RU2379312C2 - Lysine compounds, pharmaceutical composition containing these compounds, application of said compounds for treatment or prevention of hiv-infection - Google Patents
Lysine compounds, pharmaceutical composition containing these compounds, application of said compounds for treatment or prevention of hiv-infection Download PDFInfo
- Publication number
- RU2379312C2 RU2379312C2 RU2007107798/04A RU2007107798A RU2379312C2 RU 2379312 C2 RU2379312 C2 RU 2379312C2 RU 2007107798/04 A RU2007107798/04 A RU 2007107798/04A RU 2007107798 A RU2007107798 A RU 2007107798A RU 2379312 C2 RU2379312 C2 RU 2379312C2
- Authority
- RU
- Russia
- Prior art keywords
- group
- compounds
- compound
- formula
- carbon atoms
- Prior art date
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 186
- 239000008194 pharmaceutical composition Substances 0.000 title claims abstract description 25
- 208000031886 HIV Infections Diseases 0.000 title claims abstract description 19
- 208000037357 HIV infectious disease Diseases 0.000 title claims abstract description 17
- 208000033519 human immunodeficiency virus infectious disease Diseases 0.000 title claims abstract description 17
- 238000011282 treatment Methods 0.000 title abstract description 24
- 230000002265 prevention Effects 0.000 title abstract description 9
- 125000004432 carbon atom Chemical group C* 0.000 claims abstract description 81
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 47
- 150000003839 salts Chemical class 0.000 claims abstract description 33
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 26
- 108010017640 Aspartic Acid Proteases Proteins 0.000 claims abstract description 23
- 102000004580 Aspartic Acid Proteases Human genes 0.000 claims abstract description 23
- 229910052731 fluorine Inorganic materials 0.000 claims abstract description 13
- 125000005982 diphenylmethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])(*)C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 claims abstract description 11
- 229910052794 bromium Inorganic materials 0.000 claims abstract description 10
- 229910052801 chlorine Inorganic materials 0.000 claims abstract description 10
- 229910052740 iodine Inorganic materials 0.000 claims abstract description 10
- 125000003545 alkoxy group Chemical group 0.000 claims abstract description 6
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 claims abstract description 4
- -1 4-aminobenzenesulfonyl Chemical group 0.000 claims description 86
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 claims description 23
- 238000000034 method Methods 0.000 claims description 17
- 239000003696 aspartic proteinase inhibitor Substances 0.000 claims description 12
- 230000002401 inhibitory effect Effects 0.000 claims description 8
- KJAMZCVTJDTESW-UHFFFAOYSA-N tiracizine Chemical compound C1CC2=CC=CC=C2N(C(=O)CN(C)C)C2=CC(NC(=O)OCC)=CC=C21 KJAMZCVTJDTESW-UHFFFAOYSA-N 0.000 claims description 8
- 239000003937 drug carrier Substances 0.000 claims description 6
- 125000006316 iso-butyl amino group Chemical group [H]N(*)C([H])([H])C([H])(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 6
- 241000124008 Mammalia Species 0.000 claims description 5
- 150000004702 methyl esters Chemical class 0.000 claims description 5
- 229910052783 alkali metal Inorganic materials 0.000 claims description 4
- 150000001340 alkali metals Chemical class 0.000 claims description 4
- KXDHJXZQYSOELW-UHFFFAOYSA-N carbonic acid monoamide Natural products NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 claims description 3
- 159000000000 sodium salts Chemical class 0.000 claims description 3
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 claims description 2
- 150000001715 carbamic acids Chemical class 0.000 claims 1
- 239000003814 drug Substances 0.000 abstract description 17
- 239000000126 substance Substances 0.000 abstract description 15
- 230000000694 effects Effects 0.000 abstract description 6
- 239000003112 inhibitor Substances 0.000 abstract description 3
- 239000002184 metal Substances 0.000 abstract 1
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 57
- 239000000203 mixture Substances 0.000 description 25
- 125000000753 cycloalkyl group Chemical group 0.000 description 23
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 23
- 239000000243 solution Substances 0.000 description 23
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 22
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 21
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 19
- 241000725303 Human immunodeficiency virus Species 0.000 description 19
- 239000004480 active ingredient Substances 0.000 description 19
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 19
- 241000700605 Viruses Species 0.000 description 18
- 239000002904 solvent Substances 0.000 description 16
- QAHLFXYLXBBCPS-IZEXYCQBSA-N methyl n-[(2s)-1-[[(5s)-5-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-6-hydroxyhexyl]amino]-1-oxo-3,3-diphenylpropan-2-yl]carbamate Chemical compound C=1C=CC=CC=1C([C@H](NC(=O)OC)C(=O)NCCCC[C@@H](CO)N(CC(C)C)S(=O)(=O)C=1C=CC(N)=CC=1)C1=CC=CC=C1 QAHLFXYLXBBCPS-IZEXYCQBSA-N 0.000 description 15
- 238000002360 preparation method Methods 0.000 description 15
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 15
- 229940079593 drug Drugs 0.000 description 14
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 13
- 229910019142 PO4 Inorganic materials 0.000 description 13
- 239000003795 chemical substances by application Substances 0.000 description 13
- 235000021317 phosphate Nutrition 0.000 description 13
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 12
- 239000004030 hiv protease inhibitor Substances 0.000 description 12
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 12
- 239000010452 phosphate Substances 0.000 description 12
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 11
- 150000002148 esters Chemical class 0.000 description 11
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 11
- 229910052727 yttrium Inorganic materials 0.000 description 11
- 241000713772 Human immunodeficiency virus 1 Species 0.000 description 10
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 9
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 9
- 239000004472 Lysine Substances 0.000 description 9
- 235000018977 lysine Nutrition 0.000 description 9
- 239000000047 product Substances 0.000 description 9
- 230000003612 virological effect Effects 0.000 description 9
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 8
- 208000030507 AIDS Diseases 0.000 description 8
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 8
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 8
- 230000015572 biosynthetic process Effects 0.000 description 8
- 238000006243 chemical reaction Methods 0.000 description 8
- 235000019439 ethyl acetate Nutrition 0.000 description 8
- 238000001727 in vivo Methods 0.000 description 8
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 8
- 239000000546 pharmaceutical excipient Substances 0.000 description 8
- 239000011734 sodium Substances 0.000 description 8
- 238000003786 synthesis reaction Methods 0.000 description 8
- 230000001225 therapeutic effect Effects 0.000 description 8
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 7
- 150000003863 ammonium salts Chemical class 0.000 description 7
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 7
- 239000000543 intermediate Substances 0.000 description 7
- 238000012545 processing Methods 0.000 description 7
- 208000024891 symptom Diseases 0.000 description 7
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 208000036142 Viral infection Diseases 0.000 description 6
- 125000003277 amino group Chemical group 0.000 description 6
- 239000003443 antiviral agent Substances 0.000 description 6
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 description 6
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 6
- 238000004519 manufacturing process Methods 0.000 description 6
- GTCAXTIRRLKXRU-UHFFFAOYSA-N methyl carbamate Chemical compound COC(N)=O GTCAXTIRRLKXRU-UHFFFAOYSA-N 0.000 description 6
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 6
- 239000012074 organic phase Substances 0.000 description 6
- 230000010076 replication Effects 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 6
- 230000009385 viral infection Effects 0.000 description 6
- 239000007864 aqueous solution Substances 0.000 description 5
- IYYIVELXUANFED-UHFFFAOYSA-N bromo(trimethyl)silane Chemical compound C[Si](C)(C)Br IYYIVELXUANFED-UHFFFAOYSA-N 0.000 description 5
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 5
- 210000004027 cell Anatomy 0.000 description 5
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 5
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 5
- 208000015181 infectious disease Diseases 0.000 description 5
- 239000012071 phase Substances 0.000 description 5
- 230000000069 prophylactic effect Effects 0.000 description 5
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 5
- 238000011269 treatment regimen Methods 0.000 description 5
- 229960005486 vaccine Drugs 0.000 description 5
- HBOMLICNUCNMMY-XLPZGREQSA-N zidovudine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](N=[N+]=[N-])C1 HBOMLICNUCNMMY-XLPZGREQSA-N 0.000 description 5
- 229960002555 zidovudine Drugs 0.000 description 5
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 4
- 125000000389 2-pyrrolyl group Chemical group [H]N1C([*])=C([H])C([H])=C1[H] 0.000 description 4
- 125000004208 3-hydroxyphenyl group Chemical group [H]OC1=C([H])C([H])=C([H])C(*)=C1[H] 0.000 description 4
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 4
- 125000004203 4-hydroxyphenyl group Chemical group [H]OC1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 4
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 229940122440 HIV protease inhibitor Drugs 0.000 description 4
- WREGKURFCTUGRC-POYBYMJQSA-N Zalcitabine Chemical compound O=C1N=C(N)C=CN1[C@@H]1O[C@H](CO)CC1 WREGKURFCTUGRC-POYBYMJQSA-N 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 125000005530 alkylenedioxy group Chemical group 0.000 description 4
- 230000000840 anti-viral effect Effects 0.000 description 4
- 210000004369 blood Anatomy 0.000 description 4
- 239000008280 blood Substances 0.000 description 4
- 239000000969 carrier Substances 0.000 description 4
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 4
- LGTLXDJOAJDFLR-UHFFFAOYSA-N diethyl chlorophosphate Chemical compound CCOP(Cl)(=O)OCC LGTLXDJOAJDFLR-UHFFFAOYSA-N 0.000 description 4
- 239000012467 final product Substances 0.000 description 4
- 238000009472 formulation Methods 0.000 description 4
- 238000004128 high performance liquid chromatography Methods 0.000 description 4
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 4
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 4
- 239000003921 oil Substances 0.000 description 4
- 235000019198 oils Nutrition 0.000 description 4
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 4
- 229920000728 polyester Polymers 0.000 description 4
- 150000003138 primary alcohols Chemical class 0.000 description 4
- 238000011321 prophylaxis Methods 0.000 description 4
- 125000004307 pyrazin-2-yl group Chemical group [H]C1=C([H])N=C(*)C([H])=N1 0.000 description 4
- 125000004076 pyridyl group Chemical group 0.000 description 4
- 229920005989 resin Polymers 0.000 description 4
- 239000011347 resin Substances 0.000 description 4
- 229910000104 sodium hydride Inorganic materials 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 4
- 238000004809 thin layer chromatography Methods 0.000 description 4
- DQWPFSLDHJDLRL-UHFFFAOYSA-N triethyl phosphate Chemical compound CCOP(=O)(OCC)OCC DQWPFSLDHJDLRL-UHFFFAOYSA-N 0.000 description 4
- SCWIUHICGSPBKM-VIFPVBQESA-N (3s)-3-(2-methylpropylamino)azepan-2-one Chemical compound CC(C)CN[C@H]1CCCCNC1=O SCWIUHICGSPBKM-VIFPVBQESA-N 0.000 description 3
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 3
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 description 3
- GVNVAWHJIKLAGL-UHFFFAOYSA-N 2-(cyclohexen-1-yl)cyclohexan-1-one Chemical compound O=C1CCCCC1C1=CCCCC1 GVNVAWHJIKLAGL-UHFFFAOYSA-N 0.000 description 3
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 description 3
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- 101710110426 Aspartyl protease inhibitor Proteins 0.000 description 3
- 0 CC(C)CN(*)S(c(cc1)ccc1NC(OC(C)(C)C)=O)(=O)=O Chemical compound CC(C)CN(*)S(c(cc1)ccc1NC(OC(C)(C)C)=O)(=O)=O 0.000 description 3
- JBKVHLHDHHXQEQ-UHFFFAOYSA-N Caprolactam Natural products O=C1CCCCCN1 JBKVHLHDHHXQEQ-UHFFFAOYSA-N 0.000 description 3
- 101150065749 Churc1 gene Proteins 0.000 description 3
- 241000598436 Human T-cell lymphotropic virus Species 0.000 description 3
- 206010020460 Human T-cell lymphotropic virus type I infection Diseases 0.000 description 3
- 241000714260 Human T-lymphotropic virus 1 Species 0.000 description 3
- 102000006992 Interferon-alpha Human genes 0.000 description 3
- 108010047761 Interferon-alpha Proteins 0.000 description 3
- 102100038239 Protein Churchill Human genes 0.000 description 3
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 3
- 210000001744 T-lymphocyte Anatomy 0.000 description 3
- 238000010521 absorption reaction Methods 0.000 description 3
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 3
- 150000001342 alkaline earth metals Chemical class 0.000 description 3
- 235000001014 amino acid Nutrition 0.000 description 3
- 229940024606 amino acid Drugs 0.000 description 3
- 150000001413 amino acids Chemical class 0.000 description 3
- 238000013459 approach Methods 0.000 description 3
- 239000004305 biphenyl Substances 0.000 description 3
- 235000010290 biphenyl Nutrition 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 238000002648 combination therapy Methods 0.000 description 3
- 239000012043 crude product Substances 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 239000003085 diluting agent Substances 0.000 description 3
- 239000012153 distilled water Substances 0.000 description 3
- 238000003818 flash chromatography Methods 0.000 description 3
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 3
- 239000005457 ice water Substances 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 229910052749 magnesium Inorganic materials 0.000 description 3
- 239000011777 magnesium Substances 0.000 description 3
- 238000002844 melting Methods 0.000 description 3
- 230000008018 melting Effects 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 239000003419 rna directed dna polymerase inhibitor Substances 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 239000012312 sodium hydride Substances 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 238000002560 therapeutic procedure Methods 0.000 description 3
- 230000000699 topical effect Effects 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- AZJJNORIBURVFN-HNNXBMFYSA-N (2s)-2-(methoxycarbonylamino)-3,3-diphenylpropanoic acid Chemical compound C=1C=CC=CC=1C([C@H](NC(=O)OC)C(O)=O)C1=CC=CC=C1 AZJJNORIBURVFN-HNNXBMFYSA-N 0.000 description 2
- KBPLFHHGFOOTCA-UHFFFAOYSA-N 1-Octanol Chemical compound CCCCCCCCO KBPLFHHGFOOTCA-UHFFFAOYSA-N 0.000 description 2
- GRDXCFKBQWDAJH-UHFFFAOYSA-N 4-acetamidobenzenesulfonyl chloride Chemical compound CC(=O)NC1=CC=C(S(Cl)(=O)=O)C=C1 GRDXCFKBQWDAJH-UHFFFAOYSA-N 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- 229920002261 Corn starch Polymers 0.000 description 2
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 2
- BXZVVICBKDXVGW-NKWVEPMBSA-N Didanosine Chemical compound O1[C@H](CO)CC[C@@H]1N1C(NC=NC2=O)=C2N=C1 BXZVVICBKDXVGW-NKWVEPMBSA-N 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 241000714259 Human T-lymphotropic virus 2 Species 0.000 description 2
- 241000713340 Human immunodeficiency virus 2 Species 0.000 description 2
- 102100020873 Interleukin-2 Human genes 0.000 description 2
- 108010002350 Interleukin-2 Proteins 0.000 description 2
- AMIMRNSIRUDHCM-UHFFFAOYSA-N Isopropylaldehyde Chemical compound CC(C)C=O AMIMRNSIRUDHCM-UHFFFAOYSA-N 0.000 description 2
- KJHKTHWMRKYKJE-SUGCFTRWSA-N Kaletra Chemical compound N1([C@@H](C(C)C)C(=O)N[C@H](C[C@H](O)[C@H](CC=2C=CC=CC=2)NC(=O)COC=2C(=CC=CC=2C)C)CC=2C=CC=CC=2)CCCNC1=O KJHKTHWMRKYKJE-SUGCFTRWSA-N 0.000 description 2
- 150000008545 L-lysines Chemical group 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 102000035195 Peptidases Human genes 0.000 description 2
- 108091005804 Peptidases Proteins 0.000 description 2
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 239000004365 Protease Substances 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- XNKLLVCARDGLGL-JGVFFNPUSA-N Stavudine Chemical compound O=C1NC(=O)C(C)=CN1[C@H]1C=C[C@@H](CO)O1 XNKLLVCARDGLGL-JGVFFNPUSA-N 0.000 description 2
- 102100036011 T-cell surface glycoprotein CD4 Human genes 0.000 description 2
- 108700010756 Viral Polyproteins Proteins 0.000 description 2
- CSCPPACGZOOCGX-WFGJKAKNSA-N acetone d6 Chemical compound [2H]C([2H])([2H])C(=O)C([2H])([2H])[2H] CSCPPACGZOOCGX-WFGJKAKNSA-N 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- YMARZQAQMVYCKC-OEMFJLHTSA-N amprenavir Chemical compound C([C@@H]([C@H](O)CN(CC(C)C)S(=O)(=O)C=1C=CC(N)=CC=1)NC(=O)O[C@@H]1COCC1)C1=CC=CC=C1 YMARZQAQMVYCKC-OEMFJLHTSA-N 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 230000000798 anti-retroviral effect Effects 0.000 description 2
- 229940088710 antibiotic agent Drugs 0.000 description 2
- 229940124522 antiretrovirals Drugs 0.000 description 2
- 239000003903 antiretrovirus agent Substances 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 239000007900 aqueous suspension Substances 0.000 description 2
- 239000002585 base Substances 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 235000019445 benzyl alcohol Nutrition 0.000 description 2
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 2
- 150000001649 bromium compounds Chemical class 0.000 description 2
- 229910052792 caesium Inorganic materials 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 239000008120 corn starch Substances 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 2
- 239000002270 dispersing agent Substances 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- NLFBCYMMUAKCPC-KQQUZDAGSA-N ethyl (e)-3-[3-amino-2-cyano-1-[(e)-3-ethoxy-3-oxoprop-1-enyl]sulfanyl-3-oxoprop-1-enyl]sulfanylprop-2-enoate Chemical compound CCOC(=O)\C=C\SC(=C(C#N)C(N)=O)S\C=C\C(=O)OCC NLFBCYMMUAKCPC-KQQUZDAGSA-N 0.000 description 2
- 230000029142 excretion Effects 0.000 description 2
- 235000019197 fats Nutrition 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 150000004665 fatty acids Chemical class 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 125000005456 glyceride group Chemical group 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 210000000936 intestine Anatomy 0.000 description 2
- 150000004694 iodide salts Chemical class 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 238000011068 loading method Methods 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 230000004060 metabolic process Effects 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 125000001624 naphthyl group Chemical group 0.000 description 2
- QAGYKUNXZHXKMR-HKWSIXNMSA-N nelfinavir Chemical compound CC1=C(O)C=CC=C1C(=O)N[C@H]([C@H](O)CN1[C@@H](C[C@@H]2CCCC[C@@H]2C1)C(=O)NC(C)(C)C)CSC1=CC=CC=C1 QAGYKUNXZHXKMR-HKWSIXNMSA-N 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 239000000346 nonvolatile oil Substances 0.000 description 2
- ZRSNZINYAWTAHE-UHFFFAOYSA-N p-methoxybenzaldehyde Chemical compound COC1=CC=C(C=O)C=C1 ZRSNZINYAWTAHE-UHFFFAOYSA-N 0.000 description 2
- 239000002953 phosphate buffered saline Substances 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N phosphoric acid Substances OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 230000004962 physiological condition Effects 0.000 description 2
- 125000003386 piperidinyl group Chemical group 0.000 description 2
- 229920001451 polypropylene glycol Polymers 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- 235000018102 proteins Nutrition 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 2
- 238000005956 quaternization reaction Methods 0.000 description 2
- 108010043277 recombinant soluble CD4 Proteins 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 230000001177 retroviral effect Effects 0.000 description 2
- 238000010839 reverse transcription Methods 0.000 description 2
- NCDNCNXCDXHOMX-XGKFQTDJSA-N ritonavir Chemical compound N([C@@H](C(C)C)C(=O)N[C@H](C[C@H](O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1SC=NC=1)CC=1C=CC=CC=1)C(=O)N(C)CC1=CSC(C(C)C)=N1 NCDNCNXCDXHOMX-XGKFQTDJSA-N 0.000 description 2
- 239000000523 sample Substances 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 239000012363 selectfluor Substances 0.000 description 2
- 238000009097 single-agent therapy Methods 0.000 description 2
- 238000013222 sprague-dawley male rat Methods 0.000 description 2
- 230000001629 suppression Effects 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 230000002195 synergetic effect Effects 0.000 description 2
- 238000011200 topical administration Methods 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 210000002845 virion Anatomy 0.000 description 2
- 239000007966 viscous suspension Substances 0.000 description 2
- 239000001993 wax Substances 0.000 description 2
- 229960000523 zalcitabine Drugs 0.000 description 2
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical compound CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- KHYOYKLXGIPSRF-UHFFFAOYSA-N (2,5-dioxopyrrolidin-3-yl) methyl carbonate Chemical compound COC(=O)OC1CC(=O)NC1=O KHYOYKLXGIPSRF-UHFFFAOYSA-N 0.000 description 1
- FMCAFXHLMUOIGG-JTJHWIPRSA-N (2s)-2-[[(2r)-2-[[(2s)-2-[[(2r)-2-formamido-3-sulfanylpropanoyl]amino]-3-methylbutanoyl]amino]-3-(4-hydroxy-2,5-dimethylphenyl)propanoyl]amino]-4-methylsulfanylbutanoic acid Chemical compound O=CN[C@@H](CS)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(=O)N[C@@H](CCSC)C(O)=O)CC1=CC(C)=C(O)C=C1C FMCAFXHLMUOIGG-JTJHWIPRSA-N 0.000 description 1
- KRFUEXGPDYTGGI-INIZCTEOSA-N (2s)-2-acetamido-3,3-diphenylpropanoic acid Chemical compound C=1C=CC=CC=1C([C@H](NC(=O)C)C(O)=O)C1=CC=CC=C1 KRFUEXGPDYTGGI-INIZCTEOSA-N 0.000 description 1
- PECGVEGMRUZOML-AWEZNQCLSA-N (2s)-2-amino-3,3-diphenylpropanoic acid Chemical compound C=1C=CC=CC=1C([C@H](N)C(O)=O)C1=CC=CC=C1 PECGVEGMRUZOML-AWEZNQCLSA-N 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- VFWCMGCRMGJXDK-UHFFFAOYSA-N 1-chlorobutane Chemical class CCCCCl VFWCMGCRMGJXDK-UHFFFAOYSA-N 0.000 description 1
- VUQPJRPDRDVQMN-UHFFFAOYSA-N 1-chlorooctadecane Chemical class CCCCCCCCCCCCCCCCCCCl VUQPJRPDRDVQMN-UHFFFAOYSA-N 0.000 description 1
- FDCJDKXCCYFOCV-UHFFFAOYSA-N 1-hexadecoxyhexadecane Chemical class CCCCCCCCCCCCCCCCOCCCCCCCCCCCCCCCC FDCJDKXCCYFOCV-UHFFFAOYSA-N 0.000 description 1
- DFPYXQYWILNVAU-UHFFFAOYSA-N 1-hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1.C1=CC=C2N(O)N=NC2=C1 DFPYXQYWILNVAU-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- CHHHXKFHOYLYRE-UHFFFAOYSA-M 2,4-Hexadienoic acid, potassium salt (1:1), (2E,4E)- Chemical compound [K+].CC=CC=CC([O-])=O CHHHXKFHOYLYRE-UHFFFAOYSA-M 0.000 description 1
- LEACJMVNYZDSKR-UHFFFAOYSA-N 2-octyldodecan-1-ol Chemical compound CCCCCCCCCCC(CO)CCCCCCCC LEACJMVNYZDSKR-UHFFFAOYSA-N 0.000 description 1
- WMPPDTMATNBGJN-UHFFFAOYSA-N 2-phenylethylbromide Chemical class BrCCC1=CC=CC=C1 WMPPDTMATNBGJN-UHFFFAOYSA-N 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- DYPMSRIXASMFIF-POYBYMJQSA-N 3-[(2R,5S)-5-(hydroxymethyl)oxolan-2-yl]-6-imino-1,3-thiazin-2-one Chemical compound O1[C@H](CO)CC[C@@H]1N1C(=O)SC(=N)C=C1 DYPMSRIXASMFIF-POYBYMJQSA-N 0.000 description 1
- ZRPLANDPDWYOMZ-UHFFFAOYSA-N 3-cyclopentylpropionic acid Chemical compound OC(=O)CCC1CCCC1 ZRPLANDPDWYOMZ-UHFFFAOYSA-N 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical class [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- CIUUIPMOFZIWIZ-UHFFFAOYSA-N Bropirimine Chemical compound NC1=NC(O)=C(Br)C(C=2C=CC=CC=2)=N1 CIUUIPMOFZIWIZ-UHFFFAOYSA-N 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- 108010041397 CD4 Antigens Proteins 0.000 description 1
- HJJVHNKOXFYNTJ-UHFFFAOYSA-N COCc1cccnc1 Chemical compound COCc1cccnc1 HJJVHNKOXFYNTJ-UHFFFAOYSA-N 0.000 description 1
- MBGSSZLPFNHEHX-UHFFFAOYSA-N COCc1ccncc1 Chemical compound COCc1ccncc1 MBGSSZLPFNHEHX-UHFFFAOYSA-N 0.000 description 1
- QDTSINVSGKAPBV-UHFFFAOYSA-N COCc1ncccc1 Chemical compound COCc1ncccc1 QDTSINVSGKAPBV-UHFFFAOYSA-N 0.000 description 1
- QAGYKUNXZHXKMR-UHFFFAOYSA-N CPD000469186 Natural products CC1=C(O)C=CC=C1C(=O)NC(C(O)CN1C(CC2CCCCC2C1)C(=O)NC(C)(C)C)CSC1=CC=CC=C1 QAGYKUNXZHXKMR-UHFFFAOYSA-N 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- 108020004414 DNA Proteins 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 1
- 108050009340 Endothelin Proteins 0.000 description 1
- 102000002045 Endothelin Human genes 0.000 description 1
- 102000003951 Erythropoietin Human genes 0.000 description 1
- 108090000394 Erythropoietin Proteins 0.000 description 1
- 108090000371 Esterases Proteins 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-M Glycolate Chemical compound OCC([O-])=O AEMRFAOFKBGASW-UHFFFAOYSA-M 0.000 description 1
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 102000004457 Granulocyte-Macrophage Colony-Stimulating Factor Human genes 0.000 description 1
- 108010010369 HIV Protease Proteins 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 102000008100 Human Serum Albumin Human genes 0.000 description 1
- 108091006905 Human Serum Albumin Proteins 0.000 description 1
- 108010016183 Human immunodeficiency virus 1 p16 protease Proteins 0.000 description 1
- 108010016191 Human immunodeficiency virus 2 p16 protease Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 206010061598 Immunodeficiency Diseases 0.000 description 1
- 208000029462 Immunodeficiency disease Diseases 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 229920001214 Polysorbate 60 Polymers 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 102000007327 Protamines Human genes 0.000 description 1
- 108010007568 Protamines Proteins 0.000 description 1
- 208000035415 Reinfection Diseases 0.000 description 1
- 108090000783 Renin Proteins 0.000 description 1
- 102100028255 Renin Human genes 0.000 description 1
- IWUCXVSUMQZMFG-AFCXAGJDSA-N Ribavirin Chemical compound N1=C(C(=O)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 IWUCXVSUMQZMFG-AFCXAGJDSA-N 0.000 description 1
- NCDNCNXCDXHOMX-UHFFFAOYSA-N Ritonavir Natural products C=1C=CC=CC=1CC(NC(=O)OCC=1SC=NC=1)C(O)CC(CC=1C=CC=CC=1)NC(=O)C(C(C)C)NC(=O)N(C)CC1=CSC(C(C)C)=N1 NCDNCNXCDXHOMX-UHFFFAOYSA-N 0.000 description 1
- 241000713311 Simian immunodeficiency virus Species 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-M Thiocyanate anion Chemical compound [S-]C#N ZMZDMBWJUHKJPS-UHFFFAOYSA-M 0.000 description 1
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical class [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 1
- 102100022563 Tubulin polymerization-promoting protein Human genes 0.000 description 1
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 1
- 108020005202 Viral DNA Proteins 0.000 description 1
- 108010046377 Whey Proteins Proteins 0.000 description 1
- YGSFNCRAZOCNDJ-TXHXQZCNSA-N [2H]C(C(=O)C([2H])([2H])[2H])([2H])[2H].CC(=O)C Chemical class [2H]C(C(=O)C([2H])([2H])[2H])([2H])[2H].CC(=O)C YGSFNCRAZOCNDJ-TXHXQZCNSA-N 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 229960004150 aciclovir Drugs 0.000 description 1
- YBCVMFKXIKNREZ-UHFFFAOYSA-N acoh acetic acid Chemical compound CC(O)=O.CC(O)=O YBCVMFKXIKNREZ-UHFFFAOYSA-N 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 239000012670 alkaline solution Substances 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- CEGOLXSVJUTHNZ-UHFFFAOYSA-K aluminium tristearate Chemical compound [Al+3].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CEGOLXSVJUTHNZ-UHFFFAOYSA-K 0.000 description 1
- 229940063655 aluminum stearate Drugs 0.000 description 1
- 229960001830 amprenavir Drugs 0.000 description 1
- 230000000202 analgesic effect Effects 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 239000002259 anti human immunodeficiency virus agent Substances 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 239000013011 aqueous formulation Substances 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- FZIZEIAMIREUTN-UHFFFAOYSA-N azane;cerium(3+) Chemical compound N.[Ce+3] FZIZEIAMIREUTN-UHFFFAOYSA-N 0.000 description 1
- 235000013871 bee wax Nutrition 0.000 description 1
- 239000012166 beeswax Substances 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 229940050390 benzoate Drugs 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 229920001400 block copolymer Polymers 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 229950009494 bropirimine Drugs 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 239000012876 carrier material Substances 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 108091092356 cellular DNA Proteins 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 229940081733 cetearyl alcohol Drugs 0.000 description 1
- 229920001429 chelating resin Polymers 0.000 description 1
- 238000000451 chemical ionisation Methods 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 238000011260 co-administration Methods 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 239000008119 colloidal silica Substances 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000013329 compounding Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000011443 conventional therapy Methods 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000002274 desiccant Substances 0.000 description 1
- 150000008050 dialkyl sulfates Chemical class 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- UCQFCFPECQILOL-UHFFFAOYSA-N diethyl hydrogen phosphate Chemical class CCOP(O)(=O)OCC UCQFCFPECQILOL-UHFFFAOYSA-N 0.000 description 1
- LMBWSYZSUOEYSN-UHFFFAOYSA-N diethyldithiocarbamic acid Chemical compound CCN(CC)C(S)=S LMBWSYZSUOEYSN-UHFFFAOYSA-N 0.000 description 1
- IJKVHSBPTUYDLN-UHFFFAOYSA-N dihydroxy(oxo)silane Chemical compound O[Si](O)=O IJKVHSBPTUYDLN-UHFFFAOYSA-N 0.000 description 1
- GXGAKHNRMVGRPK-UHFFFAOYSA-N dimagnesium;dioxido-bis[[oxido(oxo)silyl]oxy]silane Chemical compound [Mg+2].[Mg+2].[O-][Si](=O)O[Si]([O-])([O-])O[Si]([O-])=O GXGAKHNRMVGRPK-UHFFFAOYSA-N 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- GAFRWLVTHPVQGK-UHFFFAOYSA-N dipentyl sulfate Chemical class CCCCCOS(=O)(=O)OCCCCC GAFRWLVTHPVQGK-UHFFFAOYSA-N 0.000 description 1
- KCIDZIIHRGYJAE-YGFYJFDDSA-L dipotassium;[(2r,3r,4s,5r,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl] phosphate Chemical compound [K+].[K+].OC[C@H]1O[C@H](OP([O-])([O-])=O)[C@H](O)[C@@H](O)[C@H]1O KCIDZIIHRGYJAE-YGFYJFDDSA-L 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 229950004394 ditiocarb Drugs 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 229940043264 dodecyl sulfate Drugs 0.000 description 1
- VLCYCQAOQCDTCN-UHFFFAOYSA-N eflornithine Chemical compound NCCCC(N)(C(F)F)C(O)=O VLCYCQAOQCDTCN-UHFFFAOYSA-N 0.000 description 1
- 229960002759 eflornithine Drugs 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 239000008387 emulsifying waxe Substances 0.000 description 1
- ZUBDGKVDJUIMQQ-UBFCDGJISA-N endothelin-1 Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(O)=O)NC(=O)[C@H]1NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@@H](CC=2C=CC(O)=CC=2)NC(=O)[C@H](C(C)C)NC(=O)[C@H]2CSSC[C@@H](C(N[C@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N2)=O)NC(=O)[C@@H](CO)NC(=O)[C@H](N)CSSC1)C1=CNC=N1 ZUBDGKVDJUIMQQ-UBFCDGJISA-N 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 229940105423 erythropoietin Drugs 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 150000002168 ethanoic acid esters Chemical class 0.000 description 1
- OCLXJTCGWSSVOE-UHFFFAOYSA-N ethanol etoh Chemical compound CCO.CCO OCLXJTCGWSSVOE-UHFFFAOYSA-N 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- OJCSPXHYDFONPU-UHFFFAOYSA-N etoac etoac Chemical compound CCOC(C)=O.CCOC(C)=O OJCSPXHYDFONPU-UHFFFAOYSA-N 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 229960002963 ganciclovir Drugs 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 229960002449 glycine Drugs 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- UBHWBODXJBSFLH-UHFFFAOYSA-N hexadecan-1-ol;octadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO.CCCCCCCCCCCCCCCCCCO UBHWBODXJBSFLH-UHFFFAOYSA-N 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000000589 high-performance liquid chromatography-mass spectrometry Methods 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N hydrogen thiocyanate Natural products SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 230000007813 immunodeficiency Effects 0.000 description 1
- 239000002955 immunomodulating agent Substances 0.000 description 1
- 229940121354 immunomodulator Drugs 0.000 description 1
- 229960001936 indinavir Drugs 0.000 description 1
- CBVCZFGXHXORBI-PXQQMZJSSA-N indinavir Chemical compound C([C@H](N(CC1)C[C@@H](O)C[C@@H](CC=2C=CC=CC=2)C(=O)N[C@H]2C3=CC=CC=C3C[C@H]2O)C(=O)NC(C)(C)C)N1CC1=CC=CN=C1 CBVCZFGXHXORBI-PXQQMZJSSA-N 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 229940102223 injectable solution Drugs 0.000 description 1
- 229940102213 injectable suspension Drugs 0.000 description 1
- 229940124524 integrase inhibitor Drugs 0.000 description 1
- 239000002850 integrase inhibitor Substances 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- JTEGQNOMFQHVDC-NKWVEPMBSA-N lamivudine Chemical compound O=C1N=C(N)C=CN1[C@H]1O[C@@H](CO)SC1 JTEGQNOMFQHVDC-NKWVEPMBSA-N 0.000 description 1
- 229960001627 lamivudine Drugs 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 229960004525 lopinavir Drugs 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000004324 lymphatic system Anatomy 0.000 description 1
- 239000000391 magnesium silicate Substances 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229910000386 magnesium trisilicate Inorganic materials 0.000 description 1
- 229940099273 magnesium trisilicate Drugs 0.000 description 1
- 235000019793 magnesium trisilicate Nutrition 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 230000035800 maturation Effects 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 1
- 229930182817 methionine Natural products 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 230000003020 moisturizing effect Effects 0.000 description 1
- MEFBJEMVZONFCJ-UHFFFAOYSA-N molybdate Chemical compound [O-][Mo]([O-])(=O)=O MEFBJEMVZONFCJ-UHFFFAOYSA-N 0.000 description 1
- 125000001421 myristyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- PEECTLLHENGOKU-UHFFFAOYSA-N n,n-dimethylpyridin-4-amine Chemical compound CN(C)C1=CC=NC=C1.CN(C)C1=CC=NC=C1 PEECTLLHENGOKU-UHFFFAOYSA-N 0.000 description 1
- DQCKKXVULJGBQN-XFWGSAIBSA-N naltrexone Chemical compound N1([C@@H]2CC3=CC=C(C=4O[C@@H]5[C@](C3=4)([C@]2(CCC5=O)O)CC1)O)CC1CC1 DQCKKXVULJGBQN-XFWGSAIBSA-N 0.000 description 1
- 229960003086 naltrexone Drugs 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 229960000884 nelfinavir Drugs 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- FEMOMIGRRWSMCU-UHFFFAOYSA-N ninhydrin Chemical compound C1=CC=C2C(=O)C(O)(O)C(=O)C2=C1 FEMOMIGRRWSMCU-UHFFFAOYSA-N 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- LVSJDHGRKAEGLX-UHFFFAOYSA-N oxolane;2,2,2-trifluoroacetic acid Chemical compound C1CCOC1.OC(=O)C(F)(F)F LVSJDHGRKAEGLX-UHFFFAOYSA-N 0.000 description 1
- YBVNFKZSMZGRAD-UHFFFAOYSA-N pentamidine isethionate Chemical compound OCCS(O)(=O)=O.OCCS(O)(=O)=O.C1=CC(C(=N)N)=CC=C1OCCCCCOC1=CC=C(C(N)=N)C=C1 YBVNFKZSMZGRAD-UHFFFAOYSA-N 0.000 description 1
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- VLTRZXGMWDSKGL-UHFFFAOYSA-M perchlorate Inorganic materials [O-]Cl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-M 0.000 description 1
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 235000019271 petrolatum Nutrition 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- 235000011007 phosphoric acid Nutrition 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 150000003016 phosphoric acids Chemical class 0.000 description 1
- 229940075930 picrate Drugs 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-M picrate anion Chemical compound [O-]C1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-M 0.000 description 1
- 229950010765 pivalate Drugs 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 1
- 230000036470 plasma concentration Effects 0.000 description 1
- 229920000058 polyacrylate Polymers 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 239000001818 polyoxyethylene sorbitan monostearate Substances 0.000 description 1
- 235000010989 polyoxyethylene sorbitan monostearate Nutrition 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 229940113124 polysorbate 60 Drugs 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000004302 potassium sorbate Substances 0.000 description 1
- 235000010241 potassium sorbate Nutrition 0.000 description 1
- 229940069338 potassium sorbate Drugs 0.000 description 1
- OXCMYAYHXIHQOA-UHFFFAOYSA-N potassium;[2-butyl-5-chloro-3-[[4-[2-(1,2,4-triaza-3-azanidacyclopenta-1,4-dien-5-yl)phenyl]phenyl]methyl]imidazol-4-yl]methanol Chemical compound [K+].CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C2=N[N-]N=N2)C=C1 OXCMYAYHXIHQOA-UHFFFAOYSA-N 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- 238000004237 preparative chromatography Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 235000013772 propylene glycol Nutrition 0.000 description 1
- 229950008679 protamine sulfate Drugs 0.000 description 1
- 230000002797 proteolythic effect Effects 0.000 description 1
- 238000003908 quality control method Methods 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 229940100618 rectal suppository Drugs 0.000 description 1
- 239000006215 rectal suppository Substances 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 229960000329 ribavirin Drugs 0.000 description 1
- HZCAHMRRMINHDJ-DBRKOABJSA-N ribavirin Natural products O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1N=CN=C1 HZCAHMRRMINHDJ-DBRKOABJSA-N 0.000 description 1
- 229960000311 ritonavir Drugs 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 239000012266 salt solution Substances 0.000 description 1
- 238000011894 semi-preparative HPLC Methods 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- AWUCVROLDVIAJX-GSVOUGTGSA-N sn-glycerol 3-phosphate Chemical compound OC[C@@H](O)COP(O)(O)=O AWUCVROLDVIAJX-GSVOUGTGSA-N 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- JJICLMJFIKGAAU-UHFFFAOYSA-M sodium;2-amino-9-(1,3-dihydroxypropan-2-yloxymethyl)purin-6-olate Chemical compound [Na+].NC1=NC([O-])=C2N=CN(COC(CO)CO)C2=N1 JJICLMJFIKGAAU-UHFFFAOYSA-M 0.000 description 1
- RMLUKZWYIKEASN-UHFFFAOYSA-M sodium;2-amino-9-(2-hydroxyethoxymethyl)purin-6-olate Chemical compound [Na+].O=C1[N-]C(N)=NC2=C1N=CN2COCCO RMLUKZWYIKEASN-UHFFFAOYSA-M 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 1
- MLDFGFYWCJHWLS-VWLOTQADSA-N tert-butyl n-[4-[[(2s)-1-diethoxyphosphoryloxy-6-[(2-methylpropan-2-yl)oxycarbonylamino]hexan-2-yl]-(2-methylpropyl)sulfamoyl]phenyl]carbamate Chemical compound CC(C)(C)OC(=O)NCCCC[C@@H](COP(=O)(OCC)OCC)N(CC(C)C)S(=O)(=O)C1=CC=C(NC(=O)OC(C)(C)C)C=C1 MLDFGFYWCJHWLS-VWLOTQADSA-N 0.000 description 1
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 238000011287 therapeutic dose Methods 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- YFNGWGVTFYSJHE-UHFFFAOYSA-K trisodium;phosphonoformate Chemical compound [Na+].[Na+].[Na+].OP(O)(=O)C([O-])=O.OP(O)(=O)C([O-])=O.OP(O)(=O)C([O-])=O YFNGWGVTFYSJHE-UHFFFAOYSA-K 0.000 description 1
- 102000003390 tumor necrosis factor Human genes 0.000 description 1
- 238000007039 two-step reaction Methods 0.000 description 1
- ZDPHROOEEOARMN-UHFFFAOYSA-N undecanoic acid Chemical compound CCCCCCCCCCC(O)=O ZDPHROOEEOARMN-UHFFFAOYSA-N 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 230000029812 viral genome replication Effects 0.000 description 1
- 235000021119 whey protein Nutrition 0.000 description 1
- 239000003871 white petrolatum Substances 0.000 description 1
- 210000002268 wool Anatomy 0.000 description 1
- 150000003751 zinc Chemical class 0.000 description 1
Landscapes
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract

Description
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕFIELD OF THE INVENTION
Данное изобретение относится к соединениям на основе лизина, которые демонстрируют хорошую растворимость и биологическую доступность. Более конкретно настоящее изобретение относится к соединениям на основе лизина, содержащим физиологически отщепляемое звено, посредством чего при отщеплении данного звена соединение способно высвобождать ингибитор протеазы ВИЧ. Соединения и фармацевтические композиции по настоящему изобретению особенно хорошо подходят для снижения лекарственной нагрузки и улучшения соблюдения больным режима и схемы лечения.This invention relates to lysine-based compounds that exhibit good solubility and bioavailability. More specifically, the present invention relates to lysine-based compounds containing a physiologically cleavable unit, whereby when the unit is cleaved, the compound is capable of releasing an HIV protease inhibitor. The compounds and pharmaceutical compositions of the present invention are particularly well suited to reduce drug loading and improve patient compliance with treatment regimens and regimens.
УРОВЕНЬ ТЕХНИКИBACKGROUND
Ингибиторы вирусной протеазы ВИЧ были разработаны относительно недавно, и их использование началось только в 1996 году. В настоящее время они рассматриваются как наиболее эффективные лекарственные препараты против ВИЧ-инфекции. К сожалению, большинство имеющихся в настоящее время ингибиторов протеаз представляют собой относительно крупные гидрофобные молекулы, которые обладают довольно низкой биологической доступностью. Поэтому для достижения терапевтической дозы у пациента требуется высокая лекарственная нагрузка. Это является сдерживающим фактором, который слишком часто приводит к несоблюдению больным режима и схемы лечения и к недостаточным результатам лечения. Данная ситуация ведет к субоптимальной терапевтической концентрации лекарственного препарата, что в свою очередь ведет к развитию устойчивых штаммов ВИЧ.HIV viral protease inhibitors were developed relatively recently, and their use only began in 1996. They are currently regarded as the most effective anti-HIV drugs. Unfortunately, most of the currently available protease inhibitors are relatively large hydrophobic molecules that have fairly low bioavailability. Therefore, to achieve a therapeutic dose for a patient, a high drug load is required. This is a deterrent, which too often leads to non-compliance with the treatment regimen and regimen and insufficient treatment results. This situation leads to a suboptimal therapeutic concentration of the drug, which in turn leads to the development of resistant strains of HIV.
Следовательно, существует неотложная потребность улучшения растворимости и биологической доступности ингибиторов протеазы.Therefore, there is an urgent need to improve the solubility and bioavailability of protease inhibitors.
Примеры улучшенных соединений были разработаны в форме пролекарств ингибиторов аспартил-протеазы, таких как соединения, описанные, например, в патенте США № 6436989 Hale et al., полное содержание которого включено в описание в виде ссылки. Данный патент показывает новый класс молекул, характеризующихся подходящей растворимостью в воде, высокой пероральной биологической доступностью и легким генерированием in vivo активного ингредиента. Однако хорошо известно, что ВИЧ обладает способностью развивать устойчивость к имеющимся в настоящее время лекарственным препаратам. Таким образом, существует необходимость в альтернативных ингибиторах ВИЧ-протеазы, активных по отношению к диким типам вирусных штаммов и устойчивым вирусным штаммам. Так, для борьбы с устойчивыми вирусными штаммами желательными являются молекулы, полученные из имеющихся в настоящее время ингибиторов ВИЧ-протеазы, показывающих улучшенную растворимость и биологическую доступность.Examples of improved compounds have been developed in the form of prodrugs of aspartyl protease inhibitors, such as those described, for example, in US Pat. No. 6,436,989 to Hale et al. , the full contents of which are incorporated into the description by reference. This patent shows a new class of molecules characterized by suitable solubility in water, high oral bioavailability and easy in vivo generation of the active ingredient. However, it is well known that HIV has the ability to develop resistance to currently available drugs. Thus, there is a need for alternative HIV protease inhibitors active against wild types of viral strains and resistant viral strains. So, to combat resistant viral strains, molecules derived from currently available HIV protease inhibitors showing improved solubility and bioavailability are desirable.
Уникальный класс ароматических производных, которые являются ингибиторами аспартил-протеаз, описан в патенте США № 6632816 Stranix et al., полное содержание которого включается здесь ссылкой. Более конкретно данный патент включает производные L-лизина, замещенные N,-синтетической аминокислотой, обладающие эффективными ингибирующими свойствами по отношению к аспартил-протеазе. Однако было бы выгодно улучшить данные производные, увеличивая растворимость в воде и биологическую доступность, чтобы снизить лекарственную нагрузку и поддерживать соблюдение больным режима и схемы лечения. Поскольку является многообещающим создание активных ингибиторов протеазы, в особенности к диким типам штаммов и устойчивым штаммам, то образование производных оригинальных ингибиторов ВИЧ-протеазы, таких как ингибиторы, описанные в патенте США № 6632816 Stranix et al., которые, как известно, являются активными по отношению к устойчивым штаммам, представляет собой жизнеспособный путь со значительными преимуществами. Более конкретно при разработке эффективного лекарственного препарата является желательным получение соединений с улучшенными растворимостью в воде, биологической доступностью, продолжительностью действия и свойствами рецептуры вместе с другими преимуществами.A unique class of aromatic derivatives that are aspartyl protease inhibitors is described in US Pat. No. 6,632,816 to Stranix et al ., The entire contents of which are incorporated herein by reference. More specifically, this patent includes derivatives of L-lysine substituted with N, a synthetic amino acid, having effective inhibitory properties against aspartyl protease. However, it would be beneficial to improve these derivatives by increasing water solubility and bioavailability in order to reduce drug loading and maintain patient compliance with treatment regimen and regimen. Since it is promising to create active protease inhibitors, especially to wild types of strains and resistant strains, the formation of derivatives of the original HIV protease inhibitors, such as those described in US Pat. No. 6,632,816 to Stranix et al ., Which are known to be active in in relation to resistant strains, is a viable path with significant advantages. More specifically, in the development of an effective drug, it is desirable to provide compounds with improved water solubility, bioavailability, duration of action and formulation properties, along with other advantages.
СУЩНОСТЬ ИЗОБРЕТЕНИЯSUMMARY OF THE INVENTION
Настоящее изобретение предлагает новые соединения на основе лизина, источником которых является класс производных, являющихся эффективными ингибиторами аспартил-протеазы, и их фармацевтически приемлемые производные. Данные соединения могут легко расщепляться in vivo, высвобождая активный ингредиент. Активный ингредиент имеет сродство к аспартил-протеазам, в частности к аспартил-протеазе ВИЧ-1 (патент США № 6632816). Активные ингредиенты также демонстрируют эффективную антивирусную активность при тестировании на немутантном вирусном штамме ВИЧ-1 (NL4.3 в качестве дикого типа вируса), а также нескольких мутантных штаммах. Поэтому соединения по настоящему изобретению могут быть применены в качестве средства для увеличения растворимости и улучшения биологической доступности активного ингредиента (ингибитора протеазы). Соединения по настоящему изобретению можно использовать в одиночку или в комбинации с другими терапевтическими или профилактическими средствами для лечения или профилактики ВИЧ-инфекции. Соединения по настоящему изобретению обладают хорошей растворимостью и биологической доступностью и могут вводиться перорально в виде водного раствора.The present invention provides new compounds based on lysine, the source of which is a class of derivatives that are effective aspartyl protease inhibitors, and their pharmaceutically acceptable derivatives. These compounds can be readily cleaved in vivo , releasing the active ingredient. The active ingredient has an affinity for aspartyl proteases, in particular HIV-1 aspartyl protease (US Pat. No. 6,632,816). The active ingredients also show effective antiviral activity when tested on the non-mutant viral strain of HIV-1 (NL4.3 as the wild-type virus), as well as several mutant strains. Therefore, the compounds of the present invention can be used as a means to increase solubility and improve the bioavailability of the active ingredient (protease inhibitor). The compounds of the present invention can be used alone or in combination with other therapeutic or prophylactic agents for treating or preventing HIV infection. The compounds of the present invention have good solubility and bioavailability and can be administered orally in the form of an aqueous solution.
Основная цель данного изобретения состоит в предложении улучшенного класса соединений на основе лизина, которые способны высвобождать ингибитор аспартил-протеазы и, особенно, ингибиторы ВИЧ аспартил-протеазы. Соединения на основе лизина по настоящему изобретению могут содержать отщепляемое звено, посредством чего при отщеплении данного звена соединение способно высвобождать ингибитор ВИЧ-протеазы. Настоящее изобретение также предлагает фармацевтические композиции, включающие описанные здесь соединения на основе лизина.The main objective of this invention is to provide an improved class of lysine-based compounds that are capable of releasing an aspartyl protease inhibitor and especially HIV aspartyl protease inhibitors. The lysine-based compounds of the present invention may contain a cleavable unit, whereby when the unit is cleaved, the compound is capable of releasing an HIV protease inhibitor. The present invention also provides pharmaceutical compositions comprising the lysine-based compounds described herein.
Следовательно, настоящее изобретение в одном аспекте предлагает соединения на основе лизина, которые при in vivo физиологических условиях (например, метаболических, кишечных, желудочно-кишечных и т.д.) позволяют высвобождаться ингибитору протеазы (например, ингибитору аспартил-протеазы). Соединения по настоящему изобретению могут служить в качестве средства для улучшения растворимости и/или биологической доступности ингибиторов протеазы, и, поэтому, могут снижать лекарственную нагрузку, и могут благоприятствовать соблюдению больным режима и схемы лечения.Therefore, the present invention, in one aspect, provides lysine-based compounds which, under in vivo physiological conditions (e.g., metabolic, intestinal, gastrointestinal, etc.) allow a protease inhibitor (e.g., an aspartyl protease inhibitor) to be released. The compounds of the present invention can serve as a means to improve the solubility and / or bioavailability of protease inhibitors, and, therefore, can reduce the drug load, and may favor patient compliance with the treatment regimen and regimen.
Соединения по настоящему изобретению могут содержать, например, (например, физиологически) расщепляющуюся (например, гидролизующуюся) связь или звено, которые при отщеплении расщепляющейся связи или звена дают ингибитор протеазы (например, активный ингибитор протеазы).The compounds of the present invention may contain, for example, (for example, physiologically) a cleavable (for example, hydrolyzable) bond or link, which upon cleavage of the cleavable link or link give a protease inhibitor (for example, an active protease inhibitor).
Ингибитор протеазы может действовать на аспартил-протеазу ВИЧ-1, включая мутантные или немутантные вирусные штаммы ВИЧ-1 (например, NL4.3), или на протеазу ВИЧ-2 (мутантного или немутантного) или даже на протеазу родственных вирусов (SIV и т.д.). Соединения по настоящему изобретению можно использовать в одиночку или в комбинации с другими терапевтическими или профилактическими средствами для лечения или профилактики, например, ВИЧ-инфекции.A protease inhibitor can act on HIV-1 aspartyl protease, including mutant or non-mutant HIV-1 viral strains (e.g., NL4.3), or on HIV-2 protease (mutant or non-mutant), or even a protease of related viruses (SIV and t .d.). The compounds of the present invention can be used alone or in combination with other therapeutic or prophylactic agents for the treatment or prophylaxis of, for example, HIV infection.
Соединения и фармацевтические композиции по настоящему изобретению могут высвобождать ингибитор протеазы (активный ингредиент) in vivo и, посредством этого, могут подавлять (например, in vivo) активность ВИЧ аспартил-протеазы, фермента, существенного для развития и инфективности вируса. Соединения и фармацевтические композиции по настоящему изобретению могут обладать более высокой биологической доступностью, а также могут подходить для снижения необходимых для подавления дозировок и, следовательно, могут улучшать лечение ВИЧ-инфицированных пациентов.The compounds and pharmaceutical compositions of the present invention can release a protease inhibitor (active ingredient) in vivo and, thereby, can inhibit (for example, in vivo ) the activity of HIV aspartyl protease, an enzyme essential for the development and infectivity of the virus. The compounds and pharmaceutical compositions of the present invention may have higher bioavailability, and may also be suitable for lowering the dosage required to suppress and, therefore, may improve the treatment of HIV-infected patients.
Настоящее изобретение согласно своему одному аспекту предлагает соединение (например, соединение, способное генерировать ингибитор ВИЧ-протеазы) формулы I:The present invention, in one aspect, provides a compound (for example, a compound capable of generating an HIV protease inhibitor) of formula I:

и его фармацевтически приемлемые соли и производные (например, когда соединение по настоящему изобретению включает аминогруппу, фармацевтически приемлемая соль может представлять собой соль аммония),and pharmaceutically acceptable salts and derivatives thereof (for example, when the compound of the present invention includes an amino group, the pharmaceutically acceptable salt may be an ammonium salt),
где n может быть равно, например, 3 или 4,where n may be equal to, for example, 3 or 4,
где X и Y, одинаковые или различные, могут быть выбраны, например, из группы, состоящей из H, неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, циклоалкильной группы, содержащей от 3 до 6 атомов углерода, F, Cl, Br, I, -CF3, -OCF3, -CN, -NO2, -NR4R5, -NHCOR4, -OR4, -SR4, -COOR4, -COR4 и -CH2OH, или X и Y вместе определяют алкилендиоксигруппу, выбранную из группы, состоящей из метилендиоксигруппы формулы -OCH2O- и этилендиоксигруппы формулы -OCH2CH2O-, где R6 может быть выбран, например, из группы, состоящей из неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, циклоалкилалкильной группы, содержащей от 3 до 6 атомов углерода в своей циклоалкильной части и от 1 до 3 атомов углерода в своей алкильной части,where X and Y, the same or different, can be selected, for example, from the group consisting of H, an unbranched alkyl group containing from 1 to 6 carbon atoms, a branched alkyl group containing from 3 to 6 carbon atoms, a cycloalkyl group containing from 3 to 6 carbon atoms, F, Cl, Br, I, —CF 3 , —OCF 3 , —CN, —NO 2 , —NR 4 R 5 , —NHCOR 4 , —OR 4 , —SR 4 , —COOR 4 , —COR 4 and —CH 2 OH, or X and Y together define an alkylenedioxy group selected from the group consisting of a methylenedioxy group of the formula —OCH 2 O— and an ethylenedioxy group of the formula —OCH 2 CH 2 O—, where R 6 can be selected, e.g. from gro PP, consisting of an unbranched alkyl group containing from 1 to 6 carbon atoms, a branched alkyl group containing from 3 to 6 carbon atoms, a cycloalkylalkyl group containing from 3 to 6 carbon atoms in its cycloalkyl part and from 1 to 3 carbon atoms in its alkyl part,
где R3 может быть выбран, например, из группы, состоящей из H, неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, циклоалкильной группы, содержащей от 3 до 6 атомов углерода, и группы формулы R3A-CO-, где R3A может быть выбран, например, из группы, состоящей из неразветвленной или разветвленной алкильной группы, содержащей от 1 до 6 атомов углерода (например, метильной, этильной, пропильной, изопропильной, бутильной, изобутильной, трет-бутильной, трет-бутил-CH2 и т.д.), циклоалкильной группы, содержащей от 3 до 6 атомов углерода (например, циклопропильной, циклогексильной и т.д.), циклоалкилалкильной группы, содержащей от 3 до 6 атомов углерода в своей циклоалкильной части и от 1 до 3 атомов углерода в своей алкильной части (например, циклопропил-CH2-, циклогексил-CH2- и т.д.), алкилоксигруппы, содержащей от 1 до 6 атомов углерода (например, CH3O-, CH3CH2O-, изобутил-O-, трет-бутил-O- (Boc) и т.д.), тетрагидро-3-фуранилокси, -CH2OH, -CF3, -CH2CF3, -CH2CH2CF3, пирролидинила, пиперидинила, 4-морфолинила, CH3O2C-, CH3O2CCH2-, ацетил-OCH2CH2-, HO2CCH2-, 3-гидроксифенила, 4-гидроксифенила, 4-CH3OC6H4CH2-, CH3NH-, (CH3)2N-, (CH3CH2)2N-, (CH3CH2CH2)2N-, HOCH2CH2NH-, CH3OCH2O-, CH3OCH2CH2O-, C6H5CH2O-, 2-пирролила, 2-пиридила, 3-пиридила, 4-пиридила, 2-пиразинила, 2-хинолила, 3-хинолила, 4-хинолила, 1-изохинолила, 3-изохинолила, 2-хиноксалинила, фенильной группы формулыwhere R 3 can be selected, for example, from the group consisting of H, an unbranched alkyl group containing from 1 to 6 carbon atoms, a branched alkyl group containing from 3 to 6 carbon atoms, a cycloalkyl group containing from 3 to 6 carbon atoms and groups of the formula R 3A —CO—, where R 3A can be selected, for example, from the group consisting of a straight or branched alkyl group containing from 1 to 6 carbon atoms (for example, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tert- butyl, tert- butyl-CH 2 and t etc.), a cycloalkyl group containing from 3 to 6 carbon atoms (for example, cyclopropyl, cyclohexyl, etc.), a cycloalkylalkyl group containing from 3 to 6 carbon atoms in its cycloalkyl part and from 1 to 3 carbon atoms in its alkyl part (for example, cyclopropyl-CH 2 -, cyclohexyl-CH 2 -, etc.), alkyloxy group containing from 1 to 6 carbon atoms (for example, CH 3 O-, CH 3 CH 2 O-, isobutyl- O-, tert- butyl-O- (Boc), etc.), tetrahydro-3-furanyloxy, -CH 2 OH, -CF 3 , -CH 2 CF 3 , -CH 2 CH 2 CF 3 , pyrrolidinyl, piperidinyl, 4-morpholinyl, CH 3 O 2 C-, CH 3 O 2 CCH 2 -, acetyl-OCH 2 CH 2 -, HO 2 CCH 2 -, 3-hydroxyphenyl, 4-hydroxyphenyl, 4-CH 3 OC 6 H 4 CH 2 -, CH 3 NH-, (CH 3 ) 2 N-, (CH 3 CH 2 ) 2 N-, ( CH 3 CH 2 CH 2 ) 2 N-, HOCH 2 CH 2 NH-, CH 3 OCH 2 O-, CH 3 OCH 2 CH 2 O-, C 6 H 5 CH 2 O-, 2-pyrrolyl, 2-pyridyl , 3-pyridyl, 4-pyridyl, 2-pyrazinyl, 2-quinolyl, 3-quinolyl, 4-quinolyl, 1-isoquinolyl, 3-isoquinolyl, 2-quinoxalinyl, a phenyl group of the formula

пиколильной группы, выбранной из группы, состоящей изpicolyl group selected from the group consisting of

пиколилоксигруппы, выбранной из группы, состоящей изpicolyloxy group selected from the group consisting of

замещенной пиридильной группы, выбранной из группы, состоящей изsubstituted pyridyl group selected from the group consisting of

и группы формулыand groups of the formula

где X' и Y', одинаковые или различные, могут быть выбраны, например, из группы, состоящей из H, неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, циклоалкильной группы, содержащей от 3 до 6 атомов углерода, F, Cl, Br, I, -CF3, -NO2, -NR4R5, -NHCOR4, -OR4, -SR4, -COOR4, -COR4 и -CH2OH,where X 'and Y', the same or different, can be selected, for example, from the group consisting of H, an unbranched alkyl group containing from 1 to 6 carbon atoms, a branched alkyl group containing from 3 to 6 carbon atoms, a cycloalkyl group containing from 3 to 6 carbon atoms, F, Cl, Br, I, —CF 3 , —NO 2 , —NR 4 R 5 , —NHCOR 4 , —OR 4 , —SR 4 , —COOR 4 , —COR 4 and -CH 2 OH,
где R4 и R5, одинаковые или различные, могут быть выбраны, например, из группы, состоящей из H, неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, циклоалкильной группы, содержащей от 3 до 6 атомов углерода,where R 4 and R 5 , the same or different, can be selected, for example, from the group consisting of H, an unbranched alkyl group containing from 1 to 6 carbon atoms, a branched alkyl group containing from 3 to 6 carbon atoms, a cycloalkyl group containing from 3 to 6 carbon atoms,
где R2 может быть выбран, например, из группы, состоящей из дифенилметильной группы, формулы IVwhere R 2 can be selected, for example, from the group consisting of diphenylmethyl group of the formula IV

нафтил-1-CH2-группы формулы Vnaphthyl-1-CH 2 groups of formula V

нафтил-2-CH2-группы формулы VInaphthyl-2-CH 2 groups of formula VI

бифенилметильной группы формулы VIIbiphenylmethyl group of formula VII

и антрил-9-CH2-группы формулы VIIIand antryl-9-CH 2 groups of formula VIII

и где R1 может представлять собой отщепляемое звено (например, физиологически отщепляемое звено), посредством чего при отщеплении данного звена соединение высвобождает ингибитор протеазы (ингибитор протеазы ВИЧ), при условии, что R1 не является H. Например, R1 может представлять собой ферментно или метаболически отщепляемое звено или гидролизующуюся связь, которые могут быть отщеплены в условиях, существующих в кишечнике и/или желудочно-кишечном тракте (рН) или других физиологических условиях.and where R 1 may be a cleavable unit (for example, a physiologically cleavable unit), whereby when the unit is cleaved, the compound releases a protease inhibitor (HIV protease inhibitor), provided that R 1 is not H. For example, R 1 may be an enzyme or metabolically cleavable unit or a hydrolyzable bond that can be cleaved under conditions existing in the intestine and / or gastrointestinal tract (pH) or other physiological conditions.
По настоящему изобретению R1 можно выбрать, например, из группы, состоящей из (HO)2P(O) и (MO)2P(O), где M представляет собой щелочной металл (например, Na, K, Cs и т.д.) или щелочноземельный металл (Ca, Mg и т.д.).According to the present invention, R 1 can be selected, for example, from the group consisting of (HO) 2 P (O) and (MO) 2 P (O), where M is an alkali metal (e.g., Na, K, Cs, etc.). e.) or alkaline earth metal (Ca, Mg, etc.).
Далее по настоящему изобретению R1 может представлять собой группу формулы R1A-CO-, где R1A может быть выбран, например, из группы, состоящей из неразветвленной или разветвленной алкильной группы, содержащей от 1 до 6 атомов углерода (например, метильной, этильной, пропильной, изопропильной, бутильной, изобутильной, трет-бутильной, трет-бутил-CH2- и т.д.), циклоалкильной группы, содержащей от 3 до 6 атомов углерода (например, циклопропильной, циклогексильной и т.д.), циклоалкилалкильной группы, содержащей от 3 до 6 атомов углерода в своей циклоалкильной части и от 1 до 3 атомов углерода в своей алкильной части (например, циклопропил-CH2-, циклогексил-CH2- и т.д.), алкилоксигруппы, содержащей от 1 до 6 атомов углерода (например, CH3O-, CH3CH2O-, изобутил-O-, трет-бутил-O- (Boc) и т.д.), -CH2OH, CH3O2C-, CH3O2CCH2-, ацетил-OCH2CH2-, HO2CCH2-, 2-гидроксифенила, 3-гидроксифенила, 4-гидроксифенила, (CH3)2NCH2-, (CH3)2CHCH(NH2)-, HOCH2CH2NH-, CH3OCH2O-, CH3OCH2CH2O-, 2-пирролила, 2-пиридила, 3-пиридила, 4-пиридила, 1-метил-1,4-дигидро-3-пиридила, 2-пиразинила, 2-хинолила, 3-хинолила, 4-хинолила, 1-изохинолила, 3-изохинолила, 2-хиноксалинила, фенильной группы формулыFurther, according to the present invention, R 1 can be a group of the formula R 1A —CO—, where R 1A can be selected, for example, from the group consisting of a straight or branched alkyl group containing from 1 to 6 carbon atoms (for example, methyl, ethyl propyl, isopropyl, butyl, isobutyl, tert- butyl, tert- butyl-CH 2 - etc.), a cycloalkyl group containing from 3 to 6 carbon atoms (e.g. cyclopropyl, cyclohexyl, etc.), cycloalkylalkyl group containing from 3 to 6 carbon atoms in its cycloalkyl the second part and from 1 to 3 carbon atoms in its alkyl moiety (e.g. cyclopropyl-CH 2 -, cyclohexyl-CH 2 -, etc.), an alkyloxy group containing from 1 to 6 carbon atoms (e.g., CH 3 O- , CH 3 CH 2 O-, isobutyl-O-, tert- butyl-O- (Boc) etc.), -CH 2 OH, CH 3 O 2 C-, CH 3 O 2 CCH 2 -, acetyl -OCH 2 CH 2 -, HO 2 CCH 2 -, 2-hydroxyphenyl, 3-hydroxyphenyl, 4-hydroxyphenyl, (CH 3 ) 2 NCH 2 -, (CH 3 ) 2 CHCH (NH 2 ) -, HOCH 2 CH 2 NH-, CH 3 OCH 2 O-, CH 3 OCH 2 CH 2 O-, 2-pyrrolyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 1-methyl-1,4-dihydro-3-pyridyl, 2 -pyrazinyl, 2-quinolyl, 3-quinolyl, 4-quinolyl, 1-isoquinolyl, 3-isoquinolyl, 2-quinoxalinyl, phenyl group pp formulas

пиколильной группы, выбранной из группы, состоящей изpicolyl group selected from the group consisting of

пиколилоксигруппы, выбранной из группы, состоящей изpicolyloxy group selected from the group consisting of

замещенной пиридильной группы, выбранной из группы, состоящей изsubstituted pyridyl group selected from the group consisting of

и группы формулыand groups of the formula

где X', Y', R4 и R5 являются такими, как определено в настоящем описании.where X ', Y', R 4 and R 5 are as defined herein.
В другом аспекте настоящее изобретение далее предлагает соединение формулы II,In another aspect, the present invention further provides a compound of formula II,

и его фармацевтически приемлемые соли и производные (например, когда соединение по настоящему изобретению включает аминогруппу, фармацевтически приемлемая соль может представлять собой соль аммония),and pharmaceutically acceptable salts and derivatives thereof (for example, when the compound of the present invention includes an amino group, the pharmaceutically acceptable salt may be an ammonium salt),
где n может быть равно 3 или 4,where n may be 3 or 4,
где X и Y, одинаковые или различные, могут быть выбраны, например, из группы, состоящей из H, неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, циклоалкильной группы, содержащей от 3 до 6 атомов углерода, F, Cl, Br, I, -CF3, -OCF3, -CN, -NO2, -NR4R5, -NHCOR4, -OR4, -SR4, -COOR4, -COR4 и -CH2OH, или X и Y вместе определяют алкилендиоксигруппу, выбранную из группы, состоящей из метилендиоксигруппы формулы -OCH2O- и этилендиоксигруппы формулы -OCH2CH2O-,where X and Y, the same or different, can be selected, for example, from the group consisting of H, an unbranched alkyl group containing from 1 to 6 carbon atoms, a branched alkyl group containing from 3 to 6 carbon atoms, a cycloalkyl group containing from 3 to 6 carbon atoms, F, Cl, Br, I, —CF 3 , —OCF 3 , —CN, —NO 2 , —NR 4 R 5 , —NHCOR 4 , —OR 4 , —SR 4 , —COOR 4 , —COR 4 and —CH 2 OH, or X and Y together define an alkylenedioxy group selected from the group consisting of a methylenedioxy group of the formula —OCH 2 O— and an ethylenedioxy group of the formula —OCH 2 CH 2 O—,
где R6 может быть выбран, например, из группы, состоящей из неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, циклоалкилалкильной группы, содержащей от 3 до 6 атомов углерода в своей циклоалкильной части и от 1 до 3 атомов углерода в своей алкильной части,where R 6 can be selected, for example, from the group consisting of an unbranched alkyl group containing from 1 to 6 carbon atoms, a branched alkyl group containing from 3 to 6 carbon atoms, a cycloalkylalkyl group containing from 3 to 6 carbon atoms in its cycloalkyl part and from 1 to 3 carbon atoms in its alkyl part,
где R3 может быть выбран, например, из группы, состоящей из H, неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, циклоалкильной группы, содержащей от 3 до 6 атомов углерода, и группы формулы R3A-CO-, где R3A может быть выбран, например, из группы, состоящей из неразветвленной или разветвленной алкильной группы, содержащей от 1 до 6 атомов углерода (например, метильной, этильной, пропильной, изопропильной, бутильной, изобутильной, трет-бутильной, трет-бутил-CH2- и т.д.), циклоалкильной группы, содержащей от 3 до 6 атомов углерода (например, циклопропильной, циклогексильной и т.д.), циклоалкилалкильной группы, содержащей от 3 до 6 атомов углерода в своей циклоалкильной части и от 1 до 3 атомов углерода в своей алкильной части (например, циклопропил-CH2-, циклогексил-CH2- и т.д.), алкилоксигруппы, содержащей от 1 до 6 атомов углерода (например, CH3O-, CH3CH2O-, изобутил-O-, трет-бутил-O- (Boc) и т.д.), тетрагидро-3-фуранилокси, -CH2OH, -CF3, -CH2CF3, -CH2CH2CF3, пирролидинила, пиперидинила, 4-морфолинила, CH3O2C-, CH3O2CCH2-, ацетил-OCH2CH2-, HO2CCH2-, 3-гидроксифенила, 4-гидроксифенила, 4-CH3OC6H4CH2-, CH3NH-, (CH3)2N-, (CH3CH2)2N-, (CH3CH2CH2)2N-, HOCH2CH2NH-, CH3OCH2O-, CH3OCH2CH2O-, C6H5CH2O-, 2-пирролила, 2-пиридила, 3-пиридила, 4-пиридила, 2-пиразинила, 2-хинолила, 3-хинолила, 4-хинолила, 1-изохинолила, 3-изохинолила, 2-хиноксалинила, фенильной группы формулыwhere R 3 can be selected, for example, from the group consisting of H, an unbranched alkyl group containing from 1 to 6 carbon atoms, a branched alkyl group containing from 3 to 6 carbon atoms, a cycloalkyl group containing from 3 to 6 carbon atoms and groups of the formula R 3A —CO—, where R 3A can be selected, for example, from the group consisting of a straight or branched alkyl group containing from 1 to 6 carbon atoms (for example, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tert- butyl, tert- butyl-CH 2 - and etc.), a cycloalkyl group containing from 3 to 6 carbon atoms (e.g., cyclopropyl, cyclohexyl, etc.), a cycloalkylalkyl group containing from 3 to 6 carbon atoms in its cycloalkyl part and from 1 to 3 carbon atoms in its alkyl part (for example, cyclopropyl-CH 2 -, cyclohexyl-CH 2 - etc.), an alkyloxy group containing from 1 to 6 carbon atoms (for example, CH 3 O-, CH 3 CH 2 O-, isobutyl -O-, tert- butyl-O- (Boc), etc.), tetrahydro-3-furanyloxy, -CH 2 OH, -CF 3 , -CH 2 CF 3 , -CH 2 CH 2 CF 3 , pyrrolidinyl , piperidinyl, 4-morpholinyl, CH 3 O 2 C-, CH 3 O 2 CCH 2 -, acetyl-OCH 2 CH 2 - , HO 2 CCH 2 -, 3-hydroxyphenyl, 4-hydroxyphenyl, 4-CH 3 OC 6 H 4 CH 2 -, CH 3 NH-, (CH 3 ) 2 N-, (CH 3 CH 2 ) 2 N-, (CH 3 CH 2 CH 2 ) 2 N-, HOCH 2 CH 2 NH-, CH 3 OCH 2 O-, CH 3 OCH 2 CH 2 O-, C 6 H 5 CH 2 O-, 2-pyrrolyl, 2- pyridyl, 3-pyridyl, 4-pyridyl, 2-pyrazinyl, 2-quinolyl, 3-quinolyl, 4-quinolyl, 1-isoquinolyl, 3-isoquinolyl, 2-quinoxalinyl, phenyl group

пиколильной группы, выбранной из группы, состоящей изpicolyl group selected from the group consisting of

пиколилоксигруппы, выбранной из группы, состоящей изpicolyloxy group selected from the group consisting of

замещенной пиридильной группы, выбранной из группы, состоящей изsubstituted pyridyl group selected from the group consisting of

и группы формулыand groups of the formula

где X' и Y', одинаковые или различные, могут быть выбраны, например, из группы, состоящей из H, неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, циклоалкильной группы, содержащей от 3 до 6 атомов углерода, F, Cl, Br, I, -CF3, -NO2, -NR4R5, -NHCOR4, -OR4, -SR4, -COOR4, -COR4 и -CH2OH,where X 'and Y', the same or different, can be selected, for example, from the group consisting of H, an unbranched alkyl group containing from 1 to 6 carbon atoms, a branched alkyl group containing from 3 to 6 carbon atoms, a cycloalkyl group containing from 3 to 6 carbon atoms, F, Cl, Br, I, —CF 3 , —NO 2 , —NR 4 R 5 , —NHCOR 4 , —OR 4 , —SR 4 , —COOR 4 , —COR 4 and -CH 2 OH,
где R4 и R5, одинаковые или различные, могут быть выбраны, например, из группы, состоящей из H, неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, и циклоалкильной группы, содержащей от 3 до 6 атомов углерода,where R 4 and R 5 , the same or different, can be selected, for example, from the group consisting of H, an unbranched alkyl group containing from 1 to 6 carbon atoms, a branched alkyl group containing from 3 to 6 carbon atoms, and cycloalkyl a group containing from 3 to 6 carbon atoms,
где R2 может быть выбран, например, из группы, состоящей из дифенилметильной группы, формулы IVwhere R 2 can be selected, for example, from the group consisting of diphenylmethyl group of the formula IV

нафтил-1-CH2-группы формулы Vnaphthyl-1-CH 2 groups of formula V

нафтил-2-CH2-группы формулы VInaphthyl-2-CH 2 groups of formula VI

бифенилметильной группы формулы VIIbiphenylmethyl group of formula VII

и антрил-9-CH2-группы формулы VIIIand antryl-9-CH 2 groups of formula VIII
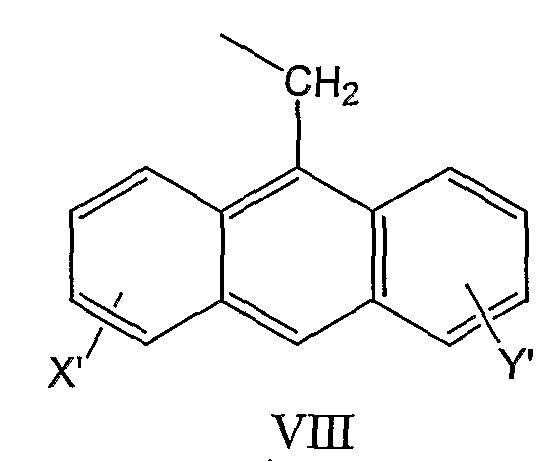
и где R1 может представлять собой физиологически отщепляемое звено, посредством чего при отщеплении данного звена соединение может быть способно высвобождать ингибитор протеазы при условии, что R1 не является H.and where R 1 may be a physiologically cleavable unit, whereby when the unit is cleaved, the compound may be capable of releasing a protease inhibitor, provided that R 1 is not H.
По настоящему изобретению R1 можно выбрать, например, из группы, состоящей из (HO)2P(O) и (MO)2P(O), где M представляет собой щелочной металл (например, Na, K, Cs и т.д.) или щелочноземельный металл (Ca, Mg и т.д.).According to the present invention, R 1 can be selected, for example, from the group consisting of (HO) 2 P (O) and (MO) 2 P (O), where M is an alkali metal (e.g., Na, K, Cs, etc.). e.) or alkaline earth metal (Ca, Mg, etc.).
Далее по настоящему изобретению R1 может представлять собой группу формулы R1A-CO-, где R1A может быть выбран из группы, состоящей из неразветвленной или разветвленной алкильной группы, содержащей от 1 до 6 атомов углерода (например, метильной, этильной, пропильной, изопропильной, бутильной, изобутильной, трет-бутильной, трет-бутил-CH2- и т.д.), циклоалкильной группы, содержащей от 3 до 6 атомов углерода (например, циклопропильной, циклогексильной и т.д.), циклоалкилалкильной группы, содержащей от 3 до 6 атомов углерода в своей циклоалкильной части и от 1 до 3 атомов углерода в своей алкильной части (например, циклопропил-CH2-, циклогексил-CH2- и т.д.), алкилоксигруппы, содержащей от 1 до 6 атомов углерода (например, CH3O-, CH3CH2O-, изобутил-O-, трет-бутил-O- (Boc) и т.д.), -CH2OH, CH3O2C-, CH3O2CCH2-, ацетил-OCH2CH2-, HO2CCH2-, 2-гидроксифенила, 3-гидроксифенила, 4-гидроксифенила, (CH3)2NCH2-, (CH3)2CHCH(NH2)-, HOCH2CH2NH-, CH3OCH2O-, CH3OCH2CH2O-, 2-пирролила, 2-пиридила, 3-пиридила, 4-пиридила, 1-метил-1,4-дигидро-3-пиридила, 2-пиразинила, 2-хинолила, 3-хинолила, 4-хинолила, 1-изохинолила, 3-изохинолила, 2-хиноксалинила, фенильной группы формулыFurther, according to the present invention, R 1 can be a group of the formula R 1A —CO—, where R 1A can be selected from the group consisting of an unbranched or branched alkyl group containing from 1 to 6 carbon atoms (e.g. methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tert- butyl, tert- butyl-CH 2 - etc.), a cycloalkyl group containing from 3 to 6 carbon atoms (for example, cyclopropyl, cyclohexyl, etc.), cycloalkylalkyl group, containing from 3 to 6 carbon atoms in its cycloalkyl part and from 1 to 3 carbon atoms in its alkyl part (for example, cyclopropyl-CH 2 -, cyclohexyl-CH 2 -, etc.), alkyloxy group containing from 1 to 6 carbon atoms (for example, CH 3 O-, CH 3 CH 2 O-, isobutyl-O-, tert- butyl-O- (Boc), etc.), -CH 2 OH, CH 3 O 2 C-, CH 3 O 2 CCH 2 -, acetyl-OCH 2 CH 2 -, HO 2 CCH 2 -, 2-hydroxyphenyl, 3-hydroxyphenyl, 4-hydroxyphenyl, (CH 3 ) 2 NCH 2 -, (CH 3 ) 2 CHCH (NH 2 ) -, HOCH 2 CH 2 NH-, CH 3 OCH 2 O-, CH 3 OCH 2 CH 2 O-, 2-pyrrolyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 1-methyl-1,4-dihydro-3-pyridyl, 2-pyrazinyl, 2-quinolyl, 3-quinolyl, 4-quinolyl, 1-isoquinolyl, 3-isoquinolyl, 2-quinoxalinyl, phenyl group of the formulas s

пиколильной группы, выбранной из группы, состоящей изpicolyl group selected from the group consisting of

пиколилоксигруппы, выбранной из группы, состоящей изpicolyloxy group selected from the group consisting of

замещенной пиридильной группы, выбранной из группы, состоящей изsubstituted pyridyl group selected from the group consisting of

и группы формулыand groups of the formula
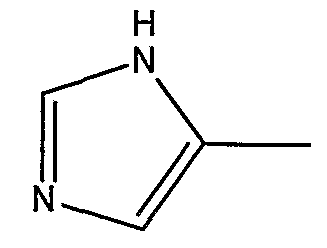
где X', Y', R4 и R5 являются такими, как определено в настоящем описании.where X ', Y', R 4 and R 5 are as defined herein.
В дальнейшем аспекте настоящее изобретение предлагает соединение формулы IIa:In a further aspect, the present invention provides a compound of formula IIa:

его фармацевтически приемлемые соли и производные (например, когда соединение по настоящему изобретению включает аминогруппу, фармацевтически приемлемая соль может представлять собой соль аммония),its pharmaceutically acceptable salts and derivatives (for example, when the compound of the present invention includes an amino group, the pharmaceutically acceptable salt may be an ammonium salt),
где X и Y, одинаковые или различные, могут быть выбраны, например, из группы, состоящей из H, неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, циклоалкильной группы, содержащей от 3 до 6 атомов углерода, F, Cl, Br, I, -CF3, -OCF3, -CN, -NO2, -NR4R5, -NHCOR4, -OR4, -SR4, -COOR4, -COR4 и -CH2OH, или X и Y вместе определяют алкилендиоксигруппу, выбранную из группы, состоящей из метилендиоксигруппы формулы -OCH2O- и этилендиоксигруппы формулы -OCH2CH2O-,where X and Y, the same or different, can be selected, for example, from the group consisting of H, an unbranched alkyl group containing from 1 to 6 carbon atoms, a branched alkyl group containing from 3 to 6 carbon atoms, a cycloalkyl group containing from 3 to 6 carbon atoms, F, Cl, Br, I, —CF 3 , —OCF 3 , —CN, —NO 2 , —NR 4 R 5 , —NHCOR 4 , —OR 4 , —SR 4 , —COOR 4 , —COR 4 and —CH 2 OH, or X and Y together define an alkylenedioxy group selected from the group consisting of a methylenedioxy group of the formula —OCH 2 O— and an ethylenedioxy group of the formula —OCH 2 CH 2 O—,
где X' и Y', одинаковые или различные, могут быть выбраны, например, из группы, состоящей из H, неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, циклоалкильной группы, содержащей от 3 до 6 атомов углерода, F, Cl, Br, I, -CF3, -NO2, -NR4R5, -NHCOR4, -OR4, -SR4, -COOR4, -COR4 и -CH2OH,where X 'and Y', the same or different, can be selected, for example, from the group consisting of H, an unbranched alkyl group containing from 1 to 6 carbon atoms, a branched alkyl group containing from 3 to 6 carbon atoms, a cycloalkyl group containing from 3 to 6 carbon atoms, F, Cl, Br, I, —CF 3 , —NO 2 , —NR 4 R 5 , —NHCOR 4 , —OR 4 , —SR 4 , —COOR 4 , —COR 4 and -CH 2 OH,
и где n, R1, R3, R4, R5 и R6 являются такими, как определено в настоящем описании.and where n, R 1 , R 3 , R 4 , R 5 and R 6 are as defined herein.
В дополнительном аспекте настоящее изобретение предлагает соединение формулы IIb:In an additional aspect, the present invention provides a compound of formula IIb:

его фармацевтически приемлемые соли и производные (например, когда соединение по настоящему изобретению включает аминогруппу, фармацевтически приемлемая соль может представлять собой соль аммония),its pharmaceutically acceptable salts and derivatives (for example, when the compound of the present invention includes an amino group, the pharmaceutically acceptable salt may be an ammonium salt),
где X и Y, одинаковые или различные, могут быть выбраны, например, из группы, состоящей из H, неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, циклоалкильной группы, содержащей от 3 до 6 атомов углерода, F, Cl, Br, I, -CF3, -OCF3, -CN, -NO2, -NR4R5, -NHCOR4, -OR4, -SR4, -COOR4, -COR4 и -CH2OH, или X и Y вместе определяют алкилендиоксигруппу, выбранную из группы, состоящей из метилендиоксигруппы формулы -OCH2O- и этилендиоксигруппы формулы -OCH2CH2O-,where X and Y, the same or different, can be selected, for example, from the group consisting of H, an unbranched alkyl group containing from 1 to 6 carbon atoms, a branched alkyl group containing from 3 to 6 carbon atoms, a cycloalkyl group containing from 3 to 6 carbon atoms, F, Cl, Br, I, —CF 3 , —OCF 3 , —CN, —NO 2 , —NR 4 R 5 , —NHCOR 4 , —OR 4 , —SR 4 , —COOR 4 , —COR 4 and —CH 2 OH, or X and Y together define an alkylenedioxy group selected from the group consisting of a methylenedioxy group of the formula —OCH 2 O— and an ethylenedioxy group of the formula —OCH 2 CH 2 O—,
где X' и Y', одинаковые или различные, могут быть выбраны, например, из группы, состоящей из H, неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода, циклоалкильной группы, содержащей от 3 до 6 атомов углерода, F, Cl, Br, I, -CF3, -NO2, -NR4R5, -NHCOR4, -OR4, -SR4, -COOR4, -COR4 и -CH2OH,where X 'and Y', the same or different, can be selected, for example, from the group consisting of H, an unbranched alkyl group containing from 1 to 6 carbon atoms, a branched alkyl group containing from 3 to 6 carbon atoms, a cycloalkyl group containing from 3 to 6 carbon atoms, F, Cl, Br, I, —CF 3 , —NO 2 , —NR 4 R 5 , —NHCOR 4 , —OR 4 , —SR 4 , —COOR 4 , —COR 4 and -CH 2 OH,
и где n, R1, R3, R4, R5 и R6 являются такими, как определено в настоящем описании.and where n, R 1 , R 3 , R 4 , R 5 and R 6 are as defined herein.
В еще одном дополнительном аспекте настоящее изобретение предлагает соединение формулы IIc:In yet a further aspect, the present invention provides a compound of formula IIc:

его фармацевтически приемлемые соли и производные (например, когда соединение по настоящему изобретению включает аминогруппу, фармацевтически приемлемая соль может представлять собой соль аммония),its pharmaceutically acceptable salts and derivatives (for example, when the compound of the present invention includes an amino group, the pharmaceutically acceptable salt may be an ammonium salt),
и где n, X, Y, X', Y', R1, R3, R4, R5 и R6 являются такими, как определено в настоящем описании.and where n, X, Y, X ', Y', R 1 , R 3 , R 4 , R 5 and R 6 are as defined herein.
В другом аспекте настоящее изобретение относится к соединению формулы IIAIn another aspect, the present invention relates to a compound of formula IIA

где Y, n, R1, R2, R3, X' и Y' являются такими, как определено в настоящем описании.where Y, n, R 1 , R 2 , R 3 , X 'and Y' are as defined herein.
По настоящему изобретению R1 может представлять собой, например, (HO)2P(O) или (NaO)2P(O). Далее по настоящему изобретению n может быть равно 4. Y может, например, представлять собой H. R3 может, например, представлять собой CH3O-CO. R2 может, например, представлять собой дифенилметильную группу формулы IV, где X' и Y' могут представлять собой, например, H,According to the present invention, R 1 may be, for example, (HO) 2 P (O) or (NaO) 2 P (O). Further, in the present invention, n may be 4. Y may, for example, be H. R 3 may, for example, be CH 3 O-CO. R 2 may, for example, be a diphenylmethyl group of formula IV, where X 'and Y' may be, for example, H,

Следовательно, соединения формулы IIA', а также их фармацевтически приемлемые соли и производные охватываются настоящим изобретением,Therefore, the compounds of formula IIA ', as well as their pharmaceutically acceptable salts and derivatives are encompassed by the present invention,

такие как, например, соединение формулы IIA', где R1 представляет собой (HO)2P(O), или соединение формулы IIA', где R1 представляет собой (NaO)2P(O).such as, for example, a compound of formula IIA ', where R 1 is (HO) 2 P (O), or a compound of formula IIA', where R 1 is (NaO) 2 P (O).
В еще одном отличающемся аспекте настоящее изобретение относится к фармацевтической композиции, включающей, по меньшей мере, одно соединение формулы I, II, IIa, IIb, IIc, IIA, IIA' или комбинацию соединений формулы I, II, IIa, IIb, IIc, IIA и/или IIA'. Фармацевтическая композиция может включать фармацевтически приемлемый носитель. Фармацевтическая композиция может включать, например, фармацевтически эффективное количество такого одного или нескольких соединений или, в соответствующих случаях, их фармацевтически приемлемых солей аммония.In another distinct aspect, the present invention relates to a pharmaceutical composition comprising at least one compound of formula I, II, IIa, IIb, IIc, IIA, IIA 'or a combination of compounds of formula I, II, IIa, IIb, IIc, IIA and / or IIA '. The pharmaceutical composition may include a pharmaceutically acceptable carrier. The pharmaceutical composition may include, for example, a pharmaceutically effective amount of such one or more compounds or, where appropriate, their pharmaceutically acceptable ammonium salts.
Например, фармацевтическая композиция по настоящему изобретению может включать одно или несколько из следующих ниже соединений:For example, the pharmaceutical composition of the present invention may include one or more of the following compounds:
- соединение формулы IIa, где n равно 4, R1 представляет собой (HO)2P(O), X представляет собой 4-NH2, Y представляет собой H, X' представляет собой H, Y' представляет собой H, R6 представляет собой изобутил и R3 представляет собой CH3O-CO,- a compound of formula IIa, where n is 4, R 1 is (HO) 2 P (O), X is 4-NH 2 , Y is H, X 'is H, Y' is H, R 6 represents isobutyl and R 3 represents CH 3 O-CO,
- соединение формулы IIa, где n равно 4, R1 представляет собой (NaO)2P(O), X представляет собой 4-NH2, Y представляет собой H, X' представляет собой H, Y' представляет собой H, R6 представляет собой изобутил и R3 представляет собой CH3O-CO,- a compound of formula IIa, where n is 4, R 1 represents (NaO) 2 P (O), X represents 4-NH 2 , Y represents H, X 'represents H, Y' represents H, R 6 represents isobutyl and R 3 represents CH 3 O-CO,
- соединение формулы IIa, где n равно 4, R1 представляет собой (HO)2P(O), X представляет собой 4-NH2, Y представляет собой H, X' представляет собой H, Y' представляет собой H, R6 представляет собой изобутил и R3 представляет собой CH3CO,- a compound of formula IIa, where n is 4, R 1 is (HO) 2 P (O), X is 4-NH 2 , Y is H, X 'is H, Y' is H, R 6 represents isobutyl and R 3 represents CH 3 CO,
- соединение формулы IIa, где n равно 4, R1 представляет собой (HO)2P(O), X представляет собой 4-NH2, Y представляет собой 3-F, X' представляет собой H, Y' представляет собой H, R6 представляет собой изобутил и R3 представляет собой CH3O-CO,- a compound of formula IIa, where n is 4, R 1 is (HO) 2 P (O), X is 4-NH 2 , Y is 3-F, X 'is H, Y' is H, R 6 represents isobutyl and R 3 represents CH 3 O-CO,
- соединение формулы IIa, где n равно 4, R1 представляет собой CH3CO, X представляет собой 4-NH2, Y представляет собой H, X' представляет собой H, Y' представляет собой H, R6 представляет собой изобутил и R3 представляет собой CH3O-CO,- a compound of formula IIa, where n is 4, R 1 represents CH 3 CO, X represents 4-NH 2 , Y represents H, X 'represents H, Y' represents H, R 6 represents isobutyl and R 3 represents CH 3 O-CO,
- соединение формулы IIa, где n равно 4, R1 представляет собой 3-пиридил-CO, X представляет собой 4-NH2, Y представляет собой H, X' представляет собой H, Y' представляет собой H, R6 представляет собой изобутил и R3 представляет собой CH3O-CO,- a compound of formula IIa, where n is 4, R 1 represents 3-pyridyl-CO, X represents 4-NH 2 , Y represents H, X 'represents H, Y' represents H, R 6 represents isobutyl and R 3 represents CH 3 O-CO,
- соединение формулы IIa, где n равно 4, R1 представляет собой (CH3)2NCH2CO, X представляет собой 4-NH2, Y представляет собой H, X' представляет собой H, Y' представляет собой H, R6 представляет собой изобутил и R3 представляет собой CH3O-CO,- a compound of formula IIa, where n is 4, R 1 represents (CH 3 ) 2 NCH 2 CO, X represents 4-NH 2 , Y represents H, X 'represents H, Y' represents H, R 6 represents isobutyl and R 3 represents CH 3 O-CO,
- соединение формулы IIa, где n равно 4, R1 представляет собой (CH3)2CHCH(NH2)CO, X представляет собой 4-NH2, Y представляет собой H, X' представляет собой H, Y' представляет собой H, R6 представляет собой изобутил и R3 представляет собой CH3O-CO,- a compound of formula IIa, where n is 4, R 1 represents (CH 3 ) 2 CHCH (NH 2 ) CO, X represents 4-NH 2 , Y represents H, X 'represents H, Y' represents H , R 6 represents isobutyl and R 3 represents CH 3 O-CO,
- соединение формулы IIb, где n равно 4, R1 представляет собой (HO)2P(O), X представляет собой 4-NH2, Y представляет собой H, X' представляет собой H, Y' представляет собой H, R6 представляет собой изобутил и R3 представляет собой CH3O-CO, и где нафтильная группа представляет собой нафтил-2-CH2 группу,- a compound of formula IIb, where n is 4, R 1 is (HO) 2 P (O), X is 4-NH 2 , Y is H, X 'is H, Y' is H, R 6 represents isobutyl and R 3 represents CH 3 O-CO, and where the naphthyl group is a naphthyl-2-CH 2 group,
- соединение формулы IIb, где n равно 4, R1 представляет собой (HO)2P(O), X представляет собой 4-NH2, Y представляет собой H, X' представляет собой H, Y' представляет собой H, R6 представляет собой изобутил и R3 представляет собой 4-морфолин-CO, и где нафтильная группа представляет собой нафтил-1-CH2 группу, или- a compound of formula IIb, where n is 4, R 1 is (HO) 2 P (O), X is 4-NH 2 , Y is H, X 'is H, Y' is H, R 6 represents isobutyl and R 3 represents 4-morpholine-CO, and where the naphthyl group is a naphthyl-1-CH 2 group, or
- комбинацию любого из вышеуказанных соединений.- a combination of any of the above compounds.
В дополнительном аспекте настоящее изобретение относится к использованию, по меньшей мере, одного соединения формулы I, II, IIa, IIb, IIc, IIA, IIA' или комбинации соединений формулы I, II, IIa, IIb, IIc, IIA и/или IIA' или его фармацевтически приемлемых солей или производных (а также их комбинаций) при изготовлении лекарственного препарата (или фармацевтической композиции) для лечения или профилактики ВИЧ-инфекции.In an additional aspect, the present invention relates to the use of at least one compound of formula I, II, IIa, IIb, IIc, IIA, IIA 'or a combination of compounds of formula I, II, IIa, IIb, IIc, IIA and / or IIA' or its pharmaceutically acceptable salts or derivatives (and combinations thereof) in the manufacture of a medicament (or pharmaceutical composition) for the treatment or prevention of HIV infection.
В дальнейшем аспекте настоящее изобретение относится к использованию, по меньшей мере, одного соединения формулы I, II, IIa, IIb, IIc, IIA, IIA' или комбинации соединений формулы I, II, IIa, IIb, IIc, IIA и/или IIA' или его фармацевтически приемлемых солей или производных при лечении или профилактике ВИЧ-инфекции у млекопитающих, нуждающихся в этом, или для задержки возникновения СПИДа.In a further aspect, the present invention relates to the use of at least one compound of formula I, II, IIa, IIb, IIc, IIA, IIA 'or a combination of compounds of formula I, II, IIa, IIb, IIc, IIA and / or IIA' or its pharmaceutically acceptable salts or derivatives in the treatment or prevention of HIV infection in mammals in need thereof, or to delay the onset of AIDS.
В еще одном дополнительном аспекте настоящее изобретение относится к способу лечения или профилактики ВИЧ-инфекции (или для задержки возникновения СПИДа), включающему введение, по меньшей мере, одного соединения формулы I, II, IIa, IIb, IIc, IIA, IIA' или комбинации соединений формулы I, II, IIa, IIb, IIc, IIA и/или IIA' или его фармацевтически приемлемых солей или производных млекопитающему, нуждающемуся в этом.In yet a further aspect, the present invention relates to a method for treating or preventing HIV infection (or for delaying the onset of AIDS), comprising administering at least one compound of formula I, II, IIa, IIb, IIc, IIA, IIA ', or a combination compounds of formula I, II, IIa, IIb, IIc, IIA and / or IIA 'or its pharmaceutically acceptable salts or derivatives thereof to a mammal in need thereof.
В другом аспекте настоящее изобретение относится к соединению формулы I, II, IIa, IIb, IIc, IIA или IIA', его фармацевтически приемлемым солям или производным, для использования при лечении или профилактике ВИЧ-инфекции.In another aspect, the present invention relates to a compound of formula I, II, IIa, IIb, IIc, IIA or IIA ', its pharmaceutically acceptable salts or derivatives thereof, for use in the treatment or prevention of HIV infection.
В еще одном дополнительном аспекте настоящее изобретение относится к способу изготовления соединения на основе лизина, использующему любое из соединений, описанных в патенте США № 6632816, выданном Stranix et al., или к способу изготовления соединения, способного образовывать любое из соединений, описанных в патенте США № 6632816, выданном Stranix et al., при отщеплении (in vivo) отщепляемого звена.In yet a further aspect, the present invention relates to a method for the manufacture of a lysine-based compound using any of the compounds described in US Pat. No. 6,632,816 to Stranix et al. or to a method for manufacturing a compound capable of forming any of the compounds described in US Pat. No. 6,632,816 to Stranix et al. , when cleaving ( in vivo ) cleavable link.
Соединения, перечисленные в настоящем описании, являются иллюстративными вариантами осуществления настоящего изобретения на практике и необходимо понимать, что настоящее изобретение не ограничивается только данными соединениями.The compounds listed in the present description are illustrative embodiments of the present invention in practice and it should be understood that the present invention is not limited to these compounds.
Термин ″фармацевтически эффективное количество″ относится к количеству, эффективному для лечения или профилактики ВИЧ-инфекции у пациента или для уменьшения или исключения симптомов СПИД. Также здесь необходимо понимать, что ″фармацевтически эффективное количество″ может быть истолковано как количество, дающее желаемый терапевтический эффект, принимаемое в виде однократной дозы либо многократных доз, или в любой дозировке или маршрутом, или принимаемое в одиночку или в комбинации с другими терапевтическими средствами. В случае настоящего изобретения ″фармацевтически эффективное количество″ можно понимать как количество, обладающее ингибирующим действием на инфекционный цикл ВИЧ (ВИЧ-1 и ВИЧ-2, а также родственных вирусов (например, HTLV-I и HTLV-II и обезьяньего вируса иммунодефицита (SIV))) (например, подавление размножения, реинфекции, созревания, репликации и т.д.) и на любой организм, жизненный цикл которого зависит от аспартил-протеаз. Ингибирующее действие в контексте настоящего описания необходимо понимать как снижение способности организма (например, ВИЧ) воспроизводить себя (реплицировать), реинфицировать окружающие клетки и т.д. или даже полное подавление (или уничтожение) организма.The term “pharmaceutically effective amount” refers to an amount effective to treat or prevent HIV infection in a patient or to reduce or eliminate the symptoms of AIDS. It is also to be understood here that a “pharmaceutically effective amount” can be interpreted as an amount giving the desired therapeutic effect, taken as a single dose or multiple doses, or in any dosage or route, or taken alone or in combination with other therapeutic agents. In the case of the present invention, a "pharmaceutically effective amount" can be understood as an amount having an inhibitory effect on the HIV infection cycle (HIV-1 and HIV-2, as well as related viruses (e.g., HTLV-I and HTLV-II and simian immunodeficiency virus (SIV ))) (for example, suppression of reproduction, reinfection, maturation, replication, etc.) and on any organism whose life cycle depends on aspartyl proteases. The inhibitory effect in the context of the present description should be understood as a decrease in the body's ability (for example, HIV) to reproduce itself (replicate), reinfect the surrounding cells, etc. or even complete suppression (or destruction) of the body.
Термины ″ВИЧ-протеаза″ и ″ВИЧ аспартил-протеаза″ используются взаимозаменяемо и включают аспартил-протеазу, кодированную вирусом иммунодефицита человека типа 1 или 2.The terms "HIV protease" and "HIV aspartyl protease" are used interchangeably and include aspartyl protease encoded by type 1 or 2 human immunodeficiency virus.
Термин ″профилактически эффективное количество″ относится к количеству, эффективному для профилактики ВИЧ-инфекции у пациента. Используемый здесь термин ″пациент″ относится к млекопитающему, включая человека.The term "prophylactically effective amount" refers to an amount effective to prevent HIV infection in a patient. As used herein, the term “patient” refers to a mammal, including humans.
Термины ″фармацевтически приемлемый носитель″, ″фармацевтически приемлемое вспомогательное вещество″ и ″физиологически приемлемый растворитель″ относятся к нетоксичному носителю или вспомогательному веществу, которые можно вводить пациенту вместе с одним или несколькими соединениями по настоящему изобретению, и которые не разрушают их фармакологическую активность.The terms "pharmaceutically acceptable carrier", "pharmaceutically acceptable excipient" and "physiologically acceptable solvent" refer to a non-toxic carrier or excipient that can be administered to a patient together with one or more compounds of the present invention and which do not destroy their pharmacological activity.
Настоящее изобретение предлагает фармацевтически приемлемые производные соединений формулы I (такие как соединения формулы II, IIa, IIb, IIc, IIA и IIA') и, где применимо, их фармацевтически приемлемые соли, такие как, например, соли аммония. ″Фармацевтически приемлемое производное″ обозначает фармацевтически приемлемые соль, сложный эфир или соль такого сложного эфира, соединения по данному изобретению или любое другое соединение, которое, при введении реципиенту, способно предоставить (прямо или косвенно) соединение по данному изобретению или его антивирусно активный метаболит или остаток.The present invention provides pharmaceutically acceptable derivatives of compounds of formula I (such as compounds of formula II, IIa, IIb, IIc, IIA and IIA ′) and, where applicable, their pharmaceutically acceptable salts, such as, for example, ammonium salts. “Pharmaceutically acceptable derivative” means a pharmaceutically acceptable salt, ester or salt of such an ester, a compound of this invention or any other compound which, when administered to a recipient, is capable of providing (directly or indirectly) a compound of this invention or an antiviral active metabolite thereof or the remainder.
Необходимо понимать, что ″неразветвленная алкильная группа, содержащая от 1 до 6 атомов углерода″, включает, например, метил, этил, пропил, бутил, пентил, гексил.You must understand that the "unbranched alkyl group containing from 1 to 6 carbon atoms ″ includes, for example, methyl, ethyl, propyl, butyl, pentyl, hexyl.
Необходимо понимать, что ″разветвленная алкильная группа, содержащая от 3 до 6 атомов углерода″, включает, например, без ограничения, изобутил, трет-бутил, 2-пентил, 3-пентил и т.д.It should be understood that a “branched alkyl group containing from 3 to 6 carbon atoms” includes, for example, without limitation, isobutyl, tert- butyl, 2-pentyl, 3-pentyl, etc.
Необходимо понимать, что ″циклоалкильная группа, содержащая от 3 до 6 атомов углерода″, включает, например, без ограничения, циклопропил, циклобутил, циклопентил, циклоциклогексил (т.е. C6H11).It should be understood that the “cycloalkyl group containing from 3 to 6 carbon atoms” includes, for example, without limitation, cyclopropyl, cyclobutyl, cyclopentyl, cyclocyclohexyl (i.e., C 6 H 11 ).
Соли, полученные из соответствующих оснований, включают соли щелочных металлов (например, натрия), щелочноземельных металлов (например, магния), аммония и N-(C1-4алкил)4 +.Salts derived from the appropriate bases include salts of alkali metals (e.g. sodium), alkaline earth metals (e.g. magnesium), ammonium and N- (C 1-4 alkyl) 4 + .
Соединения по данному изобретению содержат один или более асимметричных атомов углерода и, таким образом, могут находиться в виде рацематов и рацемических смесей, отдельного энантиомера, диастереомерных смесей и индивидуальных диастереоизомеров. Все такие изомерные формы данных соединений определенно включены в настоящее изобретение. Каждый стереогенный атом углерода может иметь R или S конфигурацию.The compounds of this invention contain one or more asymmetric carbon atoms and, therefore, can be in the form of racemates and racemic mixtures, a single enantiomer, diastereomeric mixtures and individual diastereoisomers. All such isomeric forms of these compounds are specifically included in the present invention. Each stereogenic carbon atom may have an R or S configuration.
Фармацевтически приемлемые соли соединений по данному изобретению включают соли, полученные из фармацевтически приемлемых неорганических и органических кислот и оснований. Примеры таких солей кислот включают: ацетат, адипат, альгинат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, цитрат, камфорат, камфорсульфонат, циклопентанпропионат, диглюконат, додецилгидросульфат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептаноат, глицерофосфат, гликолят, гемисульфат, гептаноат, гексаноат, гидрохлорид, гидробромид, гидройодид, 2-гидроксиэтансульфонат, лактат, малеат, малонат, метансульфонат, 2-нафтилсульфонат, никотинат, нитрат, оксалат, памоат, пектинат, перхлорат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, салицилат, сукцинат, сульфат, тартрат, тиоцианат, тозилат и ундеканоат.Pharmaceutically acceptable salts of the compounds of this invention include salts derived from pharmaceutically acceptable inorganic and organic acids and bases. Examples of such acid salts include: acetate, adipate, alginate, aspartate, benzoate, benzenesulfonate, bisulfate, butyrate, citrate, camphorate, camphorsulfonate, cyclopentanepropionate, digluconate, dodetsilgidrosulfat, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptanoate, glycerophosphate, glycolate, hemisulfate, heptanoate, hexanoate, hydrochloride, hydrobromide, hydroiodide, 2-hydroxyethanesulfonate, lactate, maleate, malonate, methanesulfonate, 2-naphthyl sulfonate, nicotinate, nitrate, oxalate, pamoate, pectinate, perchlorate, persulfosate, 3-phenyl Fat, picrate, pivalate, propionate, salicylate, succinate, sulfate, tartrate, thiocyanate, tosylate and undecanoate.
Данное изобретение также охватывает кватернизацию любых содержащих основной азот групп соединений, раскрытых в настоящем описании. Основной азот можно кватернизировать любыми реагентами, известными специалисту в данной области, включая, например, низшие алкилгалогениды, такие как метил-, этил-, пропил- и бутил- хлориды, бромиды и йодиды; диалкилсульфаты, включая диметил-, диэтил-, дибутил- и диамилсульфаты; длинноцепочечные галогениды, такие как, децил-, лаурил-, миристил- и стеарил- хлориды, бромиды и йодиды, и аралкилгалогениды, включая бензил- и фенэтилбромиды. Такой кватернизацией можно получить водо- или маслорастворимые или дисперсные продукты.The invention also encompasses the quaternization of any groups of compounds containing basic nitrogen disclosed herein. Basic nitrogen can be quaternized with any reagents known to one skilled in the art, including, for example, lower alkyl halides such as methyl, ethyl, propyl and butyl chlorides, bromides and iodides; dialkyl sulfates, including dimethyl, diethyl, dibutyl and diamyl sulfates; long chain halides such as decyl, lauryl, myristyl and stearyl chlorides, bromides and iodides, and aralkyl halides, including benzyl and phenethyl bromides. By such quaternization, water- or oil-soluble or dispersible products can be obtained.
Здесь необходимо понимать, что если указывается ″диапазон″ или ″группа веществ″ относительно конкретной характеристики (например, температуры, концентрации, времени или аналогичного) настоящего изобретения, то настоящее изобретение относится и недвусмысленно включает все до единого конкретные члены и комбинации поддиапазонов или подгрупп в настоящем описании. Таким образом, любые указанные диапазон или группу необходимо понимать как стенографический путь ссылки ко всем до единого членам диапазона или группы индивидуально, а также ко всем до единого возможным поддиапазонам или подгруппам, охватываемым настоящим описанием; и аналогично относительно любых поддиапазонов или подгрупп настоящего описания. Таким образом, например,It is necessary to understand here that if a ″ range ″ or ″ group of substances ″ is indicated with respect to a specific characteristic (for example, temperature, concentration, time or the like) of the present invention, the present invention relates and expressly includes all and all specific members and combinations of subranges or subgroups in the present description. Thus, any specified range or group must be understood as a shorthand way to refer to all members of the range or group individually, as well as to all possible common subbands or subgroups covered by this description; and similarly with respect to any subbands or subgroups of the present description. So for example
- относительно числа атомов углерода, указание диапазона от 1 до 6 атомов углерода в настоящем описании необходимо понимать как включающее все до единого индивидуальные количества атомов углерода, а также поддиапазоны, такие как, например, 1 атом углерода, 3 атома углерода, от 4 до 6 атомов углерода и т.д.;- with respect to the number of carbon atoms, an indication of the range from 1 to 6 carbon atoms in the present description should be understood as including all individual individual amounts of carbon atoms, as well as subranges, such as, for example, 1 carbon atom, 3 carbon atoms, from 4 to 6 carbon atoms, etc .;
- относительно времени реакции, время, равное 1 минуте или более, необходимо понимать, как конкретно включающее здесь все до единого индивидуальные времена, а также поддиапазон, больше 1 минуты, такие как, например, 1 минуту, от 3 до 15 минут, от 1 минуты до 20 часов, от 1 до 3 часов, 16 часов, от 3 часов до 20 часов и т.д.;- relative to the reaction time, a time of 1 minute or more, it is necessary to understand how specifically including here all the same individual times, as well as a sub-range of more than 1 minute, such as, for example, 1 minute, from 3 to 15 minutes, from 1 minutes to 20 hours, from 1 to 3 hours, 16 hours, from 3 hours to 20 hours, etc .;
- и аналогично относительно других параметров, таких как концентрации, элементы и т.д.- and similarly with respect to other parameters, such as concentrations, elements, etc.
В частности здесь необходимо понимать, что каждая формула соединения включает все до единого индивидуальные соединения, описанные таким образом, а также все до единого возможные классы, или подгруппы, или подклассы соединений, независимо от того, определен ли такой класс или подкласс, как определенно включающий конкретные соединения, как исключающий конкретные соединения или как комбинация этого; например, исключающее определение для формулы (например, I) можно читать следующим образом: ″при условии, что когда один из A и B представляет собой -COOH, а другой представляет собой H, -COOH может не присутствовать в положении 4'″.In particular, it is necessary to understand here that each compound formula includes each and every individual compound described in this way, as well as all possible classes or subgroups or subclasses of compounds, regardless of whether such a class or subclass is defined as specifically including specific compounds, as excluding specific compounds or as a combination of this; for example, an exclusive definition for a formula (eg, I) can be read as follows: ″ provided that when one of A and B is —COOH and the other is H, —COOH may not be present at position 4 ′ ″.
Также необходимо понимать, что ″г″ или ″гм″ является ссылкой к граммовым единицам массы, а ″C″ или ″°C″ является ссылкой к единице температуры по Цельсию.You also need to understand that ″ g ″ or ″ um ″ is a reference to gram units of mass, and ″ C ″ or ″ ° C ″ is a reference to a unit temperature Celsius.
Соединения по настоящему изобретению можно легко получить, используя обычные методы из легкодоступных исходных веществ. Детальные описания данных подходов представлены, например, в схемах 1-5, обсужденных ниже.The compounds of the present invention can be easily prepared using conventional methods from readily available starting materials. Detailed descriptions of these approaches are presented, for example, in schemes 1-5, discussed below.
Схема 1 иллюстрирует характерный пример получения фосфатного сложного моноэфира III , полученного из первичного спирта (смотри I ), соединения ингибиторов ВИЧ-протеазы (смотри пример 1 (стадии G и H) в экспериментальной части данного документа в качестве конкретного примера данного синтеза).Scheme 1 illustrates a typical example of the preparation of a phosphate monoester III obtained from a primary alcohol (see I ), a compound of HIV protease inhibitors (see example 1 (stages G and H) in the experimental part of this document as a specific example of this synthesis).
Следует отметить:It should be noted:
a) R2 и R3 являются такими, как определено в настоящем описании.a) R 2 and R 3 are as defined herein.
При синтезе фосфатного сложного моноэфира III в качестве исходного вещества можно использовать ингибитор ВИЧ аспартил-протеазы ( I , смотри патент США № 6632816). Эфир диэтилфосфорной кислоты II получали с хорошим выходом при обработке диэтилхлорфосфатом и гидридом натрия в смеси тетрагидрофурана и триэтилфосфата. Затем, добавление триметилсилилбромида в дихлорметане (ДХМ) дало соединение III с выходами от хорошего до превосходного.When synthesizing a phosphate monoester III , an aspartyl protease inhibitor ( I , see US Pat. No. 6,632,816) can be used as a starting material. The diethylphosphoric ester II was obtained in good yield by treatment with diethyl chlorophosphate and sodium hydride in a mixture of tetrahydrofuran and triethyl phosphate. Then, the addition of trimethylsilyl bromide in dichloromethane (DCM) afforded compound III in good to excellent yields.

Схема 1A представляет другой характерный пример получения фосфатного сложного моноэфира IIIA , полученного из первичного спирта (смотри IA ), соединения ингибиторов ВИЧ-протеазы.Scheme 1A is another representative example of the preparation of a IIIA phosphate monoester derived from a primary alcohol (see IA ), a compound of HIV protease inhibitors.
Следует отметить:It should be noted:
a) n, X, Y, R2, R3 и R6 являются такими, как определено в настоящем описании.a) n, X, Y, R 2 , R 3 and R 6 are as defined herein.

Синтез фосфатного сложного моноэфира IIIA осуществляют, как описано для получения III (схема 1).The synthesis of the phosphate monoester IIIA is carried out as described for preparation III (Scheme 1).
Схема 2 иллюстрирует характерный пример получения фосфатного сложного моноэфира III , соединения ингибиторов ВИЧ-протеазы, с другим подходом, используя в качестве исходного вещества (3S)-3-изобутиламиноазепан-2-он ( IV ).Scheme 2 illustrates a typical example of the preparation of a phosphate monoester III , a compound of HIV protease inhibitors, with a different approach, using (3S) -3-isobutylaminoazepan-2-one ( IV ) as the starting material.
Следует отметить:It should be noted:
a) R2 и R3 являются такими, как определено в настоящем описании.a) R 2 and R 3 are as defined herein.
Как показано на схеме 2, производное, представляющее собой фосфатный сложный моноэфир III , получали из (3S)-3-изобутиламиноазепан-2-она ( IV ) в семистадийной последовательности реакций. В начале, (2S)-3-изобутиламиноазепан-2-он ( IV ) сульфонировали 4-ацетамидобензолсульфонилхлоридом в присутствии триэтиламина в дихлорметане, получая соединение V с превосходными выходами. Производное VI получали количественно при обработке V ди-трет-бутилпирокарбонатом и ДМАП в ацетонитриле. Восстановительное раскрытие цикла боргидридом натрия в этаноле приводит к ключевым промежуточным продуктам VII с хорошим выходом. Эфир диэтилфосфорной кислоты VIII получали с хорошим выходом при обработке диэтилхлорфосфатом и гидридом натрия в смеси тетрагидрофурана и триэтилфосфата. Boc защитные группы удаляли обработкой HCl в этаноле, получая соединение IX количественно (T.W. Greene and P.G.M. Wuts, Protective groups in Organic Synthesis, 3rd Edition, John Wiley & Sons, Inc. 1999). Затем сочетание свободной аминогруппы, присутствующей на промежуточном соединении IX , с рядом синтетических аминокислот в присутствии 1-гидроксибензотриазола (HOBt) и 1-[3-(диметиламино)пропил]-3-этилкарбодиимид гидрохлорида (EDAC) привело к производному II с выходами от хорошего до превосходного. Наконец, добавление триметилсилилбромида в дихлорметане (ДХМ) дало соединение III с выходами от хорошего до превосходного.As shown in Scheme 2, the phosphate monoester III derivative was prepared from (3S) -3-isobutylaminoazepan-2-one ( IV ) in a seven-step reaction sequence. At the beginning, (2S) -3-isobutylaminoazepan-2-one ( IV ) was sulfonated with 4-acetamidobenzenesulfonyl chloride in the presence of triethylamine in dichloromethane to give compound V in excellent yields. Derivative VI was obtained quantitatively by treating V with di- tert-butyl pyrocarbonate and DMAP in acetonitrile. The reductive opening of the cycle with sodium borohydride in ethanol leads to key intermediates VII in good yield. The diethylphosphoric ester VIII was obtained in good yield by treatment with diethyl chlorophosphate and sodium hydride in a mixture of tetrahydrofuran and triethyl phosphate. Boc deprotected by treatment with HCl in ethanol to give compound IX quantitatively (TW Greene and PGM Wuts, Protective groups in Organic Synthesis, 3 rd Edition, John Wiley & Sons, Inc. 1999). Then, the combination of the free amino group present on intermediate IX with a number of synthetic amino acids in the presence of 1-hydroxybenzotriazole (HOBt) and 1- [3- (dimethylamino) propyl] -3-ethylcarbodiimide hydrochloride (EDAC) led to derivative II in good yields to excellent. Finally, the addition of trimethylsilyl bromide in dichloromethane (DCM) afforded compound III in good to excellent yields.

Схема 3 представляет превращение дифенилметильного производного; метилового эфира (1S,5S)-(1-{5-[(4-аминобензолсульфонил)изобутиламино]-6-гидроксигексилкарбамоил}-2,2-дифенилэтил)карбаминовой кислоты (PL-100) в его аналог XI , представляющий собой натриевую соль фторированного фосфатного сложного моноэфира. Данную последовательность реакций можно использовать для получения любых других аналогичных соединений, полученных из незамещенных (или замещенных) дифенилметильных, 1-нафтильных, 2-нафтильных, бифенильных и 9-антрильных групп, описанных в данном изобретении.Scheme 3 represents the conversion of a diphenylmethyl derivative; (1S, 5S) methyl ester - (1- {5 - [(4-aminobenzenesulfonyl) isobutylamino] -6-hydroxyhexylcarbamoyl} -2,2-diphenylethyl) carbamic acid ( PL-100 ) in its analogue XI , which is a sodium salt fluorinated phosphate monoester. This reaction sequence can be used to obtain any other similar compounds derived from unsubstituted (or substituted) diphenylmethyl, 1-naphthyl, 2-naphthyl, biphenyl and 9-antryl groups described in this invention.
Так, обработка PL-100 с помощью Selectfluor™ в ацетонитриле дала производное X с выходом 38%. Введение группы фосфатного сложного моноэфира осуществляли, как описано ранее в схемах 1 и 2. Сначала промежуточное соединение, представляющее собой эфир диэтилтрифосфорной кислоты, получали с хорошим выходом при обработке диэтилхлорфосфатом и гидридом натрия в смеси тетрагидрофурана и триэтилфосфата. Затем, добавление триметилсилилбромида в дихлорметане (ДХМ) дало соединение фосфатного сложного моноэфира с выходами от хорошего до превосходного. Конечный продукт XI легко получали при обработке фосфатного сложного моноэфира раствором гидроксида натрия с хорошими выходами.Thus, treatment of PL-100 with Selectfluor ™ in acetonitrile gave derivative X in 38% yield. The introduction of the phosphate monoester group was carried out as described previously in Schemes 1 and 2. First, an intermediate compound representing diethyl triphosphoric acid ester was obtained in good yield by treatment with diethyl chlorophosphate and sodium hydride in a mixture of tetrahydrofuran and triethyl phosphate. Then, the addition of trimethylsilyl bromide in dichloromethane (DCM) afforded the phosphate monoester compound in good to excellent yields. The final product XI was easily obtained by treating the phosphate monoester with sodium hydroxide solution in good yields.

Схема 4 иллюстрирует типичный пример превращения фосфотриэфира II в его фторированный аналог XIII в двухстадийной последовательности реакций. Данный типичный пример представляет второй подход к синтезу фторированных соединений по данному изобретению. В данном случае атом фтора добавляют к фосфотриэфиру II , а не к производному общей формулы I , представляющему собой первичный спирт, или более конкретно PL-100, как показано на схеме 3. Данную альтернативную последовательность реакций можно использовать для синтеза любых других аналогичных соединений, полученных из незамещенных (или замещенных) дифенилметильных, 1-нафтильных, 2-нафтильных, бифенильных и 9-антрильных групп, описанных в данном изобретении.Scheme 4 illustrates a typical example of the conversion of phosphotriether II to its fluorinated analogue XIII in a two-step reaction sequence. This typical example represents a second approach to the synthesis of fluorinated compounds of this invention. In this case, the fluorine atom is added to the phosphotriether II , and not to the derivative of the general formula I , which is a primary alcohol, or more specifically PL-100 , as shown in Scheme 3. This alternative reaction sequence can be used to synthesize any other similar compounds obtained from unsubstituted (or substituted) diphenylmethyl, 1-naphthyl, 2-naphthyl, biphenyl and 9-antryl groups described in this invention.
Следует отметить:It should be noted:
a) R2 и R3 являются такими, как определено в настоящем описании.a) R 2 and R 3 are as defined herein.
Кратко, обработка производного II с помощью Selectfluor™ в ацетонитриле дала производное XII с хорошим выходом. Затем добавление триметилсилилбромида в дихлорметане (ДХМ) дала соединение XIII , представляющее собой фосфатный сложный моноэфир, с выходом от хорошего до превосходного. Если желательно, конечный продукт XIII можно легко превратить в аналог, представляющий собой натриевую соль фосфатного сложного моноэфира, как описано ранее в схеме 3.Briefly, treatment of derivative II with Selectfluor ™ in acetonitrile gave derivative XII in good yield. The addition of trimethylsilyl bromide in dichloromethane (DCM) then gave compound XIII , which is a phosphate monoester, in a yield from good to excellent. If desired, the final product XIII can easily be converted into an analogue, which is a sodium salt of a phosphate complex monoester, as described previously in scheme 3.

Схема 5 иллюстрирует синтез различных соединений сложных эфиров XVI по изобретению. Известно, что соединения сложных эфиров легко расщепляются in vivo ферментами эстеразы и в результате могут высвобождать активный ингредиент. В данной схеме R2 определен как дифенилметильная группа. Однако данную последовательность реакций можно использовать для синтеза любых других аналогичных соединений, полученных из незамещенных (или замещенных) дифенилметильных, 1-нафтильных, 2-нафтильных, бифенильных и 9-антрильных групп, описанных в данном изобретении.Scheme 5 illustrates the synthesis of various compounds of esters XVI according to the invention. It is known that ester compounds are readily cleaved in vivo by esterase enzymes and, as a result, can release the active ingredient. In this scheme, R 2 is defined as a diphenylmethyl group. However, this reaction sequence can be used to synthesize any other similar compounds derived from unsubstituted (or substituted) diphenylmethyl, 1-naphthyl, 2-naphthyl, biphenyl and 9-antryl groups described in this invention.
Следует отметить:It should be noted:
a) R1A представляет ″остаток″ молекулы кислоты, который связан со свободной группой первичного спирта, присутствующей на промежуточном соединении XV , и является таким, как определено в настоящем описании.a) R 1A represents a “residue” of an acid molecule that is bound to the free group of a primary alcohol present on intermediate XV and is as defined herein.
Соединения XVI обычно получают в трехстадийной последовательности реакций с высокими выходами. Этерификация трет-бутилового эфира (1S)-{4-[(5-трет-бутоксикарбониламино-1-гидроксиметилпентил)изобутилсульфамоил]фенил}карбаминовой кислоты ( VII ) различной кислотой в присутствии 1-гидроксибензотриазола (HOBt) и 1-[3-(диметиламино)пропил]-3-этилкарбодиимид гидрохлорида (EDAC) привела к желаемым сложным эфирам XIV с превосходными выходами. Сложный уксуснокислый эфир получали количественно, используя уксусный ангидрид в присутствии N,N-диметиламинопиридина (ДМАП) в дихлорметане (ДХМ). Отрыв Boc защитной группы достигали количественно при обработке трифторуксусной кислотой (ТФУ) в ДХМ. Второе сочетание с (2S)-2-метоксикарбониламино-3,3-дифенилпропионовой кислотой осуществляют на первичной аминогруппе промежуточного соединения XV с HOBt и EDAC, получая желаемые соединения XVI с выходами от хорошего до превосходного. Если необходимо, осуществляют каталитическое гидрирование бензилоксикарбонильной группы, используя 10% палладий на угле, получая конечное соединение XVII .Compounds XVI are usually prepared in a three-step reaction sequence in high yields. The esterification of (1S) tert-butyl ester - {4 - [(5-tert-butoxycarbonylamino-1-hydroxymethylpentyl) isobutylsulfamoyl] phenyl} carbamic acid ( VII ) with a different acid in the presence of 1-hydroxybenzotriazole (HOBt) and 1- [3- ( dimethylamino) propyl] -3-ethylcarbodiimide hydrochloride (EDAC) yielded the desired esters XIV in excellent yields. Acetic acid ester was quantified using acetic anhydride in the presence of N, N- dimethylaminopyridine (DMAP) in dichloromethane (DCM). Detachment of the Boc protecting group was achieved quantitatively by treatment with trifluoroacetic acid (TFA) in DCM. A second combination with (2S) -2-methoxycarbonylamino-3,3-diphenylpropionic acid is carried out on the primary amino group of intermediate XV with HOBt and EDAC to give the desired compounds XVI in good to excellent yields. If necessary, catalytic hydrogenation of the benzyloxycarbonyl group is carried out using 10% palladium-carbon to give the final compound XVII .


Специалист в данной области поймет, что вышеуказанные синтетические схемы не имеют намерения представлять собой полный список всех средств, которыми можно синтезировать соединение, описанное и заявленное в данной заявке на изобретение, а представляют собой лишь иллюстрацию методов синтеза среди прочих. Дальнейшие методы будут очевидны специалистам в данной области.One skilled in the art will understand that the above synthetic schemes are not intended to constitute a complete list of all the means by which the compound described and claimed in this application for synthesis can be synthesized, but are merely an illustration of synthetic methods, among others. Further methods will be apparent to those skilled in the art.
Соединения по данному изобретению можно модифицировать, добавляя соответствующие функциональности для усиления селективных биологических свойств. Такие модификации известны из уровня техники и включают модификации, которые увеличивают биологическую проницаемость в данную биологическую систему (например, кровь, лимфатическую систему, центральную нервную систему), увеличивают пероральную доступность, увеличивают растворимость, чтобы дать возможность введения инъекцией, изменяют метаболизм и изменяют скорость выделения.The compounds of this invention can be modified by adding appropriate functionality to enhance selective biological properties. Such modifications are known in the art and include modifications that increase the biological permeability of a given biological system (e.g., blood, lymphatic system, central nervous system), increase oral availability, increase solubility to allow injection, alter metabolism and alter excretion rate .
Как обсуждено выше, новые соединения могут высвобождать активные ингредиенты, которые являются превосходными лигандами по отношению к аспартил-протеазам, например, ВИЧ-1 протеазе. Соответственно, данные соединения, посредством высвобождения активного ингредиента, способны нацеливаться и подавлять события поздней фазы при репликации, т.е. процессинг вирусных полипротеинов ВИЧ-кодированной протеазой. Соединения по данному изобретению преимущественно подавляют способность ВИЧ-1 вируса инфицировать иммортализованные Т-клетки человека в течение периода, составляющего дни, как определено анализом, измеряющим количество внеклеточного p24 антигена; специфичного маркера репликации вируса (смотри, Meek et al., Nature, 343, pp.90-92 (1990)).As discussed above, the novel compounds can release active ingredients that are excellent ligands for aspartyl proteases, for example, HIV-1 protease. Accordingly, these compounds, by releasing the active ingredient, are able to target and suppress late-phase events during replication, i.e. processing of viral polyproteins by an HIV-encoded protease. The compounds of this invention advantageously inhibit the ability of the HIV-1 virus to infect immortalized human T cells for a period of days, as determined by analysis measuring the amount of extracellular p24 antigen; a specific marker of virus replication (see, Meek et al., Nature, 343, pp.90-92 (1990)).
Кроме их использования при профилактике или лечении ВИЧ или HTLV-инфекции, соединения по данному изобретению можно также использовать в качестве ингибирующих или прерывающих средств для других вирусов, которые используют аспартил-протеазы, аналогичные ВИЧ или HTLV аспартил-протеазам, в своем жизненном цикле. Такие соединения подавляют протеолитический процессинг предшественников вирусных полипротеинов, подавляя аспартил-протеазу. Поскольку аспартил-протеаза является существенной для продукции зрелых вирионов, подавление данного процессинга эффективно блокирует распространение вируса, подавляя продукцию и репродукцию инфекционных вирионов, в частности из остро и хронически инфицированных клеток. Соединения по данному изобретению преимущественно подавляют аспартил-протеазы, таким образом блокируя способность аспартил-протеаз катализировать гидролиз пептидных связей.In addition to their use in the prevention or treatment of HIV or HTLV infection, the compounds of this invention can also be used as inhibitory or interrupting agents for other viruses that use aspartyl proteases similar to HIV or HTLV aspartyl proteases in their life cycle. Such compounds inhibit the proteolytic processing of viral polyprotein precursors by inhibiting aspartyl protease. Since aspartyl protease is essential for the production of mature virions, the inhibition of this processing effectively blocks the spread of the virus, inhibiting the production and reproduction of infectious virions, in particular from acutely and chronically infected cells. The compounds of this invention advantageously inhibit aspartyl proteases, thereby blocking the ability of aspartyl proteases to catalyze the hydrolysis of peptide bonds.
Соединения по данному изобретению можно применять обычным способом для лечения или профилактики ВИЧ, HTLV или других вирусных инфекций, которые включают в свой жизненный цикл (репликацию) аспартил-протеазы. Такие методы лечения, их уровни дозировок и требования могут быть выбраны специалистом в данной области из имеющихся методов и методик. Например, соединение по данному изобретению можно объединить с фармацевтически приемлемым вспомогательным веществом для введения инфицированному вирусом пациенту в фармацевтически приемлемой манере и в количестве, эффективном для снижения тяжести вирусной инфекции.The compounds of this invention can be used in the usual way for the treatment or prevention of HIV, HTLV or other viral infections, which include aspartyl proteases in their life cycle (replication). Such treatment methods, their dosage levels and requirements can be selected by a person skilled in the art from the available methods and techniques. For example, the compound of this invention can be combined with a pharmaceutically acceptable excipient for administration to a virus infected patient in a pharmaceutically acceptable manner and in an amount effective to reduce the severity of the viral infection.
Альтернативно, соединения по данному изобретению можно использовать в вакцинах и способах защиты особей от вирусной инфекции в течение продолжительного периода времени. Соединения можно применять в таких вакцинах либо в одиночку, либо вместе с другими соединениями по данному изобретению в манере, согласующейся с традиционным использованием ингибиторов протеазы или производных ингибиторов протеазы в вакцинах. Например, соединение по данному изобретению можно объединить с фармацевтически приемлемыми вспомогательными веществами, или системами доставки, обычно используемыми в вакцинах, и ввести в профилактически эффективных количествах для защиты особей в течение продолжительного периода времени от вирусных инфекций, таких как ВИЧ-инфекция. По существу, новые соединения по настоящему изобретению (при отщеплении физиологически расщепляющегося звена) можно вводить в качестве агентов для лечения или профилактики вирусных инфекций, включая ВИЧ-инфекцию, у млекопитающих.Alternatively, the compounds of this invention can be used in vaccines and methods of protecting individuals from viral infection for an extended period of time. The compounds can be used in such vaccines either alone or together with other compounds of this invention in a manner consistent with the traditional use of protease inhibitors or derived protease inhibitors in vaccines. For example, the compound of this invention can be combined with pharmaceutically acceptable excipients, or delivery systems commonly used in vaccines, and administered in prophylactically effective amounts to protect individuals for an extended period of time from viral infections such as HIV infection. As such, the novel compounds of the present invention (by cleaving a physiologically cleavable unit) can be administered as agents for the treatment or prophylaxis of viral infections, including HIV infection, in mammals.
Соединения по данному изобретению можно вводить здоровому или ВИЧ-инфицированному пациенту (до или после появления симптомов СПИД) в виде одиночного агента или в комбинации с другими противовирусными средствами, которые мешают циклу репликации ВИЧ. Вводя соединения по данному изобретению с другими противовирусными средствами, которые нацелены на другие события в жизненном цикле вируса, терапевтический эффект данных соединений усиливается. Например, совместно вводимое противовирусное средство может представлять собой средство, которое нацелено на ранние события жизненного цикла вируса, такие как присоединение к клеточному рецептору или входу клетки, обратную транскрипцию и интеграцию вирусной ДНК в клеточную ДНК. Противовирусные агенты, нацеленные на такие ранние события жизненного цикла, включают, среди прочего, полисульфированные полисахариды, sT4 (растворимый CD4)и другие соединения, которые блокируют связывание вируса с CD4 рецептором на несущих CD4 Т-лимфоцитах и других CD4(+) клетках, или подавляют слияние оболочки вируса с цитоплазматической мембраной, и диданозин (ddI), зальцитабин (ddC), ставудин (d4T), зидовудин (AZT) и ламивудин (3TC), которые ингибируют обратную транскрипцию. Например, с соединениями по настоящему изобретению можно использовать другой ингибитор протеазы. Другие антриретровирусные и противовирусные лекарственные средства также можно совместно вводить с соединениями по данному изобретению, чтобы обеспечить терапевтическое лечение для существенного снижения или исключения вирусной инфекционности и связанных с ней симптомов. Примеры других противовирусных средств включают ганцикловир, дидеоксицитидин, тринатрия фосфоноформат, эфлорнитин, рибавирин, ацикловир, альфа-интерферон и трименотрексат. Кроме того, для усиления действия соединений по данному изобретению можно использовать другие типы лекарственных средств, такие как ингибиторы утраты вирусом оболочки, ингибиторы Tat или Rev трансактивирующих белков, антисмысловые молекулы или ингибиторы интегразы вируса. Данные соединения также можно совместно вводить с другими ингибиторами ВИЧ аспартил-протеазы. Кроме того, может оказаться полезным вводить соединения по настоящему изобретению с другим лекарственным средством (другими противовирусными соединениями, антибиотиками, аналгизирующим средством и т.д.).The compounds of this invention can be administered to a healthy or HIV-infected patient (before or after the onset of AIDS symptoms) as a single agent or in combination with other antiviral agents that interfere with the HIV replication cycle. By administering the compounds of this invention with other antiviral agents that target other events in the life cycle of the virus, the therapeutic effect of these compounds is enhanced. For example, a co-administered antiviral agent may be one that targets early events in the life cycle of a virus, such as attachment to a cell receptor or cell entry, reverse transcription, and integration of viral DNA into cellular DNA. Antiviral agents targeting such early life-cycle events include, but are not limited to, polysulfonated polysaccharides, sT4 (soluble CD4) and other compounds that block the binding of the virus to the CD4 receptor on CD4-bearing T lymphocytes and other CD4 (+) cells, or inhibit the fusion of the envelope of the virus with the cytoplasmic membrane, and didanosine (ddI), zalcitabine (ddC), stavudine (d4T), zidovudine (AZT) and lamivudine (3TC), which inhibit reverse transcription. For example, another protease inhibitor can be used with the compounds of the present invention. Other antiretroviral and antiviral drugs can also be co-administered with the compounds of this invention to provide therapeutic treatment to substantially reduce or eliminate viral infectivity and its associated symptoms. Examples of other antiviral agents include ganciclovir, dideoxycytidine, trisodium phosphonoformate, eflornithine, ribavirin, acyclovir, interferon alpha, and trimenotrexate. In addition, other types of drugs can be used to enhance the effects of the compounds of this invention, such as envelope virus loss inhibitors, Tat or Rev inhibitors of transactivating proteins, antisense molecules, or virus integrase inhibitors. These compounds can also be co-administered with other HIV aspartyl protease inhibitors. In addition, it may be useful to administer the compounds of the present invention with another drug (other antiviral compounds, antibiotics, an analgesic, etc.).
Комбинированные терапии по данному изобретению оказывают синергическое действие при подавлении репликации ВИЧ, поскольку каждый компонентный агент комбинации действует на различные участки ВИЧ-репликации. Использование таких комбинаций также преимущественно снижает дозировки заданного традиционного антриретровирусного средства, которые потребовались бы для желаемого терапевтического или профилактического действия, по сравнению со случаем, когда такой агент вводят в виде монотерапии. Данные комбинации могут уменьшить или исключить побочные эффекты традиционных терапий одиночным антиретровирусным средством, в то же время не мешая антиретровирусной активности данных средств. Данные комбинации снижают потенциал резистентности к терапиям одиночным средством, в то же время сводя к минимуму сопутствующую токсичность. Данные комбинации также могут увеличить эффективность традиционного средства, не увеличивая сопутствующую токсичность. Комбинированные терапии, охватываемые настоящим изобретением, включают, например, введение соединения по изобретению с AZT, 3TC, ddI, ddC, d4T или другими ингибиторами обратной транскриптазы.The combination therapies of this invention have a synergistic effect in suppressing HIV replication, as each component combination agent acts on different sites of HIV replication. The use of such combinations also advantageously reduces the dosage of a given conventional antiretroviral agent that would be required for the desired therapeutic or prophylactic action, compared with the case when such an agent is administered as monotherapy. These combinations can reduce or eliminate the side effects of conventional therapies with a single antiretroviral agent, while not interfering with the antiretroviral activity of these agents. These combinations reduce the potential for resistance to single-agent therapies, while minimizing concomitant toxicity. These combinations can also increase the effectiveness of a traditional product without increasing concomitant toxicity. Combination therapies encompassed by the present invention include, for example, administering a compound of the invention with AZT, 3TC, ddI, ddC, d4T or other reverse transcriptase inhibitors.
Альтернативно, соединения по данному изобретению можно также совместно вводить с другими ингибиторами ВИЧ-протеазы, такими как Ro 31-8959 (Cаквинавир; Roche), L-735,524 (Индинавир; Merck), AG-1343 (Нелфинавир; Agouron), A-84538 (Ритонавир; Abbott), ABT-378/r (Лопинавир; Abbott) и VX-478 (Ампренавир; Glaxo), для увеличения эффекта терапии или профилактики против различных вирусных мутантов или членов других ВИЧ-квазивидов.Alternatively, the compounds of this invention can also be co-administered with other HIV protease inhibitors, such as Ro 31-8959 (Squinavir; Roche), L-735,524 (Indinavir; Merck), AG-1343 (Nelfinavir; Agouron), A-84538 (Ritonavir; Abbott), ABT-378 / r (Lopinavir; Abbott) and VX-478 (Amprenavir; Glaxo), to increase the effect of therapy or prophylaxis against various viral mutants or members of other HIV quasivids.
Ведение соединений по настоящему изобретению можно осуществить, например, в виде одиночных средств или в комбинации с ингибиторами ретровирусной обратной транскриптазы или другими ингибиторами ВИЧ аспартил-протеазы. Совместное введение соединений по данному изобретению с ингибиторами ретровирусной обратной транскриптазы или ингибиторами ВИЧ аспартил-протеазы может оказать значительное синергическое действие, таким образом предотвращая, существенно уменьшая или полностью исключая инфекционность вируса и сопутствующие симптомы.The administration of the compounds of the present invention can be carried out, for example, in the form of single agents or in combination with retroviral reverse transcriptase inhibitors or other HIV aspartyl protease inhibitors. Co-administration of the compounds of this invention with retroviral reverse transcriptase inhibitors or HIV aspartyl protease inhibitors can have a significant synergistic effect, thereby preventing, substantially reducing or completely eliminating viral infectivity and associated symptoms.
Соединения по настоящему изобретению можно вводить таким образом или в форме, которая может допускать отщепление звена R1 для высвобождения ингибитора протеазы. Соединения по данному изобретению также можно вводить, например, в комбинации с иммуномодуляторами (например, бропиримином, античеловеческими антителами к альфа-интерферону, IL-2, GM-CSF, метионин-энцефалином, интерфероном альфа, диэтилдитиокарбаматом натрия, фактором некроза опухоли, налтрексоном и rEPO), антибиотиками (например, пентамидина изотионатом) или вакцинами для профилактики или борьбы с инфекцией или болезнью, связанными с ВИЧ-инфекцией, такими как СПИД и СПИД-ассоциированный комплекс.The compounds of the present invention can be administered in this way or in a form that can allow cleavage of the R 1 unit to release a protease inhibitor. The compounds of this invention can also be administered, for example, in combination with immunomodulators (e.g., bropirimine, anti-human antibodies to alpha interferon, IL-2, GM-CSF, methionine encephalin, interferon alpha , sodium diethyldithiocarbamate, tumor necrosis factor, naltrexone and rEPO), antibiotics (e.g., pentamidine isothionate) or vaccines for the prevention or control of an infection or illness associated with HIV infection, such as AIDS and the AIDS-associated complex.
Когда соединения по данному изобретению вводят в комбинированных терапиях с другими средствами, их можно вводить пациенту последовательно или параллельно. Альтернативно, фармацевтические или профилактические композиции по данному изобретению могут содержать комбинацию одного или нескольких соединений по данному изобретению и другого терапевтического или профилактического средства.When the compounds of this invention are administered in combination therapies with other agents, they can be administered to a patient sequentially or in parallel. Alternatively, the pharmaceutical or prophylactic compositions of this invention may comprise a combination of one or more compounds of this invention and another therapeutic or prophylactic agent.
Хотя данное изобретение фокусируется на использовании соединений, раскрытых в настоящем описании, для профилактики и лечения ВИЧ-инфекции соединения по данному изобретению также можно использовать в качестве ингибирующих средств для других вирусов, которые зависят от аналогичных аспартил-протеаз для обязательных событий в их жизненном цикле. Данные вирусы включают, но не ограничиваются этим, ретровирусы, вызывающие подобные СПИД заболевания, такие как обезьяньи вирусы иммунодефицита, ВИЧ-2, HTLV-I и HTLV-II. Кроме того, соединения по данному изобретению также можно использовать для подавления других аспартил-протеаз и, в частности, других аспартил-протеаз человека, включая ренин и аспартил-протеазы, которые участвуют в процессинге предшественников эндотелина.Although the invention focuses on the use of the compounds disclosed herein, for the prevention and treatment of HIV infection, the compounds of this invention can also be used as inhibitory agents for other viruses that depend on similar aspartyl proteases for binding events in their life cycle. These viruses include, but are not limited to, retroviruses that cause AIDS-like diseases, such as monkey immunodeficiency viruses, HIV-2, HTLV-I, and HTLV-II. In addition, the compounds of this invention can also be used to inhibit other aspartyl proteases and, in particular, other human aspartyl proteases, including renin and aspartyl proteases, which are involved in the processing of endothelin precursors.
Фармацевтические композиции по данному изобретению включают любое соединение по настоящему изобретению и его фармацевтически приемлемые соли с любым фармацевтически приемлемым носителем, вспомогательным веществом или растворителем. Фармацевтически приемлемые носители, вспомогательные вещества и растворители, которые можно использовать в фармацевтических композициях по данному изобретению, включают, но не ограничиваются этим, ионообменники, оксид алюминия, стеарат алюминия, лецитин, сывороточные белки, такие как человеческий альбумин, буферные вещества, такие как фосфаты, глицин, сорбиновую кислоту, сорбат калия, смеси неполных глицеридов насыщенных растительных жирных кислот, воду, соли или электролиты, такие как протамин сульфат, кислый фосфат динатрия, кислый фосфат дикалия, хлорид натрия, соли цинка, коллоидный кремнезем, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, натрий карбоксиметилцеллюлозу, полиакрилаты, воски, блок-сополимеры полиэтилен-полиоксипропилен, полиэтиленгликоль и ланолин.The pharmaceutical compositions of this invention include any compound of the present invention and its pharmaceutically acceptable salts with any pharmaceutically acceptable carrier, excipient or solvent. Pharmaceutically acceptable carriers, excipients, and solvents that can be used in the pharmaceutical compositions of this invention include, but are not limited to, ion exchangers, alumina, aluminum stearate, lecithin, whey proteins such as human albumin, buffering agents such as phosphates , glycine, sorbic acid, potassium sorbate, mixtures of incomplete glycerides of saturated vegetable fatty acids, water, salts or electrolytes such as protamine sulfate, disodium hydrogen phosphate, acid phosphate SFAT dipotassium, sodium chloride, zinc salts, colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, cellulose-based substances, polyethylene glycol, sodium carboxymethylcellulose, polyacrylates, waxes, block copolymers of polyethylene-polyoxypropylene, polyethylene glycol and wool fat.
Фармацевтические композиции по данному изобретению можно вводить перорально, парентерально аэрозолем для ингаляции, местно, ректально, назально, трансбукально, вагинально или посредством имплантированного резервуара. Следовательно, здесь понятно, что пероральное введение или введение инъекцией охватывается настоящим изобретением. Например, соединения по настоящему изобретению можно вводить, например, перорально в водном растворе. Фармацевтические композиции по данному изобретению могут содержать любые обычные нетоксичные фармацевтически приемлемые носители, вспомогательные вещества или растворители. Используемый здесь термин ″парентеральное″ включает методику подкожной, внутрикожной, внутривенной, внутримышечной, интраартикулярной, внутрисуставной, подложечной, интратекальной, непосредственно в пораженную ткань, интракриниальной инъекции или инфузии.The pharmaceutical compositions of this invention can be administered orally, parenterally with an aerosol for inhalation, topically, rectally, nasally, buccally, vaginally or through an implanted reservoir. Therefore, it is understood that oral administration or injection is encompassed by the present invention. For example, the compounds of the present invention can be administered, for example, orally in an aqueous solution. The pharmaceutical compositions of this invention may contain any conventional non-toxic pharmaceutically acceptable carriers, excipients or solvents. The term “parenteral” as used herein includes the technique of subcutaneous, intradermal, intravenous, intramuscular, intraarticular, intraarticular, epigastric, intrathecal, directly into the affected tissue, intracrinal injection or infusion.
Фармацевтические композиции могут быть в форме стерильного препарата для инъекций, например, в виде стерильной впрыскиваемой водной и масляной суспензии. Рецептуру данной суспензии можно составить согласно методикам, известным из уровня техники, используя подходящие диспергирующие или увлажняющие средства (такие как, например, Tween 80) и суспендирующие вещества. Стерильный впрыскиваемый препарат также может представлять собой стерильный впрыскиваемый раствор или суспензию в нетоксичном парентерально приемлемом разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. Среди приемлемых носителей и растворителей, которые можно использовать, находятся аминокислоты, вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно используют стерильные, нелетучие масла. Для данной цели можно использовать любое легкое нелетучее масло, включая синтетические моно- и диглицериды. Жирные кислоты, такие как олеиновая кислота и ее глицеридные производные применимы при получении препаратов для инъекций, как, например, природные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, особенно в своих полиоксиэтилированных вариантах. Данные масляные растворы или суспензии также могут содержать разбавители или диспергирующие вещества, представляющие собой спирты с длинной цепью, такие как Ph. Helv. или аналогичный спирт.The pharmaceutical compositions may be in the form of a sterile injectable preparation, for example, in the form of a sterile injectable aqueous and oily suspension. The formulation of this suspension can be formulated according to methods known in the art using suitable dispersing or moisturizing agents (such as, for example, Tween 80) and suspending agents. The sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally acceptable diluent or solvent, for example, as a solution in 1,3-butanediol. Among the acceptable carriers and solvents that can be used are amino acids, water, Ringer's solution, and isotonic sodium chloride solution. In addition, sterile, fixed oils are usually used as a solvent or suspending medium. For this purpose, you can use any light non-volatile oil, including synthetic mono - and diglycerides. Fatty acids such as oleic acid and its glyceride derivatives are useful in the manufacture of injectable preparations, such as, for example, natural pharmaceutically acceptable oils, such as olive oil or castor oil, especially in their polyoxyethylene versions. These oil solutions or suspensions may also contain diluents or dispersants, which are long chain alcohols, such as Ph. Helv. or similar alcohol.
Фармацевтические композиции по данному изобретению можно вводить перорально в любой перорально приемлемой лекарственной форме, включая, но, не ограничиваясь этим, капсулы, таблетки и водные суспензии и растворы. В случае таблеток для перорального использования носители, которые обычно используют, включают лактозу и кукурузный крахмал. Также типично добавляют смазывающие вещества, такие как стеарат магния. Для перорального введения в форме капсулы, применимые разбавители включают лактозу и сухой кукурузный крахмал. Когда водные суспензии вводят перорально, активный ингредиент объединяют с эмульгирующими и суспендирующими веществами. Если желательно, можно добавить определенные подслащивающие вещества, и/или корригенты, и/или красители.The pharmaceutical compositions of this invention can be administered orally in any orally acceptable dosage form, including, but not limited to, capsules, tablets, and aqueous suspensions and solutions. In the case of tablets for oral use, carriers that are commonly used include lactose and corn starch. Lubricants such as magnesium stearate are also typically added. For oral administration in capsule form, useful diluents include lactose and dry corn starch. When aqueous suspensions are administered orally, the active ingredient is combined with emulsifying and suspending agents. If desired, certain sweetening agents and / or flavoring agents and / or coloring agents may be added.
Фармацевтические композиции по данному изобретению также можно вводить в форме суппозиториев для ректального введения. Данные композиции можно получить, смешивая соединение по изобретению с подходящим нераздражающим наполнителем, который является твердым при комнатной температуре, но жидким при ректальной температуре и, следовательно, будет плавиться в прямой кишке, высвобождая активные компоненты. Такие материалы включают, но не ограничиваются этим, масло какао, пчелиный воск и полиэтиленгликоли.The pharmaceutical compositions of this invention can also be administered in the form of suppositories for rectal administration. These compositions can be prepared by mixing the compound of the invention with a suitable non-irritating excipient that is solid at room temperature but liquid at rectal temperature and therefore will melt in the rectum, releasing the active components. Such materials include, but are not limited to, cocoa butter, beeswax, and polyethylene glycols.
Местное введение фармацевтических композиций по данному изобретению особенно применимо, когда желаемая терапия включает области или органы, легко доступные местным нанесением. Для нанесения местно на кожу фармацевтическая композиция должна быть составлена в виде подходящей мази, содержащей активные компоненты, суспендированные или растворенные в носителе. Носители для местного введения соединений по данному изобретению включают, но не ограничиваются этим, минеральное масло, жидкую нефть, белый вазелин, пропиленгликоль, соединение полиоксиэтилена или полиоксипропилена, эмульгирующий воск и воду. Альтернативно, фармацевтическую композицию можно составить в виде подходящего лосьона или крема, содержащего активное соединение, суспендированное или растворенное в носителе. Подходящие носители включают, но не ограничиваются этим, минеральное масло, сорбит моностеарат, полисорбат 60, цетиловые эфиры, воск, цетеариловый спирт, 2-октилдодеканол, бензиловый спирт и воду. Фармацевтические композиции по данному изобретению можно также наносить местно в нижнюю часть кишечника рецептурой ректального суппозитория или в подходящей чистой рецептуре. Местные трансдермальные пластыри также включаются в данное изобретение.Topical administration of the pharmaceutical compositions of this invention is particularly applicable when the desired therapy includes areas or organs readily accessible by topical application. For topical application to the skin, the pharmaceutical composition must be formulated as a suitable ointment containing the active ingredients suspended or dissolved in a carrier. Carriers for topical administration of the compounds of this invention include, but are not limited to, mineral oil, liquid oil, white petrolatum, propylene glycol, a polyoxyethylene or polyoxypropylene compound, emulsifying wax and water. Alternatively, the pharmaceutical composition may be formulated as a suitable lotion or cream containing the active compound, suspended or dissolved in a carrier. Suitable carriers include, but are not limited to, mineral oil, sorbitol monostearate, polysorbate 60, cetyl ethers, wax, cetearyl alcohol, 2-octyldodecanol, benzyl alcohol and water. The pharmaceutical compositions of this invention can also be applied topically to the lower intestine with a rectal suppository formulation or in a suitable, pure formulation. Topical transdermal patches are also included in this invention.
Фармацевтические композиции по данному изобретению можно вводить назальным аэрозолем или ингаляцией. Такие композиции получают методиками, хорошо известными из уровня техники фармацевтических процедур, и их можно приготовить в виде растворов в насыщенных растворах соли, используя бензиловый спирт или другие подходящие консерванты, усилители абсорбции для увеличения биологической доступности, фторуглероды и/или другие солюбилизаторы или диспергирующие агенты, известные из уровня техники.The pharmaceutical compositions of this invention can be administered by nasal aerosol or inhalation. Such compositions are prepared by methods well known in the art of pharmaceutical procedures and can be prepared as solutions in saturated salt solutions using benzyl alcohol or other suitable preservatives, absorption enhancers to increase bioavailability, fluorocarbons and / or other solubilizers or dispersing agents, known from the prior art.
Для профилактики или лечения вирусной инфекции, включая ВИЧ-инфекцию, пригодны уровни дозировок примерно от 0,01 до 25 мг/кг массы тела в день, например, приблизительно от 0,5 до 25 мг/кг массы тела в день соединения активного ингредиента. Типично фармацевтические композиции по данному изобретению будут вводить примерно от 1 до 5 раз в сутки или, альтернативно, в виде непрерывного вливания. Такое введение можно использовать в виде хронической или острой терапии. Количество активного ингредиента, которое можно объединить с веществами носителя для получения монолитной лекарственной формы, будет различаться в зависимости от пациента, которого лечат, и конкретного режима введения. Типичный препарат будет содержать примерно от 5% до 95% активного ингредиента (мас./мас.). Например, такие препараты могут содержать примерно от 20% до 80% активного ингредиента.Dosage levels of from about 0.01 to 25 mg / kg body weight per day, for example from about 0.5 to 25 mg / kg body weight per day of compounding of the active ingredient, are suitable for the prophylaxis or treatment of viral infection, including HIV infection. Typically, the pharmaceutical compositions of this invention will be administered from about 1 to 5 times per day, or, alternatively, as a continuous infusion. Such an introduction can be used in the form of chronic or acute therapy. The amount of active ingredient that can be combined with carrier materials to form a monolithic dosage form will vary depending on the patient being treated and the particular mode of administration. A typical preparation will contain from about 5% to 95% of the active ingredient (w / w). For example, such preparations may contain from about 20% to 80% of the active ingredient.
При улучшении состояния пациента, можно вводить поддерживающую дозу соединения, композиции или комбинации по данному изобретению, если это необходимо. Затем дозировку или частоту введения, или то и другое, можно понизить в виде функции симптомов до уровня, при котором сохраняется улучшенное состояние. Когда симптомы облегчили до желаемого уровня, лечение приостанавливают. Однако пациенты могут требовать периодического лечения на долговременной основе при рецидиве симптомов заболевания.If the patient's condition improves, a maintenance dose of the compound, composition or combination of this invention may be administered, if necessary. Then, the dosage or frequency of administration, or both, can be reduced as a function of symptoms to a level at which an improved condition persists. When symptoms have eased to the desired level, treatment is stopped. However, patients may require periodic treatment on a long-term basis for relapse of the symptoms of the disease.
Специалисту в данной области понятно, что могут быть желательны более низкие или более высокие дозы, чем изложенные выше. Конкретные дозировки и режимы лечения для конкретного пациента могут зависеть от разнообразных факторов, включая активность конкретного применяемого соединения, возраст, массу тела, общее состояние здоровья, пол, режим питания, время введения, скорость выведения, комбинацию лекарственных препаратов, тяжесть и течение инфекции, склонность пациента к инфекции и мнение лечащего врача.One skilled in the art will recognize that lower or higher doses than those set forth above may be desirable. The specific dosage and treatment regimen for a particular patient may depend on a variety of factors, including the activity of the particular compound used, age, body weight, general health, gender, diet, time of administration, excretion rate, combination of drugs, severity and course of infection, tendency patient to infection and the opinion of the attending physician.
В данном описании используют следующие сокращения:The following abbreviations are used in this description:
ПРИМЕРЫEXAMPLES
Данный раздел описывает синтез соединений на основе лизина, способных высвобождать ингибиторы ВИЧ аспартил-протеазы, как описано в настоящей заявке. Данные примеры даны только для иллюстративных целей и их не следует рассматривать как ограничивающие каким-либо образом объем изобретения. Данный раздел предоставляет детальный синтез соединений № 1-10 по данному изобретению.This section describes the synthesis of lysine-based compounds capable of releasing HIV aspartyl protease inhibitors as described herein. These examples are given for illustrative purposes only and should not be construed as limiting in any way the scope of the invention. This section provides a detailed synthesis of compounds No. 1-10 of this invention.
Вещества и методыSubstances and Methods
Аналитическую тонкослойную хроматографию (ТСХ) проводили на 0,25 мм силикагелевых пластинах E. Merck 60 F254 и элюировали показанными системами растворителей. Препаративную хроматографию осуществляли флэш-хроматографией, используя силикагель 60 (EM Science) с показанными системами растворителей и избыточным давлением воздуха, чтобы обеспечить надлежащую скорость элюирования. Определение соединений осуществляли, воздействуя на подвергнутые элюированию пластины (аналитические или препаративные) йодом, УФ-излучением и/или обрабатывая аналитические пластины 2% раствором п-анизальдегида в этаноле, содержащем 3% серной кислоты и 1% уксусной кислоты, после чего следовало нагревание. Альтернативно, аналитические пластины можно обработать 0,3% раствором нингидрина в этаноле, содержащем 3% уксусной кислоты, и/или САМ раствором, изготовленным из 20 г (NH4)6Mo7O24 и 8,3 г Ce(SO4)2 полигидрата в воде (750 мл), содержащей концентрированную серную кислоту (90 мл).Analytical thin layer chromatography (TLC) was performed on 0.25 mm silica gel plates of E. Merck 60 F 254 and was eluted with the solvent systems shown. Preparative chromatography was performed by flash chromatography using silica gel 60 (EM Science) with the solvent systems shown and excess air pressure to ensure proper elution rate. The compounds were determined by exposing the eluted (analytical or preparative) plates to iodine, UV radiation and / or treating the analytical plates with a 2% solution of p-anisaldehyde in ethanol containing 3% sulfuric acid and 1% acetic acid, followed by heating. Alternatively, assay plates can be treated with a 0.3% solution of ninhydrin in ethanol containing 3% acetic acid and / or a CAM solution made from 20 g (NH 4 ) 6 Mo 7 O 24 and 8.3 g Ce (SO 4 ) 2 polyhydrates in water (750 ml) containing concentrated sulfuric acid (90 ml).
Препаративную ВЭЖХ проводили на приборе Gilson, оборудованном С18 колонкой, модулем подачи жидкости 215 и напорными насосами производительностью 25 мл/мин. ВЭЖХ работает с программным обеспечением Gilson UniPoint System.Preparative HPLC was performed on a Gilson instrument equipped with a C18 column, a 215 fluid delivery module and pressure pumps with a capacity of 25 ml / min. HPLC works with the Gilson UniPoint System software.
Условия полупрепаративной ВЭЖХ для очистки тестируемых соединенийSemi-preparative HPLC conditions for purification of test compounds
Система ВЭЖХ: 2 насоса Gilson #305-25 мл, устройство Gilson #215 для ввода и сбора жидкости и детектор УФ-видимого поглощения Gilson #155, все устройства контролируются программным обеспечением Gilson UniPoint V1.91.HPLC system: 2 Gilson # 305-25 ml pumps, Gilson # 215 liquid inlet and collector, and Gilson # 155 UV-Vis detector, all devices are monitored by Gilson UniPoint V1.91 software.
Колонка: Alltech (#96053) Hyperprep PEP, C-18, 100 Еα, 8 мкм, 22х250 ммColumn: Alltech (# 96053) Hyperprep PEP, C-18, 100 Еα, 8 μm, 22x250 mm
Расход: 15 мл/минFlow rate: 15 ml / min
Растворители: A: H2O, B: CH3CNSolvents: A: H 2 O, B: CH 3 CN
Градиент: от 25% до 80% B в течение 40 минGradient: 25% to 80% B for 40 min
Детектор: поглощение; λ:210 & 265 нмDetector: absorption; λ: 210 & 265 nm
Неочищенное вещество, растворенное в ацетонитриле до концентрации примерно от 50 до 80 мг/2 мл, инжектировали в каждом цикле. Фракции собирали в количестве 9 мл, имеющее отношение поглощение определяли УФ-детектором.The crude substance, dissolved in acetonitrile to a concentration of about 50 to 80 mg / 2 ml, was injected in each cycle. Fractions were collected in an amount of 9 ml, the relevant absorption was determined by a UV detector.
Если не указано иным образом, все исходные вещества получали из коммерческих источников, таких как Aldrich Co. или Sigma Co.Unless otherwise indicated, all starting materials were obtained from commercial sources such as Aldrich Co. or Sigma Co.
Температуры плавления (Тпл.) определяли на приборе для измерения температуры плавления Bϋchi 530 в капиллярных трубках, и они не корректировались.Melting points (mp.) Were determined on the device 530 for measuring Bϋchi melting point capillary tubes and are uncorrected.
Масс-спектры записывали на системе Hewlett Packard LC/MSD 1100 с использованием источника APCI или электрораспылительного источника в отрицательном режиме или положительном режиме.Mass spectra were recorded on a Hewlett Packard LC / MSD 1100 system using an APCI source or an electrospray source in negative or positive mode.
Спектры ядерного магнитного резонанса (ЯМР) записывали на Bruker AMX-II-500, оборудованном реверсивным или QNP зондом. Образцы растворяли в дейтерированном хлороформе (CDCl3), дейтерированном ацетоне (ацетон-d6), дейтерированном метаноле (CD3OD) или дейтерированном диметилсульфоксиде (ДМСО-d6), используя тетраметилсилан в качестве внутреннего стандарта для получения данных. Химические сдвиги (*) выражены в миллионных долях (м.д.), константы взаимодействия (J) выражены в Герцах (Гц), тогда как мультиплетности обозначают как с для синглета, д для дублета, 2д для двух дублетов, дд для дублета дублетов, т для триплета, кв для квартета, квинт для квинтета, м для мультиплета и ушир. с для уширенного синглета.Nuclear Magnetic Resonance Spectra (NMR) were recorded on a Bruker AMX-II-500 equipped with a reversible or QNP probe. Samples were dissolved in deuterated chloroform (CDCl 3 ), deuterated acetone (acetone-d 6 ), deuterated methanol (CD 3 OD), or deuterated dimethyl sulfoxide (DMSO-d 6 ) using tetramethylsilane as the internal standard for obtaining data. Chemical shifts (*) are expressed in parts per million (ppm), interaction constants ( J ) are expressed in Hertz (Hz), while the multiplicities are denoted as c for singlet, d for doublet, 2d for two doublets, dd for doublet of doublets , t for triplet, q for quartet, quint for quintet, m for multiplet and broad. with for an extended singlet.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯDETAILED DESCRIPTION OF THE INVENTION
ПРИМЕРЫ:EXAMPLES
Конкретные примеры получения производных общей формулы ISpecific Examples of Derivatives of General Formula I
Следующие ниже соединения получают из производных L-лизина, используя методики, суммированные в схемах 1, 1A, 2, 3, 4 и 5 данного изобретения.The following compounds are prepared from L-lysine derivatives using the procedures summarized in Schemes 1, 1A, 2, 3, 4, and 5 of the present invention.
Пример 1. Получение метилового эфира (1S,5S)-(1-{5-[(4-аминобензолсульфонил)изобутиламино]-6-фосфоноксигексилкарбамоил}-2,2-дифенилэтил)карбаминовой кислоты (PL-461)Example 1. Preparation of (1S, 5S) - (1- {5 - [(4-aminobenzenesulfonyl) isobutylamino] -6-phosphonoxyhexylcarbamoyl} -2,2-diphenylethyl) carbamic acid methyl ester (PL-461)
Получение указанного в заголовке соединения основано на схемах 1 и 2 данного изобретения.The preparation of the title compound is based on Schemes 1 and 2 of the present invention.
Стадия A. Получение (3S)-3-изобутиламиноазепан-2-она ( IV )Stage A. Obtaining (3S) -3-isobutylaminoazepan-2-one ( IV )
L-α-амино-,-капролактам (22,0 г) растворяют в холодном дихлорэтане (ДХМ, 200 мл). Медленно добавляют изобутиральдегид (12,6 г) и перемешивают, пока выделившееся тепло не рассеется (на поверхности образуется вода). Холодный раствор добавляют к 46,5 г порошкообразного NaBH(OAc)3 в ДХМ (0,5 л). К данному раствору добавляют AcOH (70 мл). Слегка мутную смесь перемешивают при 20°C в течение 4 час. К мутной смеси медленно добавляют 500 мл раствора 2М NaOH, рН доводят до 11, используя концентрированный раствор NaOH, и затем смесь перемешивают в течение дополнительных 20 мин. После экстракции слой ДХМ сушат над MgSO4, фильтруют и выпаривают. Полученное таким образом маслянистое вещество медленно кристаллизуют в неподвижном состоянии (27,8 г, 85%) и используют без дополнительной очистки в следующей стадии.L-α-amino -, - caprolactam (22.0 g) was dissolved in cold dichloroethane (DCM, 200 ml). Isobutyraldehyde (12.6 g) is added slowly and stirred until the heat released is dissipated (water forms on the surface). A cold solution is added to 46.5 g of powdered NaBH (OAc) 3 in DCM (0.5 L). AcOH (70 ml) was added to this solution. The slightly turbid mixture was stirred at 20 ° C for 4 hours. 500 ml of a 2M NaOH solution was slowly added to the turbid mixture, the pH was adjusted to 11 using a concentrated NaOH solution, and then the mixture was stirred for an additional 20 minutes. After extraction, the DCM layer was dried over MgSO 4 , filtered and evaporated. The oily substance thus obtained was slowly crystallized in a stationary state (27.8 g, 85%) and was used without further purification in the next step.
1Н ЯМР (CDCl3): 1Н ЯМР (CDCl3): δ 0,93 (д, J=6,5, 3H), 0,97 (д, J=6,5, 3H), 1,39 (т, J=9,8, 1H), 1,47 (м, 1H), 1,78-1,65 (м, 2H), 2,00-1,93 (м, 2H), 2,32-2,2 (м, 2H), 2,38 (т, J=9,7, 1H), 3,16 (м, 3H), 6,62 (с, 1H (NH)). Тпл. 52-54°C (гексан). 1 H NMR (CDCl 3 ): 1 H NMR (CDCl 3 ): δ 0.93 (d, J = 6.5, 3H), 0.97 (d, J = 6.5, 3H), 1.39 (t, J = 9.8, 1H), 1.47 (m, 1H), 1.78-1.65 (m, 2H), 2.00-1.93 (m, 2H), 2.32 -2.2 (m, 2H), 2.38 (t, J = 9.7, 1H), 3.16 (m, 3H), 6.62 (s, 1H (NH)). T pl. 52-54 ° C (hexane).
Небольшой образец превращают в S-метилбензилмочевину, добавляя твердое вещество к раствору S-метилбензилизоцианата в MeCN. ЯМР дает 98% эи.A small sample is converted to S-methylbenzylurea by adding a solid to a solution of S-methylbenzylisocyanate in MeCN. NMR gives 98% ee.
Стадия B. Получение Nα-изобутил-Nα-(4-ацетамидобензолсульфонил)-L-α-амино-,-капролактама ( V )Stage B. Obtaining N α-isobutyl- N α- (4-acetamidobenzenesulfonyl) -L-α-amino -, - caprolactam ( V )
Nα-изобутил-L-α-амино-,-капролактам ( IV ) (4,1 г свободного основания) растворяют в ДХМ (200 мл) и обрабатывают 4,0 г триэтиламина, после чего 4-ацетамидобензолсульфонилхлоридом (5,2 г). Добавляют 0,1 г часть диметиламинопиридина и смесь перемешивают в течение 5 час. Полученную в результате вязкую суспензию выливают в 500 мл 0,5 М HCl и энергично встряхивают. Твердое вещество в двухфазном растворе отфильтровывают и промывают холодным ацетоном, получая 7,3 г (87%) чистого продукта. N α-isobutyl-L-α-amino -, - caprolactam ( IV ) (4.1 g of the free base) was dissolved in DCM (200 ml) and treated with 4.0 g of triethylamine, followed by 4-acetamidobenzenesulfonyl chloride (5.2 g ) 0.1 g of dimethylaminopyridine is added and the mixture is stirred for 5 hours. The resulting viscous suspension was poured into 500 ml of 0.5 M HCl and shaken vigorously. The solid in a two-phase solution is filtered off and washed with cold acetone to give 7.3 g (87%) of pure product.
1Н ЯМР (ДМСО-d6): * 0,93 (д, J=6,0, 3H), 0,96 (д, J=6,0, 3H), 1,39 (т, J=12,0, 1H), 1,85-1,65 (м, 3H), 2,08-2,18 (м и с, 6H), 2,90-2,97 (м, 1Н), 3,00-3,06 (м, 2H), 3,35 (дд, J=14,2, 8,5, 1H), 4,65 (д, J=8,7, 1H), 6,3 (с, 1H), 7,42 (д, J=8,8, 2H), 7,6 (д, J=8,8, 2H). Тпл. 230-233°C (EtOH). 1 H NMR (DMSO-d 6 ): * 0.93 (d, J = 6.0, 3H), 0.96 (d, J = 6.0, 3H), 1.39 (t, J = 12 , 0, 1H), 1.85-1.65 (m, 3H), 2.08-2.18 (m s, 6H), 2.90-2.97 (m, 1H), 3.00 -3.06 (m, 2H), 3.35 (dd, J = 14.2, 8.5, 1H), 4.65 (d, J = 8.7, 1H), 6.3 (s, 1H), 7.42 (d, J = 8.8, 2H), 7.6 (d, J = 8.8, 2H). T pl. 230-233 ° C (EtOH).
Стадия C. Получение трет-бутилового эфира (3S)-3-{[4-ацетил-трет-бутоксикарбониламино)бензолсульфонил]изобутиламино}-2-оксоазепан-1-карбоновой кислоты (Boc активация) ( VI )Step C. Preparation of (3S) -3 - {[4-acetyl-tert-butoxycarbonylamino) benzenesulfonyl] isobutylamino} -2-oxoazepane-1-carboxylic acid tert-butyl ester (Boc activation) ( VI )
4,2 г Nα-изобутил-Nα-(4-ацетамидобензолсульфонил)-L-α-амино-,-капролактама ( V ) суспендируют в 30 мл MeCN и кратковременно обрабатывают ультразвуком для измельчения крупных кусков. К данной белой суспензии добавляют 6,7 г (3 экв.) ди-трет-бутилпирокарбоната в 10 мл MeCN. Суспензию перемешивают магнитной мешалкой и добавляют 120 мг порцию ДМАП. Раствор становится ярко-желтого цвета после нескольких минут. ТСХ (EtOAc) обнаруживает 1 продукт Rf 0,9 (исходное вещество Rf 0,4). Раствор выливают в 20 мл дистиллированной воды и экстрагируют эфиром, сушат над Na2SO4 и выпаривают, получая 6,90 г. Образец перекристаллизовывают из гексана.4.2 g of N α-isobutyl- N α- (4-acetamidobenzenesulfonyl) -L-α-amino -, - caprolactam ( V ) are suspended in 30 ml of MeCN and sonicated briefly to grind large pieces. To this white suspension was added 6.7 g (3 equiv.) Of di- tert-butyl pyrocarbonate in 10 ml of MeCN. The suspension is stirred with a magnetic stirrer and a 120 mg portion of DMAP is added. The solution turns bright yellow after a few minutes. TLC (EtOAc) detects 1 product Rf 0.9 (starting material Rf 0.4). The solution was poured into 20 ml of distilled water and extracted with ether, dried over Na 2 SO 4 and evaporated to give 6.90 g. A sample was recrystallized from hexane.
1Н ЯМР (ДМСО-d6): * 0,68 (д, J=6,0, 3H), 0,85 (д, J=6,0, 3H), 1,39 (с, 10H), 1,47 (с, 9H), 1,85-1,65 (м, 3H), 2,15 (с, 3H), 2,80 (кв, J=4, 1Н), 3,10-3,36 (м, 2H), 4,01 (д, J=8,0, 1Н), 4,85 (д, J=8,7, 1H), 7,32 (д, J=8,8, 2H), 7,87 (д, J=8,8, 2H) Тпл. 123-124°C. 1 H NMR (DMSO-d 6 ): * 0.68 (d, J = 6.0, 3H), 0.85 (d, J = 6.0, 3H), 1.39 (s, 10H), 1.47 (s, 9H), 1.85-1.65 (m, 3H), 2.15 (s, 3H), 2.80 (q, J = 4, 1H), 3.10-3, 36 (m, 2H), 4.01 (d, J = 8.0, 1H), 4.85 (d, J = 8.7, 1H), 7.32 (d, J = 8.8, 2H ), 7.87 (d, J = 8.8, 2H) T pl. 123-124 ° C.
Стадия D. Получение (1S)-4-амино-N-(5-амино-1-гидроксиметилпентил)-N-изобутилбензолсульфонамида ( VII-без защиты) (восстановительное раскрытие цикла и снятие защиты)Step D. Preparation of (1S) -4-amino- N - (5-amino-1-gidroksimetilpentil) - N -izobutilbenzolsulfonamida (VII- no protection) (reductive ring opening and deprotection)
3,0 г порцию трет-бутилового эфира (3S)-3-{[4-ацетил-трет-бутоксикарбониламино)бензолсульфонил]изобутиламино}-2-оксоазепан-1-карбоновой кислоты ( VI , стадия C) растворяют в 40 мл EtOH, после чего добавляют 750 мг NaBH4. Кратковременное нагревание феном дает прозрачный раствор. ТСХ обнаруживает только полосчатое пятно через 20 мин (EtOAc). Раствор концентрируют до пасты, выливают в 40 мл 1 н. NaOH и экстрагируют этилацетатом, органическую фазу сушат над Na2SO4 и выпаривают, получая 2,8 г промежуточного продукта ( VII ); трет-бутилового эфира (1S)-{4-[(5-трет-бутоксикарбониламино-1-гидроксиметилпентил)изобутилсульфамоил]фенил}карбаминовой кислоты ( VII )A 3.0 g portion of (3S) -3 - {[4-acetyl-tert-butoxycarbonylamino) benzenesulfonyl] isobutylamino} -2-oxoazepan-1-carboxylic acid tert-butyl ester ( VI , step C) is dissolved in 40 ml of EtOH, then add 750 mg of NaBH 4 . Short-term heating with a hairdryer gives a clear solution. TLC only detected a streaky spot after 20 minutes (EtOAc). The solution is concentrated to a paste, poured into 40 ml of 1 N. NaOH and extracted with ethyl acetate, the organic phase is dried over Na 2 SO 4 and evaporated, obtaining 2.8 g of intermediate ( VII ); (1S) tert-butyl ester - {4 - [(5-tert-butoxycarbonylamino-1-hydroxymethylpentyl) isobutylsulfamoyl] phenyl} carbamic acid ( VII )
Вышеуказанный промежуточный продукт растворяют в 5 мл EtOH и добавляют 5 мл 12 н. HCl. В течение нескольких минут наблюдается энергичное выделение газа. Через 2 ч раствор выпаривают, делают щелочным концентрированным KOH и экстрагируют EtOAc, получая 1,75 г белого порошка.The above intermediate was dissolved in 5 ml EtOH and 5 ml 12 N was added. HCl. Vigorous gas evolution is observed for several minutes. After 2 hours, the solution was evaporated, concentrated alkaline KOH was made and extracted with EtOAc to obtain 1.75 g of a white powder.
1Н ЯМР (ДМСО-d6): * 0,82 (м, 6H), 0,97-1,12 (м, 2H), 1,15-1,30 (м, 3H), 1,57 (м, 1Н), 1,84 (м, 1H), 2,40 (т, J=7,8, 2H), 2,75 (м, 1H), 2,85 (м, 1H), 3,21 (м, 1H), 3,44 (д, J=6,4, 2H), 5,92 (ушир.с, 2H), 6,59 (д, J=8,0, 2H), 7,39 (д, J=8,0, 2H). 1 H NMR (DMSO-d 6 ): * 0.82 (m, 6H), 0.97-1.12 (m, 2H), 1.15-1.30 (m, 3H), 1.57 ( m, 1H), 1.84 (m, 1H), 2.40 (t, J = 7.8, 2H), 2.75 (m, 1H), 2.85 (m, 1H), 3.21 (m, 1H), 3.44 (d, J = 6.4, 2H), 5.92 (br s, 2H), 6.59 (d, J = 8.0, 2H), 7.39 (d, J = 8.0, 2H).
Стадия E. Получение (2S)-2-метоксикарбониламино-3,3-дифенилпропионовой кислотыStage E. Obtaining (2S) -2-methoxycarbonylamino-3,3-diphenylpropionic acid
К раствору L-дифенилаланина (241 мг, 1,0 ммоль) (Peptech Corp.) в 5 мл 1 н. NaOH и 0,5 мл насыщенного Na2CO3 (полученный в результате раствор при рН 10) добавляют метоксикарбонилоксисукцинимид ((2,5-диоксопирролидин-1-ил)метиловый эфир углекислоты) (180 мг, 1,1 ммоль), растворенный в 5 мл. После этого реакционную смесь перемешивают при комнатной температуре в течение 2 час. Щелочной раствор один раз экстрагируют эфиром (10 мл) и водную фазу подкисляют 1 н. HCl. Данную фазу дважды экстрагируют 20 мл EtOAc и объединенные органические фазы промывают 50 мл 1 н. HCl. Органическую фазу сушат над Na2SO4, фильтруют и выпаривают до маслянистого вещества, которое отверждают, получая 250 мг (83%) желаемого вещества. Данное производное используют без дополнительной обработки в следующей стадии.To a solution of L-diphenylalanine (241 mg, 1.0 mmol) (Peptech Corp.) in 5 ml of 1 N. NaOH and 0.5 ml saturated Na 2 CO 3 (the resulting solution at pH 10) add methoxycarbonyloxysuccinimide ((2,5-dioxopyrrolidin-1-yl) carbonic acid methyl ester) (180 mg, 1.1 mmol) dissolved in 5 ml After that, the reaction mixture was stirred at room temperature for 2 hours. The alkaline solution is extracted once with ether (10 ml) and the aqueous phase is acidified with 1 N. HCl. This phase is extracted twice with 20 ml of EtOAc and the combined organic phases are washed with 50 ml of 1 N. HCl. The organic phase was dried over Na 2 SO 4 , filtered and evaporated to an oily substance, which was solidified, yielding 250 mg (83%) of the desired substance. This derivative is used without further processing in the next step.
Стадия F. Получение метилового эфира (1S,5S)-(1-{5-[(4-аминобензолсульфонил)изобутиламино]-6-гидроксигексилкарбамоил}-2,2-дифенилэтил)карбаминовой кислоты (PL-100)Step F. Preparation of (1S, 5S) - (1- {5 - [(4-aminobenzenesulfonyl) isobutylamino] -6-hydroxyhexylcarbamoyl} -2,2-diphenylethyl) carbamic acid methyl ester (PL-100)
Указанное в заголовке соединение получают из (1S)-4-амино-N-(5-амино-1-гидроксиметилпентил)-N-изобутилбензолсульфонамида ( VII-без защиты) (стадия D) и (2S)-2-метоксикарбониламино-3,3-дифенилпропионовой кислоты (стадия E), используя процедуру сочетания с HOBt и EDAC, описанную в примере 3 (стадия D). Конечный продукт получают с выходом 67% (121 мг).The title compound was prepared from (1S) -4-amino- N - (5-amino-1-gidroksimetilpentil) - N -izobutilbenzolsulfonamida (VII- no protection) (step D) and (2S) -2-methoxycarbonylamino-3, 3-diphenylpropionic acid (step E) using the coupling procedure with HOBt and EDAC described in Example 3 (step D). The final product was obtained in 67% yield (121 mg).
ЖХ-МС: 625,3 (M+H)+, 95% чист.LC-MS: 625.3 (M + H) + , 95% pure.
1Н ЯМР (CD3OD): δ 0,71-0,85 (м, 2H), 0,88 (д, J=6,3, 5H), 0,91-0,96 (м, 2H), 1,29-1,34 5 (м, 1H), 1,41-1,52 (м, 1H) 1,82-1,92 (м, 1H), 2,61-2,68 (м, 1H), 2,81-2,85 (м, 2H), 2,94-3,05 (м, 2H), 3,38-3,40 (т, J=5,0, 1Н), 3,50-3,51 (м, 1H), 3,52 (с, 3H), 4,28 (д, J=11,0 1Н), 4,87 (д, J=11,0, 1H), 6,69 (д, J=8,0, 2H), 7,15-718 (м, 2H), 7,20-7,31 (м, 6H), 7,33 (д, J=7,9, 2H), 7,47 (д, J=7,5, 1H). 1 H NMR (CD 3 OD): δ 0.71-0.85 (m, 2H), 0.88 (d, J = 6.3, 5H), 0.91-0.96 (m, 2H) , 1.29-1.34 5 (m, 1H), 1.41-1.52 (m, 1H) 1.82-1.92 (m, 1H), 2.61-2.68 (m, 1H), 2.81-2.85 (m, 2H), 2.94-3.05 (m, 2H), 3.38-3.40 (t, J = 5.0, 1H), 3, 50-3.51 (m, 1H), 3.52 (s, 3H), 4.28 (d, J = 11.0 1H), 4.87 (d, J = 11.0, 1H), 6 69 (d, J = 8.0, 2H), 7.15-718 (m, 2H), 7.20-7.31 (m, 6H), 7.33 (d, J = 7.9, 2H), 7.47 (d, J = 7.5, 1H).
13С ЯМР (CD3OD): δ 20,0, 20,1, 23,3, 25,4, 28,1, 28,5, 28,9, 38,1, 40,0, 51,2, 51,6, 53,1, 57,2, 57,4, 59,5, 61,9, 62,4, 112,6, 125,7, 126,2, 126,3, 127,9, 128,1, 128,15, 128,2, 128,4, 128,7, 141,3, 141,9, 152,4, 155,9, 169,9, 172,5. 13 C NMR (CD 3 OD): δ 20.0, 20.1, 23.3, 25.4, 28.1, 28.5, 28.9, 38.1, 40.0, 51.2, 51.6, 53.1, 57.2, 57.4, 59.5, 61.9, 62.4, 112.6, 125.7, 126.2, 126.3, 127.9, 128, 1, 128.15, 128.2, 128.4, 128.7, 141.3, 141.9, 152.4, 155.9, 169.9, 172.5.
Стадия G. Получение метилового эфира (1S,5S)-{1-[5-[(4-аминобензолсульфонил)изобутиламино]-6-(диэтоксифосфорилокси)гексилкарбамоил]-2,2-дифенилэтил}карбаминовой кислотыStep G. Preparation of (1S, 5S) - {1- [5 - [(4-aminobenzenesulfonyl) isobutylamino] -6- (diethoxyphosphoryloxy) hexylcarbamoyl] -2,2-diphenylethyl} carbamic acid methyl ester
Соединение PL-100 (продукт стадии F, 203 мг, 0,325 ммоль) растворяют в смеси сухого тетрагидрофурана (3 мл) и 0,2 мл триэтилфосфата под атмосферой N2. Смесь перемешивают при данной температуре в течение 15 мин, после чего добавляют диэтилхлорфосфат (0,061 мл, 0,423 ммоль). При 0°C добавляют гидрид натрия (60% в минеральном масле) (17 мг, 0,423 ммоль). Раствор перемешивают в течение 1 часа при 0°C и 12 ч при комнатной температуре. К данному раствору добавляют 20 мл Amberlite XAD-2 и гранулы тщательно перемешивают с растворителем. К смеси добавляют воду со льдом 2 мл и ТГФ выпаривают. Затем гранулы 6 раз промывают дистиллированной водой 100 мл, затем три раза экстрагируют этилацетатом (30 мл). Объединенную фазу выпаривают и остаток сушат под высоким вакуумом. Неочищенный продукт очищают флэш-хроматографией, используя смесь этилацетат/гексан (8/2), затем EtOAc 100% в качестве элюента. Выход данной реакции составляет 152 мг 61%.Compound PL-100 (product of step F, 203 mg, 0.325 mmol) was dissolved in a mixture of dry tetrahydrofuran (3 ml) and 0.2 ml of triethyl phosphate under an atmosphere of N 2 . The mixture was stirred at this temperature for 15 minutes, after which diethyl chlorophosphate (0.061 ml, 0.423 mmol) was added. Sodium hydride (60% in mineral oil) (17 mg, 0.423 mmol) was added at 0 ° C. The solution was stirred for 1 hour at 0 ° C and 12 hours at room temperature. To this solution, 20 ml of Amberlite XAD-2 is added and the granules are thoroughly mixed with the solvent. 2 ml of ice-water was added to the mixture, and THF was evaporated. Then the granules are washed 6 times with distilled water 100 ml, then extracted three times with ethyl acetate (30 ml). The combined phase was evaporated and the residue was dried under high vacuum. The crude product was purified by flash chromatography using ethyl acetate / hexane (8/2), then EtOAc 100% as eluent. The yield of this reaction is 152 mg 61%.
ЖХ-МС: 761,2 (M+H)+, 90% чист.LC-MS: 761.2 (M + H) + , 90% pure.
1Н ЯМР (CD3OD): δ 0,68-0,75 (м, 1H), 0,75-0,84 (м, 1Н), 0,84-1,10 (м, 9H), 1,21-1,50 (м, 8H), 1,88 (м, 1H), 2,58-2,71 (м, 1Н), 2,80-2,89 (м, 1Н), 2,89-3,08 (м, 2H), 3,49-3,60 (с, 3H), 3,65-3,74 (м, 1H), 3,85-3,95 (м, 1H), 3,97-4,02 (м, 1Н), 4,07-4,21 (м, 4H), 4,29 (д, J=10,8, 1H), 6,71 (д, J=8,0, 2H), 7,10-7,20 (м, 2H), 7,20-7,32 (м, 5H), 7,32-7,45 (м, 3H), 7,50 (д, J=7,5,2H), 7,86 (ушир.с, 1Н). 1 H NMR (CD 3 OD): δ 0.68-0.75 (m, 1H), 0.75-0.84 (m, 1H), 0.84-1.10 (m, 9H), 1 , 21-1.50 (m, 8H), 1.88 (m, 1H), 2.58-2.71 (m, 1H), 2.80-2.89 (m, 1H), 2.89 -3.08 (m, 2H), 3.49-3.60 (s, 3H), 3.65-3.74 (m, 1H), 3.85-3.95 (m, 1H), 3 97-4.02 (m, 1H), 4.07-4.21 (m, 4H), 4.29 (d, J = 10.8, 1H), 6.71 (d, J = 8, 0, 2H), 7.10-7.20 (m, 2H), 7.20-7.32 (m, 5H), 7.32-7.45 (m, 3H), 7.50 (d, J = 7.5.2H), 7.86 (br s, 1H).
31P ЯМР (CD3OD): δ 1,62. 31 P NMR (CD 3 OD): δ 1.62.
Стадия H. Получение метилового эфира (1S,5S)-(1-{5-[(4-аминобензолсульфонил)изобутиламино]-6-фосфоноксигексилкарбамоил}-2,2-дифенилэтил)карбаминовой кислоты (PL-461)Step H. Preparation of (1S, 5S) - (1- {5 - [(4-aminobenzenesulfonyl) isobutylamino] -6-phosphonoxyhexylcarbamoyl} -2,2-diphenylethyl) carbamic acid methyl ester (PL-461)
Продукт из стадии G, приготовленный выше, (152 мг) растворяют в безводном дихлорметане (3,0 мл). При 0°C добавляют триметилсилилбромид (0,5 мл). Смесь перемешивают в течение 1 ч при данной температуре и в течение ночи при комнатной температуре. Растворитель выпаривают и к остатку добавляют 0,2 мл воды. Добавляют 3 мл EtOH, перемешивают и выпаривают. Данную стадию повторяют три раза и остаток сушат под вакуумом. Выход 98 мг 70% указанных в заголовке производных из данного первого примера.The product from step G prepared above (152 mg) was dissolved in anhydrous dichloromethane (3.0 ml). Trimethylsilyl bromide (0.5 ml) was added at 0 ° C. The mixture was stirred for 1 h at this temperature and overnight at room temperature. The solvent was evaporated and 0.2 ml of water was added to the residue. Add 3 ml EtOH, mix and evaporate. This step is repeated three times and the residue is dried under vacuum. Yield 98 mg, 70% of the title derivatives of this first example.
ЖХ-МС: 705,2 (M+H)+, 95% чист.LC-MS: 705.2 (M + H) + , 95% pure.
1Н ЯМР (CD3OD): δ 0,65-0,73 (м, 1H), 0,75-0,83 (м, 1H), 0,89 (д, J=5,6, 8H), 1,27-1,38, (м, 1H), 1,42-4,55 (м, 1H), 1,82-1,94 (м, 1Н), 2,57-2,68 (м, 1H), 2,78-2,90 (м, 1H), 2,91-20 3,09 (м, 2H), 3,54 (с, 3H), 3,60-3,72 (м, 1H), 3,87-4,05 (м, 1H), 4,00 (м, 1Н), 4,29 (д, J=11,3, 1Н), 4,90 (д, J=11,4, 1H), 6,73 (д, J=8,0, 2H), 7,13-7,22 (м, 2H), 7,22-7,33 (м, 6H), 7,33-7,45 (м, 2H), 7,51 (д, J=7,5, 2H). 1 H NMR (CD 3 OD): δ 0.65-0.73 (m, 1H), 0.75-0.83 (m, 1H), 0.89 (d, J = 5.6, 8H) , 1.27-1.38, (m, 1H), 1.42-4.55 (m, 1H), 1.82-1.94 (m, 1H), 2.57-2.68 (m , 1H), 2.78-2.90 (m, 1H), 2.91-20 3.09 (m, 2H), 3.54 (s, 3H), 3.60-3.72 (m, 1H), 3.87-4.05 (m, 1H), 4.00 (m, 1H), 4.29 (d, J = 11.3, 1H), 4.90 (d, J = 11, 4, 1H), 6.73 (d, J = 8.0, 2H), 7.13-7.22 (m, 2H), 7.22-7.33 (m, 6H), 7.33- 7.45 (m, 2H); 7.51 (d, J = 7.5, 2H).
31P ЯМР (CD3OD): δ 2,80. 31 P NMR (CD 3 OD): δ 2.80.
Пример 2. Получение натриевой соли метилового эфира (1S,5S)-(1-{5-[(4-аминобензолсульфонил)изобутиламино]-6-фосфоноксигексилкарбамоил}-2,2-дифенилэтил)карбаминовой кислоты (PL-462)Example 2. Obtaining the sodium salt of methyl ester (1S, 5S) - (1- {5 - [(4-aminobenzenesulfonyl) isobutylamino] -6-phosphonoxyhexylcarbamoyl} -2,2-diphenylethyl) carbamic acid (PL-462)
70,7 мг конечного продукта из примера 1 добавляют к 1 мл 0,1 н. NaOH и разбавляют 1 мл дистиллированной воды. Данный раствор затем замораживают и подвергают лиофилизации. Выход 67,2 мг (92%) желаемого вещества с чистотой 95%.70.7 mg of the final product from example 1 is added to 1 ml of 0.1 N. NaOH and diluted with 1 ml of distilled water. This solution is then frozen and lyophilized. Yield 67.2 mg (92%) of the desired substance with a purity of 95%.
1Н ЯМР (CD3OD): δ 0,72-0,83 (м, 1H), 0,90 (д, J=5,8, 9H), 1,26-1,38 (м, 1Н), 1,53-1,65 (м, 1Н), 1,88-2,00 (м, 1Н), 2,60-2,70 (м, 1Н), 2,79-2,89 (м, 1Н), 2,98-3,00 (м, 1H), 3,00-3,08 (м, 1H), 3,54 (с, 3H), 3,58-3,71 (м, 1H), 3,72-3,83 (м, 1H), 3,84-3,95 (м, 1H), 4,28 (д, J=11,1, 1H), 4,91 (д, J=11,0, 1H), 6,70 (д, J=7,6, 2H), 7,12-7,22 (м, 2H), 7,22-7,32 (м, 6H), 7,33-7,40 (м, 2H), 7,50 (д, J=7,7, 2H). 1 H NMR (CD 3 OD): δ 0.72-0.83 (m, 1H), 0.90 (d, J = 5.8, 9H), 1.26-1.38 (m, 1H) 1.53-1.65 (m, 1H), 1.88-2.00 (m, 1H), 2.60-2.70 (m, 1H), 2.79-2.89 (m, 1H), 2.98-3.00 (m, 1H), 3.00-3.08 (m, 1H), 3.54 (s, 3H), 3.58-3.71 (m, 1H) 3.72-3.83 (m, 1H); 3.84-3.95 (m, 1H); 4.28 (d, J = 11.1; 1H); 4.91 (d, J = 11.0, 1H), 6.70 (d, J = 7.6, 2H), 7.12-7.22 (m, 2H), 7.22-7.32 (m, 6H), 7, 33-7.40 (m, 2H); 7.50 (d, J = 7.7, 2H).
31P ЯМР (CD3OD): δ 3,13. 31 P NMR (CD 3 OD): δ 3.13.
Пример 3. ПолучениеExample 3. Getting метилового эфира (1S,5S)-(1-{5-[(4-аминобензолсульфонил)изобутиламино]-6-фосфоноксигексилкарбамоил}-2-нафталин-2-илэтил)карбаминовой кислоты (PL-507)(1S, 5S) methyl ester - (1- {5 - [(4-aminobenzenesulfonyl) isobutylamino] -6-phosphonoxyhexylcarbamoyl} -2-naphthalen-2-yl-ethyl) carbamic acid (PL-507)
Получение указанного в заголовке соединения основано на схеме 2 данного изобретения.The preparation of the title compound is based on Scheme 2 of the present invention.
Схема A. Получение трет-бутилового эфира (1S)-(4-{[5-трет-бутоксикарбониламино-1-(диэтоксифосфорилоксиметил)пентил]изобутилсульфамоил}фенил)-карбаминовой кислоты ( VIII )Scheme A. Preparation of (1S) - (4 - {[5-tert-butoxycarbonylamino-1- (diethoxyphosphoryloxymethyl) pentyl] isobutylsulfamoyl} phenyl) carbamic acid tert-butyl ester ( VIII )
2,00 г (3,7 ммоль) трет-бутилового эфира (1S)-{4-[(5-трет-бутоксикарбониламино-1-гидроксиметилпентил)изобутилсульфамоил]фенил}карбаминовой кислоты ( VII ) (пример 1, стадия D) растворяют в смеси 0,63 мл триэтилфосфата и 10 мл ТГФ при 0°C в инертной атмосфере аргона. Добавляют 0,63 мл (4,44 ммоль) диэтилхлорфосфата и затем порциями добавляют 0,25 г (6,2 ммоль) NaH 60% в масле. Смеси дают нагреться до комнатной температуры и оставляют перемешиваться в течение 2 ч (ЖХ-МС показывает завершение после 1 ч). К данному раствору добавляют 20 мл смолы Amberlite XAD-2, суспензию тщательно перемешивают и добавляют к 200 мл воды со льдом. После перемешивания в течение 15 мин суспензию смолы отфильтровывают и смолу несколько раз промывают дистиллированной водой (500 мл). Желаемый продукт десорбируют из смолы ацетоном (5 Х 50 мл), EtOAc (5 Х 50 мл), затем органическую фазу сушат над Na2SO4. После выпаривания растворителя получают 2,66 г (89%) прозрачного маслянистого вещества. Неочищенный продукт содержит фракцию с двумя диэтилфосфатами и используют без дополнительной обработки в следующей стадии.2.00 g (3.7 mmol) of (1S) tert-butyl ether - {4 - [(5-tert-butoxycarbonylamino-1-hydroxymethylpentyl) isobutylsulfamoyl] phenyl} carbamic acid ( VII ) (Example 1, step D) are dissolved in a mixture of 0.63 ml of triethyl phosphate and 10 ml of THF at 0 ° C in an inert atmosphere of argon. 0.63 ml (4.44 mmol) of diethyl chlorophosphate is added and then 0.25 g (6.2 mmol) of 60% NaH in oil is added portionwise. The mixture was allowed to warm to room temperature and allowed to mix for 2 hours (LC-MS shows completion after 1 hour). To this solution, 20 ml of Amberlite XAD-2 resin is added, the suspension is thoroughly mixed and added to 200 ml of ice water. After stirring for 15 minutes, the resin suspension is filtered off and the resin washed several times with distilled water (500 ml). The desired product is stripped from the resin with acetone (5 X 50 ml), EtOAc (5 X 50 ml), then the organic phase is dried over Na 2 SO 4 . After evaporation of the solvent, 2.66 g (89%) of a clear oily substance are obtained. The crude product contains a fraction with two diethyl phosphates and is used without further processing in the next step.
1Н ЯМР (CD3OD): δ 0,91 (д, J=6,3, 6H), 1,11-1,21 (м, 2H), 1,33 (т, J=6,9, 10Н), 1,43 (с, 9H), 1,53 (с, 10H), 1,90-1,97 (м, 1H), 2,88-2,96 (м, 3H), 2,96-3,04 (м, 1H), 3,81-3,90 (м, 1H), 3,91-3,99 (м, 1H), 4,01-4,14 (м, 4H), 7,61 (д, J=8,3, 2H), 7,72 (д, J=8,4,2H). 1 H NMR (CD 3 OD): δ 0.91 (d, J = 6.3, 6H), 1.11-1.21 (m, 2H), 1.33 (t, J = 6.9, 10H), 1.43 (s, 9H), 1.53 (s, 10H), 1.90-1.97 (m, 1H), 2.88-2.96 (m, 3H), 2.96 -3.04 (m, 1H), 3.81-3.90 (m, 1H), 3.91-3.99 (m, 1H), 4.01-4.14 (m, 4H), 7 61 (d, J = 8.3, 2H); 7.72 (d, J = 8.4.2H).
31P ЯМР (CD3OD): δ 1,59. 31 P NMR (CD 3 OD): δ 1.59.
Стадия B. Получение (диэтил)(6-амино-2-[(4-аминобензолсульфонил)изобутиламино]гексилового) эфира (2S)-фосфорной кислоты ( IX )Stage B. Obtaining (diethyl) (6-amino-2 - [(4-aminobenzenesulfonyl) isobutylamino] hexyl) ether (2S) -phosphoric acid ( IX )
Неочищенный продукт, полученный в предыдущей стадии ( VIII , 2,66 г) растворяют в 12 мл EtOH. Добавляют 4 мл HClконц. и кратковременно нагревают до 70°C, затем оставляют при комнатной температуре в течение 3 ч. Растворитель выпаривают и остаток растирают в 50 мл эфира. Затем вязкий остаток растворяют в 3 мл воды со льдом и рН корректируют до 12 50% NaOH. Полученную вязкую суспензию экстрагируют EtOAc (3 Х 50 мл) и органическую фазу сушат над Na2SO4. После отфильтровывания осушающего агента органическую фазу откачивают, получая 1,84 г (98%) желаемого продукта ( IX ).The crude product obtained in the previous step ( VIII , 2.66 g) was dissolved in 12 ml of EtOH. 4 ml of HCl conc. and briefly heated to 70 ° C, then left at room temperature for 3 hours. The solvent was evaporated and the residue was triturated with 50 ml of ether. Then the viscous residue is dissolved in 3 ml of ice water and the pH is adjusted to 12 to 50% NaOH. The resulting viscous suspension was extracted with EtOAc (3 X 50 ml) and the organic phase was dried over Na 2 SO 4 . After filtering off the drying agent, the organic phase is evacuated, yielding 1.84 g (98%) of the desired product ( IX ).
ЖХ-МС: 480,2 (M+H)+, 95% чист.LC-MS: 480.2 (M + H) + , 95% pure.
1H ЯМР (CD3OD): δ 0,91 (с, 6H), 1,11-1,26 (м, 3H), 1,28-1,43 (м, 8H), 1,45-1,51 (м, 1Н), 1,52-1,61 (м, 1H), 1,89-1,96 (м, 1Н), 2,56 (т, J=6,7, 2H), 2,85-2,91 (м, 1Н), 2,98-3,11 (м, 1H), 3,79-3,99 (м, 1H), 3,94 (д, J=5,3, 1Н), 4,09-4,11 (м, 4H), 6,69 (д, J=7,9, 2H), 7,50 (д, J=7,9, 2H). 1 H NMR (CD 3 OD): δ 0.91 (s, 6H), 1.11-1.26 (m, 3H), 1.28-1.43 (m, 8H), 1.45-1 51 (m, 1H), 1.52-1.61 (m, 1H), 1.89-1.96 (m, 1H), 2.56 (t, J = 6.7, 2H), 2 85-2.91 (m, 1H), 2.98-3.11 (m, 1H), 3.79-3.99 (m, 1H), 3.94 (d, J = 5.3, 1H), 4.09-4.11 (m, 4H), 6.69 (d, J = 7.9, 2H), 7.50 (d, J = 7.9, 2H).
31P ЯМР (CD3OD): δ 1,61 31 P NMR (CD 3 OD): δ 1.61
Стадия C. Получение (2S)-2-метоксикарбониламино-3-нафталин-2-илпропионовой кислоты (или L-Moc-2-нафтилаланина)Step C. Preparation of (2S) -2-methoxycarbonylamino-3-naphthalen-2-yl-propionic acid (or L-Moc-2-naphthylalanine)
К раствору L-2-нафтилаланина (215 мг, 1 ммоль) (Peptech Corp.) в 5 мл 1 н. NaOH и 0,5 мл насыщенного Na2CO3 (pH полученного в результате раствора 10) добавляют метоксикарбонилоксисукцинимид (187 мг, 1,1 ммоль), растворенный в 5 мл. После этого реакционную смесь перемешивают при комнатной температуре в течение 2 ч. Щелочной раствор один раз экстрагируют эфиром (10 мл) и водную фазу подкисляют 1 н. HCl. Данную фазу дважды экстрагируют 20 мл EtOAc и объединенные органические фазы промывают 50 мл 1 н. HCl. Органическую фазу сушат над Na2SO4, фильтруют и выпаривают до маслянистого вещества, которое отверждают, получая 200 мг (73%) желаемого вещества. Данное промежуточное вещество (называемое N-замещенной аминокислотой) используют без дополнительной обработки в следующей стадии.To a solution of L-2-naphthylalanine (215 mg, 1 mmol) (Peptech Corp.) in 5 ml of 1 N. NaOH and 0.5 ml of saturated Na 2 CO 3 (pH of the resulting solution 10) add methoxycarbonyloxysuccinimide (187 mg, 1.1 mmol) dissolved in 5 ml. After that, the reaction mixture was stirred at room temperature for 2 hours. The alkaline solution was extracted once with ether (10 ml) and the aqueous phase was acidified with 1N HCl. This phase is extracted twice with 20 ml of EtOAc and the combined organic phases are washed with 50 ml of 1 N. HCl. The organic phase was dried over Na 2 SO 4 , filtered and evaporated to an oily substance, which was solidified, yielding 200 mg (73%) of the desired substance. This intermediate (called an N- substituted amino acid) is used without further processing in the next step.
Стадия D. Получение метилового эфира (1S,5S)-(1-{5-[(4-аминобензолсульфонил)изобутиламино]-6-фосфоноксигексилкарбамоил}-2-нафталин-2-илэтил)карбаминовой кислоты (PL-507)Step D. Preparation of (1S, 5S) - (1- {5 - [(4-aminobenzenesulfonyl) isobutylamino] -6-phosphonoxyhexylcarbamoyl} -2-naphthalen-2-yl-ethylcarbamic acid methyl ester (PL-507)
100 мг L-Moc-2-нафтилаланина (стадия C) активируют 100 мг EDAC и 57 мг HOBt в 1,5 мл ДМФА в течение 30 минут. Затем добавляют 100 мг (6-амино-2-[(4-аминобензолсульфонил)изобутиламино]гексил)(диэтилового) эфира фосфорной кислоты (стадия B) и оставляют перемешиваться при комнатной температуре в течение 1 ч. К раствору ДМФА добавляют 40 мл 1М K2CO3 и оставляют в течение 10 мин. Затем добавляют 50 мл EtOAc и затем смесь энергично перемешивают. Осуществляют отделение фазы EtOAc, после чего следует экстракция один раз 5% лимонной кислотой (50 мл), затем 3 раза водой (50 мл) и, наконец, насыщенным раствором соли. Органическую фазу отделяют и выпаривают. Остаток отбирают в 50 мл ДХМ и повторно выпаривают. Остаток снова отбирают в 50 мл ДХМ и добавляют 0,5 мл TMSBr. Раствор оставляют на ночь (16 ч). ДХМ откачивают и добавляют охлажденный на ледяной бане раствор MeOH:Вода 1:1, кратковременно перемешивают и откачивают. Остаток растирают в эфире, затем растворяют в 1 н. NaOH. Прозрачный раствор экстрагируют эфиром и водную фазу подкисляют 6 н. HCl. Затем фильтрованием собирают белый осадок и сушат под вакуумом в течение ночи. Выход 88 мг указанного в заголовке соединения.100 mg of L-Moc-2-naphthylalanine (step C) activate 100 mg of EDAC and 57 mg of HOBt in 1.5 ml of DMF for 30 minutes. Then, 100 mg of (6-amino-2 - [(4-aminobenzenesulfonyl) isobutylamino] hexyl) (diethyl) phosphoric acid ester (step B) was added and allowed to stir at room temperature for 1 h. 40 ml of 1M K was added to the DMF solution 2 CO 3 and left for 10 minutes Then add 50 ml EtOAc and then the mixture is vigorously stirred. The EtOAc phase is separated, followed by extraction once with 5% citric acid (50 ml), then 3 times with water (50 ml) and, finally, with brine. The organic phase is separated and evaporated. The residue was taken up in 50 ml of DCM and re-evaporated. The residue was again taken up in 50 ml of DCM and 0.5 ml of TMSBr was added. The solution was left overnight (16 hours). DXM is pumped out and a solution of MeOH: Water 1: 1 cooled in an ice bath is added, it is briefly mixed and pumped out. The residue is triturated in ether, then dissolved in 1 N. NaOH. The clear solution was extracted with ether and the aqueous phase was acidified with 6 N HCl. Then a white precipitate was collected by filtration and dried in vacuo overnight. Yield 88 mg of the title compound.
ЖХ-МС: 679,8 (M+H)+, 95% чист.LC-MS: 679.8 (M + H) + , 95% pure.
1H ЯМР (CD3OD): δ 0,89-0,98 (м, 8H), 1,15 (м, 2H), 1,35 (м, 1Н), 1,45 (м, 1H), 1,88 (м, 1Н), 2,84 (м, 2H), 2,98 (м, 1Н), 3,01 (м, 2H), 3,24 (м, 1Н), 3,56 (с, 3H), 3,60 (м, 1Н), 3,81 (м, 1Н), 3,99 (м, 1Н), 4,39 (т, J=6,8, 1Н), 6,91 (д, J=8,0, 2H), 7,34 (д, J=8,0, 1Н), 7,45 (м, 2H), 7,58 (д, J=8,0,2H), 7,66 (с, 1H), 7,70-7,82 (м, 3H). 1 H NMR (CD 3 OD): δ 0.89-0.98 (m, 8H), 1.15 (m, 2H), 1.35 (m, 1H), 1.45 (m, 1H), 1.88 (m, 1H), 2.84 (m, 2H), 2.98 (m, 1H), 3.01 (m, 2H), 3.24 (m, 1H), 3.56 (s , 3H), 3.60 (m, 1H), 3.81 (m, 1H), 3.99 (m, 1H), 4.39 (t, J = 6.8, 1H), 6.91 ( d, J = 8.0, 2H), 7.34 (d, J = 8.0, 1H), 7.45 (m, 2H), 7.58 (d, J = 8.0.2H), 7.66 (s, 1H); 7.70-7.82 (m, 3H).
31P ЯМР (CD3OD): δ 2,56 31 P NMR (CD 3 OD): δ 2.56
Пример 4. Получение моно-(2-[(4-аминобензолсульфонил)изобутиламино]-6-{2-[(морфолин-4-карбонил)амино]-3-нафталин-1-илпропиониламино}гексилового эфира (2S,2S)фосфорной кислоты (PL-498)Example 4. Obtaining mono- (2 - [(4-aminobenzenesulfonyl) isobutylamino] -6- {2 - [(morpholine-4-carbonyl) amino] -3-naphthalene-1-ylpropionylamino} hexyl ester (2S, 2S) phosphoric acids (PL-498)
Стадия A. Получение (2S)-2-[(морфолин-4-карбонил)амино]-3-нафталин-1-илпропионовой кислотыStep A. Preparation of (2S) -2 - [(morpholine-4-carbonyl) amino] -3-naphthalene-1-yl-propionic acid
К раствору L-1-нафтилаланина (215 мг, 1 ммоль) (Peptech Corp.) в 5 мл 1 н. NaOH и 0,5 мл насыщенного Na2CO3 (pH полученного в результате раствора 10) добавляют морфолин-4-карбонилхлорид (150 мг, 1,0 ммоль), растворенный в 5 мл. После этого реакционную смесь перемешивают при комнатной температуре в течение 2 ч. Щелочной раствор один раз экстрагируют эфиром (10 мл) и водную фазу подкисляют 1 н. HCl. Данную фазу дважды экстрагируют 20 мл EtOAc и объединенные органические фазы промывают 50 мл 1 н. HCl. Органическую фазу сушат над Na2SO4, фильтруют и выпаривают до маслянистого вещества, которое отверждают, получая 125 мг (38%) желаемого вещества. Данное промежуточное соединение используют без дополнительной обработки в следующей стадии.To a solution of L-1-naphthylalanine (215 mg, 1 mmol) (Peptech Corp.) in 5 ml of 1 N. NaOH and 0.5 ml of saturated Na 2 CO 3 (pH of the resulting solution 10) add morpholine-4-carbonyl chloride (150 mg, 1.0 mmol) dissolved in 5 ml. After that, the reaction mixture was stirred at room temperature for 2 hours. The alkaline solution was extracted once with ether (10 ml) and the aqueous phase was acidified with 1N HCl. This phase is extracted twice with 20 ml of EtOAc and the combined organic phases are washed with 50 ml of 1 N. HCl. The organic phase was dried over Na 2 SO 4 , filtered and evaporated to an oily substance, which was solidified, yielding 125 mg (38%) of the desired substance. This intermediate is used without further processing in the next step.
Стадия B. Получение моно-(2-[(4-аминобензолсульфонил)изобутиламино]-6-{2-[(морфолин-4-карбонил)амино]-3-нафталин-1-илпропиониламино}гексилового эфира (2S,2S)фосфорной кислоты (PL-498)Step B. Preparation of Mono- (2 - [(4-aminobenzenesulfonyl) isobutylamino] -6- {2 - [(morpholine-4-carbonyl) amino] -3-naphthalene-1-ylpropionylamino} hexyl ester (2S, 2S) phosphoric acids (PL-498)
Данное соединение синтезируют, так же как в методе получения продукта из примера 3 (стадия D) с использованием 100 мг (2S)-2-[(морфолин-4-карбонил)амино]-3-нафталин-1-илпропионовой кислоты (стадия A данного примера). Полученный в результате осажденный остаток дополнительно очищают препаративной ВЭЖХ с обращенной фазой. Выход 41 мг конечного соединения.This compound is synthesized in the same way as in the method of obtaining the product from example 3 (stage D) using 100 mg of (2S) -2 - [(morpholine-4-carbonyl) amino] -3-naphthalene-1-yl-propionic acid (stage A of this example). The resulting precipitated residue was further purified by preparative reverse phase HPLC. Yield 41 mg of the final compound.
ЖХ-МС: 734,8 (M+H)+, 95% чист.LC-MS: 734.8 (M + H) + , 95% pure.
1H ЯМР (CD3OD): δ 0,83-0,98 (м, 8H), 1,00-1,25 (м, 4H), 1,45-1,52 (м, 1H), 1,52-1,66 (м, 1H), 1,88-1,99 (м, 1H), 2,77-2,92 (м, 2H), 2,98-3,16 (м, 3H), 3,40-3,49 (м, 1H), 3,50-3,56 (м, 6H), 3,67-3,69 (м, 1H), 3,81-3,89 (м, 1H), 3,99-4,05 (м, 1H), 4,59 (т, J=6,0, 1Н), 6,75 (д, J=8,0, 2H), 7,30-7,60 (м, 7H), 7,75 (д, J=8,0, 1Н), 7,90 (д, J=7,8, 1H), 8,23 (д, J=7,8 2H). 1 H NMR (CD 3 OD): δ 0.83-0.98 (m, 8H), 1.00-1.25 (m, 4H), 1.45-1.52 (m, 1H), 1 52-1.66 (m, 1H), 1.88-1.99 (m, 1H), 2.77-2.92 (m, 2H), 2.98-3.16 (m, 3H) 3.40-3.49 (m, 1H), 3.50-3.56 (m, 6H), 3.67-3.69 (m, 1H), 3.81-3.89 (m, 1H), 3.99-4.05 (m, 1H), 4.59 (t, J = 6.0, 1H), 6.75 (d, J = 8.0, 2H), 7.30- 7.60 (m, 7H), 7.75 (d, J = 8.0, 1H), 7.90 (d, J = 7.8, 1H), 8.23 (d, J = 7.8 2H).
31P ЯМР (CD3OD): δ 2,71 31 P NMR (CD 3 OD): δ 2.71
Пример 5. Получение моно-{6-(2-ацетиламино-3,3-дифенилпропиониламино)-2-[(4-аминобензолсульфонил)изобутиламино]гексилового}эфира (2S,2S)-фосфорной кислоты (PL-504)Example 5. Obtaining mono {6- (2-acetylamino-3,3-diphenylpropionylamino) -2 - [(4-aminobenzenesulfonyl) isobutylamino] hexyl} ether (2S, 2S) -phosphoric acid (PL-504)
Стадия A. Получение (2S)-2-ацетиламино-3,3-дифенилпропионовой кислотыStep A. Preparation of (2S) -2-acetylamino-3,3-diphenylpropionic acid
К раствору L-дифенилаланина (100 мг, 0,4 ммоль) (Peptech Corp.) в 5 мл 1 н. NaOH и 0,5 мл насыщенного Na2CO3 (pH полученного в результате раствора 10) добавляют ацетилхлорид (0,5 ммоль), растворенный в 5 мл. После этого реакционную смесь перемешивают при комнатной температуре в течение 2 ч. Щелочной раствор один раз экстрагируют эфиром (10 мл) и водную фазу подкисляют 1 н. HCl. Данную фазу дважды экстрагируют 20 мл EtOAc и объединенные органические фазы промывают 50 мл 1 н. HCl. Органическую фазу сушат над Na2SO4, фильтруют и выпаривают до маслянистого вещества, которое отверждают, получая 70 мг (60%) желаемого вещества. Данное неочищенное промежуточное соединение используют без дополнительной обработки в следующей стадии.To a solution of L-diphenylalanine (100 mg, 0.4 mmol) (Peptech Corp.) in 5 ml of 1 N. NaOH and 0.5 ml saturated Na 2 CO 3 (pH of the resulting solution 10) add acetyl chloride (0.5 mmol) dissolved in 5 ml. After that, the reaction mixture was stirred at room temperature for 2 hours. The alkaline solution was extracted once with ether (10 ml) and the aqueous phase was acidified with 1N HCl. This phase is extracted twice with 20 ml of EtOAc and the combined organic phases are washed with 50 ml of 1 N. HCl. The organic phase was dried over Na 2 SO 4 , filtered and evaporated to an oily substance, which was solidified, yielding 70 mg (60%) of the desired substance. This crude intermediate is used without further processing in the next step.
Стадия B. Получение моно-{6-(2-ацетиламино-3,3-дифенилпропиониламино)-2-[(4-аминобензолсульфонил)изобутиламино]гексилового} эфира (2S,2S)-фосфорной кислоты (PL-504)Stage B. Obtaining mono {6- (2-acetylamino-3,3-diphenylpropionylamino) -2 - [(4-aminobenzenesulfonyl) isobutylamino] hexyl} ether (2S, 2S) -phosphoric acid (PL-504)
Данное соединение синтезируют, так же как в методе получения продукта из примера 3 (стадия D) с использованием 100 мг (2S)-2-ацетиламино-3,3-дифенилпропионовой кислоты (стадия A данного примера). Конечный продукт получают с выходом 30% (30 мг).This compound is synthesized in the same way as in the method of obtaining the product from example 3 (step D) using 100 mg of (2S) -2-acetylamino-3,3-diphenylpropionic acid (step A of this example). The final product is obtained in 30% yield (30 mg).
ЖХ-МС: 689,3 (M+H)+, 95% чист.LC-MS: 689.3 (M + H) + , 95% pure.
1H ЯМР (CD3OD): δ 0,77-1,04 (м, 9H), 1,10-1,17 (м, 1H), 1,23-1,49 (м, 1H), 1,46-1,57 (м, 1H), 1,78 (с, 3H), 1,88-1,99 (м, 1H), 2,80-2,92 (м, 2H), 2,92-3,08 (м, 2H), 3,63-3,75 (м, 1H), 3,79-3,95 (м, 1Н), 4,00 (м, 1H), 4,34 (д, J=11,3, 1H), 5,19-5,28 (м, 1H), 6,77-6,85 (м, 2H), 7,10-7,20 (м, 2H), 7,27-7,33 (м, 6H), 7,32-7,41 (м, 2H), 7,49-7,62 (м, 2H). 1 H NMR (CD 3 OD): δ 0.77-1.04 (m, 9H), 1.10-1.17 (m, 1H), 1.23-1.49 (m, 1H), 1 46-1.57 (m, 1H), 1.78 (s, 3H), 1.88-1.99 (m, 1H), 2.80-2.92 (m, 2H), 2.92 -3.08 (m, 2H), 3.63-3.75 (m, 1H), 3.79-3.95 (m, 1H), 4.00 (m, 1H), 4.34 (d , J = 11.3, 1H), 5.19-5.28 (m, 1H), 6.77-6.85 (m, 2H), 7.10-7.20 (m, 2H), 7 27-7.33 (m, 6H); 7.32-7.41 (m, 2H); 7.49-7.62 (m, 2H).
31P ЯМР (CD3OD): δ 2,70 31 P NMR (CD 3 OD): δ 2.70
Пример 6. Получение метилового эфира (1S,5S)-(1-{5-[(4-амино-3-фторбензолсульфонил)изобутиламино]-6-фосфоноксигексилкарбамоил}-2,2-дифенилэтил)карбаминовой кислоты (PL-515)Example 6. Preparation of (1S, 5S) - (1- {5 - [(4-amino-3-fluorobenzenesulfonyl) isobutylamino] -6-phosphonoxyhexylcarbamoyl} -2,2-diphenylethyl) carbamic acid methyl ester (PL-515)
Первая методика: Получение указанного в заголовке соединения основано на схеме 3 данного изобретения. First Procedure: The preparation of the title compound is based on Scheme 3 of the present invention.
Стадия A. Получение метилового эфира (1-{5-[(4-амино-3-фторбензолсульфонил)изобутиламино]-6-гидроксигексилкарбамоил}-2,2-дифенилэтил)карбаминовой кислоты ( X )(PL-337)Step A. Preparation of (1- {5 - [(4-amino-3-fluorobenzenesulfonyl) isobutylamino] -6-hydroxyhexylcarbamoyl} -2,2-diphenylethyl) carbamic acid methyl ester ( X ) (PL-337)
Продукт из примера 1, стадия F (0,624 г, 1 ммоль) растворяют в 5 мл MeCN при 24°C. Одной порцией добавляют SelectFluor 0,35 г (1 ммоль) и перемешивают в течение 1 ч. Добавляют 1 мл воды и раствор впрыскивают непосредственно в препаративную ВЭЖХ с обращенной фазой. Продукт собирают и подвергают лиофилизации, получая выход белого твердого вещества 250 мг (38%).The product from example 1, step F (0.624 g, 1 mmol) was dissolved in 5 ml of MeCN at 24 ° C. 0.35 g (1 mmol) of SelectFluor was added in one portion and stirred for 1 h. 1 ml of water was added and the solution was injected directly into preparative reverse phase HPLC. The product was collected and lyophilized to give a white solid yield of 250 mg (38%).
ЖХ-МС: 643,3 (M+H)+, 99% чист.LC-MS: 643.3 (M + H) + , 99% pure.
1H ЯМР (MeOD): δ 0,71-0,85 (м 2H), 0,88 (д, J=6,3, 6H), 0,91-0,96 (м, 2H), 1,21-1,29 (м, 1H), 1,41-1,52 (м, 1H) 1,82-1,92 (м, 1H), 2,61-2,68 (м, 1H), 2,81-2,85 (м, 2H), 2,94-3,05 (м, 2H), 3,38-3,40 (т, J=5, 1H), 3,49-3,52 (м, 5H), 4,28 (д, J=10, 1Н), 4,87 (д, J=10, 1H) 6,90 (т, J=8,3, 1Н), 7,20 (м, 2H), 7,28 (м, 3H), 7,33 (м, 3H), 7,39 (м, 4H). 1 H NMR (MeOD): δ 0.71-0.85 (m 2H), 0.88 (d, J = 6.3, 6H), 0.91-0.96 (m, 2H), 1, 21-1.29 (m, 1H), 1.41-1.52 (m, 1H) 1.82-1.92 (m, 1H), 2.61-2.68 (m, 1H), 2 81-2.85 (m, 2H), 2.94-3.05 (m, 2H), 3.38-3.40 (t, J = 5, 1H), 3.49-3.52 ( m, 5H), 4.28 (d, J = 10, 1H), 4.87 (d, J = 10, 1H) 6.90 (t, J = 8.3, 1H), 7.20 (m , 2H), 7.28 (m, 3H), 7.33 (m, 3H), 7.39 (m, 4H).
Стадия B. Получение метилового эфира (1S,5S)-{1-[5-[(4-амино-3-фторбензолсульфонил)изобутиламино]-6-(диэтоксифосфорилокси)гексилкарбамоил]-2,2-дифенилэтил}карбаминовой кислотыStep B. Preparation of (1S, 5S) - {1- [5 - [(4-amino-3-fluorobenzenesulfonyl) isobutylamino] -6- (diethoxyphosphoryloxy) hexylcarbamoyl] -2,2-diphenylethyl} carbamic acid methyl ester
Продукт из стадии A фосфорилируют хлордиэтилфосфатом, следуя процедуре, описанной в примере 1, стадия G. Выходы 157 мг, 68%.The product from step A is phosphorylated with chlordiethyl phosphate following the procedure described in example 1, step G. Yields 157 mg, 68%.
ЖХ-МС: 779,3 (M+H)+, 95% чист.LC-MS: 779.3 (M + H) + , 95% pure.
1H ЯМР (CD3OD): δ 0,82 (м, 1H), 0,92 (д, J=6,2, 8H), 0,96 (м, 3H), 1,36 (д, J=3,7, 6H), 1,90 (м, 1H), 2,69 (м, 1Н), 2,89 (м, 1Н), 2,98 (м, 2H), 3,56 (с, 3H), 3,74 (м, 1H), 3,93 (м, 1H), 4,03 (м, 1H), 4,12 (кв, J=7,5 и 14,8,4H), 4,32 (д, J=11,4, 1H), 4,92 (д, J=11,4, 1H), 6,90 (т, J=8,3, 1H), 7,20 (м, 2H), 7,28 (м, 3H), 7,33 (м, 3H), 7,39 (м, 4H). 1 H NMR (CD 3 OD): δ 0.82 (m, 1H), 0.92 (d, J = 6.2, 8H), 0.96 (m, 3H), 1.36 (d, J = 3.7, 6H), 1.90 (m, 1H), 2.69 (m, 1H), 2.89 (m, 1H), 2.98 (m, 2H), 3.56 (s, 3H), 3.74 (m, 1H), 3.93 (m, 1H), 4.03 (m, 1H), 4.12 (q, J = 7.5 and 14.8.4H), 4 32 (d, J = 11.4, 1H), 4.92 (d, J = 11.4, 1H), 6.90 (t, J = 8.3, 1H), 7.20 (m, 2H), 7.28 (m, 3H), 7.33 (m, 3H), 7.39 (m, 4H).
31P ЯМР (CD3OD): δ 1,65 31 P NMR (CD 3 OD): δ 1.65
Стадия C. Получение метилового эфира (1S,5S)-(1-{5-[(4-амино-3-фторбензолсульфонил)изобутиламино]-6-фосфоноксигексилкарбамоил}-2,2-дифенилэтил)карбаминовой кислоты ( XI ) (PL-515)Step C. Preparation of (1S, 5S) - (1- {5 - [(4-amino-3-fluorobenzenesulfonyl) isobutylamino] -6-phosphonoxyhexylcarbamoyl} -2,2-diphenylethyl) carbamic acid methyl ester ( XI ) (PL- 515)
Снятие защиты осуществляют, используя процедуру, описанную в примере 1, стадия G. Выходы 101 мг.Deprotection is carried out using the procedure described in example 1, step G. Yields 101 mg.
ЖХ-МС: 723,2 (M+H)+, 95% чист.LC-MS: 723.2 (M + H) + , 95% pure.
1H ЯМР (CD3OD): 0,65-0,77 (м, 1H), 0,77-0,85 (м, 1H), 0,85-1,05 (м, 9H), 1,25-1,39 (м, 1H), 1,40-1,52 (м, 1H), 1,82-1,98 (м, 1Н), 2,58-2,72 (м, 1H), 2,82-2,92 (м, 1H), 2,92-3,05 (м, 2H), 3,54 (с, 3H), 3,64-3,75 (м, 1H), 3,80-3,92 (м, 1H), 3,91-4,04 (м, 1Н), 4,29 (д, J=11,4, 1H), 7,19 (т, J=6,6, 1H), 7,13-7,21 (м, 2H), 7,22-7,33 (м, 6H), 7,34-7,38 (м, 2H), 7,39-7,48 (м, 2H) 1 H NMR (CD 3 OD): 0.65-0.77 (m, 1H), 0.77-0.85 (m, 1H), 0.85-1.05 (m, 9H), 1, 25-1.39 (m, 1H), 1.40-1.52 (m, 1H), 1.82-1.98 (m, 1H), 2.58-2.72 (m, 1H), 2.82-2.92 (m, 1H), 2.92-3.05 (m, 2H), 3.54 (s, 3H), 3.64-3.75 (m, 1H), 3, 80-3.92 (m, 1H), 3.91-4.04 (m, 1H), 4.29 (d, J = 11.4, 1H), 7.19 (t, J = 6.6 , 1H), 7.13-7.21 (m, 2H), 7.22-7.33 (m, 6H), 7.34-7.38 (m, 2H), 7.39-7.48 (m, 2H)
31P ЯМР (CD3OD): δ 2,74 31 P NMR (CD 3 OD): δ 2.74
Вторая методика: Получение указанного в заголовке соединения основано на схеме 4 данного изобретения. Second method: The preparation of the title compound is based on Scheme 4 of the present invention.
Стадия A. Получение метилового эфира (1S,5S)-(1-{5-[(4-аминобензолсульфонил)изобутиламино]-6-фосфоноксигексилкарбамоил}-2,2-дифенилэтил)карбаминовой кислоты (PL-461).Step A. Preparation of (1S, 5S) - (1- {5 - [(4-aminobenzenesulfonyl) isobutylamino] -6-phosphonoxyhexylcarbamoyl} -2,2-diphenylethyl) carbamic acid methyl ester (PL-461).
(2S)-2-метоксикарбониламино-3,3-дифенилпропионовую кислоту ((пример 1, стадия E) 0,9 г, 3 ммоль) активируют в ДМФА (5 мл) посредством EDAC (1,7 г, 9 ммоль) и HOBt (1,2 г, 9 ммоль). К данному раствору добавляют 1,17 г (диэтил)(6-амино-2-[(4-аминобензолсульфонил)изобутиламино]гексилового) эфира (2S)-фосфорной кислоты ( IX ) (пример 3, стадия B) и смесь перемешивают в течение 3 ч. Затем добавляют 20 г Amberlite XAD-2 и гранулы оставляют намокать в течение 10 мин. Смолу переносят на стеклянный фильтр и тщательно промывают дистиллированной водой (400 мл) и 200 мл 1М NaHCO3. Затем гранулы промывают 4 х 50 мл порциями MeOH, затем EtOAc 200 мл. Органическую фазу выпаривают. Остаток адсорбируют на силикагеле и пропускают через короткую силикагелевую колонку (EtOAc), получая 2,4 г (83%) белого твердого вещества после выпаривания.(2S) -2-methoxycarbonylamino-3,3-diphenylpropionic acid ((example 1, step E) 0.9 g, 3 mmol) is activated in DMF (5 ml) by EDAC (1.7 g, 9 mmol) and HOBt (1.2 g, 9 mmol). To this solution was added 1.17 g of (diethyl) (6-amino-2 - [(4-aminobenzenesulfonyl) isobutylamino] hexyl) ether (2S) -phosphoric acid ( IX ) (Example 3, step B) and the mixture was stirred for 3 hours. Then add 20 g of Amberlite XAD-2 and the granules are left to wet for 10 minutes. The resin was transferred to a glass filter and washed thoroughly with distilled water (400 ml) and 200 ml of 1M NaHCO 3 . Then the granules are washed with 4 x 50 ml portions of MeOH, then EtOAc 200 ml. The organic phase is evaporated. The residue was adsorbed onto silica gel and passed through a short silica gel column (EtOAc) to give 2.4 g (83%) of a white solid after evaporation.
1H ЯМР идентичен примеру 1, стадия H. 1 H NMR identical to Example 1, step H.
Стадия B. Получение метилового эфира (1S,5S)-{1-[5-[(4-амино-3-фторбензолсульфонил)изобутиламино]-6-(диэтоксифосфорилокси)гексилкарбамоил]-2,2-дифенилэтил}карбаминовой кислоты ( XII )Step B. Preparation of (1S, 5S) - {1- [5 - [(4-amino-3-fluorobenzenesulfonyl) isobutylamino] -6- (diethoxyphosphoryloxy) hexylcarbamoyl] -2,2-diphenylethyl} carbamic acid methyl ester ( XII )
Продукт из приведенной выше стадии A, метиловый эфир (1S,5S)-(1-{5-[(4-аминобензолсульфонил)изобутиламино]-6-фосфоноксигексилкарбамоил}-2,2-дифенилэтил)карбаминовой кислоты (0,555 г, 0,73 ммоль) растворяют в 5 мл MeCN. Добавляют Selectfluor (0,26 г, 0,7 ммоль) и смесь перемешивают в течение 30 мин. Смесь очищают препаративной ВЭЖХ с обращенной фазой и подвергают лиофилизации, получая 278 мг (выход 48%) белого твердого вещества.Product from Step A above, (1S, 5S) - (1- {5 - [(4-aminobenzenesulfonyl) isobutylamino] -6-phosphonoxyhexylcarbamoyl} -2,2-diphenylethyl) carbamic acid methyl ester (0.555 g, 0.73 mmol) is dissolved in 5 ml of MeCN. Selectfluor (0.26 g, 0.7 mmol) was added and the mixture was stirred for 30 minutes. The mixture was purified by reverse phase preparative HPLC and lyophilized to obtain 278 mg (48% yield) of a white solid.
1H ЯМР идентичен предшествующему описанию, смотри первую методику выше. 1 H NMR identical to the previous description, see first procedure above.
Стадия C. Получение метилового эфира (1S,5S)-(1-{5-[(4-амино-3-фторбензолсульфонил)изобутиламино]-6-фосфоноксигексилкарбамоил}-2,2-дифенилэтил)карбаминовой кислоты ( XIII , в данном конкретном случае представляет собой соединение XI )(PL-515)Step C. Preparation of (1S, 5S) - (1- {5 - [(4-amino-3-fluorobenzenesulfonyl) isobutylamino] -6-phosphonoxyhexylcarbamoyl} -2,2-diphenylethyl) carbamic acid methyl ester ( XIII , in this particular case is a compound XI ) (PL-515)
Процедура, дающая данное производное, является такой, как описано в стадии снятия защиты для описанной выше методики. Выходы 139 мг 70% после ВЭЖХ с обращенной фазой.The procedure giving this derivative is as described in the deprotection step for the above procedure. Yields 139 mg 70% after reverse phase HPLC.
1H ЯМР идентичен предшествующему описанию, смотри первую методику выше. 1 H NMR identical to the previous description, see first procedure above.
Пример 7. Получение 2-[(4-аминобензолсульфонил)изобутиламино]-6-(2-метоксикарбониламино-3,3-дифенилпропиониламино)гексилового эфира (2S,2S)уксусной кислоты (PL-521)Example 7. Obtaining 2 - [(4-aminobenzenesulfonyl) isobutylamino] -6- (2-methoxycarbonylamino-3,3-diphenylpropionylamino) hexyl ester (2S, 2S) acetic acid (PL-521)
Получение указанного в заголовке производного основано на схеме 5 данного изобретения.The preparation of the title derivative is based on Scheme 5 of the present invention.
Стадия A. Получение 6-трет-бутоксикарбониламино-2-[(4-трет-бутоксикарбониламинобензолсульфонил)изобутиламино]гексилового эфира (2S)уксусной кислоты ( XIV , R1A=CH3)Step A. Preparation of 6-tert-butoxycarbonylamino-2 - [(4-tert-butoxycarbonylaminobenzenesulfonyl) isobutylamino] hexyl ester (2S) acetic acid ( XIV , R 1A = CH 3 )
К перемешиваемому раствору трет-бутилового эфира (1S)-{4-[(5-трет-бутоксикарбониламино-1-гидроксиметилпентил)изобутилсульфамоил]фенил}карбаминовой кислоты (промежуточный продукт ( VII ) из примера 1, стадия D, 97 мг, 0,18 ммоль) в безводном CH2Cl2 (3 мл) добавляют N,N-диметиламинопиридин (22 мг, 0,18 ммоль) и уксусный ангидрид (0,014 мл, 0,18 ммоль). Смесь перемешивают при комнатной температуре в течение 1 часа. Растворитель выпаривают. Добавляют этилацетат (50 мл) и органический слой промывают водой (30 мл), затем сушат над Na2SO4 и концентрируют. Остаток очищают флэш-хроматографией, элюируя этилацетатом. Полученный выход является количественным (100 мг).To a stirred solution of (1S) tert-butyl ether - {4 - [(5-tert-butoxycarbonylamino-1-hydroxymethylpentyl) isobutylsulfamoyl] phenyl} carbamic acid (intermediate ( VII ) from Example 1, step D, 97 mg, 0, 18 mmol) in anhydrous CH 2 Cl 2 (3 ml) add N, N- dimethylaminopyridine (22 mg, 0.18 mmol) and acetic anhydride (0.014 ml, 0.18 mmol). The mixture was stirred at room temperature for 1 hour. The solvent was evaporated. Ethyl acetate (50 ml) was added and the organic layer was washed with water (30 ml), then dried over Na 2 SO 4 and concentrated. The residue was purified by flash chromatography, eluting with ethyl acetate. The resulting yield is quantitative (100 mg).
ЖХ-МС: 586,2 (M+H)+, 95% чист.LC-MS: 586.2 (M + H) + , 95% pure.
Стадия B. Получение 6-амино-2-[(4-аминобензолсульфонил)изобутиламино]гексилового эфира (2S)-уксусной кислоты ( XV , R1A=CH3)Step B. Preparation of 6-amino-2 - [(4-aminobenzenesulfonyl) isobutylamino] hexyl ester of (2S) -acetic acid ( XV , R 1A = CH 3 )
Данное производное получают из 6-трет-бутоксикарбониламино-2-[(4-трет-бутоксикарбониламинобензолсульфонил)изобутиламино]гексилового эфира (2S)-уксусной кислоты, как описано в примере 15, стадия B. Твердое вещество желтого цвета (66 мг) используют в следующей реакции без очистки.This derivative is prepared from 6-tert-butoxycarbonylamino-2 - [(4-tert-butoxycarbonylaminobenzenesulfonyl) isobutylamino] hexyl ether (2S) -acetic acid as described in Example 15, step B. A yellow solid (66 mg) was used in next reaction without purification.
ЖХ-МС: 386,2 (M+H)+, 95% чист.LC-MS: 386.2 (M + H) + , 95% pure.
Стадия C. Получение 2-[(4-аминобензолсульфонил)изобутиламино]-6-(2-метоксикарбониламино-3,3-дифенилпропиониламино)гексилового эфира (2S,2S)-уксусной кислоты ( XVI, R1A=CH3) (PL-521)Step C. Preparation of 2 - [(4-aminobenzenesulfonyl) isobutylamino] -6- (2-methoxycarbonylamino-3,3-diphenylpropionylamino) hexyl ester (2S, 2S) -acetic acid ( XVI, R 1A = CH 3 ) (PL- 521)
Данное производное получают из 6-амино-2-[(4-аминобензолсульфонил)изобутиламино]гексилового эфира (2S)-уксусной кислоты (продукт из стадии B), как описано в примере 15, стадия B. Конечный продукт очищают флэш-хроматографией смесью элюентов гексан/этилацетат (2/8). Получают желтое твердое вещество с выходом 70% (70 мг).This derivative is prepared from 6-amino-2 - [(4-aminobenzenesulfonyl) isobutylamino] hexyl ester of (2S) -acetic acid (product from step B) as described in Example 15, step B. The final product is purified by flash chromatography using a mixture of eluents hexane / ethyl acetate (2/8). A yellow solid is obtained with a yield of 70% (70 mg).
ЖХ-МС: 667,3 (M+H)+, 95% чист.LC-MS: 667.3 (M + H) + , 95% pure.
1H ЯМР (ацетон-d6): δ 0,85-0,97 (м, 12H), 1,21-1,41 (м, 2H), 1,88-2,00 (с, 3H), 2,59-2,69 (м, 1H), 2,83-2,90 (м, 1H), 2,90-3,01 (м, 1H), 3,01-3,10 (ушир.с, 1H), 3,45-3,60 (с, 3H), 3,70-3,80 (м, 1H), 3,93-4,00 (м, 1Н), 4,00-4,11 (м, 1H), 4,38-4,45 (д, J=11,0, 1H), 4,89-4,98 (т, J=10,0, 1H), 5,43-5,58 (ушир.с, 1H), 6,28-6,48 (д, J=8,9, 1H), 6,72-6,83 (д, J=8,0, 2H), 6,85-6,93 (ушир.с, 1Н), 7,12-7,22 (т, J=7,4, 1H), 7,21-7,31 (д, J=7,0, 4H), 7,31-7,45 (м, 5H), 7,48-7,57 (д, J=8,0, 2H). 1 H NMR (acetone-d 6 ): δ 0.85-0.97 (m, 12H), 1.21-1.41 (m, 2H), 1.88-2.00 (s, 3H), 2.59-2.69 (m, 1H), 2.83-2.90 (m, 1H), 2.90-3.01 (m, 1H), 3.01-3.10 (br s , 1H), 3.45-3.60 (s, 3H), 3.70-3.80 (m, 1H), 3.93-4.00 (m, 1H), 4.00-4.11 (m, 1H), 4.38-4.45 (d, J = 11.0, 1H), 4.89-4.98 (t, J = 10.0, 1H), 5.43-5, 58 (br s, 1H), 6.28-6.48 (d, J = 8.9, 1H), 6.72-6.83 (d, J = 8.0, 2H), 6.85 -6.93 (br s, 1H), 7.12-7.22 (t, J = 7.4, 1H), 7.21-7.31 (d, J = 7.0, 4H), 7.31-7.45 (m, 5H); 7.48-7.57 (d, J = 8.0, 2H).
Пример 8. Получение 2-[(4-аминобензолсульфонил)изобутиламино]-6-(2-метоксикарбониламино-3,3-дифенилпропиониламино)гексилового эфира (2S,2S)-никотиновой кислоты (PL-520)Example 8. Obtaining 2 - [(4-aminobenzenesulfonyl) isobutylamino] -6- (2-methoxycarbonylamino-3,3-diphenylpropionylamino) hexyl ester (2S, 2S) -nicotinic acid (PL-520)
Стадия A. Получение 6-трет-бутоксикарбониламино-2-[(4-трет-бутоксикарбониламинобензолсульфонил)изобутиламино]гексилового эфира (2S)-никотиновой кислоты ( XIV , R1A=3-пиридил)Step A. Preparation of 6-tert-butoxycarbonylamino-2 - [(4-tert-butoxycarbonylaminobenzenesulfonyl) isobutylamino] hexyl ester (2S) -nicotinic acid ( XIV , R 1A = 3-pyridyl)
Трет-бутиловый эфир (1S)-{4-[(5-трет-бутоксикарбониламино-1-гидроксиметилпентил)изобутилсульфамоил]фенил}карбаминовой кислоты (промежуточный продукт ( VII ) из примера 1, стадия D, 130 мг, 0,24 ммоль) растворяют в безводном ДМФА (1 мл) и обрабатывают 0,066 мл (0,48 ммоль) триэтиламина, поле чего EDC (120 мг, 0,65 ммоль), HOBt (88 мг, 0,65 ммоль) и никотиновой кислотой (27 мг, 0,22 ммоль). Смесь перемешивают в течение ночи при комнатной температуре. Продукт экстрагируют смесью этилацетата (40 мл) и воды (40 мл). Органическую фазу отделяют и сушат над Na2SO4, затем выпаривают, получая 200 мг неочищенного продукта. Данное соединение очищают флэш-хроматографией с этилацетатом в качестве элюента. Получают прозрачное маслянистое вещество с выходом 100% (150 мг).(1S) tert-butyl ether - {4 - [(5-tert-butoxycarbonylamino-1-hydroxymethylpentyl) isobutylsulfamoyl] phenyl} carbamic acid (intermediate ( VII ) from Example 1, step D, 130 mg, 0.24 mmol) dissolved in anhydrous DMF (1 ml) and treated with 0.066 ml (0.48 mmol) of triethylamine, the field of which is EDC (120 mg, 0.65 mmol), HOBt (88 mg, 0.65 mmol) and nicotinic acid (27 mg, 0.22 mmol). The mixture was stirred overnight at room temperature. The product was extracted with a mixture of ethyl acetate (40 ml) and water (40 ml). The organic phase was separated and dried over Na 2 SO 4 , then evaporated to give 200 mg of a crude product. This compound was purified by flash chromatography with ethyl acetate as eluent. A clear oily substance is obtained in a yield of 100% (150 mg).
ЖХ-МС: 649,3 (M+H)+, 95% чист.LC-MS: 649.3 (M + H) + , 95% pure.
1H ЯМР (ацетон-d6): δ 0,90-1,14 (д, J=5,9, 6H), 1,31-1,42 (м, 2H), 1,48 (с, 9H), 1,51-1,55 (м, 2H), 1,59 (с, 9H), 1,62-1,69 (м, 1H), 1,72-1,83 (м, 1Н), 3,00-3,11 (м, 2H), 3,11-30 3,17 (м, 1Н), 3,19-3,27 (м, 1H), 4,15-4,24 (м, 1H), 4,35-4,44 (т, J=9,1, 1H), 4,50-4,58 (дд, J=4,4 и 11,5, 1H), 5,89-5,99 (ушир.с, 1Н), 7,53-7,60 (м, 1H), 7,70-7,77 (д, J=8,2, 2H), 7,80-7,87 (д, J=8,2, 2H), 8,24-8,31 (д, J=7,3, 1H), 8,75-8,82 (м, 1H), 8,82-8,88 (м, 1H), 9,12-9,18 (ушир.с, 1H). 1 H NMR (acetone-d 6 ): δ 0.90-1.14 (d, J = 5.9, 6H), 1.31-1.42 (m, 2H), 1.48 (s, 9H ), 1.51-1.55 (m, 2H), 1.59 (s, 9H), 1.62-1.69 (m, 1H), 1.72-1.83 (m, 1H), 3.00-3.11 (m, 2H), 3.11-30 3.17 (m, 1H), 3.19-3.27 (m, 1H), 4.15-4.24 (m, 1H), 4.35-4.44 (t, J = 9.1, 1H), 4.50-4.58 (dd, J = 4.4 and 11.5, 1H), 5.89-5 99 (br s, 1H), 7.53-7.60 (m, 1H), 7.70-7.77 (d, J = 8.2, 2H), 7.80-7.87 ( d, J = 8.2, 2H), 8.24-8.31 (d, J = 7.3, 1H), 8.75-8.82 (m, 1H), 8.82-8.88 (m, 1H), 9.12-9.18 (br s, 1H).
Стадия B. Получение 6-амино-2-[(4-аминобензолсульфонил)изобутиламино]гексилового эфира (2S)-никотиновой кислоты ( XV , R1A=3-пиридил)Step B. Preparation of 6-amino-2 - [(4-aminobenzenesulfonyl) isobutylamino] hexyl ester of (2S) -nicotinic acid ( XV , R 1A = 3-pyridyl)
Продукт из стадии A, 6-трет-бутоксикарбониламино-2-[(4-трет-бутоксикарбониламинобензолсульфонил)изобутиламино]гексиловый эфир (2S)-никотиновой кислоты (150 мг, 0,23 ммоль) растворяют в CH2Cl2 (5 мл) и добавляют трифторуксусную кислоту (1 мл). Смесь перемешивают в течение 2 часов при комнатной температуре. Растворитель выпаривают и остаток экстрагируют смесью этилацетата (40 мл) и NaOH 1М (40 мл) (pH 10). Органическую часть отделяют, сушат над Na2SO4 и затем выпаривают. Остаток (100 мг) используют для следующей реакции без дополнительной очистки. Выход количественный.The product from step A, 6-tert-butoxycarbonylamino-2 - [(4-tert-butoxycarbonylaminobenzenesulfonyl) isobutylamino] hexyl ether (2S) -nicotinic acid (150 mg, 0.23 mmol) was dissolved in CH 2 Cl 2 (5 ml) and trifluoroacetic acid (1 ml) was added. The mixture was stirred for 2 hours at room temperature. The solvent was evaporated and the residue was extracted with a mixture of ethyl acetate (40 ml) and NaOH 1M (40 ml) (pH 10). The organic portion was separated, dried over Na 2 SO 4 and then evaporated. The residue (100 mg) was used for the next reaction without further purification. The output is quantitative.
ЖХ-МС: 449,3 (M+H)+, 95% чист.LC-MS: 449.3 (M + H) + , 95% pure.
Стадия C. Получение 2-[(4-аминобензолсульфонил)изобутиламино]-6-(2-метоксикарбониламино-3,3-дифенилпропиониламино)гексилового эфира (2S,2S)-никотиновой кислоты (PL-520)Step C. Preparation of 2 - [(4-aminobenzenesulfonyl) isobutylamino] -6- (2-methoxycarbonylamino-3,3-diphenylpropionylamino) hexyl ester (2S, 2S) -nicotinic acid (PL-520)
Продукт из стадии B, 6-амино-2-[(4-аминобензолсульфонил)изобутиламино]гексиловый эфир (2S)-никотиновой кислоты (100 мг, 0,22 ммоль) растворяют в безводном ДМФА (2 мл) и обрабатывают 0,062 мл (0,45 ммоль) триэтиламина, а затем EDC (100 мг, 0,56 ммоль), HOBt (75 мг, 0,56 ммоль) и (2S)-2-метоксикарбониламино-3,3-дифенилпропионовой кислотой (56 мг, 0,19 ммоль). Данную смесь перемешивают в течение ночи при комнатной температуре. Продукт экстрагируют смесью этилацетата (40 мл) и воды (40 мл). Органический слой отделяют, сушат над Na2SO4 и затем выпаривают, получая 160 мг неочищенного маслянистого вещества. Остаток очищают флэш-хроматографией со смесью элюентов гексан/этилацетат (2/8). Указанное в заголовке соединение получают в виде прозрачного маслянистого вещества с выходом 20% (25 мг).The product from stage B, 6-amino-2 - [(4-aminobenzenesulfonyl) isobutylamino] hexyl ether (2S) -nicotinic acid (100 mg, 0.22 mmol) was dissolved in anhydrous DMF (2 ml) and treated with 0.062 ml (0 , 45 mmol) of triethylamine, followed by EDC (100 mg, 0.56 mmol), HOBt (75 mg, 0.56 mmol) and (2S) -2-methoxycarbonylamino-3,3-diphenylpropionic acid (56 mg, 0, 19 mmol). This mixture was stirred overnight at room temperature. The product was extracted with a mixture of ethyl acetate (40 ml) and water (40 ml). The organic layer was separated, dried over Na 2 SO 4 and then evaporated to give 160 mg of a crude oily substance. The residue was purified by flash chromatography with hexanes / ethyl acetate (2/8). The title compound was obtained as a clear oily substance in 20% yield (25 mg).
ЖХ-МС: 730,2 (M+H)+, 95% чист.LC-MS: 730.2 (M + H) + , 95% pure.
1H ЯМР (ацетон-d6): δ 0,80-0,97 (м, 9H), 0,97-1,13 (м, 2H), 1,26-1,40 (м, 1Н), 1,40-1,57 (м, 1H), 2,61-2,73 (м, 1H), 2,86-2,98 (м, 2H), 3,00-3,17 (м, 2H), 3,45-3,59 (с, 3H), 3,91-4,00 (м, 1H), 4,24-4,34 (м, 1H), 4,34-4,47 (м, 2H), 4,90-4,99 (т, J=9,7, 1Н), 6,35-6,44 (м, 1H), 6,68-6,79 (д, J=7,9, 1H), 6,91-7,00 (ушир.с, 1H), 7,13-7,22 (м, 2H), 7,22-7,31 (м, 3H), 7,35-7,48 (м, 4H), 7,49-7,64 (м, 2H), 7,75-7,84 (м, 1H), 8,25-8,36 (м, 1H), 8,76-8,88 (ушир.с, 1H), 9,12-9,26 (ушир.с, 1H). 1 H NMR (acetone-d 6 ): δ 0.80-0.97 (m, 9H), 0.97-1.13 (m, 2H), 1.26-1.40 (m, 1H), 1.40-1.57 (m, 1H), 2.61-2.73 (m, 1H), 2.86-2.98 (m, 2H), 3.00-3.17 (m, 2H) ), 3.45-3.59 (s, 3H), 3.91-4.00 (m, 1H), 4.24-4.34 (m, 1H), 4.34-4.47 (m , 2H), 4.90-4.99 (t, J = 9.7, 1H), 6.35-6.44 (m, 1H), 6.68-6.79 (d, J = 7, 9, 1H), 6.91-7.00 (br s, 1H), 7.13-7.22 (m, 2H), 7.22-7.31 (m, 3H), 7.35- 7.48 (m, 4H), 7.49-7.64 (m, 2H), 7.75-7.84 (m, 1H), 8.25-8.36 (m, 1H), 8, 76-8.88 (br s, 1H); 9.12 - 9.26 (br s, 1H).
Пример 9. Получение 2-[(4-аминобензолсульфонил)изобутиламино]-6-(2-метоксикарбониламино-3,3-дифенилпропиониламино)гексилового эфира (2S,2S)-диметиламиноуксусной кислоты (PL-534)Example 9. Obtaining 2 - [(4-aminobenzenesulfonyl) isobutylamino] -6- (2-methoxycarbonylamino-3,3-diphenylpropionylamino) hexyl ester (2S, 2S) -dimethylaminoacetic acid (PL-534)
Стадия A. Получение 6-трет-бутоксикарбониламино-2-[(4-трет-бутоксикарбониламинобензолсульфонил)изобутиламино]гексилового эфира (2S)-диметиламиноуксусной кислоты ( XIV , R1A=(CH3)2NCH2-)Step A. Preparation of 6-tert-butoxycarbonylamino-2 - [(4-tert-butoxycarbonylaminobenzenesulfonyl) isobutylamino] hexyl ester (2S) -dimethylaminoacetic acid ( XIV , R 1A = (CH 3 ) 2 NCH 2 -)
Данное указанное в заголовке соединение получают из трет-бутилового эфира (1S)-{4-[(5-трет-бутоксикарбониламино-1-гидроксиметилпентил)изобутилсульфамоил]фенил}карбаминовой кислоты (промежуточный продукт ( VII ) из примера 1, стадия D), как описано в примере 15, стадия A, с использованием N,N-диметилглицина. Получают прозрачное масло с выходом 100% (150 мг).The title compound was prepared from (1S) - {4 - [(5-tert-butoxycarbonylamino-1-hydroxymethylpentyl) isobutylsulfamoyl] phenyl} carbamic acid tert-butyl ester (intermediate ( VII ) from Example 1, step D), as described in example 15, step A, using N, N- dimethylglycine. Get a clear oil with a yield of 100% (150 mg).
ЖХ-МС: 629,3 (M+H)+, 95% чист.LC-MS: 629.3 (M + H) + , 95% pure.
1H ЯМР (ацетон-d6): δ 0,81-0,95 (д, J=6,1, 6H), 1,18-1,30 (м, 2H), 1,32-1,43 (с, 9H), 1,43-1,52 (с, 8H), 1,52-1,62 (м, 1H), 1,93-2,00 (м, 1H), 2,19-2,29 (с, 4H), 2,69-2,80 (м,,4H), 2,90-3,05 (м, 6H), 3,60-3,65 (м, 1H), 3,85-3,97 (м, 1Н), 3,98-4,08 (м, 1H), 4,08-4,14 (м, 1H), 5,78-5,88 (м, 1Н), 7,68-7,80 (м, 3H), 8,80-8,88 (ушир. с, 1Н). 1 H NMR (acetone-d 6 ): δ 0.81-0.95 (d, J = 6.1, 6H), 1.18-1.30 (m, 2H), 1.32-1.43 (s, 9H), 1.43-1.52 (s, 8H), 1.52-1.62 (m, 1H), 1.93-2.00 (m, 1H), 2.19-2 29 (s, 4H), 2.69-2.80 (m, 4H), 2.90-3.05 (m, 6H), 3.60-3.65 (m, 1H), 3, 85-3.97 (m, 1H), 3.98-4.08 (m, 1H), 4.08-4.14 (m, 1H), 5.78-5.88 (m, 1H), 7.68-7.80 (m, 3H); 8.80-8.88 (br s, 1H).
Стадия B. Получение 6-амино-2-[(4-аминобензолсульфонил)изобутиламино]гексилового эфира (2S)-диметиламиноуксусной кислоты ( XV , R1A=(CH3)2NCH2-)Step B. Preparation of 6-amino-2 - [(4-aminobenzenesulfonyl) isobutylamino] hexyl ester of (2S) -dimethylaminoacetic acid ( XV , R 1A = (CH 3 ) 2 NCH 2 -)
Указанное в заголовке производное получают из 6-трет-бутоксикарбониламино-2-[(4-трет-бутоксикарбониламинобензолсульфонил)изобутиламино]гексилового эфира (2S)-диметиламиноуксусной кислоты, как описано в примере 15, стадия B. Конечный продукт (100 мг) используют без дополнительной обработки в следующей стадии.The title derivative was prepared from 6-tert-butoxycarbonylamino-2 - [(4-tert-butoxycarbonylaminobenzenesulfonyl) isobutylamino] hexyl ester of (2S) -dimethylaminoacetic acid as described in Example 15, step B. The final product (100 mg) was used without additional processing in the next stage.
ЖХ-МС: 429,3 (M+H)+, 90% чист.LC-MS: 429.3 (M + H) + , 90% pure.
Стадия C. Получение 2-[(4-аминобензолсульфонил)изобутиламино]-6-(2-метоксикарбониламино-3,3-дифенилпропиониламино)гексилового эфира (2S,2S)-диметиламиноуксусной кислоты (PL-534)Step C. Preparation of 2 - [(4-aminobenzenesulfonyl) isobutylamino] -6- (2-methoxycarbonylamino-3,3-diphenylpropionylamino) hexyl ester (2S, 2S) -dimethylaminoacetic acid (PL-534)
Данное указанное в заголовке соединение получают из 6-амино-2-[(4-аминобензолсульфонил)изобутиламино]гексилового эфира (2S)-диметиламиноуксусной кислоты, как описано в примере 15, стадия C. Неочищенный продукт очищают препаративной ЖХ. Конечное соединение получают с выходом 10% (10 мг).The title compound was prepared from 6-amino-2 - [(4-aminobenzenesulfonyl) isobutylamino] hexyl ester of (2S) dimethylaminoacetic acid as described in Example 15, Step C. The crude product was purified by preparative LC. The final compound was obtained in 10% yield (10 mg).
ЖХ-МС: 710,3 (M+H)+, 92% чист.LC-MS: 710.3 (M + H) + , 92% pure.
1H ЯМР (ацетон-d6): δ 0,81-0,98 (м, 12H), 1,14-1,30 (м, 2H), 1,31-1,45 (м, 1Н), 2,58-2,77 (м, 2H), 2,79-2,90 (м, 2H), 3,42-3,56 (с, 3H), 3,75-3,85 (м, 1H), 3,99-4,17 (м, 3H), 4,23-4,35 (м, 1H), 4,36-4,45 (м, 1H), 4,86-4,96 (м, 1H), 6,33-6,42 (м, 1Н), 6,74-6,83 (м, 1H), 6,85-6,90 (м, 1H), 7,12-7,22 (м, 3H), 7,23-7,31 (м, 4H), 7,31-7,44 (м, 5H), 7,47-7,55 (м, 1Н), 7,73-7,80 (м, 1Н). 1 H NMR (acetone-d 6 ): δ 0.81-0.98 (m, 12H), 1.14-1.30 (m, 2H), 1.31-1.45 (m, 1H), 2.58-2.77 (m, 2H), 2.79-2.90 (m, 2H), 3.42-3.56 (s, 3H), 3.75-3.85 (m, 1H ), 3.99-4.17 (m, 3H), 4.23-4.35 (m, 1H), 4.36-4.45 (m, 1H), 4.86-4.96 (m , 1H), 6.33-6.42 (m, 1H), 6.74-6.83 (m, 1H), 6.85-6.90 (m, 1H), 7.12-7.22 (m, 3H), 7.23-7.31 (m, 4H), 7.31-7.44 (m, 5H), 7.47-7.55 (m, 1H), 7.73-7 80 (m, 1H).
Пример 10. Получение 2-[(4-аминобензолсульфонил)изобутиламино]-6-(2-метоксикарбониламино-3,3-дифенилпропиониламино)гексилового эфира (2S,2S)-2-амино-3-метилмасляной кислоты (PL-530)Example 10. Obtaining 2 - [(4-aminobenzenesulfonyl) isobutylamino] -6- (2-methoxycarbonylamino-3,3-diphenylpropionylamino) hexyl ester (2S, 2S) -2-amino-3-methylbutyric acid (PL-530)
Стадия A. Получение 6-трет-бутоксикарбониламино-2-[(4-трет-бутоксикарбониламинобензолсульфонил)изобутиламино]гексилового эфира (2S)-2-бензилоксикарбониламино-3-метилмасляной кислоты ( XIV , R1A=(CH3)2CHCH(NH2)-)Step A. Preparation of 6-tert-butoxycarbonylamino-2 - [(4-tert-butoxycarbonylaminobenzenesulfonyl) isobutylamino] hexyl ester (2S) -2-benzyloxycarbonylamino-3-methylbutyric acid ( XIV , R 1A = (CH 3 ) 2 CHCH (NH 2 ) -)
Данное указанное в заголовке соединение получают из трет-бутилового эфира (1S)-{4-[(5-трет-бутоксикарбониламино-1-гидроксиметилпентил)изобутилсульфамоил]фенил}карбаминовой кислоты (промежуточный продукт ( VII ) из примера 1, стадия D), как описано в примере 15, стадия A с использованием (2S)-2-бензилоксикарбониламино-3-метилмасляной кислоты. Неочищенный продукт очищают флэш-хроматографией, элюируя смесью гексан/этилацетат (1/1). Полученный выход равен 100% (150 мг).The title compound was prepared from (1S) - {4 - [(5-tert-butoxycarbonylamino-1-hydroxymethylpentyl) isobutylsulfamoyl] phenyl} carbamic acid tert-butyl ester (intermediate ( VII ) from Example 1, step D), as described in example 15, step A using (2S) -2-benzyloxycarbonylamino-3-methylbutyric acid. The crude product was purified by flash chromatography, eluting with hexane / ethyl acetate (1/1). The resulting yield is 100% (150 mg).
ЖХ-МС: 777,3 (M+H)+, 95% чист.LC-MS: 777.3 (M + H) + , 95% pure.
1H ЯМР (ацетон-d6): δ 0,80-1,00 (м,14), 1,13-1,28 (с, 2H), 1,30-1,44 (с, 11Н), 1,45-1,56 (с, 10), 1,58-1,67 (м, 1H), 2,87-3,04 (м, 4H), 3,84-3,97 (м, 1Н), 3,97-4,12 (м, 2H), 4,12-4,21 (м, 1Н), 4,99-5,14 (м, 2H), 5,78-5,89 (м, 1H), 6,38-6,52 (м, 1Н), 7,24-7,34 (м, 1H), 7,34-7,41 (м, 2H), 7,65-7,83 (м, 4H), 8,77-8,86 (м, 1Н). 1 H NMR (acetone-d 6 ): δ 0.80-1.00 (m, 14), 1.13-1.28 (s, 2H), 1.30-1.44 (s, 11H), 1.45-1.56 (s, 10), 1.58-1.67 (m, 1H), 2.87-3.04 (m, 4H), 3.84-3.97 (m, 1H) ), 3.97-4.12 (m, 2H), 4.12-4.21 (m, 1H), 4.99-5.14 (m, 2H), 5.78-5.89 (m , 1H), 6.38-6.52 (m, 1H), 7.24-7.34 (m, 1H), 7.34-7.41 (m, 2H), 7.65-7.83 (m, 4H), 8.77-8.86 (m, 1H).
Стадия B. Получение 6-амино-2-[(4-аминобензолсульфонил)изобутиламино]гексилового эфира (2S)-бензилоксикарбониламино-3-метилмасляной кислоты ( XV , R1A=(CH3)2CHCH(NH2)-)Step B. Preparation of 6-amino-2 - [(4-aminobenzenesulfonyl) isobutylamino] hexyl ester of (2S) benzyloxycarbonylamino-3-methylbutyric acid ( XV , R 1A = (CH 3 ) 2 CHCH (NH 2 ) -)
Данное производное получают из 6-трет-бутоксикарбониламино-2-[(4-трет-бутоксикарбониламинобензолсульфонил)изобутиламино]гексилового эфира (2S)-2-бензилоксикарбониламино-3-метилмасляной кислоты (продукт из стадии A), как описано в примере 15, стадия B. Конечное соединение получают с количественным выходом (110 мг) и используют в следующей стадии без очистки.This derivative is prepared from 6-tert-butoxycarbonylamino-2 - [(4-tert-butoxycarbonylaminobenzenesulfonyl) isobutylamino] hexyl ester (2S) -2-benzyloxycarbonylamino-3-methylbutyric acid (the product from Step A) as described in Example 15, step B. The final compound is obtained in quantitative yield (110 mg) and used in the next step without purification.
ЖХ-МС: 577,3 (M+H)+, 90% чист.LC-MS: 577.3 (M + H) + , 90% pure.
Стадия C. Получение 2-[(4-аминобензолсульфонил)изобутиламино]-6-(2-метоксикарбониламино-3,3-дифенилпропиониламино)гексилового эфира (2S,2S)-2-бензилоксикарбониламино-3-метилмасляной кислотыStep C. Preparation of 2 - [(4-aminobenzenesulfonyl) isobutylamino] -6- (2-methoxycarbonylamino-3,3-diphenylpropionylamino) hexyl ester (2S, 2S) -2-benzyloxycarbonylamino-3-methylbutyric acid
Указанное в заголовке соединение получают из 6-амино-2-[(4-аминобензолсульфонил)изобутиламино]гексилового эфира (2S)-бензилоксикарбониламино-3-метилмасляной кислоты (продукт из стадии B), как описано в примере 15, стадия C. Прозрачное маслянистое вещество получают с выходом 86% (120 мг).The title compound was prepared from 6-amino-2 - [(4-aminobenzenesulfonyl) isobutylamino] hexyl ester of (2S) -benzyloxycarbonylamino-3-methylbutyric acid (product from step B) as described in Example 15, step C. Transparent oily the substance is obtained in 86% yield (120 mg).
ЖХ-МС: 858,3 (M+H)+, 95% чист.LC-MS: 858.3 (M + H) + , 95% pure.
Стадия D. Получение 2-[(4-аминобензолсульфонил)изобутиламино]-6-(2-метоксикарбониламино-3,3-дифенилпропиониламино)гексилового эфира (2S,2S)-2-амино-3-метилмасляной кислоты (PL-530)Step D. Preparation of 2 - [(4-aminobenzenesulfonyl) isobutylamino] -6- (2-methoxycarbonylamino-3,3-diphenylpropionylamino) hexyl ester (2S, 2S) -2-amino-3-methylbutyric acid (PL-530)
К перемешиваемому раствору 2-[(4-аминобензолсульфонил)изобутиламино]-6-(2-метоксикарбониламино-3,3-дифенилпропиониламино)гексилового эфира (2S,2S)-2-бензилоксикарбониламино-3-метилмасляной кислоты (стадия C, 120 мг, 0,14 ммоль) в безводном ТГФ (8 мл) в атмосфере азота добавляют палладий 10% мас. на активированном угле (160 мг). Смесь подвергают взаимодействию в атмосфере водорода в течение ночи при комнатной температуре. Раствор фильтруют и палладий на угле промывают ТГФ (50 мг). Растворитель выпаривают и остаток (110 мг) очищают флэш-хроматографией, используя этилацетат в качестве элюента. Получают прозрачное маслянистое вещество с выходом 47% (47 мг).To a stirred solution of 2 - [(4-aminobenzenesulfonyl) isobutylamino] -6- (2-methoxycarbonylamino-3,3-diphenylpropionylamino) hexyl ester (2S, 2S) -2-benzyloxycarbonylamino-3-methylbutyric acid (step C, 120 mg, 0.14 mmol) in anhydrous THF (8 ml) in a nitrogen atmosphere add palladium 10% wt. on activated carbon (160 mg). The mixture is reacted in an atmosphere of hydrogen overnight at room temperature. The solution was filtered and the palladium-carbon was washed with THF (50 mg). The solvent was evaporated and the residue (110 mg) was purified by flash chromatography using ethyl acetate as an eluent. A clear oily substance is obtained with a yield of 47% (47 mg).
ЖХ-МС: 796,4 (M+H)+, 95% чист.LC-MS: 796.4 (M + H) + , 95% pure.
1H ЯМР (ацетон-d6): 0,84-0,97 (м, 12H), 0,97-1,08 (м, 2H), 1,27-1,43 (м, 3H), 1,49-1,62 (м, 4H), 1,80-1,93 (м, 1H), 1,94-2,00 (м, 1Н), 2,36-2,46 (м, 1Н), 2,58-2,74 (м, 2H), 2,86-2,96 (м, 3H), 2,99-3,10 (м, 2H), 3,46-3,52 (с, 3H), 3,52-3,60 (м, 2H), 3,75-3,87 (м, 2H), 3,95-4,04 (м, 1Н), 4,10-4,18 (м, 1H), 4,37-4,44 (м, 1H), 4,89-4,97 (м, 1Н), 5,40-5,48 (м, 1H), 6,30-6,40 (м, 1Н), 6,76-6,83 (д, J=8,2, 1H), 6,87-7,03 (м, 2H), 7,14-7,22 (м, 1H), 7,23-7,34 (м, 3H), 7,35-7,45 (м, 4H), 7,50-7,56 (м, 1H), 7,57-7,65 (м, 1H). 1 H NMR (acetone-d 6 ): 0.84-0.97 (m, 12H), 0.97-1.08 (m, 2H), 1.27-1.43 (m, 3H), 1 49-1.62 (m, 4H), 1.80-1.93 (m, 1H), 1.94-2.00 (m, 1H), 2.36-2.46 (m, 1H) 2.58-2.74 (m, 2H), 2.86-2.96 (m, 3H), 2.99-3.10 (m, 2H), 3.46-3.52 (s, 3H), 3.52-3.60 (m, 2H), 3.75-3.87 (m, 2H), 3.95-4.04 (m, 1H), 4.10-4.18 ( m, 1H), 4.37-4.44 (m, 1H), 4.89-4.97 (m, 1H), 5.40-5.48 (m, 1H), 6.30-6, 40 (m, 1H), 6.76-6.83 (d, J = 8.2, 1H), 6.87-7.03 (m, 2H), 7.14-7.22 (m, 1H ), 7.23-7.34 (m, 3H), 7.35-7.45 (m, 4H), 7.50-7.56 (m, 1H), 7.57-7.65 (m , 1H).
Биологическая доступность соединенийThe bioavailability of compounds
Чтобы оценить степень отщепления фосфатной группы in vivo от предполагаемых соединений, соединения PL-100, PL-462 (на основе PL-100), PL-337 и PL-515 (на основе PL-337) вводят перорально (50 мг/кг) самцам крыс Спрег-Доули и их концентрации в плазме измеряют при различных временных интервалах после введения.In order to evaluate the degree of in vivo removal of the phosphate group from the intended compounds, compounds PL-100, PL-462 (based on PL-100), PL-337 and PL-515 (based on PL-337) are orally administered (50 mg / kg) male Sprague-Dawley rats and their plasma concentrations are measured at various time intervals after administration.
PL-100 представляет собой активный ингредиент (ингибитор протеазы) следующей формулы:PL-100 is an active ingredient (protease inhibitor) of the following formula:

PL-337 представляет собой активный ингредиент (ингибитор протеазы) следующей формулы:PL-337 is an active ingredient (protease inhibitor) of the following formula:

Как было показано, данный активный ингредиент является эффективным по отношению к ВИЧ-1 аспартил-протеазе (патент США № 6632816). Активные ингредиенты также показывают эффективную антивирусную активность при тестировании на немутантном ВИЧ-1 вирусном штамме (NL4.3 в качестве вируса дикого типа), а также на нескольких мутантных штаммах.This active ingredient has been shown to be effective against HIV-1 aspartyl protease (US Pat. No. 6,632,816). The active ingredients also show effective antiviral activity when tested on a non-mutant HIV-1 virus strain (NL4.3 as wild-type virus), as well as on several mutant strains.
Все тестируемые продукты (PL-100, PL-462, PL-337 и PL-515) готовят в различных растворителях с конечной концентрацией 25 мг/мл. Композиция растворителя является следующей: (1) 20% этанола, 50% пропиленгликоля, 0,05% мас./об. Tween 20 и вода (Смесь); (2) забуференный фосфатом физиологический раствор (PBS).All test products (PL-100, PL-462, PL-337 and PL-515) are prepared in various solvents with a final concentration of 25 mg / ml. The solvent composition is as follows: (1) 20% ethanol, 50% propylene glycol, 0.05% w / v. Tween 20 and water (Mixture); (2) phosphate buffered saline (PBS).
Тестируемые продукты вводят самцам крыс Спрег-Доули при однократной пероральной дозе 50 мг/кг. Каждый продукт тестируют для трех крыс. Образцы крови (0,2-0,3 мл) собирают через 10, 20, 40, 60, 120, 180 и 360 мин после введения дозы. Собранную кровь центрифугируют, чтобы выделить плазму. Полученную в результате плазму отделяют и хранят при -70°C.Test products are administered to male Sprague-Dawley rats at a single oral dose of 50 mg / kg. Each product is tested for three rats. Blood samples (0.2-0.3 ml) are collected 10, 20, 40, 60, 120, 180 and 360 minutes after the dose. The collected blood is centrifuged to isolate the plasma. The resulting plasma is separated and stored at -70 ° C.
Образцы плазмы вместе со стандартами и образцами контроля качества обрабатывают, чтобы осадить белки, затем анализируют ВЭЖХ-МС на присутствие PL-462, PL-100, PL-515 и PL-337.Plasma samples, together with standards and quality control samples, are processed to precipitate the proteins, then HPLC-MS is analyzed for the presence of PL-462, PL-100, PL-515 and PL-337.
(Прим. №2) PL-462
(Note No. 2)
(Прим. №1-F) PL-100
(Note No. 1-F)
(Прим. №13) PL-515
(Note No. 13)
(Прим. №13-A)PL-337
(Note No. 13-A)
(PL-100, обнаружен)0.816 ± 0.295
(PL-100 detected)
(PL-337, обнаружен)1,075 ± 0,625
(PL-337, detected)
50 мг/кг PL-515 = 43 мг/кг PL-337
50 mg / kg PL-462 = 43 mg / kg PL-100
50 mg / kg PL-515 = 43 mg / kg PL-337
Результаты демонстрируют, что соединения PL-462 и PL-515 можно доставить перорально в водных растворах. Ни одно из соединений PL-462 и PL-515, доставленное в виде водного раствора, не обнаруживается в образцах крови, что говорит о быстром метаболизме исходных лекарственных веществ в PL-100 и PL-337.The results demonstrate that compounds PL-462 and PL-515 can be delivered orally in aqueous solutions. None of the compounds PL-462 and PL-515, delivered in the form of an aqueous solution, are detected in blood samples, which indicates a rapid metabolism of the starting drugs in PL-100 and PL-337.
Установлено, что водные дозировки растворов PL-462 и PL-515 слегка превосходят доставку PL-100 и PL-337 по сравнению с неводными рецептурами PL-100 и PL-337.It was found that aqueous dosages of PL-462 and PL-515 solutions slightly exceed the delivery of PL-100 and PL-337 compared to non-aqueous formulations of PL-100 and PL-337.
Основываясь на данных результатах все фосфорилированные соединения, описанные в настоящем изобретении, будут демонстрировать аналогичные фармакокинетические свойства.Based on these results, all phosphorylated compounds described in the present invention will exhibit similar pharmacokinetic properties.
Коэффициенты распределения (LogP) выбранных соединений и соответствующих ингибиторов ВИЧ-протеазы (лекарственных средств) являются следующими:Distribution coefficients (LogP) of selected compounds and corresponding HIV protease inhibitors (drugs) are as follows:
LogP измеряют стандартно, растворяя 1 мг соединения в 0,8 мл октанола или фосфатного буфера рН 7,4 (0,04 М KHPO4). Концентрацию соединений в фазах определяют ЖХ-МС. Данный тест демонстрирует растворимость соединений при физиологическом значении рН. Полученные LogP показывают, что соединения хорошо растворимы по сравнению с соответствующими лекарственными средствами.LogP is measured standardly by dissolving 1 mg of the compound in 0.8 ml of octanol or phosphate buffer pH 7.4 (0.04 M KHPO 4 ). The concentration of compounds in the phases is determined by LC-MS. This test demonstrates the solubility of the compounds at physiological pH. The resulting LogPs show that the compounds are readily soluble in comparison with the corresponding drugs.
Соединения, перечисленные в таблице 3, готовят по следующим схемам 1, 1A, 2, 3, 4 или 5; и более конкретно, как описано в каждом примере, перечисленном выше. Номера соединений, перечисленных в таблице 3 (Прим. №), соответствуют номерам примеров, представленных выше.The compounds listed in table 3 are prepared according to the following schemes 1, 1A, 2, 3, 4, or 5; and more specifically, as described in each example listed above. The numbers of the compounds listed in table 3 (Note No.) correspond to the numbers of the examples presented above.
Таблица 3Table 3
Структуры соединений на основе лизина по настоящему изобретениюStructures of lysine-based compounds of the present invention

Claims (15)

или его фармацевтически приемлемая соль,
где n равно 3 или 4,
где Х и Y, одинаковые или различные, выбраны из группы, состоящей из Н, F, Cl, Br, I и -NR4R5,
где R6 выбран из группы, состоящей из неразветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, и разветвленной алкильной группы, содержащей от 3 до 6 атомов углерода,
где R3 выбран из группы, состоящей из группы формулы R3A-CO-, причем R3A выбран из группы, состоящей из неразветвленной или разветвленной алкильной группы, содержащей от 1 до 6 атомов углерода, алкилоксигруппы, содержащей от 1 до 6 атомов углерода, и 4-морфолинила,
где R4 и R5, одинаковые, представляют собой Н,
где R2 выбран из группы, состоящей из дифенилметильной группы формулы IV

нафтил-1-СН2-группы формулы V

и нафтил-2-СН2-группы формулы VI

где X' и Y' одинаковые, представляют собой Н,
и где R1 выбран из группы, состоящей из (НО)2Р(O) и (МО)2Р(O), где М представляет собой щелочной металл.1. The compound of formula I
or a pharmaceutically acceptable salt thereof,
where n is 3 or 4,
where X and Y, identical or different, are selected from the group consisting of H, F, Cl, Br, I and —NR 4 R 5 ,
where R 6 selected from the group consisting of an unbranched alkyl group containing from 1 to 6 carbon atoms, and a branched alkyl group containing from 3 to 6 carbon atoms,
where R 3 selected from the group consisting of a group of the formula R 3A -CO-, and R 3A selected from the group consisting of a straight or branched alkyl group containing from 1 to 6 carbon atoms, alkyloxy group containing from 1 to 6 carbon atoms, and 4-morpholinyl,
where R 4 and R 5 are the same, represent H,
where R 2 selected from the group consisting of diphenylmethyl group of formula IV
naphthyl-1-CH 2 groups of formula V
and naphthyl-2-CH 2 groups of formula VI
where X 'and Y' are the same, represent H,
and where R 1 is selected from the group consisting of (HO) 2 P (O) and (MO) 2 P (O), where M is an alkali metal.

или его фармацевтически приемлемая соль,
где значения R1, R2, R3, R6, n, Х и Y определены в п.1.2. The compound according to claim 1, which is a compound of formula II,
or a pharmaceutically acceptable salt thereof,
where the values of R 1 , R 2 , R 3 , R 6 , n, X and Y are defined in claim 1.

или его фармацевтически приемлемая соль,
где значения R1, R3, R6, n, X, Y, X' и Y' определены в п.1.8. The compound according to claim 2, which is a compound of formula IIa
or a pharmaceutically acceptable salt thereof,
where the values of R 1 , R 3 , R 6 , n, X, Y, X 'and Y' are defined in claim 1.

Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU2007107798/04A RU2379312C2 (en) | 2004-08-02 | 2004-08-02 | Lysine compounds, pharmaceutical composition containing these compounds, application of said compounds for treatment or prevention of hiv-infection |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU2007107798/04A RU2379312C2 (en) | 2004-08-02 | 2004-08-02 | Lysine compounds, pharmaceutical composition containing these compounds, application of said compounds for treatment or prevention of hiv-infection |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2009139259/04A Division RU2009139259A (en) | 2004-08-02 | 2009-10-23 | LYSINE COMPOUNDS |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU2007107798A RU2007107798A (en) | 2008-09-10 |
| RU2379312C2 true RU2379312C2 (en) | 2010-01-20 |
Family
ID=39866532
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2007107798/04A RU2379312C2 (en) | 2004-08-02 | 2004-08-02 | Lysine compounds, pharmaceutical composition containing these compounds, application of said compounds for treatment or prevention of hiv-infection |
Country Status (1)
| Country | Link |
|---|---|
| RU (1) | RU2379312C2 (en) |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6436989B1 (en) * | 1997-12-24 | 2002-08-20 | Vertex Pharmaceuticals, Incorporated | Prodrugs of aspartyl protease inhibitors |
| US6506786B2 (en) * | 2001-02-13 | 2003-01-14 | Pharmacor Inc. | HIV protease inhibitors based on amino acid derivatives |
| US6632816B1 (en) * | 2002-12-23 | 2003-10-14 | Pharmacor Inc. | Aromatic derivatives as HIV aspartyl protease inhibitors |
| RU2003130227A (en) * | 2001-03-26 | 2005-04-10 | Новартис АГ (CH) | 2-AMINOPROPANOL DERIVATIVES |
-
2004
- 2004-08-02 RU RU2007107798/04A patent/RU2379312C2/en not_active IP Right Cessation
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6436989B1 (en) * | 1997-12-24 | 2002-08-20 | Vertex Pharmaceuticals, Incorporated | Prodrugs of aspartyl protease inhibitors |
| US6506786B2 (en) * | 2001-02-13 | 2003-01-14 | Pharmacor Inc. | HIV protease inhibitors based on amino acid derivatives |
| RU2003130227A (en) * | 2001-03-26 | 2005-04-10 | Новартис АГ (CH) | 2-AMINOPROPANOL DERIVATIVES |
| US6632816B1 (en) * | 2002-12-23 | 2003-10-14 | Pharmacor Inc. | Aromatic derivatives as HIV aspartyl protease inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| RU2007107798A (en) | 2008-09-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8008297B2 (en) | Lysine based compounds | |
| US6559137B1 (en) | Sulphonamide derivatives as prodrugs of aspartyl protease inhibitors | |
| US8580995B2 (en) | Lysine-based prodrugs of aspartyl protease inhibitors and processes for their preparation | |
| US20120053139A1 (en) | Method for improving pharmacokinetics of protease inhibitors and protease inhibitor precursors | |
| KR20010033595A (en) | Prodrugs of aspartyl protease inhibitors | |
| US6632816B1 (en) | Aromatic derivatives as HIV aspartyl protease inhibitors | |
| AU2009230800A1 (en) | Lysine based compounds | |
| EP2109461A2 (en) | Method for improving pharmacokinetics | |
| RU2379312C2 (en) | Lysine compounds, pharmaceutical composition containing these compounds, application of said compounds for treatment or prevention of hiv-infection | |
| CN102093411A (en) | Lysine-based compounds | |
| HK1105406B (en) | Lysine based compounds | |
| NZ552853A (en) | Lysine based compounds | |
| KR20070057815A (en) | Lysine compounds | |
| ZA200607352B (en) | Lysine based compounds | |
| HK1124861B (en) | Lysme-based prodrugs of aspartyl protease inhibitors and processes for their preparation |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| MM4A | The patent is invalid due to non-payment of fees |
Effective date: 20110803 |