CN111574511A - Synthesis method and application of sulfuryl pyraflufen - Google Patents
Synthesis method and application of sulfuryl pyraflufen Download PDFInfo
- Publication number
- CN111574511A CN111574511A CN202010600009.1A CN202010600009A CN111574511A CN 111574511 A CN111574511 A CN 111574511A CN 202010600009 A CN202010600009 A CN 202010600009A CN 111574511 A CN111574511 A CN 111574511A
- Authority
- CN
- China
- Prior art keywords
- add
- solvent
- reactant
- synthetic method
- reaction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- -1 sulfuryl pyraflufen Chemical compound 0.000 title abstract description 18
- 238000001308 synthesis method Methods 0.000 title abstract description 6
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims abstract description 83
- 239000012074 organic phase Substances 0.000 claims abstract description 63
- 239000000376 reactant Substances 0.000 claims abstract description 62
- 239000002904 solvent Substances 0.000 claims abstract description 57
- 230000007704 transition Effects 0.000 claims abstract description 30
- 238000002360 preparation method Methods 0.000 claims abstract description 23
- 238000005660 chlorination reaction Methods 0.000 claims abstract description 16
- 239000007800 oxidant agent Substances 0.000 claims abstract description 13
- 230000001590 oxidative effect Effects 0.000 claims abstract description 7
- 238000005799 fluoromethylation reaction Methods 0.000 claims abstract description 6
- 239000000575 pesticide Substances 0.000 claims abstract description 6
- 238000006243 chemical reaction Methods 0.000 claims description 101
- HHLFWLYXYJOTON-UHFFFAOYSA-N glyoxylic acid Chemical compound OC(=O)C=O HHLFWLYXYJOTON-UHFFFAOYSA-N 0.000 claims description 86
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 claims description 58
- 239000000203 mixture Substances 0.000 claims description 52
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 51
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 39
- 238000000034 method Methods 0.000 claims description 38
- 239000003153 chemical reaction reagent Substances 0.000 claims description 36
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 claims description 34
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 claims description 31
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 claims description 28
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 claims description 26
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 24
- 238000010189 synthetic method Methods 0.000 claims description 24
- 230000002194 synthesizing effect Effects 0.000 claims description 20
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 claims description 17
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Natural products NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 claims description 15
- 230000008569 process Effects 0.000 claims description 14
- QWMFKVNJIYNWII-UHFFFAOYSA-N 5-bromo-2-(2,5-dimethylpyrrol-1-yl)pyridine Chemical compound CC1=CC=C(C)N1C1=CC=C(Br)C=N1 QWMFKVNJIYNWII-UHFFFAOYSA-N 0.000 claims description 13
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 claims description 10
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 claims description 9
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 claims description 9
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 claims description 9
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 claims description 8
- 239000002994 raw material Substances 0.000 claims description 7
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 6
- PHTQWCKDNZKARW-UHFFFAOYSA-N isoamylol Chemical compound CC(C)CCO PHTQWCKDNZKARW-UHFFFAOYSA-N 0.000 claims description 6
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 claims description 6
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 claims description 5
- OCJKUQIPRNZDTK-UHFFFAOYSA-N ethyl 4,4,4-trifluoro-3-oxobutanoate Chemical compound CCOC(=O)CC(=O)C(F)(F)F OCJKUQIPRNZDTK-UHFFFAOYSA-N 0.000 claims description 4
- 239000012467 final product Substances 0.000 claims description 4
- 150000003839 salts Chemical class 0.000 claims description 4
- XMVONEAAOPAGAO-UHFFFAOYSA-N sodium tungstate Chemical compound [Na+].[Na+].[O-][W]([O-])(=O)=O XMVONEAAOPAGAO-UHFFFAOYSA-N 0.000 claims description 4
- OCJBOOLMMGQPQU-UHFFFAOYSA-N 1,4-dichlorobenzene Chemical compound ClC1=CC=C(Cl)C=C1 OCJBOOLMMGQPQU-UHFFFAOYSA-N 0.000 claims description 3
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 claims description 3
- DKPFZGUDAPQIHT-UHFFFAOYSA-N Butyl acetate Natural products CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 claims description 3
- 239000012425 OXONE® Substances 0.000 claims description 3
- YGYAWVDWMABLBF-UHFFFAOYSA-N Phosgene Chemical compound ClC(Cl)=O YGYAWVDWMABLBF-UHFFFAOYSA-N 0.000 claims description 3
- 125000003545 alkoxy group Chemical group 0.000 claims description 3
- 125000003277 amino group Chemical group 0.000 claims description 3
- PASDCCFISLVPSO-UHFFFAOYSA-N benzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1 PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 claims description 3
- 229910052794 bromium Inorganic materials 0.000 claims description 3
- CDQSJQSWAWPGKG-UHFFFAOYSA-N butane-1,1-diol Chemical compound CCCC(O)O CDQSJQSWAWPGKG-UHFFFAOYSA-N 0.000 claims description 3
- 239000002131 composite material Substances 0.000 claims description 3
- 229940117389 dichlorobenzene Drugs 0.000 claims description 3
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 claims description 3
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 3
- RWSOTUBLDIXVET-UHFFFAOYSA-M hydrosulfide Chemical compound [SH-] RWSOTUBLDIXVET-UHFFFAOYSA-M 0.000 claims description 3
- HJKYXKSLRZKNSI-UHFFFAOYSA-I pentapotassium;hydrogen sulfate;oxido sulfate;sulfuric acid Chemical compound [K+].[K+].[K+].[K+].[K+].OS([O-])(=O)=O.[O-]S([O-])(=O)=O.OS(=O)(=O)O[O-].OS(=O)(=O)O[O-] HJKYXKSLRZKNSI-UHFFFAOYSA-I 0.000 claims description 3
- FAIAAWCVCHQXDN-UHFFFAOYSA-N phosphorus trichloride Chemical compound ClP(Cl)Cl FAIAAWCVCHQXDN-UHFFFAOYSA-N 0.000 claims description 3
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 claims description 3
- 125000004400 (C1-C12) alkyl group Chemical group 0.000 claims description 2
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 claims 1
- 150000001335 aliphatic alkanes Chemical class 0.000 claims 1
- 239000000047 product Substances 0.000 abstract description 61
- 239000006227 byproduct Substances 0.000 abstract description 9
- 238000002156 mixing Methods 0.000 abstract description 3
- 239000002699 waste material Substances 0.000 abstract description 3
- 239000005605 Pyraflufen-ethyl Substances 0.000 abstract 1
- 238000004064 recycling Methods 0.000 abstract 1
- 239000000543 intermediate Substances 0.000 description 155
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 75
- 238000003756 stirring Methods 0.000 description 71
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 60
- 239000007864 aqueous solution Substances 0.000 description 59
- 239000000243 solution Substances 0.000 description 56
- 238000006467 substitution reaction Methods 0.000 description 29
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 26
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 26
- 235000011121 sodium hydroxide Nutrition 0.000 description 25
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 24
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 24
- 239000007789 gas Substances 0.000 description 23
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 22
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 16
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 15
- 229910000039 hydrogen halide Inorganic materials 0.000 description 15
- 239000012433 hydrogen halide Substances 0.000 description 15
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 14
- 239000012141 concentrate Substances 0.000 description 14
- VQTUBCCKSQIDNK-UHFFFAOYSA-N Isobutene Chemical compound CC(C)=C VQTUBCCKSQIDNK-UHFFFAOYSA-N 0.000 description 13
- 150000007529 inorganic bases Chemical class 0.000 description 13
- 229910000027 potassium carbonate Inorganic materials 0.000 description 13
- 235000011181 potassium carbonates Nutrition 0.000 description 13
- 229910000029 sodium carbonate Inorganic materials 0.000 description 13
- 235000017550 sodium carbonate Nutrition 0.000 description 13
- VOPWNXZWBYDODV-UHFFFAOYSA-N Chlorodifluoromethane Chemical compound FC(F)Cl VOPWNXZWBYDODV-UHFFFAOYSA-N 0.000 description 12
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 12
- 239000003960 organic solvent Substances 0.000 description 12
- 239000008098 formaldehyde solution Substances 0.000 description 11
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 11
- 235000017557 sodium bicarbonate Nutrition 0.000 description 11
- 238000012360 testing method Methods 0.000 description 11
- QAOWNCQODCNURD-UHFFFAOYSA-N sulfuric acid Substances OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 10
- 241000196324 Embryophyta Species 0.000 description 9
- 239000000126 substance Substances 0.000 description 8
- 239000003513 alkali Substances 0.000 description 7
- 238000007254 oxidation reaction Methods 0.000 description 7
- 238000010792 warming Methods 0.000 description 7
- 239000007791 liquid phase Substances 0.000 description 6
- 239000012071 phase Substances 0.000 description 6
- 230000015572 biosynthetic process Effects 0.000 description 5
- 230000003647 oxidation Effects 0.000 description 5
- 235000011118 potassium hydroxide Nutrition 0.000 description 5
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- CASLETQIYIQFTQ-UHFFFAOYSA-N 3-[[5-(difluoromethoxy)-1-methyl-3-(trifluoromethyl)pyrazol-4-yl]methylsulfonyl]-5,5-dimethyl-4h-1,2-oxazole Chemical compound CN1N=C(C(F)(F)F)C(CS(=O)(=O)C=2CC(C)(C)ON=2)=C1OC(F)F CASLETQIYIQFTQ-UHFFFAOYSA-N 0.000 description 3
- OMKAMTCDOWAJAA-UHFFFAOYSA-N 3-bromo-5,5-dimethyl-4h-1,2-oxazole Chemical compound CC1(C)CC(Br)=NO1 OMKAMTCDOWAJAA-UHFFFAOYSA-N 0.000 description 3
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 241001148683 Zostera marina Species 0.000 description 3
- 125000000218 acetic acid group Chemical group C(C)(=O)* 0.000 description 3
- 239000012065 filter cake Substances 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 238000003032 molecular docking Methods 0.000 description 3
- 238000011084 recovery Methods 0.000 description 3
- HYHCSLBZRBJJCH-UHFFFAOYSA-M sodium hydrosulfide Chemical compound [Na+].[SH-] HYHCSLBZRBJJCH-UHFFFAOYSA-M 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 238000013517 stratification Methods 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 2
- 230000001476 alcoholic effect Effects 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 230000032823 cell division Effects 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 239000007806 chemical reaction intermediate Substances 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 230000002363 herbicidal effect Effects 0.000 description 2
- 239000004009 herbicide Substances 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- AWBKQZSYNWLCMW-UHFFFAOYSA-N n-(dibromomethylidene)hydroxylamine Chemical compound ON=C(Br)Br AWBKQZSYNWLCMW-UHFFFAOYSA-N 0.000 description 2
- ZOCLAPYLSUCOGI-UHFFFAOYSA-M potassium hydrosulfide Chemical compound [SH-].[K+] ZOCLAPYLSUCOGI-UHFFFAOYSA-M 0.000 description 2
- 150000003384 small molecules Chemical class 0.000 description 2
- 238000000967 suction filtration Methods 0.000 description 2
- 230000014616 translation Effects 0.000 description 2
- 239000002351 wastewater Substances 0.000 description 2
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- ZNBNBTIDJSKEAM-UHFFFAOYSA-N 4-[7-hydroxy-2-[5-[5-[6-hydroxy-6-(hydroxymethyl)-3,5-dimethyloxan-2-yl]-3-methyloxolan-2-yl]-5-methyloxolan-2-yl]-2,8-dimethyl-1,10-dioxaspiro[4.5]decan-9-yl]-2-methyl-3-propanoyloxypentanoic acid Chemical compound C1C(O)C(C)C(C(C)C(OC(=O)CC)C(C)C(O)=O)OC11OC(C)(C2OC(C)(CC2)C2C(CC(O2)C2C(CC(C)C(O)(CO)O2)C)C)CC1 ZNBNBTIDJSKEAM-UHFFFAOYSA-N 0.000 description 1
- 244000291564 Allium cepa Species 0.000 description 1
- 235000002732 Allium cepa var. cepa Nutrition 0.000 description 1
- 240000002234 Allium sativum Species 0.000 description 1
- 240000001592 Amaranthus caudatus Species 0.000 description 1
- 235000009328 Amaranthus caudatus Nutrition 0.000 description 1
- 244000105624 Arachis hypogaea Species 0.000 description 1
- 235000007320 Avena fatua Nutrition 0.000 description 1
- 241001647031 Avena sterilis Species 0.000 description 1
- 235000004535 Avena sterilis Nutrition 0.000 description 1
- 240000002791 Brassica napus Species 0.000 description 1
- 235000004977 Brassica sinapistrum Nutrition 0.000 description 1
- UGVAQFVQIHNCJV-UHFFFAOYSA-N CN1N=C(C(F)(F)F)C=C1O Chemical compound CN1N=C(C(F)(F)F)C=C1O UGVAQFVQIHNCJV-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 241000233838 Commelina Species 0.000 description 1
- 229920000742 Cotton Polymers 0.000 description 1
- 241000207901 Cuscuta Species 0.000 description 1
- 240000004585 Dactylis glomerata Species 0.000 description 1
- 235000001602 Digitaria X umfolozi Nutrition 0.000 description 1
- 240000003176 Digitaria ciliaris Species 0.000 description 1
- 235000017898 Digitaria ciliaris Nutrition 0.000 description 1
- 235000005476 Digitaria cruciata Nutrition 0.000 description 1
- 235000006830 Digitaria didactyla Nutrition 0.000 description 1
- 235000005804 Digitaria eriantha ssp. eriantha Nutrition 0.000 description 1
- 235000010823 Digitaria sanguinalis Nutrition 0.000 description 1
- 240000003173 Drymaria cordata Species 0.000 description 1
- 244000058871 Echinochloa crus-galli Species 0.000 description 1
- 235000014716 Eleusine indica Nutrition 0.000 description 1
- 244000140063 Eragrostis abyssinica Species 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 description 1
- 241000219146 Gossypium Species 0.000 description 1
- 244000020551 Helianthus annuus Species 0.000 description 1
- 235000003222 Helianthus annuus Nutrition 0.000 description 1
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 1
- 241000289581 Macropus sp. Species 0.000 description 1
- 244000061176 Nicotiana tabacum Species 0.000 description 1
- 235000002637 Nicotiana tabacum Nutrition 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- 244000046052 Phaseolus vulgaris Species 0.000 description 1
- 235000010627 Phaseolus vulgaris Nutrition 0.000 description 1
- 108010064851 Plant Proteins Proteins 0.000 description 1
- 235000004443 Ricinus communis Nutrition 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 244000061456 Solanum tuberosum Species 0.000 description 1
- 235000002595 Solanum tuberosum Nutrition 0.000 description 1
- 244000152045 Themeda triandra Species 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 235000015278 beef Nutrition 0.000 description 1
- 230000031709 bromination Effects 0.000 description 1
- 238000005893 bromination reaction Methods 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 239000007810 chemical reaction solvent Substances 0.000 description 1
- 239000012320 chlorinating reagent Substances 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 238000012937 correction Methods 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 230000027721 electron transport chain Effects 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- 238000005755 formation reaction Methods 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 235000004611 garlic Nutrition 0.000 description 1
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 1
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 1
- 229910000040 hydrogen fluoride Inorganic materials 0.000 description 1
- 229910000378 hydroxylammonium sulfate Inorganic materials 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical compound C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- HDZGCSFEDULWCS-UHFFFAOYSA-N monomethylhydrazine Chemical compound CNN HDZGCSFEDULWCS-UHFFFAOYSA-N 0.000 description 1
- 150000002923 oximes Chemical class 0.000 description 1
- 235000020232 peanut Nutrition 0.000 description 1
- 230000029553 photosynthesis Effects 0.000 description 1
- 238000010672 photosynthesis Methods 0.000 description 1
- 235000021118 plant-derived protein Nutrition 0.000 description 1
- 239000011736 potassium bicarbonate Substances 0.000 description 1
- 235000015497 potassium bicarbonate Nutrition 0.000 description 1
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 1
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 1
- 235000012015 potatoes Nutrition 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 238000001243 protein synthesis Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000029058 respiratory gaseous exchange Effects 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 230000002786 root growth Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 210000002435 tendon Anatomy 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- 150000003585 thioureas Chemical class 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01N—PRESERVATION OF BODIES OF HUMANS OR ANIMALS OR PLANTS OR PARTS THEREOF; BIOCIDES, e.g. AS DISINFECTANTS, AS PESTICIDES OR AS HERBICIDES; PEST REPELLANTS OR ATTRACTANTS; PLANT GROWTH REGULATORS
- A01N43/00—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds
- A01N43/72—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds having rings with nitrogen atoms and oxygen or sulfur atoms as ring hetero atoms
- A01N43/80—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds having rings with nitrogen atoms and oxygen or sulfur atoms as ring hetero atoms five-membered rings with one nitrogen atom and either one oxygen atom or one sulfur atom in positions 1,2
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Agronomy & Crop Science (AREA)
- Pest Control & Pesticides (AREA)
- Plant Pathology (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Dentistry (AREA)
- General Health & Medical Sciences (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Environmental Sciences (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description
技术领域technical field
本发明涉及农药技术领域,具体的更涉及一种砜吡草唑的合成方法及其应用。The invention relates to the technical field of pesticides, in particular to a method for synthesizing sulfofenapyr and its application.
背景技术Background technique
砜吡草唑(pyroxasulfone),分子式C12H14F5N3O4S,化学名称为3-[5-(二氟甲氧基)-1-甲基-3-(三氟甲氧基)吡唑-4-基甲基磺酰基]-4,5-二氢-5,5-二甲基-1,2-异噁唑。砜吡草唑是异噁唑类除草剂,具有抑制植物细胞分裂的作用。该除草剂最早由日本K-I化学研究所发现,以后由日本组合化学公司与日本庵原化学公司联合实现了产业化。砜吡草唑主要用于防除部分禾本科杂草及阔叶杂草。砜吡草唑主要通过单子叶植物的胚芽鞘或双子叶植物的下胚轴吸收,吸收后向上传导,通过阻碍蛋白质合成而抑制细胞生长,也抑制植物的呼吸作用,使杂草幼芽、幼根生长停止,进而死亡;也作为电子传递链的抑制剂和解偶联剂可抑制植物的光合作用,并能干扰植物体蛋白质的生物合成,影响细胞分裂,影响膜的生物合成及完整性。广泛用于玉米、棉花、豆类、花生、马铃薯、油菜、大蒜、烟草、向日葵、蓖麻、大葱等田防除一年生禾本科杂草和部分小粒种子的阔叶杂草。对马唐、狗尾草、牛筋草、稗草、千金子、看麦娘、野燕麦、早熟禾、硬草、画眉草等一年生禾本科杂草有特效,对藜科、苋科、蓼科、鸭跖草、牛繁缕、莬丝子等阔叶杂草也有一定的防效。但是现有关于砜吡草唑的合成方法存在明显的产率低、副产物含量高等缺点。Pyroxasulfone (pyroxasulfone), molecular formula C 12 H 14 F 5 N 3 O 4 S, chemical name is 3-[5-(difluoromethoxy)-1-methyl-3-(trifluoromethoxy) ) pyrazol-4-ylmethylsulfonyl]-4,5-dihydro-5,5-dimethyl-1,2-isoxazole. Trifenapyr is an isoxazole herbicide that inhibits plant cell division. The herbicide was first discovered by Japan's KI Chemical Research Institute, and later industrialized by Japan Combination Chemical Company and Japan's Anhara Chemical Company. Sufenapyr is mainly used to control some grass weeds and broadleaf weeds. Sufenapyr is mainly absorbed through the coleoptiles of monocotyledonous plants or the hypocotyls of dicotyledonous plants. After absorption, it conducts upwards, inhibits cell growth by hindering protein synthesis, and also inhibits plant respiration, so that weed shoots, young Root growth stops and then dies; it also acts as an inhibitor and uncoupler of the electron transport chain, which can inhibit plant photosynthesis, interfere with plant protein biosynthesis, affect cell division, and affect membrane biosynthesis and integrity. Widely used in corn, cotton, beans, peanuts, potatoes, rapeseed, garlic, tobacco, sunflower, castor, green onions and other fields to control annual grass weeds and some broad-leaved weeds with small seeds. It has special effects on annual grass weeds such as crabgrass, foxtail, beef tendon, barnyardgrass, chinensis, kangaroo, wild oat, bluegrass, hard grass, teff, etc. Broadleaf weeds such as Commelina, Niu chickweed, and Cuscuta also have certain control effects. However, the existing synthetic methods for fenfenpyr have obvious shortcomings such as low yield and high content of by-products.
发明内容SUMMARY OF THE INVENTION
为了解决上述的技术问题,本发明的第一个方面提供了一种砜吡草唑的合成方法,制备步骤包括:In order to solve the above-mentioned technical problems, a first aspect of the present invention provides a method for synthesizing sulfofenazone, and the preparation steps include:
步骤(1):对反应体(Ⅰ)进行羟烷基化、氟甲基化、氯化反应制得中间体2;Step (1): reaction body (I) is subjected to hydroxyalkylation, fluoromethylation and chlorination to obtain intermediate 2;
步骤(2):将中间体1、中间体2进行混合,加水分离出有机相,得到过渡中间体,加入溶剂、氧化剂进行反应,即得;Step (2): mix the intermediate 1 and the intermediate 2, add water to separate the organic phase, obtain a transition intermediate, add a solvent and an oxidizing agent to react, to obtain the final product;
所述反应体(Ⅰ)的结构为式1所示;所述中间体1的结构式为式2;The structure of the reactant (I) is shown in formula 1; the structural formula of the intermediate 1 is formula 2;

作为一种优选的技术方案,本发明中所述R1的结构选自-SH、式3中的一种;所述式3如下所示,其中所述R2表示氨基、氢原子、取代或未取代的C1-C12的烷基、烷氧基中的一种;As a preferred technical solution, the structure of R1 in the present invention is selected from one of -SH and formula 3; the formula 3 is as follows, wherein the R2 represents an amino group, a hydrogen atom, substituted or unsubstituted One of the C1-C12 alkyl and alkoxy groups;

作为一种优选的技术方案,本发明中所述步骤(1)中氯化反应过程中包含氯化试剂;所述氯化试剂包含二氯亚砜、三氯化磷、磺酰氯、氯气、碳酰氯、苯甲酰氯中的至少一种。As a preferred technical solution, a chlorination reagent is included in the chlorination reaction process in step (1) in the present invention; the chlorination reagent includes thionyl chloride, phosphorus trichloride, sulfonyl chloride, chlorine, carbon At least one of acid chloride and benzoyl chloride.
作为一种优选的技术方案,本发明中所述步骤(2)中的溶剂包含C1-C8的小分子醇、二氯甲烷、1,2-二氯乙烷、氯苯、二氯苯、乙酸乙酯、乙酸丁酯中的至少一种。As a preferred technical solution, the solvent in step (2) described in the present invention comprises C1-C8 small molecular alcohol, dichloromethane, 1,2-dichloroethane, chlorobenzene, dichlorobenzene, acetic acid At least one of ethyl ester and butyl acetate.
作为一种优选的技术方案,本发明中所述步骤(2)的氧化剂包含双氧水、间氯过氧苯甲酸、单过硫酸氢钾复合盐中的至少一种。As a preferred technical solution, the oxidant of step (2) in the present invention comprises at least one of hydrogen peroxide, m-chloroperoxybenzoic acid, and potassium monopersulfate composite salt.
作为一种优选的技术方案,本发明中所述C1-C8的小分子醇选自甲醇、乙醇、叔丁醇、丁二醇、丙二醇、异丁醇、异戊醇、异丙醇、乙二醇、丙三醇中的至少一种中的至少一种。As a preferred technical solution, the C1-C8 small molecular alcohol in the present invention is selected from methanol, ethanol, tert-butanol, butanediol, propylene glycol, isobutanol, isoamyl alcohol, isopropanol, ethylene glycol At least one of at least one of alcohol and glycerol.
作为一种优选的技术方案,本发明中所述中间体1的纯度≥90%;所述中间体2的纯度≥85%。As a preferred technical solution, the purity of the intermediate 1 in the present invention is greater than or equal to 90%; the purity of the intermediate 2 is greater than or equal to 85%.
作为一种优选的技术方案,本发明中所述氧化剂中还包含钨酸钠;所述双氧水、二水合钨酸钠之间的摩尔比为1:(0.005-0.35)。As a preferred technical solution, the oxidant in the present invention also includes sodium tungstate; the molar ratio between the hydrogen peroxide and sodium tungstate dihydrate is 1:(0.005-0.35).
作为一种优选的技术方案,本发明中所述反应体(Ⅰ)的制备原料包括三氟乙酰乙酸乙酯;所述中间体1的制备原料包括乙醛酸、硫脲、硫氢化盐、溴中的至少一种。As a preferred technical solution, the raw materials for the preparation of the reactant (I) in the present invention include ethyl trifluoroacetoacetate; the raw materials for the preparation of the intermediate 1 include glyoxylic acid, thiourea, hydrosulfide, bromine at least one of them.
本发明的第二个方面提供了一种砜吡草唑的应用,应用于农药;所述砜吡草唑由所述合成方法制备得到。The second aspect of the present invention provides an application of fenfenpyr, which is applied to pesticides; the fenfenazole is prepared by the synthetic method.
与现有技术相比,本发明具有如下优异的有益效果:Compared with the prior art, the present invention has the following excellent beneficial effects:
本发明提供了一种砜吡草唑的新合成方法,通过对生产路线的改进,首先通过对反应体(Ⅰ)进行O-二氟甲基化合成中间体2,然后再与中间体1对接,最后在经过氧化反应生成目标产物。这种生产方法避免了反应体(Ⅰ)与中间体1先反应在进行氟甲基化过程中带来的副产物,提高了最终产物的纯度以及收率;发明人意外发现,在中间体1、中间体2反应的过程中选用小分子醇作为反应溶剂可以减少反应过程中副产物的产生,避免现存方法中使用乙酸做溶剂,难以回收溶剂且产生大量废水。此外,整个生产路线过程中,合成中间体1和2过程中都不需要出料,可以直接进行下一步反应的合成工艺,在这个过程中,本发明可以做到不仅能够提高了目标产物的整体产率,而且能够降低副产物的生成,非常完善的降低了三废的产生以及实现溶剂回收再用。The present invention provides a new method for synthesizing sulfofenazone. By improving the production route, firstly, the intermediate 2 is synthesized by performing O-difluoromethylation on the reactant (I), and then the intermediate 1 is docked. , and finally generate the target product through the oxidation reaction. This production method avoids the by-products caused by the first reaction of the reactant (I) and the intermediate 1 during the fluoromethylation process, and improves the purity and yield of the final product; the inventor unexpectedly found that in the intermediate 1 , In the process of the reaction of intermediate 2, selecting small molecular alcohol as the reaction solvent can reduce the generation of by-products in the reaction process, avoid using acetic acid as the solvent in the existing method, and it is difficult to recover the solvent and generate a large amount of waste water. In addition, in the process of the whole production route, the synthesis process of the intermediates 1 and 2 does not need to be discharged, and the synthesis process of the next step reaction can be directly carried out. In this process, the present invention can not only improve the overall quality of the target product It can also reduce the generation of by-products, perfectly reduce the generation of three wastes and realize solvent recovery and reuse.
附图说明Description of drawings
图1为对实施例1所述的砜吡草唑合成方法得到的砜吡草唑进行液相测试结果。Fig. 1 is the liquid phase test result of the sulfofenapyr obtained by the sulfofenazone synthesis method described in Example 1.
具体实施方式Detailed ways
下面结合具体实施方式对本发明提供技术方案中的技术特征作进一步清楚、完整的描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。The technical features in the technical solutions provided by the present invention will be further clearly and completely described below with reference to the specific embodiments. Obviously, the described embodiments are only a part of the embodiments of the present invention, rather than all the embodiments. Based on the embodiments of the present invention, all other embodiments obtained by those of ordinary skill in the art without creative efforts shall fall within the protection scope of the present invention.
本技术领域技术人员可以理解,除非另外定义,这里使用的所有术语(包括技术术语和科学术语)具有与本发明所属领域中的普通技术人员的一般理解相同的意义。还应该理解的是,诸如通用字典中定义的那些术语应该被理解为具有与现有技术的上下文中的意义一致的意义,并且除非像这里一样定义,不会用理想化或过于正式的含义来解释。It will be understood by those skilled in the art that, unless otherwise defined, all terms (including technical and scientific terms) used herein have the same meaning as commonly understood by one of ordinary skill in the art to which this invention belongs. It should also be understood that terms such as those defined in general dictionaries should be understood to have meanings consistent with their meanings in the context of the prior art and, unless defined as herein, are not to be taken in an idealized or overly formal sense. explain.
本发明中的词语“优选的”、“优选地”、“更优选的”等是指,在某些情况下可提供某些有益效果的本发明实施方案。然而,在相同的情况下或其他情况下,其他实施方案也可能是优选的。此外,对一个或多个优选实施方案的表述并不暗示其他实施方案不可用,也并非旨在将其他实施方案排除在本发明的范围之外。The words "preferred", "preferably", "more preferred" and the like in the present invention refer to embodiments of the invention which, under certain circumstances, may provide certain benefits. However, other embodiments may also be preferred, under the same or other circumstances. Furthermore, the recitation of one or more preferred embodiments does not imply that other embodiments are not available, nor is it intended to exclude other embodiments from the scope of the present invention.
本发明的第一个方面提供了一种砜吡草唑的合成方法,制备步骤包括:A first aspect of the present invention provides a method for synthesizing sulfofenazone, the preparation step comprising:
步骤(1):对反应体(Ⅰ)进行羟烷基化、氟甲基化、氯化反应制得中间体2;Step (1): reaction body (I) is subjected to hydroxyalkylation, fluoromethylation and chlorination to obtain intermediate 2;
步骤(2):将中间体1、中间体2进行混合,加水分离出有机相,得到过渡中间体,加入溶剂、氧化剂进行反应,即得;Step (2): mix the intermediate 1 and the intermediate 2, add water to separate the organic phase, obtain a transition intermediate, add a solvent and an oxidizing agent to react, to obtain the final product;
所述反应体(Ⅰ)的结构为式1所示;所述中间体1的结构式为式2;The structure of the reactant (I) is shown in formula 1; the structural formula of the intermediate 1 is formula 2;

在一些实施方式中,所述砜吡草唑的合成方法中的反应通式,如下所示:In some embodiments, the general reaction formula in the method for synthesizing sulfofenapyr is as follows:

中间体1Intermediate 1
在一些实施方式中,所述R1的结构选自-SH、式3中的一种;所述式3如下所示,其中所述R2表示氨基、氢原子、取代或未取代的C1-C12的烷基、烷氧基中的一种;In some embodiments, the structure of the R1 is selected from one of -SH and formula 3; the formula 3 is as follows, wherein the R2 represents an amino group, a hydrogen atom, a substituted or unsubstituted C1-C12 One of alkyl and alkoxy;
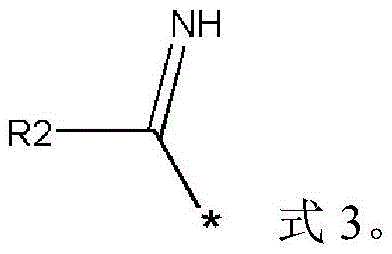
在一些优选的实施方式中,所述中间体1的结构式为式2-1、式2-2中的一种;优选的,所述中间体1的结构式为式2-1;其中,所述式2-1、式2-2的结构如下所示:In some preferred embodiments, the structural formula of the intermediate 1 is one of formula 2-1 and formula 2-2; preferably, the structural formula of the intermediate 1 is formula 2-1; wherein, the The structures of formula 2-1 and formula 2-2 are as follows:

在一些实施方式中,所述中间体1的来源不做特殊的限定,优选的,本发明中所述中间体1为通过如下所述的中间体1的合成方法制备得到。In some embodiments, the source of the intermediate 1 is not particularly limited. Preferably, the intermediate 1 in the present invention is prepared by the synthesis method of the intermediate 1 as described below.
在一些实施方式中,所述中间体1的合成方法,步骤至少包括:以乙醛酸原料,经成肟、溴化和环化反应制得3-溴-5,5-二甲基二氢异噁唑,之后与取代试剂反应,得到中间体1。In some embodiments, the method for synthesizing Intermediate 1 includes at least the steps of: using glyoxylic acid as a raw material to prepare 3-bromo-5,5-dimethyldihydrogen through oxime formation, bromination and cyclization reactions Isoxazole, followed by reaction with a substitution reagent, affords intermediate 1.
在一些实施方式中,所述取代试剂选自硫脲、硫氢化钠、硫氢化钾中的一种;优选的,所述取代试剂为硫脲。In some embodiments, the substitution reagent is selected from one of thiourea, sodium hydrosulfide, and potassium hydrosulfide; preferably, the substitution reagent is thiourea.
在一些优选的实施方式中,所述中间体1的合成方法,步骤至少包括:In some preferred embodiments, the synthetic method of described intermediate 1, the step comprises at least:
1)在第一反应容器中,加入乙醛酸水溶液、羟胺化合物、水,搅拌混合,升温至60-80℃,搅拌1-5小时;降低温度至0℃以下,加入第一无机碱、有机溶剂,滴加液溴,结束后回温至20-35℃,并保温5-10小时,分离有机相,作为第一产物;1) In the first reaction vessel, add glyoxylic acid aqueous solution, hydroxylamine compound, and water, stir and mix, heat up to 60-80 ° C, and stir for 1-5 hours; lower the temperature to below 0 ° C, add the first inorganic base, organic Solvent, add liquid bromine dropwise, return the temperature to 20-35 ° C after the end, and keep the temperature for 5-10 hours, separate the organic phase, and use it as the first product;
2)向第一产物中加入第二无机碱,搅拌升温至70-90℃,通入异丁烯气体,反应1-5小时,加入水,分离出有机相,除溶剂,得到第二产物;2) adding the second inorganic base to the first product, stirring and warming up to 70-90 ° C, feeding isobutene gas, reacting for 1-5 hours, adding water, separating the organic phase, removing the solvent, and obtaining the second product;
3)将取代试剂、醇类溶剂、卤化氢溶液加入到第二反应容器中混合,将第二产物滴加至第二反应容器中,结束后,除溶剂,叔丁醇重结晶,即得。3) The substitution reagent, alcohol solvent and hydrogen halide solution are added into the second reaction vessel and mixed, and the second product is added dropwise to the second reaction vessel.
在一些更优选的实施方式中,所述中间体1的合成方法,步骤至少包括:In some more preferred embodiments, the synthetic method of described intermediate 1, the step comprises at least:
1)在第一反应容器中,加入80g乙醛酸水溶液(50%水溶液,0.50mol)、羟胺化合物(0.55mol,1.1eq)、120g水,搅拌溶解,然后升温至60-80℃并搅拌3小时;将反应温度降至0℃,加入第一无机碱(1.10mol,2.2eq)和有机溶剂,滴加80g液溴(0.50mol,1.0eq),结束后回温至20-35℃,并保温5-10小时,分离有机相,作为第一产物;1) In the first reaction vessel, add 80 g of glyoxylic acid aqueous solution (50% aqueous solution, 0.50 mol), hydroxylamine compound (0.55 mol, 1.1 eq), 120 g of water, stir to dissolve, then heat up to 60-80 ° C and stir for 3 hour; the reaction temperature was lowered to 0 °C, the first inorganic base (1.10 mol, 2.2 eq) and an organic solvent were added, 80 g of liquid bromine (0.50 mol, 1.0 eq) was added dropwise, and the temperature was returned to 20-35 ° C after completion, and Incubate for 5-10 hours, separate the organic phase as the first product;
2)向第一产物中加入第二无机碱(1.44mol,3.0eq),搅拌升温至70-90℃,最后以10g/min速率通入异丁烯气体,反应2小时。反应结束后加入600g水,分离出有机相,除溶剂,得到第二产物;2) The second inorganic base (1.44mol, 3.0eq) was added to the first product, the temperature was raised to 70-90°C with stirring, and finally isobutylene gas was introduced at a rate of 10g/min to react for 2 hours. After the reaction finishes, add 600g of water, separate out the organic phase, remove the solvent, and obtain the second product;
3)将取代试剂(0.33mol,1.1eq)、50-200mL的醇类溶剂、卤化氢溶液加入到第二反应容器中混合,再加入0.5-2.5mL卤化氢溶液,将第二产物滴加至第二反应容器中,结束后,除溶剂,叔丁醇重结晶,即得。3) Add the substitution reagent (0.33mol, 1.1eq), 50-200mL of alcohol solvent and hydrogen halide solution into the second reaction vessel and mix, then add 0.5-2.5mL of hydrogen halide solution, and add the second product dropwise to In the second reaction vessel, after the end, the solvent is removed, and the tert-butanol is recrystallized to obtain.
在一些实施方式中,所述羟胺化合物选自盐酸羟胺、硫酸羟胺、羟胺水溶液中的至少一种;优选的,所述羟胺化合物为盐酸羟胺。In some embodiments, the hydroxylamine compound is selected from at least one of hydroxylamine hydrochloride, hydroxylamine sulfate, and hydroxylamine aqueous solution; preferably, the hydroxylamine compound is hydroxylamine hydrochloride.
在一些实施方式中,步骤1)中所述羟胺化合物、乙醛酸水溶液中的乙醛酸溶质之间的摩尔比为(1-3):1;优选的,所述羟胺化合物、乙醛酸水溶液中的乙醛酸溶质之间的摩尔比为2.2:1;所述乙醛酸水溶液的质量浓度为50%。In some embodiments, the molar ratio between the hydroxylamine compound and glyoxylic acid solute in the glyoxylic acid aqueous solution in step 1) is (1-3): 1; preferably, the hydroxylamine compound, glyoxylic acid The molar ratio between the glyoxylic acid solutes in the aqueous solution is 2.2:1; the mass concentration of the glyoxylic acid aqueous solution is 50%.
在一些实施方式中,步骤1)中水、乙醛酸水溶液的质量比为(1-2):1;优选的,水、乙醛酸水溶液的质量比为1.5:1。In some embodiments, the mass ratio of water and glyoxylic acid aqueous solution in step 1) is (1-2):1; preferably, the mass ratio of water and glyoxylic acid aqueous solution is 1.5:1.
在一些实施方式中,步骤1)中所述第一无机碱选自碳酸钠、碳酸钾、碳酸氢钠、氢氧化钠、氢氧化钾中的至少一种;优选的,所述第一无机碱为碳酸钠。In some embodiments, the first inorganic base in step 1) is selected from at least one of sodium carbonate, potassium carbonate, sodium bicarbonate, sodium hydroxide, and potassium hydroxide; preferably, the first inorganic base for sodium carbonate.
在一些实施方式中,步骤1)中所述第一无机碱、乙醛酸水溶液中的乙醛酸溶质之间的摩尔比为(2-2.5):1;优选的,步骤1)中所述第一无机碱、乙醛酸水溶液中的乙醛酸溶质之间的摩尔比为2.2:1。In some embodiments, the molar ratio between the first inorganic base and the glyoxylic acid solute in the glyoxylic acid aqueous solution described in step 1) is (2-2.5): 1; preferably, the molar ratio described in step 1) The molar ratio between the first inorganic base and the glyoxylic acid solute in the glyoxylic acid aqueous solution is 2.2:1.
在一些实施方式中,步骤1)中所述有机溶剂选自1,2-二氯乙烷、二氯甲烷、氯仿、甲苯、二甲苯、氯苯中的至少一种;优选的,步骤1)中所述有机溶剂为1,2-二氯乙烷。In some embodiments, the organic solvent in step 1) is selected from at least one of 1,2-dichloroethane, dichloromethane, chloroform, toluene, xylene, and chlorobenzene; preferably, step 1) The organic solvent described in the above is 1,2-dichloroethane.
在一些实施方式中,步骤1)中所述有机溶剂、乙醛酸水溶液的质量比为(1.5-2):1;优选的,步骤1)中所述有机溶剂、乙醛酸水溶液的质量比为1.8:1。In some embodiments, the mass ratio of the organic solvent and the glyoxylic acid aqueous solution in step 1) is (1.5-2): 1; preferably, the mass ratio of the organic solvent and glyoxylic acid aqueous solution in step 1) 1.8:1.
在一些实施方式中,步骤1)中所述液溴、乙醛酸水溶液中的乙醛酸溶质之间的摩尔比为(0.1-1.5):1;优选的,所述液溴、乙醛酸水溶液中的乙醛酸溶质之间的摩尔比为1:1。In some embodiments, the molar ratio between the liquid bromine and the glyoxylic acid solute in the glyoxylic acid aqueous solution in step 1) is (0.1-1.5): 1; preferably, the liquid bromine and glyoxylic acid are The molar ratio between the glyoxylic acid solutes in the aqueous solution is 1:1.
在一些实施方式中,步骤2)中所述第二无机碱选自碳酸氢钠、碳酸钠、氢氧化钠、碳酸钾、氢氧化钾中的至少一种;优选的,所述第二无机碱为碳酸氢钠。In some embodiments, the second inorganic base in step 2) is selected from at least one of sodium bicarbonate, sodium carbonate, sodium hydroxide, potassium carbonate and potassium hydroxide; preferably, the second inorganic base for sodium bicarbonate.
在一些实施方式中,步骤2)中所述第二无机碱、第一产物中二溴甲醛肟的含量之间的摩尔比为(2.5-4):1;优选的,步骤2)中所述第二无机碱、第一产物中二溴甲醛肟的含量之间的摩尔比为3:1。In some embodiments, the molar ratio between the second inorganic base described in step 2) and the content of dibromoformaldoxime in the first product is (2.5-4): 1; preferably, described in step 2) The molar ratio between the second inorganic base and the content of dibromoformaloxime in the first product is 3:1.
在一些实施方式中,步骤3)中的醇类溶剂包含甲醇、乙醇、异丙醇、叔丁醇中的至少一种;优选的,步骤3)中的醇类溶剂为叔丁醇。In some embodiments, the alcoholic solvent in step 3) includes at least one of methanol, ethanol, isopropanol, and tert-butanol; preferably, the alcoholic solvent in step 3) is tert-butanol.
在一些实施方式中,步骤3)中所述取代试剂选自硫脲、硫氢化钠、硫氢化钾中的一种;优选的,所述取代试剂为硫脲。In some embodiments, the substitution reagent in step 3) is selected from one of thiourea, sodium hydrosulfide, and potassium hydrosulfide; preferably, the substitution reagent is thiourea.
在一些实施方式中,步骤3)中所述取代试剂的用量、第二产物中的3-溴-5,5-二甲基-4,5-二氢异噁唑之间的摩尔比为(0.5-1.5):1;优选的,所述取代试剂的用量、第二产物中的3-溴-5,5-二甲基-4,5-二氢异噁唑之间的摩尔比为1.1:1。In some embodiments, the molar ratio between the amount of the substitution reagent described in step 3) and the 3-bromo-5,5-dimethyl-4,5-dihydroisoxazole in the second product is ( 0.5-1.5): 1; preferably, the molar ratio between the amount of the substitution reagent and the 3-bromo-5,5-dimethyl-4,5-dihydroisoxazole in the second product is 1.1 :1.
在一些实施方式中,步骤3)中所述卤化氢溶液选自溴化氢溶液、氯化氢溶液、氟化氢溶液中的一种;优选的,所述卤化氢溶液为溴化氢溶液。In some embodiments, the hydrogen halide solution in step 3) is selected from a hydrogen bromide solution, a hydrogen chloride solution, and a hydrogen fluoride solution; preferably, the hydrogen halide solution is a hydrogen bromide solution.
在一些实施方式中,步骤1)中所述第一产物中二溴甲醛肟的含量和产率通过HPLC检测。In some embodiments, the content and yield of dibromoformaldoxime in the first product described in step 1) are detected by HPLC.
在一些实施方式中,步骤2)中所述第二产物3-溴-5,5-二甲基-4,5-二氢异恶唑的含量和产率通过HPLC检测。In some embodiments, the content and yield of the second product 3-bromo-5,5-dimethyl-4,5-dihydroisoxazole in step 2) are detected by HPLC.
在一些实施方式中,步骤3)中所述中间体1的纯度HPLC检测。In some embodiments, the purity of the intermediate 1 in step 3) is detected by HPLC.
本发明中所述中间体1为盐,因此鉴定方法是用质谱,HRMS(ESI):calcd.ForC6H12BrN3OS:[Μ]+m/z 253.0。The intermediate 1 described in the present invention is a salt, so the identification method is mass spectrometry, HRMS (ESI): calcd. ForC 6 H 12 BrN 3 OS: [M] + m/z 253.0.
中间体2Intermediate 2
在一些实施方式中,所述步骤(1)为对反应体(Ⅰ)进行羟烷基化、氟甲基化、氯化反应制得中间体2。In some embodiments, the step (1) is to perform hydroxyalkylation, fluoromethylation and chlorination on the reactant (I) to obtain the intermediate 2.
在一些优选的实施方式中,所述步骤(1)如下所示:取反应体(Ⅰ)、第一无机碱溶液进行混合,滴加甲醛溶液,结束后加入第二无机碱溶液、第一有机溶剂,通入一氯二氟甲烷气体,并搅拌,静置分层,分离出的上层有机相,浓缩得到初反应物;将初反应物溶解在第二有机溶剂中,在0℃以下滴加氯化试剂,回温至20-30℃,反应至少30min,加水混合,分离出下层有机相,浓缩即得中间体中间体2。In some preferred embodiments, the step (1) is as follows: mixing the reactant (I) and the first inorganic alkali solution, adding formaldehyde solution dropwise, and adding the second inorganic alkali solution, the first organic alkali solution after the end Solvent, introduce chlorodifluoromethane gas, stir, stand for stratification, separate the upper organic phase, concentrate to obtain the initial reactant; dissolve the initial reactant in the second organic solvent, add dropwise below 0 °C Chlorinating reagent, return the temperature to 20-30 °C, react for at least 30 min, add water and mix, separate the lower organic phase, and concentrate to obtain Intermediate Intermediate 2.
在一些更优选的实施方式中,所述步骤(1)如下所示:取50g(0.30mol)反应体(Ⅰ)、36g氢氧化钠水溶液(50%,0.45mol,1.5eq)进行混合,滴加33g甲醛溶液(30%,0.33mol),结束后加入36g氢氧化钠水溶液(50%,0.45mol,1.5eq)、乙腈,通入一氯二氟甲烷气体,并搅拌,静置分层,分离出的上层有机相,浓缩得到初反应物;将初反应物溶解在1,2-二氯乙烷中,在0℃以下滴加35.7g二氯亚砜(0.30mol),回温至20-30℃,反应至少30min,加水混合,分离出下层有机相,浓缩即得中间体2。In some more preferred embodiments, the step (1) is as follows: take 50g (0.30mol) reactant (I), 36g aqueous sodium hydroxide solution (50%, 0.45mol, 1.5eq) and mix, dropwise Add 33g formaldehyde solution (30%, 0.33mol), add 36g aqueous sodium hydroxide solution (50%, 0.45mol, 1.5eq) and acetonitrile after the end, pass into chlorodifluoromethane gas, stir, stand for stratification, The separated upper organic phase was concentrated to obtain the initial reactant; the initial reactant was dissolved in 1,2-dichloroethane, and 35.7g of thionyl chloride (0.30mol) was added dropwise below 0°C, and the temperature was returned to 20 -30°C, react for at least 30 min, add water and mix, separate the lower organic phase, and concentrate to obtain Intermediate 2.
在本发明中,对砜吡草唑的制备工艺进行改进,发明人发现,先通过O-二氟甲基化合成中间物质的中间体2,然后再与中间体1进行对接反应,最后再氧化成目标产物,可以在最后得到的砜吡草唑的反应产物的纯度和收率得到明显的提高,发明人认为可能是由于,在本发明中通过先通过O-二氟甲基化合成中间物质的中间体2再与中间体1对接反应,可以避免先对中间体1与反应体(Ⅰ)进行反应之后再进行O-二氟甲基化时带来的N-二氟甲基化副产物,发明人认为,如果中间体1在先与反应体(Ⅰ)进行反应之后,会使得该路线制备的反应中间产物中含有碳氮双键和碳硫键,该结构性质非常的不稳定,但是该路线时的反应中间物还需要经过各种的氯化氧化等各种活性较高的反应过程,容易形成的N-二氟甲基化副产物不容易去除,造成最终制备的砜吡草唑的含量和收率都比较低,而采用先通过O-二氟甲基化合成中间物质的中间体2再与中间体1对接反应,则可以很好的避免带来的N-二氟甲基化副产物。In the present invention, the preparation process of sulfofenazone is improved. The inventor finds that the intermediate 2 of the intermediate substance is first synthesized by O-difluoromethylation, and then the docking reaction is carried out with the intermediate 1, and finally the oxidation is carried out again. The target product can be obtained, and the purity and yield of the reaction product of sulfometazone obtained at the end can be significantly improved. The inventor believes that it may be because, in the present invention, the intermediate substance is synthesized through O-difluoromethylation first. The intermediate 2 is then docked with the intermediate 1, which can avoid the by-product of N-difluoromethylation when the intermediate 1 is reacted with the reactant (I) first and then the O-difluoromethylation is carried out. , the inventor believes that if the intermediate 1 is reacted with the reactant (I) first, the reaction intermediate prepared by this route will contain carbon-nitrogen double bonds and carbon-sulfur bonds, and the structural properties are very unstable, but The reaction intermediate in this route also needs to undergo various reaction processes with high activity such as various chlorination and oxidation, and the easily formed N-difluoromethylation by-product is not easy to remove, resulting in the final preparation of fenflufenac The content and yield of N-difluoromethyl are relatively low, and the use of intermediate 2, which first synthesizes the intermediate substance through O-difluoromethylation, and then docking with intermediate 1, can well avoid the resulting N-difluoromethyl. by-products.
本发明中,所述中间体2核磁测试结果为:1H NMR(400MHz,CDCl3):6.69(1H,t,J=71.5Hz),4.51(2H,s),3.82(3H,s)。In the present invention, the nuclear magnetic test results of the intermediate 2 are: 1 H NMR (400MHz, CDCl3): 6.69 (1H, t, J=71.5Hz), 4.51 (2H,s), 3.82 (3H,s).
在一些实施方式中,所述中间体1的纯度≥90%;所述中间体2的纯度≥85%;优选的,所述中间体1的纯度为95%;所述中间体2的纯度为90%。In some embodiments, the purity of the intermediate 1 is ≥90%; the purity of the intermediate 2 is ≥85%; preferably, the purity of the intermediate 1 is 95%; the purity of the intermediate 2 is 90%.
在一些优选的实施方式中,所述步骤(1)中第一无机碱溶液、第二无机碱溶液独立的选自氢氧化钠水溶液、氢氧化钾、碳酸钾、碳酸氢钾中的至少一种;优选的,所述步骤(1)中第一无机碱溶液、第二无机碱溶液分别为氢氧化钠水溶液;更优选的,所述氢氧化钠水溶液质量浓度为40-60%;更优选的,氢氧化钠水溶液质量浓度为50%。In some preferred embodiments, in the step (1), the first inorganic alkali solution and the second inorganic alkali solution are independently selected from at least one of sodium hydroxide aqueous solution, potassium hydroxide, potassium carbonate and potassium bicarbonate Preferably, the first inorganic alkali solution and the second inorganic alkali solution in the step (1) are respectively an aqueous sodium hydroxide solution; more preferably, the mass concentration of the aqueous sodium hydroxide solution is 40-60%; more preferably , the mass concentration of sodium hydroxide aqueous solution is 50%.
在一些优选的实施方式中,所述步骤(1)中的第一有机溶剂、第二有机溶剂独立的选自乙腈、1,2-二氯乙烷、二氯甲烷、氯苯中的至少一种;更有选的,所述步骤(1)中的第一有机溶剂为乙腈;所述步骤(1)中的第二有机溶剂为1,2-二氯乙烷。In some preferred embodiments, the first organic solvent and the second organic solvent in the step (1) are independently selected from at least one of acetonitrile, 1,2-dichloroethane, dichloromethane, and chlorobenzene more preferably, the first organic solvent in the step (1) is acetonitrile; the second organic solvent in the step (1) is 1,2-dichloroethane.
在一些实施方式中,所述步骤(1)中氯化反应过程中包含氯化试剂;所述氯化试剂包含二氯亚砜、三氯化磷、磺酰氯、氯气、碳酰氯、苯甲酰氯中的至少一种;优选的,所述氯化试剂为二氯亚砜。In some embodiments, a chlorination reagent is included during the chlorination reaction in the step (1); the chlorination reagent includes thionyl chloride, phosphorus trichloride, sulfonyl chloride, chlorine, phosgene, and benzoyl chloride At least one of; preferably, the chlorination reagent is thionyl chloride.
在一些实施方式中,所述反应体(Ⅰ)的制备原料包括三氟乙酰乙酸乙酯;所述中间体1的制备原料包括乙醛酸、硫脲、硫氢化盐、溴中的至少一种。In some embodiments, the raw materials for the preparation of the reactant (I) include ethyl trifluoroacetoacetate; the raw materials for the preparation of the intermediate 1 include at least one of glyoxylic acid, thiourea, hydrosulfide, and bromine .
在一些实施方式中,所述反应体(Ⅰ)得制备方法如下所示:In some embodiments, the preparation method of the reactant (I) is as follows:
取92g三氟乙酰乙酸乙酯(0.50mol)和150mL溶剂(可用乙醇、甲醇等溶剂替代),在0℃下,滴加64g甲基肼(40%,0.55mol,1.1eq),然后将反应升温至80℃并搅拌5小时。反应结束后,向反应中加入200mL水并搅拌30分钟,有白色固体析出,抽滤,滤饼用水洗涤三遍,即得反应体(Ⅰ)78g;所述反应体(Ⅰ)的含量为97%,产率:96%。Take 92 g of ethyl trifluoroacetoacetate (0.50 mol) and 150 mL of solvent (which can be replaced by ethanol, methanol and other solvents), at 0 °C, add 64 g of methylhydrazine (40%, 0.55 mol, 1.1 eq) dropwise, and then react The temperature was raised to 80°C and stirred for 5 hours. After the reaction was completed, 200 mL of water was added to the reaction and stirred for 30 minutes, a white solid was precipitated, suction filtration, and the filter cake was washed three times with water to obtain 78 g of reaction body (I); the content of the reaction body (I) was 97 g. %, yield: 96%.
在一些实施方式中,反应体(Ⅰ)得制备方法中所述溶剂选自乙酸、乙醇、甲醇、叔丁醇中的至少一种;优选的,所述溶剂为乙酸。In some embodiments, the solvent in the method for preparing the reactant (I) is selected from at least one of acetic acid, ethanol, methanol, and tert-butanol; preferably, the solvent is acetic acid.
所述反应体(Ⅰ)为N-甲基-3-三氟甲基-5-羟基吡唑。The reactant (I) is N-methyl-3-trifluoromethyl-5-hydroxypyrazole.
在一些实施方式中,所述步骤(2):将中间体1、中间体2进行混合,加水分离出有机相,得到过渡中间体,加入溶剂、氧化剂进行反应,即得;In some embodiments, the step (2): mix the intermediate 1 and the intermediate 2, add water to separate the organic phase, obtain a transition intermediate, add a solvent and an oxidant to react, and then obtain;
在一些优选的实施方式中,所述步骤(2):将中间体1、第一无机碱、第一有机溶剂搅拌混合,继续滴加中间体2,搅拌2-6小时,加水静置混合分离出有机相,浓缩得过渡中间体;将过渡中间体、溶剂混合,加入二水合钨酸钠、0.5-5mL浓硫酸、氧化剂,继续搅拌1-10小时后,加入水搅拌,过滤干燥即得。In some preferred embodiments, the step (2): stir and mix the intermediate 1, the first inorganic base and the first organic solvent, continue to drop the intermediate 2, stir for 2-6 hours, add water and stand for mixing and separation The organic phase is taken out and concentrated to obtain the transition intermediate; the transition intermediate and solvent are mixed, sodium tungstate dihydrate, 0.5-5 mL concentrated sulfuric acid and oxidizing agent are added, and after stirring for 1-10 hours, water is added, stirring, and filtration and drying.
在一些更优选的实施方式中,所述步骤(2):将42g中间体1(0.20mol)、41.4g碳酸钾(0.30mol)、乙腈搅拌混合,继续滴加58.7g中间体2(0.20mol,90%),搅拌4小时,加水静置混合分离出有机相,浓缩得过渡中间体;将过渡中间体、300mL溶剂混合,加入1.25g二水合钨酸钠(3.80mmol)、1mL浓硫酸(98%)、64.6g双氧水(0.57mol,30%),继续搅拌6小时后,加入水搅拌,过滤干燥即得。In some more preferred embodiments, the step (2): 42g of intermediate 1 (0.20mol), 41.4g of potassium carbonate (0.30mol), and acetonitrile were stirred and mixed, and 58.7g of intermediate 2 (0.20mol) was added dropwise. , 90%), stir for 4 hours, add water and stand to mix to separate out the organic phase, and concentrate to obtain the transition intermediate; mix the transition intermediate and 300 mL of solvent, add 1.25 g of sodium tungstate dihydrate (3.80 mmol), 1 mL of concentrated sulfuric acid ( 98%), 64.6g hydrogen peroxide (0.57mol, 30%), continue stirring for 6 hours, add water, stir, filter and dry.
本发明中所述步骤(2)制备得到的砜吡草唑核磁测试为:1H NMR(400MHz,CDCl3)δ:6.83(t,J=71.9Hz,1H,),4.60(s,2H),3.88(s,3H),3.11(s,2H),1.52(s,6H)。The nuclear magnetic test of sulfofenazone prepared by step (2) in the present invention is: 1 H NMR (400MHz, CDCl3) δ: 6.83(t, J=71.9Hz, 1H, ), 4.60(s, 2H), 3.88 (s, 3H), 3.11 (s, 2H), 1.52 (s, 6H).
在一些实施方式中,所述步骤(2)的氧化剂包含双氧水、间氯过氧苯甲酸、单过硫酸氢钾复合盐中的至少一种;优选的,所述步骤(2)的氧化剂为双氧水。In some embodiments, the oxidant in the step (2) comprises at least one of hydrogen peroxide, m-chloroperoxybenzoic acid, and potassium monopersulfate composite salt; preferably, the oxidant in the step (2) is hydrogen peroxide .
在一些实施方式中,所述氧化剂中还包含二水合钨酸钠;所述双氧水、二水合钨酸钠之间的摩尔比为1:(0.005-0.35);优选的,所述双氧水、二水合钨酸钠之间的摩尔比为1:0.007。In some embodiments, the oxidizing agent further comprises sodium tungstate dihydrate; the molar ratio between the hydrogen peroxide and sodium tungstate dihydrate is 1:(0.005-0.35); The molar ratio between sodium tungstate is 1:0.007.
在一些实施方式中,所述步骤(2)中的溶剂包含C1-C8的小分子醇、二氯甲烷、1,2-二氯乙烷、氯苯、二氯苯、乙酸乙酯、乙酸丁酯中的至少一种;优选的,所述步骤(2)中的溶剂包含C1-C8的小分子醇。In some embodiments, the solvent in the step (2) comprises C1-C8 small molecular alcohol, dichloromethane, 1,2-dichloroethane, chlorobenzene, dichlorobenzene, ethyl acetate, butyl acetate At least one of esters; preferably, the solvent in the step (2) contains C1-C8 small molecular alcohol.
在一些实施方式中,所述C1-C8的小分子醇选自甲醇、乙醇、叔丁醇、丁二醇、丙二醇、异丁醇、异戊醇、异丙醇、乙二醇、丙三醇中的至少一种;优选的,所述C1-C8的小分子醇选自甲醇、乙醇、叔丁醇;更有选的,所述C1-C8的小分子醇为甲醇。In some embodiments, the C1-C8 small molecule alcohol is selected from methanol, ethanol, tert-butanol, butanediol, propylene glycol, isobutanol, isoamyl alcohol, isopropanol, ethylene glycol, glycerol At least one of; preferably, the C1-C8 small molecular alcohol is selected from methanol, ethanol, and tert-butanol; more preferably, the C1-C8 small molecular alcohol is methanol.
本发明中在先通过O-二氟甲基化合成中间物质的中间体2,然后再与中间体1进行对接反应,制备得到的过渡中间体还需要进一步的进行氧化反应,发明人发现,在该过程中间体1的纯度和该氧化过程的条件对于制备得到的目标产物的纯度和收率影响非常大,而发明人意外发现,在该氧化过程中使用C1-C8的小分子醇做溶剂,配合使用双氧水和钨酸钠催化剂,其中中间体1的纯度需要≥90%的条件下,最后对制备得到目标产物的纯度可以达到99%,收率达到92%,发明人认为可能是使用C1-C8的小分子醇做溶剂作为溶剂时,尤其是选用甲醇作为溶剂时,可以降低该氧化过程中副产的生成,二是最后一步的溶剂使用小分子醇替换常规的甲酸做溶剂,可以利于回收溶剂,并且避免了乙酸做溶剂时的难以回收溶剂的问题且产生大量废水,造成污染的缺陷。此外,在本发明中,在合成中间体1和中间体2过程中都不需要采进行出料纯化,可以直接进行下一步反应的合成工艺,提高了目标产物的整体产率,降低了三废的产生以及实现溶剂回收套用等优异的效果。In the present invention, the intermediate 2 of the intermediate substance is first synthesized by O-difluoromethylation, and then the docking reaction is carried out with the intermediate 1, and the prepared transition intermediate needs further oxidation reaction. The purity of the process intermediate 1 and the conditions of the oxidation process have a great influence on the purity and yield of the prepared target product, and the inventor unexpectedly found that in the oxidation process, a C1-C8 small molecule alcohol is used as a solvent, Combined use of hydrogen peroxide and sodium tungstate catalyst, where the purity of intermediate 1 needs to be ≥90%, the final purity of the prepared target product can reach 99%, and the yield can reach 92%. The inventor believes that it may be the use of C1- When the small molecular alcohol of C8 is used as the solvent, especially when methanol is used as the solvent, the generation of by-products in the oxidation process can be reduced. Second, the solvent in the last step uses small molecular alcohol instead of conventional formic acid as the solvent, which can be beneficial to recovery. solvent, and avoids the problem of being difficult to recover the solvent when acetic acid is used as the solvent, and generates a large amount of waste water, causing the defect of pollution. In addition, in the present invention, in the process of synthesizing intermediate 1 and intermediate 2, it is not necessary to extract and purify the material, and the synthesis process of the next reaction can be directly carried out, which improves the overall yield of the target product and reduces the three wastes. Produce and realize excellent effects such as solvent recovery and application.
本发明的第二个方面提供了一种砜吡草唑的应用,应用于农药;所述砜吡草唑由所述砜吡草唑的合成方法制备得到。The second aspect of the present invention provides an application of fenfenapyr, which is applied to a pesticide; the fenfenapyr is prepared by the method for synthesizing the fenfenpyr.
下面通过实施例对本发明进行具体的描述,以下实施例只能用于本发明做进一步说明,并不能理解为本发明保护的限制,该领域的专业技术人员根据上述发明的内容作出的非本质的改正和调整,仍属于本发明的保护的范围。The present invention will be specifically described by the following examples. The following examples can only be used to further illustrate the present invention, and should not be construed as a limitation of the protection of the present invention. Corrections and adjustments still fall within the protection scope of the present invention.
实施例1Example 1
一种砜吡草唑的合成方法,制备步骤包括:A method for synthesizing sulfofenazone, the preparation steps comprising:
步骤(1):取50g(0.30mol)反应体(Ⅰ)、36g氢氧化钠水溶液(50%,0.45mol,1.5eq)进行混合,滴加33g甲醛溶液(30%,0.33mol),结束后加入36g氢氧化钠水溶液(50%,0.45mol,1.5eq)、100mL乙腈,通入一氯二氟甲烷气体,并搅拌,静置分层,分离出的上层有机相,浓缩得到初反应物;将初反应物溶解在1,2-二氯乙烷中,在0℃滴加35.7g二氯亚砜(0.3mol),回温至25℃,反应至少30min,加水混合,分离出下层有机相,浓缩即得中间体2。Step (1): Mix 50g (0.30mol) reactant (I) and 36g aqueous sodium hydroxide solution (50%, 0.45mol, 1.5eq), dropwise add 33g formaldehyde solution (30%, 0.33mol), after the end Add 36g sodium hydroxide aqueous solution (50%, 0.45mol, 1.5eq), 100mL acetonitrile, pass into chlorodifluoromethane gas, stir, stand for layering, the separated upper organic phase is concentrated to obtain the initial reactant; The initial reactant was dissolved in 1,2-dichloroethane, 35.7 g of thionyl chloride (0.3 mol) was added dropwise at 0 °C, the temperature was returned to 25 °C, the reaction was performed for at least 30 min, water was added to mix, and the lower organic phase was separated. , and concentrated to obtain intermediate 2.
所述步骤(2):将42g中间体1(0.20mol)、41.4g碳酸钾(0.30mol)、200mL乙腈搅拌混合,继续滴加58.7g中间体2(0.20mol,90%),搅拌4小时,加水静置混合分离出有机相,浓缩得过渡中间体;将过渡中间体、300mL甲醇混合,加入1.25g二水合钨酸钠(3.80mmol)、1mL浓硫酸(98%)、64.6g双氧水(0.57mol,30%),继续搅拌6小时后,加入水搅拌,过滤干燥即得。The step (2): 42g of intermediate 1 (0.20mol), 41.4g of potassium carbonate (0.30mol), and 200mL of acetonitrile were stirred and mixed, and 58.7g of intermediate 2 (0.20mol, 90%) was added dropwise, and stirred for 4 hours , add water to stand and mix to separate the organic phase, and concentrate to obtain the transition intermediate; mix the transition intermediate and 300 mL of methanol, add 1.25 g of sodium tungstate dihydrate (3.80 mmol), 1 mL of concentrated sulfuric acid (98%), 64.6 g of hydrogen peroxide ( 0.57mol, 30%), continue stirring for 6 hours, add water, stir, filter and dry.
所述中间体1的纯度为95%;所述中间体2的纯度为90%。The purity of the intermediate 1 is 95%; the purity of the intermediate 2 is 90%.
所述中间体1的合成方法,步骤包括:The synthetic method of described intermediate 1, the step comprises:
1)在第一反应容器中,加入80g乙醛酸水溶液(50%水溶液,0.50mol)、盐酸羟胺(0.55mol,1.1eq)、120g水,搅拌溶解,然后升温至70℃并搅拌3小时;将反应温度降至0℃,加入117g碳酸钠(1.10mol,2.2eq)和150g1,2-二氯乙烷,滴加80g液溴(0.50mol,1eq),结束后回温至25℃,并保温8小时,分离有机相,作为第一产物;步骤1)中水、乙醛酸水溶液的质量比为1.5:1。1) In the first reaction vessel, add 80 g of glyoxylic acid aqueous solution (50% aqueous solution, 0.50 mol), hydroxylamine hydrochloride (0.55 mol, 1.1 eq), 120 g of water, stir to dissolve, then be warmed to 70 ° C and stirred for 3 hours; The reaction temperature was lowered to 0 °C, 117 g of sodium carbonate (1.10 mol, 2.2 eq) and 150 g of 1,2-dichloroethane were added, 80 g of liquid bromine (0.50 mol, 1 eq) was added dropwise, and the temperature was returned to 25 ° C after completion, and Incubate for 8 hours, separate the organic phase, and use it as the first product; the mass ratio of water and glyoxylic acid aqueous solution in step 1) is 1.5:1.
2)向第一产物中加入120g碳酸氢钠(1.44mol,3eq),搅拌升温至80℃,最后以10g/min速率通入异丁烯气体,反应2小时。反应结束后加入600g水,分离出有机相,除溶剂,得到第二产物;2) 120 g of sodium bicarbonate (1.44 mol, 3 eq) was added to the first product, the temperature was raised to 80° C. with stirring, and finally isobutylene gas was introduced at a rate of 10 g/min to react for 2 hours. After the reaction finishes, add 600g of water, separate out the organic phase, remove the solvent, and obtain the second product;
3)将取代试剂(0.33mol,1.1eq)、100mL叔丁醇、卤化氢溶液加入到第二反应容器中混合,再加入1mL溴化氢(48wt%),将第二产物滴加至第二反应容器中,结束后,除溶剂,叔丁醇重结晶,即得;所述取代试剂为硫脲。3) Add the substitution reagent (0.33mol, 1.1eq), 100mL tert-butanol and hydrogen halide solution into the second reaction vessel and mix, then add 1mL hydrogen bromide (48wt%), and add the second product dropwise to the second reaction vessel. In the reaction vessel, after the end, the solvent is removed, and the tert-butanol is recrystallized to obtain; the substitution reagent is thiourea.
所述反应体(Ⅰ)的结构为式1所示;所述中间体1的结构式为式2-1;The structure of the reactant (I) is shown in formula 1; the structural formula of the intermediate 1 is formula 2-1;


实施例2Example 2
一种砜吡草唑的合成方法,制备步骤包括:A method for synthesizing sulfofenazone, the preparation steps comprising:
步骤(1):取50g(0.30mol)反应体(Ⅰ)、50g氢氧化钾水溶液(50%,0.45mol,1.5eq)进行混合,滴加33g甲醛溶液(30%,0.33mol),结束后加入50g氢氧化钾水溶液(50%,0.45mol,1.5eq)、100mL1,2-二氯乙烷,通入一氯二氟甲烷气体,并搅拌,静置分层,分离出的上层有机相,浓缩得到初反应物;将初反应物溶解在1,2-二氯乙烷中,在0℃滴加29.7g碳酰氯(0.30mol),回温至25℃,反应30min,加水混合,分离出下层有机相,浓缩即得中间体2。Step (1): get 50g (0.30mol) reactant (I), 50g potassium hydroxide aqueous solution (50%, 0.45mol, 1.5eq) and mix, dropwise add 33g formaldehyde solution (30%, 0.33mol), after finishing Add 50 g of potassium hydroxide aqueous solution (50%, 0.45 mol, 1.5 eq), 100 mL of 1,2-dichloroethane, pass chlorodifluoromethane gas, stir, stand for stratification, and separate the upper organic phase, Concentrate to obtain the initial reactant; dissolve the initial reactant in 1,2-dichloroethane, add 29.7 g of phosgene (0.30 mol) dropwise at 0 °C, return the temperature to 25 °C, react for 30 min, add water and mix, and separate out The lower organic phase was concentrated to obtain Intermediate 2.
所述步骤(2):将42g中间体1(0.20mol)、41.4g碳酸钾(0.30mol)、200mL二氯甲烷搅拌混合,继续滴加58.7g中间体2(0.20mol,90%),搅拌4小时,加水静置混合分离出有机相,浓缩得过渡中间体;将过渡中间体、300mL叔丁醇混合,加入1.25g二水合钨酸钠(3.80mmol)、1mL浓硫酸(98%)、64.6g双氧水(0.57mol,30%),继续搅拌6小时后,加入水搅拌,过滤干燥即得。The step (2): 42g of intermediate 1 (0.20mol), 41.4g of potassium carbonate (0.30mol), and 200mL of dichloromethane were stirred and mixed, and 58.7g of intermediate 2 (0.20mol, 90%) was added dropwise, and stirred For 4 hours, add water to stand and mix to separate the organic phase, and concentrate to obtain the transition intermediate; mix the transition intermediate and 300 mL of tert-butanol, add 1.25 g of sodium tungstate dihydrate (3.80 mmol), 1 mL of concentrated sulfuric acid (98%), 64.6 g of hydrogen peroxide (0.57 mol, 30%) was continuously stirred for 6 hours, then added with water, stirred, filtered and dried.
所述中间体1的纯度为95%;所述中间体2的纯度为90%。The purity of the intermediate 1 is 95%; the purity of the intermediate 2 is 90%.
所述中间体1的合成方法,步骤包括:The synthetic method of described intermediate 1, the step comprises:
1)在第一反应容器中,加入80g乙醛酸水溶液(50%水溶液,0.5mol)、盐酸羟胺(0.55mol,1.1eq)、120g水,搅拌溶解,然后升温至70℃并搅拌3小时;将反应温度降至0℃,加入117g碳酸钠(1.10mol,2.2eq)和150g1,2-二氯乙烷,滴加80g液溴(0.50mol,1eq),结束后回温至25℃,并保温8小时,分离有机相,作为第一产物;步骤1)中水、乙醛酸水溶液的质量比为1.5:1。1) In the first reaction vessel, add 80g glyoxylic acid aqueous solution (50% aqueous solution, 0.5mol), hydroxylamine hydrochloride (0.55mol, 1.1eq), 120g water, stir to dissolve, then be warming up to 70 ° C and stir for 3 hours; The reaction temperature was lowered to 0 °C, 117 g of sodium carbonate (1.10 mol, 2.2 eq) and 150 g of 1,2-dichloroethane were added, 80 g of liquid bromine (0.50 mol, 1 eq) was added dropwise, and the temperature was returned to 25 ° C after completion, and Incubate for 8 hours, separate the organic phase, and use it as the first product; the mass ratio of water and glyoxylic acid aqueous solution in step 1) is 1.5:1.
2)向第一产物中加入120g碳酸氢钠(1.44mol,3eq),搅拌升温至80℃,最后以10g/min速率通入异丁烯气体,反应2小时。反应结束后加入600g水,分离出有机相,除溶剂,得到第二产物;2) 120 g of sodium bicarbonate (1.44 mol, 3 eq) was added to the first product, the temperature was raised to 80° C. with stirring, and finally isobutylene gas was introduced at a rate of 10 g/min to react for 2 hours. After the reaction finishes, add 600g of water, separate out the organic phase, remove the solvent, and obtain the second product;
3)将取代试剂(0.33mol,1.1eq)、100mL叔丁醇、卤化氢溶液加入到第二反应容器中混合,再加入1mL溴化氢(48wt%),将第二产物滴加至第二反应容器中,结束后,除溶剂,叔丁醇重结晶,即得;所述取代试剂为硫脲。3) Add the substitution reagent (0.33mol, 1.1eq), 100mL tert-butanol and hydrogen halide solution into the second reaction vessel and mix, then add 1mL hydrogen bromide (48wt%), and add the second product dropwise to the second reaction vessel. In the reaction vessel, after the end, the solvent is removed, and the tert-butanol is recrystallized to obtain; the substitution reagent is thiourea.
所述反应体(Ⅰ)的结构为式1所示;所述中间体1的结构式为式2-1;The structure of the reactant (I) is shown in formula 1; the structural formula of the intermediate 1 is formula 2-1;

实施例3Example 3
一种砜吡草唑的合成方法,制备步骤包括:A method for synthesizing sulfofenazone, the preparation steps comprising:
步骤(1):取50g(0.30mol)反应体(Ⅰ)、36g氢氧化钠水溶液(50%,0.45mol,1.5eq)进行混合,滴加33g甲醛溶液(30%,0.33mol),结束后加入36g氢氧化钠水溶液(50%,0.45mol,1.5eq)、100mL乙腈,通入一氯二氟甲烷气体,并搅拌,静置分层,分离出的上层有机相,浓缩得到初反应物;将初反应物溶解在1,2-二氯乙烷中,在0℃滴加35.7g二氯亚砜(0.30mol),回温至25℃,反应至少30min,加水混合,分离出下层有机相,浓缩即得中间体2。Step (1): Mix 50g (0.30mol) reactant (I) and 36g aqueous sodium hydroxide solution (50%, 0.45mol, 1.5eq), dropwise add 33g formaldehyde solution (30%, 0.33mol), after the end Add 36g sodium hydroxide aqueous solution (50%, 0.45mol, 1.5eq), 100mL acetonitrile, pass into chlorodifluoromethane gas, stir, stand for layering, the separated upper organic phase is concentrated to obtain the initial reactant; Dissolve the initial reactant in 1,2-dichloroethane, add 35.7 g of thionyl chloride (0.30 mol) dropwise at 0 °C, return the temperature to 25 °C, react for at least 30 min, add water and mix, and separate the lower organic phase , and concentrated to obtain intermediate 2.
所述步骤(2):将42g中间体1(0.20mol)、41.4g碳酸钾(0.30mol)、200mL乙腈搅拌混合,继续滴加58.7g中间体2(0.20mol,90%),搅拌4小时,加水静置混合分离出有机相,浓缩得过渡中间体;将过渡中间体、300mL二氯甲烷混合,98.36g间氯过氧苯甲酸(0.57mol),继续搅拌6小时后,加入水搅拌,过滤干燥即得。The step (2): 42g of intermediate 1 (0.20mol), 41.4g of potassium carbonate (0.30mol), and 200mL of acetonitrile were stirred and mixed, and 58.7g of intermediate 2 (0.20mol, 90%) was added dropwise, and stirred for 4 hours , add water to stand and mix to separate out the organic phase, and concentrate to obtain the transition intermediate; mix the transition intermediate and 300 mL of dichloromethane, 98.36 g of m-chloroperoxybenzoic acid (0.57 mol), continue stirring for 6 hours, add water and stir, Filter and dry.
所述中间体1的纯度为95%;所述中间体2的纯度为90%。The purity of the intermediate 1 is 95%; the purity of the intermediate 2 is 90%.
所述中间体1的合成方法,步骤包括:The synthetic method of described intermediate 1, the step comprises:
1)在第一反应容器中,加入80g乙醛酸水溶液(50%水溶液,0.50mol)、盐酸羟胺(0.55mol,1.1eq)、120g水,搅拌溶解,然后升温至70℃并搅拌3小时;将反应温度降至0℃,加入117g碳酸钠(1.10mol,2.2eq)和150g1,2-二氯乙烷,滴加80g液溴(0.50mol,1eq),结束后回温至25℃,并保温8小时,分离有机相,作为第一产物;步骤1)中水、乙醛酸水溶液的质量比为1.5:1。1) In the first reaction vessel, add 80 g of glyoxylic acid aqueous solution (50% aqueous solution, 0.50 mol), hydroxylamine hydrochloride (0.55 mol, 1.1 eq), 120 g of water, stir to dissolve, then be warmed to 70 ° C and stirred for 3 hours; The reaction temperature was lowered to 0 °C, 117 g of sodium carbonate (1.10 mol, 2.2 eq) and 150 g of 1,2-dichloroethane were added, 80 g of liquid bromine (0.50 mol, 1 eq) was added dropwise, and the temperature was returned to 25 ° C after completion, and Incubate for 8 hours, separate the organic phase, and use it as the first product; the mass ratio of water and glyoxylic acid aqueous solution in step 1) is 1.5:1.
2)向第一产物中加入120g碳酸氢钠(1.44mol,3eq),搅拌升温至80℃,最后以10g/min速率通入异丁烯气体,反应2小时。反应结束后加入600g水,分离出有机相,除溶剂,得到第二产物;2) 120 g of sodium bicarbonate (1.44 mol, 3 eq) was added to the first product, the temperature was raised to 80° C. with stirring, and finally isobutylene gas was introduced at a rate of 10 g/min to react for 2 hours. After the reaction finishes, add 600g of water, separate out the organic phase, remove the solvent, and obtain the second product;
3)将取代试剂(0.33mol,1.1eq)、100mL叔丁醇、卤化氢溶液加入到第二反应容器中混合,再加入1mL溴化氢(48wt%),将第二产物滴加至第二反应容器中,结束后,除溶剂,叔丁醇重结晶,即得;所述取代试剂为硫氢化钠。3) Add the substitution reagent (0.33mol, 1.1eq), 100mL tert-butanol and hydrogen halide solution into the second reaction vessel and mix, then add 1mL hydrogen bromide (48wt%), and add the second product dropwise to the second reaction vessel. In the reaction vessel, after the end, the solvent is removed and the tert-butanol is recrystallized to obtain; the substitution reagent is sodium hydrosulfide.
所述反应体(Ⅰ)的结构为式1所示;所述中间体1的结构式为式2-2;The structure of the reactant (I) is shown in formula 1; the structural formula of the intermediate 1 is formula 2-2;

实施例4Example 4
一种砜吡草唑的合成方法,制备步骤包括:A method for synthesizing sulfofenazone, the preparation steps comprising:
步骤(1):取50g(0.30mol)反应体(Ⅰ)、36g氢氧化钠水溶液(50%,0.45mol,1.5eq)进行混合,滴加33g甲醛溶液(30%,0.33mol),结束后加入36g氢氧化钠水溶液(50%,0.45mol,1.5eq)、100mL乙腈,通入一氯二氟甲烷气体,并搅拌,静置分层,分离出的上层有机相,浓缩得到初反应物;将初反应物溶解在1,2-二氯乙烷中,在0℃滴加35.7g二氯亚砜(0.30mol),回温至25℃,反应至少30min,加水混合,分离出下层有机相,浓缩即得中间体2。Step (1): Mix 50g (0.30mol) reactant (I) and 36g aqueous sodium hydroxide solution (50%, 0.45mol, 1.5eq), dropwise add 33g formaldehyde solution (30%, 0.33mol), after the end Add 36g sodium hydroxide aqueous solution (50%, 0.45mol, 1.5eq), 100mL acetonitrile, pass into chlorodifluoromethane gas, stir, stand for layering, the separated upper organic phase is concentrated to obtain the initial reactant; Dissolve the initial reactant in 1,2-dichloroethane, add 35.7 g of thionyl chloride (0.30 mol) dropwise at 0 °C, return the temperature to 25 °C, react for at least 30 min, add water and mix, and separate the lower organic phase , and concentrated to obtain intermediate 2.
所述步骤(2):将42g中间体1(0.20mol)、41.4g碳酸钾(0.30mol)、200mL乙腈搅拌混合,继续滴加58.7g中间体2(0.20mol,90%),搅拌4小时,加水静置混合分离出有机相,浓缩得过渡中间体;将过渡中间体、300mL甲醇混合,加入1mL浓硫酸(98%)、64.6g双氧水(0.57mol,30%),继续搅拌6小时后,加入水搅拌,过滤干燥即得。The step (2): 42g of intermediate 1 (0.20mol), 41.4g of potassium carbonate (0.30mol), and 200mL of acetonitrile were stirred and mixed, and 58.7g of intermediate 2 (0.20mol, 90%) was added dropwise, and stirred for 4 hours , add water to stand and mix to separate the organic phase, and concentrate to obtain the transition intermediate; mix the transition intermediate and 300 mL of methanol, add 1 mL of concentrated sulfuric acid (98%), 64.6 g of hydrogen peroxide (0.57 mol, 30%), and continue to stir for 6 hours. , add water, stir, filter and dry.
所述中间体1的纯度为95%;所述中间体2的纯度为90%。The purity of the intermediate 1 is 95%; the purity of the intermediate 2 is 90%.
所述中间体1的合成方法,步骤包括:The synthetic method of described intermediate 1, the step comprises:
1)在第一反应容器中,加入80g乙醛酸水溶液(50%水溶液,0.5mol)、盐酸羟胺(0.55mol,1.1eq)、120g水,搅拌溶解,然后升温至70℃并搅拌3小时;将反应温度降至0℃,加入117g碳酸钠(1.1mol,2.2eq)和150g1,2-二氯乙烷,滴加80g液溴(0.5mol,1eq),结束后回温至25℃,并保温8小时,分离有机相,作为第一产物;步骤1)中水、乙醛酸水溶液的质量比为1.5:1。1) In the first reaction vessel, add 80g glyoxylic acid aqueous solution (50% aqueous solution, 0.5mol), hydroxylamine hydrochloride (0.55mol, 1.1eq), 120g water, stir to dissolve, then be warming up to 70 ° C and stir for 3 hours; The reaction temperature was lowered to 0 °C, 117 g of sodium carbonate (1.1 mol, 2.2 eq) and 150 g of 1,2-dichloroethane were added, 80 g of liquid bromine (0.5 mol, 1 eq) was added dropwise, and the temperature was returned to 25 ° C after completion, and Incubate for 8 hours, separate the organic phase, and use it as the first product; the mass ratio of water and glyoxylic acid aqueous solution in step 1) is 1.5:1.
2)向第一产物中加入120g碳酸氢钠(1.44mol,3eq),搅拌升温至80℃,最后以10g/min速率通入异丁烯气体,反应2小时。反应结束后加入600g水,分离出有机相,除溶剂,得到第二产物;2) 120 g of sodium bicarbonate (1.44 mol, 3 eq) was added to the first product, the temperature was raised to 80° C. with stirring, and finally isobutylene gas was introduced at a rate of 10 g/min to react for 2 hours. After the reaction finishes, add 600g of water, separate out the organic phase, remove the solvent, and obtain the second product;
3)将取代试剂(0.33mol,1.1eq)、100mL叔丁醇、卤化氢溶液加入到第二反应容器中混合,再加入1mL溴化氢(48wt%),将第二产物滴加至第二反应容器中,结束后,除溶剂,叔丁醇重结晶,即得;所述取代试剂为硫脲。3) Add the substitution reagent (0.33mol, 1.1eq), 100mL tert-butanol and hydrogen halide solution into the second reaction vessel and mix, then add 1mL hydrogen bromide (48wt%), and add the second product dropwise to the second reaction vessel. In the reaction vessel, after the end, the solvent is removed, and the tert-butanol is recrystallized to obtain; the substitution reagent is thiourea.
所述反应体(Ⅰ)的结构为式1所示;所述中间体1的结构式为式2-1;The structure of the reactant (I) is shown in formula 1; the structural formula of the intermediate 1 is formula 2-1;

实施例5Example 5
一种砜吡草唑的合成方法,制备步骤包括:A method for synthesizing sulfofenazone, the preparation steps comprising:
步骤(1):取50g(0.30mol)反应体(Ⅰ)、36g氢氧化钠水溶液(50%,0.45mol,1.5eq)进行混合,滴加33g甲醛溶液(30%,0.33mol),结束后加入36g氢氧化钠水溶液(50%,0.45mol,1.5eq)、100mL乙腈,通入一氯二氟甲烷气体,并搅拌,静置分层,分离出的上层有机相,浓缩得到初反应物;将初反应物溶解在1,2-二氯乙烷中,在0℃滴加35.7g二氯亚砜(0.30mol),回温至25℃,反应至少30min,加水混合,分离出下层有机相,浓缩即得中间体2。Step (1): Mix 50g (0.30mol) reactant (I) and 36g aqueous sodium hydroxide solution (50%, 0.45mol, 1.5eq), dropwise add 33g formaldehyde solution (30%, 0.33mol), after the end Add 36g sodium hydroxide aqueous solution (50%, 0.45mol, 1.5eq), 100mL acetonitrile, pass into chlorodifluoromethane gas, stir, stand for layering, the separated upper organic phase is concentrated to obtain the initial reactant; Dissolve the initial reactant in 1,2-dichloroethane, add 35.7 g of thionyl chloride (0.30 mol) dropwise at 0 °C, return the temperature to 25 °C, react for at least 30 min, add water and mix, and separate the lower organic phase , and concentrated to obtain intermediate 2.
所述步骤(2):将42g中间体1(0.20mol)、41.4g碳酸钾(0.30mol)、200mL乙腈搅拌混合,继续滴加58.7g中间体2(0.20mol,90%),搅拌4小时,加水静置混合分离出有机相,浓缩得过渡中间体;将过渡中间体、300mL乙酸混合,加入1.25g二水合钨酸钠(3.80mmol)、1mL浓硫酸(98%)、64.6g双氧水(0.57mol,30%),继续搅拌6小时后,加入水搅拌,过滤干燥即得。The step (2): 42g of intermediate 1 (0.20mol), 41.4g of potassium carbonate (0.30mol), and 200mL of acetonitrile were stirred and mixed, and 58.7g of intermediate 2 (0.20mol, 90%) was added dropwise, and stirred for 4 hours , add water to stand and mix to separate the organic phase, and concentrate to obtain the transition intermediate; mix the transition intermediate and 300 mL of acetic acid, add 1.25 g of sodium tungstate dihydrate (3.80 mmol), 1 mL of concentrated sulfuric acid (98%), 64.6 g of hydrogen peroxide ( 0.57mol, 30%), continue stirring for 6 hours, add water, stir, filter and dry.
所述中间体1的纯度为95%;所述中间体2的纯度为90%。The purity of the intermediate 1 is 95%; the purity of the intermediate 2 is 90%.
所述中间体1的合成方法,步骤包括:The synthetic method of described intermediate 1, the step comprises:
1)在第一反应容器中,加入80g乙醛酸水溶液(50%水溶液,0.50mol)、盐酸羟胺(0.55mol,1.1eq)、120g水,搅拌溶解,然后升温至70℃并搅拌3小时;将反应温度降至0℃,加入117g碳酸钠(1.10mol,2.2eq)和150g1,2-二氯乙烷,滴加80g液溴(0.50mol,1eq),结束后回温至25℃,并保温8小时,分离有机相,作为第一产物;步骤1)中水、乙醛酸水溶液的质量比为1.5:1。1) In the first reaction vessel, add 80 g of glyoxylic acid aqueous solution (50% aqueous solution, 0.50 mol), hydroxylamine hydrochloride (0.55 mol, 1.1 eq), 120 g of water, stir to dissolve, then be warmed to 70 ° C and stirred for 3 hours; The reaction temperature was lowered to 0 °C, 117 g of sodium carbonate (1.10 mol, 2.2 eq) and 150 g of 1,2-dichloroethane were added, 80 g of liquid bromine (0.50 mol, 1 eq) was added dropwise, and the temperature was returned to 25 ° C after completion, and Incubate for 8 hours, separate the organic phase, and use it as the first product; the mass ratio of water and glyoxylic acid aqueous solution in step 1) is 1.5:1.
2)向第一产物中加入120g碳酸氢钠(1.44mol,3eq),搅拌升温至80℃,最后以10g/min速率通入异丁烯气体,反应2小时。反应结束后加入600g水,分离出有机相,除溶剂,得到第二产物;2) 120 g of sodium bicarbonate (1.44 mol, 3 eq) was added to the first product, the temperature was raised to 80° C. with stirring, and finally isobutylene gas was introduced at a rate of 10 g/min to react for 2 hours. After the reaction finishes, add 600g of water, separate out the organic phase, remove the solvent, and obtain the second product;
3)将取代试剂(0.33mol,1.1eq)、100mL叔丁醇、卤化氢溶液加入到第二反应容器中混合,再加入1mL溴化氢(48wt%),将第二产物滴加至第二反应容器中,结束后,除溶剂,叔丁醇重结晶,即得;所述取代试剂为硫脲。3) Add the substitution reagent (0.33mol, 1.1eq), 100mL tert-butanol and hydrogen halide solution into the second reaction vessel and mix, then add 1mL hydrogen bromide (48wt%), and add the second product dropwise to the second reaction vessel. In the reaction vessel, after the end, the solvent is removed, and the tert-butanol is recrystallized to obtain; the substitution reagent is thiourea.
所述反应体(Ⅰ)的结构为式1所示;所述中间体1的结构式为式2-1;The structure of the reactant (I) is shown in formula 1; the structural formula of the intermediate 1 is formula 2-1;

实施例6Example 6
一种砜吡草唑的合成方法,制备步骤包括:A method for synthesizing sulfofenazone, the preparation steps comprising:
步骤(1):取50g(0.30mol)反应体(Ⅰ)、36g氢氧化钠水溶液(50%,0.45mol,1.5eq)进行混合,滴加33g甲醛溶液(30%,0.33mol),结束后加入36g氢氧化钠水溶液(50%,0.45mol,1.5eq)、100mL乙腈,通入一氯二氟甲烷气体,并搅拌,静置分层,分离出的上层有机相,浓缩得到初反应物;将初反应物溶解在1,2-二氯乙烷中,在0℃滴加35.7g二氯亚砜(0.30mol),回温至25℃,反应至少30min,加水混合,分离出下层有机相,浓缩即得中间体2。Step (1): Mix 50g (0.30mol) reactant (I) and 36g aqueous sodium hydroxide solution (50%, 0.45mol, 1.5eq), dropwise add 33g formaldehyde solution (30%, 0.33mol), after the end Add 36g sodium hydroxide aqueous solution (50%, 0.45mol, 1.5eq), 100mL acetonitrile, pass into chlorodifluoromethane gas, stir, stand for layering, the separated upper organic phase is concentrated to obtain the initial reactant; Dissolve the initial reactant in 1,2-dichloroethane, add 35.7 g of thionyl chloride (0.30 mol) dropwise at 0 °C, return the temperature to 25 °C, react for at least 30 min, add water and mix, and separate the lower organic phase , and concentrated to obtain intermediate 2.
所述步骤(2):将42g中间体1(0.20mol)、41.4g碳酸钾(0.30mol)、200mL乙腈搅拌混合,继续滴加58.7g中间体2(0.20mol,90%),搅拌4小时,加水静置混合分离出有机相,浓缩得过渡中间体;将过渡中间体、300mL甲醇混合,98.36g间氯过氧苯甲酸(0.57mol),继续搅拌6小时后,加入水搅拌,过滤干燥即得。The step (2): 42g of intermediate 1 (0.20mol), 41.4g of potassium carbonate (0.30mol), and 200mL of acetonitrile were stirred and mixed, and 58.7g of intermediate 2 (0.20mol, 90%) was added dropwise, and stirred for 4 hours , add water to stand and mix to separate out the organic phase, and concentrate to obtain the transition intermediate; mix the transition intermediate and 300 mL of methanol, 98.36 g m-chloroperoxybenzoic acid (0.57 mol), continue stirring for 6 hours, add water and stir, filter and dry That's it.
所述中间体1的纯度为95%;所述中间体2的纯度为90%。The purity of the intermediate 1 is 95%; the purity of the intermediate 2 is 90%.
所述中间体1的合成方法,步骤包括:The synthetic method of described intermediate 1, the step comprises:
1)在第一反应容器中,加入80g乙醛酸水溶液(50%水溶液,0.5mol)、盐酸羟胺(0.55mol,1.1eq)、120g水,搅拌溶解,然后升温至70℃并搅拌3小时;将反应温度降至0℃,加入117g碳酸钠(1.1mol,2.2eq)和150g1,2-二氯乙烷,滴加80g液溴(0.5mol,1eq),结束后回温至25℃,并保温8小时,分离有机相,作为第一产物;步骤1)中水、乙醛酸水溶液的质量比为1.5:1。1) In the first reaction vessel, add 80g glyoxylic acid aqueous solution (50% aqueous solution, 0.5mol), hydroxylamine hydrochloride (0.55mol, 1.1eq), 120g water, stir to dissolve, then be warming up to 70 ° C and stir for 3 hours; The reaction temperature was lowered to 0 °C, 117 g of sodium carbonate (1.1 mol, 2.2 eq) and 150 g of 1,2-dichloroethane were added, 80 g of liquid bromine (0.5 mol, 1 eq) was added dropwise, and the temperature was returned to 25 ° C after completion, and Incubate for 8 hours, separate the organic phase, and use it as the first product; the mass ratio of water and glyoxylic acid aqueous solution in step 1) is 1.5:1.
2)向第一产物中加入120g碳酸氢钠(1.44mol,3eq),搅拌升温至80℃,最后以10g/min速率通入异丁烯气体,反应2小时。反应结束后加入600g水,分离出有机相,除溶剂,得到第二产物;2) 120 g of sodium bicarbonate (1.44 mol, 3 eq) was added to the first product, the temperature was raised to 80° C. with stirring, and finally isobutylene gas was introduced at a rate of 10 g/min to react for 2 hours. After the reaction finishes, add 600g of water, separate out the organic phase, remove the solvent, and obtain the second product;
3)将取代试剂(0.33mol,1.1eq)、100mL叔丁醇、卤化氢溶液加入到第二反应容器中混合,再加入1mL溴化氢(48wt%),将第二产物滴加至第二反应容器中,结束后,除溶剂,叔丁醇重结晶,即得;所述取代试剂为硫脲。3) Add the substitution reagent (0.33mol, 1.1eq), 100mL tert-butanol and hydrogen halide solution into the second reaction vessel and mix, then add 1mL hydrogen bromide (48wt%), and add the second product dropwise to the second reaction vessel. In the reaction vessel, after the end, the solvent is removed, and the tert-butanol is recrystallized to obtain; the substitution reagent is thiourea.
所述反应体(Ⅰ)的结构为式1所示;所述中间体1的结构式为式2-1;The structure of the reactant (I) is shown in formula 1; the structural formula of the intermediate 1 is formula 2-1;

实施例7Example 7
一种砜吡草唑的合成方法,制备步骤包括:A method for synthesizing sulfofenazone, the preparation steps comprising:
步骤(1):取50g(0.3mol)反应体(Ⅰ)、36g氢氧化钠水溶液(50%,0.45mol,1.5eq)进行混合,滴加33g甲醛溶液(30%,0.33mol),结束后加入36g氢氧化钠水溶液(50%,0.45mol,1.5eq)、100mL乙腈,通入一氯二氟甲烷气体,并搅拌,静置分层,分离出的上层有机相,浓缩得到初反应物;将初反应物溶解在1,2-二氯乙烷中,在0℃滴加35.7g二氯亚砜(0.3mol),回温至25℃,反应至少30min,加水混合,分离出下层有机相,浓缩即得中间体2。Step (1): Mix 50g (0.3mol) of reactant (I) and 36g of aqueous sodium hydroxide solution (50%, 0.45mol, 1.5eq), dropwise add 33g of formaldehyde solution (30%, 0.33mol), after the end Add 36g sodium hydroxide aqueous solution (50%, 0.45mol, 1.5eq), 100mL acetonitrile, pass into chlorodifluoromethane gas, stir, stand for layering, the separated upper organic phase is concentrated to obtain the initial reactant; The initial reactant was dissolved in 1,2-dichloroethane, 35.7 g of thionyl chloride (0.3 mol) was added dropwise at 0 °C, the temperature was returned to 25 °C, the reaction was performed for at least 30 min, water was added to mix, and the lower organic phase was separated. , and concentrated to obtain intermediate 2.
所述步骤(2):将42g中间体1(0.20mol)、41.4g碳酸钾(0.30mol)、200mL乙腈搅拌混合,继续滴加58.7g中间体2(0.20mol,90%),搅拌4小时,加水静置混合分离出有机相,浓缩得过渡中间体;将过渡中间体、300mL乙醇混合,加入1.25g二水合钨酸钠(3.80mmol)、1mL浓硫酸(98%)、64.6g双氧水(0.57mol,30%),继续搅拌6小时后,加入水搅拌,过滤干燥即得。The step (2): 42g of intermediate 1 (0.20mol), 41.4g of potassium carbonate (0.30mol), and 200mL of acetonitrile were stirred and mixed, and 58.7g of intermediate 2 (0.20mol, 90%) was added dropwise, and stirred for 4 hours , add water to stand and mix to separate the organic phase, and concentrate to obtain the transition intermediate; mix the transition intermediate and 300 mL of ethanol, add 1.25 g of sodium tungstate dihydrate (3.80 mmol), 1 mL of concentrated sulfuric acid (98%), 64.6 g of hydrogen peroxide ( 0.57mol, 30%), continue stirring for 6 hours, add water, stir, filter and dry.
所述中间体1的纯度为90%;所述中间体2的纯度为90%。The purity of the intermediate 1 is 90%; the purity of the intermediate 2 is 90%.
所述中间体1的合成方法,步骤包括:The synthetic method of described intermediate 1, the step comprises:
1)在第一反应容器中,加入80g乙醛酸水溶液(50%水溶液,0.5mol)、羟胺化合物(0.55mol,1.1eq)、120g水,搅拌溶解,然后升温至70℃并搅拌3小时;将反应温度降至0℃,加入117g碳酸钠(1.1mol,2.2eq)和150g1,2-二氯乙烷,滴加80g液溴(0.5mol,1eq),结束后回温至25℃,并保温8小时,分离有机相,作为第一产物;步骤1)中水、乙醛酸水溶液的质量比为1.5:1。1) In the first reaction vessel, add 80g glyoxylic acid aqueous solution (50% aqueous solution, 0.5mol), hydroxylamine compound (0.55mol, 1.1eq), 120g water, stir to dissolve, then be warming up to 70 ° C and stir for 3 hours; The reaction temperature was lowered to 0 °C, 117 g of sodium carbonate (1.1 mol, 2.2 eq) and 150 g of 1,2-dichloroethane were added, 80 g of liquid bromine (0.5 mol, 1 eq) was added dropwise, and the temperature was returned to 25 ° C after completion, and Incubate for 8 hours, separate the organic phase, and use it as the first product; the mass ratio of water and glyoxylic acid aqueous solution in step 1) is 1.5:1.
2)向第一产物中加入120g碳酸氢钠(1.44mol,3eq),搅拌升温至80℃,最后以10g/min速率通入异丁烯气体,反应2小时。反应结束后加入600g水,分离出有机相,除溶剂,得到第二产物;2) 120 g of sodium bicarbonate (1.44 mol, 3 eq) was added to the first product, the temperature was raised to 80° C. with stirring, and finally isobutylene gas was introduced at a rate of 10 g/min to react for 2 hours. After the reaction finishes, add 600g of water, separate out the organic phase, remove the solvent, and obtain the second product;
3)将取代试剂(0.33mol,1.1eq)、100mL叔丁醇、卤化氢溶液加入到第二反应容器中混合,再加入1mL溴化氢(48wt%),将第二产物滴加至第二反应容器中,结束后,除溶剂,叔丁醇重结晶,即得;所述取代试剂为硫脲。3) Add the substitution reagent (0.33mol, 1.1eq), 100mL tert-butanol and hydrogen halide solution into the second reaction vessel and mix, then add 1mL hydrogen bromide (48wt%), and add the second product dropwise to the second reaction vessel. In the reaction vessel, after the end, the solvent is removed, and the tert-butanol is recrystallized to obtain; the substitution reagent is thiourea.
所述反应体(Ⅰ)的结构为式1所示;所述中间体1的结构式为式2-1;The structure of the reactant (I) is shown in formula 1; the structural formula of the intermediate 1 is formula 2-1;

实施例8Example 8
一种砜吡草唑的合成方法,制备步骤包括:A method for synthesizing sulfofenazone, the preparation steps comprising:
步骤(1):取50g(0.3mol)反应体(Ⅰ)、36g氢氧化钠水溶液(50%,0.45mol,1.5eq)进行混合,滴加33g甲醛溶液(30%,0.33mol),结束后加入36g氢氧化钠水溶液(50%,0.45mol,1.5eq)、100mL乙腈,通入一氯二氟甲烷气体,并搅拌,静置分层,分离出的上层有机相,浓缩得到初反应物;将初反应物溶解在1,2-二氯乙烷中,在0℃滴加35.7g二氯亚砜(0.3mol),回温至25℃,反应至少30min,加水混合,分离出下层有机相,浓缩即得中间体2。Step (1): Mix 50g (0.3mol) of reactant (I) and 36g of aqueous sodium hydroxide solution (50%, 0.45mol, 1.5eq), dropwise add 33g of formaldehyde solution (30%, 0.33mol), after the end Add 36g sodium hydroxide aqueous solution (50%, 0.45mol, 1.5eq), 100mL acetonitrile, pass into chlorodifluoromethane gas, stir, stand for layering, the separated upper organic phase is concentrated to obtain the initial reactant; The initial reactant was dissolved in 1,2-dichloroethane, 35.7 g of thionyl chloride (0.3 mol) was added dropwise at 0 °C, the temperature was returned to 25 °C, the reaction was performed for at least 30 min, water was added to mix, and the lower organic phase was separated. , and concentrated to obtain intermediate 2.
所述步骤(2):将42g中间体1(0.20mol)、41.4g碳酸钾(0.30mol)、200mL乙腈搅拌混合,继续滴加58.7g中间体2(0.20mol,90%),搅拌4小时,加水静置混合分离出有机相,浓缩得过渡中间体;将过渡中间体、300mL二氯甲烷混合,加入1.25g二水合钨酸钠(3.80mmol)、1mL浓硫酸(98%)、64.6g双氧水(0.57mol,30%),继续搅拌6小时后,加入水搅拌,过滤干燥即得。The step (2): 42g of intermediate 1 (0.20mol), 41.4g of potassium carbonate (0.30mol), and 200mL of acetonitrile were stirred and mixed, and 58.7g of intermediate 2 (0.20mol, 90%) was added dropwise, and stirred for 4 hours , add water to stand and mix to separate the organic phase, and concentrate to obtain the transition intermediate; mix the transition intermediate and 300 mL of dichloromethane, add 1.25 g of sodium tungstate dihydrate (3.80 mmol), 1 mL of concentrated sulfuric acid (98%), 64.6 g Hydrogen peroxide (0.57mol, 30%), after stirring for 6 hours, adding water, stirring, filtering and drying.
所述中间体1的纯度为95%;所述中间体2的纯度为90%。The purity of the intermediate 1 is 95%; the purity of the intermediate 2 is 90%.
所述中间体1的合成方法,步骤包括:The synthetic method of described intermediate 1, the step comprises:
1)在第一反应容器中,加入80g乙醛酸水溶液(50%水溶液,0.5mol)、羟胺化合物(0.55mol,1.1eq)、120g水,搅拌溶解,然后升温至70℃并搅拌3小时;将反应温度降至0℃,加入117g碳酸钠(1.1mol,2.2eq)和150g1,2-二氯乙烷,滴加80g液溴(0.5mol,1eq),结束后回温至25℃,并保温8小时,分离有机相,作为第一产物;步骤1)中水、乙醛酸水溶液的质量比为1.5:1。1) In the first reaction vessel, add 80g glyoxylic acid aqueous solution (50% aqueous solution, 0.5mol), hydroxylamine compound (0.55mol, 1.1eq), 120g water, stir to dissolve, then be warming up to 70 ° C and stir for 3 hours; The reaction temperature was lowered to 0 °C, 117 g of sodium carbonate (1.1 mol, 2.2 eq) and 150 g of 1,2-dichloroethane were added, 80 g of liquid bromine (0.5 mol, 1 eq) was added dropwise, and the temperature was returned to 25 ° C after completion, and Incubate for 8 hours, separate the organic phase, and use it as the first product; the mass ratio of water and glyoxylic acid aqueous solution in step 1) is 1.5:1.
2)向第一产物中加入120g碳酸氢钠(1.44mol,3eq),搅拌升温至80℃,最后以10g/min速率通入异丁烯气体,反应2小时。反应结束后加入600g水,分离出有机相,除溶剂,得到第二产物;2) 120 g of sodium bicarbonate (1.44 mol, 3 eq) was added to the first product, the temperature was raised to 80° C. with stirring, and finally isobutylene gas was introduced at a rate of 10 g/min to react for 2 hours. After the reaction finishes, add 600g of water, separate out the organic phase, remove the solvent, and obtain the second product;
3)将取代试剂(0.33mol,1.1eq)、100mL叔丁醇、卤化氢溶液加入到第二反应容器中混合,再加入1mL溴化氢(48wt%),将第二产物滴加至第二反应容器中,结束后,除溶剂,叔丁醇重结晶,即得;所述取代试剂为硫脲。3) Add the substitution reagent (0.33mol, 1.1eq), 100mL tert-butanol and hydrogen halide solution into the second reaction vessel and mix, then add 1mL hydrogen bromide (48wt%), and add the second product dropwise to the second reaction vessel. In the reaction vessel, after the end, the solvent is removed, and the tert-butanol is recrystallized to obtain; the substitution reagent is thiourea.
所述反应体(Ⅰ)的结构为式1所示;所述中间体1的结构式为式2-1;The structure of the reactant (I) is shown in formula 1; the structural formula of the intermediate 1 is formula 2-1;


实施例9Example 9
一种砜吡草唑的合成方法,制备步骤包括:A method for synthesizing sulfofenazone, the preparation steps comprising:
步骤(1):取50g(0.3mol)反应体(Ⅰ)、36g氢氧化钠水溶液(50%,0.45mol,1.5eq)进行混合,滴加33g甲醛溶液(30%,0.33mol),结束后加入36g氢氧化钠水溶液(50%,0.45mol,1.5eq)、100mL乙腈,通入一氯二氟甲烷气体,并搅拌,静置分层,分离出的上层有机相,浓缩得到初反应物;将初反应物溶解在1,2-二氯乙烷中,在0℃滴加35.7g二氯亚砜(0.3mol),回温至25℃,反应至少30min,加水混合,分离出下层有机相,浓缩即得中间体2。Step (1): Mix 50g (0.3mol) of reactant (I) and 36g of aqueous sodium hydroxide solution (50%, 0.45mol, 1.5eq), dropwise add 33g of formaldehyde solution (30%, 0.33mol), after the end Add 36g sodium hydroxide aqueous solution (50%, 0.45mol, 1.5eq), 100mL acetonitrile, pass into chlorodifluoromethane gas, stir, stand for layering, the separated upper organic phase is concentrated to obtain the initial reactant; The initial reactant was dissolved in 1,2-dichloroethane, 35.7 g of thionyl chloride (0.3 mol) was added dropwise at 0 °C, the temperature was returned to 25 °C, the reaction was performed for at least 30 min, water was added to mix, and the lower organic phase was separated. , and concentrated to obtain intermediate 2.
所述步骤(2):将42g中间体1(0.20mol)、41.4g碳酸钾(0.30mol)、200mL乙腈搅拌混合,继续滴加58.7g中间体2(0.20mol,90%),搅拌4小时,加水静置混合分离出有机相,浓缩得过渡中间体;将过渡中间体、300mL甲醇混合,加入1.25g二水合钨酸钠(3.80mmol)、1mL浓硫酸(98%)、64.6g双氧水(0.57mol,30%),继续搅拌6小时后,加入水搅拌,过滤干燥即得。The step (2): 42g of intermediate 1 (0.20mol), 41.4g of potassium carbonate (0.30mol), and 200mL of acetonitrile were stirred and mixed, and 58.7g of intermediate 2 (0.20mol, 90%) was added dropwise, and stirred for 4 hours , add water to stand and mix to separate the organic phase, and concentrate to obtain the transition intermediate; mix the transition intermediate and 300 mL of methanol, add 1.25 g of sodium tungstate dihydrate (3.80 mmol), 1 mL of concentrated sulfuric acid (98%), 64.6 g of hydrogen peroxide ( 0.57mol, 30%), continue stirring for 6 hours, add water, stir, filter and dry.
所述中间体1的纯度为89%;所述中间体2的纯度为90%。The purity of the intermediate 1 is 89%; the purity of the intermediate 2 is 90%.
所述中间体1的合成方法,步骤包括:The synthetic method of described intermediate 1, the step comprises:
1)在第一反应容器中,加入80g乙醛酸水溶液(50%水溶液,0.5mol)、羟胺化合物(0.55mol,1.1eq)、120g水,搅拌溶解,然后升温至70℃并搅拌3小时;将反应温度降至0℃,加入117g碳酸钠(1.1mol,2.2eq)和150g1,2-二氯乙烷,滴加80g液溴(0.5mol,1eq),结束后回温至25℃,并保温8小时,分离有机相,作为第一产物;步骤1)中水、乙醛酸水溶液的质量比为1.5:1。1) In the first reaction vessel, add 80g glyoxylic acid aqueous solution (50% aqueous solution, 0.5mol), hydroxylamine compound (0.55mol, 1.1eq), 120g water, stir to dissolve, then be warming up to 70 ° C and stir for 3 hours; The reaction temperature was lowered to 0 °C, 117 g of sodium carbonate (1.1 mol, 2.2 eq) and 150 g of 1,2-dichloroethane were added, 80 g of liquid bromine (0.5 mol, 1 eq) was added dropwise, and the temperature was returned to 25 ° C after completion, and Incubate for 8 hours, separate the organic phase, and use it as the first product; the mass ratio of water and glyoxylic acid aqueous solution in step 1) is 1.5:1.
2)向第一产物中加入120g碳酸氢钠(1.44mol,3eq),搅拌升温至80℃,最后以10g/min速率通入异丁烯气体,反应2小时。反应结束后加入600g水,分离出有机相,除溶剂,得到第二产物;2) 120 g of sodium bicarbonate (1.44 mol, 3 eq) was added to the first product, the temperature was raised to 80° C. with stirring, and finally isobutylene gas was introduced at a rate of 10 g/min to react for 2 hours. After the reaction finishes, add 600g of water, separate out the organic phase, remove the solvent, and obtain the second product;
3)将取代试剂(0.27mol,0.9eq)、100mL叔丁醇、卤化氢溶液加入到第二反应容器中混合,再加入1mL溴化氢(48wt%),将第二产物滴加至第二反应容器中,结束后,除溶剂,叔丁醇重结晶,即得;所述取代试剂为硫脲。3) The substitution reagent (0.27mol, 0.9eq), 100mL of tert-butanol and hydrogen halide solution were added to the second reaction vessel and mixed, then 1mL of hydrogen bromide (48wt%) was added, and the second product was added dropwise to the second reaction vessel. In the reaction vessel, after the end, the solvent is removed, and the tert-butanol is recrystallized to obtain; the substitution reagent is thiourea.
所述反应体(Ⅰ)的结构为式1所示;所述中间体1的结构式为式2-1;The structure of the reactant (I) is shown in formula 1; the structural formula of the intermediate 1 is formula 2-1;

实施例10Example 10
一种砜吡草唑的合成方法,制备步骤包括:A method for synthesizing sulfofenazone, the preparation steps comprising:
所述步骤(1):取50.0g(0.16mol)反应体(Ⅱ)溶解在200mL的N,N-二甲基甲酰胺中,再向反应体系中加入66.2g的K2CO3(0.48mol)并将该溶液搅拌1小时。之后,在半小时内向溶液中通入81g(0.8mol)一氯二氟甲烷。将反应置于25℃条件下搅拌6小时。通过液相中控反应,反应结束之后,向反应液中加入300mL水和100mL1,1-二氯甲烷,再用200mL水和200mL饱和氯化钠溶液洗涤,将分离出的有机相进行干燥,减压蒸馏除去溶剂,得到46g过渡中间体。将过渡中间体、200mL甲醇混合,加入二水合钨酸钠1.0g(3.04mmol)、浓硫酸1mL(98%)、双氧水51.7g(0.46mol,30%),继续搅拌6小时后,加入水搅拌,过滤干燥即得。The step (1): take 50.0g (0.16mol) of the reactant (II) and dissolve it in 200mL of N,N-dimethylformamide, and then add 66.2g of K 2 CO 3 (0.48mol) to the reaction system. ) and the solution was stirred for 1 hour. After that, 81 g (0.8 mol) of monochlorodifluoromethane was passed into the solution within half an hour. The reaction was stirred at 25°C for 6 hours. Control the reaction in the liquid phase. After the reaction is completed, add 300 mL of water and 100 mL of 1,1-dichloromethane to the reaction solution, wash with 200 mL of water and 200 mL of saturated sodium chloride solution, and dry the separated organic phase. The solvent was distilled off under pressure to obtain 46 g of the transition intermediate. Mix the transition intermediate and 200 mL of methanol, add 1.0 g (3.04 mmol) of sodium tungstate dihydrate, 1 mL (98%) of concentrated sulfuric acid, and 51.7 g (0.46 mol, 30%) of hydrogen peroxide, continue stirring for 6 hours, then add water and stir , filtered and dried.
所述反应体(Ⅱ)的纯度为95%。The purity of the reactant (II) was 95%.
所述反应体(Ⅱ)的合成方法,步骤包括:The synthetic method of described reactant (II), the step comprises:
1)在第一反应容器中,加入80.0g乙醛酸水溶液(50%水溶液,0.54mol)、羟胺化合物(0.55mol,1.1eq)、120g水,搅拌溶解,然后升温至70℃并搅拌3小时;将反应温度降至0℃,加入117g碳酸钠(1.1mol,2.2eq)和150g1,2-二氯乙烷,滴加80g液溴(0.5mol,1eq),结束后回温至25℃,并保温8小时,分离有机相,作为第一产物;步骤1)中水、乙醛酸水溶液的质量比为1.5:1。1) In the first reaction vessel, add 80.0 g of glyoxylic acid aqueous solution (50% aqueous solution, 0.54 mol), hydroxylamine compound (0.55 mol, 1.1 eq), 120 g of water, stir to dissolve, then heat up to 70 ° C and stir for 3 hours ; Reduce the reaction temperature to 0°C, add 117g of sodium carbonate (1.1mol, 2.2eq) and 150g of 1,2-dichloroethane, dropwise add 80g of liquid bromine (0.5mol, 1eq), return to 25°C after the end, And keep the temperature for 8 hours, separate the organic phase as the first product; the mass ratio of water and glyoxylic acid aqueous solution in step 1) is 1.5:1.
2)向第一产物中加入120g碳酸氢钠(1.44mol,3eq),搅拌升温至80℃,最后以10g/min速率通入异丁烯气体,反应2小时。反应结束后加入600g水,分离出有机相,除溶剂,得到第二产物;2) 120 g of sodium bicarbonate (1.44 mol, 3 eq) was added to the first product, the temperature was raised to 80° C. with stirring, and finally isobutylene gas was introduced at a rate of 10 g/min to react for 2 hours. After the reaction finishes, add 600g of water, separate out the organic phase, remove the solvent, and obtain the second product;
3)将取代试剂(0.27mol,0.9eq)、100mL叔丁醇、卤化氢溶液加入到第二反应容器中混合,再加入1mL溴化氢(48wt%),将第二产物滴加至第二反应容器中,结束后,除溶剂,叔丁醇重结晶,即得;所述取代试剂为硫脲盐。3) The substitution reagent (0.27mol, 0.9eq), 100mL of tert-butanol and hydrogen halide solution were added to the second reaction vessel and mixed, then 1mL of hydrogen bromide (48wt%) was added, and the second product was added dropwise to the second reaction vessel. In the reaction vessel, after the end, the solvent is removed and the tert-butanol is recrystallized to obtain; the substitution reagent is thiourea salt.
4)将40g反应体(Ⅰ)(0.24mol)和34g NaOH(0.84mol)加入到200mL水中,待其完全溶解,再滴加52g甲醛(31%,0.48mol)溶液。滴加半小时。结束之后在25℃下搅拌1个小时。将上述得到的50g(0.24mol)硫脲盐溶解在150mL水中,然后滴加到反应之中,滴加20分钟。反应在25℃条件下继续搅拌5个小时。通过液相中控反应,反应结束之后,加入20mL盐酸(35%,0.2mol)调解反应液pH=6,反应液中有固体析出,继续搅拌1小时,将所有固体都变成粉末状分散在水中之后,再进行抽滤,用水洗涤滤饼,将滤饼烘干,得到淡黄色固体的反应体(Ⅱ)。4) Add 40 g of reactant (I) (0.24 mol) and 34 g of NaOH (0.84 mol) into 200 mL of water, until it is completely dissolved, and then dropwise add 52 g of formaldehyde (31%, 0.48 mol) solution. Add dropwise for half an hour. After completion, it was stirred at 25°C for 1 hour. 50 g (0.24 mol) of the thiourea salt obtained above was dissolved in 150 mL of water, and then added dropwise to the reaction for 20 minutes. The reaction was continued to stir at 25°C for 5 hours. The reaction was controlled in the liquid phase. After the reaction was completed, 20 mL of hydrochloric acid (35%, 0.2 mol) was added to adjust the pH of the reaction solution to 6. There was solid precipitation in the reaction solution, and stirring was continued for 1 hour. After water, suction filtration is carried out, the filter cake is washed with water, and the filter cake is dried to obtain the reaction body (II) as a light yellow solid.
所述反应体(Ⅱ)的结构式2所示;The structural formula 2 of the reactant (II) is shown;

性能测试Performance Testing
1、砜吡草唑的含量、收率测试:对通过实施例1-实施例10所述的砜吡草唑的合成方法得到的砜吡草唑测试其含量和收率。1. Test of the content and yield of fenfenapyr: The content and yield of fenfenapyr obtained by the synthesis method of fenfenapyr described in Example 1-Example 10 were tested.
测试方法:等梯度扫描,A流动相是pH=4的乙酸水溶液,B流动相是甲醇。进样体积为10μL;运行时间为30.0minutes;条件如表3所示;测试结果如表1所示。Test method: isocratic scanning, A mobile phase is acetic acid aqueous solution with pH=4, B mobile phase is methanol. The injection volume is 10 μL; the running time is 30.0 minutes; the conditions are shown in Table 3; the test results are shown in Table 1.
2、液相谱图测试:对实施例1所述的砜吡草唑的合成方法得到的砜吡草唑进行液相测试,测试的色谱峰结果如表2所示,液相谱图结果如图1所示。2. Liquid phase chromatogram test: liquid phase test was carried out on the sulfofenapyr obtained by the synthetic method of sulfofenapyr described in Example 1, the chromatographic peak results of the test were as shown in Table 2, and the liquid phase chromatogram results were as shown in Table 2. Figure 1.
液相谱图测试条件:等梯度扫描,A流动相是pH=4的乙酸水溶液,B流动相是甲醇。进样体积为10μL;运行时间为30.0minutes;条件如表3所示液相谱图测试结果:实施例1所述的砜吡草唑的合成方法得到的砜吡草唑在17.308分钟有一个杂质峰;方法是等梯度扫描,A流动相是pH=4的乙酸水溶液,B流动相是甲醇。Liquid chromatography test conditions: isocratic scanning, A mobile phase is acetic acid aqueous solution with pH=4, B mobile phase is methanol. The sample injection volume is 10 μL; the running time is 30.0 minutes; the conditions are as shown in Table 3. The liquid chromatogram test results: the sulfofenapyr obtained by the synthetic method of sulfofenapyr described in Example 1 has an impurity in 17.308 minutes Peak; method is isocratic scan, A mobile phase is aqueous acetic acid pH=4, B mobile phase is methanol.
表1Table 1
表2Table 2
表3table 3


以上所述仅是本发明的较佳实施例而已,并非是对发明作其他形式的限制,任何熟悉本专业的技术人员可能利用上述揭示的技术内容加以变更或更改为等同变化的等效实施例,但凡是未脱离本发明技术方案内容,依据本发明的技术实质对以上实施例所作的任何简单修改,等同变化与改型,仍属于本发明技术方案的保护范围。The above descriptions are only preferred embodiments of the present invention, and are not intended to limit the invention in other forms. Any person skilled in the art may use the technical contents disclosed above to make changes or change to equivalent embodiments with equivalent changes. However, any simple modification, equivalent change and modification made to the above embodiments according to the technical essence of the present invention without departing from the content of the technical solution of the present invention still belong to the protection scope of the technical solution of the present invention.
Claims (10)


Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010600009.1A CN111574511A (en) | 2020-06-28 | 2020-06-28 | Synthesis method and application of sulfuryl pyraflufen |
| PCT/CN2020/103174 WO2022000603A1 (en) | 2020-06-28 | 2020-07-21 | Synthesis method for pyroxasulfone, and application of pyroxasulfone |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010600009.1A CN111574511A (en) | 2020-06-28 | 2020-06-28 | Synthesis method and application of sulfuryl pyraflufen |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN111574511A true CN111574511A (en) | 2020-08-25 |
Family
ID=72122122
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202010600009.1A Pending CN111574511A (en) | 2020-06-28 | 2020-06-28 | Synthesis method and application of sulfuryl pyraflufen |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN111574511A (en) |
| WO (1) | WO2022000603A1 (en) |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112969697A (en) * | 2019-10-31 | 2021-06-15 | 组合化学工业株式会社 | Process for producing herbicide and intermediate thereof |
| CN113336716A (en) * | 2021-06-10 | 2021-09-03 | 江苏省农用激素工程技术研究中心有限公司 | Preparation method of topramezone intermediate |
| CN113372288A (en) * | 2021-06-02 | 2021-09-10 | 安徽久易农业股份有限公司 | Synthetic method of topramezone pesticide intermediate |
| CN113735791A (en) * | 2021-10-14 | 2021-12-03 | 杭州欧晨科技有限公司 | Synthetic method of 3-bromo-5, 5-dimethyl-4, 5-dihydroisoxazole |
| CN113979944A (en) * | 2021-11-29 | 2022-01-28 | 杭州欧晨科技有限公司 | Synthesis method of high-selectivity 1-methyl-3- (trifluoromethyl) -1H-pyrazole-5-alcohol |
| WO2022137370A1 (en) * | 2020-12-23 | 2022-06-30 | クミアイ化学工業株式会社 | Sulfone derivative production method |
| CN114716428A (en) * | 2022-05-06 | 2022-07-08 | 山东潍坊润丰化工股份有限公司 | Method for preparing sulpyrazoxazole intermediate |
| WO2022170871A1 (en) * | 2021-02-11 | 2022-08-18 | Rotam Agrochem International Company Limited | Novel crystalline form of pyroxasulfone, methods for its preparation and use of the same |
| WO2022191292A1 (en) * | 2021-03-11 | 2022-09-15 | クミアイ化学工業株式会社 | Method for producing sulfone derivative as herbicide |
| CN115073441A (en) * | 2021-03-11 | 2022-09-20 | 山东润博生物科技有限公司 | Synthetic method of topramezone intermediate |
| CN115850254A (en) * | 2022-12-20 | 2023-03-28 | 山东润博生物科技有限公司 | Synthetic method of sulfuryl pyraflufen |
| CN116102510A (en) * | 2021-11-10 | 2023-05-12 | 江西仰立新材料有限公司 | Preparation method of 3-bromo-5, 5-dimethyl-4, 5-dihydro-isoxazole |
| CN117327016A (en) * | 2023-09-12 | 2024-01-02 | 湖北泰盛化工有限公司 | Preparation method of fenpyrad intermediate |
| CN117567452A (en) * | 2023-11-22 | 2024-02-20 | 江苏快达农化股份有限公司 | A kind of preparation method of sulfonazole |
| CN119241524A (en) * | 2024-09-05 | 2025-01-03 | 启农生物科技(北京)有限公司 | A kind of preparation method of sulfonepyraclostrobin |
| TWI921615B (en) | 2021-10-29 | 2026-04-11 | 日商組合化學工業股份有限公司 | Disulfide compounds, polysulfide compounds and their uses, and the uses of pyrazole derivatives. |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN121311473A (en) * | 2023-04-03 | 2026-01-09 | 格哈达化工有限公司 | Preparation method of sulfonylpyrazole |
| CN117924265B (en) * | 2024-03-22 | 2024-06-18 | 潍坊新绿化工有限公司 | Synthesis method of pyrifos |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006038657A1 (en) * | 2004-10-06 | 2006-04-13 | Ihara Chemical Industry Co., Ltd. | 3-bromo-5,5-dimethyl-4,5-dihydroisoxazole and method for producing same |
| CN1938278A (en) * | 2004-03-31 | 2007-03-28 | 庵原化学工业株式会社 | Process for producing 5-hydroxy-4-thiomethylpyrazole compound |
| WO2007071900A1 (en) * | 2005-12-21 | 2007-06-28 | Syngenta Limited | Novel herbicides |
| CN101384556A (en) * | 2006-02-14 | 2009-03-11 | 庵原化学工业株式会社 | Process for producing 5-alkoxy-4-hydroxymethylpyrazole compound |
| CN102666502A (en) * | 2009-11-26 | 2012-09-12 | 巴斯夫欧洲公司 | Method for producing 5,5-disubstituted 4,5-dihydroisoxazol-3-thiocarboxamidine salts |
| CN109574945A (en) * | 2017-09-28 | 2019-04-05 | 东莞东阳光科研发有限公司 | Isoxazoline derivative and its application in agricultural |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020240392A1 (en) * | 2019-05-24 | 2020-12-03 | Pi Industries Limited | Process for preparation of pyroxasulfone |
-
2020
- 2020-06-28 CN CN202010600009.1A patent/CN111574511A/en active Pending
- 2020-07-21 WO PCT/CN2020/103174 patent/WO2022000603A1/en not_active Ceased
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1938278A (en) * | 2004-03-31 | 2007-03-28 | 庵原化学工业株式会社 | Process for producing 5-hydroxy-4-thiomethylpyrazole compound |
| WO2006038657A1 (en) * | 2004-10-06 | 2006-04-13 | Ihara Chemical Industry Co., Ltd. | 3-bromo-5,5-dimethyl-4,5-dihydroisoxazole and method for producing same |
| WO2007071900A1 (en) * | 2005-12-21 | 2007-06-28 | Syngenta Limited | Novel herbicides |
| CN101384556A (en) * | 2006-02-14 | 2009-03-11 | 庵原化学工业株式会社 | Process for producing 5-alkoxy-4-hydroxymethylpyrazole compound |
| CN102666502A (en) * | 2009-11-26 | 2012-09-12 | 巴斯夫欧洲公司 | Method for producing 5,5-disubstituted 4,5-dihydroisoxazol-3-thiocarboxamidine salts |
| CN109574945A (en) * | 2017-09-28 | 2019-04-05 | 东莞东阳光科研发有限公司 | Isoxazoline derivative and its application in agricultural |
Non-Patent Citations (1)
| Title |
|---|
| 无: "砜吡草唑", 《农药科学与管理》 * |
Cited By (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112969697A (en) * | 2019-10-31 | 2021-06-15 | 组合化学工业株式会社 | Process for producing herbicide and intermediate thereof |
| WO2022138781A1 (en) * | 2020-12-23 | 2022-06-30 | クミアイ化学工業株式会社 | Sulfone derivative production method |
| JP7846022B2 (en) | 2020-12-23 | 2026-04-14 | クミアイ化学工業株式会社 | Method for producing sulfone derivatives |
| CN116761802A (en) * | 2020-12-23 | 2023-09-15 | 组合化学工业株式会社 | Method for producing sulfone derivatives |
| WO2022137370A1 (en) * | 2020-12-23 | 2022-06-30 | クミアイ化学工業株式会社 | Sulfone derivative production method |
| JPWO2022138781A1 (en) * | 2020-12-23 | 2022-06-30 | ||
| CN114920735A (en) * | 2021-02-11 | 2022-08-19 | 龙灯农业化工国际有限公司 | Novel crystalline forms of xaflufen, processes for their preparation and their use |
| WO2022170871A1 (en) * | 2021-02-11 | 2022-08-18 | Rotam Agrochem International Company Limited | Novel crystalline form of pyroxasulfone, methods for its preparation and use of the same |
| CN115073441A (en) * | 2021-03-11 | 2022-09-20 | 山东润博生物科技有限公司 | Synthetic method of topramezone intermediate |
| WO2022191292A1 (en) * | 2021-03-11 | 2022-09-15 | クミアイ化学工業株式会社 | Method for producing sulfone derivative as herbicide |
| JPWO2022191292A1 (en) * | 2021-03-11 | 2022-09-15 | ||
| CN115073441B (en) * | 2021-03-11 | 2024-04-02 | 山东润博生物科技有限公司 | Synthesis method of topiramate intermediate |
| CN115776978A (en) * | 2021-03-11 | 2023-03-10 | 组合化学工业株式会社 | Process for producing sulfone derivative as herbicide |
| CN113372288A (en) * | 2021-06-02 | 2021-09-10 | 安徽久易农业股份有限公司 | Synthetic method of topramezone pesticide intermediate |
| CN113336716B (en) * | 2021-06-10 | 2022-04-08 | 江苏省农用激素工程技术研究中心有限公司 | Preparation method of topramezone intermediate |
| CN113336716A (en) * | 2021-06-10 | 2021-09-03 | 江苏省农用激素工程技术研究中心有限公司 | Preparation method of topramezone intermediate |
| CN113735791A (en) * | 2021-10-14 | 2021-12-03 | 杭州欧晨科技有限公司 | Synthetic method of 3-bromo-5, 5-dimethyl-4, 5-dihydroisoxazole |
| TWI921615B (en) | 2021-10-29 | 2026-04-11 | 日商組合化學工業股份有限公司 | Disulfide compounds, polysulfide compounds and their uses, and the uses of pyrazole derivatives. |
| CN116102510A (en) * | 2021-11-10 | 2023-05-12 | 江西仰立新材料有限公司 | Preparation method of 3-bromo-5, 5-dimethyl-4, 5-dihydro-isoxazole |
| CN116102510B (en) * | 2021-11-10 | 2025-03-25 | 江西仰立新材料有限公司 | A preparation method of 3-bromo-5,5-dimethyl-4,5-dihydroisoxazole |
| CN113979944B (en) * | 2021-11-29 | 2023-08-22 | 杭州欧晨科技有限公司 | A kind of synthetic method of highly selective 1-methyl-3-(trifluoromethyl)-1H-pyrazol-5-ol |
| CN113979944A (en) * | 2021-11-29 | 2022-01-28 | 杭州欧晨科技有限公司 | Synthesis method of high-selectivity 1-methyl-3- (trifluoromethyl) -1H-pyrazole-5-alcohol |
| CN114716428B (en) * | 2022-05-06 | 2024-02-06 | 山东潍坊润丰化工股份有限公司 | Method for preparing metazopyr intermediate |
| CN114716428A (en) * | 2022-05-06 | 2022-07-08 | 山东潍坊润丰化工股份有限公司 | Method for preparing sulpyrazoxazole intermediate |
| CN115850254A (en) * | 2022-12-20 | 2023-03-28 | 山东润博生物科技有限公司 | Synthetic method of sulfuryl pyraflufen |
| CN115850254B (en) * | 2022-12-20 | 2024-06-25 | 山东润博生物科技有限公司 | Synthesis method of pyrifos |
| CN117327016A (en) * | 2023-09-12 | 2024-01-02 | 湖北泰盛化工有限公司 | Preparation method of fenpyrad intermediate |
| CN117567452A (en) * | 2023-11-22 | 2024-02-20 | 江苏快达农化股份有限公司 | A kind of preparation method of sulfonazole |
| CN119241524A (en) * | 2024-09-05 | 2025-01-03 | 启农生物科技(北京)有限公司 | A kind of preparation method of sulfonepyraclostrobin |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2022000603A1 (en) | 2022-01-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN111574511A (en) | Synthesis method and application of sulfuryl pyraflufen | |
| EP3133061B1 (en) | Florfenicol synthesizing method | |
| US11820724B2 (en) | Method for preparing 2-ethyl-4-fluoro-1-nitrobenzene | |
| CN113735791A (en) | Synthetic method of 3-bromo-5, 5-dimethyl-4, 5-dihydroisoxazole | |
| CN106986838A (en) | A kind of preparation method of prothioconazoles | |
| CN108017578A (en) | A kind of chloro- N of 2-, the preparation method of N- dimethyl nicotinamides | |
| CN107698578A (en) | A kind of preparation method of Diacloden | |
| KR101127814B1 (en) | Novel intermediate and process for preparing sildenafil or its salt using the same | |
| McManus et al. | Tetrazole analogs of plant auxins1 | |
| CN107188875B (en) | Preparation method and intermediate of substituted phthalide compound | |
| CN113801039B (en) | A kind of method for preparing sodium N-methyl taurate | |
| CN116102510A (en) | Preparation method of 3-bromo-5, 5-dimethyl-4, 5-dihydro-isoxazole | |
| CN113861097A (en) | Synthesis method of multi-configuration 1-Boc-N-Fmoc tryptophan compound | |
| CN106317042B (en) | A kind of synthetic method of the fluoro- 6- amino -4- of 7- (2- propargyls) -1,4- benzoxazines -3 (4H) -one derivative | |
| CN115784992A (en) | Preparation method and application of pyrazole mercaptomethylated derivative | |
| CN111807997B (en) | Synthesis method of N- (4-methoxycarbonyl-3-aminosulfonylbenzyl) methanesulfonamide | |
| CN103214386B (en) | New method for synthesizing amide compounds | |
| JP3135011B2 (en) | Method for producing bisimide compound | |
| CN110386889B (en) | A kind of synthetic method of NSC128981 | |
| WO2022262768A1 (en) | Hybutimibe intermediate and preparation method therefor | |
| CN111320570A (en) | Preparation method of lansoprazole key intermediate | |
| CN117658866B (en) | Synthesis method of azido amino acid derivative | |
| CN111704604A (en) | Preparation method of pyridine quinazoline | |
| CN110256350B (en) | Pyrazole ligand, zero-dimensional functional complex, preparation method and application thereof | |
| CN112778197A (en) | Preparation method of novel abscisic acid agonist AM1 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| RJ01 | Rejection of invention patent application after publication | ||
| RJ01 | Rejection of invention patent application after publication |
Application publication date: 20200825 |