CN112812129A - Novel crystalline form of midostaurin, process for its preparation and its use - Google Patents
Novel crystalline form of midostaurin, process for its preparation and its use Download PDFInfo
- Publication number
- CN112812129A CN112812129A CN202011614974.0A CN202011614974A CN112812129A CN 112812129 A CN112812129 A CN 112812129A CN 202011614974 A CN202011614974 A CN 202011614974A CN 112812129 A CN112812129 A CN 112812129A
- Authority
- CN
- China
- Prior art keywords
- midostaurin
- crystal form
- preparation
- ethanol
- volume ratio
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images






Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/22—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains four or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Steroid Compounds (AREA)
- Peptides Or Proteins (AREA)
Abstract
本发明涉及米哚妥林的新晶型A、B和C以及它们的制备方法和用途。所述晶型在物理化学稳定性和加工适应性方面均具有优异的性质;本发明结晶工艺简单、便于操作、可实现工业化生产。The present invention relates to new crystal forms A, B and C of midostaurin and their preparation methods and uses. The crystal form has excellent properties in terms of physical and chemical stability and processing adaptability; the crystallization process of the invention is simple, easy to operate, and can realize industrial production.
Description
Technical Field
The invention relates to the field of chemical pharmacy. More particularly, the present invention relates to a new crystalline form of midostaurin, to processes for the preparation of the new crystalline form and to their medical uses.
Technical Field
Protein kinase C (PKC is an abbreviation) is one of the key enzymes in the cell signal transduction pathway, and it has a key role in the control of cell proliferation and differentiation. PKC is a family of serine/threonine kinases.
Midostaurin shows high antiproliferative and antitumor activity and its highly selective and potent inhibition of PKC brings better clinical results for the patient, i.e. delays or inhibits the development of the disease, than equally tolerated treatment regimens, which is extremely useful for cancer treatment, especially for breast cancer, colon cancer, ovarian cancer and leukemia.
The drug midostaurin is used as an antineoplastic agent, and in general the preparation of midostaurin is known in the art. However, it is known that different crystalline forms of the same drug may differ substantially in some important properties of the drug, and there is therefore a continuing need for new solid forms of midostaurin and new processes for the preparation thereof.
The chemical name of midostaurin is: [ (9 α,10 β,11 β,13 α) -N- (2,3,10,11,12, 13-hexahydro-10-methoxy-9-methyl-1-oxo-9, 13-epoxy-1H, 9H-diindole [1',2',3' -gh:3,2,1-1m ] pyrrolo [3,4-j ] [1,7] benzodiazepin-11-yl ] -N-methylbenzamide

Midostaurin [ international nonprotected drug name ] is also known as N-benzoylstar bract or PKC 412.
Midostaurin is a naturally occurring derivative of the alkaloid starbracin and has been described in detail in european patent 0296110, us patent 5093330.
Form ii and the amorphous form of midostaurin are disclosed in CN 101048416A.
In CN102639538 there are disclosed crystalline form iii and crystalline form iv of midostaurin, the melting point of form iii is 206 ± 10 ℃ and contains about 3.2% of residual solvent or water, the melting point of form iv is 215 ± 10 ℃ and contains about 6.2% of residual solvent or water, the preparation process of both forms is GAS (GAS antisolvent) recrystallization.
WO2018165071A discloses midostaurin crystal forms v, vi, vii, viii, ix, x, xi, xii, xiii, xiv, xv, xvi, a total of 12 crystal forms, all solvates being ethyl acetate solvate, hydrate, methyl isobutyl ketone solvate, 4-heptanone solvate, methyl acetate solvate, acetone solvate, ethyl formate solvate, isopropyl acetate solvate, diethyl carbonate solvate, benzonitrile solvate, butyl acetate solvate, tert-butyl alcohol solvate, respectively.
The crystal form used by the currently marketed midostaurin preparation is the crystal form II, and according to the patent description, a seed crystal needs to be added in the preparation process to initiate crystallization, so that spontaneous crystallization is difficult to occur, and the preparation technology has limitations and is difficult to popularize. The preparation methods of the crystal form III and the crystal form IV are supercritical fluid methods, are suitable for laboratory research, and are difficult to realize industrial operation. The crystal forms V, VI, VII, VIII, IX, X, XI, XII, XIII, XIV, XV, XVI and the like are all solvates and are difficult to apply to preparations.
Disclosure of Invention
In view of the drawbacks of the prior art, it is an object of the present invention to provide crystalline form A, B and C of midostaurin which is chemically and physically stable, and which has excellent properties with respect to physical and chemical stability; and the preparation method of the crystal form is simple and convenient to operate and is easy for industrial production.
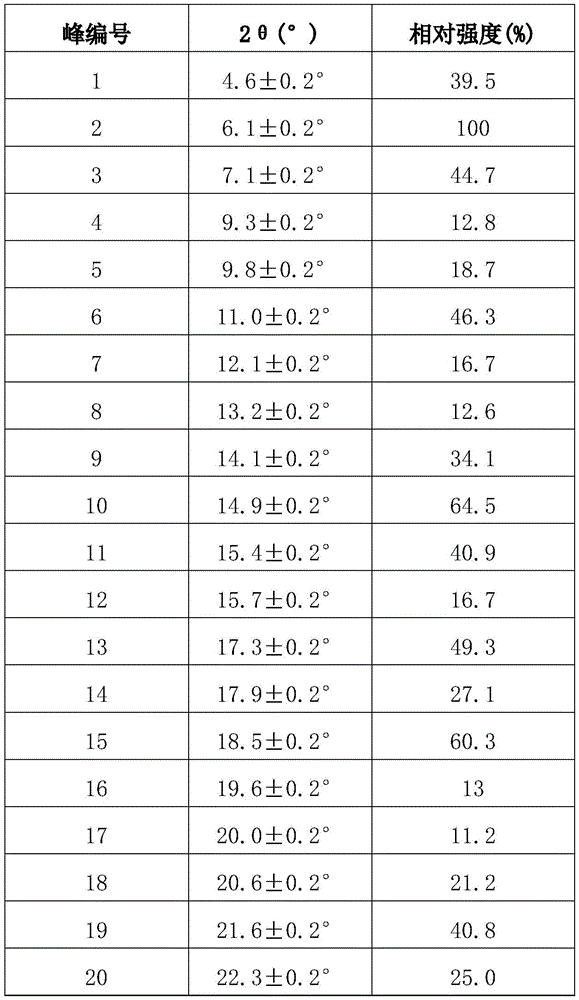
The X-ray powder diffraction (XRD) pattern of the midostaurin crystal form A has characteristic peaks at the following diffraction angles 2 theta: 4.6 +/-0.2 degrees, 6.1 +/-0.2 degrees, 7.1 +/-0.2 degrees, 11.0 +/-0.2 degrees, 14.1 +/-0.2 degrees, 14.9 +/-0.2 degrees, 15.4 +/-0.2 degrees, 17.3 +/-0.2 degrees, 18.5 +/-0.2 degrees and 21.6 +/-0.2 degrees.
Preferably, the X-ray powder diffraction pattern of crystalline form a of midostaurin according to the invention further has characteristic peaks at the following 2 Θ: 9.3 +/-0.2 degrees, 9.8 +/-0.2 degrees, 12.1 +/-0.2 degrees, 13.2 +/-0.2 degrees, 15.7 +/-0.2 degrees, 17.9 +/-0.2 degrees, 19.6 +/-0.2 degrees, 20.0 +/-0.2 degrees, 22.3 +/-0.2 degrees, 23.4 +/-0.2 degrees, 23.9 +/-0.2 degrees, 25.5 +/-0.2 degrees, 26.5 +/-0.2 degrees and 29.2 +/-0.2 degrees.
Preferably, crystalline form a of midostaurin according to the invention has substantially the same X-ray powder diffraction pattern as shown in figure 1.
The X-ray powder diffraction pattern has 2 θ and relative intensity data as shown in table 1 below:
TABLE 1

Preferably, the crystalline form a of midostaurin according to the invention has a melting point of 196 ℃ as measured by a melting point apparatus.
Preferably, crystalline form a of midostaurin according to the invention has substantially the same TGA profile as shown in figure 2.
Another object of the present invention is to provide a process for the preparation of crystalline form a of midostaurin comprising the steps of:
(1) dissolving midostaurin in benzyl alcohol, heating to 40-80 ℃ for dissolving, and filtering after dissolving;
(2) and (2) rapidly adding the poor solvent into the filtrate obtained in the step (1) under stirring, cooling to 0-20 ℃, stirring and crystallizing to obtain the midostaurin crystal form A.
Preferably, the weight to volume ratio (g/mL) of midostaurin to benzyl alcohol in step (1) is 1: 2-10.
Preferably, the poor solvent in the step (2) is a mixed solvent of ethanol and water, and the volume ratio (mL/mL) of the ethanol to the water is 1: 1.
Preferably, the volume ratio (mL/mL) of the benzyl alcohol in the step (1) to the poor solvent in the step (2) is 1: 6-50.
Preferably, the duration of the poor solvent addition in the step (2) is 5-30min, and the stirring crystallization time is 1-48 h.
The present invention also relates to a pharmaceutical composition containing crystalline form a of midostaurin comprising a therapeutically effective amount of crystalline form a of midostaurin, together with one or more pharmaceutically acceptable carriers.
The invention also relates to the use of a pharmaceutical composition containing crystalline form a of midostaurin and crystalline form a for the preparation of a medicament for the treatment of neoplastic diseases, preferably breast cancer, colon cancer, ovarian cancer or leukemia.
The X-ray powder diffraction (XRD) pattern of the midostaurin crystal form B has characteristic peaks at the following diffraction angles 2 theta: 4.9 +/-0.2 degrees, 6.5 +/-0.2 degrees, 7.3 +/-0.2 degrees, 12.0 +/-0.2 degrees, 12.9 +/-0.2 degrees, 14.6 +/-0.2 degrees, 15.7 +/-0.2 degrees, 17.5 +/-0.2 degrees, 18.7 +/-0.2 degrees, 20.6 +/-0.2 degrees and 21.9 +/-0.2 degrees
Preferably, crystalline form B of midostaurin according to the invention has substantially the same X-ray powder diffraction pattern as shown in figure 3.
The X-ray powder diffraction pattern has 2 θ and relative intensity data as shown in table 2 below:
TABLE 2


Preferably, the crystalline form B of midostaurin according to the invention has a melting point measured by a melting point apparatus of 198 ℃.
Preferably, crystalline form B of midostaurin according to the invention has substantially the same TGA profile as shown in figure 4.
Another object of the present invention is to provide a process for the preparation of crystalline form B of midostaurin, comprising the steps of:
(1) dissolving midostaurin in a mixed solution of acetic acid and ethanol, heating to 70-80 ℃ for dissolving, and filtering after dissolving;
(2) and (2) adding water into the filtrate obtained in the step (1) while stirring, and crystallizing to obtain the midostaurin crystal form B.
Preferably, the mass-to-volume ratio (g/mL) of the midostaurin to the mixed solution of acetic acid and ethanol in the step (1) is 1:50-100, and the volume ratio (mL/mL) of the acetic acid and the ethanol is 1: 15-30.
Preferably, the volume ratio (mL/mL) of the mixed solution of acetic acid and ethanol in the step (1) to the water in the step (2) is 1: 1-5.
Preferably, the crystallization temperature in the step (2) is 0-20 ℃, and the crystallization time is 1-12 h.
The invention also relates to a pharmaceutical composition containing crystalline form B of midostaurin comprising a therapeutically effective amount of crystalline form B of midostaurin, together with one or more pharmaceutically acceptable carriers.
The invention also relates to the use of a pharmaceutical composition containing crystalline form B of midostaurin and crystalline form B for the preparation of a medicament for the treatment of neoplastic diseases, preferably breast cancer, colon cancer, ovarian cancer or leukemia.
The X-ray powder diffraction (XRD) pattern of the midostaurin crystal form C has characteristic peaks at the following diffraction angles 2 theta: 5.4 +/-0.2 degrees, 5.9 +/-0.2 degrees, 6.3 +/-0.2 degrees, 6.9 +/-0.2 degrees, 7.4 +/-0.2 degrees, 8.6 +/-0.2 degrees, 12.4 +/-0.2 degrees, 16.9 +/-0.2 degrees and 18.5 +/-0.2 degrees.
Preferably, the X-ray powder diffraction pattern of crystalline form C of midostaurin according to the invention further has characteristic peaks at the following 2 Θ: 3.6 +/-0.2 degrees, 4.1 +/-0.2 degrees, 13.8 +/-0.2 degrees, 17.7 +/-0.2 degrees, 19.1 +/-0.2 degrees, 19.9 +/-0.2 degrees, 22.4 +/-0.2 degrees and 23.2 +/-0.2 degrees.
Preferably, crystalline form C of midostaurin according to the invention has substantially the same X-ray powder diffraction pattern as shown in figure 5.
The X-ray powder diffraction pattern has 2 θ and relative intensity data as shown in table 3 below:
TABLE 3
| Peak numbering | 2θ(°) | Relative Strength (%) |
| 1 | 3.6±0.2° | 1.4 |
| 2 | 4.1±0.2° | 2.6 |
| 3 | 5.4±0.2° | 2.0 |
| 4 | 5.9±0.2° | 60.9 |
| 5 | 6.3±0.2° | 5.8 |
| 6 | 6.9±0.2° | 100 |
| 7 | 7.4±0.2° | 1.8 |
| 8 | 8.6±0.2° | 14.0 |
| 9 | 12.4±0.2° | 13.2 |
| 10 | 13.8±0.2° | 11.5 |
| 11 | 16.9±0.2° | 18.9 |
| 12 | 17.7±0.2° | 9.0 |
| 13 | 18.5±0.2° | 16.5 |
| 14 | 19.1±0.2° | 12.5 |
| 15 | 19.9±0.2° | 8.8 |
| 16 | 22.4±0.2° | 11.6 |
| 17 | 23.2±0.2° | 13.3 |
Preferably, the crystalline form C of midostaurin according to the invention has a melting point of 127 ℃ measured with a melting point meter.
Preferably, crystalline form C of midostaurin according to the invention has substantially the same TGA profile as shown in figure 6.
Another object of the present invention is to provide a process for the preparation of crystalline form C of midostaurin comprising the steps of:
(1) dissolving midostaurin in benzyl alcohol, heating to 40-80 ℃ for dissolving, and filtering after dissolving;
(2) and (2) cooling the filtrate obtained in the step (1) to 0-10 ℃, slowly dropwise adding a poor solvent while stirring, and crystallizing to obtain the midostaurin crystal form C.
Preferably, the mass-to-volume ratio (g/mL) of midostaurin to benzyl alcohol in step (1) is 1: 2-10.
Preferably, the volume ratio (mL/mL) of the benzyl alcohol in the step (1) to the poor solvent in the step (2) is 1:6-50, the poor solvent in the step (2) is a mixed solution of ethanol and water, and the volume ratio (mL/mL) of the ethanol to the water is 1: 1.
Preferably, the crystallization temperature in the step (2) is 0-10 ℃, and the dripping duration of the poor solvent is 3-5 h; the crystallization time is 1-12 h.
The present invention also relates to a pharmaceutical composition containing crystalline form C of midostaurin comprising a therapeutically effective amount of crystalline form C of midostaurin, together with one or more pharmaceutically acceptable carriers.
The invention also relates to the use of a pharmaceutical composition containing crystalline form C of midostaurin, and crystalline form C, for the preparation of a medicament for the treatment of neoplastic diseases, preferably breast cancer, colon cancer, ovarian cancer or leukemia.
The inventor of the invention discovers a new crystal form A, a new crystal form B or a new crystal form C of midostaurin through a great deal of research, and the novel crystal form A, the novel crystal form B or the novel crystal form C of midostaurin has the advantages of simple crystallization process, convenient operation, small pollution, high yield and capability of realizing industrial production; the crystal form medicament provided by the invention has the advantages of high product purity, excellent physicochemical properties and good chemical stability.
Drawings
Figure 1 is an X-ray powder diffraction pattern of crystalline form a of midostaurin obtained in example 1.
Figure 2 is a TGA profile of crystalline form a of midostaurin obtained in example 1.
Figure 3 is an X-ray powder diffraction pattern of crystalline form B of midostaurin obtained in example 5.
Figure 4 is a TGA profile of crystalline form B of midostaurin obtained in example 5.
Figure 5 is an X-ray powder diffraction pattern of crystalline form C of midostaurin obtained in example 9.
Figure 6 is a TGA profile of crystalline form C of midostaurin obtained in example 9.
Detailed Description
The following examples are intended to further illustrate the present invention, but they are not intended to limit or restrict the scope of the invention.
The crude midostaurin used in the process of the present invention is commercially available or obtained by reacting starbractein with benzoyl chloride under alkaline conditions according to known methods, all prepared according to the reaction method of patent CN 106083830A.
The solvent used in the present invention is not particularly limited, and a commercially available conventional solvent can be used, and for example, the ethanol may be a commercially available ethanol including industrial ethanol, anhydrous ethanol, and the like.
Unless otherwise indicated, "stirring" as used herein in the method of the present invention may be performed by methods conventional in the art, for example, by means of stirring including magnetic stirring, mechanical stirring, at a stirring speed of 50-300rpm/min, preferably 100-200 rpm/min.
The X-ray powder diffraction instrument and the test conditions related by the invention are as follows: x-ray diffraction apparatus model Rigaku D/max-2200Cu target; the operation method comprises the following steps: the scanning speed is 4 degrees/min, and the scanning step width is 0.01 degrees.
The type of the melting point instrument related by the invention is as follows: optimal time MPA 100.
The thermogravimetric analyzer (TGA) and the test conditions related to the present invention are: TGA model PerkinElmer TGA 400; the test conditions are that the temperature rising rate is 10 ℃/min, and the temperature range is 30-300 ℃.
The purity detection conditions of the midostaurin HPLC related to the invention are as follows: a chromatographic column: waters, ACE PFP C184.6X 150mm, 3 μm; mobile phase A: 0.1% phosphoric acid aqueous solution, mobile phase B: acetonitrile: methanol 300:200, diluent: 75% acetonitrile; detection wavelength: 240 nm; the flow rate is 1.0 mL/min; sample introduction amount: 10 mu L of the solution; column temperature: 35 ℃; the detection method is shown in table 4:
TABLE 4
| Time(min) | A | B% | |
| 0 | 70 | 30 | |
| 30 | 20 | 80 | |
| 40 | 20 | 80 | |
| 40.1 | 70 | 30 | |
| 46 | 70 | 30 |
The detection conditions of soluble residues (GC) of midostaurin related by the invention are as follows: the instrument model is as follows: agilent 6890; the detection methods and parameters are shown in table 5:
TABLE 5

It should be emphasized that the values or numerical end-points referred to in the claims are not limited to the numbers per se, and those skilled in the art will appreciate that they include the allowable error ranges that are well accepted in the art, such as experimental errors, measurement errors, statistical errors, random errors, etc., and that such error ranges are included in the scope of the invention.
Example 1 preparation of crystalline form A of midostaurin
Dissolving 1.0g of crude midostaurin in 4mL of benzyl alcohol, heating to 80 ℃, dissolving, filtering, adding 50mL of poor solvent (the volume ratio of ethanol to water is 1:1) while stirring, adding within 10min, cooling to 0 ℃, stirring, crystallizing for 16h, filtering, and drying in vacuum at 40 ℃ to obtain 0.95g of crystals which are easy to filter and have the purity of 99.72% by HPLC (high performance liquid chromatography).
The crystal has an X-ray powder diffraction pattern as shown in figure 1 and a TGA pattern as shown in figure 2, and is named as midostaurin crystal form A in the invention.
Example 2 preparation of crystalline form A of midostaurin
Dissolving 0.5g of crude midostaurin in 1mL of benzyl alcohol, heating to 80 ℃, dissolving, filtering, adding 6mL of poor solvent (the volume ratio of ethanol to water is 1:1) while stirring, adding within 5min, cooling to 0 ℃, stirring, crystallizing for 48h, filtering, and drying in vacuum at 40 ℃ to obtain 0.47g of crystals which are easy to filter and have the purity of 99.71 percent by HPLC (high performance liquid chromatography). The crystalline form A of midostaurin is confirmed by measuring an X-ray powder diffraction pattern (XRD).
Example 3 preparation of crystalline form A of midostaurin
Dissolving 0.5g of crude midostaurin in 5mL of benzyl alcohol, heating to 40 ℃, dissolving, filtering, adding 250mL of poor solvent (the volume ratio of ethanol to water is 1:1) while stirring, adding within 30min, cooling to 20 ℃, stirring, crystallizing for 24h, filtering, and drying in vacuum at 40 ℃ to obtain 0.47g of crystals which are easy to filter and have the purity of 99.71 percent by HPLC (high performance liquid chromatography). The crystalline form A of midostaurin is confirmed by measuring an X-ray powder diffraction pattern (XRD).
Example 4 preparation of crystalline form A of midostaurin
Dissolving 0.5g of crude midostaurin in 2mL of benzyl alcohol, heating to 60 ℃, dissolving, filtering, adding 20mL of poor solvent (the volume ratio of ethanol to water is 1:1) while stirring, adding within 10min, cooling to 10 ℃, stirring, crystallizing for 20h, filtering, and drying in vacuum at 40 ℃ to obtain 0.46g of crystals which are easy to filter and have the purity of 99.69% by HPLC (high performance liquid chromatography). The crystalline form A of midostaurin is confirmed by measuring an X-ray powder diffraction pattern (XRD).
Example 5 preparation of crystalline form B of midostaurin
Dissolving 1.0g of crude midostaurin in 63mL of mixed solution of acetic acid and ethanol (the volume ratio of acetic acid to ethanol is 1:20), heating to 70 ℃, dissolving, filtering, adding 65mL of water while stirring, crystallizing at 20 ℃ for 12h, filtering, and drying in vacuum at 40 ℃ to obtain 0.95g of crystals which are easy to filter and have the purity of 99.62% by HPLC (high performance liquid chromatography).
The crystal has an X-ray powder diffraction pattern as shown in figure 3 and a TGA pattern as shown in figure 4, and is named as midostaurin crystal form B in the invention.
Example 6 preparation of crystalline form B of midostaurin
Dissolving 0.5g of crude midostaurin in 25mL of mixed solution of acetic acid and ethanol (the volume ratio of acetic acid to ethanol is 3:47), heating to 80 ℃, dissolving, filtering, adding 125mL of water while stirring, crystallizing for 4 hours at 0 ℃, filtering, and drying in vacuum at 40 ℃ to obtain 0.46g of crystals which are easy to filter and have the purity of 99.60 percent by HPLC (high performance liquid chromatography). The crystal form B is confirmed by measuring an X-ray powder diffraction pattern (XRD).
Example 7 preparation of crystalline form B of midostaurin
Dissolving 0.5g of crude midostaurin in 32mL of mixed solution of acetic acid and ethanol (the volume ratio of acetic acid to ethanol is 1:15), heating to 70 ℃, dissolving, filtering, adding 160mL of water while stirring, crystallizing for 10h at 10 ℃, filtering, and drying in vacuum at 40 ℃ to obtain 0.45g of crystals which are easy to filter and have the purity of 99.60% by HPLC (high performance liquid chromatography). The crystal form B is confirmed by measuring an X-ray powder diffraction pattern (XRD).
Example 8 preparation of crystalline form B of midostaurin
Dissolving 0.5g of crude midostaurin in 50mL of mixed solution of acetic acid and ethanol (the volume ratio of acetic acid to ethanol is 1:15), heating to 80 ℃, dissolving, filtering, adding 50mL of water while stirring, crystallizing for 8 hours at 0 ℃, filtering, and drying in vacuum at 40 ℃ to obtain 0.45g of crystals which are easy to filter and have the purity of 99.61% by HPLC (high performance liquid chromatography). The crystal form B is confirmed by measuring an X-ray powder diffraction pattern (XRD).
Example 9 preparation of crystalline form C of midostaurin
Dissolving 1.0g of crude midostaurin in 4mL of benzyl alcohol, heating to 70 ℃, dissolving, filtering, cooling the filtrate to 10 ℃, slowly dropwise adding 50mL of poor solvent (the volume ratio of ethanol to water is 1:1) while stirring, dropwise adding for about 4h, stirring and crystallizing for 3h at 10 ℃ after dropwise adding, filtering, and drying in vacuum at 40 ℃ to obtain 0.96g of crystals, wherein the crystals are easy to filter, and the purity is 99.80% by HPLC detection. The crystal has an X-ray powder diffraction pattern as shown in figure 5 and a TGA pattern as shown in figure 6, and is named as midostaurin crystal form C in the invention.
Example 10 preparation of crystalline form C of midostaurin
Dissolving 1.0g of crude midostaurin product in 2mL of benzyl alcohol, heating to 80 ℃, dissolving, filtering, cooling the filtrate to 0 ℃, slowly dropwise adding 12mL of poor solvent (the volume ratio of ethanol to water is 1:1) while stirring, dropwise adding after about 3h, stirring at 0 ℃ after dropwise adding for crystallization for 1h, filtering, and drying in vacuum at 40 ℃ to obtain 0.94g of crystals, wherein the crystals are easy to filter, and the purity is 99.74% by HPLC detection. The crystal form C is confirmed by measuring an X-ray powder diffraction pattern (XRD).
Example 11 preparation of crystalline form C of midostaurin
Dissolving 1.0g of crude midostaurin in 10mL of benzyl alcohol, heating to 40 ℃, dissolving, filtering, cooling the filtrate to 10 ℃, slowly dropwise adding 100mL of poor solvent (the volume ratio of ethanol to water is 1:1) while stirring, dropwise adding after 4h, stirring at 10 ℃ for crystallization for 10h after dropwise adding, filtering, and drying in vacuum at 40 ℃ to obtain 0.96g of crystals, wherein the crystals are easy to filter, and the purity is 99.76% by HPLC detection. The crystal form C is confirmed by measuring an X-ray powder diffraction pattern (XRD).
Example 12 preparation of crystalline form C of midostaurin
Dissolving 1.0g of crude midostaurin product in 3mL of benzyl alcohol, heating to 60 ℃, dissolving, filtering, cooling the filtrate to 0 ℃, slowly dropwise adding 150mL of poor solvent (the volume ratio of ethanol to water is 1:1) while stirring, dropwise adding after about 5h, stirring at 0 ℃ after dropwise adding for crystallization for 12h, filtering, and drying in vacuum at 40 ℃ to obtain 0.95g of crystals, wherein the crystals are easy to filter, and the purity is 99.77% by HPLC detection. The crystal form C is confirmed by measuring an X-ray powder diffraction pattern (XRD).
Preparation example
Reference is made to patent CN101048416A, example 3 for the preparation of midostaurin amorphous form, example 5 for the preparation of midostaurin crystalline form II; reference is made to patent CN102639538A, example 1 for the preparation of crystalline midostaurin form iii, example 2 for the preparation of crystalline midostaurin form iv; reference is made to patent WO2018165071A for preparation of crystalline midostaurin form V by the process of example 3, for preparation of crystalline midostaurin form vi by the process of example 5, for preparation of crystalline midostaurin form vii by the process of example 6, for preparation of crystalline midostaurin form viii by the process of example 7, for preparation of crystalline midostaurin form ix by the process of example 8, for preparation of crystalline midostaurin form x by the process of example 9, for preparation of crystalline midostaurin form xi by the process of example 10, for preparation of crystalline midostaurin form xii by the process of example 11, for preparation of crystalline midostaurin form xiii by the process of example 12, for preparation of crystalline midostaurin form xiv by the process of example 13, for preparation of crystalline midostaurin form xiv by the process of example 15 and for preparation of crystalline midostaurin form xvi by the process of example 16.
Comparative example 1
Compared with the preparation method of the crystal form II in the patent CN101048416A, the preparation of the crystal form II can be stably obtained only by adding crystal seeds of the crystal form II, which shows that the crystal form II is difficult to prepare by spontaneous crystallization and has certain risk in industrial preparation; in patent CN102639538A, a supercritical fluid method is needed for preparing crystal forms III and IV, which cannot be applied to conventional production, and the crystal form III contains 3.2% of residual solvent or water, the crystal form IV contains 6.2% of residual solvent or water, and the residual solvent is tetrahydrofuran solvent through detection; in the patent WO2018165071A, crystal forms V, VI, VII, VIII, IX, X, XI, XII, XIII, XIV, XV and XVI are ethyl acetate solvate, hydrate, methyl isobutyl ketone solvate, 4-heptanone solvate, methyl acetate solvate, acetone solvate, ethyl formate solvate, isopropyl acetate solvate, diethyl carbonate solvate, benzonitrile solvate, butyl acetate solvate and tert-butyl alcohol solvate respectively, and except the hydrate crystal form VI, other crystal forms are solvates, the dissolved residues of which exceed the standard and cannot be directly applied to preparations. The preparation of the crystal forms A, B and C is simple in operation, high in yield, capable of being obtained by spontaneous crystallization, low in industrial preparation difficulty, and qualified in solvent residue, and the results are shown in Table 6.
TABLE 6


Comparative example 2
Taking the crystal form A obtained in example 1, the crystal form B obtained in example 5, the crystal form C obtained in example 9 and the amorphous form, the crystal form II and the crystal form VI obtained in the preparation example, a stability experiment of standing for 180 days at 40 ℃ is carried out. The crystal form, HPLC purity and maximum single impurity content of the compound before and after standing were examined and the results are shown in table 7 below:
TABLE 7

As can be seen from the above table, the stability data of 180 days at 40 ℃ can be seen: the HPLC purities and the maximum single impurity contents of the crystal form A, the crystal form B and the crystal form C are less changed, and the crystal forms are not changed; the HPLC purity of the crystal form VI is obviously reduced, and the maximum single impurity content is also obviously increased; the amorphous crystal transformation phenomenon appears after 180 days, and the crystal form is unstable; the comparison result shows that the stability of the crystal form A, the crystal form B and the crystal form C obtained by the invention is superior to that of the amorphous crystal form and the crystal form VI at the temperature of 40 ℃.
Comparative example 3
As described in patent CN102639538A, midostaurin has poor water solubility, whereas melt extrusion (mixing of the therapeutic compound with an inert carrier using a twin screw extruder) can form solid dispersions with improved solubility and dissolution, whereas heating of the twin screw extruder can facilitate mixing of the therapeutic compound with the carrier. Crystalline form II of midostaurin has a rather high melting point (260 ℃) close to its decomposition temperature, which makes it difficult to apply it to melt extrusion for the preparation of pharmaceutical compositions. The melting point of form III (206 ℃) and the melting point of form IV (215 ℃) is significantly lower than the melting point of form II (260 ℃), in other words, for form III and form IV the decomposition temperature is significantly higher than its melting temperature, so that during melt extrusion the intimate mixing of form III and form IV with pharmaceutically acceptable excipients can be done at lower temperatures while reducing the risk of decomposition; whereas crystal form a, crystal form B and crystal form C of the present invention have lower melting points than crystal form III, crystal form IV, crystal form a having a melting point of 196 ℃, crystal form B having a melting point of 198 ℃, and crystal form C having a melting point of 127 ℃, during melt extrusion intimate mixing of crystal form a, crystal form B and crystal form C with pharmaceutically acceptable excipients can be accomplished at lower temperatures than crystal form II while reducing the risk of decomposition.
Claims (31)
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202011614974.0A CN112812129A (en) | 2020-12-31 | 2020-12-31 | Novel crystalline form of midostaurin, process for its preparation and its use |
| PCT/CN2021/133470 WO2022142914A1 (en) | 2020-12-31 | 2021-11-26 | Novel crystal form of midostaurin, preparation method therefor and use thereof |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202011614974.0A CN112812129A (en) | 2020-12-31 | 2020-12-31 | Novel crystalline form of midostaurin, process for its preparation and its use |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN112812129A true CN112812129A (en) | 2021-05-18 |
Family
ID=75855497
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202011614974.0A Pending CN112812129A (en) | 2020-12-31 | 2020-12-31 | Novel crystalline form of midostaurin, process for its preparation and its use |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN112812129A (en) |
| WO (1) | WO2022142914A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022142914A1 (en) * | 2020-12-31 | 2022-07-07 | 浙江海正药业股份有限公司 | Novel crystal form of midostaurin, preparation method therefor and use thereof |
| CN119930649A (en) * | 2023-11-02 | 2025-05-06 | 福建科瑞药业有限公司 | MTL-II crystal form of midostaurin and its preparation method and application |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH05247055A (en) * | 1992-03-03 | 1993-09-24 | Meiji Seika Kaisha Ltd | Staurosporine derivative and antiulcer effect enhancer containing the same derivative |
| WO2019215759A1 (en) * | 2018-05-09 | 2019-11-14 | Alaparthi Lakshmi Prasad | An improved process for preparation of midostaurin |
| CN111393454A (en) * | 2020-05-07 | 2020-07-10 | 奥锐特药业(天津)有限公司 | Novel crystalline form of midostaurin and process for its preparation |
| WO2020200945A1 (en) * | 2019-03-29 | 2020-10-08 | Procos S.P.A. | Process for the preparation of midostaurin with high purity |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5093330A (en) * | 1987-06-15 | 1992-03-03 | Ciba-Geigy Corporation | Staurosporine derivatives substituted at methylamino nitrogen |
| IL86632A0 (en) * | 1987-06-15 | 1988-11-30 | Ciba Geigy Ag | Derivatives substituted at methyl-amino nitrogen |
| RU2410098C2 (en) * | 2004-08-31 | 2011-01-27 | Новартис Аг | Administration of midostaurin for treating gastrointestinal stromal tumours |
| CN101665498A (en) * | 2004-11-05 | 2010-03-10 | 诺瓦提斯公司 | Organic compounds |
| EP2327706A1 (en) * | 2009-11-30 | 2011-06-01 | Novartis AG | Polymorphous forms III and IV of N-benzoyl-staurosporine |
| EP3592749A1 (en) * | 2017-03-06 | 2020-01-15 | Teva Pharmaceutical Works Ltd. | Solid state forms of midostaurin |
| WO2020261293A1 (en) * | 2019-06-24 | 2020-12-30 | Dr. Reddy's Laboratories Limited | Process for preparation of midostaurin |
| CN112812129A (en) * | 2020-12-31 | 2021-05-18 | 浙江海正药业股份有限公司 | Novel crystalline form of midostaurin, process for its preparation and its use |
-
2020
- 2020-12-31 CN CN202011614974.0A patent/CN112812129A/en active Pending
-
2021
- 2021-11-26 WO PCT/CN2021/133470 patent/WO2022142914A1/en not_active Ceased
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH05247055A (en) * | 1992-03-03 | 1993-09-24 | Meiji Seika Kaisha Ltd | Staurosporine derivative and antiulcer effect enhancer containing the same derivative |
| WO2019215759A1 (en) * | 2018-05-09 | 2019-11-14 | Alaparthi Lakshmi Prasad | An improved process for preparation of midostaurin |
| WO2020200945A1 (en) * | 2019-03-29 | 2020-10-08 | Procos S.P.A. | Process for the preparation of midostaurin with high purity |
| CN111393454A (en) * | 2020-05-07 | 2020-07-10 | 奥锐特药业(天津)有限公司 | Novel crystalline form of midostaurin and process for its preparation |
Non-Patent Citations (2)
| Title |
|---|
| 王立平 等: "PKC-412 卤代衍生物的合成及细胞毒活性研究" * |
| 王金英 等, 人民卫生出版社 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022142914A1 (en) * | 2020-12-31 | 2022-07-07 | 浙江海正药业股份有限公司 | Novel crystal form of midostaurin, preparation method therefor and use thereof |
| CN119930649A (en) * | 2023-11-02 | 2025-05-06 | 福建科瑞药业有限公司 | MTL-II crystal form of midostaurin and its preparation method and application |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2022142914A1 (en) | 2022-07-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN111187253B (en) | Novel crystal form of acitinib | |
| EP4306171A2 (en) | Formulation comprising a mek inhibitor | |
| WO2020053660A1 (en) | Solid forms of a bet inhibitor | |
| NO329618B1 (en) | New crystalline forms of the compound ZD1839, solvate thereof, processes for the preparation thereof and pharmaceutical preparations containing such | |
| CN112142679B (en) | Gefitinib and vanilloid eutectic methanol solvate and preparation method thereof | |
| EP3805229B1 (en) | Salt of fused ring pyrimidine compound, crystal form thereof and preparation method therefor and use thereof | |
| US11028069B2 (en) | Salt of substituted piperidine compound | |
| CN112812129A (en) | Novel crystalline form of midostaurin, process for its preparation and its use | |
| CN113651770B (en) | Epalrestat crystal form, and preparation method and application thereof | |
| CN113966332A (en) | Polymorphic substance of CDK9 inhibitor and preparation method and application thereof | |
| CN118660873A (en) | Salts and crystal forms of dipeptidyl peptidase inhibitor compounds | |
| JP4015954B2 (en) | Suplatast tosilate crystal | |
| CN111484489B (en) | Amorphous B-RAF Kinase Dimer Inhibitor | |
| JP2026508943A (en) | Crystal morphology | |
| EP3007700B1 (en) | New polymorphic forms of icotinib phosphate and uses thereof | |
| CN109705090B (en) | Tartaric acid addition salts of 3, 4-disubstituted 1H-pyrazole compounds and crystalline forms thereof | |
| CN108822054B (en) | Odaterol hydrochloride crystal form C and preparation method thereof | |
| CN110291071B (en) | Crystal form of SB-939 salt, preparation method and application thereof | |
| CN110642854A (en) | Polycrystalline form of fused ring compound, composition, preparation method and application thereof | |
| EP4509502A1 (en) | Crystal form of fgfr4 selective inhibitor compound, and preparation method therefor and use thereof | |
| CN108997248B (en) | Odaterol hydrochloride crystal form B and preparation method thereof | |
| CN114401957B (en) | Preparation method of quinazoline derivative and crystal thereof | |
| CN109096218B (en) | Odaterol hydrochloride crystal form A and preparation method thereof | |
| WO2022253261A1 (en) | Hydrate crystal form of lazertinib methanesulfonate, preparation method therefor and use thereof | |
| HK40040064B (en) | Salt of fused ring pyrimidine compound, crystal form thereof and preparation method therefor and use thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| WD01 | Invention patent application deemed withdrawn after publication | ||
| WD01 | Invention patent application deemed withdrawn after publication |
Application publication date: 20210518 |