrophenyl)propionamide (11.0 g, 0.0322 mol)and 3% platinum on carbon in N,N- dimethylformamide (100 ml) was hydrogenated at 15 bar for 6 hours at room temperature. The catalyst was removed by filtration and the filtrate evaporated in vacuo to give N-(4- chloro-3-methylisothiazol-5-yl)-2-(3-amino-4-hydroxyphenyl)propionamide (9.0 g) as an off- white solid, m.p. 222-223 °C. 1H NMR ( -DMSO / CDCI3) δ: 1.08 (d, 3H); 1.96 (s, 3H); 2.90 (b, 2H); 3.62 (q, IH); 6.14 (dd, IH); 6.30 (m, 2H); 8.33 (b, IH); 10.1 (b, IH) Step 7
Preparation of N-(4-chloro-3-methylisothiazol-5-yl)-2-[3-(3,3,3-trifluoropropionamido)- 4-hydroxyphenyl]propionamide
A mixture of N-(4-chloro-3-methylisothiazol-5-yl)-2-(3-amino-4- hydroxyphenyl)propionamide (0.65 g, 0.002 mol), 3,3,3-trifluoropropionic acid (0.267 g, 0.002 mol) and l-(3-dimethyl-aminopropyl)-3-ethylcarbodiimide hydrochloride (0.40 g, 0.002 mol) were stirred together in N,N-dimethylacetamide (8 ml) at room temperature for 7 hours, and then allowed to stand at room temperature overnight. The reaction was quenched with water, and extracted with ethyl acetate. The organic extract was washed with water, dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. The residue was purified by flash column chromatography on silica gel, eluting with ethyl acetate : hexane 1 : 1 to give N-(4-chloro-3-methylisothiazol-5-yl)-2-[3-(3,3,3- trifluoropropionamido)-4-hydroxyphenyl]propionamide ( 0.62g) as a white solid.
Η NMR (CDC13) δ: 1.54 (d, 3H); 2.37 (d, 3H); 3.36 (q, 2H); 3.96 (IH); 6.98 (d, IH); 7.04 (dd, IH); 7.86 (d, IH)
- o -
Step 8
Preparation of N-(4-chloro-3-methylisothiazol-5-yl)-2-[2-(2,2,2-trifluoroethyl)- benzoxazoI-5-yl]propionamide
A mixture of N-(4-chloro-3-methylisothiazol-5-yl)-2-[3-(3,3,3- trifluoropropionamido)-4-hydroxyphenyl]propionamide (0.58 g, 0.0014 mol) and para- toluenesulfonic acid (0.02 g) in 1,1,2,2-tetrachloroethane was heated at reflux for 26 hours. The mixture was cooled to room temperature and the solvent evaporated in vacuo. The residue was purified by flash column chromatography on silica gel, eluting with ethyl acetate : hexane 35 : 65 to give N-(4-chloro-3-methylisothiazol-5-yl)-2-[2-(2,2,2- trifluoroethyl)benzoxazol-5-yl]propionamide (0.255 g) as a pale yellow solid, m.p. 148- 150°C
1H NMR (CDC13) δ: 1.70 (d, 3H); 2.38 (s, 3H); 3.84 (q, 2H); 4.04 (q, IH); 7.40 (dd, IH); 7.62 (d, IH); 7.76 (d, IH); 8.05 (b, IH). Step 9 Preparation of N-(4-chloro-2-ethoxymethyl-3-methylisothiazolin-5-yIidene)-2-[2-(2,2,2- trifluoroethyl)benzoxazol-5-yl]propionamide
A solution of N,O-bis(trimethylsilyl)acetamide (0.141 g, 0.0007 mol) in dichloromethane (1 ml) was added to a solution of N-(4-chloro-3-methylisothiazol-5-yl)-2- [2-(2,2,2-trifluoroethyl)benzoxazol-5-yl]propionamide (0.236 g, 0.0006 mol) in dichloromethane (4 ml) and the mixture stirred at room temperature for 15 minutes.
Chloromethylethyl ether (0.11 g, 0.001 mol) was added and stirring was continued for 24 h. Further quantities of N,O-bis(trimethylsilyl)acetamide (0.141 g, 0.0007 mol) and chloromethylethyl ether (0.11 g, 0.001 mol) were added and stirring continued for 6 hours. The reaction was quenched with saturated aqueous sodium bicarbonate solution and extracted with ethyl acetate. The organic extract was washed with brine, dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. Purification by flash column chromatography on silica gel, eluting initially with ethyl acetate : hexane 1 : 4 and then with ethyl acetate : hexane 45 : 55 gave the desired product (0.14 g) as a pale yellow oil.
EXAMPLE 6 This Example illustrates the preparation of Compound No. 70 of Table No. 3. Step 1 Preparation of methyl (3-heptafluorobutyramido-4-hydroxyphenyl)acetate Sodium bicarbonate (51 g, 0.607 mol) was suspended in 1,2-dimethoxyethane
(180ml), and methyl (3-amino-4-hydroxyphenyl)acetate (58.26 g, 0.32 mol) added, followed by a further quantity of 1,2-dimethoxyethane (75ml) was added. To this mixture was added, dropwise, a solution of heptafluorobutyryl chloride (112.5g, 0.48mol) in 1,2-dimethoxyethane (140ml), at such a rate that the reaction temperature was maintained at 19-20°C. Once the addition was complete the mixture was stirred at room temperature for 90minutes. The reaction mixture was filtered, the solid taken up in with ethyl acetate and washed with saturated aqueous bicarbonate solution and brine. The organic phase was dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. The residue was taken up in acetone (450 ml) and water (450 ml) added. The mixture was stirred for 1 Vi hours, then the solvent decanted from an oil and a further quantity (250 ml) of water added. The mixture was stirred for 1 hour, during which time the product desired product crystallised. The product was collected by filtration, washed with water and dried to give methyl [3-heptafluorobutyramido)-4-hydroxyphenyl]acetate (87.0g).
1H NMR (^-DMSO / CDC13) δ: 3.56 (s, 2H); 3.69 (s, 3H); 6.95 (m, 2H); 8.1 (d, IH); 8.83 (b, IH); 9.51 (s, IH) Step 2 Preparation of methyl (2-heptafluoropropyIbenzoxazol-5-yl)acetate
A mixture of methyl [3-heptafluorobutyramido)-4-hydroxyphenyl]acetate (20.0 g, 0.053 mol) and αrα-toluenesulfonic acid (1.71 g) in toluene (150 ml) was heated at reflux (using a Dean-Stark apparatus) for 24 hours. The mixture was cooled to room temperature and the mixture diluted with ethyl acetate (150 ml). The mixture was washed with saturated aqueous sodium bicarbonate solution and brine, dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. The residue was purified by column chromatography on silica gel, eluting with dichloromethane to give methyl (2- heptafluoropropylbenzoxazol-5-yl)acetate (12.9 g) as an off-white solid.
Η NMR (CDCI3) δ: 3.71 (s,3H); 3.8 (s,2H); 7.48 (dd,lH); 7.63 (d,lH); 7.8 (d,lH).
Step 3
Preparation of methyl 2-(2-heptafluoropropylbenzoxazol-5-yl)propionate
A solution of lithium diisopropylamide (2.0 M solution in tetrahydrofuran /ethyl benzene / heptane, 15.32 ml, 0.0306 mol) was added dropwise to a solution of methyl (2- heptafluoropropylbenzoxazol-5-yl)acetate (11.0 g, 0.0306 mol) in tetrahydrofuran (175 ml) at -70 °C under a nitrogen atmosphere and the mixture stirred at -70 °C for 1 hour. A solution of methyl iodide (39.1 g, 0.275 mol) in tetrahydrofuran (25 ml) was added dropwise and the mixture was stirred at -70 °C for a further 1 hour. The cooling bath was removed and the mixture allowed to warm to room temperature over a period of 3 hours. The reaction was quenched with water, acidified with dilute aqueous hydrochloric acid and extracted with ethyl acetate. The organic extract was washed with brine, dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo to give methyl 2- (2-heptafluoropropylbenzoxazol-5-yl)propionate (11.6 g) as a reddish oil, used without further purification in the next step. 1H NMR (CDC13) δ: 1.56 (d, 3H); 3.67 (s, 3H); 3.9 (q, IH); 7.5 (dd, IH); 7.62 (d,
IH); 7.82 (d, IH). Step 4 Preparation of 2-(2-heptafluoropropylbenzoxazol-5-yl)propionic acid
A mixture of methyl 2-(2-heptafluoropropylbenzoxazol-5-yl)propionate (11.6 g, 0.0311 mol), hexamethyldisilane (6.81 g, 0.047 mol) and iodine ( 11.85 g, 0.047 mol) were stirred together in refluxing toluene (110 ml) for 6 hours. The mixture was cooled to room temperature, diluted with ethyl acetate and washed sequentially with water, saturated aqueous sodium thiosulfate solution and brine. The organic phase was dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. The residue was purified by column chromatography on silica gel, eluting initially with dichloromethane and then with ethyl acetate : dichloromethane 1 : 4 to give 2-(2-heptafluoropropylbenzoxazol-5-yl)propionic acid (6.3 g).
1H NMR (CDCI3) δ: 1.6 (d, 3H); 3.9 (q, IH); 7.52 (dd, IH); 7.64 (d, IH); 7.87 (d, IH) Step 5
Preparation of N-(4-chloro-3-methylisothiazol-5-yl)-2-[2-(heptafluoropropyl)- benzoxazol-5-yI]propionamide
_ ,
- 86 -
Oxalyl chloride (4.46 g, 0.035 mol) was added dropwise to a solution of 2-(2- heptafluoro-propylbenzoxazol-5-yl)propionic acid (6.3 g, 0.0176 mol) in dichloromethane (60 ml) and the mixture stirred at room temperature overnight. The solvent was evaporated in vacuo, and the residue dissolved in 1,2-dichloroethane (70 ml) and warmed to reflux. A solution of 5-amino-4-chloro-3-methylisothiazole (3.12 g, 0.021 mol) in 1,2-dichloroethane (30 ml) was added dropwise over 30 minutes to the refluxing mixture, and the mixture was heated at reflux for a further 5 hours. The mixture was cooled to room temperature, the solid collected by filtration, washed with 1 ,2-dichloroethane and diethyl ether and dried to give N- (4-chloro-3-methyl-isothiazol-5-yl)-2-[2-(heptafluoropropyl)benzoxazol-5-yl]propionamide (6.4 g)
1H NMR (CDC13) δ: 1.72 (d, 3H); 2.39 (s, 3H); 4.06 (m, IH); 7.58 (dd, IH); 7.71 (d, IH); 7.91 (d, IH); 8.1 (b, IH) Step 6
Preparation of N-(4-chloro-2-ethoxymethyl-3-methylisothiazolin-5-ylidene)-2-[2- heptafluoropropy lbenzoxazol-5-y 1] propionamide
A solution of N,O-bis(trimethylsilyl)acetamide (0.50 g, 0.00245 mol) was added to a solution of N-(4-chloro-3-methylisothiazol-5-yl)-2-[2-heptafluoropropylbenzoxazol-5- yl]propionamide (1.00 g, 0.002 mol) in dichloromethane (10 ml) and the mixture stirred at room temperature for 15 minutes. Chloromethylethyl ether (0.386 g, 0.004 mol) was added and stirring was continued for 48hours. The reaction mixture was diluted with dichloromethane and poured into water. The organic phase was washed with brine, dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. The residue was purified by flash column chromatography on silica gel, eluting initially with ethyl acetate : hexane 4: 1 and subsequently with a gradient elution to ethyl acetate : hexane 1 : 1 to give the desired product (0.159 g), m.p. 101-102°C.
EXAMPLE 7 This Example illustrates the preparation of Compound No. 67 of Table No. 10. Step l Preparation of methyl [2-(2,2-dimethylpropyl)benzoxazol-5-yl]fluoroacetate A solution of lithium diisopropylamide (2.0 M solution in tetrahydrofuran /ethyl benzene / heptane, 3.85 ml, 0.0077 mol) was added dropwise to a solution of methyl [2-(2,2- dimethylpropyl)benzoxazol-5-yl]acetate (2.0 g, 0.0077 mol) in tetrahydrofuran (40 ml) at -78
__
- 87 -
°C under a nitrogen atmosphere and the mixture stirred at -78 °C for 1 hour. A solution of N- fluorobenzenesulfonimide (2.42 g, 0.0077 mol) in tetrahydrofuran (10 ml) was added dropwise and the mixture was stirred at -70 °C for a further 1 hour. The cooling bath was removed and the mixture allowed to warm to room temperature over a period of 20hours. The reaction mixture was diluted with water, acidified with dilute aqueous hydrochloric acid and extracted with ethyl acetate. The organic extracts were combined, washed with water, dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. The residue was purified by flash column chromatography on silica gel, eluting with ethyl acetate : dichloromethane 5 : 95, and further purified by column chromatography on silica gel, eluting with ethyl acetate : hexane 1 : 3 to give methyl [2-(2,2-dimethylpropyl)benzoxazol-5- yl]-fluoroacetate (1.4 g).
1H NMR (CDC13) δ: 1.08 (s, 9H); 2.83 (s, 2H); 3.79 (s, 3H); 5.90 (d, IH); 7.43 (dd, IH); 7.53 (d, IH); 7.80 (d, IH) Step 2 Preparation of N-(4-chloro-3-methylisothiazol-5-yl)-[2-(2,2-dimethylpropyl)- benzoxazol-5-yI]fluoroacetamide
5-Amino-4-chloro-3-methylisothiazole (0.41 g, 0.0028 mol) was added to a suspension of sodium methoxide (0.34 g, 0.0063 mol) in tetrahydrofuran (5 ml) and the mixture stirred at room temperature form V2 hour. A solution of methyl [2-(2,2- dimethylpropyl)-benzoxazol-5-yl]fluoroacetate (0.70 g, 0.0025 mol) in tetrahydrofuran (3 ml) was added dropwise, and the mixture stirred at room temperature for 3 days. The mixture was diluted with water, neutralised with saturated aqueous ammonium chloride solution and extracted with ethyl acetate. The organic extracts were combined, dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. The residue was purified by flash column chromatography on silica gel, eluting with ethyl acetate : hexane 1 : 1 to give N-(4-chloro-3-methylisothiazol-5-yl)-[2-(2,2-dimethylpropyl)benzoxazol-5- yl]fluoro- acetamide (0.48 g).
1H NMR (CDCI3) δ: 1.08 (s, 9H); 2.43 (s, 3H); 2.83 (s, 2H); 6.18 (d, IH); 7.43 (dd, IH); 7.56 (d, IH); 7.82 (d, IH); 9.08 (b, IH) Step 3
Preparation of N-(4-chloro-2-ethoxymethyI-3-methylisothiazolin-5-ylidene)- [2-(2,2- dimethylpropyl)benzoxazoI-5-yl]fluoroacetamide
O 01 55139
A solution of N,O-bis(trimethylsilyl)acetamide (0.219 g, 0.00108 mol) was added to a solution of N-(4-chloro-3-methylisothiazol-5-yl)-[2-(2,2-dimethylpropylbenzoxazol-5-yl]- fluoroacetamide (0.35 g, 0.0009 mol) in dichloromethane (4 ml) and the mixture stirred at room temperature for 20 minutes. Chloromethylethyl ether (0.167 g, 0.0018 mol) was added and stirring was continued for 72hours. The reaction mixture was quenched by addition of saturated aqueous sodium bicarbonate solution and extracted with ethyl acetate. The organic phase was washed with brine, dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. T.l.c analysis the reaction to be incomplete, so the above procedure repeated, with stirring for 48 (rather than 72) hours. The reaction mixture was worked up as before, and the residue purified by flash column chromatography on silica gel, eluting initially with ethyl acetate : hexane 9 : 31 and subsequently with ethyl acetate : hexane 1 : 1 to give the desired product (0.065 g) as a colourless solid, m.p. 145-146 °C
EXAMPLE 8 This Example illustrates the preparation of Compound No. 94 of Table No. 1. Di-tert-amyl dicarbonate (0.184 ml, 0.00075 mol) was added to a solution of N-(4- chloro-3-methylisothiazol-5-yl)-(2-[2,2-dimethylpropyl]benzoxazol-5-yl)acetamide (0.189 g, 0.0005 mol) and 4-dimethylaminopyridine (0.006 g) in acetonitrile and the mixture stirred at room temperature for 3 h. The solvent was removed in vacuo, and the residue taken up in dichloromethane and washed sequentially with saturated aqueous potassium hydrogensulfate solution, saturated aqueous sodium bicarbonate solution and brine. The organic phase was dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. Trituration with ether gave the desired product (0.1158 g) as a white solid, m.p. 146.8- 147.1°C.
EXAMPLE 9 This Example illustrates the preparation of Compound No. 67 of Table No. 22.
Step 1 - Preparation of N-(2-hydroxy-5-bromophenyl)-3,3-dimethylbutyramide
A solution of tert-butylacetyl chloride (2.7 g, 0.020 mol) in diethyl ether (20 ml) was added dropwise to a solution of 2ramino-4-bromophenol (3.8 g , 0.020 mol) and triethylamine (2.1 g, 0.020 mol) in diethyl ether (160 ml) and the mixture stirred at room temperature for 3 h. The mixture was filtered and the filtrate evaporated in vacuo.
Purification by flash column chromatography on silica gel, eluting initially with ethyl acetate
: dichloromethane 1.5 : 98.5 and then with ethyl acetate : dichloromethane 2.5 : 97.5 gave N- (2-hydroxy-5-bromophenyl)-3,3-dimethylbutyramide (1.8g).
1H NMR (CDC13) δ: 1.12 (s, 9H); 2.32 (s, 2H); 6.90 (d, IH); 7.17 (d, IH); 7.22 (dd, IH); 7.32 (b, IH); 8.69 (s, IH). Step 2 - Preparation of 2-(2,2-dimethylpropyl)-5-bromobenzoxazole
A mixture of N-(2-hydroxy-5-bromophenyl)-3,3-dimethylbutyramide (1.75 g, 0.006 mol) and pαra-toluenesulfonic acid (0.05 g) were heated together in refluxing 1,1,2,2- tetrachloroethane (40 ml) for 24 h. The mixture was cooled to room temperature, the solvent removed in vacuo and the residue purified by flash column chromatography on silica gel, eluting with ethyl acetate : hexane 5 : 95 to give 2-(2,2-dimethylpropyl)-5- bromobenzoxazole (1.38 g).
1H NMR (CDCI3) δ: 1.08 (s, 9H); 2.22 (s, 2H); 7.38 (d, IH); 7.41 (dd, IH); 7.82 (d, IH) Step 3 Preparation of ethyl 3-[2-(2,2-dimethylpropyl)benzoxazol-5-yI]propenoate
A mixture of 2-(2,2-dimethylpropyl)-5-bromobenzoxazole (0.60 g, 0.00225 mol), ethyl acrylate (1.08 g, 0.0108 mol), palladium acetate (0.051 g, 0.00023 mol), tή-o- tolylphosphine (0.135 g, 0.0045 mol) and N,N-diisopropylethylamine (0.585 g, 0.0045 mol) were heated together at 100 °C for 6 h. The mixture was cooled to room temperature, diluted with water and extracted with ethyl acetate. The organic extracts were dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. The residue was purified by flash column chromatography on silica gel, eluting with ethyl acetate : dichloromethane 3 : 97 to give ethyl 3-[2-(2,2-dimethylpropyl)benzoxazol-5-yl]propenoate (0.634 g) 1H ΝMR (CDC13) δ: 1.10 s, 9H); 1.35 (t, 3H); 2.83 (s, 2H); 4.29 (q, 2H); 6.45 (d, IH); 7.50 (s, 2H); 7.80 (d, IH); 7.85 (s, IH). Step 4 Preparation of ethyl 3-[2-(2,2-dimethylpropyl)benzoxazoI-5-yl]propanoate
A solution of ethyl 3-[2-(2,2-dimethylpropyl)benzoxazol-5-yl]propenoate (0.500 g, 0.00174 mol) in ethanol (30 ml) was hydrogenated at 4.0 bar over 5% Pd on C for 4 hours at room temperature and then at 5.5 bar for 6 hours. The catalyst was removed by filtration and the filtrate evaporated in vacuo. The residue was purified by flash column chromatography
on silica gel, eluting with ethyl acetate : dichloromethane 3 : 97 to give ethyl 3-[2-(2,2- dimethylpropyl)benzoxazol-5-yl]propanoate (0.465 g).
1H NMR (CDC13) δ: 1.08 (s, 9H); 1.24 (t, 3H); 2.67 (t, 2H); 2.80 (s, 2H); 3.06 (t, 2H); 4.14 (q, 2H); 7.14 (dd, IH); 7.40 (d, IH); 7.51 (d, IH) Step 5 - Preparation of N-(4-chloro-3-methylisothiazol-5-yl)-3-[2-(2,2-dimethyl- propyl)benzoxazol-5-yl]propionamide
5-Amino-4-chloro-3-methylisothiazole (0.225 g, 0.00152 mol) was added to a suspension of sodium methoxide (0197 g, 0.00365 mol) in tetrahydrofuran (8 ml) and the mixture stirred at room temperature form 20 minutes. A solution of ethyl 3-[2-(2,2- dimethylpropyl)benzoxazol-5-yl]propanoate (0.400 g, 0.00138 mol) in tetrahydrofuran (2 ml) was added dropwise, and the mixture stirred at room temperature for 20hours. The mixture was diluted with water, acidified with saturated aqueous ammonium chloride solution and extracted with ethyl acetate. The organic extracts were combined, washed with brine, dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. The residue was purified by flash column chromatography on silica gel, eluting with ethyl acetate : dichloromethane 15 : 85 to give N-(4-chloro-3-methylisothiazol-5-yl)-3-[2-(2,2-dimethyl- propyl)benzoxazol-5-yl]propionamide (0.49 g) as a white solid, m.p. 170-171 °C
1H NMR (CDCI3) δ: 1.08 (s, 9H); 2.38 (s, 3H); 2.80 (s, 2H); 2.90 (t, 2H); 3.29 (t, 2H); 7.17 (dd, IH); 7.41 (d, IH); 7.52 (d, IH); 8.34 (b, IH). Step 6
Preparation of N-(4-chIoro-2-ethoxymethyl-3-methylisothiazolin-5-ylidene)-3-[2-(2,2- dimethylpropyl)benzoxazol-5-yl]propionamide
A solution of N,O-bis(trimethylsilyl)acetamide (0.27 g, 0.00135 mol) in dichloromethane (1 ml) was added to a solution of N-(4-chloro-3-methylisothiazol-5-yl)-3- [2-(2,2-dimethyl-propyl)benzoxazol-5-yl]propionamide (0.44 g, 0.00112 mol) in dichloromethane (6 ml) and the mixture stirred at room temperature for 15 minutes. Chloromethylethyl ether (0.212 g, 0.00224 mol) was added and stirring was continued for 20hours. The reaction mixture was partitioned between saturated aqueous sodium bicarbonate solution and ethyl acetate. The organic phase was washed with brine, dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. The residue was purified by flash column chromatography on silica gel, eluting initially with ethyl acetate :
O 01/55139 _ 91 _ PCT/GBOl/00301
dichloromethane 1 : 9 and then with ethyl acetate : dichloromethane 3 : 7 to give the desired product (0.045 g) as a white solid, m.p. 118-119°C.
EXAMPLE 10 This Example illustrates the preparation of Compound No. 67 of Table No. 19. Step 1 - Preparation of N-(2-hydroxy-5-methoxyphenyl)-3,3-dimethylbutyramide
A solution of tert-butylacetyl chloride (2.9 g, 0.0216 mol) in diethyl ether (10 ml) was added dropwise to a solution of 2-amino-4-methoxyphenol (3.0 g , 0.0216 mol) and triethylamine (2.18 g, 0.0216 mol) in diethyl ether (165 ml) and the mixture stirred at room temperature for 2 h. The mixture was filtered and the filtrate evaporated in vacuo. Recrystallisation (ethyl acetate) gave N-(2-hydroxy-5-methoxyphenyl)-3,3- dimethylbutyramide.
1H NMR (CDC13) δ: 1.12 (s, 9H); 2.22 (s, 2H); 3.76 (s, 3H); 6.62 (d, IH); 6.72 (dd, IH); 6.96 (d, IH); 7.40 (b, IH); 8.03 (b, IH)
Step 2 - Preparation of 2-(2,2-dimethyIpropyl)-5-methoxybenzoxazole A mixture of N-(2-hydroxy-5-methoxyphenyl)-3,3-dimethylbutyramide (2.85 g, 0.012 mol) and pαrα-toluenesulfonic acid (0.05 g) were heated together in refluxing 1,1,2,2- tetrachloroethane (50 ml) for 6 h. The mixture was cooled to room temperature, the solvent removed in vacuo and the residue purified by column chromatography on silica gel, eluting with ethyl acetate : hexane 12.5 : 87.5 to give 2-(2,2-dimethylpropyl)-5-bromobenzoxazole (2.40 g).
1H NMR (CDCI3) δ: 1.08 (s, 9H); 2.79 (s, 2H); 3.83 (s, 3H); 6.89 (dd, IH); 7.18 (d, IH); 7.37 (d, IH) Step 3 - Preparation of 2-(2,2-dimethylpropyl)-5-hydroxybenzoxazole
Boron tribromide (1.0 M solution in dichloromethane, 35 ml, 0.035 mol) was added dropwise to a solution of 2-(2,2-dimethylpropyl)-5-methoxybenzoxazole (2.2 g, 0.010 mol) in dichloromethane at -78 °C and the mixture stirred at -78 °C for 1 hour, and then allowed to warm to room temperature overnight. The reaction mixture was quenched by careful addition of methanol, the solvent evaporated in vacuo and the residue partitioned between ethyl acetate and brine. The organic phase was dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo to give 2-(2,2-dimethylpropyl)-5- hydroxybenzoxazole (2.05 g) as a pale brown solid.
1H NMR (CDCI3) δ: 1.07 (s, 9H); 2.77 (s, 2H); 6.83 (dd, IH); 7.13 (d, lh); 7.28 (d, lh); 8.79 (s, IH)
Step 4 - Preparation of methyl [2-(2,2-dimethylpropyI)benzoxazol-5-yl]oxyacetate and [2-(2,2-dimethylpropyl)benzoxazol-5-yl]oxyacetic acid A mixture of 2-(2,2-dimethylpropyl)-5-hydroxybenzoxazole (1.94 g, 0.00946 mol), potassium carbonate (1.31 g, 0.00946 mol) and methyl bromoacetate (1.45 g, 0.00946 mol) in N,N-dimethylformamide (20 ml) were heated together at 110 °C overnight. The solvent was removed in vacuo, the residue taken up in ethyl acetate and washed with saturated aqueous sodium bicarbonate solution. The organic phase was dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vαcwø..The residue was triturated with ethyl acetate : hexane 1 : 3, the solid removed by filtration and the filtrate concentrated in vacuo to give methyl [2-(2,2-dimethylpropyl)benzoxazol-5-yl]oxyacetate (0.50 g).
1H NMR (CDCI3) δ: 1.08 (s, 9H); 2.80 (s, 2H); 3.82 (s, 3H); 4.68 (s, 2H(; 6.97 (dd, IH); 7.15 (d, lH); 7.39 (d, lH) The aqueous washings were acidified using concentrated hydrochloric acid and extracted with ethyl acetate. The organic extract was dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo to give [2-(2,2-dimethylpropyl)benzoxazol-5-yl]oxyacetic acid (0.80 g) as a dark brown solid.
1H NMR (CDCI3) δ 1.08 (s, 9H); 2.82 (s, 2H); 4.74 (s, 2H); 7.03 (dd, IH); 7.25 (d, IH); 7.40 (d, IH); 11.58 (b, IH).
Step 5 - Preparation of N-(4-chloro-3-methyIisothiazol-5-yl)-[2-(2,2- dimethylpropyl)benzoxazol-5-yl]oxyacetamide
A solution of oxalyl chloride (0.28g, 0.0022 mol) in tetrahydrofuran (3 ml) was added dropwise to a mixture of [2-(2,2-dimethylpropyl)benzoxazol-5-yl]oxyacetic acid (0.326 g, 0.0022 mol) and N,N-dimethylformamide (1 drop) in tetrahydrofuran (25 ml) and the mixture stirred at room temperature for 2 h. To this mixture was added carefully a solution of 5-amino-4-chloro-3-methylisothiazole (0.326 g, 0.0022 mol) and triethylamine (0.225 g, 0.0022 mol) in tetrahydrofuran and stirring was continued at room temperature for a further 21/2hours. The mixture was filtered and the filtrate evaporated in vacuo. The residue was purified by column chromatography on silca gel, eluting with ethyl acetate : dichloromethane 1 : 9 to give N-(4-chloro-3-methylisothiazol-5-yl)-[2-(2,2-dimethylpropyl)benzoxazol-5- yl]oxyacetamide as a white solid, m.p. 179-181 °C
1H NMR (CDCI3) δ: 1.09 (s, 9H); 2.45 (s, 3H); 2.81 (s, 2H); 4.81 (s, 2H); 7.03 (dd, IH); 7.28 (d, IH); 7.47 (d, lh); 9.26 (b, IH).
Step 6 - Preparation of N-(4-chloro-2-ethoxymethyl-3-methylisothiazolin-5-ylidene)-3- [2-(2,2-dimethylpropyl)benzoxazol-5-yl]oxyacetamide A solution of N,O-bis(trimethylsilyl)acetamide (0.155 g, 0.00135 mol) in dichloromethane (1 ml) was added to a solution of N-(4-chloro-3-methylisothiazol-5-yl)-[2- (2,2-dimethylpropyl)-benzoxazol-5-yl]oxyacetamide (0.250 g, 0.000635 mol) in dichloromethane (4 ml) and the mixture stirred at room temperature for 20 minutes. A solution of chloromethylethyl ether (0.120 g, 0.00127 mol) in dichloromethane (1 ml) was added and stirring was continued for 22hours. The reaction mixture was partitioned between saturated aqueous sodium bicarbonate solution and ethyl acetate, and the organic phase dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. The residue was taken up in dichloromethane (4 ml), a solution of N,O-bis(trimethylsilyl)acetamide (0.155 g, 0.00135 mol) in dichloromethane (1 ml) added and the mixture stirred for 20 minutes at room temperature. A solution of chloro-methylethyl ether (0.120 g, 0.00127 mol) was added and stirring was continued for 24hours. The reaction mixture was partitioned between saturated aqueous sodium bicarbonate solution and ethyl acetate, and the organic phase dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. The residue was purified by flash column chromatography on silica gel, eluting initially with ethyl acetate : hexane 1 : 3 and then with ethyl acetate : hexane 1 : 1 to the desired product (0.100 g) as a white solid, m.p. 122-124°C.
EXAMPLE 11 This Example illustrates the preparation of Compound No. 66 of Table No. 51. Step 1 - Preparation of methyl 4-hydroxy-3-iodophenylacetate Potassium iodate (3.9 g, 0.018 mol) and iodine (9.2 g, 0.036 mol) were added to a solution of methyl 4-hydroxyphenylacetate (15.0 g, 0.090 mol) in glacial acetic acid (340 ml) and water (90 ml) at 5 °C and the mixture stirred at 5 °C for 1 h. The cooling bath was removed and the mixture allowed to warm to room temperature and stirred for a further 20 h. The solvent was evaporated in vacuo and the residue partitioned between diethyl ether and water. The organic phase was washed sequentially with saturated aqueous sodium bicarbonate solution and saturated sdium hydrogensulfate solution, dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. The residue was purified by
flash column chromatography on silica gel, eluting initially with ethyl acetate : dichloromethane 1 : 99 and then with ethyl acetate : dichloromethane 3 : 97 to give methyl 4- hydroxy-3-iodophenyl-acetate (11.6g).
1H NMR (CDC13) δ 3.52 (s, 2H); 3.70 (s, 3H); 5.44 (s, IH); 6.90 (d, IH); 7.14 (dd, IH); 7.58 (d, IH).
Step 2 - Preparation of methyl [2-(2-methylpropyl)benzofuran-5-yl]acetate
A mixture of methyl 4-hydroxy-3-iodophenyl-acetate (0.600 g, 0.002055 mol), 4- methylpent-1-yne (0.169 g, 0.002055 mol), cuprous oxide (0.185 g, 0.00128 mol) and pyridine (7 ml) was placed in a sealed tube under an atmosphere of nitrogen and heated to 120 °C for 20hours. The mixture was cooled to room temperature, filtered and the filtrate evaporated in vacuo. The residue was purified by flash column chromatography on silica gel, eluting with ethyl acetate : hexane 1 : 9 to give methyl [2-(2-methylpropyl)benzofuran-5- yl] acetate (0.43g).
1H NMR (CDCI3) δ 0.98 (d, 6H); 2.10 (m, IH); 2.62 (d, 2H); 3.68 (s, 3H); 3.68 (s, 2H); 6.33 (s, IH); 7.10 (dd, IH); 7.35 (d, IH; 7.38 (d, IH)
Step 3 - Preparation of N-(4-chloro-3-methylisothiazol-5-yl)-[2-(2- methylpropyl)benzofuran-5-yl]acetamide
5-Amino-4-chloro-3-methylisothiazole (0.246 g, 0.00165 mol) was added to a suspension of sodium methoxide (0.210 g, 0.0039 mol) in tetrahydrofuran (6 ml) and the mixture stirred at room temperature for 20 minutes. A solution of methyl [2-(2- methylpropyl)benzofuran-5-yl]acetate (0.370 g, 0.0015 mol) in tetrahydrofuran (4 ml) was added dropwise and the mixture stirred at room temperature for 20 h. The mixture was diluted with water, acidifed with saturated aqueous ammonium chloride solution and extracted with ethyl acetate. The organic extract was dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. The residue was purified by flash column chromatography on silica gel, eluting with ethyl acetate : dichloromethane 2 : 98 to give N-(4-chloro-3-methylisothiazol-5-yl)-[2-(2-methylpropyl)benzofuran-5-yl]acetamide (0.535 g) as a white solid, m.ρ. 106-107°C.
1H NMR (CDCI3) δ 1.00 (d, 6H); 2.12 (m, IH); 2.35 (s, 3H); 2.65 (d, 2H); 3.93 (s, 2H); 6.39 (s, IH); 7.14 (dd, IH); 7.43 (d, IH); 7.45 (d, IH); 8.18 (b, IH)
Step 4 - Preparation of N-(4-chloro-2-ethoxymethyl-3-methylisothiazolin-5-yIidene)-3- [2-(2-methylpropyI)benzofuran-5-yl]acetamide
A solution of N,O-bis(trimethylsilyl)acetamide (0.325 g, 0.00160 mol) in dichloromethane (1 ml) was added to a solution of N-(4-chloro-3-methylisothiazol-5-yl)-[2- (2,2-dimethylpropyl)-benzoxazol-5-yl]oxyacetamide (0.475 g, 0.00132 mol) in dichloromethane (4 ml) and the mixture stirred at room temperature for 20 minutes. A solution of chloromethylethyl ether (0.250 g, 0.00264 mol) in dichloromethane (1 ml) was added and stirring was continued for 22hours. The reaction mixture was partitioned between saturated aqueous sodium bicarbonate solution and ethyl acetate, and the organic phase dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. The residue was purified by flash column chromatography on silica gel, eluting initially with ethyl acetate : hexane 15 : 85 and then with ethyl acetate : hexane 2 : 3 to give the desired product (0.185 g) as a white solid, m.p. 73-75°C.
1H NMR (CDC13) δ 0.96 (d, 6H); 1.15 (t, 3H); 2.08 (m, IH); 2.53 (s, 3H); 2.60 (d, 2H); 3.48 (q, 2H); 4.14 (s, 2H); 5.17 (s, 2H); 6.32 (s, IH); 7.21 (dd, IH); 7.33 (d, IH); 7.48 (d, IH). EXAMPLE 12
This Example illustrates the preparation of Compound No. 136 of Table No. 25. Step 1 - Preparation of methyl 4-fluoro-3-nitrophenylacetate
4-Fluoro-3-nitrophenylacetic acid (31.0 g, 0.156 mol) was added to a mixture of concentrated sulfuric acid (1 .6 ml) and methanol (160 ml) and the mixture was stirred for 3 days at room temperature. Most of the solvent was removed in vacuo, and the residue was partitioned between diethyl ether and water. The organic phase was washed with saturated aqueous sodium bicarbonate solution and brine, dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo to give methyl 4-fluoro-3-nitrophenylacetate (27.2 g) as a yellow oil. 1H NMR (CDCI3) δ 3.72(s,2H); 3.75(s,3H); 7.27(dd,lH); 7.58(m,lH); 8.01(dd,lH).
Step 2 - Preparation of methyl 3-amino-4-fluorophenylacetate
Iron powder (8.1 g) was added to a solution of methyl 4-fluoro-3-nitrophenylacetate (27. 24 g, 0.128 mol) in a mixture of concentrated hydrochloric acid (1.5 ml), isopropanol (265 ml) and water (26.5 ml) and the mixture stirred at room temperature for 1 hour. A second portion of iron powder (8.1 g) was added, and the mixture heated to reflux for 1 hour. A further quantity of concentrated hydrochloric acid was added and the mixture refluxed for a further 1 hour. The mixture was cooled to room temperature, filtered through a plug of
01/55139
- 96 -
Hyflo® diatomaceous earth and the filtrate evaporated in vacuo to give methyl 3-amino-4- fluorophenylacetate (22.98 g) used without further purification in the next step. Step 3 - Preparation of methyl 4-fluoro-3-(3-methylbutyramido)phenyIacetate
Isobutyryl chloride (2.93 ml, 2.90 g, 0.024 mol) was added, dropwise, to a chilled (ice-bath) solution of methyl 3-amino-4-fluorophenylacetate (4.00 g, 0.022 mol) in pyridine (16 ml) and once the addition was complete the cooling bath was removed and the reaction mixture was stirred at room temperature for 30 minutes. The mixture was diluted with water and extracted with ethyl acetate. The organic extract was washed with water, dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. Trituration with dichloromethane / hexane gave methyl 4-fluoro-3-(3-methylbutyramido)phenylacetate (4.88 g) as a pale brown solid.
1H NMR (CDC13) δ 1.03 (d, 6H); 2.22 (m, IH); 2.26 (s, 2H); 3.6 (s, 2H); 3.69 (s, 3H); 6.98 (m, IH); 7.03 (dd, IH); 7.33 (b, IH); 8.39 (d, IH) Step 4 - Preparation of methyl [2-(2-methylpropyl)benzothiazol-5-yI]acetate [2,4-bis(4-Methoxyphenyl)-l,3-dithia-2,4-diphosρhetane-2,4-disulfide (7.39 g, 0.018 mol) was added, portionwise, to a solution of methyl 4-fluoro-3-(3- methylbutyramido)phenyl-acetate (4.88 g, 0.018 mol) in refluxing 1,2-dimethoxyethane (70 ml) and once the addition was complete the mixture was refluxed for 3 h. The mixture was cooled to room temperature, poured into water and extracted with dichloromethane. The organic phase was dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. The residue was passed through a plug of silica gel, eluting with ethyl acetate : hexane 1 : 1 and fractions containing the desired product were combined and evaporated in vacuo. The residue (5.92 g) was taken up in N.N-dimethylacetamide (48 ml), potassium carbonate (5.776 g, 0.042 mol) added and the mixture heated to 110 °C for 2 h. The mixture was cooled to room temperature, poured into water and extracted with dichloromethane. The organic extract was washed with brine, dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. The residue was further purified by flash column chromatography on silica gel, eluting with ethyl acetate : hexane 1 : 4 to give methyl [2-(2-methylpropyl)-benzothiazol-5-yl]acetate (2.01 g). 1H NMR (CDCI3) δ 1.07 (d, 6H); 2.23 (m, IH); 3.00 (d, 2H); 3.71 (s, 3H); 3.78 (s,
2H); 7.29 (dd, IH); 7.79 (d, IH); 7.88 (d, IH) Step 5 - Preparation of methyl 2-[2-(2-methylpropyI)benzothiazol-5-yl]propionate
01/55139
A solution of lithium diisopropylamide (2.0 M solution in tetrahydrofuran /ethyl benzene / heptane, 2.09 ml, 0.0042 mol) was added dropwise to a solution of methyl [2-(2,- methyl-propyl)benzothiazol-5-yl]acetate (1.1 g, 0.0042 mol) in tetrahydrofuran (27 ml) at -78 °C under a nitrogen atmosphere and the mixture stirred at below -60 °C for 1 hour. Methyl iodide (2.29 ml, 0.037 mol) was added dropwise and the mixture was stirred at below -60 °C for a further 1 hour. The cooling bath was removed and the mixture allowed to warm to room temperature over a period of 1 h. The mixture was diluted with water, acidified with 2M aqueous hydrochloric acid and extracted with ethyl acetate. The organic extract was dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo to give methyl 2-[2-(2-methylpropyl)benzothiazol-5-yl]propionate (1.15 g) used without further purification in the next step.
1H NMR (CDC13) δ 1.04 (d, 6H); 1.58 (d, 3H); 2.22 (m, IH); 2.98 (d, 2H); 3.67 (s, 3H); 3.87 (q, IH); 7.31 (dd, IH); 7.79 (d, IH); 7.91 (d, IH) Step 6 - Preparation of N-(4-chIoro-3-ethyIisothiazoI-5-yl)-2-[2-(2- methylpropyl)benzothiazol-5-yI]propionamide
5-Amino-4-chloro-3-ethylisothiazole (0.81 g, 0.005 mol) was added to a suspension of sodium methoxide (0.56 g, 0.010 mol) in tetrahydrofuran (5 ml) and the mixture stirred at room temperature for 25 minutes. A solution of give methyl 2-[2-(2-methylpropyl)benzo- thiazol-5-yl]proρionate (1.15 g, 0.004 mol) in tetrahydrofuran (4 ml) was added dropwise and the mixture stirred at room temperature for 1 ¥2 h. The mixture was diluted with saturated aqueous ammonium chloride solution and extracted with dichloromethane. The organic extract was dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. Trituration with diethyl ether gave N-(4-chloro-3-ethylisothiazol-5-yl)- 2-[2-(2-methylpropyl)benzothiazol-5-yl]propionamide (1.06 g). 1H NMR (CDCI3) δ 1.05 (d, 6H); 1.25 (t, 3H); 1.72 (d, 3H); 2.24 (m, IH); 2.72 (q,
2H); 3.01 (d, 2H); 4.03 (q, IH); 7.36 (dd, IH); 7.88 (d, IH); 7.97 (d, IH) Step 7 - Preparation of N-(4-chloro-2-ethoxymethyI-3-ethylisothiazolin-5-ylidene)-2-[2- (2-methyIpropyl)benzothiazol-5-yI]propionamide
A mixture of N,O-bis(trimethylsilyl)acetamide (0.533 ml, 0.439 g, 0.0022 mol), N- (4-chloro-3-ethylisothiazol-5-yl)-2-[2-(2-methylpropyl)benzothiazol-5-yl]propionamide (0.800 g, 0.002 mol) and chloromethylethyl ether (0.365 ml, 0.358 g, 0.0038 mol) in dichloromethane (10 ml) was stirred at room temperature for 5 V2 h. The reaction mixture
01/55139 gg
was diluted with dichloromethane and washed with saturated aqueous sodium bicarbonate solution. The organic phase was dried over anhydrous magnesium sulfate, filtered and the filtrate evaporated in vacuo. The residue was purified by column chromatography on silica gel, eluting with ethyl acetate : hexane 1: 4 to give the desired product (0.183 g). The following compounds were prepared individually using similar methods from appropriate starting materials: In Table 1 compound Nos 11, 13, 17, 21, 23, 26, 31, 33, 41, 46, 51, 53, 57, 64, 66, 67, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125, 126, 127, 128 and 129; From table 3 compound Nos 3, 67 and 140; Compound 67 from table 4, Compound 67 from table 5, Compound 137 from table 8, Compound 140 from table 10, Compound 7 from table 13, Compound 67 from table 21, Compound 67 from table 24, Compound 137 from table 24, Compound 63 from table 51, and Compound 67 from table 72. EXAMPLE 13
This Example illustrates the pesticidal/insecticidal properties of compounds of formula (I). The activities of individual compounds of formula (I) were determined using a variety of pests. The pests were treated with a liquid composition containing 500 parts per million (ppm) by weight of a compound of formula (I). Each composition was made by dissolving the compound in an acetone and ethanol (50:50 by volume) mixture and diluting the solution with water containing 0.05% by volume of a wetting agent, S YNPERONIC NP8, until the liquid composition contained the required concentration of the compound. SYNPERONIC is a registered trade mark.
The test procedure adopted with regard to each pest was essentially the same and comprised supporting a number of the pests on a medium, which was usually a substrate, a host plant or a foodstuff on which the pests feed, and treating either or both the medium and the pests with a composition. Pest mortality was assessed usually between two and five days after treatment.
In each test against peach potato aphids (Myzus persicae), Chinese cabbage leaves were infested with aphids, the infested leaves were sprayed with a test composition and pest mortality was assessed after three days.
Similar tests were conducted against, independently, two-spotted spider mites (Tetranychus urticae), fruit flies (Drosophila melanogaster), tobacco budworms (Heliothis virescens), diamond back moth (Plutella xylostella) and corn root worm (Diabrotica balteata).
Tests were also conducted against root knot nematodes (Meloidogyne incognita) using an in vitro test in which nematodes were suspended in a liquid composition which had been prepared as described above except that it contained a concentration of 12.5ppm by weight of a compound of formula (I) and it contained no SYNPERONIC NP8.
Results from these tests are displayed in Table 76, in which each mortality (score) is designated as 9, 5 or 0 wherein 9 indicates 80-100%) mortality, 5 indicates 40-79% mortality and 0 indicates less than 40% mortality; and Dm represents Drosophila melanogaster; Mp represents Myzus persicae; Hv represents Heliothis virescens; Px represents Plutella xylostella; Tu represents Tetranychus urticae; Db represents Diabrotica balteata; and Mi represents Meloidogyne incognita.
Table 76
EXAMPLE 14 This Example illustrates the fungicidal properties of compounds of formula (I). The compounds were tested against a variety of foliar fungal diseases of plants. The technique employed was as follows. Plants were grown in John Innes Potting Compost (No.l or 2) in 4cm diameter, 3.5cm depth minipots. The test compounds were individually formulated as a solution either in acetone or acetone/ethanol (1:1 by volume) which was diluted in deionised water to a concentration of lOOppm (that is, lmg of compound in a final volume of 10ml) immediately before use. When foliar sprays were applied to monocotyledonous crops, TWEEN 20 (0.1% by volume) was added. TWEEN is a registered trade mark.
Individual compounds of formula (I) were applied as a foliar (Folr) application (where the chemical solution was applied to the foliage of the test plants by spraying the plant to maximum droplet retention.)
These tests were carried out against Uncinula necator (UNCINE), on vines; Phytophthora infestans lycopersici (PHYTIN) on tomatoes; Puccinia recondita (PUCCRT), on wheat; and Pyricularia oryzae (PYRIOR) on rice. Each treatment was applied to two or more replicate plants for Phytophthora infestans lycopersici and Uncinula necator. For tests on Puccinia recondita and Pyricularia oryzae two replicate pots each containing 6 to 10 plants were used for each treatment. The plants were inoculated one day before (Erad) or one day after (Prot) chemical application. The Phytophthora infestans lycopersici, Puccinia recondita and Pyricularia oryzae plants were inoculated with a calibrated fungal spore suspension. The Uncinula necator plants were inoculated using a 'blowing' inoculation technique.
After chemical application and inoculation, the plants were incubated under high humidity conditions and then put into an appropriate environment to allow infection to proceed, until the disease was ready for assessment. The time period between chemical application and assessment varied from five to fourteen days according to the disease and environment. However, each individual disease was assessed after the same time period for all compounds. Assessments were performed on each of two leaves on each of the replicate plants for
Phytophthora infestans lycopersici. Assessments were performed on a single leaf of each of
the replicate plants for Uncinula necator. For Puccinia recondita and Pyricularia recondita assessments were carried out collectively on the plants in each replicate pot.
The disease level present (that is, the percentage leaf area covered by actively sporulating disease) was assessed visually. For each treatment, the assessed values for all its replicates were meaned to provide mean disease values. Untreated control plants were assessed in the same manner. The data were then processed by the method, described hereinafter, to provide PRCO (Percentage Reduction from Control) values.
An example of a typical calculation is as follows:
Mean disease level for treatment A = 25% Mean disease level on untreated controls = 85%
PRCO = 100 - { Mean disease level for treatment A } x 100 {Mean disease level on untreated controls}
= 100 - (25 x 100) = 70.6 85
The PRCO is then rounded to the nearest whole number; therefore, in this particular example, the PRCO result is 71. If no test data were available this is indicated in the table by
It is possible for negative PRCO values to be obtained. The PRCO results are set out in Table77.
TABLE 77
Key to Table 77
PHYTIN = Phytophthora infestans lycopersici
PYRIOR = Pyricularia oryzae
UNCINE = Uncinula necator
PUCCRT = Puccinia triticina