CN115304613A - Preparation method of heterocyclic SHP2 inhibitor - Google Patents
Preparation method of heterocyclic SHP2 inhibitor Download PDFInfo
- Publication number
- CN115304613A CN115304613A CN202210491240.0A CN202210491240A CN115304613A CN 115304613 A CN115304613 A CN 115304613A CN 202210491240 A CN202210491240 A CN 202210491240A CN 115304613 A CN115304613 A CN 115304613A
- Authority
- CN
- China
- Prior art keywords
- compound
- formula
- salt
- solvate
- hydrate
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
- C07D491/107—Spiro-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description
技术领域technical field
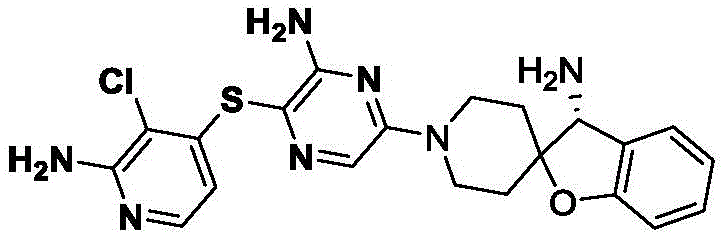
本发明属于医药化学领域,具体涉及(R)-6-氨基-2-(3-氨基-3H-螺[苯并呋喃-2,4’-哌啶]-1’-基)-3-甲基-5-((2-(三氟甲基)吡啶-3-基)硫基)嘧啶-4(3H)-酮或其盐、水合物、溶剂合物或结晶的制备方法。The invention belongs to the field of medicinal chemistry, in particular to (R)-6-amino-2-(3-amino-3H-spiro[benzofuran-2,4'-piperidin]-1'-yl)-3-methanol A method for producing yl-5-((2-(trifluoromethyl)pyridin-3-yl)thio)pyrimidin-4(3H)-one or a salt, hydrate, solvate or crystal thereof.
背景技术Background technique
SHP2磷酸酶是PTPN11基因编码的非受体PTP(protein tyrosine phosphatase)。其中包括了两个N端的SRC(肉瘤基因)同源结构域(SH2),一个PTP结构域和一个C端尾巴。X衍射结果表明SHP2通过N端的SH2与PTP结构域相互作用并阻断了ATP进入催化位点,该激酶处于一个自抑制的构型。一些可结合于SH2结构域的小肽或蛋白,可以激活该酶的磷酸化,促使癌症的发展。在细胞中,SHP2的功能与细胞质中下游的受体酪氨酸激酶有关,包括RAS-ERK,PI3K-AKT,JAK-STAT。首先,SHP2可结合于RAS并使RAS去磷酸化,从而增加效应蛋白RAF的作用而激活RAS/ERK/MAPK促进增殖的信号通路。其次,SHP2参与PD-1/PD-L1信号通路,并促进免疫逃逸。PD-1/SHP2/STAT1/T-bet信号轴介导了PD-1对Th1细胞的免疫抑制作用。因此,抑制PD-1或SHP2可以恢复Th1的免疫作用和T细胞的激活,解除肿瘤微环境中的免疫抑制。SHP2 phosphatase is a non-receptor PTP (protein tyrosine phosphatase) encoded by PTPN11 gene. It includes two N-terminal SRC (sarcoma gene) homology domains (SH2), a PTP domain and a C-terminal tail. The results of X-ray diffraction showed that SHP2 interacted with the PTP domain through the N-terminal SH2 and blocked ATP from entering the catalytic site. The kinase was in a self-inhibited configuration. Some small peptides or proteins that can bind to the SH2 domain can activate the phosphorylation of this enzyme and promote the development of cancer. In cells, the function of SHP2 is related to the downstream receptor tyrosine kinases in the cytoplasm, including RAS-ERK, PI3K-AKT, and JAK-STAT. First, SHP2 can bind to RAS and dephosphorylate RAS, thereby increasing the effect of the effector protein RAF and activating the RAS/ERK/MAPK signaling pathway to promote proliferation. Second, SHP2 participates in the PD-1/PD-L1 signaling pathway and promotes immune escape. The PD-1/SHP2/STAT1/T-bet signaling axis mediates the immunosuppressive effect of PD-1 on Th1 cells. Therefore, inhibition of PD-1 or SHP2 can restore the immune function of Th1 and the activation of T cells, and relieve the immune suppression in the tumor microenvironment.
SHP2与多种疾病的发生相关,如努南综合征(Noonan syndrome)、乳腺癌、黑色素瘤、胃癌、食道癌、肺癌、结肠癌、头癌、成神经细胞瘤、头颈的鳞状细胞癌、间变性大细胞淋巴瘤和成胶质细胞瘤等。SHP2 is associated with the occurrence of various diseases, such as Noonan syndrome, breast cancer, melanoma, gastric cancer, esophageal cancer, lung cancer, colon cancer, head cancer, neuroblastoma, squamous cell carcinoma of the head and neck, Anaplastic large cell lymphoma and glioblastoma.
针对其催化位点的抑制剂一般选择性和成药性都较差,近年来,研究者发现通过变构位点抑制SHP2的活性可以提高活性和选择性,药物研究也取得了一些进展。不过,仍然需要开发更优异的SHP2抑制剂,以便获得活性优、药代性质更好的药物,从而用于治疗SHP2介导的相关疾病。Inhibitors targeting its catalytic site are generally poor in selectivity and druggability. In recent years, researchers have found that inhibiting the activity of SHP2 through the allosteric site can improve the activity and selectivity, and some progress has been made in drug research. However, there is still a need to develop better SHP2 inhibitors in order to obtain drugs with superior activity and better pharmacokinetic properties for the treatment of SHP2-mediated related diseases.
发明内容Contents of the invention
本发明的发明人发现了一种杂环类SHP2抑制剂,该抑制剂的化合物结构如下式(I)所示,其化学名称为(R)-6-氨基-2-(3-氨基-3H-螺[苯并呋喃-2,4’-哌啶]-1’-基)-3-甲基-5-((2-(三氟甲基)吡啶-3-基)硫基)嘧啶-4(3H)-酮(以下简称“式(I)化合物”):The inventors of the present invention have discovered a heterocyclic SHP2 inhibitor, the compound structure of which is shown in the following formula (I), and its chemical name is (R)-6-amino-2-(3-amino-3H -Spiro[benzofuran-2,4'-piperidin]-1'-yl)-3-methyl-5-((2-(trifluoromethyl)pyridin-3-yl)thio)pyrimidine- 4(3H)-ketone (hereinafter referred to as "compound of formula (I)"):

本发明的发明人研究发现,式(I)化合物或其水合物、溶剂合物或结晶对SHP2表现出了显著的抑制活性,非常有希望成为SHP2相关疾病的治疗剂。The inventors of the present invention have found that the compound of formula (I) or its hydrate, solvate or crystal has significant inhibitory activity on SHP2, and is very promising as a therapeutic agent for SHP2-related diseases.
众所周知,对于人类用药,出于安全性因素要求,国内和国际管理机构对原料药(API)中未确证或毒性未定杂质的限度规定非常低。原料药中的杂质可能是由于自身的降解而产生的,也可能来源于制备方法,例如,包括未反应的起始原料、起始原料中包含的杂质的化学衍生物、合成副产物等。因此,需要对式(I)化合物或其衍生物的制备方法进行研究,以获得反应条件温和,工艺稳定,纯化容易,易于操作,有利于工业化大生产的制备式(I)化合物或其药学可接受的盐、异构体、溶剂合物或结晶的方法。It is well known that for human medicinal products, domestic and international regulatory agencies have very low limits for unidentified or undetermined toxic impurities in APIs due to safety concerns. Impurities in APIs may be produced due to their own degradation, or may originate from the preparation process, for example, including unreacted starting materials, chemical derivatives of impurities contained in starting materials, synthetic by-products, etc. Therefore, it is necessary to study the preparation method of the compound of formula (I) or its derivatives in order to obtain a compound of formula (I) or its pharmaceutically acceptable preparation with mild reaction conditions, stable process, easy purification, and easy operation, which is conducive to large-scale industrial production. Accepted salts, isomers, solvates or methods of crystallization.
本发明的一个目的是提供式(I)所示的化合物或其盐、水合物、溶剂合物或结晶的制备方法,包括使式(II)化合物在酸性试剂作用下脱除氨基保护基生成式(I)化合物的步骤,其中,R为氨基保护基,One object of the present invention is to provide a preparation method of the compound shown in formula (I) or its salt, hydrate, solvate or crystal, comprising making the compound of formula (II) remove the amino protecting group under the action of an acidic reagent to generate formula (1) the step of compound, wherein, R is an amino protecting group,

在一些优选的实施方案中,本发明提供的本发明的式(I)化合物或其盐、水合物、溶剂合物或结晶的制备方法,其中R选自烷基三硅烷基、芳基三硅烷基、烷基、烷基酰基、芳基酰基、烷基磺酰基、芳基磺酰基、烷氧基羰基、芳基氧基羰基、烷氧基和芳基氧基,所述烷基三硅烷基、芳基三硅烷基、烷基、烷基酰基、芳基酰基、烷基磺酰基、芳基磺酰基、烷氧基羰基、芳基氧基羰基、烷氧基和芳基氧基任选被一个或多个卤素、羟基、氨基、羧基、氰基、硝基、烷基取代;进一步优选地,R选自C1-10烷基三硅烷基、C6-10芳基三硅烷基、C1-10烷基、C1-10烷基酰基、C6-10芳基酰基、C1-6烷基磺酰基、C6-10芳基磺酰基、C1-6烷氧基羰基、C6-10芳基氧基羰基C1-6烷氧基和C6-10芳基氧基,所述烷基三硅烷基、芳基三硅烷基、烷基、烷基酰基、芳基酰基、烷基磺酰基、芳基磺酰基、烷氧基羰基、芳基氧基羰基、烷氧基和芳基氧基任选被一个或多个卤素、羟基、氨基、羧基、氰基、硝基、C1-6烷基取代;更进一步优选地,R选自三甲基硅基、三乙基硅基、三异丙基硅基、叔丁基二甲基硅基、叔丁基二苯基硅基、甲基、叔丁基、烯丙基、三苯甲基、苄基、甲氧基甲基、乙氧基乙基、2-四氢吡喃基(THP)、甲酰基、乙酰基、三氟乙酰基、苯甲酰基、甲磺酰基、乙磺酰基、丙基磺酰基、丁基磺酰基、叔丁基磺酰基、苯甲磺酰基、甲氧羰基、乙氧羰基、苄氧羰基、2-联苯基-2-丙氧羰基、笏甲氧羰基、苯氧羰基、叔丁氧羰基(t-butyloxy carbonyl,Boc)、甲氧基、乙氧基、苯氧基和三甲基硅基乙氧基;更进一步优选地,R选自三甲基硅基、三甲基硅基乙氧基、苄氧羰基、叔丁氧羰基、2-联苯基-2-丙氧羰基、笏甲氧羰基、叔丁基磺酰基、甲酰基、三氟乙酰基、三苯甲基和苄基。In some preferred embodiments, the present invention provides the preparation method of the compound of formula (I) or its salt, hydrate, solvate or crystal of the present invention, wherein R is selected from alkyl trisilyl, aryl trisilane group, alkyl, alkylacyl, arylacyl, alkylsulfonyl, arylsulfonyl, alkoxycarbonyl, aryloxycarbonyl, alkoxy and aryloxy, the alkyltrisilyl , aryltrisilyl, alkyl, alkylacyl, arylacyl, alkylsulfonyl, arylsulfonyl, alkoxycarbonyl, aryloxycarbonyl, alkoxy and aryloxy are optionally One or more halogen, hydroxyl, amino, carboxyl, cyano, nitro, alkyl substitution; more preferably, R is selected from C 1-10 alkyl trisilyl, C 6-10 aryl trisilyl, C 1-10 alkyl, C 1-10 alkyl acyl, C 6-10 aryl acyl, C 1-6 alkylsulfonyl, C 6-10 arylsulfonyl, C 1-6 alkoxycarbonyl, C 6-10 aryloxycarbonyl C 1-6 alkoxy and C 6-10 aryloxy, the alkyl trisilyl, aryl trisilyl, alkyl, alkyl acyl, aryl acyl, Alkylsulfonyl, arylsulfonyl, alkoxycarbonyl, aryloxycarbonyl, alkoxy and aryloxy are optionally replaced by one or more of halogen, hydroxy, amino, carboxyl, cyano, nitro, C 1-6 alkyl substitution; more preferably, R is selected from trimethylsilyl, triethylsilyl, triisopropylsilyl, tert-butyldimethylsilyl, tert-butyldiphenyl Silyl, methyl, tert-butyl, allyl, trityl, benzyl, methoxymethyl, ethoxyethyl, 2-tetrahydropyranyl (THP), formyl, acetyl , trifluoroacetyl, benzoyl, methanesulfonyl, ethylsulfonyl, propylsulfonyl, butylsulfonyl, tert-butylsulfonyl, phenylmethylsulfonyl, methoxycarbonyl, ethoxycarbonyl, benzyloxycarbonyl , 2-biphenyl-2-propoxycarbonyl, Wat methoxycarbonyl, phenoxycarbonyl, t-butyloxycarbonyl (t-butyloxycarbonyl, Boc), methoxy, ethoxy, phenoxy and trimethyl Silylethoxy; More preferably, R is selected from trimethylsilyl, trimethylsilylethoxy, benzyloxycarbonyl, tert-butoxycarbonyl, 2-biphenyl-2-propoxycarbonyl, Watt methoxycarbonyl, tert-butylsulfonyl, formyl, trifluoroacetyl, trityl and benzyl.
在一些优选的实施方案中,根据本发明的式(I)所示的化合物的制备方法,其进一步包括使式(II)化合物在酸性试剂作用下脱除氨基保护基生成式(I)化合物的盐,式(I)化合物的盐再与碱性试剂发生酸碱中和反应生成式(I)化合物的的步骤。In some preferred embodiments, according to the preparation method of the compound represented by the formula (I) of the present invention, it further comprises making the compound of the formula (II) remove the amino protecting group under the action of an acidic reagent to generate the compound of the formula (I) Salt, a step in which the salt of the compound of formula (I) reacts with an alkaline reagent to generate the compound of formula (I) through an acid-base neutralization reaction.
在一些优选的实施方案中,本发明提供本发明的式(I)化合物的制备方法,其中所述的酸性试剂为无机酸或有机酸;更进一步优选地,所述的酸性试剂选自盐酸、硫酸、甲磺酸、磷酸、HCl-乙醇和HCl-二氧六环;更进一步优选地,所述酸性试剂选自HCl-二氧六环。In some preferred embodiments, the present invention provides a method for preparing the compound of formula (I) of the present invention, wherein the acidic reagent is an inorganic acid or an organic acid; more preferably, the acidic reagent is selected from hydrochloric acid, Sulfuric acid, methanesulfonic acid, phosphoric acid, HCl-ethanol and HCl-dioxane; more preferably, the acidic reagent is selected from HCl-dioxane.
在一些优选的实施方案中,本发明提供本发明的式(I)化合物的制备方法,其中所述的式(I)化合物的盐为式(I)化合物的无机酸盐或有机酸盐;更进一步优选地,所述的式(I)化合物的盐选自式(I)化合物的盐酸盐、硫酸盐、甲磺酸盐和磷酸盐。In some preferred embodiments, the present invention provides a method for preparing the compound of formula (I) of the present invention, wherein the salt of the compound of formula (I) is an inorganic acid or organic acid salt of the compound of formula (I); more Further preferably, the salt of the compound of formula (I) is selected from hydrochloride, sulfate, methanesulfonate and phosphate of the compound of formula (I).
本发明的发明人对式(II)所示的化合物及酸性试剂的摩尔当量配比进行了进一步地考察,本发明的发明人发现酸性试剂摩尔当量偏低,物料转化不完全;酸性试剂摩尔当量偏高,对主反应进行没有明显的改善。综合数据,在一些优选的实施方案中,本发明提供本发明的式(I)化合物的制备方法,其中式(II)化合物与酸性试剂的摩尔比为约1:1至约1:10;更进一步优选地,式(II)化合物与酸性试剂的摩尔比为约1:4至约1:8;更进一步优选地,式(II)化合物与酸性试剂的摩尔比为约1:6。The inventor of the present invention further investigated the molar equivalent ratio of the compound shown in formula (II) and the acidic reagent, and the inventor of the present invention found that the molar equivalent of the acidic reagent was low, and the material conversion was incomplete; the molar equivalent of the acidic reagent On the high side, there is no significant improvement on the main reaction. Comprehensive data, in some preferred embodiments, the present invention provides the preparation method of the compound of formula (I) of the present invention, wherein the molar ratio of compound of formula (II) and acidic reagent is about 1:1 to about 1:10; More More preferably, the molar ratio of the compound of formula (II) to the acidic reagent is about 1:4 to about 1:8; even more preferably, the molar ratio of the compound of formula (II) to the acidic reagent is about 1:6.
在一些具体的实施方案中,本发明的式(I)化合物或其盐、水合物、溶剂合物或结晶的制备方法,其进一步包括使式(III)化合物与式(IV)化合物发生反应生成式(II)化合物的步骤,其中X为离去基团,R为氨基保护基,In some specific embodiments, the preparation method of the compound of formula (I) or its salt, hydrate, solvate or crystal of the present invention further comprises reacting the compound of formula (III) with the compound of formula (IV) to form The step of the compound of formula (II), wherein X is a leaving group, R is an amino protecting group,

在一些具体的实施方案中,本发明提供本发明的式(II)化合物的制备方法,其中所述离去基团X选自卤素、羟基、烷氧基、酰氧基、芳氧基、杂芳氧基、磺酰氧基、任选取代的烷基磺酰氧基、任选取代的烯基磺酰氧基、任选取代的芳基磺酰氧基、酰基和羟基的活性酯;优选地,X选自卤素和羟基的活性酯;进一步优选地,X选自氟、氯、溴、碘、羧酸酯基、磺酸酯基、磷酸酯基和硼酸酯基。在一些具体的实施方案中,所述离去基团X选自氟、氯、溴、碘、甲磺酸酯基、三氟甲磺酸酯基、苯磺酸酯基、甲苯磺酸酯基、对溴苯磺酸酯基和对硝基苯磺酸酯基。In some specific embodiments, the present invention provides a method for preparing the compound of formula (II) of the present invention, wherein the leaving group X is selected from halogen, hydroxyl, alkoxy, acyloxy, aryloxy, hetero Active esters of aryloxy, sulfonyloxy, optionally substituted alkylsulfonyloxy, optionally substituted alkenylsulfonyloxy, optionally substituted arylsulfonyloxy, acyl and hydroxy; preferably Preferably, X is selected from active esters of halogen and hydroxyl; further preferably, X is selected from fluorine, chlorine, bromine, iodine, carboxylate, sulfonate, phosphate and borate. In some specific embodiments, the leaving group X is selected from fluorine, chlorine, bromine, iodine, mesylate, triflate, benzenesulfonate, tosylate , brosylate group and p-nitrobenzenesulfonate group.
在一些具体的实施方案中,本发明提供本发明的式(II)化合物的制备方法,其中所述的制备方法包括催化剂,优选地,所述催化剂为酸性试剂;进一步优选地,所述催化剂为有机酸;更进一步优选地,所述催化剂选自甲酸、乙酸、盐酸、磷酸、磷酸二氢钠、二水磷酸二氢钠、HCl-乙醇和HCl-二氧六环;更进一步优选地,所述催化剂选自乙酸。In some specific embodiments, the present invention provides a method for preparing the compound of formula (II) of the present invention, wherein the preparation method includes a catalyst, preferably, the catalyst is an acidic reagent; more preferably, the catalyst is Organic acid; more preferably, the catalyst is selected from formic acid, acetic acid, hydrochloric acid, phosphoric acid, sodium dihydrogen phosphate, sodium dihydrogen phosphate dihydrate, HCl-ethanol and HCl-dioxane; still more preferably, the The catalyst is selected from acetic acid.
本发明的发明人发现使式(III)化合物与式(IV)化合物发生反应生成式(II)化合物的步骤中,溶剂对该反应影响较大,对反应溶剂进行了筛选,见表1。在一些优选的实施方案中,本发明提供本发明的式(II)化合物的制备方法,其中反应溶剂为高沸点溶剂,进一步优选地,所述反应溶剂选自乙腈、乙酸乙酯、四氢呋喃、N,N-二甲基甲酰胺和二氧六环;更进一步优选地,所述反应溶剂选自二氧六环。The inventors of the present invention found that in the step of reacting the compound of formula (III) with the compound of formula (IV) to generate the compound of formula (II), the solvent had a great influence on the reaction, and the reaction solvent was screened, see Table 1. In some preferred embodiments, the present invention provides the preparation method of the compound of formula (II) of the present invention, wherein the reaction solvent is a high boiling point solvent, more preferably, the reaction solvent is selected from acetonitrile, ethyl acetate, tetrahydrofuran, N , N-dimethylformamide and dioxane; more preferably, the reaction solvent is selected from dioxane.
表1Table 1

在一些具体的实施方案中,本发明的式(I)化合物或其盐、水合物、溶剂合物或结晶的制备方法,其进一步包括使式(V)化合物或其盐与式(VI)化合物在碱性试剂的作用下发生反应生成式(IV)化合物的步骤,其中X为离去基团,R为氨基保护基,In some specific embodiments, the preparation method of the compound of formula (I) or its salt, hydrate, solvate or crystal of the present invention further comprises making the compound of formula (V) or its salt and the compound of formula (VI) Under the action of basic reagent, reaction occurs to generate the step of compound of formula (IV), wherein X is a leaving group, R is an amino protecting group,

在一些优选的实施方案中,本发明提供本发明的式(IV)化合物的制备方法,其中,式(V)化合物或其盐与式(VI)化合物在碱性试剂的作用下发生缩合或取代反应。In some preferred embodiments, the present invention provides the preparation method of the compound of formula (IV) of the present invention, wherein, the compound of formula (V) or its salt and the compound of formula (VI) are condensed or substituted under the action of basic reagent reaction.
本发明的发明人对碱性试剂的选择进行了考察,大量实验数据显示,式(V)化合物与式(VI)化合物在碱性试剂的作用下发生反应生成式(IV)化合物的步骤中,碱的种类对反应起关键作用,数据表明,有机碱要优于无机碱,部分实验数据见表2。在一些优选的实施方案中,本发明提供本发明的式(IV)化合物的制备方法,其中碱性试剂选自有机碱,进一步优选选自DBU(1,8-二氮杂二环[5.4.0]十一碳-7-烯)、DBN(1,5-二氮杂双环[4.3.0]壬-5-烯)、N,N-二异丙基乙胺、三乙胺、2,4,6-三甲基吡啶和N-甲基吗啉,更进一步优选为DBU(1,8-二氮杂二环[5.4.0]十一碳-7-烯)。The inventors of the present invention have investigated the selection of alkaline reagents, and a large number of experimental data show that in the step of reacting the compound of formula (V) with the compound of formula (VI) under the action of alkaline reagents to generate the compound of formula (IV), The type of base plays a key role in the reaction. The data show that organic bases are better than inorganic bases. Some experimental data are shown in Table 2. In some preferred embodiments, the present invention provides a method for preparing the compound of formula (IV) of the present invention, wherein the basic reagent is selected from organic bases, more preferably selected from DBU (1,8-diazabicyclo[5.4. 0]undec-7-ene), DBN(1,5-diazabicyclo[4.3.0]non-5-ene), N,N-diisopropylethylamine, triethylamine, 2, 4,6-collidine and N-methylmorpholine, more preferably DBU (1,8-diazabicyclo[5.4.0]undec-7-ene).
表2Table 2

在一些具体的实施方案中,本发明提供本发明的式(IV)化合物的制备方法,其进一步包含催化剂,所述催化剂优选选自苯并三氮唑-1-基氧基三(二甲氨基)磷鎓六氟磷酸盐(BOP)。In some specific embodiments, the present invention provides a method for preparing the compound of formula (IV) of the present invention, which further comprises a catalyst, preferably selected from the group consisting of benzotriazol-1-yloxytris(dimethylamino) ) Phosphonium hexafluorophosphate (BOP).
本发明的发明人发现式(V)化合物与式(VI)化合物的反应,反应机理较为复杂,其中催化剂不仅可以活化底物催化主反应,同时也可以加速副产物的生成,本发明的发明人发现当分批加入催化剂BOP的时候,能够较好的控制主反应,抑制副反应。在一些具体的实施方案中,本发明提供本发明的式(IV)化合物的制备方法,其中BOP采用分批次加入的方法。The inventors of the present invention have found that the reaction mechanism between the compound of formula (V) and the compound of formula (VI) is relatively complicated, wherein the catalyst can not only activate the substrate to catalyze the main reaction, but also accelerate the formation of by-products. The inventor of the present invention It is found that when the catalyst BOP is added in batches, the main reaction can be better controlled and the side reaction can be suppressed. In some specific embodiments, the present invention provides a method for preparing the compound of formula (IV) of the present invention, wherein BOP is added in batches.
此外,物料配比对反应进程起关键作用,在结合投料方式后,本发明的发明人对物料配比进行了筛选,本发明的发明人发现当式(VI)化合物的配比过高,会与催化剂产生杂质,配比过低,式(V)化合物残留增大。因此,在一些优选的实施方案中,本发明提供本发明的式(IV)化合物的制备方法,其中式(V)化合物与式(VI)化合物的摩尔比选自1:1。根据数据可知,催化体系BOP和DBU,当DBU配比偏低,起始物料会有较大残留,同时副产物产生也会减少,当BOP配比偏低,物料转化不完全,当BOP配比偏高,对主反应影响有限,但是副产物增加。因此,在一些优选的实施方案中,本发明提供本发明的式(IV)化合物的制备方法,其中BOP和DBU的摩尔比优选选自1.5:5至2.0:5。In addition, the ratio of materials plays a key role in the reaction process. After combining the feeding method, the inventors of the present invention screened the ratio of materials. The inventors of the present invention found that when the ratio of the compound of formula (VI) is too high, it will Impurities are produced with the catalyst, the proportioning ratio is too low, and the residue of the compound of formula (V) increases. Therefore, in some preferred embodiments, the present invention provides a preparation method of the compound of formula (IV) of the present invention, wherein the molar ratio of the compound of formula (V) to the compound of formula (VI) is selected from 1:1. According to the data, the catalyst system BOP and DBU, when the ratio of DBU is low, there will be a large residue of starting materials, and the generation of by-products will also be reduced. When the ratio of BOP is low, the conversion of materials is incomplete. On the high side, the influence on the main reaction is limited, but the by-products increase. Therefore, in some preferred embodiments, the present invention provides a process for the preparation of the compound of formula (IV) of the present invention, wherein the molar ratio of BOP and DBU is preferably selected from 1.5:5 to 2.0:5.
在一些优选的实施方案中,本发明提供本发明的式(IV)化合物的制备方法,其中式(V)化合物与式(VI)化合物、BOP、DBU的摩尔比为约1:0.9:1:5至1:1.05:10:5,进一步优选为约1:1:1.5:5至1:1:6:5,更进一步优选为约1:1:1.5:5至1:1:2:5。In some preferred embodiments, the present invention provides the preparation method of compound of formula (IV) of the present invention, wherein the mol ratio of compound of formula (V) and compound of formula (VI), BOP, DBU is about 1:0.9:1: 5 to 1:1.05:10:5, more preferably about 1:1:1.5:5 to 1:1:6:5, more preferably about 1:1:1.5:5 to 1:1:2:5 .
在一些优选的实施方案中,本发明提供本发明的式(IV)化合物的制备方法,反应溶剂选自高沸点溶剂,优选选自乙腈、乙酸乙酯、四氢呋喃、N,N-二甲基甲酰胺、二氧六环、DMSO和DMF,更进一步地优选选自DMF。In some preferred embodiments, the present invention provides the preparation method of the compound of formula (IV) of the present invention, the reaction solvent is selected from high boiling point solvents, preferably selected from acetonitrile, ethyl acetate, tetrahydrofuran, N,N-dimethylformaldehyde Amide, dioxane, DMSO and DMF are further preferably selected from DMF.
在一些具体的实施方案中,本发明提供式(I)化合物或其盐、水合物、溶剂合物或结晶的制备方法,其中所述方法包括如下步骤:In some specific embodiments, the present invention provides a method for preparing a compound of formula (I) or a salt, hydrate, solvate or crystal thereof, wherein the method comprises the following steps:

1)式(V-1)化合物与式(VI-1)化合物在碱性试剂的作用下发生反应生成式(IV-1)化合物;1) The compound of formula (V-1) reacts with the compound of formula (VI-1) under the action of an alkaline reagent to generate the compound of formula (IV-1);
2)式(III)化合物与式(IV-1)化合物发生反应生成式(II-1)化合物;2) The compound of formula (III) reacts with the compound of formula (IV-1) to generate the compound of formula (II-1);
3)式(II-1)化合物在酸性试剂作用下发生反应生成式(I-1)化合物,式(I-1)化合物再与碱性试剂发生酸碱中和反应生成式(I)化合物;3) The compound of formula (II-1) reacts under the action of an acidic reagent to generate a compound of formula (I-1), and the compound of formula (I-1) reacts with an alkaline reagent to generate a compound of formula (I);
其中,x为1、2、3或4。Wherein, x is 1, 2, 3 or 4.
在一些优选的实施方案中,本发明提供式(I)化合物或其盐、水合物、溶剂合物或结晶的一种精制方法,其中所述方法包括将式(I)化合物或其盐、水合物、溶剂合物或结晶溶解在溶剂中,降温析晶;进一步优选地,本发明提供式(I)化合物或其盐、水合物、溶剂合物或结晶的一种精制方法,其中所述方法包括将式(I)化合物或其盐、水合物、溶剂合物或结晶溶解在溶剂中,加入不良溶剂析晶;其中所述溶剂优选选自极性较大的溶剂,进一步优选选自N,N-二甲基甲酰胺(DMF)和二甲基亚砜(DMSO);所述不良溶剂选自甲烷、乙烷、丙烷、丁烷、戊烷、庚烷、甲醇、乙醇和乙酸乙酯。In some preferred embodiments, the present invention provides a purification method of the compound of formula (I) or its salt, hydrate, solvate or crystallization, wherein said method comprises the compound of formula (I) or its salt, hydration Substances, solvates or crystals are dissolved in a solvent, and the temperature is reduced to crystallize; further preferably, the present invention provides a refining method for the compound of formula (I) or its salt, hydrate, solvate or crystals, wherein the method Including dissolving the compound of formula (I) or its salt, hydrate, solvate or crystal in a solvent, adding a poor solvent to crystallize; wherein the solvent is preferably selected from a more polar solvent, more preferably selected from N, N-dimethylformamide (DMF) and dimethylsulfoxide (DMSO); the poor solvent is selected from methane, ethane, propane, butane, pentane, heptane, methanol, ethanol and ethyl acetate.
本发明的发明人发现,本发明提供的式(I)化合物或其盐、水合物、溶剂合物或结晶的制备方法反应路线步骤少、操作简便、更加环境友好、有较高的收率及纯度,反应条件温和,纯化容易,工艺稳定,易于操作,能够满足工业规模的生产和应用。The inventors of the present invention have found that the preparation method of the compound of formula (I) provided by the present invention or its salt, hydrate, solvate or crystal has fewer steps in the reaction route, is easy to operate, is more environmentally friendly, has higher yield and Purity, mild reaction conditions, easy purification, stable process, easy operation, and can meet industrial scale production and application.
术语说明Glossary
除非另外定义,本文使用的所有技术和科学术语具有与本发明所属领域的普通技术人员通常理解的相同的含义。Unless defined otherwise, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which this invention belongs.
本发明的“盐”可以是任何盐,尤其是指药学上可接受的盐。在本文中,“药学上可接受的盐”是指本发明所述的化合物与本发明的“酸”或“酸性试剂”形成的药学上可接受的盐,本发明的“酸”或“酸性试剂”可选自盐酸、氢溴酸、磷酸、氨基磺酸、硝酸、对甲基苯磺酸、苯磺酸、对氨基苯磺酸、硫酸、乙酸、乙二酸、苯乙酸、丙酸、丙二酸、三氟乙酸、琥珀酸、乙醇酸、硬脂酸、抗坏血酸、双羟萘酸、羟基马来酸、谷氨酸、苯甲酸、水杨酸、2-乙酰氧基苯甲酸、反丁烯二酸、乙烷二磺酸、草酸、羟乙磺酸、柠檬酸、D-葡萄糖酸、乳酸、L-苹果酸、琥珀酸、L-酒石酸、富马酸、α-酮戊二酸、马尿酸、马来酸、D-酒石酸、甲烷磺酸或其类似物。本发明化合物的“药学上可接受的盐”能由包含酸性或碱性部分的本发明化合物经常规的化学方法合成,通常,碱性化合物的盐类能通过例子交换色谱法制备,或将游离碱与化学计量或过量的所期望的成盐无机或有机酸在适宜的溶剂或溶剂的各种组合中进行反应来制备。类似的,酸性化合物的盐类可通过与适宜的无机或有机碱进行反应来形成。The "salt" of the present invention may be any salt, especially a pharmaceutically acceptable salt. Herein, "pharmaceutically acceptable salt" refers to a pharmaceutically acceptable salt formed by the compound of the present invention and the "acid" or "acidic agent" of the present invention, and the "acid" or "acidic agent" of the present invention The "reagent" may be selected from hydrochloric acid, hydrobromic acid, phosphoric acid, sulfamic acid, nitric acid, p-toluenesulfonic acid, benzenesulfonic acid, p-sulfanilic acid, sulfuric acid, acetic acid, oxalic acid, phenylacetic acid, propionic acid, Malonic acid, trifluoroacetic acid, succinic acid, glycolic acid, stearic acid, ascorbic acid, pamoic acid, hydroxymaleic acid, glutamic acid, benzoic acid, salicylic acid, 2-acetoxybenzoic acid, trans Butenedioic acid, ethanedisulfonic acid, oxalic acid, isethionic acid, citric acid, D-gluconic acid, lactic acid, L-malic acid, succinic acid, L-tartaric acid, fumaric acid, α-ketoglutaric acid , hippuric acid, maleic acid, D-tartaric acid, methanesulfonic acid or their analogs. "Pharmaceutically acceptable salts" of compounds of the present invention can be synthesized by conventional chemical methods from compounds of the present invention that contain acidic or basic moieties. Usually, salts of basic compounds can be prepared by example exchange chromatography, or the free Bases are prepared by reacting stoichiometric or excess amounts of the desired salt-forming inorganic or organic acid in a suitable solvent or various combinations of solvents. Similarly, salts of acidic compounds may be formed by reaction with a suitable inorganic or organic base.
本发明“碱性试剂”是指能够使羟基或氨基去质子化的化合物。碱的实例包括但不限于,与醇溶剂组合的(C1-6烷基)氧化物((C1-6烷基)OM),其中(C1-6烷基)氧化物包括但不限于MeO-、EtO-、n-PrO-、i-PrO-、t-BuO-、i-AmO-(异戊氧基)等,且其中M是碱金属阳离子,例如Li+、Na+、K+等。醇溶剂包括(C1-6烷基)OH,例如,诸如甲醇、乙醇、正丙醇、异丙醇、叔丁醇、异戊醇等。还可以使用非烷氧基碱,例如氢氧化钠、氢氧化钾、氢化钠、六甲基二甲硅基胺钠、六甲基二甲硅基胺锂、二异丙基酰胺锂、氢化钙、碳酸钠、碳酸氢钠、碳酸钾、碳酸氢钾、碳酸铯、DBU(1,8-二氮杂二环[5.4.0]十一碳-7-烯)、DBN(1,5-二氮杂双环[4.3.0]壬-5-烯)、格氏试剂例如(C1-6烷基)Mg(卤素),其包括但不限于甲基氯化镁、甲基溴化镁、叔丁基氯化镁、叔丁基溴化镁等。The "basic reagent" in the present invention refers to a compound capable of deprotonating a hydroxyl group or an amino group. Examples of bases include, but are not limited to, (C 1-6 alkyl)oxides ((C 1-6 alkyl)OM) in combination with alcohol solvents, where (C 1-6 alkyl)oxides include, but are not limited to MeO-, EtO-, n-PrO-, i-PrO-, t-BuO-, i-AmO-(isoamyloxy), etc., and wherein M is an alkali metal cation, such as Li + , Na + , K + Wait. Alcoholic solvents include (C 1-6 alkyl)OH such as, for example, methanol, ethanol, n-propanol, isopropanol, tert-butanol, isoamyl alcohol, and the like. Non-alkoxy bases such as sodium hydroxide, potassium hydroxide, sodium hydride, sodium hexamethyldisilazide, lithium hexamethyldisilazide, lithium diisopropylamide, calcium hydride can also be used , sodium carbonate, sodium bicarbonate, potassium carbonate, potassium bicarbonate, cesium carbonate, DBU (1,8-diazabicyclo [5.4.0] undec-7-ene), DBN (1,5-di Azabicyclo[4.3.0]non-5-ene), Grignard reagents such as (C 1-6 alkyl)Mg(halogen), which include but not limited to methylmagnesium chloride, methylmagnesium bromide, tert-butyl Magnesium chloride, tert-butylmagnesium bromide, etc.
术语“溶剂合物”是指通过与溶剂分子配位形成固态或液态的配合物的本发明化合物的形式。水合物是溶剂合物的特殊形式,其中与水发生配位。在本发明范围内,溶剂合物优选是水合物。The term "solvate" refers to a form of a compound of the present invention that forms a solid or liquid complex by coordination with solvent molecules. Hydrates are a special form of solvates in which coordination with water occurs. Within the scope of the present invention, solvates are preferably hydrates.
术语“结晶”是指本发明所述的化合物形成的各种固体形态,包括晶型、无定形。The term "crystal" refers to various solid forms formed by the compounds of the present invention, including crystalline forms and amorphous forms.
本发明化合物中的“氢”、“碳”、“氧”包括其所有同位素。同位素应理解为包括具有相同原子数但具有不同质量数的那些原子。举例来说,氢的同位素包括氕、氚和氘,碳的同位素包括13C和14C,氧的同位素包括16O和18O等。"Hydrogen", "carbon", and "oxygen" in the compounds of the present invention include all isotopes thereof. Isotopes are understood to include those atoms having the same atomic number but different mass numbers. For example, isotopes of hydrogen include protium, tritium and deuterium, isotopes of carbon include 13 C and 14 C, and isotopes of oxygen include 16 O and 18 O, etc.
具体实施方式Detailed ways
下面代表性的实施例是为了更好地说明本发明,而非用于限制本发明的保护范围。以下实施例中使用的材料如无特殊说明均为商购获得。The following representative examples are for better illustrating the present invention, but not for limiting the protection scope of the present invention. Materials used in the following examples are commercially available unless otherwise specified.
实施例1:(R)-6-氨基-2-(3-氨基-3H-螺[苯并呋喃-2,4'-哌啶]-1'-基)-3-甲基-5-((2-(三氟甲基)吡啶-3-基)硫基)嘧啶-4(3H)-酮的制备Example 1: (R)-6-amino-2-(3-amino-3H-spiro[benzofuran-2,4'-piperidin]-1'-yl)-3-methyl-5-( Preparation of (2-(trifluoromethyl)pyridin-3-yl)thio)pyrimidin-4(3H)-one

步骤1:(R)-N-((R)-1'-(4-氨基-5-溴-1-甲基-6-氧代-1,6-二氢嘧啶-2-基)-3H-螺[苯并呋喃-2,4'-哌啶]-3-基)-2-甲基丙烷-2-亚磺酰胺的制备Step 1: (R)-N-((R)-1'-(4-amino-5-bromo-1-methyl-6-oxo-1,6-dihydropyrimidin-2-yl)-3H - Preparation of spiro[benzofuran-2,4'-piperidin]-3-yl)-2-methylpropane-2-sulfinamide

在50L玻璃反应釜中加入N,N-二甲基甲酰胺(DMF,9.45kg),室温下开启搅拌,然后加入(R)-2-甲基-N-((R)-3H-螺[苯并呋喃-2,4'-哌啶]-3-基)丙烷-2-亚磺酰胺(1.0kg,3.24mol)和6-氨基-5-溴-3-甲基嘧啶-2,4(1H,3H)-二酮(0.71kg,3.23mol),搅拌均匀。将1,8-二氮杂二环十一碳-7-烯(DBU,2.47kg,14.6mol)缓慢加入到反应釜中,将苯并三唑-1-三(三甲氨基)-六氟磷酸酯(BOP试剂,2.16kg,4.89mol)分成4份,每份0.4~0.6kg,加入至反应液,每隔10-15min加入一次。反应完毕,室温下向100L的玻璃反应釜中加入乙酸乙酯(15.0kg),开启搅拌,然后将上述反应液投入到搅拌的乙酸乙酯中,加入纯化水(50.0kg),搅拌10min。分出有机相,减压浓缩,干燥,得0.93kg标题化合物,收率56.2%Add N,N-dimethylformamide (DMF, 9.45kg) into a 50L glass reactor, start stirring at room temperature, and then add (R)-2-methyl-N-((R)-3H-spiro[ Benzofuran-2,4'-piperidin]-3-yl)propane-2-sulfinamide (1.0kg, 3.24mol) and 6-amino-5-bromo-3-methylpyrimidine-2,4( 1H,3H)-diketone (0.71kg, 3.23mol), stir well. 1,8-diazabicycloundec-7-ene (DBU, 2.47kg, 14.6mol) was slowly added to the reaction kettle, and benzotriazole-1-tris(trimethylamino)-hexafluorophosphoric acid The ester (BOP reagent, 2.16kg, 4.89mol) was divided into 4 parts, each 0.4-0.6kg, and added to the reaction solution every 10-15min. After the reaction was complete, ethyl acetate (15.0 kg) was added to a 100 L glass reactor at room temperature, and stirring was started, then the above reaction solution was put into the stirred ethyl acetate, purified water (50.0 kg) was added, and stirred for 10 min. The organic phase was separated, concentrated under reduced pressure, and dried to obtain 0.93kg of the title compound, yield 56.2%
步骤2:(R)-N-((R)-1'-(4-氨基-1-甲基-6-氧代-5-((2-(三氟甲基)吡啶-3-基)硫基)-1,6-二氢嘧啶-2-基)-3H-螺[苯并呋喃-2,4'-哌啶]-3-基)-2-甲基丙烷-2-亚磺酰胺的制备Step 2: (R)-N-((R)-1'-(4-amino-1-methyl-6-oxo-5-((2-(trifluoromethyl)pyridin-3-yl) Thio)-1,6-dihydropyrimidin-2-yl)-3H-spiro[benzofuran-2,4'-piperidin]-3-yl)-2-methylpropane-2-sulfinamide preparation of

取1,4-二氧六环(19.0kg)加入到50L玻璃反应釜中,开启搅拌,加入(R)-N-((R)-1'-(4-氨基-5-溴-1-甲基-6-氧代-1,6-二氢嘧啶-2-基)-3H-螺[苯并呋喃-2,4'-哌啶]-3-基)-2-甲基丙烷-2-亚磺酰胺(0.92kg,1.8mol)、2-(三氟甲基)哌啶-3-硫醇钠(0.51kg,2.52mol)和乙酸(0.11kg,1.8mol),升温,维持反应液温度60~70℃反应。将反应液抽滤,减压浓缩,得标题化合物粗品1.07kg,收率97.5%Take 1,4-dioxane (19.0kg) and add it to a 50L glass reactor, start stirring, add (R)-N-((R)-1'-(4-amino-5-bromo-1- Methyl-6-oxo-1,6-dihydropyrimidin-2-yl)-3H-spiro[benzofuran-2,4'-piperidin]-3-yl)-2-methylpropane-2 - sulfinamide (0.92kg, 1.8mol), 2-(trifluoromethyl)piperidine-3-thiolate sodium (0.51kg, 2.52mol) and acetic acid (0.11kg, 1.8mol), warming up, maintaining the reaction solution The reaction temperature is 60-70°C. The reaction solution was suction filtered and concentrated under reduced pressure to obtain 1.07 kg of the crude product of the title compound, with a yield of 97.5%.
步骤3:(R)-6-氨基-2-(3-氨基-3H-螺[苯并呋喃-2,4'-哌啶]-1'-基)-3-甲基-5-((2-(三氟甲基)吡啶-3-基)硫基)嘧啶-4(3H)-酮粗品的制备Step 3: (R)-6-amino-2-(3-amino-3H-spiro[benzofuran-2,4'-piperidin]-1'-yl)-3-methyl-5-(( Preparation of crude 2-(trifluoromethyl)pyridin-3-yl)thio)pyrimidin-4(3H)-one

称取二氯甲烷(30.0kg)加入到50L玻璃反应釜中,开启搅拌,加入(R)-N-((R)-1'-(4-氨基-1-甲基-6-氧代-5-((2-(三氟甲基)吡啶-3-基)硫基)-1,6-二氢嘧啶-2-基)-3H-螺[苯并呋喃-2,4'-哌啶]-3-基)-2-甲基丙烷-2-亚磺酰胺(1.07kg),降温,保持反应液温度15~25℃,滴加氯化氢-二氧六环溶液(2.63L),搅拌2-3小时。离心。Weigh dichloromethane (30.0kg) into a 50L glass reactor, start stirring, add (R)-N-((R)-1'-(4-amino-1-methyl-6-oxo- 5-((2-(trifluoromethyl)pyridin-3-yl)thio)-1,6-dihydropyrimidin-2-yl)-3H-spiro[benzofuran-2,4'-piperidine ]-3-yl)-2-methylpropane-2-sulfinamide (1.07kg), lower the temperature, keep the temperature of the reaction solution at 15-25°C, add hydrogen chloride-dioxane solution (2.63L) dropwise, and stir for 2 -3 hours. centrifugal.
然后在20~30℃下缓慢加NaOH水溶液(氢氧化钠0.3kg,纯化水0.6kg)调节体系pH8-9,继续搅拌2-4小时。将物料离心,纯化水淋洗,收集得到的游离碱固体转移至50L玻璃反应釜中,加入纯化水/无水乙醇(20:1)溶液(3.37kg),打浆。将物料离心,固体依次用纯化水和无水乙醇洗涤。干燥,得0.51kg标题化合物粗品,收率57.5%。Then slowly add NaOH aqueous solution (sodium hydroxide 0.3kg, purified water 0.6kg) at 20-30°C to adjust the pH of the system to 8-9, and continue stirring for 2-4 hours. The material was centrifuged, rinsed with purified water, and the collected free base solid was transferred to a 50L glass reactor, and purified water/absolute ethanol (20:1) solution (3.37kg) was added to make a slurry. The material was centrifuged, and the solid was washed successively with purified water and absolute ethanol. After drying, 0.51 kg of the crude product of the title compound was obtained, with a yield of 57.5%.
步骤4:(R)-6-氨基-2-(3-氨基-3H-螺[苯并呋喃-2,4'-哌啶]-1'-基)-3-甲基-5-((2-(三氟甲基)吡啶-3-基)硫基)嘧啶-4(3H)-酮精品的制备Step 4: (R)-6-amino-2-(3-amino-3H-spiro[benzofuran-2,4'-piperidin]-1'-yl)-3-methyl-5-(( Preparation of 2-(trifluoromethyl)pyridin-3-yl)thio)pyrimidin-4(3H)-one

在50L玻璃反应釜中加入二甲基亚砜(3.37kg),开启搅拌,然后加入上述反应中制得的粗品(0.51kg),升温至75~85℃搅拌溶清,保温0.5~1.0小时,滴加无水乙醇(1.58kg)析出晶体,关闭加热,自然降温至60℃左右时继续滴加无水乙醇(2.04kg),并控制反应温度在40~60℃。加入完毕后自然降至室温20~30℃继续搅拌析晶1-2小时。抽滤,用无水乙醇(0.76kg)洗涤固体,干燥,得0.37kg固体,收率72.5%。1H NMR(400MHz,DMSO-d6)δ8.40(d,J=4.0Hz,1H),7.52-7.50(m,1H),7.43(d,J=8.5Hz,1H),7.33(d,J=7.5Hz,1H),7.14(t,J=7.5Hz,1H),6.87(t,J=7.5Hz,1H),6.77(d,J=8.0Hz,1H),4.12(s,1H),3.63-3.53(m,2H),3.31-3.24(m,5H),2.09-2.03(m,1H),1.92-1.89(m,2H),1.86-1.82(m,2H),1.77-1.74(m,1H)。ESI-MS m/z:505.2[M+H]+。Add dimethyl sulfoxide (3.37kg) into a 50L glass reactor, start stirring, then add the crude product (0.51kg) obtained in the above reaction, heat up to 75-85°C, stir to dissolve, and keep warm for 0.5-1.0 hours. Add absolute ethanol (1.58kg) dropwise to precipitate crystals, turn off the heating, and continue to add absolute ethanol (2.04kg) dropwise when the temperature is naturally cooled to about 60°C, and control the reaction temperature at 40-60°C. After the addition, it was naturally lowered to room temperature at 20-30°C and continued to stir and crystallize for 1-2 hours. After suction filtration, the solid was washed with absolute ethanol (0.76 kg) and dried to obtain 0.37 kg of solid with a yield of 72.5%. 1 H NMR (400MHz, DMSO-d 6 )δ8.40(d, J=4.0Hz, 1H), 7.52-7.50(m, 1H), 7.43(d, J=8.5Hz, 1H), 7.33(d, J=7.5Hz, 1H), 7.14(t, J=7.5Hz, 1H), 6.87(t, J=7.5Hz, 1H), 6.77(d, J=8.0Hz, 1H), 4.12(s, 1H) ,3.63-3.53(m,2H),3.31-3.24(m,5H),2.09-2.03(m,1H),1.92-1.89(m,2H),1.86-1.82(m,2H),1.77-1.74( m, 1H). ESI-MS m/z: 505.2 [M+H] + .
比较例1Comparative example 1
参照WO2018/172984(PCT/IB2018/051973)中化合物45公开的方法制备下式代表的化合物(化合物A),并通过氢谱和质谱鉴定,The compound (compound A) represented by the following formula was prepared according to the method disclosed in compound 45 in WO2018/172984 (PCT/IB2018/051973), and was identified by hydrogen spectrum and mass spectrum,
实验例1:细胞增殖抑制实验Experimental Example 1: Cell Proliferation Inhibition Experiment
1.实验材料1. Experimental materials
受试化合物:本发明式(I)的化合物和比较例制备的化合物,每个化合物用DMSO配制成20mM。作用于NCI-H358细胞的化合物浓度依次为100μM、25μM、6.25μM、1.56μM、0.391μM、0.098μM、0.024μM、0.006μM、0.0015μM、0.00038μM;Test compound: the compound of formula (I) of the present invention and the compound prepared in the comparative example, each compound was prepared in 20 mM with DMSO. The concentrations of the compounds acting on NCI-H358 cells were 100 μM, 25 μM, 6.25 μM, 1.56 μM, 0.391 μM, 0.098 μM, 0.024 μM, 0.006 μM, 0.0015 μM, 0.00038 μM;
人非小细胞肺癌细胞NCI-H358购于美国典型培养物保藏中心(ATCC)。Human non-small cell lung cancer cells NCI-H358 were purchased from the American Type Culture Collection (ATCC).
试剂:CCK-8增殖抑制检测试剂盒,购自于中国江苏凯基生物技术股份有限公司仪器:CKX41倒置显微镜,购自于日本Olympus公司;多功能读板机,购自于美国MolecularDevices公司;细胞培养箱,购自于美国Thermo公司。Reagents: CCK-8 Proliferation Inhibition Detection Kit, purchased from China Jiangsu Kaiji Biotechnology Co., Ltd. Instrument: CKX41 Inverted Microscope, purchased from Olympus Company, Japan; Multi-functional plate reader, purchased from Molecular Devices Company, USA; The incubator was purchased from Thermo Company, USA.
2.实验方法2. Experimental method
2.1细胞培养:2.1 Cell culture:
细胞复苏:从液氮罐中取出NCI-H358细胞冻存管置于37℃水浴锅中,轻轻摇动使其尽快解冻。解冻后取出冻存管,用酒精棉球消毒后旋开盖子,吸出细胞液注入离心管,并加入1mL含血清的完全培养基,混匀后置于离心机中,1000rpm,离心5min。之后弃上清液,加入完全培养基反复吹打至细胞完全吹散、重悬。以适宜浓度接种于培养皿中。置37℃,5%CO2、95%潮湿空气的CO2培养箱中培养。Cell recovery: Take out the NCI-H358 cell cryopreservation tube from the liquid nitrogen tank and place it in a 37°C water bath, shake it gently to thaw as soon as possible. After thawing, take out the cryopreservation tube, unscrew the cap after disinfection with alcohol cotton ball, suck out the cell solution and pour it into the centrifuge tube, add 1mL of complete medium containing serum, mix well and place in a centrifuge at 1000rpm for 5min. Then discard the supernatant, add complete medium and pipette repeatedly until the cells are completely dispersed and resuspended. Inoculate in a petri dish at an appropriate concentration. Place them in a CO 2 incubator with 5% CO 2 and 95% humid air at 37°C.
细胞传代:细胞生长至约80-90%融合,吸弃原有培养液(1640培养基+10%FBS+1%青链霉素+1mM丙酮酸钠),加入1mL的PBS将残余培养基洗净后吸弃,加入1mL胰蛋白酶消化液,消化1-2min,镜下观察细胞伪足回缩变圆但细胞还未成片脱落,此时吸弃胰酶并用1-2mL完全培养基终止消化,轻轻吹打并收集细胞悬液,1000rpm,离心5min。去除上清,用完全培养基重悬细胞,按所需密度接种于培养皿中,置于37℃、5%CO2、95%潮湿空气的CO2培养箱中培养,视细胞生长情况每2-3天换一次培养液或进行传代。Cell passage: Cells grow to about 80-90% confluence, discard the original culture medium (1640 medium + 10% FBS + 1% penicillin + 1mM sodium pyruvate), add 1mL of PBS to wash the remaining medium After cleaning, aspirate and discard, add 1mL trypsin digestion solution, digest for 1-2min, observe under the microscope that the pseudopods of the cells retract and become round, but the cells have not fallen off in pieces, at this time, aspirate the trypsin and stop digestion with 1-2mL complete medium. Gently pipette and collect the cell suspension, centrifuge at 1000rpm for 5min. Remove the supernatant, resuspend the cells with complete medium, inoculate them in culture dishes according to the required density, and culture them in a CO 2 incubator at 37°C, 5% CO 2 , and 95% humid air. -Change the culture medium every 3 days or carry out subculture.
2.2实验步骤:2.2 Experimental steps:
NCI-H358细胞传代后用新鲜培养基(1640培养基+3%FBS+1%青链霉素+1mM丙酮酸钠)重悬,细胞计数之后按照1.5X104个细胞/mL的密度接种到96孔细胞培养板中,每孔加入100μL(即为1.5X103个细胞/孔)。24h后,在原有旧培养基的基础上,加入100μL含不同浓度(2×)药物的新鲜培养基。化合物终浓度分别为100μM、25μM、6.25μM、1.56μM、0.391μM、0.098μM、0.024μM、0.006μM、0.0015μM、0.00038μM,每个浓度组设置两个复孔。继续放入培养箱培养168h后吸弃孔内培养基,尽量吸干,加入100μL已加入CCK-8的培养基(CCK-8:培养基=1:10)。继续放入培养箱培养一定时间后,将96孔板从培养箱中取出,置于室温中平衡5min,置于多功能读板机中,检测450nm处的吸光度(OD值),并计算细胞增殖抑制率。计算公式为Inhibition(%)=100-(OD实验孔-OD空白孔)/(OD溶剂对照孔-OD空白孔)*100,根据不同药物浓度及其所对应的抑制率,使用GraghPad 5.0软件进行IC50曲线绘制,分析数据,得出最终IC50值。实验结果见表3。After subculture, NCI-H358 cells were resuspended with fresh medium (1640 medium + 3% FBS + 1% penicillin + 1mM sodium pyruvate), and after cell counting, inoculated to 96 cells at a density of 1.5X10 cells/mL. In a well cell culture plate, add 100 μL to each well (that is, 1.5×10 3 cells/well). After 24 hours, on the basis of the original old medium, 100 μL of fresh medium containing different concentrations (2×) of drugs was added. The final concentrations of the compounds were 100 μM, 25 μM, 6.25 μM, 1.56 μM, 0.391 μM, 0.098 μM, 0.024 μM, 0.006 μM, 0.0015 μM, 0.00038 μM, and two replicate wells were set for each concentration group. After continuing to culture in the incubator for 168 hours, discard the culture medium in the wells, blot as dry as possible, and add 100 μL of the medium to which CCK-8 has been added (CCK-8: medium = 1:10). After continuing to culture in the incubator for a certain period of time, take the 96-well plate out of the incubator, place it at room temperature for 5 minutes, place it in a multi-function plate reader, detect the absorbance (OD value) at 450nm, and calculate the cell proliferation Inhibition rate. The calculation formula is Inhibition (%)=100-(OD experimental well- OD blank well )/(OD solvent control well- OD blank well )*100, according to different drug concentrations and their corresponding inhibition rates, using GraghPad 5.0 software The IC 50 curve was drawn, and the data was analyzed to obtain the final IC 50 value. The experimental results are shown in Table 3.
表3table 3

从以上实验可以看出,本发明的式(I)化合物对NCI-H358细胞表现出了良好的抑制活性,非常有希望成为非小细胞肺癌治疗剂。It can be seen from the above experiments that the compound of formula (I) of the present invention exhibits good inhibitory activity on NCI-H358 cells, and is very promising as a therapeutic agent for non-small cell lung cancer.
实验例2药物动力学实验Experimental example 2 pharmacokinetic experiment
1.实验材料1. Experimental materials
化合物:本发明式(I)的化合物。药物溶媒为Captisol/50mM sodium acetate,pH4.6(10%/90%,w/v%)。口服药物配制成0.5mg/mL,透明溶液;静脉药物配置成0.1mg/mL,透明溶液。Compound: a compound of formula (I) of the present invention. The drug vehicle is Captisol/50mM sodium acetate, pH4.6 (10%/90%, w/v%). Oral drug is formulated as 0.5mg/mL, clear solution; intravenous drug is formulated as 0.1mg/mL, clear solution.
动物:雄性BALB/c小鼠,SPF级,购自上海西普尔-必凯实验动物有限公司;18-20g。实验前给予2~3天适应期。Animals: male BALB/c mice, SPF grade, purchased from Shanghai Xipro-Bicai Laboratory Animal Co., Ltd.; 18-20g. An adaptation period of 2 to 3 days was given before the experiment.
仪器:美国AB公司API4500型三重四级杆液质联用仪,配有电喷雾离子源(ESI),LC-30AD双泵;SIL-30AC自动进样器;CTO-30AC柱温箱;DGU-20A3R脱气机;AnalystQSA01.01色谱工作站;Milli-Q超纯水器(Millipore Inc);QilinbeierVortex-5振荡器;HITACHICF16RⅩⅡ台式高速冷冻离心机。Instrument: API4500 type triple quadrupole liquid mass spectrometer of American AB Company, equipped with electrospray ion source (ESI), LC-30AD double pump; SIL-30AC automatic sampler; CTO-30AC column thermostat; DGU- 20A3R degasser; AnalystQSA01.01 chromatographic workstation; Milli-Q ultrapure water device (Millipore Inc); Qilinbeier Vortex-5 oscillator; HITACHICF16RXⅡ desktop high-speed refrigerated centrifuge.
2.实验方法2. Experimental method
(1)每组3只小鼠。灌胃(I.G.)给予本发明式(I)的化合物10mg/kg,静脉(IV)给予本发明式(I)的化合物1mg/kg;(1) 3 mice in each group. Intragastric (I.G.) administration of 10 mg/kg of the compound of formula (I) of the present invention, intravenous (IV) administration of 1 mg/kg of the compound of formula (I) of the present invention;
(2)灌胃及静脉给药后于5min、15min、30min、1h、2h、6h、10h、24h自眼眶静脉丛采血于肝素化EP管(0.6ML)中,8000rpm/min离心5min后取上层血浆,-20℃冻存,LC-MS/MS分析;(2) Blood was collected from the orbital venous plexus at 5min, 15min, 30min, 1h, 2h, 6h, 10h, and 24h after gavage and intravenous administration, and was centrifuged at 8000rpm/min for 5min to take the upper layer Plasma, stored at -20°C, analyzed by LC-MS/MS;
(3)根据上述步骤所得的血药浓度数据绘制血药浓度-时间曲线图,采用WinNonlin软件求算药代动力学参数。实验结果见表4。(3) Draw a blood drug concentration-time curve according to the blood drug concentration data obtained in the above steps, and use WinNonlin software to calculate the pharmacokinetic parameters. The experimental results are shown in Table 4.
表4Table 4

使用实验例2的方法测定比较例的化合物A的药物动力学。实验结果显示化合物A的生物利用度(F)为51.2%,明显低于本发明的式(I)化合物。The pharmacokinetics of Compound A of Comparative Example was measured using the method of Experimental Example 2. The experimental results show that the bioavailability (F) of compound A is 51.2%, which is obviously lower than that of the compound of formula (I) of the present invention.
实验结果表明,本发明的化合物具有较好的半衰期T1/2、曲线下面积AUC、生物利用度F,口服吸收暴露好,适于成药。Experimental results show that the compound of the present invention has good half-life T 1/2 , area under the curve AUC, bioavailability F, good oral absorption and exposure, and is suitable for pharmaceutical preparation.
尽管以上已经对本发明作了详细描述,但是本领域技术人员理解,在不偏离本发明的精神和范围的前提下可以对本发明进行各种修改和改变。本发明的权利范围并不限于上文所作的详细描述,而应归属于权利要求书。Although the present invention has been described in detail above, it will be understood by those skilled in the art that various modifications and changes can be made to the present invention without departing from the spirit and scope of the present invention. The scope of rights of the present invention is not limited to the detailed description above, but should be attributed to the claims.
Claims (10)



Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2021105002520 | 2021-05-08 | ||
| CN202110500252 | 2021-05-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN115304613A true CN115304613A (en) | 2022-11-08 |
Family
ID=83855450
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202210491240.0A Pending CN115304613A (en) | 2021-05-08 | 2022-05-07 | Preparation method of heterocyclic SHP2 inhibitor |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN115304613A (en) |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023172940A1 (en) | 2022-03-08 | 2023-09-14 | Revolution Medicines, Inc. | Methods for treating immune refractory lung cancer |
| WO2023240263A1 (en) | 2022-06-10 | 2023-12-14 | Revolution Medicines, Inc. | Macrocyclic ras inhibitors |
| WO2024206858A1 (en) | 2023-03-30 | 2024-10-03 | Revolution Medicines, Inc. | Compositions for inducing ras gtp hydrolysis and uses thereof |
| WO2024211712A1 (en) | 2023-04-07 | 2024-10-10 | Revolution Medicines, Inc. | Condensed macrocyclic compounds as ras inhibitors |
| WO2024211663A1 (en) | 2023-04-07 | 2024-10-10 | Revolution Medicines, Inc. | Condensed macrocyclic compounds as ras inhibitors |
| WO2024216016A1 (en) | 2023-04-14 | 2024-10-17 | Revolution Medicines, Inc. | Crystalline forms of a ras inhibitor |
| WO2024216048A1 (en) | 2023-04-14 | 2024-10-17 | Revolution Medicines, Inc. | Crystalline forms of ras inhibitors, compositions containing the same, and methods of use thereof |
| WO2024229406A1 (en) | 2023-05-04 | 2024-11-07 | Revolution Medicines, Inc. | Combination therapy for a ras related disease or disorder |
| WO2025034702A1 (en) | 2023-08-07 | 2025-02-13 | Revolution Medicines, Inc. | Rmc-6291 for use in the treatment of ras protein-related disease or disorder |
| WO2025080946A2 (en) | 2023-10-12 | 2025-04-17 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2025171296A1 (en) | 2024-02-09 | 2025-08-14 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2025240847A1 (en) | 2024-05-17 | 2025-11-20 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2025255438A1 (en) | 2024-06-07 | 2025-12-11 | Revolution Medicines, Inc. | Methods of treating a ras protein-related disease or disorder |
| WO2025265060A1 (en) | 2024-06-21 | 2025-12-26 | Revolution Medicines, Inc. | Therapeutic compositions and methods for managing treatment-related effects |
| WO2026006747A1 (en) | 2024-06-28 | 2026-01-02 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2026015790A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026015801A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026015796A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026015825A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Use of ras inhibitor for treating pancreatic cancer |
| WO2026050446A1 (en) | 2024-08-29 | 2026-03-05 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2026072904A2 (en) | 2024-09-26 | 2026-04-02 | Revolution Medicines, Inc. | Compositions and methods for treating lung cancer |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020063760A1 (en) * | 2018-09-26 | 2020-04-02 | Jacobio Pharmaceuticals Co., Ltd. | Novel heterocyclic derivatives useful as shp2 inhibitors |
| WO2020072656A1 (en) * | 2018-10-03 | 2020-04-09 | Gilead Sciences, Inc. | Imidozopyrimidine derivatives |
| CN111138412A (en) * | 2018-11-06 | 2020-05-12 | 上海奕拓医药科技有限责任公司 | Spiro aromatic ring compound and application thereof |
| CN111153901A (en) * | 2018-11-07 | 2020-05-15 | 如东凌达生物医药科技有限公司 | A class of nitrogen-containing fused heterocyclic SHP2 inhibitor compounds, preparation method and use |
| WO2020108590A1 (en) * | 2018-11-30 | 2020-06-04 | 上海拓界生物医药科技有限公司 | Pyrimidine and five-membered nitrogen heterocycle derivative, preparation method therefor, and medical uses thereof |
| WO2020156242A1 (en) * | 2019-01-31 | 2020-08-06 | 贝达药业股份有限公司 | Shp2 inhibitor and application thereof |
| CN111704611A (en) * | 2019-07-25 | 2020-09-25 | 上海凌达生物医药有限公司 | Aryl spiro SHP2 inhibitor compound, preparation method and application |
| CN112166110A (en) * | 2018-03-21 | 2021-01-01 | 传达治疗有限公司 | SHP2 phosphatase inhibitors and methods of use |
| CN112778276A (en) * | 2019-11-08 | 2021-05-11 | 南京圣和药业股份有限公司 | Compound as SHP2 inhibitor and application thereof |
-
2022
- 2022-05-07 CN CN202210491240.0A patent/CN115304613A/en active Pending
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112166110A (en) * | 2018-03-21 | 2021-01-01 | 传达治疗有限公司 | SHP2 phosphatase inhibitors and methods of use |
| WO2020063760A1 (en) * | 2018-09-26 | 2020-04-02 | Jacobio Pharmaceuticals Co., Ltd. | Novel heterocyclic derivatives useful as shp2 inhibitors |
| WO2020072656A1 (en) * | 2018-10-03 | 2020-04-09 | Gilead Sciences, Inc. | Imidozopyrimidine derivatives |
| CN111138412A (en) * | 2018-11-06 | 2020-05-12 | 上海奕拓医药科技有限责任公司 | Spiro aromatic ring compound and application thereof |
| CN111153901A (en) * | 2018-11-07 | 2020-05-15 | 如东凌达生物医药科技有限公司 | A class of nitrogen-containing fused heterocyclic SHP2 inhibitor compounds, preparation method and use |
| WO2020108590A1 (en) * | 2018-11-30 | 2020-06-04 | 上海拓界生物医药科技有限公司 | Pyrimidine and five-membered nitrogen heterocycle derivative, preparation method therefor, and medical uses thereof |
| WO2020156242A1 (en) * | 2019-01-31 | 2020-08-06 | 贝达药业股份有限公司 | Shp2 inhibitor and application thereof |
| CN111704611A (en) * | 2019-07-25 | 2020-09-25 | 上海凌达生物医药有限公司 | Aryl spiro SHP2 inhibitor compound, preparation method and application |
| CN112778276A (en) * | 2019-11-08 | 2021-05-11 | 南京圣和药业股份有限公司 | Compound as SHP2 inhibitor and application thereof |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023172940A1 (en) | 2022-03-08 | 2023-09-14 | Revolution Medicines, Inc. | Methods for treating immune refractory lung cancer |
| WO2023240263A1 (en) | 2022-06-10 | 2023-12-14 | Revolution Medicines, Inc. | Macrocyclic ras inhibitors |
| WO2024206858A1 (en) | 2023-03-30 | 2024-10-03 | Revolution Medicines, Inc. | Compositions for inducing ras gtp hydrolysis and uses thereof |
| WO2024211712A1 (en) | 2023-04-07 | 2024-10-10 | Revolution Medicines, Inc. | Condensed macrocyclic compounds as ras inhibitors |
| WO2024211663A1 (en) | 2023-04-07 | 2024-10-10 | Revolution Medicines, Inc. | Condensed macrocyclic compounds as ras inhibitors |
| WO2024216016A1 (en) | 2023-04-14 | 2024-10-17 | Revolution Medicines, Inc. | Crystalline forms of a ras inhibitor |
| WO2024216048A1 (en) | 2023-04-14 | 2024-10-17 | Revolution Medicines, Inc. | Crystalline forms of ras inhibitors, compositions containing the same, and methods of use thereof |
| WO2024229406A1 (en) | 2023-05-04 | 2024-11-07 | Revolution Medicines, Inc. | Combination therapy for a ras related disease or disorder |
| WO2025034702A1 (en) | 2023-08-07 | 2025-02-13 | Revolution Medicines, Inc. | Rmc-6291 for use in the treatment of ras protein-related disease or disorder |
| WO2025080946A2 (en) | 2023-10-12 | 2025-04-17 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2025171296A1 (en) | 2024-02-09 | 2025-08-14 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2025240847A1 (en) | 2024-05-17 | 2025-11-20 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2025255438A1 (en) | 2024-06-07 | 2025-12-11 | Revolution Medicines, Inc. | Methods of treating a ras protein-related disease or disorder |
| WO2025265060A1 (en) | 2024-06-21 | 2025-12-26 | Revolution Medicines, Inc. | Therapeutic compositions and methods for managing treatment-related effects |
| WO2026006747A1 (en) | 2024-06-28 | 2026-01-02 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2026015790A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026015801A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026015796A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026015825A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Use of ras inhibitor for treating pancreatic cancer |
| WO2026050446A1 (en) | 2024-08-29 | 2026-03-05 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2026072904A2 (en) | 2024-09-26 | 2026-04-02 | Revolution Medicines, Inc. | Compositions and methods for treating lung cancer |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN115304613A (en) | Preparation method of heterocyclic SHP2 inhibitor | |
| US11866435B2 (en) | Heterocyclic compounds as immunomodulators | |
| CN115304612A (en) | Crystalline forms of a heterocyclic SHP2 inhibitor | |
| TW202128691A (en) | Kras mutein inhibitors | |
| CN106029659B (en) | Glutaminase inhibitors | |
| US10300058B2 (en) | Tyrosine kinase inhibitor and uses thereof | |
| TW202430184A (en) | Crystalline form of kras g12d inhibitors and preparation method thereof | |
| WO2020233641A1 (en) | Compound used as ret kinase inhibitor and application thereof | |
| JP2018511634A (en) | Maleate of B-RAF kinase, crystal form, preparation method and use thereof | |
| CN119654319A (en) | Polymorphs of a CDK inhibitor and its phosphate | |
| KR20180073682A (en) | Pyrimidine Derivatives and Their Use | |
| WO2020114388A1 (en) | Substituted pyrazolo[1,5-a]pyridine compound, composition containing the same and use thereof | |
| TW201718583A (en) | New epidermal growth factor receptor inhibitor and application thereof | |
| WO2025093060A1 (en) | 3-(indolamino)methylene indoline derivatives, preparation method therefor and use thereof | |
| WO2025093059A1 (en) | 3-(amino)methylene indoline derivatives, preparation method therefor and use thereof | |
| US11713309B2 (en) | Solid forms of Cerdulatinib | |
| CN109111447A (en) | 7- substituted azole triazin compounds or its pharmaceutically available salt, and its preparation method and application | |
| CN110028509B (en) | Pyrrolopyrimidines as selective JAK2 inhibitors, their synthesis methods and uses | |
| TW201245183A (en) | Solid forms of (S)-2-amino-3-(4-(2-amino-6-((R)-1-(4-chloro-2-(3-methyl-1H-pyrazol-1-yl)phenyl)-2,2,2-trifluoroethoxy)pyrimidin-4-yl)phenyl)propanoic acid | |
| WO2024026423A1 (en) | Substituted quinoline derivatives as pi3k inhibitors | |
| WO2019015463A1 (en) | Crystal form and salt form of n-phenyl-2-aminopyrimidine compound, and preparation method therefor | |
| CN102656172A (en) | 8-oxodihydropurine derivatives | |
| TW201536771A (en) | Bicyclic nitrogen-containing aromatic heterocyclic amide compound | |
| WO2025093058A1 (en) | 2-oxoindoline derivatives, preparation method therefor and use thereof | |
| CN110054616B (en) | Preparation method of triazine IDH inhibitor |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination |