CN107497418B - 用于产生丙烯酸和丙烯酸类的包含钒、铋和钨的催化剂 - Google Patents
用于产生丙烯酸和丙烯酸类的包含钒、铋和钨的催化剂 Download PDFInfo
- Publication number
- CN107497418B CN107497418B CN201710829640.7A CN201710829640A CN107497418B CN 107497418 B CN107497418 B CN 107497418B CN 201710829640 A CN201710829640 A CN 201710829640A CN 107497418 B CN107497418 B CN 107497418B
- Authority
- CN
- China
- Prior art keywords
- bismuth
- catalyst
- vanadium
- acrylic
- tungsten
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000003054 catalyst Substances 0.000 title claims abstract description 344
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 title claims abstract description 173
- LEONUFNNVUYDNQ-UHFFFAOYSA-N vanadium atom Chemical compound [V] LEONUFNNVUYDNQ-UHFFFAOYSA-N 0.000 title claims abstract description 94
- 229910052720 vanadium Inorganic materials 0.000 title claims abstract description 93
- 229910052797 bismuth Inorganic materials 0.000 title claims abstract description 77
- JCXGWMGPZLAOME-UHFFFAOYSA-N bismuth atom Chemical compound [Bi] JCXGWMGPZLAOME-UHFFFAOYSA-N 0.000 title claims abstract description 68
- 229910052721 tungsten Inorganic materials 0.000 title claims abstract description 60
- 239000010937 tungsten Substances 0.000 title claims abstract description 58
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 title claims abstract description 57
- 238000004519 manufacturing process Methods 0.000 title claims description 20
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 title description 51
- 229920006397 acrylic thermoplastic Polymers 0.000 title description 8
- 229920003229 poly(methyl methacrylate) Polymers 0.000 title description 8
- ISXSCDLOGDJUNJ-UHFFFAOYSA-N tert-butyl prop-2-enoate Chemical compound CC(C)(C)OC(=O)C=C ISXSCDLOGDJUNJ-UHFFFAOYSA-N 0.000 title description 8
- 239000000203 mixture Substances 0.000 claims abstract description 157
- 238000000034 method Methods 0.000 claims abstract description 67
- 239000002253 acid Substances 0.000 claims abstract description 49
- 239000003795 chemical substances by application Substances 0.000 claims abstract description 38
- 239000002243 precursor Substances 0.000 claims description 28
- 229910052698 phosphorus Inorganic materials 0.000 claims description 25
- 239000011574 phosphorus Substances 0.000 claims description 25
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 claims description 24
- 150000001621 bismuth Chemical class 0.000 claims description 23
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 22
- 229910052760 oxygen Inorganic materials 0.000 claims description 22
- 239000001301 oxygen Substances 0.000 claims description 22
- 150000003657 tungsten Chemical class 0.000 claims description 17
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 claims description 15
- 238000005882 aldol condensation reaction Methods 0.000 claims description 15
- 238000001354 calcination Methods 0.000 claims description 15
- 238000001035 drying Methods 0.000 claims description 11
- 239000011230 binding agent Substances 0.000 claims description 10
- 239000003638 chemical reducing agent Substances 0.000 claims description 10
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 claims description 8
- 229910052719 titanium Inorganic materials 0.000 claims description 8
- 239000010936 titanium Substances 0.000 claims description 8
- 229910000147 aluminium phosphate Inorganic materials 0.000 claims description 7
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 claims description 6
- 125000002947 alkylene group Chemical group 0.000 abstract 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 390
- 238000006243 chemical reaction Methods 0.000 description 121
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 114
- 230000000052 comparative effect Effects 0.000 description 98
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 72
- 239000011148 porous material Substances 0.000 description 61
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 40
- 239000000243 solution Substances 0.000 description 38
- 239000000047 product Substances 0.000 description 35
- 230000008569 process Effects 0.000 description 34
- 235000019256 formaldehyde Nutrition 0.000 description 32
- 239000003607 modifier Substances 0.000 description 29
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 29
- 229910052751 metal Inorganic materials 0.000 description 25
- 239000002184 metal Substances 0.000 description 25
- 229910001868 water Inorganic materials 0.000 description 25
- BAPJBEWLBFYGME-UHFFFAOYSA-N Methyl acrylate Chemical compound COC(=O)C=C BAPJBEWLBFYGME-UHFFFAOYSA-N 0.000 description 24
- 150000002148 esters Chemical class 0.000 description 23
- -1 bismuth carboxylates Chemical class 0.000 description 21
- 239000000377 silicon dioxide Substances 0.000 description 18
- 238000010438 heat treatment Methods 0.000 description 17
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 16
- WSFSSNUMVMOOMR-NJFSPNSNSA-N methanone Chemical group O=[14CH2] WSFSSNUMVMOOMR-NJFSPNSNSA-N 0.000 description 16
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 15
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 15
- 239000007789 gas Substances 0.000 description 15
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 14
- 230000000694 effects Effects 0.000 description 14
- 239000000463 material Substances 0.000 description 14
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 14
- 150000001875 compounds Chemical class 0.000 description 13
- 238000002360 preparation method Methods 0.000 description 13
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 12
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 12
- 150000007513 acids Chemical class 0.000 description 11
- 229910052739 hydrogen Inorganic materials 0.000 description 11
- 230000002829 reductive effect Effects 0.000 description 11
- 230000015572 biosynthetic process Effects 0.000 description 10
- 239000001257 hydrogen Substances 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 9
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical class CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 9
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 9
- 230000003247 decreasing effect Effects 0.000 description 9
- 230000001965 increasing effect Effects 0.000 description 9
- 229920000609 methyl cellulose Polymers 0.000 description 9
- 239000001923 methylcellulose Substances 0.000 description 9
- 235000010981 methylcellulose Nutrition 0.000 description 9
- 239000002245 particle Substances 0.000 description 9
- 239000007787 solid Substances 0.000 description 9
- 239000002028 Biomass Substances 0.000 description 8
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 8
- 229910019142 PO4 Inorganic materials 0.000 description 8
- 239000000654 additive Substances 0.000 description 8
- 229910052799 carbon Inorganic materials 0.000 description 8
- GNTDGMZSJNCJKK-UHFFFAOYSA-N divanadium pentaoxide Chemical compound O=[V](=O)O[V](=O)=O GNTDGMZSJNCJKK-UHFFFAOYSA-N 0.000 description 8
- 238000005470 impregnation Methods 0.000 description 8
- 229910044991 metal oxide Inorganic materials 0.000 description 8
- 150000004706 metal oxides Chemical class 0.000 description 8
- 239000000126 substance Substances 0.000 description 8
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 7
- MCMNRKCIXSYSNV-UHFFFAOYSA-N Zirconium dioxide Chemical compound O=[Zr]=O MCMNRKCIXSYSNV-UHFFFAOYSA-N 0.000 description 7
- 238000000855 fermentation Methods 0.000 description 7
- 150000002739 metals Chemical class 0.000 description 7
- 229910052757 nitrogen Inorganic materials 0.000 description 7
- 239000000843 powder Substances 0.000 description 7
- 230000035484 reaction time Effects 0.000 description 7
- IKHGUXGNUITLKF-UHFFFAOYSA-N Acetaldehyde Chemical compound CC=O IKHGUXGNUITLKF-UHFFFAOYSA-N 0.000 description 6
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 6
- 230000002378 acidificating effect Effects 0.000 description 6
- 150000001299 aldehydes Chemical class 0.000 description 6
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 6
- 229910002091 carbon monoxide Inorganic materials 0.000 description 6
- 230000004151 fermentation Effects 0.000 description 6
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 description 6
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 6
- 235000019260 propionic acid Nutrition 0.000 description 6
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 6
- 150000003839 salts Chemical class 0.000 description 6
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 5
- 229910000540 VOPO4 Inorganic materials 0.000 description 5
- 239000012018 catalyst precursor Substances 0.000 description 5
- 229920002678 cellulose Polymers 0.000 description 5
- 239000001913 cellulose Substances 0.000 description 5
- 235000010980 cellulose Nutrition 0.000 description 5
- 238000009833 condensation Methods 0.000 description 5
- 230000005494 condensation Effects 0.000 description 5
- 235000014113 dietary fatty acids Nutrition 0.000 description 5
- 239000000194 fatty acid Substances 0.000 description 5
- 229930195729 fatty acid Natural products 0.000 description 5
- 150000004665 fatty acids Chemical group 0.000 description 5
- RXPAJWPEYBDXOG-UHFFFAOYSA-N hydron;methyl 4-methoxypyridine-2-carboxylate;chloride Chemical compound Cl.COC(=O)C1=CC(OC)=CC=N1 RXPAJWPEYBDXOG-UHFFFAOYSA-N 0.000 description 5
- 239000003345 natural gas Substances 0.000 description 5
- 150000007524 organic acids Chemical class 0.000 description 5
- 230000003647 oxidation Effects 0.000 description 5
- 238000007254 oxidation reaction Methods 0.000 description 5
- 238000003786 synthesis reaction Methods 0.000 description 5
- HGINCPLSRVDWNT-UHFFFAOYSA-N Acrolein Chemical compound C=CC=O HGINCPLSRVDWNT-UHFFFAOYSA-N 0.000 description 4
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 4
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 4
- 241000186660 Lactobacillus Species 0.000 description 4
- 229910003206 NH4VO3 Inorganic materials 0.000 description 4
- 235000021355 Stearic acid Nutrition 0.000 description 4
- 125000005250 alkyl acrylate group Chemical group 0.000 description 4
- 150000001735 carboxylic acids Chemical class 0.000 description 4
- 239000008119 colloidal silica Substances 0.000 description 4
- 239000008367 deionised water Substances 0.000 description 4
- 229910021641 deionized water Inorganic materials 0.000 description 4
- 238000009792 diffusion process Methods 0.000 description 4
- 238000005984 hydrogenation reaction Methods 0.000 description 4
- 229940039696 lactobacillus Drugs 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 239000000395 magnesium oxide Substances 0.000 description 4
- CPLXHLVBOLITMK-UHFFFAOYSA-N magnesium oxide Inorganic materials [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 description 4
- 238000002844 melting Methods 0.000 description 4
- 230000008018 melting Effects 0.000 description 4
- 229910017604 nitric acid Inorganic materials 0.000 description 4
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 4
- 241000894007 species Species 0.000 description 4
- 239000008117 stearic acid Substances 0.000 description 4
- DCXXMTOCNZCJGO-UHFFFAOYSA-N tristearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(OC(=O)CCCCCCCCCCCCCCCCC)COC(=O)CCCCCCCCCCCCCCCCC DCXXMTOCNZCJGO-UHFFFAOYSA-N 0.000 description 4
- 239000002699 waste material Substances 0.000 description 4
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- 241000722955 Anaerobiospirillum Species 0.000 description 3
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 3
- 241000196324 Embryophyta Species 0.000 description 3
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 3
- 241000186429 Propionibacterium Species 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- 241000186339 Thermoanaerobacter Species 0.000 description 3
- 240000008042 Zea mays Species 0.000 description 3
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 3
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 3
- IKHGUXGNUITLKF-XPULMUKRSA-N acetaldehyde Chemical compound [14CH]([14CH3])=O IKHGUXGNUITLKF-XPULMUKRSA-N 0.000 description 3
- 229910052782 aluminium Inorganic materials 0.000 description 3
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- 229910052796 boron Inorganic materials 0.000 description 3
- ZTQSAGDEMFDKMZ-UHFFFAOYSA-N butyric aldehyde Natural products CCCC=O ZTQSAGDEMFDKMZ-UHFFFAOYSA-N 0.000 description 3
- 229910052792 caesium Inorganic materials 0.000 description 3
- TVFDJXOCXUVLDH-UHFFFAOYSA-N caesium atom Chemical compound [Cs] TVFDJXOCXUVLDH-UHFFFAOYSA-N 0.000 description 3
- 238000005119 centrifugation Methods 0.000 description 3
- 239000003245 coal Substances 0.000 description 3
- 238000006482 condensation reaction Methods 0.000 description 3
- 235000005822 corn Nutrition 0.000 description 3
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- 150000004820 halides Chemical class 0.000 description 3
- 239000011964 heteropoly acid Substances 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- LYRFLYHAGKPMFH-UHFFFAOYSA-N octadecanamide Chemical compound CCCCCCCCCCCCCCCCCC(N)=O LYRFLYHAGKPMFH-UHFFFAOYSA-N 0.000 description 3
- 235000006408 oxalic acid Nutrition 0.000 description 3
- 239000008188 pellet Substances 0.000 description 3
- 239000003208 petroleum Substances 0.000 description 3
- 239000000376 reactant Substances 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- DLYUQMMRRRQYAE-UHFFFAOYSA-N tetraphosphorus decaoxide Chemical compound O1P(O2)(=O)OP3(=O)OP1(=O)OP2(=O)O3 DLYUQMMRRRQYAE-UHFFFAOYSA-N 0.000 description 3
- 239000002023 wood Substances 0.000 description 3
- BGJSXRVXTHVRSN-UHFFFAOYSA-N 1,3,5-trioxane Chemical compound C1OCOCO1 BGJSXRVXTHVRSN-UHFFFAOYSA-N 0.000 description 2
- QYIGOGBGVKONDY-UHFFFAOYSA-N 1-(2-bromo-5-chlorophenyl)-3-methylpyrazole Chemical compound N1=C(C)C=CN1C1=CC(Cl)=CC=C1Br QYIGOGBGVKONDY-UHFFFAOYSA-N 0.000 description 2
- KKMOSYLWYLMHAL-UHFFFAOYSA-N 2-bromo-6-nitroaniline Chemical compound NC1=C(Br)C=CC=C1[N+]([O-])=O KKMOSYLWYLMHAL-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- 235000003276 Apios tuberosa Nutrition 0.000 description 2
- 244000105624 Arachis hypogaea Species 0.000 description 2
- 235000010777 Arachis hypogaea Nutrition 0.000 description 2
- 235000010744 Arachis villosulicarpa Nutrition 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- HWSISDHAHRVNMT-UHFFFAOYSA-N Bismuth subnitrate Chemical compound O[NH+]([O-])O[Bi](O[N+]([O-])=O)O[N+]([O-])=O HWSISDHAHRVNMT-UHFFFAOYSA-N 0.000 description 2
- 241000193403 Clostridium Species 0.000 description 2
- 241000193171 Clostridium butyricum Species 0.000 description 2
- 241000193161 Clostridium formicaceticum Species 0.000 description 2
- QPLDLSVMHZLSFG-UHFFFAOYSA-N Copper oxide Chemical compound [Cu]=O QPLDLSVMHZLSFG-UHFFFAOYSA-N 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- RJUFJBKOKNCXHH-UHFFFAOYSA-N Methyl propionate Chemical compound CCC(=O)OC RJUFJBKOKNCXHH-UHFFFAOYSA-N 0.000 description 2
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 description 2
- 241000178985 Moorella Species 0.000 description 2
- 241000193459 Moorella thermoacetica Species 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- NBBJYMSMWIIQGU-UHFFFAOYSA-N Propionic aldehyde Chemical compound CCC=O NBBJYMSMWIIQGU-UHFFFAOYSA-N 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical class N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- 229920002125 Sokalan® Polymers 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- BOTDANWDWHJENH-UHFFFAOYSA-N Tetraethyl orthosilicate Chemical compound CCO[Si](OCC)(OCC)OCC BOTDANWDWHJENH-UHFFFAOYSA-N 0.000 description 2
- VNDYJBBGRKZCSX-UHFFFAOYSA-L Zinc bromide Inorganic materials Br[Zn]Br VNDYJBBGRKZCSX-UHFFFAOYSA-L 0.000 description 2
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical compound [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 description 2
- DYDNWZRECQKENE-UHFFFAOYSA-N [W].[Bi].[V] Chemical compound [W].[Bi].[V] DYDNWZRECQKENE-UHFFFAOYSA-N 0.000 description 2
- 230000000789 acetogenic effect Effects 0.000 description 2
- 230000000996 additive effect Effects 0.000 description 2
- 239000002154 agricultural waste Substances 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 150000001335 aliphatic alkanes Chemical class 0.000 description 2
- 229910052783 alkali metal Inorganic materials 0.000 description 2
- 150000001340 alkali metals Chemical class 0.000 description 2
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 2
- 150000004703 alkoxides Chemical class 0.000 description 2
- 150000008064 anhydrides Chemical class 0.000 description 2
- 125000003118 aryl group Chemical group 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- 229960001482 bismuth subnitrate Drugs 0.000 description 2
- ZREIPSZUJIFJNP-UHFFFAOYSA-K bismuth subsalicylate Chemical compound C1=CC=C2O[Bi](O)OC(=O)C2=C1 ZREIPSZUJIFJNP-UHFFFAOYSA-K 0.000 description 2
- JDIBGQFKXXXXPN-UHFFFAOYSA-N bismuth(3+) Chemical compound [Bi+3] JDIBGQFKXXXXPN-UHFFFAOYSA-N 0.000 description 2
- WMWLMWRWZQELOS-UHFFFAOYSA-N bismuth(iii) oxide Chemical compound O=[Bi]O[Bi]=O WMWLMWRWZQELOS-UHFFFAOYSA-N 0.000 description 2
- REKWPXFKNZERAA-UHFFFAOYSA-K bismuth;2-carboxyphenolate Chemical compound [Bi+3].OC1=CC=CC=C1C([O-])=O.OC1=CC=CC=C1C([O-])=O.OC1=CC=CC=C1C([O-])=O REKWPXFKNZERAA-UHFFFAOYSA-K 0.000 description 2
- PIMIKCFPAJSEQM-UHFFFAOYSA-N bismuth;trinitrate;hydrate Chemical compound O.[Bi+3].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O PIMIKCFPAJSEQM-UHFFFAOYSA-N 0.000 description 2
- ZQUAVILLCXTKTF-UHFFFAOYSA-H bismuth;tripotassium;2-hydroxypropane-1,2,3-tricarboxylate Chemical compound [K+].[K+].[K+].[Bi+3].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O.[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O ZQUAVILLCXTKTF-UHFFFAOYSA-H 0.000 description 2
- 239000006227 byproduct Substances 0.000 description 2
- 239000001569 carbon dioxide Substances 0.000 description 2
- 229910002092 carbon dioxide Inorganic materials 0.000 description 2
- 239000003575 carbonaceous material Substances 0.000 description 2
- 230000006315 carbonylation Effects 0.000 description 2
- 238000005810 carbonylation reaction Methods 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 238000006555 catalytic reaction Methods 0.000 description 2
- 239000000919 ceramic Substances 0.000 description 2
- 229910052593 corundum Inorganic materials 0.000 description 2
- 230000009849 deactivation Effects 0.000 description 2
- 230000001627 detrimental effect Effects 0.000 description 2
- GUJOJGAPFQRJSV-UHFFFAOYSA-N dialuminum;dioxosilane;oxygen(2-);hydrate Chemical compound O.[O-2].[O-2].[O-2].[Al+3].[Al+3].O=[Si]=O.O=[Si]=O.O=[Si]=O.O=[Si]=O GUJOJGAPFQRJSV-UHFFFAOYSA-N 0.000 description 2
- QDOXWKRWXJOMAK-UHFFFAOYSA-N dichromium trioxide Chemical compound O=[Cr]O[Cr]=O QDOXWKRWXJOMAK-UHFFFAOYSA-N 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- POULHZVOKOAJMA-UHFFFAOYSA-N dodecanoic acid Chemical compound CCCCCCCCCCCC(O)=O POULHZVOKOAJMA-UHFFFAOYSA-N 0.000 description 2
- 230000002708 enhancing effect Effects 0.000 description 2
- 230000007613 environmental effect Effects 0.000 description 2
- 230000002349 favourable effect Effects 0.000 description 2
- 239000006260 foam Substances 0.000 description 2
- 238000002309 gasification Methods 0.000 description 2
- 239000010439 graphite Substances 0.000 description 2
- 229910002804 graphite Inorganic materials 0.000 description 2
- 229910052735 hafnium Inorganic materials 0.000 description 2
- VBJZVLUMGGDVMO-UHFFFAOYSA-N hafnium atom Chemical compound [Hf] VBJZVLUMGGDVMO-UHFFFAOYSA-N 0.000 description 2
- 239000008241 heterogeneous mixture Substances 0.000 description 2
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 2
- 239000008240 homogeneous mixture Substances 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 150000002430 hydrocarbons Chemical class 0.000 description 2
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 2
- 229910052742 iron Inorganic materials 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- AXZKOIWUVFPNLO-UHFFFAOYSA-N magnesium;oxygen(2-) Chemical compound [O-2].[Mg+2] AXZKOIWUVFPNLO-UHFFFAOYSA-N 0.000 description 2
- NUJOXMJBOLGQSY-UHFFFAOYSA-N manganese dioxide Chemical compound O=[Mn]=O NUJOXMJBOLGQSY-UHFFFAOYSA-N 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 229910001463 metal phosphate Inorganic materials 0.000 description 2
- CKFGINPQOCXMAZ-UHFFFAOYSA-N methanediol Chemical compound OCO CKFGINPQOCXMAZ-UHFFFAOYSA-N 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 229940017219 methyl propionate Drugs 0.000 description 2
- 239000012022 methylating agents Substances 0.000 description 2
- 244000005700 microbiome Species 0.000 description 2
- 229910003455 mixed metal oxide Inorganic materials 0.000 description 2
- 229910052750 molybdenum Inorganic materials 0.000 description 2
- 239000011733 molybdenum Substances 0.000 description 2
- JKQOBWVOAYFWKG-UHFFFAOYSA-N molybdenum trioxide Chemical compound O=[Mo](=O)=O JKQOBWVOAYFWKG-UHFFFAOYSA-N 0.000 description 2
- 229910052901 montmorillonite Inorganic materials 0.000 description 2
- ZKATWMILCYLAPD-UHFFFAOYSA-N niobium pentoxide Chemical compound O=[Nb](=O)O[Nb](=O)=O ZKATWMILCYLAPD-UHFFFAOYSA-N 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 150000002894 organic compounds Chemical class 0.000 description 2
- UPWOEMHINGJHOB-UHFFFAOYSA-N oxo(oxocobaltiooxy)cobalt Chemical compound O=[Co]O[Co]=O UPWOEMHINGJHOB-UHFFFAOYSA-N 0.000 description 2
- 239000000123 paper Substances 0.000 description 2
- 235000021317 phosphate Nutrition 0.000 description 2
- 229910052615 phyllosilicate Inorganic materials 0.000 description 2
- 239000006069 physical mixture Substances 0.000 description 2
- 229920003023 plastic Polymers 0.000 description 2
- 239000004033 plastic Substances 0.000 description 2
- 229920005862 polyol Polymers 0.000 description 2
- 150000003077 polyols Chemical class 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 229960003975 potassium Drugs 0.000 description 2
- 238000009420 retrofitting Methods 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 229910052710 silicon Inorganic materials 0.000 description 2
- 239000010703 silicon Substances 0.000 description 2
- HBMJWWWQQXIZIP-UHFFFAOYSA-N silicon carbide Chemical compound [Si+]#[C-] HBMJWWWQQXIZIP-UHFFFAOYSA-N 0.000 description 2
- 229910010271 silicon carbide Inorganic materials 0.000 description 2
- 238000001694 spray drying Methods 0.000 description 2
- 239000010902 straw Substances 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 239000011593 sulfur Substances 0.000 description 2
- TUNFSRHWOTWDNC-UHFFFAOYSA-N tetradecanoic acid Chemical compound CCCCCCCCCCCCCC(O)=O TUNFSRHWOTWDNC-UHFFFAOYSA-N 0.000 description 2
- DZKDPOPGYFUOGI-UHFFFAOYSA-N tungsten(iv) oxide Chemical compound O=[W]=O DZKDPOPGYFUOGI-UHFFFAOYSA-N 0.000 description 2
- 150000003682 vanadium compounds Chemical class 0.000 description 2
- 125000005287 vanadyl group Chemical group 0.000 description 2
- 229910001845 yogo sapphire Inorganic materials 0.000 description 2
- 229910052726 zirconium Inorganic materials 0.000 description 2
- FRPZMMHWLSIFAZ-UHFFFAOYSA-N 10-undecenoic acid Chemical compound OC(=O)CCCCCCCCC=C FRPZMMHWLSIFAZ-UHFFFAOYSA-N 0.000 description 1
- TWJNQYPJQDRXPH-UHFFFAOYSA-N 2-cyanobenzohydrazide Chemical compound NNC(=O)C1=CC=CC=C1C#N TWJNQYPJQDRXPH-UHFFFAOYSA-N 0.000 description 1
- ZSLUVFAKFWKJRC-IGMARMGPSA-N 232Th Chemical compound [232Th] ZSLUVFAKFWKJRC-IGMARMGPSA-N 0.000 description 1
- TXQJIMNDEHCONN-UHFFFAOYSA-N 3-(2-methoxyphenoxy)propanoic acid Chemical compound COC1=CC=CC=C1OCCC(O)=O TXQJIMNDEHCONN-UHFFFAOYSA-N 0.000 description 1
- OQEBBZSWEGYTPG-UHFFFAOYSA-N 3-aminobutanoic acid Chemical compound CC(N)CC(O)=O OQEBBZSWEGYTPG-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- YPIFGDQKSSMYHQ-UHFFFAOYSA-M 7,7-dimethyloctanoate Chemical compound CC(C)(C)CCCCCC([O-])=O YPIFGDQKSSMYHQ-UHFFFAOYSA-M 0.000 description 1
- 241000186426 Acidipropionibacterium acidipropionici Species 0.000 description 1
- 241000609240 Ambelania acida Species 0.000 description 1
- 241000722954 Anaerobiospirillum succiniciproducens Species 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- 229910011255 B2O3 Inorganic materials 0.000 description 1
- 241000606125 Bacteroides Species 0.000 description 1
- 229910000014 Bismuth subcarbonate Inorganic materials 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 244000025254 Cannabis sativa Species 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 244000068645 Carya illinoensis Species 0.000 description 1
- 235000009025 Carya illinoensis Nutrition 0.000 description 1
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- RJNBTFHJYBHWCU-UHFFFAOYSA-L Cl[W](Cl)(=O)=O Chemical compound Cl[W](Cl)(=O)=O RJNBTFHJYBHWCU-UHFFFAOYSA-L 0.000 description 1
- 244000060011 Cocos nucifera Species 0.000 description 1
- 235000013162 Cocos nucifera Nutrition 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- 102000002322 Egg Proteins Human genes 0.000 description 1
- 108010000912 Egg Proteins Proteins 0.000 description 1
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- GYHNNYVSQQEPJS-UHFFFAOYSA-N Gallium Chemical compound [Ga] GYHNNYVSQQEPJS-UHFFFAOYSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 229920002488 Hemicellulose Polymers 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 239000005639 Lauric acid Substances 0.000 description 1
- 239000002841 Lewis acid Substances 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- STNJBCKSHOAVAJ-UHFFFAOYSA-N Methacrolein Chemical compound CC(=C)C=O STNJBCKSHOAVAJ-UHFFFAOYSA-N 0.000 description 1
- 240000003433 Miscanthus floridulus Species 0.000 description 1
- 235000021360 Myristic acid Nutrition 0.000 description 1
- 240000007594 Oryza sativa Species 0.000 description 1
- 235000007164 Oryza sativa Nutrition 0.000 description 1
- 235000021314 Palmitic acid Nutrition 0.000 description 1
- 229930040373 Paraformaldehyde Natural products 0.000 description 1
- 241000209504 Poaceae Species 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 244000018633 Prunus armeniaca Species 0.000 description 1
- 235000009827 Prunus armeniaca Nutrition 0.000 description 1
- 241000186336 Pseudopropionibacterium propionicum Species 0.000 description 1
- 241000606009 Ruminobacter Species 0.000 description 1
- 241000606008 Ruminobacter amylophilus Species 0.000 description 1
- KJTLSVCANCCWHF-UHFFFAOYSA-N Ruthenium Chemical compound [Ru] KJTLSVCANCCWHF-UHFFFAOYSA-N 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 240000006661 Serenoa repens Species 0.000 description 1
- 235000005318 Serenoa repens Nutrition 0.000 description 1
- 229910052581 Si3N4 Inorganic materials 0.000 description 1
- 229910020413 SiO2—MgO Inorganic materials 0.000 description 1
- 229910020442 SiO2—TiO2 Inorganic materials 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- BQCADISMDOOEFD-UHFFFAOYSA-N Silver Chemical compound [Ag] BQCADISMDOOEFD-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 241000204649 Thermoanaerobacter kivui Species 0.000 description 1
- 229910052776 Thorium Inorganic materials 0.000 description 1
- 229910010442 TiO2-SnO2 Inorganic materials 0.000 description 1
- 229910010257 TiO2—SnO2 Inorganic materials 0.000 description 1
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 1
- 229910021627 Tin(IV) chloride Inorganic materials 0.000 description 1
- 235000021307 Triticum Nutrition 0.000 description 1
- 244000098338 Triticum aestivum Species 0.000 description 1
- 229910052770 Uranium Inorganic materials 0.000 description 1
- NIJJYAXOARWZEE-UHFFFAOYSA-N Valproic acid Chemical compound CCCC(C(O)=O)CCC NIJJYAXOARWZEE-UHFFFAOYSA-N 0.000 description 1
- 235000009754 Vitis X bourquina Nutrition 0.000 description 1
- 235000012333 Vitis X labruscana Nutrition 0.000 description 1
- 240000006365 Vitis vinifera Species 0.000 description 1
- 235000014787 Vitis vinifera Nutrition 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- OFCPHUYEFORJCF-UHFFFAOYSA-E [Ti+4].[V+5].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O Chemical class [Ti+4].[V+5].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O OFCPHUYEFORJCF-UHFFFAOYSA-E 0.000 description 1
- GLMOMDXKLRBTDY-UHFFFAOYSA-A [V+5].[V+5].[V+5].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O Chemical compound [V+5].[V+5].[V+5].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O GLMOMDXKLRBTDY-UHFFFAOYSA-A 0.000 description 1
- CAUHCBDRRFADDB-UHFFFAOYSA-A [V+5].[V+5].[V+5].[V+5].[O-]P([O-])(=O)OP([O-])([O-])=O.[O-]P([O-])(=O)OP([O-])([O-])=O.[O-]P([O-])(=O)OP([O-])([O-])=O.[O-]P([O-])(=O)OP([O-])([O-])=O.[O-]P([O-])(=O)OP([O-])([O-])=O Chemical compound [V+5].[V+5].[V+5].[V+5].[O-]P([O-])(=O)OP([O-])([O-])=O.[O-]P([O-])(=O)OP([O-])([O-])=O.[O-]P([O-])(=O)OP([O-])([O-])=O.[O-]P([O-])(=O)OP([O-])([O-])=O.[O-]P([O-])(=O)OP([O-])([O-])=O CAUHCBDRRFADDB-UHFFFAOYSA-A 0.000 description 1
- 150000001241 acetals Chemical class 0.000 description 1
- 229960001138 acetylsalicylic acid Drugs 0.000 description 1
- 150000008065 acid anhydrides Chemical class 0.000 description 1
- 239000003377 acid catalyst Substances 0.000 description 1
- 150000001253 acrylic acids Chemical class 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 239000012615 aggregate Substances 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 150000001447 alkali salts Chemical class 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 125000003342 alkenyl group Chemical group 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 229910000323 aluminium silicate Inorganic materials 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 229940053200 antiepileptics fatty acid derivative Drugs 0.000 description 1
- 229910052787 antimony Inorganic materials 0.000 description 1
- WATWJIUSRGPENY-UHFFFAOYSA-N antimony atom Chemical compound [Sb] WATWJIUSRGPENY-UHFFFAOYSA-N 0.000 description 1
- ADCOVFLJGNWWNZ-UHFFFAOYSA-N antimony trioxide Inorganic materials O=[Sb]O[Sb]=O ADCOVFLJGNWWNZ-UHFFFAOYSA-N 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 229910052785 arsenic Inorganic materials 0.000 description 1
- RQNWIZPPADIBDY-UHFFFAOYSA-N arsenic atom Chemical compound [As] RQNWIZPPADIBDY-UHFFFAOYSA-N 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- UNTBPXHCXVWYOI-UHFFFAOYSA-O azanium;oxido(dioxo)vanadium Chemical compound [NH4+].[O-][V](=O)=O UNTBPXHCXVWYOI-UHFFFAOYSA-O 0.000 description 1
- 239000010905 bagasse Substances 0.000 description 1
- 229910052788 barium Inorganic materials 0.000 description 1
- DSAJWYNOEDNPEQ-UHFFFAOYSA-N barium atom Chemical compound [Ba] DSAJWYNOEDNPEQ-UHFFFAOYSA-N 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 229940050390 benzoate Drugs 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- GMWXRWTXGXBWDE-UHFFFAOYSA-L bis(sulfanylidene)tungsten;piperidine;sulfanide Chemical compound [SH-].[SH-].S=[W]=S.C1CCNCC1.C1CCNCC1 GMWXRWTXGXBWDE-UHFFFAOYSA-L 0.000 description 1
- PPJPTAQKIFHZQU-UHFFFAOYSA-N bis(tert-butylimino)tungsten;dimethylazanide Chemical compound C[N-]C.C[N-]C.CC(C)(C)N=[W]=NC(C)(C)C PPJPTAQKIFHZQU-UHFFFAOYSA-N 0.000 description 1
- 229940104825 bismuth aluminate Drugs 0.000 description 1
- 229940036348 bismuth carbonate Drugs 0.000 description 1
- JHXKRIRFYBPWGE-UHFFFAOYSA-K bismuth chloride Chemical compound Cl[Bi](Cl)Cl JHXKRIRFYBPWGE-UHFFFAOYSA-K 0.000 description 1
- 229940049676 bismuth hydroxide Drugs 0.000 description 1
- 229910000416 bismuth oxide Inorganic materials 0.000 description 1
- 229940036359 bismuth oxide Drugs 0.000 description 1
- 229940073609 bismuth oxychloride Drugs 0.000 description 1
- MGLUJXPJRXTKJM-UHFFFAOYSA-L bismuth subcarbonate Chemical compound O=[Bi]OC(=O)O[Bi]=O MGLUJXPJRXTKJM-UHFFFAOYSA-L 0.000 description 1
- 229940036358 bismuth subcarbonate Drugs 0.000 description 1
- 229960004645 bismuth subcitrate Drugs 0.000 description 1
- JAONZGLTYYUPCT-UHFFFAOYSA-K bismuth subgallate Chemical compound OC(=O)C1=CC(O)=C2O[Bi](O)OC2=C1 JAONZGLTYYUPCT-UHFFFAOYSA-K 0.000 description 1
- 229960000199 bismuth subgallate Drugs 0.000 description 1
- 229960000782 bismuth subsalicylate Drugs 0.000 description 1
- 229910000380 bismuth sulfate Inorganic materials 0.000 description 1
- TXKAQZRUJUNDHI-UHFFFAOYSA-K bismuth tribromide Chemical compound Br[Bi](Br)Br TXKAQZRUJUNDHI-UHFFFAOYSA-K 0.000 description 1
- 229940077187 bismuth tribromophenate Drugs 0.000 description 1
- JTPAGNYHZFJFGV-UHFFFAOYSA-N bismuth;1h-quinolin-2-one Chemical compound [Bi].C1=CC=CC2=NC(O)=CC=C21 JTPAGNYHZFJFGV-UHFFFAOYSA-N 0.000 description 1
- SFOQXWSZZPWNCL-UHFFFAOYSA-K bismuth;phosphate Chemical compound [Bi+3].[O-]P([O-])([O-])=O SFOQXWSZZPWNCL-UHFFFAOYSA-K 0.000 description 1
- TZSXPYWRDWEXHG-UHFFFAOYSA-K bismuth;trihydroxide Chemical compound [OH-].[OH-].[OH-].[Bi+3] TZSXPYWRDWEXHG-UHFFFAOYSA-K 0.000 description 1
- 239000001273 butane Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 235000012241 calcium silicate Nutrition 0.000 description 1
- 229910052918 calcium silicate Inorganic materials 0.000 description 1
- VNSBYDPZHCQWNB-UHFFFAOYSA-N calcium;aluminum;dioxido(oxo)silane;sodium;hydrate Chemical compound O.[Na].[Al].[Ca+2].[O-][Si]([O-])=O VNSBYDPZHCQWNB-UHFFFAOYSA-N 0.000 description 1
- OYACROKNLOSFPA-UHFFFAOYSA-N calcium;dioxido(oxo)silane Chemical compound [Ca+2].[O-][Si]([O-])=O OYACROKNLOSFPA-UHFFFAOYSA-N 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 229910002090 carbon oxide Inorganic materials 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 150000001733 carboxylic acid esters Chemical class 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 239000011111 cardboard Substances 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000012159 carrier gas Substances 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 229920002301 cellulose acetate Polymers 0.000 description 1
- 239000013626 chemical specie Substances 0.000 description 1
- 238000005229 chemical vapour deposition Methods 0.000 description 1
- 229910052804 chromium Inorganic materials 0.000 description 1
- 239000011651 chromium Substances 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 239000002734 clay mineral Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229910017052 cobalt Inorganic materials 0.000 description 1
- 239000010941 cobalt Substances 0.000 description 1
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 1
- 238000002485 combustion reaction Methods 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 239000007859 condensation product Substances 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 229910052878 cordierite Inorganic materials 0.000 description 1
- XAYGUHUYDMLJJV-UHFFFAOYSA-Z decaazanium;dioxido(dioxo)tungsten;hydron;trioxotungsten Chemical compound [H+].[H+].[NH4+].[NH4+].[NH4+].[NH4+].[NH4+].[NH4+].[NH4+].[NH4+].[NH4+].[NH4+].O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.[O-][W]([O-])(=O)=O.[O-][W]([O-])(=O)=O.[O-][W]([O-])(=O)=O.[O-][W]([O-])(=O)=O.[O-][W]([O-])(=O)=O.[O-][W]([O-])(=O)=O XAYGUHUYDMLJJV-UHFFFAOYSA-Z 0.000 description 1
- TYIXMATWDRGMPF-UHFFFAOYSA-N dibismuth;oxygen(2-) Chemical compound [O-2].[O-2].[O-2].[Bi+3].[Bi+3] TYIXMATWDRGMPF-UHFFFAOYSA-N 0.000 description 1
- GMZOPRQQINFLPQ-UHFFFAOYSA-H dibismuth;tricarbonate Chemical compound [Bi+3].[Bi+3].[O-]C([O-])=O.[O-]C([O-])=O.[O-]C([O-])=O GMZOPRQQINFLPQ-UHFFFAOYSA-H 0.000 description 1
- BEQZMQXCOWIHRY-UHFFFAOYSA-H dibismuth;trisulfate Chemical compound [Bi+3].[Bi+3].[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O BEQZMQXCOWIHRY-UHFFFAOYSA-H 0.000 description 1
- JSKIRARMQDRGJZ-UHFFFAOYSA-N dimagnesium dioxido-bis[(1-oxido-3-oxo-2,4,6,8,9-pentaoxa-1,3-disila-5,7-dialuminabicyclo[3.3.1]nonan-7-yl)oxy]silane Chemical compound [Mg++].[Mg++].[O-][Si]([O-])(O[Al]1O[Al]2O[Si](=O)O[Si]([O-])(O1)O2)O[Al]1O[Al]2O[Si](=O)O[Si]([O-])(O1)O2 JSKIRARMQDRGJZ-UHFFFAOYSA-N 0.000 description 1
- GZVBAOSNKYQKIT-UHFFFAOYSA-N dimethoxymethane Chemical compound COCOC.COCOC GZVBAOSNKYQKIT-UHFFFAOYSA-N 0.000 description 1
- NKDDWNXOKDWJAK-UHFFFAOYSA-N dimethoxymethane Chemical group COCOC NKDDWNXOKDWJAK-UHFFFAOYSA-N 0.000 description 1
- KZHJGOXRZJKJNY-UHFFFAOYSA-N dioxosilane;oxo(oxoalumanyloxy)alumane Chemical group O=[Si]=O.O=[Si]=O.O=[Al]O[Al]=O.O=[Al]O[Al]=O.O=[Al]O[Al]=O KZHJGOXRZJKJNY-UHFFFAOYSA-N 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- POULHZVOKOAJMA-UHFFFAOYSA-M dodecanoate Chemical compound CCCCCCCCCCCC([O-])=O POULHZVOKOAJMA-UHFFFAOYSA-M 0.000 description 1
- 210000003278 egg shell Anatomy 0.000 description 1
- CCGKOQOJPYTBIH-UHFFFAOYSA-N ethenone Chemical compound C=C=O CCGKOQOJPYTBIH-UHFFFAOYSA-N 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 238000001125 extrusion Methods 0.000 description 1
- 239000004744 fabric Substances 0.000 description 1
- 210000003608 fece Anatomy 0.000 description 1
- 239000000835 fiber Substances 0.000 description 1
- 239000008394 flocculating agent Substances 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 239000000446 fuel Substances 0.000 description 1
- 229910021485 fumed silica Inorganic materials 0.000 description 1
- 229910052733 gallium Inorganic materials 0.000 description 1
- 238000001879 gelation Methods 0.000 description 1
- 229910052732 germanium Inorganic materials 0.000 description 1
- GNPVGFCGXDBREM-UHFFFAOYSA-N germanium atom Chemical compound [Ge] GNPVGFCGXDBREM-UHFFFAOYSA-N 0.000 description 1
- 239000003365 glass fiber Substances 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 239000011121 hardwood Substances 0.000 description 1
- 238000003306 harvesting Methods 0.000 description 1
- 239000011874 heated mixture Substances 0.000 description 1
- 229910052734 helium Inorganic materials 0.000 description 1
- 239000001307 helium Substances 0.000 description 1
- SWQJXJOGLNCZEY-UHFFFAOYSA-N helium atom Chemical compound [He] SWQJXJOGLNCZEY-UHFFFAOYSA-N 0.000 description 1
- PDSAKIXGSONUIX-UHFFFAOYSA-N hexaaluminum;dibismuth;oxygen(2-) Chemical compound [O-2].[O-2].[O-2].[O-2].[O-2].[O-2].[O-2].[O-2].[O-2].[O-2].[O-2].[O-2].[Al+3].[Al+3].[Al+3].[Al+3].[Al+3].[Al+3].[Bi+3].[Bi+3] PDSAKIXGSONUIX-UHFFFAOYSA-N 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- 230000003403 homoacetogenic effect Effects 0.000 description 1
- 229920001519 homopolymer Polymers 0.000 description 1
- BHEPBYXIRTUNPN-UHFFFAOYSA-N hydridophosphorus(.) (triplet) Chemical compound [PH] BHEPBYXIRTUNPN-UHFFFAOYSA-N 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 239000002440 industrial waste Substances 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 229910001867 inorganic solvent Inorganic materials 0.000 description 1
- 239000003049 inorganic solvent Substances 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 239000011872 intimate mixture Substances 0.000 description 1
- UQSXHKLRYXJYBZ-UHFFFAOYSA-N iron oxide Inorganic materials [Fe]=O UQSXHKLRYXJYBZ-UHFFFAOYSA-N 0.000 description 1
- 235000013980 iron oxide Nutrition 0.000 description 1
- VBMVTYDPPZVILR-UHFFFAOYSA-N iron(2+);oxygen(2-) Chemical class [O-2].[Fe+2] VBMVTYDPPZVILR-UHFFFAOYSA-N 0.000 description 1
- JEIPFZHSYJVQDO-UHFFFAOYSA-N iron(III) oxide Inorganic materials O=[Fe]O[Fe]=O JEIPFZHSYJVQDO-UHFFFAOYSA-N 0.000 description 1
- 239000002655 kraft paper Substances 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 229910052746 lanthanum Inorganic materials 0.000 description 1
- FZLIPJUXYLNCLC-UHFFFAOYSA-N lanthanum atom Chemical compound [La] FZLIPJUXYLNCLC-UHFFFAOYSA-N 0.000 description 1
- 239000004816 latex Substances 0.000 description 1
- 229920000126 latex Polymers 0.000 description 1
- 229940070765 laurate Drugs 0.000 description 1
- 150000007517 lewis acids Chemical class 0.000 description 1
- 229930013686 lignan Natural products 0.000 description 1
- 235000009408 lignans Nutrition 0.000 description 1
- 150000005692 lignans Chemical class 0.000 description 1
- 229920005610 lignin Polymers 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 239000010871 livestock manure Substances 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- FPYJFEHAWHCUMM-UHFFFAOYSA-N maleic anhydride Chemical compound O=C1OC(=O)C=C1 FPYJFEHAWHCUMM-UHFFFAOYSA-N 0.000 description 1
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical compound [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 description 1
- 239000013335 mesoporous material Substances 0.000 description 1
- 150000002736 metal compounds Chemical class 0.000 description 1
- 239000012702 metal oxide precursor Substances 0.000 description 1
- 150000004702 methyl esters Chemical class 0.000 description 1
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 1
- 238000003801 milling Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 125000000896 monocarboxylic acid group Chemical group 0.000 description 1
- 238000000465 moulding Methods 0.000 description 1
- 229940105132 myristate Drugs 0.000 description 1
- WQEPLUUGTLDZJY-UHFFFAOYSA-N n-Pentadecanoic acid Natural products CCCCCCCCCCCCCCC(O)=O WQEPLUUGTLDZJY-UHFFFAOYSA-N 0.000 description 1
- IJDNQMDRQITEOD-UHFFFAOYSA-N n-butane Chemical compound CCCC IJDNQMDRQITEOD-UHFFFAOYSA-N 0.000 description 1
- OFBQJSOFQDEBGM-UHFFFAOYSA-N n-pentane Natural products CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 229910052758 niobium Inorganic materials 0.000 description 1
- 239000010955 niobium Substances 0.000 description 1
- GUCVJGMIXFAOAE-UHFFFAOYSA-N niobium atom Chemical compound [Nb] GUCVJGMIXFAOAE-UHFFFAOYSA-N 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 229910052755 nonmetal Inorganic materials 0.000 description 1
- 150000002843 nonmetals Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000001151 other effect Effects 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- BWOROQSFKKODDR-UHFFFAOYSA-N oxobismuth;hydrochloride Chemical compound Cl.[Bi]=O BWOROQSFKKODDR-UHFFFAOYSA-N 0.000 description 1
- RVTZCBVAJQQJTK-UHFFFAOYSA-N oxygen(2-);zirconium(4+) Chemical compound [O-2].[O-2].[Zr+4] RVTZCBVAJQQJTK-UHFFFAOYSA-N 0.000 description 1
- 239000003973 paint Substances 0.000 description 1
- 229920002866 paraformaldehyde Polymers 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000003209 petroleum derivative Substances 0.000 description 1
- 150000002989 phenols Chemical class 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- AVFBYUADVDVJQL-UHFFFAOYSA-N phosphoric acid;trioxotungsten;hydrate Chemical compound O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.OP(O)(O)=O AVFBYUADVDVJQL-UHFFFAOYSA-N 0.000 description 1
- 125000004437 phosphorous atom Chemical group 0.000 description 1
- 150000003017 phosphorus Chemical class 0.000 description 1
- 150000003018 phosphorus compounds Chemical class 0.000 description 1
- UHZYTMXLRWXGPK-UHFFFAOYSA-N phosphorus pentachloride Chemical compound ClP(Cl)(Cl)(Cl)Cl UHZYTMXLRWXGPK-UHFFFAOYSA-N 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 239000004584 polyacrylic acid Substances 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229920000137 polyphosphoric acid Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 238000003825 pressing Methods 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 1
- WYVAMUWZEOHJOQ-UHFFFAOYSA-N propionic anhydride Chemical compound CCC(=O)OC(=O)CC WYVAMUWZEOHJOQ-UHFFFAOYSA-N 0.000 description 1
- WHMDPDGBKYUEMW-UHFFFAOYSA-N pyridine-2-thiol Chemical class SC1=CC=CC=N1 WHMDPDGBKYUEMW-UHFFFAOYSA-N 0.000 description 1
- YBBJKCMMCRQZMA-UHFFFAOYSA-N pyrithione Chemical compound ON1C=CC=CC1=S YBBJKCMMCRQZMA-UHFFFAOYSA-N 0.000 description 1
- 229960002026 pyrithione Drugs 0.000 description 1
- 229940107700 pyruvic acid Drugs 0.000 description 1
- 229910052761 rare earth metal Inorganic materials 0.000 description 1
- 150000002910 rare earth metals Chemical class 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 238000002407 reforming Methods 0.000 description 1
- 239000012744 reinforcing agent Substances 0.000 description 1
- 229910052702 rhenium Inorganic materials 0.000 description 1
- WUAPFZMCVAUBPE-UHFFFAOYSA-N rhenium atom Chemical compound [Re] WUAPFZMCVAUBPE-UHFFFAOYSA-N 0.000 description 1
- 235000009566 rice Nutrition 0.000 description 1
- WBHHMMIMDMUBKC-QJWNTBNXSA-M ricinoleate Chemical compound CCCCCC[C@@H](O)C\C=C/CCCCCCCC([O-])=O WBHHMMIMDMUBKC-QJWNTBNXSA-M 0.000 description 1
- 229940066675 ricinoleate Drugs 0.000 description 1
- 229910052701 rubidium Inorganic materials 0.000 description 1
- IGLNJRXAVVLDKE-UHFFFAOYSA-N rubidium atom Chemical compound [Rb] IGLNJRXAVVLDKE-UHFFFAOYSA-N 0.000 description 1
- 229910052707 ruthenium Inorganic materials 0.000 description 1
- 229910000275 saponite Inorganic materials 0.000 description 1
- 230000009291 secondary effect Effects 0.000 description 1
- 150000004760 silicates Chemical class 0.000 description 1
- CGFYHILWFSGVJS-UHFFFAOYSA-N silicic acid;trioxotungsten Chemical compound O[Si](O)(O)O.O=[W]1(=O)O[W](=O)(=O)O[W](=O)(=O)O1.O=[W]1(=O)O[W](=O)(=O)O[W](=O)(=O)O1.O=[W]1(=O)O[W](=O)(=O)O[W](=O)(=O)O1.O=[W]1(=O)O[W](=O)(=O)O[W](=O)(=O)O1 CGFYHILWFSGVJS-UHFFFAOYSA-N 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- HQVNEWCFYHHQES-UHFFFAOYSA-N silicon nitride Chemical compound N12[Si]34N5[Si]62N3[Si]51N64 HQVNEWCFYHHQES-UHFFFAOYSA-N 0.000 description 1
- 229910052709 silver Inorganic materials 0.000 description 1
- 239000004332 silver Substances 0.000 description 1
- 239000010802 sludge Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- HELHAJAZNSDZJO-OLXYHTOASA-L sodium L-tartrate Chemical compound [Na+].[Na+].[O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O HELHAJAZNSDZJO-OLXYHTOASA-L 0.000 description 1
- 239000001433 sodium tartrate Substances 0.000 description 1
- 229960002167 sodium tartrate Drugs 0.000 description 1
- 235000011004 sodium tartrates Nutrition 0.000 description 1
- 239000011122 softwood Substances 0.000 description 1
- 238000005118 spray pyrolysis Methods 0.000 description 1
- 238000000629 steam reforming Methods 0.000 description 1
- 229940037312 stearamide Drugs 0.000 description 1
- 229940114926 stearate Drugs 0.000 description 1
- 239000010907 stover Substances 0.000 description 1
- 238000000859 sublimation Methods 0.000 description 1
- 230000008022 sublimation Effects 0.000 description 1
- 239000001384 succinic acid Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- SEEPANYCNGTZFQ-UHFFFAOYSA-N sulfadiazine Chemical compound C1=CC(N)=CC=C1S(=O)(=O)NC1=NC=CC=N1 SEEPANYCNGTZFQ-UHFFFAOYSA-N 0.000 description 1
- 229960004306 sulfadiazine Drugs 0.000 description 1
- XTQHKBHJIVJGKJ-UHFFFAOYSA-N sulfur monoxide Chemical class S=O XTQHKBHJIVJGKJ-UHFFFAOYSA-N 0.000 description 1
- 229910052815 sulfur oxide Inorganic materials 0.000 description 1
- 239000000057 synthetic resin Substances 0.000 description 1
- 229920003002 synthetic resin Polymers 0.000 description 1
- 238000009475 tablet pressing Methods 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 229910052715 tantalum Inorganic materials 0.000 description 1
- GUVRBAGPIYLISA-UHFFFAOYSA-N tantalum atom Chemical compound [Ta] GUVRBAGPIYLISA-UHFFFAOYSA-N 0.000 description 1
- PBCFLUZVCVVTBY-UHFFFAOYSA-N tantalum pentoxide Inorganic materials O=[Ta](=O)O[Ta](=O)=O PBCFLUZVCVVTBY-UHFFFAOYSA-N 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- ISIJQEHRDSCQIU-UHFFFAOYSA-N tert-butyl 2,7-diazaspiro[4.5]decane-7-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CCCC11CNCC1 ISIJQEHRDSCQIU-UHFFFAOYSA-N 0.000 description 1
- YEAUATLBSVJFOY-UHFFFAOYSA-N tetraantimony hexaoxide Chemical compound O1[Sb](O2)O[Sb]3O[Sb]1O[Sb]2O3 YEAUATLBSVJFOY-UHFFFAOYSA-N 0.000 description 1
- 239000004753 textile Substances 0.000 description 1
- 229910052716 thallium Inorganic materials 0.000 description 1
- BKVIYDNLLOSFOA-UHFFFAOYSA-N thallium Chemical compound [Tl] BKVIYDNLLOSFOA-UHFFFAOYSA-N 0.000 description 1
- 238000001248 thermal gelation Methods 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 229910052718 tin Inorganic materials 0.000 description 1
- HPGGPRDJHPYFRM-UHFFFAOYSA-J tin(iv) chloride Chemical compound Cl[Sn](Cl)(Cl)Cl HPGGPRDJHPYFRM-UHFFFAOYSA-J 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- ZIBGPFATKBEMQZ-UHFFFAOYSA-N triethylene glycol Chemical compound OCCOCCOCCO ZIBGPFATKBEMQZ-UHFFFAOYSA-N 0.000 description 1
- KOECRLKKXSXCPB-UHFFFAOYSA-K triiodobismuthane Chemical compound I[Bi](I)I KOECRLKKXSXCPB-UHFFFAOYSA-K 0.000 description 1
- SAOHCOFTVLEOCB-UHFFFAOYSA-K tris(2,4,6-tribromophenoxy)bismuthane Chemical compound [Bi+3].[O-]C1=C(Br)C=C(Br)C=C1Br.[O-]C1=C(Br)C=C(Br)C=C1Br.[O-]C1=C(Br)C=C(Br)C=C1Br SAOHCOFTVLEOCB-UHFFFAOYSA-K 0.000 description 1
- NXHILIPIEUBEPD-UHFFFAOYSA-H tungsten hexafluoride Chemical compound F[W](F)(F)(F)(F)F NXHILIPIEUBEPD-UHFFFAOYSA-H 0.000 description 1
- FZFRVZDLZISPFJ-UHFFFAOYSA-N tungsten(6+) Chemical class [W+6] FZFRVZDLZISPFJ-UHFFFAOYSA-N 0.000 description 1
- ZNOKGRXACCSDPY-UHFFFAOYSA-N tungsten(VI) oxide Inorganic materials O=[W](=O)=O ZNOKGRXACCSDPY-UHFFFAOYSA-N 0.000 description 1
- CMPGARWFYBADJI-UHFFFAOYSA-L tungstic acid Chemical compound O[W](O)(=O)=O CMPGARWFYBADJI-UHFFFAOYSA-L 0.000 description 1
- 229940075466 undecylenate Drugs 0.000 description 1
- 125000004417 unsaturated alkyl group Chemical group 0.000 description 1
- DNYWZCXLKNTFFI-UHFFFAOYSA-N uranium Chemical compound [U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U] DNYWZCXLKNTFFI-UHFFFAOYSA-N 0.000 description 1
- HGBOYTHUEUWSSQ-UHFFFAOYSA-N valeric aldehyde Natural products CCCCC=O HGBOYTHUEUWSSQ-UHFFFAOYSA-N 0.000 description 1
- 150000003681 vanadium Chemical class 0.000 description 1
- 239000012002 vanadium phosphate Substances 0.000 description 1
- 239000013598 vector Substances 0.000 description 1
- 229910052727 yttrium Inorganic materials 0.000 description 1
- VWQVUPCCIRVNHF-UHFFFAOYSA-N yttrium atom Chemical compound [Y] VWQVUPCCIRVNHF-UHFFFAOYSA-N 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J27/00—Catalysts comprising the elements or compounds of halogens, sulfur, selenium, tellurium, phosphorus or nitrogen; Catalysts comprising carbon compounds
- B01J27/14—Phosphorus; Compounds thereof
- B01J27/186—Phosphorus; Compounds thereof with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J27/195—Phosphorus; Compounds thereof with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium with vanadium, niobium or tantalum
- B01J27/198—Vanadium
- B01J27/199—Vanadium with chromium, molybdenum, tungsten or polonium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/24—Chromium, molybdenum or tungsten
- B01J23/31—Chromium, molybdenum or tungsten combined with bismuth
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/002—Mixed oxides other than spinels, e.g. perovskite
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J27/00—Catalysts comprising the elements or compounds of halogens, sulfur, selenium, tellurium, phosphorus or nitrogen; Catalysts comprising carbon compounds
- B01J27/14—Phosphorus; Compounds thereof
- B01J27/186—Phosphorus; Compounds thereof with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J27/188—Phosphorus; Compounds thereof with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium with chromium, molybdenum, tungsten or polonium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/0215—Coating
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/0215—Coating
- B01J37/0228—Coating in several steps
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/024—Multiple impregnation or coating
- B01J37/0244—Coatings comprising several layers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/03—Precipitation; Co-precipitation
- B01J37/031—Precipitation
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/03—Precipitation; Co-precipitation
- B01J37/031—Precipitation
- B01J37/035—Precipitation on carriers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/08—Heat treatment
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/08—Heat treatment
- B01J37/082—Decomposition and pyrolysis
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B25/00—Phosphorus; Compounds thereof
- C01B25/16—Oxyacids of phosphorus; Salts thereof
- C01B25/26—Phosphates
- C01B25/45—Phosphates containing plural metal, or metal and ammonium
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/347—Preparation of carboxylic acids or their salts, halides or anhydrides by reactions not involving formation of carboxyl groups
- C07C51/353—Preparation of carboxylic acids or their salts, halides or anhydrides by reactions not involving formation of carboxyl groups by isomerisation; by change of size of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/347—Preparation of carboxylic acids or their salts, halides or anhydrides by reactions not involving formation of carboxyl groups
- C07C51/377—Preparation of carboxylic acids or their salts, halides or anhydrides by reactions not involving formation of carboxyl groups by splitting-off hydrogen or functional groups; by hydrogenolysis of functional groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C57/00—Unsaturated compounds having carboxyl groups bound to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/08—Preparation of carboxylic acid esters by reacting carboxylic acids or symmetrical anhydrides with the hydroxy or O-metal group of organic compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/30—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group
- C07C67/317—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by splitting-off hydrogen or functional groups; by hydrogenolysis of functional groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/30—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group
- C07C67/333—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton
- C07C67/343—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2523/00—Constitutive chemical elements of heterogeneous catalysts
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/582—Recycling of unreacted starting or intermediate materials
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Materials Engineering (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Physics & Mathematics (AREA)
- Thermal Sciences (AREA)
- Inorganic Chemistry (AREA)
- Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
本发明涉及用于产生丙烯酸类产物的方法。该方法包括下述步骤:在有效产生丙烯酸类产物的条件下,在催化剂组合物上接触烷酸和亚烷基化剂。催化剂组合物包含钒、铋和钨。优选,催化剂在活性相中包含0.3重量%至30重量%的钒,0.1重量%至69重量%的铋和0.1重量%至61重量%的钨。
Description
本申请是申请日为2012年10月31日、申请号为201280072759.X、发明名称为“用于产生丙烯酸和丙烯酸类的包含钒、铋和钨的催化剂”的发明专利申请的分案申请。
与有关申请的交叉引用
本申请要求于2012年10月31日提交的U.S.申请号13/664,494的优先权,其要求于2012年3月13日提交的U.S.临时申请号61/610,099的优先权,通过援引将其全部内容和公开并入本文。
发明领域
本发明一般地涉及丙烯酸的制备。更具体地,本发明涉及用于经由乙酸和甲醛的醛醇缩合制备丙烯酸中的催化剂。
发明背景
α,β-不饱和酸,特别是丙烯酸和甲基丙烯酸,及其酯衍生物是化学工业中的有用有机化合物。这些酸和酯已知容易地聚合或共聚形成均聚物或共聚物。常常聚合酸用于比如超级吸附剂、分散剂、絮凝剂和增稠剂的应用当中。聚合的酯衍生物用于包衣(包括乳胶漆),纺织品,粘合剂,塑料,纤维和合成树脂当中。
因为丙烯酸及其酯具有商业价值已久,已开发出许多制备方法。一种示范性的丙烯酸酯生产过程牵涉乙炔与水和一氧化碳的反应。又一常规过程牵涉乙烯酮(常常通过裂解丙酮或乙酸获得)与甲醛的反应。这些过程出于经济、环境或其它原因已逐渐淘汰。
又一丙烯酸生产方法运用甲醛和乙酸和/或羧酸酯的缩合。该反应常常在催化剂上进行。例如,由钒、钛和/或磷的混合氧化物组成的缩合催化剂在M.Ai,J.Catal.,107,201(1987);M.Ai,J.Catal.,124,293(1990);M.Ai,Appl.Catal.,36,221(1988);和M.Ai,Shokubai,29,522(1987)中有研究和描述。这些催化剂具有1:2:x的钒:钛:磷摩尔比,其中x从4.0变化至7.0,并且传统地显示催化剂活性随磷含量增加而稳定地降低。在x是6.0的情况下,获得对醛醇缩合产物,例如丙烯酸和丙烯酸甲酯的最高选择性。用这些催化剂,钒:钛摩尔比保持在1:2或低于1:2。然而,用这些催化剂实现的乙酸转化率仍有改善余地。
即使考虑到上述参考文献,仍然需要用于产生丙烯酸的改善的方法,和能够提供高乙酸转化率和丙烯酸类产物收率的改善的催化剂。
发明概述
在一种实施方式中,本发明涉及催化剂组合物。催化剂组合物可以适用于烷酸和亚烷基化剂的醛醇缩合中以形成丙烯酸类产物。催化剂组合物包含钒、铋和钨。优选,这些组分存在于活性相中。在优选的实施方式中,活性相包含0.3重量%至30重量%的钒,0.1重量%至69重量%的铋,和0.1重量%至61重量%的钨。在一种实施方式中,本发明催化剂还包含10重量%至22重量%的磷和15重量%至50重量%的氧。
在又一实施方式中,本发明涉及用于产生丙烯酸类产物的方法。该方法包括下述步骤:在有效产生丙烯酸类产物的条件下,在上文描述的催化剂上将烷酸和亚烷基化剂接触。优选,亚烷基化剂是甲醛,烷酸是乙酸,而丙烯酸类产物是丙烯酸。在一种实施方式中,反应中的总烷酸转化率是至少15mol%,丙烯酸选择性是至少30%,而丙烯酸类产物的空时收率是至少50克每升催化剂每小时。
在又一实施方式中,本发明涉及产生上文描述的催化剂的方法。该方法包括下述步骤:将铋盐、钨盐和钒前体溶液接触以形成湿催化剂组合物和将湿催化剂组合物干燥以形成包含钒、铋和钨的干催化剂组合物。该方法可以还包括混合钒前体和还原剂溶液以形成钒前体溶液的步骤。接触步骤可以包括下述步骤:将粘合剂与铋盐、钨盐和/或钒前体溶液接触以形成湿催化剂组合物。该方法可以还包括下述步骤:将湿催化剂前体干燥以形成干催化剂组合物。
发明详述
介绍
经由绝大多数常规方法来产生不饱和羧酸比如丙烯酸和甲基丙烯酸及其酯衍生物受到经济和环境限制。用于产生这些酸和酯的更实际的方法之一牵涉甲醛和(i)乙酸和/或(ii)乙酸乙酯在催化剂上的醛醇缩合。示范性类别的常规催化剂包括二元钒-磷酸钛,钒-二氧化硅-磷酸盐,和碱金属助催化的二氧化硅例如铯或钾助催化的二氧化硅。然而,碱金属助催化的二氧化硅已知在用于醛醇缩合反应中时仅展示较低至中等的活性。
也研究了包含铋的杂多酸催化剂。U.S.7,851,397教导杂多酸催化剂用于将不饱和和/或饱和醛比如丙烯醛或异丁烯醛氧化为不饱和酸比如丙烯酸或甲基丙烯酸的用途。催化剂含有钼、磷、钒、铋和选自钾、铷、铯、铊及其混合物的第一组分。制备这些杂多酸催化剂的方法是繁复的,原因是它们牵涉将各金属溶液单独加至仲钼酸铵溶液。
现已发现某些催化剂有效地催化羧酸与亚烷基化剂例如亚甲基化剂比如甲醛的醛醇缩合,形成不饱和酸。优选,所述反应是乙酸与甲醛形成丙烯酸类产物例如丙烯酸的醛醇缩合反应。
相应地,在一种实施方式中,本发明涉及适用于乙酸和甲醛的醛醇缩合的催化剂组合物。催化剂组合物包含钒、铋和钨。令人惊讶地和出乎意料地,与不用钒-铋-钨组合的常规醛醇缩合催化剂相比,钒-铋-钨催化剂在用于醛醇缩合反应中的情况下提供高转化率、选择性和收率。
在一种实施方式中,本发明涉及用于产生丙烯酸、甲基丙烯酸和/或其盐和酯的方法。如本文所用,丙烯酸、甲基丙烯酸和/或其盐和酯集体地或单独地可以称为"丙烯酸产物"或"丙烯酸类产物"。单独地使用术语丙烯酸、甲基丙烯酸或其盐和酯并不排除其它丙烯酸类产物,并且使用术语丙烯酸类产物不要求丙烯酸、甲基丙烯酸和其盐和酯都存在。
本发明催化剂组合物具有低失活率并且在长时间段内例如在50小时内、在77小时内或在100小时内为醛醇缩合反应提供稳定效能。
在一种实施方式中,钒、铋和钨作为各自的氧化物或磷酸盐或其混合物存在。催化剂组合物可以包含活性相,其包含促进催化的组分,并且还可以包含载体或改性的载体。作为一种实例,活性相包含金属、含磷化合物和含氧化合物。在优选的实施方式中,钒、铋和钨存在于活性相中。
本发明催化剂已被发现实现出乎意料地高烷酸例如乙酸转化率。例如,取决于丙烯酸形成反应所进行的温度,用该催化剂组合物可以实现至少15mol%,例如至少25mol%,至少30mol%,例如至少40mol%,或至少50mol%的乙酸转化率。在实现乙酸转化率的这种增加的同时保持对所希望丙烯酸类产物比如丙烯酸或丙烯酸甲酯的高选择性。例如,用本发明的催化剂组合物可以实现至少35mol%,例如至少45mol%,至少60mol%或至少75mol%的对希望丙烯酸类产物的选择性。
本发明催化剂组合物中钒、铋和钨的总量可以宽泛地变化。在某些实施方式中,例如,催化剂组合物在活性相中包含至少0.3重量%的钒,例如至少0.6重量%或至少1.6重量%,基于催化剂组合物活性相的总重量。催化剂组合物可以在活性相包含至少0.1重量%的铋,例如至少0.5重量%,至少1重量%,至少3重量%或至少8重量%的铋。催化剂组合物可以在活性相中包含至少0.1重量%,例如至少0.5重量%,至少1重量%,至少2.5重量%或至少3.7重量%的钨。在范围方面,催化剂组合物可以在活性相中包含0.3重量%至30重量%,例如0.6重量%至25重量%或1.6重量%至20重量%的钒;0.1重量%至69重量%,例如3重量%至64重量%或8重量%至58重量%的铋;和0.1重量%至61重量%,例如2.5重量%至59重量%或3.7重量%至49重量%的钨。催化剂组合物可以在活性相中包含最多30重量%,例如最多25重量%或最多20重量%的钒。催化剂组合物可以在活性相中包含最多69重量%,例如最多64重量%或最多58重量%的铋。催化剂组合物可以在活性相中包含最多61重量%,例如最多59重量%或最多49重量%的钨。
在一种实施方式中,催化剂在活性相中以至少0.79重量%,例如至少1重量%,至少3重量%,至少5.6重量%或至少14重量%的量包含组合的钒和铋。在范围方面,在活性相中钒和铋组分的组合重量百分比可以是0.79重量%至70重量%,例如5.6重量%至65重量%或14重量%至61重量%。在一种实施方式中,催化剂在活性相中以至少0.76重量%,例如至少1重量%,至少3重量%,至少4.8重量%或至少8重量%的量包含组合的钒和钨。在范围方面,在活性相中钒和钨组分的组合重量百分比可以是0.76重量%至62重量%,例如4.8重量%至60重量%或8重量%至52重量%。
在一种实施方式中,在催化剂组合物的活性相中的钒:铋摩尔比是至少0.033:1,例如至少0.20:1,至少1:1,至少2:1,至少10:1,或至少50:1。在范围方面,在催化剂组合物的活性相中钒:铋摩尔比可以是0.033:1至1000:1,例如0.2:1至500:1,1:1至250:1,2:1至100:1,2:1至65:1,或10:1至65:1。在上限方面,在催化剂组合物的活性相中的钒:铋摩尔比是最高1000:1,例如最高500:1,最高250:1,最高100:1,或最高65:1。在一种实施方式中,在催化剂组合物的活性相中的铋:钨摩尔比是至少0.0033:1,例如至少0.067:1,至少0.1:1,至少0.20:1,至少0.32:1,至少0.75:1,至少1.5:1,或至少3:1。在范围方面,在催化剂组合物的活性相中的铋:钨摩尔比可以是0.0033:1至300:1,例如0.033至100:1,0.067:1至50:1,0.20:1至10:1,或0.32:1至5:1。在上限方面,在催化剂组合物的活性相中的铋:钨摩尔比是最高300:1,例如最高150:1,最高75:1,最高15:1,最高10:1,或最高5:1。在一种实施方式中,在催化剂组合物的活性相中的钒:钨摩尔比是至少0.033:1,例如至少0.067:1,至少0.20:1,至少1:1,至少10:1,或至少50:1。在范围方面,在催化剂组合物的活性相中的钒:钨摩尔比可以是0.033:1至1000:1,例如0.067:1至500:1,0.1:1至250:1,1:1至100:1,或5:1至20:1。在上限方面,在催化剂组合物的活性相中的钒:钨摩尔比是最高1000:1,例如最高500:1,最高250:1,最高100:1,最高50:1,最高15:1,或最高10:1。
在一种实施方式中,催化剂组合物基本上不含钛,例如在活性相中基本上不含钛,比如说包含小于5重量%,例如小于1重量%,或小于0.1重量%的钛。
在其它实施方式中,本发明催化剂可以还包含其它化合物或元素(金属和/或非金属)。例如,催化剂可以在活性相中还包含磷和/或氧。在这些情况中,催化剂可以在活性相中包含10重量%至22重量%,例如11重量%至20重量%或11重量%至18重量%的磷;和/或15重量%至50重量%,例如20重量%至45重量%或22重量%至38重量%的氧。
在某些实施方式中,铋以包括铋(III)和(V)盐的铋盐形式存在。例如,催化剂组合物可以在活性相中包含范围是0.1重量%至69重量%,例如3重量%至64重量%或8重量%至58重量%的量的铋盐。优选,用于本发明催化剂制备中的铋盐是铋(III)盐。铋盐可以例如选自羧酸铋,卤化铋,乙酸铋,铋磺胺嘧啶,硫酸铋,硝酸铋,碱式硝酸铋,碳酸铋,碱式碳酸铋,氧化铋,氯氧化铋,氢氧化铋,磷酸铋,铝酸铋,三溴酚铋,硫醇铋,肽铋,喹啉的铋盐及其衍生物(例如羟基喹啉铋),巯氧吡啶铋和吡啶硫醇的其它铋盐,氨基酸铋盐比如甘氨酸铋,三钾柠檬酸铋,及其混合物。在某些实施方式中,酸溶液比如硝酸或乙酸可以用来溶解铋盐以形成铋溶液。
一般来说,铋盐可以是有机或无机。它可以是碱性铋盐(碱式铋盐)比如上述的碱式盐。
适宜的羧酸铋包括铋的水杨酸盐,碱式水杨酸盐,乳酸盐,柠檬酸盐,碱式柠檬酸盐,抗坏血酸盐,乙酸盐,二丙基乙酸盐,酒石酸盐,酒石酸钠,葡糖酸盐,碱式没食子酸盐,苯甲酸盐,月桂酸盐,肉豆蔻酸盐,棕榈酸盐,丙酸盐,硬脂酸盐,十一碳烯酸盐,阿司匹林盐,新癸酸盐和蓖麻油酸盐。在它们当中,碱性水杨酸铋(碱式水杨酸)和柠檬酸铋可以是优选的。适宜的卤化物包括氯化铋,溴化铋和碘化铋。优选的铋盐可以选自卤化铋,硝酸铋,乙酸铋,和羧酸铋,比如碱式水杨酸,水杨酸铋,碱式没食子酸铋,碱式柠檬酸铋,柠檬酸铋,硝酸铋和碱式硝酸铋。
在某些实施方式中,钨以钨盐形式存在。例如,催化剂组合物可以在活性相中包含范围是0.1重量%至61重量%,例如2.5重量%至59重量%或3.7重量%至49重量%的量的钨盐。优选,用于制备本发明催化剂中的钨盐是钨(VI)盐。钨盐可以例如选自钨酸,硅钨酸,硅钨酸铵,偏钨酸铵水合物,仲钨酸铵,四硫钨酸铵,钨酸氢盐,聚合物负载的,二(叔丁基亚氨基)二(二甲基氨基)钨(VI),磷钨酸水合物,哌啶四硫钨酸盐,氯化钨(VI),二氯化二氧化钨(VI),氟化钨(VI),氧化钨(IV),氯氧化钨(VI),钨硅酸水合物。
在一种实施方式中,催化剂组合物的形成可以运用五价钒化合物的还原。在有效提供或保持钒的化合价态在+5之下的条件下,还原的五价化合物可以与磷化合物和任选地与助催化剂相组合,以形成活性的金属磷酸盐催化剂。各种还原剂和溶剂可以用来制备这些催化剂。实例包括有机酸,醇,多元醇,醛和盐酸。一般来说,金属前体、还原剂、溶剂、添加顺序、反应条件比如温度和时间、和煅烧温度的选择可以影响催化剂组成、表面积、孔隙率、结构强度和总体催化剂效能。
在一种实施方式中,充当催化剂组合物中钒来源的适宜钒化合物含有五价钒和包括但不限于五氧化二钒或钒盐比如偏钒酸铵,氧基三卤化钒,烷基羧酸钒及其混合物。
在一种实施方式中,充当催化剂中磷来源的适宜磷化合物含有五价磷和包括但不限于磷酸,五氧化二磷,多磷酸,或全卤化磷比如五氯化磷,及其混合物。
在一种实施方式中,催化剂的活性相相应于下式:
VaBibWcPdOe
其中
a是1至100,
b是0.1至30,
c是0.1至30,
d是1.0至175,和
e是5至710。
字母a、b、c、d和e(相对于1.0)是钒、铋、钨、磷和氧分别在催化剂中的相对摩尔浓度量。在这些实施方式中,a与b的比率大于1:30,例如大于1:1,大于4:1,或大于10:1。摩尔浓度变量a、b、c、d和e的优选范围示于表1。

在某些实施方式中,催化剂组合物还包含额外的金属和/或金属氧化物。这些额外的金属和/或金属氧化物可以充当助催化剂。如果存在,额外的金属和/或金属氧化物则可以选自铜、钼、镍、铌及其组合。可以包括在本发明催化剂中的其它示范性助催化剂包括锂、钠、镁、铝、铬、锰、铁、钴、钙、钇、钌、银、锡、钡、镧、稀土金属、铪、钽、铼、钍、铋、锑、锗、锆、铀、铯、锌和硅及其混合物。其它改性剂包括硼、镓、砷、硫、卤化物、路易斯酸比如BF3,ZnBr2和SnCl4。将助催化剂掺入催化剂的示范性方法描述于US专利号5,364,824,通过援引将其全部并入本文。
如果催化剂组合物包含额外的金属和/或金属氧化物,则催化剂任选地可以在活性相中以0.001重量%至30重量%,例如0.01重量%至5重量%或0.1重量%至5重量%的量包含额外的金属和/或金属氧化物。如果存在,助催化剂可以使得催化剂可以具有至少25克丙烯酸/克催化剂-h,例如至少50克丙烯酸/克催化剂-h,或至少100克丙烯酸/克催化剂-h的重量/重量空时收率。
在某些实施方式中,催化剂组合物是无载体的。在这些情况中,催化剂可以包含如上描述的均质混合物或异质混合物。在一种实施方式中,均质混合物是制备型方法比如金属醇盐或金属复合物受控水解产生的钒、铋和钨密切混合物的产物。在其它实施方式中,异质混合物是钒、铋盐和钨盐物理混合物的产物。这些混合物可以包括制备自磷酸化预先形成的含水金属氧化物的物理混合物的配制剂。在另外的情况下,上述一种或多种混合物可以包括预先形成的钒焦磷酸盐粉末的混合物。
在又一实施方式中,催化剂组合物是负载型催化剂,其以上文指出的量包含除了钒、铋和钨和任选地亚磷酸和氧之外的催化剂载体(其中指出的摩尔浓度范围无关催化剂载体的摩尔数,所述催化剂载体包括包含于催化剂载体中的任意钒、铋、钨、磷或氧)。载体(或改性载体)的总重量优选是25重量%至95重量%,例如40重量%至70重量%或50重量%至60重量%,基于催化剂的总重量。
载体可以宽泛地变化。在一种实施方式中,载体材料选自二氧化硅,氧化铝,二氧化锆,二氧化钛,硅铝酸盐,沸石化材料,混合金属氧化物(包括但不限于二元氧化物比如SiO2-Al2O3,SiO2-TiO2,SiO2-ZnO,SiO2-MgO,SiO2-ZrO2,Al2O3-MgO,Al2O3-TiO2,Al2O3-ZnO,TiO2-MgO,TiO2-ZrO2,TiO2-ZnO,TiO2-SnO2)及其混合物,其中二氧化硅是一种优选载体。其它适宜的载体材料可以包括例如稳定的基于金属氧化物的载体或基于陶瓷的载体。优选的载体包括硅质载体,比如二氧化硅,二氧化硅/氧化铝,IIA族硅酸盐比如偏硅酸钙,热解二氧化硅,高纯度二氧化硅,碳化硅,页硅酸盐或粘土矿物比如蒙脱石、贝得石、皂石、柱撑粘土,及其混合物。其它载体可以包括但不限于铁的氧化物,氧化镁,滑石,氧化镁,碳,石墨,高表面积石墨化的碳,活性碳及其混合物。其它载体可以包括包覆的结构化形式比如包覆的金属箔材,烧结的金属形式和包覆的陶瓷成形体比如成形的堇青石,板状氧化铝或针状莫来石形式。这些载体列表仅是示范性,并不意在限制本发明的范围。
在其它实施方式中,除了活性相和载体之外,本发明催化剂可以还包含载体改性剂。在一种实施方式中,改性载体涉及包括载体材料和载体改性剂的载体,其例如可以调节载体材料的化学或物理特性比如载体材料的酸度或碱性。在使用改性载体的实施方式,载体改性剂以0.1重量%至50重量%,例如0.2重量%至25重量%,0.5重量%至15重量%,或1重量%至8重量%的量存在,基于催化剂组合物总重量。
在一种实施方式中,载体改性剂是酸性载体改性剂。在某些实施方式中,催化剂载体用酸性载体改性剂改性。类似地,载体改性剂可以是具有低挥发性或几乎不具挥发性的酸性改性剂。酸性改性剂可以选自IVB族金属的氧化物,VB族金属的氧化物,VIB族金属的氧化物,铁的氧化物,铝的氧化物,及其混合物。在一种实施方式中,酸性改性剂可以选自WO3,MoO3,Fe2O3,Cr2O3,V2O5,MnO2,CuO,Co2O3,Bi2O3,TiO2,ZrO2,Nb2O5,Ta2O5,Al2O3,B2O3,P2O5和Sb2O3。
在又一实施方式中,载体改性剂是碱性载体改性剂。化学种类比如碱金属和碱土金属的存在通常被视为碱性并且可以常规地考虑视为对催化剂效能有害。然而,令人惊讶地和出乎意料地,这些种类的存在可以有助于催化剂效能。在某些实施方式中,这些种类可以充当催化剂的助催化剂或酸性催化剂结构比如层状或页硅酸盐比如蒙脱石中的必需部分。不受理论所限,假定这些阳离子与产生酸度的种类产生强偶极子。
可以包括在催化剂中的额外改性剂包括例如硼、铝、镁、锆和铪。
在其中采用载体的某些实施方式中,载体可以具有至少1m2/g,例如至少20m2/g或至少50m2/g的表面积,如BET测量所测定。催化剂载体可以包括任选具有5nm至200nm,例如5nm至50nm或10nm至25nm的平均孔径的孔。催化剂任选具有0.05cm3/g至3cm3/g,例如0.05cm3/g至0.1cm3/g或0.08cm3/g至0.1cm3/g的平均孔隙体积,如BET测量所测定。优选,具有上述直径的孔提供至少50%,例如至少70%或至少80%的孔隙体积或表面积。通过如下文讨论的孔改性剂可以形成孔和/或将之改性。在又一实施方式中,微孔度与大孔度的比率是19:1至5.67:1,例如3:1至2.33:1。微孔度是指直径小于2nm的孔,并且微孔中的运动可以通过活化的扩散来描述。中孔度是指直径大于2nm和小于50nm的孔。经过中孔的流动可以通过Knudson扩散描述。大孔度是指直径大于50nm的孔,而通过大孔的流动可以通过体相扩散描述。从而,在某些实施方式中,希望将表面积、孔径尺寸分布、催化剂或载体颗粒尺寸和形状和反应速率与反应物和产物进出孔的扩散速率加以平衡从而优化催化效能。
本领域普通技术人员应认识到,如果载体材料包括在本发明催化剂中,优选对其加以选择使得在用于形成所希望产物例如丙烯酸或丙烯酸烷基酯的方法条件下,催化剂系统是适宜地活性,选择性和耐用的。另外,包括在本发明催化剂中的活性金属可以分散于整个载体,包覆在载体外表面(蛋壳)或修饰在载体表面上。在某些实施方式中,在大孔和中孔材料的情况下,活性位点可以固定或应用至分布于整个颗粒的孔的表面,并且因此是反应物可接触的且分布于整个载体颗粒的表面位点。
本发明催化剂可以还包含其它添加剂,其实例可以包括:用于增强模压加工性的模塑助剂;用于增强催化剂强度的增强剂;用于在催化剂中形成适当孔的成孔剂或孔改性剂,和粘合剂。这些其它添加剂的实例包括硬脂酸,石墨,淀粉,甲基纤维素,二氧化硅,氧化铝,玻璃纤维,碳化硅,和氮化硅。在一种实施方式中,催化剂的活性相(非载体)包含其它添加剂。例如,活性相可以包含二氧化硅,例如胶体二氧化硅。在所述实施方式中,二氧化硅可以以0.01至50重量%,例如0.1至40重量%,0.5重量%至30重量%,1.0重量%至30重量%,或1重量%至20重量%的二氧化硅的量存在于活性相中。在下限方面,活性相可以包含至少0.01重量%,例如至少0.1重量%,至少0.5重量%或至少1重量%的二氧化硅。在上限方面,活性相可以包含小于50重量%,例如小于40重量%,小于30重量%或小于20重量%的二氧化硅。优选,这些添加剂不具有对催化效能例如转化率和/或活性的有害效果。这些各种添加剂可以以一定量添加,使得催化剂的物理强度不会容易地劣化至变得不可能将催化剂用作实际工业催化剂的程度。
在一种实施方式中,本发明催化剂组合物包含孔改性剂。在某些实施方式中,孔改性剂可以是热稳定的和具有在低于300℃例如低于250℃的温度的实质蒸气压。在一种实施方式中,孔改性剂在约150℃至约250℃,例如约150℃至约200℃的温度具有至少0.1kPa,例如至少0.5kPa的蒸气压。在其它实施方式中,孔改性剂可以热分解或灼烧以形成孔。例如,灼烧剂可以是纤维素衍生的物质比如磨碎的坚果壳。
在某些实施方式中,孔改性剂具有相对高的熔点,例如大于60℃,例如大于75℃,从而其在将催化剂前体压制为块、片或丸期间不熔化。优选,孔改性剂包含相对纯的物质而不是混合物。如此,低熔点组分在形成块或片期间将不会在压缩下液化。例如,在孔改性剂是脂肪酸的情况下,脂肪酸混合物的低熔点组分可以通过挤压作为液体除去。如果在块或片压制期间发生该现象,则液体流动可以干扰孔结构而作为孔直径的函数在催化剂组合物上产生不希望的孔隙体积分布。在其它实施方式中,孔改性剂在其熔点以下的温度具有显著蒸气压,从而它们能够通过升华为载气而除去。
例如,孔改性剂可以是相应于式CH3(CH2)xCOOH的脂肪酸,其中x>8。示范性脂肪酸包括硬脂酸(x=16),棕榈酸(x=14),月桂酸(x=10),肉豆蔻酸(x=12)。还可以使用这些酸的酯和酰胺或所述酸的其它官能化形式,例如硬脂酰胺(CH3(CH2)16CONH2)。适宜的酯可以包括甲基酯以及甘油酯比如硬脂(甘油三硬脂酸酯)。可以使用脂肪酸的混合物,但是基本上纯的酸特别是硬脂酸一般是比混合物优选的。
此外,虽然脂肪酸和脂肪酸衍生物一般是优选的,符合上述功能要求的其它组合物也适于用作孔改性剂。其它优选的孔改性剂包括但不限于多核有机化合物比如萘,石墨,天然燃烧组分比如纤维素及其纤维质衍生物,纤维素衍生的物质比如淀粉和磨碎的坚果壳,天然和合成的低聚物和聚合物比如聚乙烯、聚乙烯醇和聚丙烯酸类和酯。
催化剂制备
在催化剂是无载体的某些实施方式中,催化剂组合物经由包括下述步骤的方法形成:接触铋盐、钨盐和(预定量的)钒前体例如可溶的NH4VO3,形成湿催化剂前体。
优选,该方法还包括干燥润湿的催化剂前体以形成干燥催化剂组合物的步骤。干燥的催化剂组合物包含上文讨论的组分。确定铋盐、钨盐和钒前体的量,从而所得干催化剂组合物具有至少0.033:1,例如至少0.067:1或者至少0.20:1的钒:铋摩尔比和至少0.033:1,例如至少0.067:1或至少0.20:1的钒:钨摩尔比。
在一种实施方式中,钒前体可以形成如下:将V2O5溶于醇例如异丁醇中,与磷酸反应。此外,铋前体可以形成如下:溶于酸例如硝酸中,并与磷酸反应。两种前体可以一起混合和干燥过夜以形成VBiPO复合物。可以将偏钨酸铵加至VBiPO复合物以形成VBiWPO固体。
在一种实施方式中,方法可以还包括混合钒前体和还原剂溶液形成钒前体溶液的步骤。在一种实施方式中,还原剂溶液可以包含酸,二氧化硅,水,和/或二醇。在一种实施方式中,酸可以是可以被钒例如V5+氧化的有机酸。在一种实施方式中,酸可以选自柠檬酸,草酸,硬脂酸(steric acid),马来酸,乳酸,酒石酸,甘醇酸(glycol acid),丙酮酸,聚丙烯酸及其混合物。在一种实施方式中,还原剂溶液中所用的酸不包含不被钒例如V5+氧化的酸例如甲酸,乙酸,丁二酸及其混合物。在一种实施方式中,二醇可以选自丙二醇,乙二醇,二甘醇,三甘醇和其它多元醇。优选,还原剂溶液包含有机酸例如柠檬酸和/或草酸,胶体二氧化硅,去离子水,和乙二醇。在其它实施方式中,还原剂溶液还可以包含酮,醛,醇和酚类。
在一种实施方式中,润湿的催化剂前体的形成也包括添加粘合剂。从而,接触步骤可以包括将粘合剂例如粘合剂溶液与铋盐、钨盐和/或钒前体溶液接触以形成润湿的催化剂组合物。在一种实施方式中,粘合剂可以选自纤维素,甲基纤维素,羧甲基纤维素,醋酸纤维素,纤维素衍生的物质比如淀粉,和前述多糖中两种或多种的组合。在一种实施方式中,氧化物例如二氧化硅可以用作粘合剂。在一种实施方式中,催化剂组合物包含至少3重量%,例如至少5重量%或至少10重量%的粘合剂。在一种实施方式中,可将酸例如磷酸加至润湿的催化剂组合物。
在一种实施方式中,该方法可以还包括煅烧干燥的催化剂,优选按照温度特征曲线进行。作为一种实例,温度特征曲线包含增加的梯度步骤温度特征曲线,其包含多个增加的保持温度。温度以1℃至10℃/分钟的速度在保持温度之间增加。优选,保持温度包含第一,第二,第三和第四保持温度。第一保持温度可以是150℃至300℃,例如175℃至275℃,优选约160℃。第二保持温度可以是150℃至500℃,例如300℃至400℃,优选约250℃。第三保持温度可以是200℃至700℃,例如450℃至650℃,优选约300℃。第四保持温度可以是300℃至700℃,例如450℃至650℃,优选约450℃。当然,其它温度特征曲线也可以是适宜的。混合物的煅烧可以在惰性气氛、空气或含氧气体中于所希望的温度完成。蒸汽、烃或其它气体或蒸气可以在煅烧步骤期间或煅烧后加至气氛,以导致对物理和化学表面特性以及质地特性的希望效果比如使大孔度增加。
在一种优选的实施方式中,温度特征曲线包含:
i)从室温至160℃以10℃/分钟的速率加热干燥的催化剂;
ii)在160℃加热干燥的催化剂组合物2小时;
iii)从160℃至250℃以3℃/分钟加热干燥的催化剂组合物的速率;
iv)在250℃加热干燥的催化剂组合物2小时;
v)从250℃至300℃以3℃/分钟的速率加热干燥的催化剂组合物;
vi)在300℃加热干燥的催化剂组合物6小时;
vii)从300℃至450℃以3℃/分钟的速率加热干燥的催化剂组合物;和
viii)在450℃加热干燥的催化剂组合物6小时。
在一种实施方式中,催化剂组分例如金属氧化物和/或磷酸盐前体,可以彼此物理地组合以形成催化剂组合物。例如,可以一起研磨未煅烧的干燥催化剂组分,然后煅烧以形成活性催化剂。作为又一实例,可以混合、研磨和/或捏合催化剂组分。然后,可以煅烧形成的催化剂粉末以形成最终的干燥催化剂组合物。
在一种实施方式中,将磷酸化剂加至混合的金属氧化物前体,随后煅烧。
在一种实施方式中,在水热条件下制备催化剂,随后煅烧。
在催化剂是负载型的实施方式中,催化剂组合物通过载体(任选改性载体)的金属浸渍来形成,尽管还可以使用其它过程比如化学接枝或化学蒸气沉积。
在一种实施方式中,催化剂制备如下:用金属或其盐在适宜溶剂的溶液浸渍载体,随后干燥和任选煅烧。还可以以相似方式将改性剂或添加剂的溶液浸渍至载体上。浸渍和干燥程序可以重复多于一次以便实现金属、改性剂和/或其它添加剂的希望载量。在某些情况下,在改性剂与金属之间可以存在对载体上的活性位点的竞争。相应地,可以希望的是在金属之前掺入改性剂。如果颗粒在浸渍步骤之间完全干燥,则采用水溶液的多个浸渍步骤可以降低催化剂颗粒的强度。从而,优选的是在连续的浸渍之间允许一些水分保留在催化剂中。在一种实施方式中,在用非水溶液的情况下,首先引入改性剂和/或添加剂:用适宜的非水溶液例如改性剂金属的醇盐或乙酸盐在醇例如乙醇中的溶液进行一次或多次浸渍,随后干燥。然后,可以通过相似程序用金属化合物的适宜溶液掺入金属。
在其它实施方式中,改性剂如下掺入组合物中:改性剂元素的化合物与二氧化硅共胶凝化或共沉淀,或水解改性剂元素卤化物与卤化硅的混合物。通过溶胶凝胶处理制备二氧化硅和二氧化锆的混合氧化物的方法描述于Bosman等人的J Catalysis,第148卷,(1994),第660页和Monros等人的J Materials Science,第28卷,(1993),第5832页。另外,在从原硅酸四乙酯(TEOS)胶凝化期间用硼掺杂二氧化硅球描述于Jubb和Bowen的JMaterial Science,第22卷,(1987),第1963-1970页。制备多孔二氧化硅的方法描述于IlerR K,The Chemistry of Silica,(Wiley,New York,1979),和Academic Press(1990)出版的Brinker CJ&Scherer G W Sol-Gel Science。
在某些实施方式中,催化剂组合物用于固定床反应器中以形成希望的产物例如丙烯酸或丙烯酸烷基酯。因此,催化剂优选形成成型单元例如球、颗粒、丸、粉末、聚集体或挤出物,一般具有1至25mm,例如2至15mm范围的最大和最小尺度。在使用浸渍技术的情况下,载体可以在浸渍之前成型。另选地,组合物可以在催化剂制备的任何适宜阶段成型。催化剂的其它形式例如粉末或小珠也可以是有效的,并且可以以这些形式使用。在一种实施方式中,催化剂用于流化床反应器中。在该情况中,催化剂可以用喷雾干燥或喷雾热分解形成。优选,所得催化剂具有大于300微米,例如大于500微米的颗粒尺寸。在另外的情况下,催化剂可以经由喷雾干燥形成能够形成丸、挤出物等的粉末来制备。
丙烯酸的制备
在其它实施方式中,本发明涉及用于产生不饱和酸例如丙烯酸或其酯(丙烯酸烷基酯)的方法:在产生不饱和酸和/或丙烯酸类有效条件下,通过将烷酸与亚烷基化剂例如亚甲基化剂接触。优选,在本发明催化剂组合物存在下将乙酸与甲醛反应。烷酸或烷酸的酯可以是式R'-CH2-COOR,其中R和R'各自独立地是氢或饱和或不饱和烷基或芳基基团。例如,R和R’可以是含有例如1-4个碳原子的低级烷基。在一种实施方式中,烷酸酸酐可以用作烷酸的来源。在一种实施方式中,反应在醇存在下进行,优选对应所希望的酯的醇,例如甲醇。除了丙烯酸制备中使用的反应之外,在其它实施方式中本发明催化剂还可以用来催化其它反应。这些其它反应的实例包括但不限于丁烷氧化为马来酸酐,从甲醛和乙醛产生丙烯醛,以及从甲醛和丙酸产生甲基丙烯酸。
用于本发明方法中的原料例如乙酸可以衍生自任何适宜来源包括天然气、石油、煤、生物质等。作为实例,可以通过甲醇羰基化、乙醛氧化、乙烯氧化、氧化发酵和厌氧发酵生产乙酸。
由于石油和天然气价格波动,或多或少变得昂贵,所以由替代碳源生产乙酸和中间体例如甲醇和一氧化碳的方法已逐渐引起关注。特别地,当石油相对昂贵时,由衍生自较为可用的碳源的合成气体("合成气")生产乙酸可能变得有利。例如,美国专利No.6,232,352(通过引用将其全文并入本文)教导了改造甲醇装置用以制造乙酸的方法。通过改造甲醇装置,对于新的乙酸装置,与CO产生有关的大量资金费用得到显著降低或在很大程度上消除。使所有或部分合成气从甲醇合成环路进行分流并供给到分离器单元以回收CO,然后将其用于生产乙酸。以类似方式,用于加氢步骤的氢气可以由合成气供给。
在某些实施方式中,用于上述方法中的原料中的一些或全部可以部分或完全衍生自合成气。例如,乙酸可以由甲醇和一氧化碳形成,甲醇和一氧化碳均可以衍生自合成气。合成气可以通过部分氧化重整或蒸汽重整形成,并且可以将一氧化碳从合成气分离出。类似地,可以将用于乙酸加氢形成粗乙醇产物步骤的氢气从合成气分离出。进而,合成气可以衍生自多种碳源。碳源例如可以选自天然气、油、石油、煤、生物质和它们的组合。合成气或氢气还可以得自生物衍生的甲烷气体,例如由垃圾填埋场废物(landfill waste)或农业废物产生的生物衍生的甲烷气体。
在又一实施方式中,乙酸可以形成自生物质发酵。发酵方法优选利用产乙酸(acetogenic)方法或同型产乙酸的微生物使糖类发酵得到乙酸并产生很少(如果有的话)二氧化碳作为副产物。与通常具有约67%碳效率的常规酵母法相比,所述发酵方法的碳效率优选大于70%、大于80%或大于90%。任选地,发酵过程中使用的微生物为选自如下的属:梭菌属(Clostridium)、乳杆菌属(Lactobacillus)、穆尔氏菌属(Moorella)、热厌氧杆菌属(Thermoanaerobacter)、丙酸杆菌属(Propionibacterium)、丙酸螺菌属(Propionispera)、厌氧螺菌属(Anaerobiospirillum)和拟杆菌属(Bacteriodes),特别是选自如下的种:蚁酸醋酸梭菌(Clostridiumformicoaceticum)、丁酸梭菌(Clostridiumbutyricum)、热醋穆尔氏菌(Moorella thermoacetica)、凯伍热厌氧菌(Thermoanaerobacter kivui)、德氏乳杆菌(Lactobacillus delbrukii)、产丙酸丙酸杆菌(Propionibacterium acidipropionici)、栖树丙酸螺菌(Propionisperaarboris)、产琥珀酸厌氧螺菌(Anaerobiospirillum succinicproducens)、嗜淀粉拟杆菌(Bacteriodesamylophilus)和栖瘤胃拟杆菌(Bacteriodesruminicola)。任选地,在该过程中,可以将全部或部分的来自生物质的未发酵残余物例如木脂体气化以形成可用于本发明加氢步骤的氢气。用于形成乙酸的示范性发酵过程公开于U.S.专利号6,509,180;6,927,048;7,074,603;7,507,562;7,351,559;7,601,865;7,682,812;和7,888,082,通过援引将其全部并入本文。也可参考U.S.公开号2008/0193989和2009/0281354,通过援引将其全部并入本文。
生物质的实例包括但不限于农业废弃物、林业产品、草和其它纤维素材料、木材采伐剩余物、软木材碎片、硬木材碎片、树枝、树根、叶子、树皮、锯屑、不合格纸浆、玉米(corn)、玉米秸秆、麦秸秆、稻杆、甘蔗渣、软枝草、芒草、动物粪便、市政垃圾、市政污泥(municipalsewage)、商业废物、葡萄皮渣、杏核壳、山核桃壳、椰壳、咖啡渣、草粒、干草粒、木质颗粒、纸板、纸、塑料和布。参见例如,U.S.专利号7,884,253,通过援引将其全部并入本文。又一生物质来源是黑液(blackliquor),也即将木材转化为纸浆然后干燥造纸的Kraft过程的副产物的一种粘稠深色液体。黑液是木质素残余物、半纤维素和无机化学品的水溶液。
美国专利No.RE 35,377(也通过引用将其并入本文)提供了一种通过使含碳材料例如油、煤、天然气和生物质材料转化生产甲醇的方法。该方法包括使固体和/或液体含碳材料加氢气化以获得工艺气体,用另外的天然气将该工艺气体蒸汽热解以形成合成气。将该合成气转化为可以羰基化为乙酸的甲醇。美国专利No.5,821,111公开了一种将废生物质通过气化转化为合成气的方法,以及美国专利No.6,685,754公开了生产含氢气体组合物例如包含氢气和一氧化碳的合成气的方法,通过引用将它们全文并入本文。
给进到加氢反应器的乙酸还可以包含其它羧酸和酸酐,以及醛和/或酮例如乙醛和丙酮。优选,适宜的乙酸进料流包含选自乙酸、乙酸酐、乙醛、乙酸乙酯及其混合物的化合物中的一种或多种。在本发明的方法中还可以将这些其它化合物加氢。在一些实施方案中,在丙醇生产中羧酸例如丙酸或其酸酐的存在会是有益的。乙酸进料中还可以存在水。
适于生产乙酸的甲醇羰基化方法描述于US专利号7,208,624;7,115,772;7,005,541;6,657,078;6,627,770;6,143,930;5,599,976;5,144,068;5,026,908;5,001,259和4,994,608,通过援引将其全部并入本文。
在一种任选实施方式中,用于缩合反应中的乙酸包含乙酸和还可以包含其它羧酸例如丙酸、酯和酸酐,以及乙醛和丙酮。在一种实施方式中,进料至氢化反应的乙酸包含丙酸。例如,乙酸进料流中的丙酸可以是0.001重量%至15重量%,例如0.125重量%至12.5重量%,1.25重量%至11.25重量%,或3.75重量%至8.75重量%的范围。从而,乙酸进料流可以是更粗制的乙酸进料流,例如更不精制的乙酸进料流。
如本文所用,"亚烷基化剂"意指醛或醛的前体,其适于在醛醇缩合反应中与烷酸例如乙酸反应以形成不饱和酸例如丙烯酸或丙烯酸烷基酯。在优选的实施方式中,亚烷基化剂包含亚甲基化剂比如甲醛,其优选能够将亚甲基基团(=CH2)加至有机酸。其它亚烷基化剂可以包括例如乙醛、丙醛和丁醛。
亚烷基化剂例如甲醛可以加自任何适宜来源。示范性来源可以包括例如甲醛水溶液,衍生自甲醛干燥程序的无水甲醛,三噁烷,甲二醇的二醚和低聚甲醛。在优选的实施方式中,甲醛经由将甲醇和氧反应产生甲醛的formox设备产生。
在其它实施方式中,亚烷基化剂是作为甲醛来源的化合物。在使用并非自由或弱复合的甲醛形式的情况下,甲醛在缩合反应器中原位或者在缩合反应器之前在单独反应器中形成。例如,三噁烷可以在惰性物质上或在空管中于超过350℃的温度分解或在酸催化剂上于超过100℃的温度分解,形成甲醛。
在一种实施方式中,亚烷基化剂相应于式I。
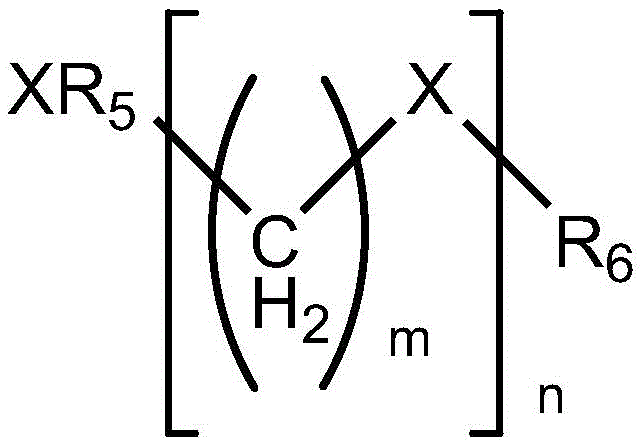
在该式中,R5和R6可以独立地选自C1-C12烃优选C1-C12烷基、烯基或芳基,或氢。优选,R5和R6独立地是C1-C6烷基或氢,最优选甲基和/或氢。X可以是氧或硫,优选氧;和n是1至10,优选1至3的整数。在某些实施方式中,m是1或2,优选1。
在一种实施方式中,式I化合物可以是在水存在下甲醛与甲醇之间平衡反应的产物。在上述情况中,式I化合物可以是适宜的甲醛来源。在一种实施方式中,甲醛来源包括任何平衡组合物。甲醛来源的实例包括但不限于甲醛缩二甲醇(1,1二甲氧基甲烷);聚甲醛-(CH2-O)i-,其中i是1至100;福尔马林;和其它平衡组合物比如甲醛、甲醇和丙酸甲酯的混合物。在一种实施方式中,甲醛的来源选自1,1二甲氧基甲烷;甲醛和甲醇的较高缩醛;和CH3-O-(CH2-O)i-CH3,其中i是2。
亚烷基化剂可以与或不与有机或无机溶剂一起使用。
术语"福尔马林"是指甲醛、甲醇和水的混合物。在一种实施方式中,福尔马林包含25重量%至85重量%的甲醛;0.01重量%至25重量%的甲醇;和15重量%至70重量%的水。在使用甲醛、甲醇和丙酸甲酯的混合物的情况下,混合物包含小于10重量%,例如小于5重量%或小于1重量%的水。
在某些实施方式中,缩合反应可以实现乙酸的有利转化率和生成丙烯酸类产物的有利选择性和生产能力。出于本发明意图,术语"转化率"是指进料中的乙酸转化为乙酸以外的化合物的量。转化率表达为基于进料中的乙酸的摩尔百分比。乙酸的总体转化率可以是至少15mol%,例如至少25mol%,至少40mol%,或至少50mol%。在又一实施方式中,可以进行反应,其中乙酸与亚烷基化剂的摩尔比是至少0.50:1,例如至少1:1。
选择性表达为基于转化的乙酸的摩尔百分比。应理解转化自乙酸的各化合物具有独立的选择性,并且该选择性独立于转化率。例如,如果30mol%的经转化乙酸转化为丙烯酸,则丙烯酸选择性将是30mol%。优选地,对丙烯酸类产物例如丙烯酸和丙烯酸甲酯的催化剂选择性是至少30mol%,例如,至少50mol%,至少60mol%,或至少70mol%。在某些实施方式中,对丙烯酸的选择性是至少30mol%,例如至少40mol%或至少50mol%;和/或对丙烯酸甲酯的选择性是至少10mol%,例如至少15mol%或至少20mol%。
术语"生产能力"如本文所用是指在缩合期间形成的指定产物例如丙烯酸类产物的克数,基于每小时所用催化剂升数。优选的是,至少20克丙烯酸类每升催化剂每小时,例如至少40克丙烯酸类每升催化剂每小时或至少100克丙烯酸类每升催化剂每小时的生产能力。在范围方面,生产能力优选是20至800克丙烯酸类每升催化剂每小时,例如100至600克丙烯酸类每千克催化剂每小时或200至400克丙烯酸类每千克催化剂每小时。
如上所述,本发明催化剂组合物提供乙酸的高转化率。有利地,实现这些高转化率并同时保持对希望丙烯酸类产物例如丙烯酸和/或丙烯酸甲酯的选择性。作为结果,与采用常规催化剂的常规生产能力相比,丙烯酸类产物生产能力得以改善。
本发明方法的优选实施方式也具有对不希望产物比如一氧化碳和二氧化碳的低选择性。对这些不期望的产物的选择性优选小于30%,例如小于20%或小于10%。更优选,这些不希望产物是不可测得的。烷烃例如乙烷的形成可以是低的,理想地,穿过催化剂的乙酸小于2%、小于1%或小于0.5%转化为烷烃,该烷烃除作为燃料外具有很小价值。
烷酸或其酯和亚烷基化剂可以独立地或在预先混合之后进料至含催化剂的反应器。反应器可以是任何适宜的反应器。优选,反应器是固定床反应器,但还可以使用其它反应器比如连续搅拌槽反应器或流化床反应器。
在某些实施方式中,将烷酸例如乙酸和亚烷基化剂例如甲醛以至少0.25:1,例如至少0.75:1或至少1:1的摩尔比进料至反应器。在范围方面,烷酸与亚烷基化剂的摩尔比可以是0.50:1至10:1或0.75:1至5:1。在某些实施方式中,烷酸和亚烷基化剂的反应用化学计量过量的烷酸进行。在这些情况中,丙烯酸类选择性可以得到改善。例如,丙烯酸类选择性可以比用过量亚烷基化剂进行反应的情况下所实现的选择性高至少10%,例如高至少20%或高至少30%。在其它实施方式中,用化学计量过量的亚烷基化剂进行烷酸和亚烷基化剂的反应。
缩合反应可以以至少250℃,例如至少300℃或至少350℃的温度进行。在范围方面,反应温度可以是200℃至500℃,例如300℃至400℃或350℃至390℃。反应压力不受特别限制,和反应一般在大气压左右进行。在一种实施方式中,反应可以在0kPa至4100kPa,例如3kPa至345kPa或6kPa至103kPa的压力进行。
水可以按反应混合物重量计以多至60重量%,例如多至50重量%或多至40重量%的量存在。然而,由于水对过程速率和分离成本的负面效果,优选将其减少。
在一种实施方式中,将惰性或反应性气体供给至反应物流。惰性气体的实例包括但不限于氮、氦、氩和甲烷。反应性气体或蒸气的实例包括但不限于氧、碳的氧化物,硫的氧化物,和烷基卤化物。在将反应性气体比如氧加至反应器的情况下,在某些实施方式中,这些气体可以以希望的水平加入整个催化剂床的各个阶段,以及在反应器开始处与其它进料组分一起进料。
在一种实施方式中,在自所希望产物充分分离之后,将未反应组分比如羧酸和甲醛以及剩余的惰性或反应性气体循环至反应器。
在所希望产物是通过将烷酸酯的酯与甲醛反应制得的不饱和酯的情况下,对应于酯的醇还可以与其它组分一起地或分开地进料至反应器。例如,在希望丙烯酸甲酯的情况下,可将甲醇进料至反应器。相比其它效果,醇尤其减少离开反应器的酸的量。不是必需在反应器开始处加入醇,其可以例如在中部或近尾部加入,以便使得酸比如丙酸、甲基丙烯酸转化为各自的酯而不抑制催化剂活性。
实施例
实施例1
用铋盐、钨盐和钒前体例如NH4VO3制备催化剂组合物。合并和混合胶体二氧化硅、去离子水和乙二醇。将有机酸例如草酸或柠檬酸加至混合物,将混合物加热至50℃。将计算量的NH4VO3加至混合物,在搅拌下将所得溶液加热至80℃。将硝酸铋和偏钨酸铵加至加热的混合物。将2重量%的甲基纤维素溶液加至铋盐/钨盐/钒前体溶液,在80℃搅拌。加入计算量的磷酸(85%)和搅拌所得溶液。然后在120℃干燥炉中蒸发最终混合物至干过夜。所得固体用下述温度特征曲线煅烧:
i)以10℃/分钟的速率从室温加热至160℃;
ii)在160℃加热2小时;
iii)以3℃/分钟的速率从160℃加热至250℃;
iv)在250℃加热2小时;
v)以3℃/分钟的速率从250℃加热至300℃;
vi)在300℃加热6小时;
vii)以3℃/分钟的速率从300℃加热至450℃;和
viii)在450℃加热6小时。
实施例2
在配有搅拌器和冷凝器的烧瓶中,将大约100mL异丁醇加热至90℃。将所希望量的V2O5(11.32g)作为粉末缓慢加入至充分搅拌的热异丁醇。一旦加入V2O5,则在搅动下将85%H3PO4(8.2g)缓慢加至热的混合物。一旦H3PO4加入完成,则将混合物的温度增至100-108℃,和在该温度搅拌混合物约14小时。
将硝酸铋水合物(30.2g)溶于10%HNO3。通过在恒定搅拌下缓慢加入稀H3PO4(8.2g-85%H3PO4)形成和沉淀BiPO4。搅拌混合物1小时,然后经由过滤或离心收集BiPO4。固体BiPO4用去离子水洗涤三次。
将BiPO4加至V2O5-H3PO4-iBuOH混合物,回流搅拌混合物1小时。让混合物冷却,经由过滤或离心分离固体形式的催化剂。固体用EtOH洗涤一次,并用去离子水再洗涤两次。固体在120℃用流动空气干燥过夜。将偏钨酸铵溶于水,加至VBiPO固体并干燥。研磨最终的VBiWPO固体以形成混合物,然后用实施例1的温度特征曲线煅烧。
实施例3
合并胶体二氧化硅(1.3g)、柠檬酸(25.7g)、乙二醇(18.5g)、去离子水(12g)和在搅拌下加热至50℃。将NH4VO3(13.6g)作为细粉末加至柠檬酸混合物,将所得溶液加热至80℃和搅拌30分钟。将偏钨酸铵溶于水,将溶液加至氧钒基溶液,在80℃搅拌15分钟。将硝酸铋水合物(0.9g)溶于10%HNO3溶液,缓慢地加至氧钒基溶液。在80℃温度搅拌混合物30分钟。
冷却混合物和将2重量%的甲基纤维素(100g)溶液加至混合物和搅拌30分钟。然后,将85%H3PO4溶液(15.7g)缓慢加至混合物和搅拌所得溶液30分钟。在干燥炉中于120℃加热最终混合物过夜,在此时间最终混合物发生热凝胶化,获得多孔泡沫状物。研磨混合所得泡沫状物,用实施例1的温度特征曲线煅烧。
表2显示经由实施例1方法制备的包含钒/铋/钨的催化剂组合物的表面积、孔隙体积和孔径尺寸。钒、钨和/或铋(以及其它制备条件)对所得催化剂的表面积、孔隙体积和孔径尺寸的效果得以研究。催化剂1具有10:1:1的V:Bi:W比率,催化剂2具有1:1:1的V:Bi:W比率,催化剂3具有6:3:1的V:Bi:W比率,而催化剂4具有10:5:1的V:Bi:W比率。如表2所示,具有10:1:1和1:1:1的V:Bi:W比率的催化剂具有相似的表面积、孔隙体积和孔径尺寸。如所显示的,在V:Bi:W比率是6:3:1或10:5:1的情况下,表面积、孔隙体积和孔径尺寸令人惊讶地且出乎意料地增加。这表明V:Bi:W:P的相对水平以及制备条件可以对催化剂表面积、孔隙体积和孔径尺寸具有显著影响。

实施例4
将包含乙酸、甲醛、甲醇、水、氧和氮的反应进料通过包含示于表2的催化剂1-4的固定床反应器。催化剂1-4的反应进行如下:370℃的反应器温度和600Hr-1的GHSV,8摩尔%的总有机物,1.5的乙酸和甲醛比率,1.0%的O2,4.4摩尔%的H2O,87摩尔%的总N2,和55%的福尔马林当量。丙烯酸和丙烯酸甲酯(统称"丙烯酸类产物")得以产生。在各反应时间点测量催化剂1-4的乙酸转化率、丙烯酸类选择性、丙烯酸类收率和丙烯酸类空时收率。也在相同条件下测试商业VPO催化剂对比例A。结果示于表3。

在各反应时间点测量催化剂1-4和对比例A的乙酸转化率、丙烯酸类选择性、丙烯酸类收率和丙烯酸类空时收率。全都含铋和钨的催化剂1-4出乎意料地优于对比例A,后者是常规不含铋和不含钨的可商购钒催化剂。催化剂1-4显示比对比例A更佳的乙酸转化率,丙烯酸类收率,和丙烯酸类STY。特别地,催化剂3和4显示分别65%和64%的高初始乙酸转化率,和分别79%和78%的高丙烯酸选择性。作为比较,对比例A具有39%的初始乙酸转化率并且在24.9小时的过程内降低至23%。对比例A的丙烯酸类收率也在24.9小时的过程内从33%降低至20%。另外,对比例A的丙烯酸类STY从31g/hr/L降低至19g/hr/L。因此,如数据所示,催化剂1-4优于可商购的VPO催化剂。
实施例5
表4显示包含钒、铋和钨的催化剂5-7的表面积、孔隙体积和孔径尺寸。实施例5-7经由实施例1的制备方法而制备,但例如在温度特征曲线的最终步骤采用不同的煅烧温度。

煅烧温度对V10Bi5WP18.4O74效果得以研究。如表4所示,催化剂5-7具有相同化学式但是各自在不同温度煅烧。催化剂对比例A、对比例B和对比例C是不含铋或钨的可商购的钒催化剂。如表4所示,随煅烧温度增加,催化剂的表面积和孔隙体积降低,而孔径尺寸稍为增加。这表明煅烧温度可以对催化剂表面积和孔隙体积具有显著影响。
实施例6
将包含乙酸、甲醛、甲醇、水、氧和氮的反应进料通过包含示于表5的催化剂5-7的固定床反应器。催化剂5-7的反应进行如下:380℃的反应器温度和2400Hr-1的GHSV,18摩尔%的总有机物,1.5的乙酸和甲醛比率,3.3%的O2,6.4摩尔%的H2O,72摩尔%的总N2,和65%的福尔马林当量。丙烯酸和丙烯酸甲酯(统称"丙烯酸类产物")得以产生。在各反应时间点测量催化剂5-7的乙酸转化率、丙烯酸类选择性、丙烯酸类收率和丙烯酸类空时收率。也在相同条件下测试商业VPO催化剂对比例A。结果示于表5。


全都含铋和钨的催化剂5-7出乎意料地优于对比例A,后者是常规不含铋和不含钨的可商购钒催化剂。例如,催化剂5-7展示分别45%、43%和35%的平均乙酸转化率,而对比例A展示仅28%的平均乙酸转化率。另外,催化剂5-7展示分别308g/hr/L、306g/hr/L和258g/hr/L的平均丙烯酸类STY,而对比例A展示仅205g/hr/L的平均收率。此外,催化剂5-7展示分别36%、36%和30%的平均丙烯酸类收率,而对比例A展示仅22%的平均收率。
令人惊讶地和出乎意料地,如所显示,催化剂6和7在长时间间隔内保持稳定的乙酸转化率、丙烯酸类选择性、丙烯酸类收率和丙烯酸类空时收率,也即催化剂6和7几乎不显示任何催化剂失活。例如,在44.4小时时间段内,催化剂6乙酸转化率保持为42%至45%。类似地,在76.1小时时间段内,催化剂7的乙酸转化率保持在33%至37%。对比例A的乙酸转化率、丙烯酸类收率和丙烯酸类空时收率在仅2.3小时之后就降低。作为比较,催化剂6和7具有42%至45%和35%-37%的乙酸转化率,81%至83%和83%至88%的丙烯酸类收率,和295g/hr/L至312g/hr/L和249g/hr/L至268g/hr/L的丙烯酸类空时收率,分别持续延长到时间段。
如所展示,在更长的时间间隔内,例如大于40小时,催化剂5-7继续展示保持稳定的反应效能;与之相比催化剂对比例A在24.9小时内显著失活。例如,对于对比例A,乙酸转化率从33%降低至26%,丙烯酸类收率从25%降低至20%,和空时收率从185g/hr/L降低至233g/hr/L。
如所显示,随催化剂煅烧温度增加,催化剂的乙酸转化率降低。例如,催化剂5具有52%的初始乙酸转化率,而催化剂6和7具有分别43%和35%的初始乙酸转化率。如上文所讨论,催化剂5具有比催化剂6和7更大的表面积。不受理论所限,表面积显得可以影响催化剂的乙酸转化率。
实施例7
表6显示包含钒、铋和钨的催化剂8和9的表面积、孔隙体积和孔径尺寸。催化剂8和9经由实施例1的制备方法而制备,但采用不同的甲基纤维素浓度。催化剂对比例B-D显示为比较实施例。甲基纤维素含量的效果得以研究。催化剂8和9分别用3%和10%甲基纤维素制备。如表6所示,甲基纤维素的增加对催化剂的表面积、孔隙体积和孔径尺寸几乎不具效果。
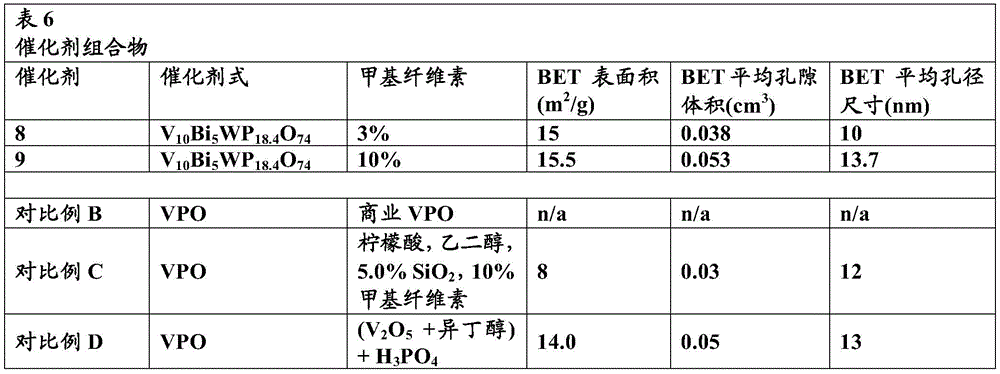
实施例8
将包含乙酸、甲醛、甲醇、水、氧和氮的反应进料通过包含示于表6的催化剂8和9的固定床反应器。催化剂8和9的反应进行如下:380℃的反应器温度和2000Hr-1的GHSV,32摩尔%的总有机物,1.5的乙酸和甲醛比率,5.7%的O2,7.2摩尔%的H2O,55摩尔%的总N2,和75%的福尔马林当量。也包括催化剂对比例B-对比例D作为比较。催化剂对比例B-对比例D的反应进行如下:375℃的反应器温度和2000Hr-1的GHSV,32摩尔%的总有机物,1.5的乙酸和甲醛比率,4.8%的O2,7.2摩尔%的H2O,56摩尔%的总N2,和75%的福尔马林当量。丙烯酸和丙烯酸甲酯(统称"丙烯酸类产物")得以产生。在各种反应时间点测量催化剂8、9、对比例B、对比例C和对比例D的乙酸转化率、丙烯酸类选择性、丙烯酸类收率和丙烯酸类空时收率。结果示于表7。

各自含铋和钨的催化剂8和9出乎意料地优于对比例B、对比例C和对比例D,后者是常规的不含铋和不含钨的可商购钒催化剂。例如,催化剂8和9展示分别34%和40%的平均乙酸转化率,而对比例B、对比例C和对比例D展示仅分别24%、22%和34%的平均乙酸转化率。另外,催化剂8和9展示分别396g/hr/L和390g/hr/L的平均丙烯酸类STY,而对比例B、对比例C和对比例D展示仅分别287g/hr/L、254g/hr/L和338g/hr/L的平均收率。此外,催化剂8和9展示分别30%和34%的平均丙烯酸类收率,而对比例B、对比例C和对比例D展示仅分别22%、19%和26%的平均收率。
此外,催化剂8和9在长时间段内显示稳定的乙酸转化率、丙烯酸类选择性、丙烯酸类收率和丙烯酸类STY。例如,催化剂8在4.6小时时间段内具有稳定的34%的乙酸转化率,87-88%的丙烯酸类选择性,稳定的30%丙烯酸类收率和393-397g/hr/L的丙烯酸类STY。催化剂9在76.5小时时间段内具有39-41%的乙酸转化率,79-88%的丙烯酸类选择性,32-35%的收率和376-400g/hr/L的丙烯酸类STY。这显示两种催化剂在长时间段内均不失活至几乎不失活。此外,较高量的甲基纤维素例如10%显得增加反应的乙酸转化率。例如,催化剂9的平均转化率是40%,与之相比催化剂8具有34%的平均转化率。与之比较,对比例B和对比例D的乙酸转化率、丙烯酸收率和丙烯酸空时收率分别在仅3.9小时和3.3小时之后就已减少。尽管催化剂对比例C在4小时时间段内显示稳定的乙酸转化率,丙烯酸类选择性,丙烯酸类收率和丙烯酸类空时收率,但是乙酸转化率是不希望的22%。作为比较,催化剂8和9在延长的时间段具有分别稳定的34%和39%-41%的乙酸转化率。
实施例9
表8显示包含钒、铋和钨的催化剂10-12的表面积、孔隙体积和孔径尺寸。催化剂10-12经由实施例1的制备方法而制备,但采用不同的磷水平。

磷水平的效果得以研究。催化剂10、11和12具有化学式中所指的不同水平的磷,其中P分别=15、16.5和18.4。如表8所示,磷水平的增加显得增加催化剂的表面积、孔隙体积和孔径尺寸。令人惊讶地和出乎意料地,在磷原子数保持在15至20.4的范围时,实现显著更高的表面积。
实施例10
将包含乙酸、甲醛、甲醇、水、氧和氮的反应进料通过包含示于表8的催化剂10、11和12的固定床反应器。催化剂8和9的反应进行如下:375℃的反应器温度和2000Hr-1的GHSV,32摩尔%的总有机物,1.5的乙酸和甲醛比率,4.8%的O2,7.0摩尔%的H2O,56摩尔%的总N2,和75%的福尔马林当量。也包括催化剂对比例B-对比例D作为比较。对比例B-对比例D的反应条件与实施例9相同。丙烯酸和丙烯酸甲酯(统称"丙烯酸类产物")得以产生。在各反应时间点测量催化剂10、11、12、对比例B、对比例C和对比例D的乙酸转化率、丙烯酸类选择性、丙烯酸类收率和丙烯酸类空时收率。结果示于表9。
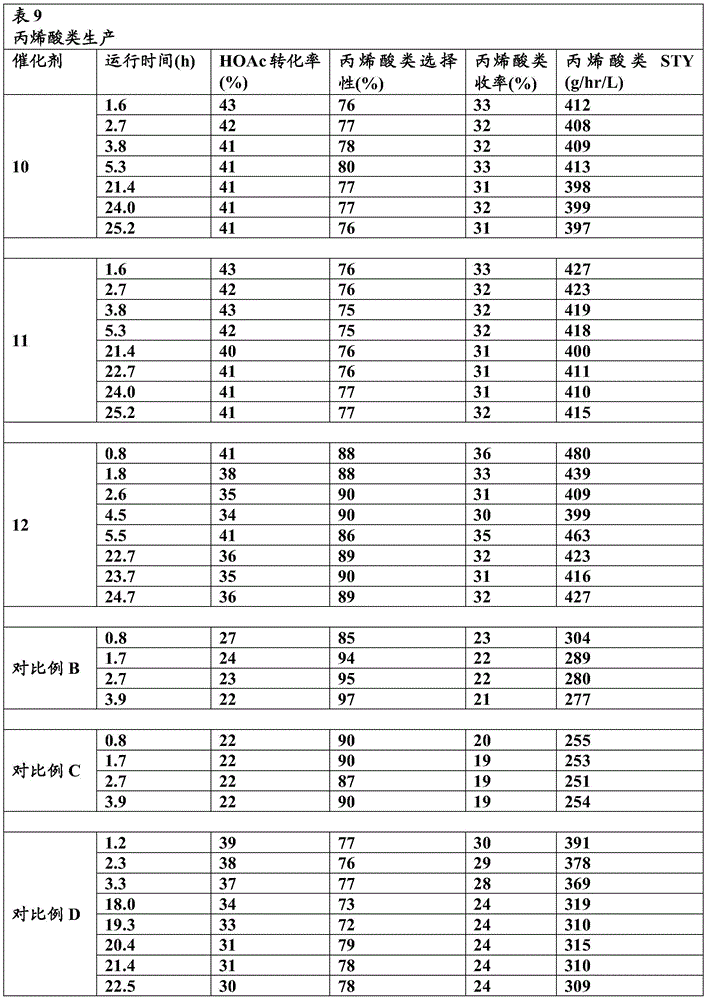
全都含铋和钨的催化剂10-12出乎意料地优于对比例B、对比例C和对比例D,后者是常规的不含铋和不含钨的可商购钒催化剂。例如,催化剂10-12展示分别41%、42%和37%的平均乙酸转化率,而对比例B、对比例C和对比例D展示仅分别24%、22%和34%的平均乙酸转化率。另外,催化剂10-12展示分别405g/hr/L、415g/hr/L和432g/hr/L的平均丙烯酸类STY,而对比例B、对比例C和对比例D展示仅分别287g/hr/L、254g/hr/L和338g/hr/L的平均收率。此外,催化剂10-12展示分别32%、32%和33%的平均丙烯酸类收率,而对比例B、对比例C和对比例D展示仅分别22%、19%和26%的平均收率。
如表9所示,催化剂10、11和12在24小时时间段内显示稳定的乙酸转化率,丙烯酸类选择性,丙烯酸类收率,和丙烯酸类STY。例如,催化剂10在25.2小时时间段内具有41-43%的乙酸转化率,76-80%的丙烯酸类选择性,31-32%的丙烯酸类收率和397-413g/hr/L的丙烯酸类STY。催化剂11在25.2小时时间段内具有40-43%的乙酸转化率,75-77%的丙烯酸类选择性,31-32%的丙烯酸类收率和400-427g/hr/L的丙烯酸类STY。催化剂12在24.7小时时间段内具有34-41%的乙酸转化率,86-90%的丙烯酸类选择性,30-36%的丙烯酸类收率和399-480g/hr/L的丙烯酸类STY。与之比较,对比例B和对比例D的乙酸转化率、丙烯酸收率和丙烯酸空时收率分别在仅3.9小时和3.3小时之后就已减少。尽管催化剂对比例D在4小时时间段内显示稳定的乙酸转化率,丙烯酸类选择性,丙烯酸类收率,和丙烯酸类空时收率,但是乙酸转化率是不希望的22%。这显示本发明的全部3种催化剂在长时间段内几乎不失活。
此外,磷水平的变化显得具有对乙酸转化率的次要效果。例如,与具有18.4磷水平的催化剂12相比,磷水平分别为15和16.5的催化剂10和11显得具有稍稍更佳的乙酸转化率。
实施例11
表10显示包含钒、铋和钨的催化剂13和14的表面积、孔隙体积和孔径尺寸。催化剂13经由实施例2的制备方法而制备,采用还原或未还原的VOPO4。催化剂14制备如下:将V2O5在稀H3PO4中回流24小时,让其冷却至室温。经由离心或过滤收集催化剂。将未还原的磷酸钒与偏钨酸铵和将43%H3PO4加入硝酸铋(10%HNO3)溶液制得的BiPO4相组合。手工激烈研磨或球磨混合物5小时,按照实施例2提供的煅烧方案煅烧混合物。

还原的VOPO4与未还原的VOPO4的效果得以对比研究。如表10所示,催化剂13和14通过物理混合粉化形式的金属前体来制备。如表10所示,与采用未还原的VOPO4的催化剂14相比,采用还原的VOPO4的催化剂13具有更大的表面积、孔隙体积和孔径尺寸。
实施例12
将包含乙酸、甲醛、甲醇、水、氧和氮的反应进料通过包含示于表10的催化剂13和14的固定床反应器。催化剂13和14的反应进行如下:375℃的反应器温度和2000Hr-1的GHSV,32摩尔%的总有机物,1.5的乙酸和甲醛比率,4.8%的O2,7.1摩尔%的H2O,56摩尔%的总N2,和75%的福尔马林当量。也包括催化剂对比例B-对比例D作为比较。对比例B-对比例D的反应条件与实施例8相同。丙烯酸和丙烯酸甲酯(统称"丙烯酸类产物")得以产生。在各反应时间点测量催化剂13、14、对比例B、对比例C和对比例D的乙酸转化率,丙烯酸类选择性,丙烯酸类收率,和丙烯酸类空时收率。结果示于表11。

全都含铋和钨的催化剂13和14出乎意料地优于对比例B、对比例C和对比例D,后者是常规的不含铋和不含钨的可商购钒催化剂。例如,催化剂13和14展示分别36%和31%的平均乙酸转化率,而对比例B、对比例C和对比例D展示仅分别24%、22%和34%的平均乙酸转化率。另外,催化剂13和14展示分别365g/hr/L和318g/hr/L的平均丙烯酸类STY,而对比例B、对比例C和对比例D展示仅分别287g/hr/L、254g/hr/L和338g/hr/L的平均收率。此外,催化剂13和14展示分别28%和26%的平均丙烯酸类收率,而对比例B、对比例C和对比例D展示仅分别22%、19%和26%的平均收率。
如表11所示,催化剂13和14显示稳定的乙酸转化率,丙烯酸类选择性,丙烯酸类收率,和丙烯酸类STY。例如,催化剂13在5.4小时时间段内具有35-38%的乙酸转化率,78-79%的丙烯酸类选择性,27-29%的丙烯酸类收率和353-371g/hr/L的丙烯酸类STY。催化剂14在23.4小时时间段内具有28-33%的乙酸转化率,83-85%的丙烯酸类选择性,24-27%的收率和298-333g/hr/L的丙烯酸类STY。这显示两种催化剂在长时间段内几乎不失活。与之比较,对比例B和对比例D的乙酸转化率、丙烯酸收率和丙烯酸空时收率分别在仅3.9小时和3.3小时之后就已减少。尽管催化剂对比例C在4小时时间段内显示稳定的乙酸转化率、丙烯酸类选择性、丙烯酸类收率和丙烯酸类空时收率,但是乙酸转化率是不希望的22%。
此外,与采用未还原的VOPO4的催化剂14相比,采用还原的VOPO4的催化剂13显得具有更高的乙酸转化率。
实施例13
表12显示包含钒和掺杂量的铋和钨的催化剂15和16的表面积、孔隙体积和孔径尺寸。催化剂15和16经由实施例3的制备方法而制备。

Bi和W掺杂的效果得以研究。如表12所示,催化剂15和16含有减少水平的Bi和W。例如,催化剂15和16具有10:0.16:0.5的V:Bi:W比率。催化剂15和16也具有不同水平的磷和氧。如表12所示,催化剂16具有比催化剂16明显更大的表面积和孔隙体积。
实施例14
将包含乙酸、甲醛、甲醇、水、氧和氮的反应进料通过包含示于表12的催化剂15和16和示于上表4的对比例B-对比例D的固定床反应器。全部催化剂的反应进行如下:375℃的反应器温度和2000Hr-1的GHSV,32摩尔%的总有机物,1.5的乙酸和甲醛比率,4.8%的O2,7.2摩尔%的H2O,56摩尔%的总N2,和75%的福尔马林当量。也包括催化剂对比例B-对比例D作为比较。对比例B-对比例D的反应条件与实施例6相同。丙烯酸和丙烯酸甲酯(统称"丙烯酸类产物")得以产生。在各反应时间点测量催化剂15、16、对比例B、对比例C和对比例D的乙酸转化率、丙烯酸类选择性、丙烯酸类收率和丙烯酸类空时收率。结果示于表13。


全都含铋和钨的催化剂15和16出乎意料地优于对比例B、对比例C和对比例D,后者是常规的不含铋和不含钨的可商购钒催化剂。例如,催化剂15和16展示分别30%和29%的平均乙酸转化率,而对比例B、对比例C和对比例D展示仅分别24%、22%和34%的平均乙酸转化率。另外,催化剂15和16展示分别307g/hr/L和335g/hr/L的平均丙烯酸类STY,而对比例B、对比例C和对比例D展示仅分别287g/hr/L、254g/hr/L和338g/hr/L的平均收率。此外,催化剂15和16展示分别24%和26%的平均丙烯酸类收率,而对比例B,对比例C,和对比例C展示仅分别22%、19%和26%的平均收率。
如表13所示,催化剂15和16显示稳定的乙酸转化率,丙烯酸类选择性,丙烯酸类收率和丙烯酸类STY。例如,催化剂15在21.8小时时间段内具有29-32%的乙酸转化率,77-84%的丙烯酸类选择性,23-26%的丙烯酸类收率和294-333g/hr/L的丙烯酸类STY。催化剂16在21.8小时时间段内具有29-31%的乙酸转化率,87-89%的丙烯酸类选择性,25-27%的收率和328-351g/hr/L的丙烯酸类STY。数据显示两种催化剂在长时间段内几乎不失活。与之比较,对比例B和对比例D的乙酸转化率、丙烯酸收率和丙烯酸空时收率分别在仅3.9小时和3.3小时之后就已减少。尽管催化剂对比例C在4小时时间段内显示稳定的乙酸转化率、丙烯酸类选择性、丙烯酸类收率和丙烯酸类空时收率,但是乙酸转化率是不希望的22%。
此外,与可商购的VPO催化剂相比,具有低水平铋和钨的催化剂显得具有相似或更佳的乙酸转化率和丙烯酸类STY。例如,催化剂15和16具有分别30%和29%的平均乙酸转化率和分别307g/hr/L和335g/hr/L的平均丙烯酸类STY。作为比较,对比例B、对比例C和对比例D具有分别24%、22%和34%的平均乙酸转化率,分别287g/hr/L、254g/hr/L和338g/hr/L的平均STY。
虽然本发明已加以详细描述,本发明主旨和范围内的修饰对于本领域技术人员来说是很明显的。鉴于前述讨论,本领域的有关知识和上文针对背景和详述讨论的参考文献,通过援引将其公开全部并入本文。此外,应理解本发明各方面和各种实施方式的各部分和下文和/或所附权利要求中所述的各种特征可以加以组合或整体或部分互换。在前述各个实施方案的描述中,如本领域技术人员所可认识到的,引用另一个实施方案的实施方案可以与其它实施方案适当地组合。此外,本领域普通技术人员将认识到前述描述仅仅是举例方式,并且不意欲限制本发明。
本发明还包括以下实施方式:
1.用于产生丙烯酸类产物的方法,该方法包括下述步骤:
在有效产生丙烯酸类产物的条件下,在催化剂上将烷酸和亚烷基化剂接触,
其中所述催化剂包含钒、铋和钨。
2.项目1的方法,其中所述亚烷基化剂包含亚甲基化剂。
3.项目1的方法,其中所述烷酸包含乙酸。
4.项目1的方法,其中烷酸与亚烷基化剂的摩尔比是至少0.50:1。
5.项目1的方法,其中总体烷酸转化率是至少15mol%。
6.项目1的方法,其中丙烯酸选择性是至少30%。
7.项目1的方法,其中丙烯酸和丙烯酸类的组合的空时收率是至少50克每升催化剂每小时。
8.项目1的方法,其中基于烷酸转化率的丙烯酸收率是至少20%。
9.用于产生催化剂组合物的方法,该方法包括下述步骤:
接触铋盐、钨盐和钒前体溶液以形成湿催化剂组合物;和
将催化剂组合物干燥以形成包含钒、铋和钨的干催化剂组合物。
10.项目9的方法,还包括下述步骤:
混合钒前体和还原剂溶液以形成钒前体溶液。
11.项目9的方法,其中所述接触包括:
将粘合剂与铋盐、钨盐和/或钒前体溶液接触以形成湿催化剂组合物。
12.项目9的方法,其中所述接触包括将磷酸与铋盐、钨盐和/或钒前体溶液接触以形成湿催化剂组合物。
13.项目9的方法,还包括按照温度特征曲线煅烧干催化剂以形成干催化剂组合物。
14.催化剂组合物,包含活性相,该活性相包含:
钒,
铋,和
钨,其中所述催化剂组合物适用于烷酸和亚烷基化剂的醛醇缩合中以形成丙烯酸类产物。
15.项目14的组合物,其中在催化剂组合物的活性相中的钒:钨摩尔比是至少0.033:1,且其中在催化剂组合物的活性相中的铋:钨摩尔比大于0.0033:1。
16.项目14的组合物,其中所述活性相还包含:
10重量%至22重量%的磷;和
15重量%至50重量%的氧。
17.项目14的组合物,其中所述活性相包含小于5重量%的钛。
18.项目14的组合物,其中所述活性相包含至少0.7重量%的组合的钒和铋。
19.项目14的组合物,其中所述活性相包含至少0.7重量%的组合的钒和钨。
20.项目14的组合物,其中所述催化剂相应于下式
VaBibWcPdOe,
其中
a是1至100,
b是0.1至30,
c是0.1至30,
d是1.0至175,和
e是5至710。
Claims (14)
1.用于产生催化剂组合物的方法,该方法包括下述步骤:
接触铋盐、钨盐和钒前体溶液以形成湿催化剂组合物;和
将催化剂组合物干燥以形成包含活性相的干催化剂组合物,所述活性相包含钒、磷、铋和钨,其中所述活性相包含至少0.7重量%组合的钒和铋,
其中所述催化剂组合物适用于烷酸和亚烷基化剂的醛醇缩合中以形成丙烯酸类产物。
2.权利要求1的方法,还包括下述步骤:
混合钒前体和还原剂溶液以形成钒前体溶液。
3.权利要求1的方法,其中所述接触包括:
将粘合剂与铋盐、钨盐和/或钒前体溶液接触以形成湿催化剂组合物。
4.权利要求1的方法,其中所述接触包括将磷酸与铋盐、钨盐和/或钒前体溶液接触以形成湿催化剂组合物。
5.权利要求1的方法,还包括按照温度特征曲线煅烧干催化剂以形成干催化剂组合物。
6.权利要求1的方法,其中所述活性相包含:
0.3重量%至30重量%的钒;
0.1重量%至69重量%的铋;和
0.1重量%至61重量%的钨。
7.催化剂组合物,包含活性相,该活性相包含:
钒,
磷,
铋,和
钨,其中所述催化剂组合物适用于烷酸和亚烷基化剂的醛醇缩合中以形成丙烯酸类产物,并且其中所述活性相包含至少0.7重量%的组合的钒和铋。
8.权利要求7的组合物,其中在催化剂组合物的活性相中的钒:钨摩尔比是至少0.033:1,且其中在催化剂组合物的活性相中的铋:钨摩尔比大于0.0033:1。
9.权利要求7的组合物,其中所述活性相还包含:
10重量%至22重量%的磷;和
15重量%至50重量%的氧。
10.权利要求7的组合物,其中所述活性相包含小于5重量%的钛。
11.权利要求7的组合物,其中所述活性相包含至少0.7重量%的组合的钒和钨。
12.权利要求7的组合物,其中所述催化剂对应于下式
VaBibWcPdOe,
其中
a是1至100,
b是0.1至30,
c是0.1至30,
d是1.0至175,和
e是5至710。
13.权利要求7的组合物,其中所述活性相包含:
0.3重量%至30重量%的钒;
0.1重量%至69重量%的铋;和
0.1重量%至61重量%的钨。
14.权利要求1的方法,其中所述活性相包含10重量%至22重量%的磷。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261610099P | 2012-03-13 | 2012-03-13 | |
| US61/610,099 | 2012-03-13 | ||
| CN201280072759.XA CN104271236B (zh) | 2012-03-13 | 2012-10-31 | 用于产生丙烯酸和丙烯酸类的包含钒、铋和钨的催化剂 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201280072759.XA Division CN104271236B (zh) | 2012-03-13 | 2012-10-31 | 用于产生丙烯酸和丙烯酸类的包含钒、铋和钨的催化剂 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN107497418A CN107497418A (zh) | 2017-12-22 |
| CN107497418B true CN107497418B (zh) | 2021-02-23 |
Family
ID=49158235
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201710829640.7A Active CN107497418B (zh) | 2012-03-13 | 2012-10-31 | 用于产生丙烯酸和丙烯酸类的包含钒、铋和钨的催化剂 |
| CN201280072759.XA Active CN104271236B (zh) | 2012-03-13 | 2012-10-31 | 用于产生丙烯酸和丙烯酸类的包含钒、铋和钨的催化剂 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201280072759.XA Active CN104271236B (zh) | 2012-03-13 | 2012-10-31 | 用于产生丙烯酸和丙烯酸类的包含钒、铋和钨的催化剂 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US9108184B2 (zh) |
| EP (1) | EP2825305A1 (zh) |
| CN (2) | CN107497418B (zh) |
| SG (1) | SG11201509242PA (zh) |
| TW (1) | TWI516472B (zh) |
| WO (1) | WO2013137936A1 (zh) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8735314B2 (en) * | 2011-09-29 | 2014-05-27 | Celanese International Corporation | Catalysts for producing acrylic acids and acrylates |
| DE102015222198A1 (de) | 2015-11-11 | 2017-05-11 | Basf Se | Oxidische Zusammensetzung |
| US9687825B1 (en) * | 2016-06-27 | 2017-06-27 | Chevron U.S.A. Inc. | Stable tungsten-phosphorus modified support for a Fischer-Tropsch catalyst |
| CN107185582A (zh) * | 2017-04-25 | 2017-09-22 | 江苏大学 | 一种w修饰sba‑15负载vpo催化剂及其制备方法和用途 |
| CN111378513B (zh) * | 2018-12-28 | 2021-04-06 | 中国石油化工股份有限公司 | 一种生物质处理用助剂及生物质气化处理方法 |
| CN119793533B (zh) * | 2023-10-10 | 2025-10-14 | 中国石油化工股份有限公司 | 新型负载催化剂及其制备方法以及制备2,2-二甲基-1,3-二氧戊环的方法 |
| CN120861126B (zh) * | 2025-09-28 | 2025-11-28 | 上海恒业微晶材料科技股份有限公司 | 一种丙烯醛合成用复合催化剂及其制备方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4866194A (en) * | 1988-05-31 | 1989-09-12 | The Standard Oil Company | Method for ammoxidation of paraffins and catalyst system therefor |
Family Cites Families (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3925464A (en) * | 1971-12-14 | 1975-12-09 | Asahi Glass Co Ltd | Process for preparing unsaturated carboxylic acids from the corresponding unsaturated aldehydes |
| US4276197A (en) | 1979-12-19 | 1981-06-30 | Atlantic Richfield Company | Preparation of a titanium promoted VO(PO3)2 oxidation catalyst |
| US5026908A (en) | 1984-05-03 | 1991-06-25 | Hoechst Celanese Corporation | Methanol carbonylation process |
| US5001259A (en) | 1984-05-03 | 1991-03-19 | Hoechst Celanese Corporation | Methanol carbonylation process |
| US5144068A (en) | 1984-05-03 | 1992-09-01 | Hoechst Celanese Corporation | Methanol carbonylation process |
| CA1299195C (en) | 1986-06-16 | 1992-04-21 | G. Paull Torrence | Addition of hydrogen to carbon monoxide feed gas in producing acetic acid by carbonylation of methanol |
| EP0293859B1 (en) | 1987-06-05 | 1992-01-22 | Nippon Shokubai Co., Ltd. | Catalyst for oxidation of acrolein and process for production thereof |
| CN1021638C (zh) * | 1990-11-05 | 1993-07-21 | 中国石油化工总公司 | 丙烯腈流化床催化剂 |
| US5821111A (en) | 1994-03-31 | 1998-10-13 | Bioengineering Resources, Inc. | Bioconversion of waste biomass to useful products |
| US5364824A (en) | 1992-12-08 | 1994-11-15 | Huntsman Specialty Chemicals Corporation | Catalysis for the production of maleic anhydride containing vanadium-phosphorus oxide with selected promoter elements |
| USRE35377E (en) | 1993-05-27 | 1996-11-12 | Steinberg; Meyer | Process and apparatus for the production of methanol from condensed carbonaceous material |
| US5599976A (en) | 1995-04-07 | 1997-02-04 | Hoechst Celanese Corporation | Recovery of acetic acid from dilute aqueous streams formed during a carbonylation process |
| CN1087653C (zh) | 1996-03-20 | 2002-07-17 | 中国科学院大连化学物理研究所 | 一种丙烷氧化制丙烯醛反应用催化剂及其反应工艺 |
| IN192600B (zh) | 1996-10-18 | 2004-05-08 | Hoechst Celanese Corp | |
| GB9807498D0 (en) | 1998-04-08 | 1998-06-10 | Ici Plc | Production of unsaturated acids therfore and catalysts therfor |
| ES2312337T3 (es) | 1999-03-11 | 2009-03-01 | Zeachem Inc. | Proceso para producir etanol. |
| US7074603B2 (en) | 1999-03-11 | 2006-07-11 | Zeachem, Inc. | Process for producing ethanol from corn dry milling |
| US6232352B1 (en) | 1999-11-01 | 2001-05-15 | Acetex Limited | Methanol plant retrofit for acetic acid manufacture |
| US6627770B1 (en) | 2000-08-24 | 2003-09-30 | Celanese International Corporation | Method and apparatus for sequesting entrained and volatile catalyst species in a carbonylation process |
| US6657078B2 (en) | 2001-02-07 | 2003-12-02 | Celanese International Corporation | Low energy carbonylation process |
| US6685754B2 (en) | 2001-03-06 | 2004-02-03 | Alchemix Corporation | Method for the production of hydrogen-containing gaseous mixtures |
| US7115772B2 (en) | 2002-01-11 | 2006-10-03 | Celanese International Corporation | Integrated process for producing carbonylation acetic acid, acetic anhydride, or coproduction of each from a methyl acetate by-product stream |
| WO2004004900A1 (ja) * | 2002-07-05 | 2004-01-15 | Mitsubishi Rayon Co., Ltd. | メタクリル酸製造用触媒の製造方法 |
| US7005541B2 (en) | 2002-12-23 | 2006-02-28 | Celanese International Corporation | Low water methanol carbonylation process for high acetic acid production and for water balance control |
| AU2005207970B2 (en) | 2004-01-29 | 2011-04-28 | Zeachem Inc. | Recovery of organic acids |
| US7208624B2 (en) | 2004-03-02 | 2007-04-24 | Celanese International Corporation | Process for producing acetic acid |
| EP1593663A1 (en) | 2004-05-05 | 2005-11-09 | Haldor Topsoe A/S | Process for the preparation of acrolein and/or acrylic acid |
| US7851397B2 (en) | 2005-07-25 | 2010-12-14 | Saudi Basic Industries Corporation | Catalyst for methacrolein oxidation and method for making and using same |
| US7449426B2 (en) * | 2005-11-29 | 2008-11-11 | Saudi Basic Industries Corporation | Catalyst composition without antimony or molybdenum for ammoxidation of alkanes, a process of making and a process of using thereof |
| WO2008093713A1 (ja) * | 2007-01-30 | 2008-08-07 | Babcock-Hitachi Kabushiki Kaisha | 排ガス浄化用触媒およびその製造方法 |
| EP2121946A4 (en) | 2007-02-09 | 2012-08-29 | Zeachem Inc | ENERGY-EFFICIENT METHOD FOR MANUFACTURING PRODUCTS |
| JP5094459B2 (ja) | 2007-03-09 | 2012-12-12 | ローム アンド ハース カンパニー | アルカンを不飽和カルボン酸に変換するための改良法 |
| AU2009244135A1 (en) | 2008-05-07 | 2009-11-12 | Zeachem Inc. | Recovery of organic acids |
| GB0809332D0 (en) | 2008-05-22 | 2008-07-02 | Lucite Int Uk Ltd | Production of ethylenically unsaturated acids or esters thereof |
| US7884253B2 (en) | 2008-12-11 | 2011-02-08 | Range Fuels, Inc. | Methods and apparatus for selectively producing ethanol from synthesis gas |
| US8481448B2 (en) * | 2010-07-19 | 2013-07-09 | Saudi Basic Industries Corporation | Catalyst for oxidation of saturated and unsaturated aldehydes to unsaturated carboxylic acid, method of making and method of using thereof |
| DE102010040921A1 (de) | 2010-09-16 | 2012-03-22 | Basf Se | Verfahren zur Herstellung von Acrylsäure aus Methanol und Essigsäure |
| DE102010040923A1 (de) | 2010-09-16 | 2012-03-22 | Basf Se | Verfahren zur Herstellung von Acrylsäure aus Ethanol und Formaldehyd |
-
2012
- 2012-10-31 CN CN201710829640.7A patent/CN107497418B/zh active Active
- 2012-10-31 EP EP12783813.4A patent/EP2825305A1/en not_active Withdrawn
- 2012-10-31 US US13/664,494 patent/US9108184B2/en active Active
- 2012-10-31 WO PCT/US2012/062714 patent/WO2013137936A1/en not_active Ceased
- 2012-10-31 SG SG11201509242PA patent/SG11201509242PA/en unknown
- 2012-10-31 CN CN201280072759.XA patent/CN104271236B/zh active Active
- 2012-11-01 TW TW101140606A patent/TWI516472B/zh not_active IP Right Cessation
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4866194A (en) * | 1988-05-31 | 1989-09-12 | The Standard Oil Company | Method for ammoxidation of paraffins and catalyst system therefor |
Also Published As
| Publication number | Publication date |
|---|---|
| US9108184B2 (en) | 2015-08-18 |
| CN104271236A (zh) | 2015-01-07 |
| SG11201509242PA (zh) | 2015-12-30 |
| CN104271236B (zh) | 2018-01-26 |
| CN107497418A (zh) | 2017-12-22 |
| TWI516472B (zh) | 2016-01-11 |
| EP2825305A1 (en) | 2015-01-21 |
| US20130245312A1 (en) | 2013-09-19 |
| WO2013137936A1 (en) | 2013-09-19 |
| TW201336820A (zh) | 2013-09-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8669201B2 (en) | Catalyst for producing acrylic acids and acrylates | |
| KR102038262B1 (ko) | 아크릴산 및 아크릴레이트 제조용 촉매 | |
| CN107497418B (zh) | 用于产生丙烯酸和丙烯酸类的包含钒、铋和钨的催化剂 | |
| US8889586B2 (en) | Process for producing acrylic acids and acrylates | |
| US8735314B2 (en) | Catalysts for producing acrylic acids and acrylates | |
| US8802585B2 (en) | Catalysts for producing acrylic acids and acrylates | |
| US20130053599A1 (en) | Catalysts for producing acrylic acids and acrylates | |
| WO2013138227A1 (en) | Catalyst for producing acrylic acids and acrylates | |
| US8962515B2 (en) | Catalyst for producing acrylic acids and acrylates |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |