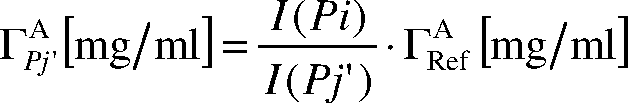-
Die vorliegende Erfindung betrifft ein Verfahren zur Identifizierung einer Substanz in einer Probe gemäß dem Oberbegriff des Anspruchs 1 sowie ein Computerprogrammprodukt, welches ein zur Ausführung eines solchen Verfahrens geeignetes Computerprogramm enthält, gemäß dem Oberbegriff des Anspruchs 9.
-
Auf dem Gebiet der kernmagnetischen Resonanzspektroskopie (NMR-Spektroskopie) existiert seit Jahrzehnten das Problem, dass es nicht möglich ist, eine automatische Identifizierung von in einer Probe enthaltenen Substanzen anhand eines gemessenen NMR-Spektrums durchzuführen. Vielmehr müssen NMR-Spektren auch heute noch aufwendig von Hand ausgewertet werden, um die mittels NMR-Spektroskopie gemessenen Substanzen identifizieren zu können. Dabei hängt der Erkennungserfolg maßgeblich von der technischen Expertise derjenigen Person ab, die die manuelle Substanzerkennung durchführt. Ferner spielt auch die Komplexität der Zusammensetzung der NMR-technisch vermessenen Probe eine Rolle. So lassen sich die einzelnen Bestandteile einer komplex zusammengesetzten Probe in aller Regel nicht eindeutig, schnell und einfach anhand eines gemeinsamen NMR-Spektrums identifizieren.
-
Grundsätzlich erzeugen Protonen und andere NMR-aktive Kerne, wie etwa 15N oder 13C in einer definierten chemischen Umgebung (also in einer spezifischen Molekülgruppe eines Moleküls) ein definiertes NMR-Signal. Folglich hat jede Substanz einen individuellen NMR-Fingerabdruck. Allerdings variieren diese NMR-Fingerabdrücke stark mit dem pH-Wert, der Temperatur, der Feldstärke, der Salzkonzentration und vielen anderen Parametern. Ferner überlagern sich die einzelnen NMR-Fingerabdrücke verschiedener in einer Probe enthaltenen Substanzen häufig zu komplexen Mustern, aus denen die einzelnen NMR-Fingerabdrücke nicht mehr ohne weiteres isoliert werden können.
-
Im Ergebnis ist es bislang nicht möglich, bei komplexen Mischungen vieler Substanzen all diese Faktoren zu berücksichtigen bzw. die unterschiedlichen Einflüsse der vorgenannten Parameter auf die einzelnen Substanzen in einer Datenbank zu hinterlegen. Denn hierfür müssten für jede einzelne Substanz umfangreiche Messreihen bei unterschiedlichen pH-Werten, Temperaturen, Salzkonzentrationen und anderen Parametern eingemessen werden.
-
Ferner müsste berücksichtigt werden, dass sich das Verhalten einer Substanz in Abhängigkeit der Anwesenheit anderer Substanzen verändern kann.
-
Aus der
DE 10 2010 038 014 A1 ist ein Verfahren zur Charakterisierung einer Probe mit den folgenden Schritten bekannt: Bereitstellung mindestens eines Analyseergebnisses mit einer Vielzahl von Werten, wobei das Analyseergebnis durch die Analyse einer Probe mittels mindestens eines Analyseverfahrens erstellt wurde; Bestimmung des Werts mindestens einer mathematischen Beziehung zwischen mindestens zwei Werten der Vielzahl von Werten; Erstellung einer charakterisierenden Signatur der Probe auf der Grundlage des Werts der mindestens einen mathematischen Beziehung. Dieses Verfahren zielt darauf ab, eine Probe – also eine komplex zusammengesetzte Mischung verschiedener Substanzen – als solche zu charakterisieren. Dabei kommt es – wie an mehreren Stellen der
DE 10 2010 038 014 A1 ausgeführt – nicht darauf an, die einzelnen in der Probe enthaltenen Substanzen zu identifizieren.
-
Aus der
EP 2 161 587 A1 ist ein Verfahren zur automatischen NMR-Spektrenauswertung bekannt, bei dem ein parameterfreies Interpretationssystem eingesetzt wird, das menschliche Logik nachahmt. Dieses Verfahren zieht Informationen aus einem NMR-Spektrum in einer ähnlichen Weise, wie ein menschlicher Experte dies tun würde. Dabei erfolgt eine Kombination von verschiedenen Expertensystemen, die bestimmte NMR-spektrale Merkmale sowie Merkmale aus einer vorgeschlagenen chemischen Struktur vorgeben. Nach mehreren iterativen Verfahrensdurchlaufen wird eine Liste von wahrscheinlich keitsgewichteten Hypothesen erstellt.
-
Aus Vu et al.: „An integrated workflow for robust alignment and simplified quantitative analysis of NMR spectrometry data”, BMC Bioinformatics 2011, 12: 405 ist ein klassisches Spektrenanpassungsverfahren bekannt, bei dem ein Referenzspektrum durch Modifikationen an ein Probenspektrum angepasst wird. Dazu wird ein Algorithmus eingesetzt, der auf einer hierarchischen Cluster-basierten Peak-Zuordnung beruht. Dieses Verfahren ist maßgeblich abhängig von den Messbedingungen, unter denen das NMR-Probenspektrum und das NMR-Referenzspektrum aufgenommen wurden.
-
Der vorliegenden Erfindung liegt die Aufgabe zugrunde, ein Verfahren anzugeben, mit dem eine automatische Substanzerkennung für ein NMR-Spektrum durchgeführt werden kann. Dieses Verfahren soll dabei für NMR-Spektren geeignet sein, die unter den unterschiedlichsten Rahmenbedingungen aufgenommen wurden.
-
Diese Aufgabe wird mit einem Verfahren mit den Merkmalen des Anspruchs 1 gelöst. Ein solches Verfahren zur Identifizierung einer Substanz in einer Probe weist die nachfolgend erläuterten Schritte auf.
-
Zunächst wird ein NMR-Spektrum einer Probe bereitgestellt, die mindestens eine Substanz enthält, welche mindestens einen NMR-aktiven Kern aufweist. Geeignete NMR-aktive Kerne sind beispielsweise 1H, 13C und 15N.
-
Anschließend erfolgt eine Linienseparation des NMR-Spektrums. Das heißt, das NMR-Spektrum wird in diskrete Spektralwerte zerlegt. Ein solcher Spektralwert kann beispielsweise eine einer einzelnen NMR-aktiven chemischen Gruppe entsprechende Spektrallinie sein. Jeder Spektralwert weist dabei einen Integralwert und einen Positionswert auf. Der Integralwert gibt die Höhe und/oder die Fläche einer einzelnen Linie des NMR-Spektrums oder aber eines einzelnen spektralen Abschnitts des NMR-Spektrums an. Der Integralwert umfasst folglich Intensitätsinformationen der betrachteten Linie oder des betrachteten spektralen Abschnitts oder besteht aus diesen Intensitätsinformationen. Jeder Positionswert gibt die Position der betrachteten Linie oder des betrachteten spektralen Abschnitts im NMR-Spektrum an. Die Positionswerte sind dabei ein Maß für die magnetische Abschirmung einzelner Molekülgruppen/Atome im Gesamtmolekül der in der Probe enthaltenen Substanz. Die Integralwerte sind ein Maß für die Anzahl einzelner Molekülgruppen/Atome im NMR-Spektrum.
-
Dabei wird als Position dabei die mittlere Position der betrachteten Linie oder des betrachteten spektralen Abschnitts verwendet. Ein derartiger spektraler Abschnitt wird fachsprachlich auch als „bin” bezeichnet. Die Separation bzw. das Zerschneiden eines NMR-Spektrums in unterschiedliche, jeweils eine oder mehrere Linien umfassende, diskrete spektrale Abschnitte ist dem Fachmann daher auch unter dem Fachbegriff „binnen” bekannt.
-
Grundsätzlich kann die Linienseparation nach dem Fachmann allgemein bekannten Verfahren erfolgen. Jedes Verfahren, das geeignet ist, Informationen zu einzelnen Molekülgruppen oder Atomen aus einem NMR-Spektrum zu extrahieren, kann grundsätzlich eingesetzt werden. Die hier beschriebene Linienseparation versucht, ein NMR-Spektrum als möglichst perfekte Überlagerung von Lorenzkurven (bzw. einer Summe von Lorenz- und Gauß-Kurven) darzustellen. Das dabei verfolgte Ziel ist es, nach der Linienseparation vorzugsweise jeder einzelnen NMR-aktiven Gruppe einen definierten Integralwert und einen entsprechenden Positionswert zuzuordnen. Das heißt, vorzugsweise wird jede Linie des NMR-Spektrums von jeder anderen Linie des NMR-Spektrums separiert.
-
Anschließend wird auf der Grundlage jeweils zweier Integralwerte des NMR-Spektrums eine Vielzahl von Integralverhältniswerten berechnet. Vorzugsweise wird jeder einzelne Integralwert des NMR-Spektrums mit jedem anderen Integralwert des NMR-Spektrums verrechnet. Jeder Integralverhältniswert gibt dabei das Höhen- und/oder Flächenverhältnis der zugrundeliegenden Spektralwerte an. Werden beispielsweise durch die Linienseparation 20 Spektralwerte mit 20 Integralwerten erhalten und nachfolgend jeweils genau zwei Integralwerte miteinander zur Bildung eines Integralverhältniswertes verrechnet, erhält man 20 × 20 = 400 Integralverhältniswerte. 20 dieser Integralverhältniswerte haben den Wert 1 (Division derselben Integralwerte durch einander) und werden vorzugsweise nicht weiter als Integralverhältniswerte berücksichtigt, da sie keine relevante Information enthalten.
-
Ferner wird eine Vielzahl von Abstandswerten aus jeweils zwei Positionswerten des NMR-Spektrums berechnet. Auch hier erfolgt vorzugsweise eine Verrechnung eines jeden einzelnen Positionswerts des NMR-Spektrums mit jedem anderen Positionswert des NMR-Spektrums. Jeder Abstandswert spiegelt dabei den spektralen Abstand zwischen den zugrundeliegenden Spektralwerten wider. Werden gemäß dem obigen Beispiel 20 Spektralwerte und dementsprechend auch 20 Positionswerte erhalten und jeweils genau zwei Positionswerte miteinander verrechnet, erhält man folglich 20 × 20 = 400 Abstandswerte. 20 dieser Abstandswerte haben den Wert 1 (Division derselben Positionswerte durch einander) und werden vorzugsweise nicht weiter als Abstandswerte berücksichtigt, da sie keine relevante Information enthalten.
-
Der spektrale Abstand ist ein Maß dafür, wie weit einzelne Linien des NMR-Spektrums voneinander entfernt sind. Ist der Abstand gering, liegen die Linien dicht beieinander. Ist der Abstand groß, liegen die Linien weit auseinander. Der spektrale Abstand kann in unterschiedlichen Einheiten, wie etwa Hz oder ppm angegeben werden.
-
Im beanspruchten Verfahren erfolgt nun ein Vergleich der berechneten Integralverhältniswerte des NMR-Spektrums mit entsprechenden Integralverhältniswerten eines NMR-Referenzspektrums. Das NMR-Referenzspektrum ist dabei ein NMR-Spektrum mindestens einer Referenzsubstanz. Zur Berechnung der Integralverhältniswerte des NMR-Referenzspektrums wird vorzugsweise analog vorgegangen wie bei der Berechnung der Integralverhältniswerte des untersuchten NMR-Spektrums. Das heißt, zunächst wird insbesondere eine Linienseparation durchgeführt, um Integralwerte mehrerer, vorzugsweise aller, Spektralwerte zu erhalten.
-
Nach dem Vergleich der Integralverhältniswerte erfolgt eine Auswahl einer ersten echten Untermenge aus den Integralverhältniswerten des NMR-Spektrums. Die erste Untermenge umfasst dabei diejenigen Integralverhältniswerte, die einem Integralverhältniswert des NMR-Referenzspektrums innerhalb einer jeweils vorgebbaren ersten Toleranzgrenze entsprechen. Ob eine Entsprechung vorliegt oder nicht, hängt grundsätzlich vom jeweiligen Einzelfall ab, wobei Faktoren wie beispielsweise die Anzahl der Linien im betrachteten NMR-Spektrum, deren Abstand voneinander und der absolute Wert der betrachteten Integralverhältniswerte zu berücksichtigen sind. Das heißt, die Qualität der miteinander verglichenen Daten ist mit ausschlaggebend, ob eine Entsprechung vorliegt oder nicht. Bei hochwertigen zu vergleichenden Daten (wenig Störungen, geringe Messfehler, gutes Signal-zu-Rauschen-Verhältnis, gut voneinander separierbare Linien, prägnante Integralverhältnisse etc.) sind die Toleranzgrenzen niedriger anzusetzen als bei weniger hochwertigen Daten. Vorzugsweise werden die Toleranzgrenzen bzw. die noch zulässigen Fehler derart gewählt, dass keine richtigen Kandidaten versehentlich ausgeschlossen werden. Das heißt, falsch negative Resultate sollen vermieden werden.
-
Liegen beispielsweise alle Integralverhältnisse bis auf einen Integralverhältniswert eines NMR-Referenzspektrums im Bereich von rund 1 und der einzige übrige Integralverhältniswert im Bereich von 10, so kann eine Entsprechung vorliegen, wenn im NMR-Spektrum der Probe ebenfalls alle Integralverhältniswerte bis auf einen Integralverhältniswert im Bereich von rund 1 liegen (und keine relevanten Abweichungen zu den Integralverhältniswerten des NMR-Referenzspektrums aufweisen) und der verbleibende Integralverhältniswert 20 beträgt. Andererseits kann eine derartige Abweichung von 100% in anderen Fallkonstellationen bei weniger prägnanten Integralverhältniswerten nicht mehr als Entsprechung gewertet werden.
-
Mit anderen Worten ausgedrückt, erfolgt die Entscheidung, ob eine Entsprechung vorliegt oder nicht, anhand individueller Kriterien. Vorzugsweise werden diese Kriterien danach bestimmt, ob eine bekannte Substanz in einem NMR-Spektrum als diese Substanz tatsächlich erkannt wird, wenn ein Vergleich des NMR-Spektrums einer diese Substanz enthaltenden Probe mit einem NMR-Referenzspektrum dieser Substanz erfolgt.
-
Vorzugsweise wird von einer Entsprechung ausgegangen, wenn ein Integralverhältniswert des NMR-Spektrums von einem Integralverhältniswert des NMR-Referenzspektrums um 100% oder weniger, insbesondere 50% oder weniger, insbesondere 40% oder weniger, insbesondere 30% oder weniger, insbesondere 20% oder weniger, insbesondere 15% oder weniger, insbesondere 10% oder weniger, insbesondere 5% oder weniger, insbesondere 4% oder weniger, insbesondere 3% oder weniger, insbesondere 2% oder weniger, insbesondere 1% oder weniger und ganz besonders 0,5% oder weniger abweicht. Für jeden zu vergleichenden Integralverhältniswert des NMR-Spektrums kann dabei eine jeweils eigene Abweichung von einem entsprechenden Integralverhältniswert des NMR-Referenzspektrums zu Grunde gelegt werden. Im Idealfall liegt eine Entsprechung dann vor, wenn ein Integralverhältniswert des NMR-Spektrums identisch zu einem Integralverhältniswert des NMR-Referenzspektrums ist. Im Rahmen der jeweiligen Messgenauigkeiten sind jedoch üblicherweise mindestens die zuvor genannten Toleranzen zu berücksichtigen.
-
Im Rahmen des beanspruchten Verfahrens erfolgt auch ein Vergleich der Abstandswerte des NMR-Spektrums mit entsprechenden Abstandswerten des NMR-Referenzspektrums. Die Abstandswerte des NMR-Referenzspektrums werden dabei vorzugsweise auf die gleiche Weise berechnet wie die Abstandswerte des NMR-Spektrums der untersuchten Probe.
-
Der Vergleich der Abstandswerte dient dazu, eine zweite echte Untermenge aus den Abstandswerten des NMR-Spektrums auszuwählen. Die zweite Untermenge umfasst dabei diejenigen Abstandswerte, die einem Abstandswert des NMR-Referenzspektrums innerhalb einer jeweils vorgebbaren zweiten Toleranzgrenze entsprechen. Dabei wird vorzugsweise unter den gleichen Voraussetzungen von einer Entsprechung ausgegangen, die in Bezug auf den Vergleich der Integralverhältniswerte erläutert wurden. In relativen Fehlern ausgedrückt, wird eine Entsprechung vorzugsweise dann angenommen, wenn die Abweichung zwischen einem Abstandswert des NMR-Spektrums und einem Abstandswert des NMR-Referenzspektrums 100% oder weniger, insbesondere 50% oder weniger, insbesondere 40% oder weniger, insbesondere 30% oder weniger, insbesondere 20% oder weniger, insbesondere 15% oder weniger, insbesondere 10% oder weniger, insbesondere 5% oder weniger, insbesondere 4% oder weniger, insbesondere 3% oder weniger, insbesondere 2% oder weniger, insbesondere 1% oder weniger und ganz besonders 0,5% oder weniger beträgt. Für jeden zu vergleichenden Abstandswert des NMR-Spektrums kann auch hier eine jeweils eigene Abweichung von einem entsprechenden Abstandswert des NMR-Referenzspektrums zu Grunde gelegt werden. Vorzugsweise liegt eine Entsprechung zwischen den Abstandswerten dann vor, wenn ein Abstandswert des NMR-Spektrums identisch zum Abstandswert des NMR-Referenzspektrums ist. Aber auch hier sind im Rahmen der Messgenauigkeit üblicherweise zumindest die vorgenannten Toleranzen zu berücksichtigen.
-
Wenn die erste Untermenge und die zweite Untermenge gebildet sind, werden diese miteinander verglichen. Anschließend werden all diejenigen Integralverhältnis- und Abstandswerte aus beiden Untermengen ausgewählt, denen dieselben Spektralwerte zugrunde liegen. Diese Auswahl stellt eine dritte echte Untermenge dar.
-
Beispielhaft wird zur näheren Erläuterung folgende Fallkonstellation angenommen: Die erste Untermenge enthält einen ersten Integralverhältniswert, der aus den Integralwerten eines ersten und eines zweiten Spektralwertes gebildet wurde. Die erste Untermenge enthält ferner einen zweiten Integralverhältniswert, der aus den Integralwerten eines dritten und eines vierten Spektralwertes gebildet wurde. Die zweite Untermenge enthält einen ersten Abstandswert, der aus dem ersten Spektralwert und dem zweiten Spektralwert gebildet wurde. Die zweite Untermenge enthält ferner einen zweiten Abstandswert, der aus einem fünften Spektralwert und einem sechsten Spektralwert gebildet wurde. Dann wären der erste Integralverhältniswert und der erste Abstandswert aus denselben Spektralwerten gebildet, während der zweite Abstandswert aus Spektralwerten gebildet wäre, aus denen kein Integralverhältniswert in der ersten Untermenge gebildet wäre. In diesem Beispiel wird die dritte Untermenge folglich aus dem ersten Integralverhältniswert oder dem ersten Abstandswert bestehen. Der zweite Integralverhältniswert und der zweite Abstandswert wären hingegen nicht in der dritten Untermenge enthalten. Die dritte Untermenge stellt folglich eine Schnittmenge zwischen der ersten Untermenge und der zweiten Untermenge dar.
-
Schließlich wird ein Gütekriterium der in der dritten Untermenge enthaltenen Werte bestimmt, das sich aus einem Vergleich dieser Werte mit den Integralverhältniswerten und/oder den Abstandswerten des NMR-Referenzspektrums ergibt. Unter Zuhilfenahme dieses Gütekriteriums kann dann eine Entscheidung getroffen werden, ob die in der Probe enthaltene Substanz als die Referenzsubstanz identifiziert wird oder nicht.
-
Das Gütekriterium kann beispielsweise ein Maß für die Abweichung der Integralverhältniswerte des NMR-Spektrums von den Integralverhältniswerten des NMR-Referenzspektrum und/oder für die Abweichung der Abstandswerte des NMR-Spektrums von den Abstandswerten des NMR-Referenzspektrums umfassen oder sein.
-
In einer weiteren Ausgestaltung kann das Gütekriterium die Anzahl der in der dritten Untermenge enthaltenen Werte im Vergleich zur Anzahl der Integralverhältniswerte oder der Abstandswerte des NMR-Referenzspektrums umfassen oder sein. Wird zur Berechnung der Integralverhältniswerte und der Abstandswerte die jeweils gleiche Anzahl von Spektralwerten herangezogen, sind die Anzahl der Integralverhältniswerte und die Anzahl der Abstandswerte identisch. Beruht die Berechnung der Integralverhältniswerte beispielsweise auf jeweils genau 2 Spektralwerten bzw. den diesen Spektralwerten zugeordneten Integralwerten, erhält man bei n Spektralwerten n2 Integralverhältniswerte. Von diesen n2 Integralverhältniswerten haben n Integralverhältniswerte genau den Wert 1 (Division zweier identischer Integralwerte durch einander). Vorzugsweise werden diese Integralverhältniswerte wie auch die entsprechenden Abstandswerte nicht als bei der Bestimmung der Anzahl der Integralverhältniswerte oder der Abstandswerte berücksichtigt. Dies gilt sowohl für das betrachtete NMR-Spektrum der analysierten Probe wie auch für das NMR-Referenzspektrum.
-
Ist die Anzahl der in der dritten Untermenge enthaltenen Werte mit der Anzahl der Integralverhältniswerte oder der Abstandswerte des NMR-Referenzspektrums identisch oder übersteigt sie zumindest eine vorgebbare untere Schwelle, kann die in der Probe enthaltene Substanz in einer Ausgestaltung als die Referenzsubstanz identifiziert werden. Sind in der dritten Untermenge hingegen signifikant weniger Werte enthalten als das NMR-Referenzspektrum Integralverhältniswerte oder Abstandswerte umfasst, kann die in der Probe enthaltene Substanz in einer Ausgestaltung nicht als die Referenzsubstanz identifiziert werden. Je nach Anzahl der in der dritten Untermenge enthaltenen Werte kann beispielsweise aber die Aussage getroffen werden, dass die in der Probe enthaltene Substanz zumindest nicht die Referenzsubstanz ist. Um die in der Probe enthaltene Substanz in einem solchen Fall eindeutig identifizieren zu können, sollten einige der vorgenannten Verfahrensschritte vorzugsweise mit einem neuen NMR-Referenzspektrum erneut durchgeführt werden.
-
Um die Aussagekraft der Anzahl der Integralverhältniswerte oder der Abstandswerte des NMR-Spektrums nicht zu verfälschen, sollten die Integralverhältniswerte und die Abstandswerte des NMR-Referenzspektrums jeweils aus der gleichen Anzahl Spektralwerte (z. B. jeweils genau 2 Spektralwerte) berechnet werden wie die Integralverhältniswerte und die Abstandswerte des NMR-Spektrums der untersuchten Probe.
-
Das Gütekriterium kann vorzugsweise unterschiedliche Parameter berücksichtigen. So ist es denkbar, dass bei einer geringen Übereinstimmung zwischen der Anzahl der Werte der dritten Untermenge mit der Anzahl der Integralverhältniswerte oder Abstandswerte des NMR-Referenzspektrums dennoch eine Substanzidentifikation erfolgt, wenn gleichzeitig nur eine geringe Abweichung zwischen den verglichenen Integralverhältniswerten und/oder Abstandswerten festgestellt wurde. Größere Abweichungen der verglichenen Integralverhältniswerte und/oder Abstandswerte können umgekehrt durch eine größere Übereinstimmung der Anzahl der Werte der dritten Untermenge mit der Anzahl der Integralverhältniswerte oder Abstandswerte des NMR-Referenzspektrums ausgeglichen werden. Die Güte der Werte der dritten Untermenge kann folglich qualitative und quantitative Aspekte der Werte berücksichtigen.
-
Das vorliegende Verfahren beruht auf der Grundlage, dass zwei voneinander unabhängige Eigenschaften eines Moleküls mit dem gleichen Messverfahren gemessen werden. Hierbei handelt es sich einerseits um die Anzahl der Molekülgruppen und deren Protonenverhältnisse im Gesamtmolekül, denen ein NMR-Signal zugeordnet werden kann. Diese Eigenschaft wird durch den Integralwert ausgedrückt. Andererseits handelt es sich dabei um die magnetische Abschirmung einzelner Molekülgruppen im Gesamtmolekül. Diese Eigenschaft wird durch den Positionswert eines jeden Spektralwerts ausgedrückt.
-
Das Besondere an dem vorliegenden Verfahren ist, dass es unabhängig von den jeweiligen Messbedingungen durchgeführt werden kann. Das heißt, sonst äußerst kritische Parameter wie etwa der pH-Wert, die Temperatur, die Salzkonzentration oder die magnetische Feldstärke, unter der eine NMR-Messung durchgeführt wird, spielen für die Auswertung praktisch keine Rolle mehr und können automatisch bearbeitet werden. Es ist auch möglich, dass das NMR-Referenzspektrum mit anderen Parametern aufgenommen wurde als das NMR-Spektrum der Probe, die die zu identifizierende Substanz enthält. Das gewährleistet eine globale Anwendbarkeit des vorliegenden Verfahrens unabhängig von einzelnen Geräten. Durch eine einmalig aufgebaute Datenbank mit NMR-Referenzspektren von Referenzsubstanzen kann in Anwendung des vorliegend beanspruchten Verfahrens folglich eine eindeutige Identifizierung einer Substanz in einer Probe mittels NMR-Spektroskopie erfolgen.
-
Alternativ kann eine Substanz auch mittels ihrer Molekülstruktur basierend auf theoretischen Grundlagen erkannt werden, sofern einzelne Molekülgruppen dieser Substanz durch das vorliegende Verfahren identifiziert wurden.
-
Ein Eingriff des Benutzers in das Verfahren ist dabei nicht erforderlich. Vielmehr kann das Verfahren vollautomatisch durchgeführt werden, so dass eine automatische bzw. automatisierte Substanzerkennung auch in komplexen Substanzgemischen möglich ist.
-
Wenngleich das NMR-Referenzspektrum ein Spektrum verschiedener Substanzen sein kann, handelt es sich in einer bevorzugten Ausgestaltung des Verfahrens beim NMR-Referenzspektrum um ein Spektrum genau einer einzelnen Referenzsubstanz.
-
In einer Variante wird die in der Probe enthaltene Substanz dann als die Referenzsubstanz identifiziert, wenn mindestens 60% der Integralverhältniswerte oder der Abstandswerte des NMR-Referenzspektrums in der dritten Untermenge enthalten sind. In weiteren bevorzugten Ausgestaltungen wird ein Wert von 70%, insbesondere 75%, insbesondere 80%, insbesondere 85%, insbesondere 90%, insbesondere 95% und ganz besonders 99% als unterer Schwellenwert für die eindeutige Identifizierung der Substanz als Referenzsubstanz gewählt. In einer weiteren bevorzugten Ausgestaltung des Verfahrens wird die Substanz eindeutig als nicht die Referenzsubstanz identifiziert, wenn weniger als 40%, insbesondere weniger als 30%, insbesondere weniger als 20%, insbesondere weniger als 10% und ganz besonders weniger als 5% der Integralverhältniswerte oder der Abstandswerte des NMR-Referenzspektrums in der dritten Untermenge enthalten sind. Wie oben erläutert, können die vorgenannten Grenzen nach oben oder unten überschritten werden, sofern beim Gütekriterium zusätzliche Informationen zu den Werten der dritten Untermenge berücksichtigt werden.
-
Die vorgenannten einzelnen Verfahrensschritte müssen dabei nicht notwendigerweise in der zuvor erläuterten Reihenfolge durchgeführt werden. Vielmehr könnten beispielsweise ein Vergleich der Integralverhältnisse und eine Auswahl der ersten Untermenge unmittelbar nach der Bestimmung der Vielzahl von Integralverhältniswerten erfolgen. Ebenso könnten beispielsweise der Vergleich der Abstandswerte und die Auswahl einer zweiten Untermenge unmittelbar nach der Bestimmung der Vielzahl von Abstandswerten erfolgen. Es versteht sich für den Fachmann von selbst, dass der Schritt des Vergleichs der ersten Untermenge mit der zweiten Untermenge erst dann durchgeführt werden kann, wenn die erste Untermenge und die zweite Untermenge gebildet wurden.
-
In einer weiteren Variante werden die oben erläuterten Schritte des Vergleichs der Integralverhältniswerte, der Auswahl einer ersten Untermenge, des Vergleichs der Abstandswerte, der Auswahl einer zweiten Untermenge, des Vergleichs der ersten Untermenge mit der zweiten Untermenge und der Entscheidung, ob die in der Probe enthaltene Substanz als Referenzsubstanz identifiziert wird, so oft wiederholt, bis alle in der Probe enthaltenen Substanzen eindeutig anhand der Referenzsubstanzen identifiziert sind oder aber keine weiteren NMR-Referenzspektren mehr vorliegen. Dabei wird in jedem Wiederholungsschritt ein anderes NMR-Referenzspektrum vorzugsweise einer anderen Referenzsubstanz für die jeweiligen Vergleichszwecke herangezogen. Auf diese Weise ist es durch ein entsprechendes iteratives Verfahren möglich, die unterschiedlichen in der Probe enthaltenen Substanzen verschiedenen Referenzsubstanzen zuzuordnen. Die einzige Voraussetzung hierfür ist, dass für die einzelnen Referenzsubstanzen bereits NMR-Referenzspektren vorliegen. Die NMR-Referenzspektren können durch Messen, durch Simulation oder durch andere Verfahren generiert werden. Diese NMR-Referenzspektren werden vorzugsweise in einer Datenbank hinterlegt.
-
Die Abstandswerte können grundsätzlich durch jedes geeignete Verfahren bestimmt werden, das eine Charakterisierung der Resonanzeigenschaften der betrachteten Spektralwerte bzw. erlaubt.
-
Vorzugsweise werden die Abstandswerte als direkte Differenzen der Positionswerte, als normierte Differenzen der Positionswerte oder als Quotient der Positionswerte berechnet. Unter Berücksichtigung absoluter Positionswerte kann mit den direkten Differenzen von Resonanzfrequenzen (va = T + Δva) gearbeitet werden. T bezeichnet dabei die Trägerfrequenz (auch als Betriebsfrequenz oder Protonenresonanzfrequenz bezeichnet) des verwendeten NMR-Spektrometers. Δva bezeichnet den Eigenfrequenzanteil des jeweiligen zu einer Substanz A gehörigen Signals.
-
Alternativ bietet sich die Verwendung normierter Differenzen von Resonanzfrequenzen zur Berechnung der Abstandswerte an, wobei die Positionswerte dann nicht mehr in Hz, sondern in ppm ausgedrückt werden. Schließlich ist eine eindeutige Abstandsbestimmung zweier Peaks bzw. Linien voneinander auch über eine Quotientenbildung möglich. Eine eindeutige Abstandszuordnung über Verhältnisse ist hier deswegen möglich, da die Trägerfrequenz sehr viel größer als die Eigenfrequenzanteile der einzelnen Signale ist (T >> Δv).
-
Die Integralverhältnisse werden aus jeweils genau zwei Integralwerten des NMR-Spektrums gebildet. Ergänzend hierzu werden auch die Abstandswerte aus jeweils genau zwei Positionswerten des NMR-Spektrums berechnet. Bei n verschiedenen Linien einer Substanz A gibt es (n2 – n)/2 relevante Integralverhältnisse zwischen jeweils zwei Linien und genauso viele relevante Abstandswerte zwischen jeweils zwei Linien. Ausgehend von n2 Integralverhältnissen bzw. Abstandswerten werden aus den oben erläuterten Überlegungen vorzugsweise n Werte, die den Wert 1 aufweisen abgezogen. Die anschließende Division der verbleibenden Werteschar wird durch 2 dividiert, da die Hälfte der jeweiligen Werte dem Kehrwert der anderen Hälfte der Werte entspricht. Damit ist die Anzahl der relevanten Integralverhältniswerte bzw. Abstandswerte nur weniger als halb so groß wie die Anzahl sämtlicher Integralverhältniswerte bzw. Abstandswerte. Die Abstandswerte können auch als Peakabstände oder Linienabstände bezeichnet werden.
-
Die Verwendung genau zweier Integralwerte bzw. genau zweier Positionswerte zur Bestimmung entsprechender Integralverhältniswerte oder Abstandswerte hat den Vorteil, dass die dann in den Integralverhältniswerten und Abstandswerten enthaltenen Informationen nicht zu komplex sind, sondern noch in verhältnismäßig einfacher Weise auf die zugrundeliegenden Spektralwerte zurückgeführt werden können. Das erleichtert die Durchführung des Verfahrens.
-
In einer weiteren Variante wird die in der Probe enthaltene Substanz nicht nur qualitativ als eine bestimmte Substanz identifiziert, sondern auch quantifiziert. Das heißt, in dieser Variante wird die Konzentration der in der Probe enthaltenen Substanz ermittelt. Um eine derartige Quantifizierung zu erlauben, muss die Substanz zunächst in qualitativer Weise identifiziert worden sein. Ist dies erfolgt, ist auch bekannt, welches NMR-Referenzspektrum zur Quantifizierung der in der Probe enthaltenen Substanz herangezogen werden kann. Nun kann beispielsweise ein Integralwert des NMR-Spektrums mit einem entsprechenden Integralwert desselben Spektralwerts des NMR-Referenzspektrums verglichen werden. Sofern die Konzentration der dem NMR-Referenzspektrum zugrundeliegenden Substanz bekannt ist, kann auf diese Weise die Konzentration der Substanz in der Probe bestimmt werden. Denn wenn die Protonenkonzentration für einen Integralwert bekannt ist, lassen sich daraus dann alle anderen Integralwerte in Protonenkonzentrationen überführen. Ist einem Integralwert bzw. dem entsprechenden Spektralwert eine Molekülgruppe zugeordnet, kann daraus deren Konzentration berechnet werden.
-
Um die Messgenauigkeit zu erhöhen, kann es sich dabei anbieten, nicht nur einen einzigen Integralwert des NMR-Spektrums mit einem entsprechenden einzigen Integralwert des NMR-Referenzspektrums zu vergleichen, sondern sämtliche für die jeweils betrachtete Substanz charakteristischen Integralwerte des NMR-Spektrums mit entsprechenden Integralwerten des NMR-Referenzspektrums. Im Idealfall sollte aus allen Spektralwerten bzw. den entsprechenden Integralwerten der Spektralwerte des NMR-Spektrums die gleiche Konzentration der jeweils betrachteten Substanz ermittelt werden können. Tatsächlich kann man jedoch aufgrund der Messgenauigkeit kleinere Abweichungen zwischen den auf unterschiedlichen Integralwerten beruhenden Konzentrationsbestimmungen ermitteln, die sich in einer normalverteilten Häufigkeitsverteilung darstellen lassen.
-
Um die gemäß dem Stand der Technik üblichen Auswerteverfahren in möglichst einfacher Weise durchführen zu können, wird üblicherweise mit entkoppelten NMR-Spektren gearbeitet. Auch das vorliegende Verfahren kann auf der Grundlage entkoppelter NMR-Spektren durchgeführt werden. Dies ist jedoch nicht erforderlich. Vielmehr können auch nicht entkoppelte NMR-Spektren verwendet werden, um eine Substanzidentifikation gemäß dem vorliegenden Verfahren durchführen zu können. In einer bevorzugten Variante wird das Verfahren zunächst unter Verwendung eines entkoppelten NMR-Spektrums durchgeführt. Falls auf diese Weise noch keine eindeutige Substanzidentifizierung der in der Probe enthaltenen Substanzen möglich war, wird das Verfahren anschließend nochmals unter Verwendung eines nicht entkoppelten NMR-Spektrums derselben Probe erneut durchgeführt. Wenngleich nicht entkoppelte NMR-Spektren komplexer und daher nach den klassischen Verfahren schwieriger auszuwerten sind, ist zu berücksichtigen, dass sie mehr Informationen als entkoppelte NMR-Spektren enthalten. Dieses Mehr an Informationen kann bei dem vorliegenden Verfahren in bevorzugter Weise dazu ausgenutzt werden, eine Substanzidentifikation auch in schwierigeren Fällen sicher und erfolgreich durchführen zu können. Wird ein entkoppeltes NMR-Spektrum analysiert, wird vorzugsweise auch ein entkoppeltes NMR-Referenzspektrum eingesetzt. Wird ein nicht entkoppeltes NMR-Spektrum analysiert, wird vorzugsweise auch ein nicht entkoppeltes NMR-Referenzspektrum eingesetzt.
-
Die der vorliegenden Erfindung zugrundeliegende Aufgabe wird auch durch ein Computerprogrammprodukt mit einem Computerprogramm gelöst, das einen Programmcode zur Durchführung eines Verfahrens gemäß den vorherigen Erläuterungen aufweist, wenn das Computerprogramm auf einen Computer ausgeführt wird. Ein solches Computerprogramm stellt die technische Lösung des bislang ungelösten technischen Problems dar, wie verschiedene in einer Probe enthaltene Substanzen auf der Grundlage eines NMR-Spektrums automatisch als bestimmte Substanzen identifiziert werden können. Da es zur Durchführung dieses Computerprogramms nicht erforderlich ist, umfangreiche Datenbanken mit NMR-Spektren vorzuhalten, die unter den unterschiedlichsten Bedingungen aufgenommen wurden, reduziert sich der rechentechnische Aufwand bei der Verwendung eines entsprechenden Computerprogramms erheblich. Dadurch können in sehr vorteilhafter Weise Ressourcen, Zeit und Geld bei einer entsprechenden Substanzidentifikation gespart werden. Darüber hinaus wird eine automatische Substanzidentifikation auf diese Weise für NMR-Spektren zugänglich, die unter unterschiedlichen Bedingungen und auf unterschiedlichen Geräten gemessen wurden.
-
Die vorliegende Erfindung wird nun anhand von Zeichnungen und Beispielen näher erläutert. Es zeigen:
-
1 ein NMR-Spektrum einer Substanz A;
-
2 eine grafische Darstellung zweier Linien eines NMR-Spektrums zur Verdeutlichung der Berechnung von Abstandswerten;
-
3 eine Gegenüberstellung eines NMR-Spektrums einer Probe und des NMR-Spektrums der Substanz A aus der 1;
-
4A ein NMR-Spektrum einer Mischprobe B1 ohne Adenosin;
-
4B ein NMR-Spektrum einer Mischprobe B1 mit Adenosin und
-
5 eine grafische Darstellung der Häufigkeitsverteilung der auf der Grundlage des NMR-Spektrums der 4B ermittelten Adenosin-Konzentration.
-
Die nachfolgende Erläuterung der Figuren ist als Ausführungsbeispiel des vorliegend beschriebenen Verfahrens zu verstehen, wobei auf einzelne Verfahrensschritte detaillierter eingegangen wird als auf andere.
-
Die
1 zeigt ein NMR-Spektrum einer als Referenzsubstanz dienenden Substanz A. Das Spektrum zeigt die über die chemische Verschiebung (gemessen in ppm) aufgetragene Intensität der einzelnen Signale. Das Spektrum der
1 weist fünf Linien oder Peaks P1, P2, P3, P4 und P5 auf. Jeder einzelne dieser Peaks ist charakteristisch für eine Molekülgruppe im Gesamtmolekül der Substanz A. Da die einzelnen Peaks des NMR-Spektrums der
1 gut voneinander getrennt sind, braucht keine besondere Linienseparation durchgeführt zu werden. Vielmehr können die Spektralwerte der einzelnen Peaks unmittelbar aus dem NMR-Spektrum herausgelesen werden. Jeder Spektralwert besteht aus einem Integralwert, der die Fläche unter den einzelnen Peaks angibt, und einem Positionswert, der die chemische Verschiebung des jeweils betrachteten Peaks angibt. Die nachfolgende Tabelle 1 listet die einzelnen Integralwerte und Positionswerte der fünf im NMR-Spektrum der Substanz A enthaltenen Spektralwerte auf. Tabelle 1: Integralwerte und Positionswerte der einzelnen im NMR-Spektrum der Substanz A enthaltenen Peaks
| Peak | Position | Integral | Breite |
| P1 P2 P3 P4 P5 | 0,151 0,200 0,251 0,362 0,481 | 0,826 1,019 1,249 1,178 0,789 | 0,0010 0,0011 0,0009 0,0009 0,0012 |
-
Wie bereits erläutert, gibt es bei n verschiedenen Peaks einer Substanz A(n2 – n)/2 relevante Integralverhältnisse und Peakabstände bzw. Abstandswerte. Die Abstandswerte können entweder (i) als direkte Differenzen von Resonanzfrequenzen oder (ii) als normierte Differenzen von Resonanzfrequenzen oder (iii) als Verhältnisse – unter Verwendung der in Relation zur Trägerfrequenz geschriebenen Resonanzfrequenzen – berechnet werden, je nachdem, welche Skalierung man verwendet.
-
Die 2 verdeutlicht die unterschiedlichen Berechnungsmethoden zur Bestimmung der Abstandswerte aus den Positionswerten der einzelnen Peaks eines NMR-Spektrums.
-
Während die Achse 1 eine Skalierung in Hertz (Hz) angibt, zeigt die Achse 2 eine Skalierung in ppm. Auf der Grundlage der Hertz-Skalierung können direkte Differenzen einzelner Resonanzfrequenzen oder Quotienten der Resonanzfrequenzen gebildet werden. Auf der Grundlage der ppm-Skala können normierte Differenzen von Resonanzfrequenzen ermittelt werden.
-
Die drei Möglichkeiten der Brechung der Abstandswerte sind nachfolgend nochmals durch entsprechende Formeln verdeutlicht:
- (i) direkte Differenzen (Achse 1): (T + Δvb) – (T + Δva) = Δvb – Δva = vb – va [Hz]
- (ii) normierte Differenzen (Achse 2):
- (iii) Abstand als Quotient (Achse 1):
-
Auf der Grundlage der Tabelle 1 werden nun zunächst alle möglichen Integralverhältnisse jeweils zweier Integralwerte gebildet. Das heißt, jeder Integralwert wird durch jeden anderen Integralwert dividiert. Die entsprechende Berechnungsgrundlage sowie die konkreten Ergebnisse für die Integralwerte der Tabelle 1 sind in der nachfolgenden Tabelle 2 dargestellt. Tabelle 2: Berechnungsgrundlage der Berechnung von Integralverhältniswerten aus Integralwerten und entsprechende Ergebnisse für die Integralwerte der Tabelle 1.
-
Nun werden ausgehend von den Positionswerten der Tabelle 1 die entsprechenden Abstände jeweils zweier Peaks als Peakabstände bzw. Abstandswerte berechnet. Dies wird nachfolgend beispielhaft gemäß der oben erläuterten zweiten Variante (Abstandswerte als normierte Differenzen von Resonanzfrequenzen) dargestellt. Tabelle 3: Berechnungsgrundlage für die Berechnung von Abstandswerten als normierte Differenz von Resonanzfrequenzen und Ergebnisse für die Positionswerte der Tabelle 1.
-
Alternativ können statt der normierten Differenzen der Resonanzfrequenzen auch Quotienten der einzelnen Resonanzfrequenzen eingesetzt werden, um die Abstände jeweils zweier Peaks zu berechnen. Die Berechnungsgrundlage hierfür sowie die entsprechenden Ergebnisse für die Positionswerte der Tabelle 1 sind in der nachfolgenden Tabelle 4 dargestellt. Tabelle 4: Berechnungsgrundlage für die Berechnung von Abstandswerten auf der Grundlage von Quotienten und konkrete Ergebnisse für die Positionswerte der Tabelle 1.
-
Die 3 zeigt das bereits in der 1 dargestellte NMR-Spektrum der Substanz A als NMR-Referenzspektrum (obere Kurve, gestrichelte Linie) sowie ein als Testspektrum bezeichnetes NMR-Spektrum einer Probe, die eine zu identifizierende Substanz enthält (untere Kurve, durchgezogene Linie). Gemäß den aus dem Stand der Technik bekannten manuellen Auswerteverfahren würde man die in dem NMR-Referenzspektrum der Substanz A enthaltenen Peaks durch optischen Vergleich im NMR-Spektrum der Probe mit der zu identifizierenden Substanz suchen. Im vorliegenden Fall wäre dies aufgrund der verhältnismäßig geringen Komplexität des NMR-Spektrums der Probe auch noch möglich.
-
Das vorliegende, beispielhaft illustrierte Verfahren geht jedoch einen anderen Weg. So erfolgt eine Zuordnung von im NMR-Spektrum der Probe enthaltenen Peaks zu entsprechenden Peaks im NMR-Referenzspektrum der Substanz A nicht durch einen Mustervergleich. Vielmehr werden die Integralverhältniswerte und die Abstandswerte der in den Tabellen 2 bis 4 dargestellten Matrizen miteinander verglichen. Zu diesem Zweck werden nun Integralverhältniswerte für alle möglichen Integralverhältnisse jeweils zweier Integralwerte des NMR-Spektrums der Probe berechnet. Außerdem werden sämtliche Abstandswerte jeweils zweier Peaks auf der Grundlage entsprechender Positionswerte für das NMR-Spektrum der Probe berechnet. Dies erfolgt auf die bereits oben für das NMR-Referenzspektrum der Substanz A dargestellte Art und Weise.
-
Anschließend werden die Integralverhältniswerte des NMR-Spektrums der Probe mit den Integralverhältniswerten des NMR-Referenzspektrums verglichen. Gleichermaßen werden die Abstandswerte des NMR-Spektrums der Probe mit den Abstandswerten des NMR-Referenzspektrums verglichen.
-
Die nachfolgende Tabelle 5 zeigt die derart berechneten Integralverhältniswerte des NMR-Spektrums der Probe, wobei diejenigen Integralverhältniswerte, die mit den Integralverhältniswerten des NMR-Referenzspektrums übereinstimmen, markiert sind. Tabelle 5: Integralverhältniswerte der Peaks des NMR-Spektrums der Probe mit markierten Übereinstimmungen zu Integralverhältniswerten des NMR-Referenzspektrums.
-
Eine Entsprechung eines Integralverhältniswertes des NMR-Spektrums der Probe mit einem entsprechenden Integralverhältniswert des NMR-Referenzspektrums wurde dann angenommen, wenn sich die jeweiligen Integralverhältniswerte um weniger als ±0,002 voneinander unterschieden. Die in der Tabelle 5 diagonal schraffiert markierten Werte stellen eine erste Untermenge aus den Integralverhältniswerten dar. Die nicht aussagekräftigen Werte, die aus einer Division durch sich selbst entstanden sind, sind in dieser Tabelle – wie auch in allen nachfolgenden Tabellen – durch eine waagerechte Schraffur markiert.
-
In der nachfolgenden Tabelle 6 sind alle möglichen Abstandswerte zwischen jeweils zwei Peaks auf der Grundlage der entsprechenden Positionswerte dieser Peaks des NMR-Spektrums der Probe dargestellt. Wie oben erläutert, kann die Berechnung der Abstandswerte auf der Grundlage von Differenzen oder Quotienten erfolgen. Abstandswerte des NMR-Spektrums der Probe, die Abstandswerten des NMR-Referenzspektrums der Substanz A entsprechen, sind wiederum markiert. Dabei wurde von einer Entsprechung ausgegangen, wenn sich die Abstandswerte des NMR-Spektrums der Probe um weniger als ±0,005 von den Abstandswerten des NMR-Referenzspektrums unterschieden. Tabelle 6: Abstandswerte zwischen jeweils zwei Peaks des NMR-Spektrums der Probe mit markierten Übereinstimmungen zu Abstandswerten des NMR-Referenzspektrums.
-
Die in der Tabelle 6 diagonal schraffiert markierten Abstandswerte stellen eine zweite Untermenge von Werten dar.
-
Vergleicht man nun die Werte der Tabelle 5 mit den Werten der Tabelle 6 und filtert nach all denjenigen Werten, die aus den gleichen Spektralwerten gebildet wurden und sowohl in der Tabelle 5 als auch in der Tabelle 6 markiert sind, erhält man das in der Tabelle 7 dargestellte Ergebnis. Tabelle 7: Visualisierung vom Werten der ersten Untermenge und der zweiten Untermenge, die aus den gleichen Spektralwerten gebildet wurden.
-
Die in der Tabelle 7 diagonal schraffiert markierten Werte stellen die Schnittmenge zwischen der ersten Untermenge und der zweiten Untermenge dar und können als dritte Untermenge bezeichnet werden. Löscht man nun alle nicht markierten Spalten und Zeilen aus der Tabelle 7, kann man die einzelnen Peaks des NMR-Spektrums der Probe unmittelbar mit den einzelnen Peaks des NMR-Referenzspektrums korrelieren. Diese Korrelation bzw. Zuordnung ist in der nachfolgenden Tabelle 8 dargestellt. Tabelle 8: Zuordnung der Peaks der Substanz A im NMR-Spektrum der Probe zu den Peaks der Substanz A im NMR-Referenzspektrum.
| Peaks der Substanz A | Peaks der Substanz A |
| im Referenzspektrum | im Testspektrum |
| P1 | P2' |
| P2 | P3' |
| P3 | P6' |
| P4 | P7' |
| P5 | P9' |
-
Im vorliegenden Ausführungsbeispiel eines Verfahrens zur Substanzidentifikation konnten also 100% der Peaks bzw. spektralen Werte der Substanz A im NMR-Referenzspektrum auch im NMR-Spektrum der Probe wiedergefunden werden. Damit kann eine eindeutige Identifikation eines Bestandteils der Probe als Substanz A erfolgen. Sofern die weitere Zusammensetzung der Probe aufgeklärt werden soll, müsste nun ein weiterer Durchlauf des zuvor beschriebenen Verfahrens erfolgen, wobei dann ein anderes NMR-Referenzspektrum zum Vergleich herangezogen würde.
-
Neben einer qualitativen Bestimmung der in der Probe enthaltenen Substanz als Substanz A ist zudem eine quantitative Aussage über die in der Probe enthaltene Konzentration der Substanz A auf der Grundlage des gemessenen NMR-Spektrums möglich.
-
Während zuvor jedem Peak der Referenzsubstanz Pi (i = 1, 2, ..., 5) ein Peak aus dem NMR-Spektrum der Probe Pj' zugeordnet wurde, lässt sich nun aus jedem einzelnen dieser zugeordneten Peaks die Konzentration Γ
A / i'j' (in mg/ml) der Substanz A im NMR-Spektrum der Probe über folgende Gleichung berechnen, wobei Γ
A / Ref (in mg/ml) die Konzentration der Substanz A im Referenzspektrum (Ref) ist:
-
Die 4A zeigt ein NMR-Spektrum einer Mischprobe B1, die aus sieben verschiedenen Einzelsubstanzen zusammengesetzt ist. Die Mischprobe B1 enthält dabei kein Adenosin. Die 4B zeigt ein NMR-Spektrum der gleichen Mischprobe B1, der jedoch als 8. Substanz 0,5 mg/ml Adenosin zugesetzt wurden. Die auf Adenosin zurückzuführenden Peaks sind schwarz dargestellt, während die auf die übrigen sieben Substanzen zurückzuführenden Peaks gestrichelt dargestellt sind. In beiden NMR-Spektren der 4 ist wiederum die Intensität über der chemischen Verschiebung (gemessen in ppm) aufgetragen.
-
Aus der Mischprobe B1 wurden die durch Adenosin hervorgerufenen Peaks mithilfe eines NMR-Referenzspektrums von Adenosin unter Durchführung des zuvor beschriebenen Substanzidentifizierungsverfahrens identifiziert. Aus jedem der Substanz Adenosin zugeordneten Peak wurde anschließend die Konzentration des Adenosins in der Mischprobe B1 bestimmt. Dazu wurde die zuvor erläuterte Formel verwendet, wobei die Adenosin-Konzentration der zur Erstellung des NMR-Referenzspektrums von Adenosin verwendeten Adenosin-Lösung bekannt war.
-
Die 5 zeigt die Häufigkeitsverteilung der auf diese Art und Weise ermittelten Adenosin-Konzentration in der Mischprobe B1. Dabei fällt auf, dass der am häufigsten ermittelte Messwert von 0,5075 mg/ml nur um 1,5% vom Sollwert von 0,5 mg/ml abwich. Das zeigt, dass eine quantitative Substanzidentifikation mit dem vorliegend beschriebenen Verfahren mit sehr hoher Genauigkeit möglich ist.
-
Um die Genauigkeit nicht nur für Adenosin ermitteln zu können, wurden sämtliche sieben Einzelsubstanzen der Mischprobe B1 mit dem zuvor beschriebenen Verfahren zunächst qualitativ und anschließend quantitativ bestimmt. Die entsprechenden Quantifizierungsergebnisse sind in der nachfolgenden Tabelle 9 dargestellt. Dabei bezeichnet die Angabe „Sollwert” die von der jeweiligen Substanz in der Mischprobe B1 tatsächlich enthaltene Konzentration. Diese wurde beim Zusammenstellen der Mischprobe B1 exakt ermittelt, um so Aussagen zur Genauigkeit des vorliegenden Quantifizierungsverfahrens treffen zu können. Tabelle 9: Quantifizierungsergebnisse aller in der Mischprobe B1 (ohne Adenosin) vorhandenen Einzelsubstanzen
| Substanz | Soll-Wert [mg/ml] | Mess-Wert [mg/ml] | Abweichung [%] |
| Adenosin | 0 | 0,000 | 0,00 |
| Leucin | 0,186 | 0,195 | 4,84 |
| Benzoat | 0,084 | 0,084 | 0,00 |
| Lactat | 0,375 | 0,403 | 7,47 |
| Uridin | 0,153 | 0,15 | 1,96 |
| Creatinin | 0,542 | 0,575 | 6,09 |
| Phenylalanin | 0,896 | 0,91 | 1,56 |
| Glucose | 0,893 | 0,852 | 4,59 |
Tabelle 10: Quantifizierungsergebnisse aller in der Mischprobe B1 (mit Adenosin) vorhandenen Einzelsubstanzen.
| Substanz | Soll-Wert [mg/ml] | Mess-Wert [mg/ml] | Abweichung [%] |
| Adenosin | 0,5 | 0,508 | 1,50 |
| Leucin | 0,186 | 0,195 | 4,84 |
| Benzoat | 0,084 | 0,084 | 0,00 |
| Lactat | 0,375 | 0,403 | 7,47 |
| Uridin | 0,153 | 0,15 | 1,96 |
| Creatinin | 0,542 | 0,575 | 6,09 |
| Phenylalanin | 0,896 | 0,91 | 1,56 |
| Glucose | 0,893 | 0,852 | 4,59 |
-
Um umfangreichere Aussagen zur Genauigkeit des vorliegend beschriebenen Quantifizierungsverfahrens treffen zu können, wurden zahlreiche Einzelsubstanzen als Lösungen mit einer Konzentration von jeweils 0,1 mg/ml bereitgestellt und NMR-spektroskopisch vermessen. Anschließend erfolgte ein Vergleich der auf diese Art und Weise ermittelten NMR-Spektren mit entsprechenden NMR-Referenzspektren der gleichen Substanzen anderer Konzentration. Der auf diese Art und Weise ermittelte Messwert wurde anschließend mit dem Sollwert verglichen, um einen Messfehler zu berechnen. Dabei fällt auf, dass der Messfehler regelmäßig im unteren einstelligen Prozentbereich liegt und stets deutlich unterhalb von 10% bleibt. Dies belegt die hohe Genauigkeit des vorliegend beschriebenen Quantifizierungsverfahrens. Tabelle 11: Quantifizierungsergebnisse verschiedener Einzelsubstanzen.
| | Soll-Wert | Mess-Wert | Unsicherheit | Abweichung |
| Substanz | [mg/ml] | [mg/ml] | [mg/ml] | [%] |
| Essigsäure | 0,1 | 0,1089 | 0,001 | 8,90 |
| Ameisensäure | 0,1 | 0,0977 | 0,001 | 2,30 |
| Propionsäure | 0,1 | 0,096 | 0,002 | 4,00 |
| Buttersäure | 0,1 | 0,099 | 0,0005 | 1,00 |
| iso-Buttersäure | 0,1 | 0,109 | 0,0005 | 9,00 |
| Valeriansäure | 0,1 | 0,105 | 0,0015 | 5,00 |
| iso-Valeriansäure | 0,1 | 0,091 | 0,0005 | 9,00 |
| Phenylessigsäure | 0,1 | 0,1043 | 0,0015 | 4,30 |
| Benzoesäure | 0,1 | 0,103 | 0,0005 | 3,00 |
| Ethanol | 0,1 | 0,096 | 0,001 | 4,00 |
| Methanol | 0,1 | 0,1033 | 0,001 | 3,30 |
| Butanol | 0,1 | 0,1 | 0,0025 | 0,00 |
| Propanol | 0,1 | 0,105 | 0,0025 | 5,00 |
| 2-Butanol | 0,1 | 0,0985 | 0,0005 | 1,50 |
| Glycerin | 0,1 | 0,1035 | 0,0015 | 3,50 |
| Aceton | 0,1 | 0,0911 | 0,0009 | 8,90 |
| Formaldehyd | 0,1 | 0,0991 | 0,001 | 0,90 |
| Acetaldehyd | 0,1 | 0,108 | 0,001 | 8,00 |
| Isopropanol | 0,1 | 0,102 | 0,003 | 2,00 |
| Bernsteinsäure | 0,1 | 0,1 | 0,001 | 0,00 |
| Citronensäure | 0,09 | 0,0975 | 0,001 | 8,33 |
| Glutamin | 0,1 | 0,101 | 0,001 | 1,00 |
| Alanin | 0,1 | 0,105 | 0,0015 | 5,00 |
| Valin | 0,1 | 0,109 | 0,001 | 9,00 |
| Glucose | 0,1 | 0,1033 | 0,0035 | 3,30 |
| Acetylcystein | 0,1 | 0,099 | 0,006 | 1,00 |
| 2-Furoesäure | 0,1 | 0,098 | 0,007 | 2,00 |
| Syringasäure | 0,1 | 0,1005 | 0,0005 | 0,50 |
| 4-Hydroxybenzaldehyd | 0,1 | 0,0985 | 0,0005 | 1,50 |
| 4-Hydroxybenzoesäure | 0,1 | 0,102 | 0,006 | 2,00 |
| Vanillinsäure | 0,1 | 0,098 | 0,001 | 2,00 |
| Indol | 0,1 | 0,1025 | 0,0025 | 2,50 |
| 2,6-Dimethoxyphenol | 0,1 | 0,099 | 0,0005 | 1,00 |
| Monomethylamin | 0,1 | 0,1006 | 0,001 | 0,60 |
| Dimethylamin | 0,1 | 0,103 | 0,001 | 3,00 |
| Trimethylamin | 0,1 | 0,0957 | 0,001 | 4,30 |
| Brenztraubensäure | 0,1 | 0,095 | 0,0005 | 5,00 |
-
Durch eine Kombination des Quantifizierungsverfahrens mit dem zuvor erläuterten Identifikationsverfahren ist es also möglich, nicht nur einzelne Substanzen eindeutig als bestimmte Substanzen identifizieren zu können, sondern auch präzise Aussagen über die Konzentration dieser Substanzen in einer Mischprobe treffen zu können.
-
Die vorliegend beispielhaft dargestellten Ausführungsbeispiele des beanspruchten Verfahrens können im Rahmen der obigen Erläuterungen beliebig variiert werden und sind nicht als Einschränkung des beanspruchten Gegenstandes zu verstehen.