CN102378635A - 重组人白蛋白-人粒细胞集落刺激因子融合蛋白的新稳定制剂 - Google Patents
重组人白蛋白-人粒细胞集落刺激因子融合蛋白的新稳定制剂 Download PDFInfo
- Publication number
- CN102378635A CN102378635A CN2010800127233A CN201080012723A CN102378635A CN 102378635 A CN102378635 A CN 102378635A CN 2010800127233 A CN2010800127233 A CN 2010800127233A CN 201080012723 A CN201080012723 A CN 201080012723A CN 102378635 A CN102378635 A CN 102378635A
- Authority
- CN
- China
- Prior art keywords
- neug
- neutropenia
- subject
- dose
- chemotherapy
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images


























Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/19—Cytokines; Lymphokines; Interferons
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/38—Albumins
- A61K38/385—Serum albumin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/19—Cytokines; Lymphokines; Interferons
- A61K38/193—Colony stimulating factors [CSF]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/38—Albumins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/64—Drug-peptide, drug-protein or drug-polyamino acid conjugates, i.e. the modifying agent being a peptide, protein or polyamino acid which is covalently bonded or complexed to a therapeutically active agent
- A61K47/643—Albumins, e.g. HSA, BSA, ovalbumin or a Keyhole Limpet Hemocyanin [KHL]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Epidemiology (AREA)
- Immunology (AREA)
- Gastroenterology & Hepatology (AREA)
- Zoology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Molecular Biology (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Diabetes (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Peptides Or Proteins (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
公开了用于治疗、预防和减轻以白细胞计数降低为特征的症状和疾病的组合物和方法。本文所述的方法和组合物包括由人血清白蛋白(“HSA”)和人粒细胞集落刺激因子(“G-CSF”)形成的融合多肽。
Description
本申请要求2009年1月16日提交的美国临时申请第61/145,440号以及2009年1月16日提交的美国临时专利申请第61/145,436号的权益。美国临时申请第61/145,440号以及美国临时专利申请第61/145,436号的全文通过引用纳入本文。
背景技术
白细胞减少症是指循环白细胞计数(WBC)的减少,通常定义为WBC计数<4000/mL。白细胞减少症中涉及的主要细胞是嗜中性粒细胞。但是,淋巴细胞、单核细胞、嗜酸性粒细胞或嗜碱性粒细胞数量减少可能对总细胞计数减少也有影响(Merck Manual(默克手册),第17版)。
嗜中性白细胞减少症的特征为血液嗜中性粒细胞计数的减少,通常引起对细菌和真菌感染易感性的提高。根据嗜中性粒细胞计数和感染的相对风险将嗜中性白细胞减少症分成:轻度(1000-1500/mL)、中度(3级,500-1000/mL)或重度(4级,<500/mL)急性和重度嗜中性白细胞减少症由于使患者易患迅速致死感染而是一种危及生命的病症(默克手册,第17版)。
嗜中性白细胞减少症可由骨髓中嗜中性粒细胞生成受损或嗜中性粒细胞加速破坏引起。当嗜中性粒细胞快速使用而其生成严重受损时,急性嗜中性白细胞减少症可能在数天内发作。慢性嗜中性白细胞减少症可能持续数月,并通常由脾脏中嗜中性粒细胞的生成减少或隔离引起。嗜中性白细胞减少症可以根据其是骨髓细胞外源因子的次生结果还是存在于骨髓祖细胞的内在缺陷进行分类(默克手册,第17版)。
嗜中性白细胞减少症及其感染性并发症是细胞毒性化疗和其它肿瘤治疗如放疗、生物治疗、靶向治疗和骨髓移植中最为常见和严重的副作用之一。细胞毒性化疗通过破坏快速生长的细胞来发挥作用,由于嗜中性粒细胞前体的高增殖率和血液嗜中性粒细胞的快速代谢回转而引发嗜中性白细胞减少症(默克手册,第17版)。化疗患者中,嗜中性白细胞减少症的最常见症状包括发热、口疮和耳感染。显著嗜中性白细胞减少症的患者常发生化脓性感染,例如败血症、皮肤蜂窝组织炎、肝脓肿、疔疮、肺炎、口腔炎、牙龈炎、围直肠炎、结肠炎、鼻窦炎和中耳炎。化疗可能被迫延迟直至身体能产生更多的嗜中性粒细胞,可能必须采用较低的剂量,导致治疗效果较低。
发明概述
本文所述方法和组合物有助于治疗、缓解或预防以白细胞计数低于正常值为特征的症状。这些症状包括但不限于白细胞减少症和嗜中性白细胞减少症。
在第一种实施方式中,描述了在人对象中治疗或预防嗜中性白细胞减少症的方法,包括对显示嗜中性白细胞减少症或有产生嗜中性白细胞减少症风险的人对象施用有效量的重组人白蛋白-人粒细胞集落刺激因子以治疗对象。在示范性实施方式中,所述人对象可能患有非骨髓恶性肿瘤并正接受至少一种骨髓抑制型抗癌药,该药物与发热性嗜中性白细胞减少症的临床显著发病率相关。
在第二种实施方式中,描述了在人对象中治疗或预防白细胞减少症的方法,包括对显示白细胞减少症或有产生白细胞减少症风险的人对象施用有效量的重组人白蛋白-人粒细胞集落刺激因子以治疗对象。
在第三种实施方式中,描述了在患有非骨髓性恶性肿瘤并在接受至少一种骨髓抑制型抗癌药的人对象中降低表现为发热性嗜中性白细胞减少症的感染发病率的方法,该抗癌药物与发热性嗜中性白细胞减少症的临床显著发病率相关,该方法包括给予对象有效剂量的人白蛋白-人粒细胞集落刺激因子以治疗对象。
在一些方法中,本文所述的复合物有助于降低感染的发病率,例如表现为发热性嗜中性白细胞减少症的感染。在一些实施方式中,所述组合物和方法包括由人血清白蛋白(“HSA”)和人粒细胞集落刺激因子(“G-CSF”)形成的融合多肽。所述融合多肽长度为759个氨基酸;融合体中的1-585位氨基酸对应于成熟形式HSA的氨基酸,而融合体的586-759位氨基酸对应于成熟形式人G-CSF的氨基酸。图1显示了融合蛋白的氨基酸序列。将名为NeugraninTM(“NEUG”)的融合多肽给予显示有白细胞减少症或嗜中性白细胞减少症风险的患者。例如,在一些实施方式中,方法包括在人对象中通过给予有效剂量的重组人白蛋白-人粒细胞集落刺激因子治疗对象的白细胞减少症或嗜中性白细胞减少症。
在一些实施方式中,嗜中性白细胞减少症是原发性嗜中性白细胞减少症、急性嗜中性白细胞减少症、重度慢性嗜中性白细胞减少症(SCN)、重度先天性嗜中性白细胞减少症(Kostmann综合征)、重度婴儿遗传性粒细胞缺乏症、良性嗜中性白细胞减少症、周期性嗜中性白细胞减少症、慢性特发性嗜中性白细胞减少症、继发性嗜中性白细胞减少症、嗜中性白细胞减少症相关综合征或免疫介导的嗜中性白细胞减少症。
在其它实施方式中,嗜中性白细胞减少症由以下原因造成或与之相关:辐射、酗酒、药物、过敏性疾病、再生障碍性贫血、自体免疫疾病、T-γ淋巴增生性疾病(T-γLPD)、脊髓发育不良、骨髓纤维化、丙种球蛋白异常血症、阵发性夜间血红蛋白尿、癌症、维生素B12缺乏、叶酸缺乏、病毒感染、细菌感染、脾脏疾病、血液透析、移植、白血病、骨髓瘤、淋巴瘤、浸润并置换骨髓的转移性实体瘤、毒素、骨髓衰竭、Schwachman-Diamond综合征、软骨-毛发发育不全、先天性角化不良、IB型糖原贮积病、各种原因导致的脾肿大、和骨髓细胞或其前体的内在缺陷。在一些实施方式中,嗜中性白细胞减少症由细胞毒性化疗造成或与之相关。
在一些实施方式中,所述人对象患有非骨髓恶性肿瘤如乳腺癌,并在接受细胞毒性化疗。例如,在一些实施方式中,患者接受至少一种与发热性嗜中性白细胞减少症的临床显著发病率有关的骨髓抑制抗癌药。在一些实施方式中,所述骨髓抑制抗癌药是多柔比星或多西他赛。在进一步的实施方式中,对至少一个疗程,通过静脉输液在同一天依次给予约50mg/m2多柔比星和约75mg/m2多西他赛。在其它实施方式中,对至少一个疗程,通过静脉输液在同一天依次给予约60mg/m2多柔比星和约75mg/m2多西他赛。
在其它实施方式中,方法包括降低人对象感染的发病率,其表现为发热性嗜中性白细胞减少症。在一些实施方式中,所述人对象患有非骨髓恶性肿瘤并正接受至少一种骨髓抑制型抗癌药,该药物与发热性嗜中性白细胞减少症的临床显著发病率相关。在一些实施方式中,给予对象有效量的重组人白蛋白-人粒细胞集落刺激因子以治疗患者的嗜中性白细胞减少症。
在一些实施方式中,降低对象中嗜中性白细胞减少症的持续时间或严重程度或消除嗜中性白细胞减少症。例如,在一些实施方式中,消除对象的4级或3级嗜中性白细胞减少症。在其它实施方式中,缩短4级或3级嗜中性白细胞减少症的持续时间。例如,在一些实施方式中,对象的4级嗜中性白细胞减少症持续时间少于5天;在一些实施方式中,对象的4级嗜中性白细胞减少症持续时间少于4天、少于3天或少于2天。在其它实施方式中,对象的3级嗜中性白细胞减少症持续得到消除,和/或对象的3级嗜中性白细胞减少症持续时间比未接受人白蛋白-人粒细胞集落刺激因子治疗的对象缩短。
在一些实施方式中,给予重组人白蛋白-人粒细胞集落刺激因子引起对象的血液白细胞计数(“WBC”)增加或降低WBC损失。例如,在一些实施方式中,对象的嗜中性粒细胞数量升高;对象的嗜中性粒细胞数量降低受到抑制,对象的最低嗜中性粒细胞绝对计数(“ANC”)增加,对象的ANC恢复提高,和/或对象的ANC恢复时间缩短。
在一些实施方式中,给予对象的重组人白蛋白-人粒细胞集落刺激因子的量从约40μg/kg到约500μg/kg;在其它实施方式中,给予对象的重组人白蛋白-人粒细胞集落刺激因子的量从约50μg/kg到约450μg/kg。在另一些实施方式中,给予对象的重组人白蛋白-人粒细胞集落刺激因子的量为约50μg/kg、约100μg/kg、约150μg/kg、约200μg/kg或约250μg/kg。在进一步的实施方式中,给予对象的重组人白蛋白-人粒细胞集落刺激因子的量为约300μg/kg、约350μg/kg或约400μg/kg。在另外的实施方式中,给予对象的重组人白蛋白-人粒细胞集落刺激因子的量为约450μg/kg。在其它实施方式中,给予对象的重组人白蛋白-人粒细胞集落刺激因子的量为约20-100mg。在进一步实施方式中,给予对象的重组人白蛋白-人粒细胞集落刺激因子的量为约30-60mg。在进一步实施方式中,给予对象的重组人白蛋白-人粒细胞集落刺激因子的量为约30mg、约40mg、约50mg或约60mg。
在一些实施方式中,重组人白蛋白-人粒细胞集落刺激因子在化疗(例如,给予骨髓抑制抗癌药)后给药。例如,在一些实施方式中,重组人白蛋白-人粒细胞集落刺激因子在化疗期间给药、或在化疗施用2小时内、4小时内、6小时内、12小时内、18小时内、24小时内或48小时内给药。
在本发明的另一实施方式中,重组人白蛋白-人粒细胞集落刺激因子可在给予骨髓抑制抗癌药后给药。例如,重组人白蛋白-人粒细胞集落刺激因子的给药时间可选自下组:(a)给予骨髓抑制抗癌药后至少2小时;(b)给予骨髓抑制抗癌药后至少4小时;(c)给予骨髓抑制抗癌药后至少6小时;(d)给予骨髓抑制抗癌药后至少12小时;(e)给予骨髓抑制抗癌药后至少18小时;(f)给予骨髓抑制抗癌药后至少24小时;(g)给予骨髓抑制抗癌药后至少48小时;或(h)在骨髓抑制抗癌药给药期间或基本与之同时。
在一些实施方式中,在化疗治疗期间或之后给予的重组人白蛋白-人粒细胞集落刺激因子引起WBC升高和/或引起ANC升高。例如,在一些实施方式中,ANC和WBC在化疗后10天内回复正常。在其它实施方式中,ANC和WBC在化疗后11天、12天、13天、14天或15天内回复正常。在一些实施方式中,化疗施用后14天,用重组人白蛋白-人粒细胞集落刺激因子治疗的患者中ANC的升高低于用等剂量聚乙二醇化非格司亭治疗的患者中ANC的升高。
在一些实施方式中,给予重组人白蛋白-人粒细胞集落刺激因子引起淋巴细胞、单核细胞、嗜酸性粒细胞或嗜碱性粒细胞升高。例如,在一些实施方式中,对象中淋巴细胞、单核细胞、嗜酸性粒细胞或嗜碱性粒细胞的数量增加。在其它实施方式中,对象中淋巴细胞、单核细胞、嗜酸性粒细胞或嗜碱性粒细胞的数量减少被抑制。
在一些实施方式中,特别是对于治疗或预防嗜中性白细胞减少症的方法,所得结果可选自下组:(a)对象的4级嗜中性白细胞减少症被消除;(b)对象的4级嗜中性白细胞减少症得以减轻;(c)对象的重度嗜中性白细胞减少症持续时间缩短;(d)对象的3级嗜中性白细胞减少症的持续被消除;(e)对象的3级嗜中性白细胞减少症持续时间缩短;或(f)其任意组合。
在一些实施方式中,特别是对于治疗或预防嗜中性白细胞减少症的方法,给予重组人白蛋白-人粒细胞集落刺激因子引起血液白细胞计数(WBC)的升高。在另一些实施方式中,特别是对于治疗或预防嗜中性白细胞减少症的方法,所得结果选自下组:(a)对象的嗜中性粒细胞数量增加;(b)对象的嗜中性粒细胞数量减少被抑制;(c)对象的最低嗜中性粒细胞绝对计数(ANC)增加;(d)对象的ANC恢复提高;(e)对象的ANC恢复时间缩短;或(f)其任意组合。
在一些实施方式中,给予对象的重组人白蛋白-人粒细胞集落刺激因子的量选自下组:(a)从约50μg/kg至约450μg/kg;(b)约50μg/kg;(c)约150μg/kg;(d)约300μg/kg;(e)约450μg/kg;(f)从约30mg至约60mg;(g)约30mg;(h)约40mg;(i)约50mg;(j)约60mg;或(k)其任意组合。
在一些实施方式中,要治疗或预防的嗜中性白细胞减少症选自原发性嗜中性白细胞减少症、急性嗜中性白细胞减少症、重度慢性嗜中性白细胞减少症(SCN)、重度先天性嗜中性白细胞减少症(Kostmann综合征)、重度婴儿遗传性粒细胞缺乏症、良性嗜中性白细胞减少症、周期性嗜中性白细胞减少症、慢性特发性嗜中性白细胞减少症、继发性嗜中性白细胞减少症、嗜中性白细胞减少症相关综合征和免疫介导的嗜中性白细胞减少症。此外,嗜中性白细胞减少症可能由以下原因造成或与之相关,例如,辐射、酗酒、药物、过敏性疾病、再生障碍性贫血、自体免疫疾病、T-γ淋巴增生性疾病(T-γLPD)、脊髓发育不良、骨髓纤维化、丙种球蛋白异常血症、阵发性夜间血红蛋白尿、癌症、维生素B12缺乏、叶酸缺乏、病毒感染、细菌感染、脾脏疾病、血液透析、移植、白血病、骨髓瘤、淋巴瘤、浸润并置换骨髓的转移性实体瘤、毒素、骨髓衰竭、Schwachman-Diamond综合征、软骨-毛发发育不全、先天性角化不良、IB型糖原贮积病、各种原因导致的脾肿大、和骨髓细胞或其前体的内在缺陷。
在本发明的实施方式中,当人对象患有非骨髓恶性肿瘤时,所述非骨髓恶性肿瘤可包括乳腺癌。
在本发明的实施方式中,当给予骨髓抑制抗癌药时,所述骨髓抑制抗癌药可包括多柔比星和多西他赛。例如,对至少一个疗程,可通过静脉输液在同一天依次给予约50mg/m2多柔比星和约75mg/m2多西他赛。或者,对至少一个疗程,可通过静脉输液在同一天依次给予约60mg/m2多柔比星和约75mg/m2多西他赛。
在本发明的一些实施方式中,ANC和WBC在治疗后选自下组的时间段内回复正常:(a)化疗后10天;(b)化疗后11天;(c)化疗后12天;(d)化疗后13天;(e)化疗后14天;或(f)化疗后15天。在另一实施方式中,化疗施用后14天,用重组人白蛋白-人粒细胞集落刺激因子治疗的患者中ANC的升高低于用等剂量聚乙二醇化非格司亭治疗的患者中ANC的升高。
在本发明的一些实施方式中,给予重组人白蛋白-人粒细胞集落刺激因子引起淋巴细胞、单核细胞、嗜酸性粒细胞或嗜碱性粒细胞或其任意组合的升高。在其它实施方式中,对象中淋巴细胞、单核细胞、嗜酸性粒细胞、嗜碱性粒细胞或其任意组合的数量增加。在本发明的其它实施方式中,对象中淋巴细胞、单核细胞、嗜酸性粒细胞或嗜碱性粒细胞的数量减少被抑制。
上述总体说明和以下的附图简要说明与详细说明都是示范性和说明性的,并旨在为本发明要求保护的范围提供进一步说明。通过本发明的以下详细描述,本发明的其它目的、优势和新特征对于本领域技术人员将是显而易见的。
附图简要说明
图1A-1C:图1A显示名为“NeugraninTM”(“NEUG”)的所述重组人白蛋白-粒细胞集落刺激因子(“rHA-G-CSF”)融合蛋白的核酸序列和氨基酸序列;图1B显示人G-CSF的氨基酸序列;图1C显示人血清白蛋白的氨基酸序列。
图2显示I期对象的嗜中性粒细胞绝对计数(“ANC”)图。对象在研究化疗后的第1疗程中接受300μg/kg NEUG(n=19),450μg/kg NEUG(n=20)或6mg聚乙二醇化非格司亭(Neulasta )(n=9)。
)(n=9)。
图3显示I期研究中人对象内NEUG的药代动力学。在没有化疗的乳腺癌患病对象中,测量指定剂量NEUG(450μg/kg、300μg/kg或150μg/kg)皮下给药的血清NEUG浓度。正方形:450μg/kg疗程0;三角形:300μg/kg疗程0;圆形:150μg/kg疗程0。
图4显示NEUG在化疗第1疗程中的药代动力学/药效学(“PK/PD”)(I期研究)。在第1疗程给药多柔比星/多西他赛后1天,患者接受450μg/kgNEUG。ANC显示为空心菱形;NEUG浓度显示为实心正方形。3级和4级嗜中性白细胞减少症的临界值用虚线显示。NEUG的定量下限(“LLOQ”)显示为6ng/ml处的点线。
图5显示在化疗第1疗程开始后一天接受30mg NEUG或6mg聚乙二醇化非格司亭(Neulasta )的患者的ANC曲线(II期研究)。3级和4级嗜中性白细胞减少症的临界值用虚线显示。
)的患者的ANC曲线(II期研究)。3级和4级嗜中性白细胞减少症的临界值用虚线显示。
图6显示I期研究化疗疗程。
图7A和7B显示对象在I期研究中的ANC和血液白细胞计数(“WBC”)计数。
图8显示用NEUG(Albugranin)或Neupogen 的NSF-60细胞增殖图。
的NSF-60细胞增殖图。
图9显示用NEUG或Neulasta的NSF-60细胞增殖图。
图10显示BDF-1小鼠在NEUG(Albugranin)和Neupogen单次SC给药后的外周血嗜中性粒细胞(Gr.1+)水平图(时间过程)。Gr.1+细胞总数表示为组平均值+/-SEM。
图11显示在NEUG(Albugranin)和Neupogen单次SC给药后,外周血造血祖细胞(c-kit+)的水平图(时间过程)。c-kit+细胞总数表示为组平均值+/-SEM。
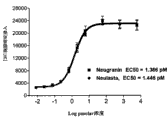
图12A和12B显示BDF-1小鼠中NEUG(Albugranin)或Neulasta 单次皮下(“SC”)给药后外周血粒细胞(Gr.1+)水平。12A显示单剂量Neulasta或NEUG后应答的时间过程,12B显示了NEUG或Neulasta的相对效力。
单次皮下(“SC”)给药后外周血粒细胞(Gr.1+)水平。12A显示单剂量Neulasta或NEUG后应答的时间过程,12B显示了NEUG或Neulasta的相对效力。
图13列表显示I期所用NEUG药物产品的组成。
图14列表显示II期所用NEUG药物产品的组成。
图15显示在NEUG(Albugranin)或Neulasta 单次皮下给药后,外周造血祖细胞(c-kit+)的水平(时间过程)。c-kit+的总数表示为各组计算所得的平均值和标准差。采用异方差t-检验分析治疗组间的差异。
单次皮下给药后,外周造血祖细胞(c-kit+)的水平(时间过程)。c-kit+的总数表示为各组计算所得的平均值和标准差。采用异方差t-检验分析治疗组间的差异。
图16显示在5-FU(150μg/kg)IP注射后1天,NEUG(Albugranin)或Neulasta单次皮下给药后的外周血嗜中性粒细胞(Gr.1+)的水平(时间过程)。每天计算GR.1+细胞的总数,表示为各组计算所得的平均值和标准差。用方差不等的双样本t-检验评估治疗组间的差异。与载剂对照相比,用任一种药剂在所有剂量水平下的治疗都引起嗜中性粒细胞数量在统计学上显著增加。
图17显示NEUG(Albugranin)对外周血嗜中性粒细胞相对百分比的影响。各研究日的嗜中性粒细胞的相对百分比表示为组平均值+/-SEM。NEUG 100μg/kg Q7的第8、9天的数据分别表示为第9、10天以便于和其它组作比较。对照为每四天一次共4次SC给予盐水或者每天一次共14次SC给予Neupogen 第1-14天视为治疗期,第15-28天为恢复期。
第1-14天视为治疗期,第15-28天为恢复期。
图18显示NEUG(Albugranin)SC、NEUG IV、或Neulasta SC重复剂量给药对猴的嗜中性粒细胞移动影响的比较。直至第22天的各研究日的嗜中性粒细胞数(K/μl)表示为组平均值+/-SEM。箭头标明剂量给药。NEUG以1.0mg/kg/剂量通过SC(n=6)或IV(n=6)给药,Neulasta以0.22mg/kg/剂量(1.0mg/kg NEUG的等摩尔剂量)通过SC(n=6)给药。NEUG载剂通过SC给药作为对照(n=2)。
SC重复剂量给药对猴的嗜中性粒细胞移动影响的比较。直至第22天的各研究日的嗜中性粒细胞数(K/μl)表示为组平均值+/-SEM。箭头标明剂量给药。NEUG以1.0mg/kg/剂量通过SC(n=6)或IV(n=6)给药,Neulasta以0.22mg/kg/剂量(1.0mg/kg NEUG的等摩尔剂量)通过SC(n=6)给药。NEUG载剂通过SC给药作为对照(n=2)。
图19A和19B列表显示体内药代动力学研究的小结。
图20列表显示提供安全性数据的体内非临床研究的小结。
图21是NEUG发酵和纯化的示范性概况的流程图。
图22显示I期A部分中(疗程0或化疗前)对象从治疗到第14天的嗜中性粒细胞绝对计数(ANC)的中位数。在第4天,图中线条由高到低分别是:300μg/kg、450μg/kg、150μg/kg和50μg/kg。
图23A和23B显示I期B部分中各治疗对象的曲线下面积(AUC),基于第0-15天的ANC数值。图23A为图表;图23A中的数据总结于表23B中。
图24显示II期治疗对象的曲线下面积(AUC),基于第1疗程第0-15天所得ANC值(固定剂量组)。对所有II期对象,计算AUCANC(第0-15天)。用Neugranin治疗的患者接受0.3-1mg/kg(根据剂量除以基线体重计算)的剂量。体重调整的剂量范围分成四组并对AUCANC作图(左图)。对所有用聚乙二醇化非格司亭治疗的对象(N=112),也计算并比较了Neugranin 30mg(N=10)、40mg(N=105)或50mg(N=105)的AUCANC。数据显示为平均值±SEM。
发明详述
本文公开了用于治疗、预防和减轻以白细胞计数减低为特征的症状和疾病的组合物和方法。本文所述的方法和组合物包括由人血清白蛋白(“HSA”)和人粒细胞集落刺激因子(“G-CSF”)形成的融合多肽。在一个优选实施方式中,所述融合多肽长度为约759个氨基酸;融合体中的1-585位氨基酸对应于成熟形式HSA的氨基酸,而融合体的586-759位氨基酸对应于成熟形式人G-CSF的氨基酸。图1显示了融合蛋白的氨基酸序列。
本发明还包括含G-CSF变体或片段的融合蛋白,和含白蛋白或白蛋白的片段或变体的融合蛋白。本发明还包括编码本发明的治疗性白蛋白融合蛋白的多核苷酸、治疗性白蛋白融合蛋白、组合物、药物组合物、制剂和试剂盒。本发明也包括转化有治疗性白蛋白融合蛋白的编码多核苷酸的宿主细胞,和采用这些多核苷酸和/或宿主细胞制备本发明的白蛋白融合蛋白的方法。
在一个实施方式中,本发明所述的白蛋白融合蛋白具有延长的保存期限。
在第二个实施方式中,本发明所述的白蛋白融合蛋白比相应未融合的G-CSF分子更稳定。
本发明还包括经修饰包含本发明所述核酸分子的转基因生物,优选修饰成表达本发明的白蛋白融合蛋白。
本发明一般涉及编码白蛋白融合蛋白的多核苷酸;白蛋白融合蛋白;和采用白蛋白融合蛋白或编码白蛋白融合蛋白的多核苷酸治疗、预防或改善疾病或失调。如本文所用,“白蛋白融合蛋白”指通过将至少一分子白蛋白(或其片段或变体)与至少一分子G-CSF(或其片段或变体)融合形成的蛋白质。本发明的白蛋白融合蛋白包含G-CSF的至少一个片段或变体和人血清白蛋白的至少一个片段或变体,两者通过基因融合(即,所述白蛋白融合蛋白通过核酸的翻译产生,该核酸中编码全部或部分G-CSF的多核苷酸与编码全部或部分白蛋白的多核苷酸在读框内连接)彼此结合。G-CSF和白蛋白一旦成为白蛋白融合蛋白的部分,就可各自称作白蛋白融合蛋白的“部分”、“区段”或“组成”(例如,“G-CSF蛋白部分”或“白蛋白蛋白部分”)。在一个高度优选的实施方式中,本发明的白蛋白融合蛋白包含至少一分子的G-CSF或其片段或变体(包括但不限于,G-CSF蛋白质的成熟形式)和至少一分子的白蛋白或其片段或变体(包括但不限于白蛋白的成熟形式)。
在进一步优选的实施方式中,本发明的白蛋白融合蛋白通过宿主细胞加工并分泌到周围的培养基中。在表达用宿主的分泌途径中发生的初生白蛋白融合蛋白的加工可包括但不限于,信号肽切割;二硫键形成;适当折叠;碳水化合物的添加和加工(例如,N-和O-连接糖基化);特异性蛋白水解切割;和装配成多聚体蛋白。本发明的白蛋白融合蛋白优选为已加工形式。在最优选的实施方式中,所述“白蛋白融合蛋白的加工形式”是指经过N末端信号肽切割的白蛋白融合蛋白产物,本文也称为“成熟的白蛋白融合蛋白”。
在一个实施方式中,本发明提供了编码白蛋白融合蛋白的多核苷酸,所述融合蛋白包含G-CSF和血清白蛋白或由其组成。在进一步实施方式中,本发明提供了包含G-CSF和血清白蛋白或由其组成的白蛋白融合蛋白。在其它实施方式中,本发明提供了包含G-CSF的生物活性和/或治疗活性片段和血清白蛋白,或由其组成的白蛋白融合蛋白。在其它实施方式中,本发明提供了包含G-CSF蛋白的生物活性和/或治疗活性变体和血清白蛋白,或由其组成的白蛋白融合蛋白。在优选的实施方式中,所述白蛋白融合蛋白的血清白蛋白组成是血清白蛋白的成熟部分。本发明还包括编码这些白蛋白融合蛋白的多核苷酸。
在进一步实施方式中,本发明提供的白蛋白融合蛋白包含G-CSF蛋白和血清白蛋白的生物活性和/或治疗活性片段,或由其组成。在进一步的实施方式中,本发明提供的白蛋白融合蛋白包含G-CSF蛋白和血清白蛋白的生物活性和/或治疗活性变体,或由其组成。在优选的实施方式中,所述白蛋白融合蛋白的G-CSF蛋白部分是G-CSF蛋白的成熟部分。在进一步优选的实施方式中,所述白蛋白融合蛋白的G-CSF蛋白部分是G-CSF蛋白的胞外可溶结构域。在替代的实施方式中,所述白蛋白融合蛋白的G-CSF蛋白部分是G-CSF蛋白的活性部分。本发明还包括编码这些白蛋白融合蛋白的多核苷酸。
在进一步实施方式中,本发明提供的白蛋白融合蛋白包含G-CSF蛋白的生物活性和/或治疗活性片段或变体和血清白蛋白的生物活性和/或治疗活性片段或变体,或由其组成。在优选的实施方式中,本发明提供的白蛋白融合蛋白包含G-CSF蛋白的成熟形式和血清白蛋白的成熟形式,或由其组成。本发明还包括编码这些白蛋白融合蛋白的多核苷酸。
I.定义
如本文所用,“多核苷酸”是具有编码融合蛋白的核苷酸序列的核酸分子,包含在读框内连接的至少一分子白蛋白(或其片段或变体)与至少一分子粒细胞集落刺激因子(G-CSF)(或其片段或变体),或由其组成。
如本文所用,“白蛋白融合构建物”指包含在读框内连接的编码至少一分子白蛋白(或其片段或变体)的多核苷酸与编码至少一分子G-CSF(或其片段或变体)的至少一个多核苷酸、或由其组成的核酸分子;以及还包含,例如一种或一种以上下列元件:(1)功能性自我复制载体(包括但不限于,穿梭载体、表达载体、整合载体和/或复制系统),(2)转录起始区(例如,启动子区如可调节或可诱导的启动子、组成型启动子),(3)转录终止区,(4)前导序列,和(5)选择性标记。编码G-CSF和白蛋白的多核苷酸一旦成为白蛋白融合构建物的部分,可各自称作白蛋白融合构建物的“部分”、“区域”或“组成”。
所述显示“治疗活性”的G-CSF多肽或具有“治疗活性”的G-CSF蛋白质是指具有一种或多种已知的G-CSF蛋白相关生物学和/或治疗活性的G-CSF多肽。作为非限制性示例,“G-CSF治疗蛋白”是可用于治疗、预防或改善疾病、症状或紊乱的G-CSF蛋白质。作为非限制性示例,“G-CSF治疗蛋白”可以与特定细胞类型(正常(如淋巴细胞)或非正常(如癌细胞))特异性结合并因此可用于将化合物(药物或细胞毒剂)特异性靶向到该细胞类型。
II.粒细胞集落刺激因子
粒细胞集落刺激因子(G-CSF)是刺激嗜中性粒细胞生成的造血生长因子。当有要刺激的存活前体细胞时,给予G-CSF可快速诱导嗜中性粒细胞白细胞增多。G-CSF的另一重要体内活性是移动造血祖细胞进入外周血液(Duhrsen等,1988,Molineux等,1999;Roberts等,1994)。该效应不但包括嗜中性粒细胞谱系,还扩展到其它单谱系与多谱系祖细胞和多能造血干细胞(Molineux等,1999)。G-CSF通过激发嗜中性粒细胞来增强作为抗感染的防御机制部分的细胞事件,从而增强其针对经调理金黄色葡萄球菌(Staphylococcus aureus)的吞噬和抗菌活性。G-CSF还引起嗜中性粒细胞与单核细胞的趋化性和嗜中性粒细胞的附着(You等,1989;Wang等,1988)。
目前已批准重组G-CSF产品用于多种临床症状以刺激嗜中性粒细胞的增殖和分化。在临床试验中,非格司亭(filgrastim,重组甲硫氨酰基人G-CSF;Neupogen Amgen,Thousand Oaks,CA)提高了外周嗜中性粒细胞数量,从而缩短了骨髓抑制化疗后嗜中性白细胞减少症的持续时间。重组G-CSF(非格司亭)通过每日皮下(SC)注射给药。
Amgen,Thousand Oaks,CA)提高了外周嗜中性粒细胞数量,从而缩短了骨髓抑制化疗后嗜中性白细胞减少症的持续时间。重组G-CSF(非格司亭)通过每日皮下(SC)注射给药。
G-CSF的另一重组形式是聚乙二醇化非格司亭,一种聚乙二醇偶联的rG-CSF(Neulasta ),用其每疗程一次来替代每日一次rG-CSF疗法以降低接受骨髓抑制行抗癌药患者的发热性嗜中性白细胞减少症发病率已被证明是安全有效的(Holmes,O’Shaughnessy等,2002;Green等,2003;NeulastaSmPC2007)。
),用其每疗程一次来替代每日一次rG-CSF疗法以降低接受骨髓抑制行抗癌药患者的发热性嗜中性白细胞减少症发病率已被证明是安全有效的(Holmes,O’Shaughnessy等,2002;Green等,2003;NeulastaSmPC2007)。
在基于年龄、医疗记录、疾病特征和化疗方案的骨髓毒性确定的高风险患者中,为预防发热性嗜中性白细胞减少症推荐用G-CSF进行初步预防。美国临床肿瘤学会(American Society of Clinical Oncology)和欧洲癌症研究和治疗组织(European Organization for Research and Treatment of Cancer)推荐当发热性嗜中性白细胞减少症风险达约20%时采用G-CSF。美国国家综合性癌症中心网络(U.S.National Comprehensive Cancer Center Network)推荐的G-CSF预防的可选指标为发热性嗜中性白细胞减少症风险为10-20%,G-CSF预防的确定性指标为发热性嗜中性白细胞减少症风险至少为20%。(Smith等,2006;Vogel等,2005;Timmer-Bonte等,2006,NCCN指南)。
推荐用集落刺激因子预防以缓解某些化疗方案的毒性。然而,这些治疗的附加成本在美国和特别是欧洲部分地区是重要考虑因素,可能引起预防性G-CSF治疗的应用不足,也可能限制患者进行强剂量化疗方案的资格(Timmer-Bonte等,2006;Adams等,2006,NCCN指南)。
G-CSF蛋白可以用野生型多核苷酸序列编码(例如,全长或成熟的),或在某些情况下所述序列可以是野生型多核苷酸序列的变体(例如编码野生型G-CSF蛋白的多核苷酸,其中该多核苷酸的DNA序列为了例如在特定种类中表达而优化;或者编码野生型G-CSF蛋白的变体的多核苷酸(即定点突变;等位基因变体))。
III.人血清白蛋白
人血清白蛋白(HSA或HA)是人循环系统中自然产生的最常见血液蛋白,测得为约40克白蛋白/升并在循环中保持逾20天。白蛋白是载体蛋白,在生理浓度下具有最小活性。尽管HSA缺乏酶或免疫功能,它在体内广泛分布,并已知是血液中各种物质的载体(例如,激素、脂肪酸、未偶联胆红素等(Yeh等,1992)。HSA和重组HA(rHA)在人体中都具有同样的长循环半衰期。
研究显示,与人白蛋白遗传融合的治疗蛋白能具有白蛋白的循环半衰期特征(Syed等,1997)。例如,在兔中,与白蛋白融合的CD4半衰期是未融合CD4的140倍(Yeh等,1992)。
人血清白蛋白负责血清渗透压的显著部分以及作为内源和外源配体载体起作用,其成熟形式是有585个氨基酸的蛋白质(如美国专利第7,592,010号中图1所示)。目前,临床使用的HA通过从人血中提取制得。在EP 330 451和EP 361 991中公开了微生物中重组HA(rHA)的生产。
IV.多肽和多核苷酸的片段和变体
A.片段
本发明还涉及本发明中G-CSF蛋白、白蛋白和/或白蛋白融合蛋白的片段。本发明还涉及本发明中编码G-CSF蛋白、白蛋白和/或白蛋白融合蛋白片段的多核苷酸。尽管从蛋白的N末端删除一个或多个氨基酸会引起本发明中G-CSF蛋白、白蛋白和/或白蛋白融合蛋白的一种或多种生物学功能的改变或丧失,其它治疗活性和/或功能活性(例如,生物活性、多聚体化的能力、结合配体的能力)仍可以保持。例如,当从N末端去除的残基少于完整多肽的主要残基时,N末端缺失多肽对识别所述多肽完整或成熟形式的抗体的诱导和/或结合能力一般仍被保留。通过本文所述的以及本领域已知晓的常规方法,可容易地确定缺少完整多肽的N末端残基的特定多肽是否保留该免疫活性。缺失大量N末端氨基酸残基的突变蛋白保留某些生物或免疫活性并非不可能。事实上,由少至6个氨基酸残基组成的肽常能引起免疫反应。
因此,对应于本发明白蛋白融合蛋白中G-CSF蛋白部分的G-CSF片段包括全长蛋白和从参比多肽(即,G-CSF蛋白、或由多核苷酸或白蛋白融合构建物编码的白蛋白融合蛋白的G-CSF蛋白部分)氨基酸序列的氨基末端缺失一个或多个残基的多肽。具体地,可以用通式m至q描述N末端缺失,其中q是表示参比多肽(例如,G-CSF蛋白、或本发明的白蛋白融合蛋白的G-CSF蛋白部分)的氨基酸残基总数的整数,m限定为从2到q减6范围内的任何整数。本发明也包括编码这些多肽的多核苷酸。
此外,对应于本发明白蛋白融合蛋白的白蛋白部分的血清白蛋白多肽片段包括全长蛋白和从参比多肽(即,血清白蛋白、或白蛋白融合蛋白的血清白蛋白部分)氨基酸序列的氨基末端缺失一个或多个残基的多肽。在优选的实施方式中,可以用通式m至585描述N末端缺失,其中585是成熟人血清白蛋白的氨基酸残基总数的整数,m限定为2到579范围内的任意整数。本发明也包括编码这些多肽的多核苷酸。在另外的实施方式中,可以用通式m至609描述N末端缺失,其中609是表示全长人血清白蛋白的氨基酸残基总数的整数,m限定为2到603范围内的任意整数。本发明也包括编码这些多肽的多核苷酸。
此外,本发明的白蛋白融合蛋白片段包括全长白蛋白融合蛋白和在所述白蛋白融合蛋白的氨基末端有一个或多个残基缺失的多肽。具体地,可以用通式m至q描述N末端缺失,其中q是表示白蛋白融合蛋白氨基酸残基总数的整数,m限定为2到q减6范围内的任意整数。本发明也包括编码这些多肽的多核苷酸。
如上所述,尽管从参比多肽(例如,G-CSF蛋白;血清白蛋白;或本发明的白蛋白融合蛋白)的N末端或C末端删除一个或多个氨基酸会引起该蛋白的一种或多种生物学功能的改变或丧失,但其它治疗活性和/或功能活性(例如,生物活性、多聚体化的能力、结合配体的能力)仍可以保持。例如,当从C末端去除的残基少于完整或成熟多肽的主要残基时,C末端缺失多肽对识别所述多肽完整或成熟形式的抗体的诱导和/或结合能力一般仍被保留。通过本文所述的和/或本领域已知晓的常规方法,可容易确定缺少参比多肽的N末端残基和/或C末端残基的特定多肽是否保留治疗活性。
本发明还提供了在对应于本发明白蛋白融合蛋白的G-CSF蛋白部分的G-CSF蛋白氨基酸序列的羧基末端有一个或多个残基缺失的多肽。具体地,可以用通式1至n描述C末端缺失,其中n是从6到q减1范围内的任何整数,而其中q是表示参比多肽(例如,G-CSF蛋白、或由多核苷酸或白蛋白融合构建物编码的白蛋白融合蛋白的G-CSF蛋白部分)的氨基酸残基总数的整数。本发明也包括编码这些多肽的多核苷酸。
此外,本发明提供了在对应于本发明的白蛋白融合蛋白中白蛋白部分的白蛋白氨基酸序列的羧基末端有一个或多个残基缺失的多肽。具体地,可用通式1至n描述C末端缺失,其中n是从6到584范围内的任意整数,其中584表示成熟人血清白蛋白的氨基酸残基总数减1的整数。本发明也包括编码这些多肽的多核苷酸。具体地,可用通式1至n描述C末端缺失,其中n是从6到608范围内的任意整数,其中608是表示人血清白蛋白的氨基酸残基总数减1的整数。本发明也包括编码这些多肽的多核苷酸。
此外,本发明提供了在本发明的白蛋白融合蛋白羧基末端有一个或一个以上残基缺失的多肽。具体地,可用通式1至n描述C末端缺失,其中n是从6到q减1范围内的任意整数,其中q是表示本发明白蛋白融合蛋白的氨基酸残基总数的整数。本发明也包括编码这些多肽的多核苷酸。
另外,上述的任何N或C末端缺失可以组合产生N和C末端缺失的参比多肽。本发明还提供了在氨基和羧基末端都有一个或一个以上氨基酸缺失的多肽,可通常表示为具有参比多肽(例如,G-CSF蛋白、或本发明白蛋白融合蛋白的G-CSF蛋白部分、或血清白蛋白、或本发明白蛋白融合蛋白的白蛋白部分、或白蛋白融合蛋白、或由本发明的多核苷酸或白蛋白融合构建物编码的白蛋白融合蛋白)的m至n残基,其中n和m为如上所述的整数。本发明也包括编码这些多肽的多核苷酸。
本申请还涉及所含多肽与本文提出的参比G-CSF多肽或参比白蛋白多肽或其片段有至少约80%、约85%、约90%、约91%、约92%、约93%、约94%、约95%、约96%、约97%、约98%或约99%相同的蛋白质。在优选的实施方式中,本申请涉及的蛋白质所含多肽与上述缺失氨基酸N和C末端的参比多肽至少有约80%、约85%、约90%、约91%、约92%、约93%、约94%、约95%、约96%、约97%、约98%或约99%相同。本发明也包括编码这些多肽的多核苷酸。
本发明的优选多肽片段包含显示G-CSF蛋白或血清白蛋白多肽序列的治疗活性和/或功能活性(例如生物活性)的氨基酸序列,或者由其组成,该氨基酸序列是片段。
其它优选的多肽片段是生物活性片段。生物活性片段是那些展示的活性与本发明多肽的活性相似,但未必相同的片段。片段的生物活性可包括所需活性的改善或不良活性的减弱。
B.变体
“变体”是指与参比核酸或多肽不同但保留其基本性质的多核苷酸或核酸。通常,变体总体上与参比核酸或多肽密切相似并在很多区段与之相同。
本文所用“变体”是指在序列上分别与G-CSF蛋白、白蛋白和/或白蛋白融合蛋白不同,但仍保留其如本文另述或本领域已知的至少一种功能和/或治疗性质的本发明的白蛋白融合蛋白的G-CSF蛋白部分、本发明的白蛋白融合蛋白的白蛋白部分、或本发明的白蛋白融合蛋白。通常,变体与白蛋白融合蛋白的G-CSF蛋白部分所对应的G-CSF蛋白、白蛋白融合蛋白的白蛋白部分所对应的白蛋白、和/或白蛋白融合蛋白的氨基酸序列在整体上非常相似,并在很多区段相同。本发明也包括编码这些变体的核酸。
本发明还涉及包含,与例如如本发明的白蛋白融合蛋白的G-CSF蛋白部分所对应的G-CSF蛋白、本发明的白蛋白融合蛋白的白蛋白部分所对应的白蛋白和/或白蛋白融合蛋白至少约80%、约85%、约90%、约91%、约92%、约93%、约94%、约95%、约96%、约97%、约98%、约99%或约100%相同的氨基酸序列,或由其组成的蛋白质。还提供了这些多肽的片段。本发明进一步包括的多肽是由在严谨杂交条件(例如,在6x氯化钠/柠檬酸钠(SSC)中以约45℃杂交从而过滤结合的DNA,然后在0.1x SSC,0.2%SDS中以约50-65℃清洗一次或多次)、在高度严谨条件(例如,在6x氯化钠/柠檬酸钠(SSC)中以约45℃杂交从而过滤结合的DNA,然后在0.1x SSC,0.2%SDS中以约68℃清洗一次或多次)或者在本领域技术人员已知的其它严谨杂交条件(参见,例如,Ausubel,F.M.等编著,1989,Current protocol in Molecular Biology(最新分子生物学实验手册),Green publishing associates,Inc.和John Wiley &Sons Inc.,New York,6.3.1-6.3.6和2.10.3各页)下与本发明白蛋白融合蛋白的编码核酸分子的补体杂交的多核苷酸编码的多肽。本发明也包括编码这些多肽的多核苷酸。
所述多肽的氨基酸序列与查询氨基酸序列至少,例如95%“相同”,意味着查询氨基酸序列的每100个氨基酸中对象多肽序列中可包括最多5个氨基酸变化,除此之外对象多肽的氨基酸序列与查询序列相同。换而言之,为得到氨基酸序列与查询氨基酸序列至少95%相同的多肽,对象序列中最多有5%的氨基酸残基可被插入、缺失或用其它氨基酸取代。参比序列的这些变化可以发生在参比氨基酸序列的氨基或羧基末端或这些末端位置之间的任何地方,可以散布于参比序列的各残基间,或者在参比序列内的一个或一个以上相邻基团中。
作为实践问题,可以采用已知的计算机程序来常规确定任何特定多肽是否与例如本发明的白蛋白融合蛋白的氨基酸序列或其片段(例如所述白蛋白融合蛋白的G-CSF蛋白部分或所述白蛋白融合蛋白的白蛋白部分)具有至少约80%、约85%、约90%、约91%、约92%、约93%、约94%、约95%、约96%、约97%、约98%或约99%相同。确定查询序列(本发明的某序列)与对象序列之间的最佳总体匹配的优选方法可以采用基于Brutlag等(Comp.App.Biosci.6:237-245(1990))的算法的FASTDB计算机程序,该方法也称为整体序列比对。在序列比对中,查询和对象序列或者都是核苷酸序列,或者都是氨基酸序列。整体序列比对的结果表示为相同百分数。FASTDB氨基酸比对中所用的优选参数为:矩阵(Matrix)=PAM 0,k-元组(k-tuple)=2,错配罚分(Mismatch Penalty)=1,连接罚分(Joining Penalty)=20,随机分组长度(Randomization GroupLength)=0,临界分值(CutoffScore)=1,窗口尺寸(Window Size)=序列长度,缺口罚分(Gap Penalty)=5,缺口尺寸罚分(Gap Size Penalty)=0.05,窗口尺寸=取500或对象氨基酸序列长度中较短者。
若对象序列比查询序列短是由于N或C末端缺失而非内部缺失,必须对结果进行人工校正。这是因为FASTDB程序在计算整体相同百分数时并不考虑对象序列的N和C末端截断。对于相对查询序列在N和C末端有截断的对象序列,通过计算查询序列中在对象序列N和C末端而不与相应的对象残基匹配/对齐的残基数占查询序列总碱基数的百分比来校正相同百分数。通过FASTDB序列比对的结果确定残基是否匹配/对齐。然后通过上述FASTDB程序采用特定参数计算所得相同百分数中减去该百分值,获得最终的相同百分数得分。该最终相同百分数得分用于本发明的目的。为了人工调整相同百分数得分,仅考虑对象序列N和C末端未与查询序列匹配/对齐的残基。即,只查询对象序列N和C末端残基最远端外侧的残基位置。
例如,90个氨基酸残基的对象序列与100个残基的查询序列比对以确定相同百分数。缺失发生在对象序列的N末端,因此FASTDB比对并未显示N末端最初10个残基的匹配/对齐。这10个未配对残基代表序列的10%(N和C末端未匹配残基数/查询序列的总残基数),因此从FASTDB程序计算得到的相同百分数得分中减去10%。若其余90个残基为理想匹配,那么最终的相同百分数将是90%。在另一示例中,比较90个残基的对象序列与100个残基的查询序列。这次缺失为内部缺失,因此对象序列的N或C末端没有与查询不匹配/对齐的残基。该情况下,FASTDB计算所得相同百分数不用人工校正。再次,只对在FASTDB比对中显示为对象序列N和C末端外侧,未与查询序列匹配/对齐的残基位置进行人工校正。没有为本发明目的进行其它人工校正。
变体通常和与变体具有相同长度的正常HA或G-CSF蛋白长度具有至少约75%(在其它实施方式中,至少约80%、约85%、约90%、约91%、约92%、约93%、约94%、约95%、约96%、约97%、约98%或约99%)的序列相同性。通过BLAST(Basic Local Alignment Search Tool(基础本地比对搜索工具))分析,采用为序列相似性搜索调整过的blastp、blastn、blastx、tblastn和tblastx所用算法(Karlin等,Proc.Natl.Acad.Sci.USA 87:2264-2268(1990)和Altschul,J.Mol.Evol.36:290-300(1993),通过引用全文纳入)确定核苷酸或氨基酸序列水平的同源性或相同性。
BLAST程序所用方法首先考虑查询序列和数据库序列之间的相似区段,然后评估所有一致匹配的统计学显著性,最后仅总结出那些满足预选的显著性阈值的匹配者。关于序列数据库相似性搜索的基本问题的讨论,参见Altschul等(Nature Genetics 6:119-129(1994)),其全文通过引用纳入本文。直方图、描述、对齐、预期(即,针对数据库序列报告匹配者的统计学显著性)、临界值、矩阵和筛选的搜索参数采用默认设置。Blastp、blastx、tblastn和tblastx采用的默认计分矩阵是BLOSUM62矩阵(Henikoff等,Proc.Natl.Acad.Sci.USA89:10915-10919(1992),通过引用全文纳入)。对于blastn,计分矩阵用M(即,匹配残基配对的奖励计分)与N(即,错配残基的罚分)的比值设定,其中M和N的默认值分别为5和-4。四项blastn参数可作如下调整:Q-10(缺口产生罚分);R=10(缺口延伸罚分);wink=1(在沿查询序列的各wink.sup.th位置产生字命中);以及gapw=16(设定产生带缺口对齐的窗口宽度)。Blastp的相应参数设定为Q=9;R=2;wink=1和gapw=32。在GCG包1.0版中可在序列间进行Bestfit比较,采用DNA参数GAP=50(缺口产生罚分)和LEN=3(缺口延伸罚分),蛋白质比较中的相应设定为GAP=8和LEN=2。
本发明的多核苷酸变体可含有在编码区、非编码区或两者内的变化。特别优选的多核苷酸变体包含产生沉默取代、添加或缺失的变化,但不改变所编码多肽的性质或活性。优选因遗传密码简并性导致的沉默取代所产生的核苷酸变体。此外,也优选多肽变体中低于50个、低于40个、低于30个、低于20个、低于10个、或5-50个、5-25个、5-10个、1-5个或1-2个氨基酸被以任意组合取代、缺失或添加。可以为多种原因产生多核苷酸变体,例如为优化特定宿主中密码子的表达(将人mRNA中的密码子变成细菌宿主如酵母或大肠杆菌中优选的密码子)。
在一个优选的实施方式中,本发明中编码白蛋白融合蛋白的白蛋白部分的多核苷酸优化以在酵母或哺乳动物细胞中表达。在进一步优选的实施方式中,本发明编码白蛋白融合蛋白的G-CSF蛋白部分的多核苷酸优化以在酵母或哺乳动物细胞中表达。在更进一步优选的实施方式中,本发明中编码白蛋白融合蛋白的多核苷酸优化以在酵母或哺乳动物细胞中表达。
在另一实施方式中,在本文所述严谨杂交条件下,编码白蛋白融合蛋白G-CSF蛋白部分的密码子优化多核苷酸不和编码G-CSF蛋白的野生型多核苷酸杂交。在进一步的实施方式中,在本文所述严谨杂交条件下,编码白蛋白融合蛋白的白蛋白部分的密码子优化多核苷酸不和编码白蛋白的野生型多核苷酸杂交。在另一个实施方式中,在本文所述严谨杂交条件下,编码白蛋白融合蛋白的密码子优化多核苷酸不和编码G-CSF蛋白部分或白蛋白部分的野生型多核苷酸杂交。
在另外的实施方式中,编码白蛋白融合蛋白的G-CSF蛋白部分的多核苷酸不包含该G-CSF蛋白的天然产生序列,或不由其组成。在进一步的实施方式中,编码白蛋白融合蛋白的白蛋白部分的多核苷酸不包含该白蛋白的天然产生序列,或不由其组成。在另外的实施方式中,编码白蛋白融合蛋白的多核苷酸不包含G-CSF蛋白部分或白蛋白部分的天然产生序列,或不由其组成。
采用已知的蛋白质工程和重组DNA技术的方法,可以产生变体以改善或改变本发明多肽的特性。例如,可以从本发明多肽的N或C末端删除一个或多个氨基酸而不显著损失生物学功能。
在优选的实施方式中,本发明的变体具有保守取代。“保守取代”是指在组内交换,例如脂族或疏水氨基酸Ala、Val、Leu和Ile的取代;羟基残基Ser和Tar的取代;酸性残基Asp和Glu的取代;酰胺残基Asn和Gln的取代;碱性残基Lys、Arg和His的取代;芳香族残基Phe、Tyr和Trp的取代;以及小尺寸氨基酸Ala、Ser、Thr、Met和Gly的取代。
提供了关于如何制备表型沉默的氨基酸取代的指南,例如,Bowie等,“Deciphering the Message in Protein Sequences:Tolerance to Amino AcidSubstitutions(解码蛋白质序列的信息:对氨基酸取代的容差)”,Science247:1306-1310(1990),其中作者表明有两种主要策略研究氨基酸序列对变化的容差。
第一种策略通过进化过程中的自然选择探索氨基酸取代的容差。通过比较不同物种间的氨基酸序列鉴别保守氨基酸。这些保守氨基酸可能对于蛋白质功能很重要。相反,其取代为自然选择容忍的氨基酸位置表明这些位置对于蛋白质功能并非关键。因此,容忍氨基酸取代的位置可被改变而仍保持所述蛋白质的生物活性。
第二种策略采用遗传工程在克隆基因的特定位置引入氨基酸变化以鉴别蛋白质功能的关键性区段。例如,可采用定点突变或丙氨酸扫描突变(在分子的各残基处引入单个丙氨酸突变)。参见Cunningham和Wells,Science244:1081-1085(1989)。然后,可测试所得突变分子的生物学活性。
如作者所指出,这两种策略揭示了蛋白质对氨基酸取代的惊人容差。作者进一步指出,在蛋白质的某些氨基酸位置哪些氨基酸变化是可能允许的。例如,大多数被包埋(在蛋白质三级结构中)的氨基酸残基要求非极性侧链,而通常表面侧链的特性极少是保守的。此外,可容忍的保守氨基酸取代涉及脂族或疏水氨基酸Ala、Val、Leu和Ile的取代;羟基残基Ser和Thr的取代;酸性残基Asp和Glu的取代;酰胺残基Asn和Gln的取代;碱性残基Lys、Arg和His的取代;芳香族残基Phe、Tyr和Trp的取代;以及小尺寸氨基酸Ala、Ser、Thr、Met和Gly的取代。除了保守氨基酸取代,本发明的变体包括(i)含一个或一个以上非保守氨基酸残基取代的多肽,其中被取代的氨基酸残基可以由或不由该遗传密码编码,或者(ii)含有一个或一个以上具有取代基团的氨基酸残基取代的多肽,或(iii)与另一化合物,例如提高多肽稳定性和/或溶解度的化合物(如聚乙二醇)融合或化学偶联的多肽,(iv)包含额外氨基酸,例如IgG Fc融合区肽的多肽。本领域技术人员在本文教导下认为这些多肽变体是在本文范围内的。
例如,包含带电氨基酸被其它带电或中性氨基酸取代的多肽变体可产生特性改善的蛋白质,例如较少聚集。药物制剂的聚集减低活性并因聚集体的免疫原活性提高了清除。参见Pinckard等,Clin.Exp.Immunol.2:331-340(1967);Robbins等,Diabetes 36:838-845(1987);Cleland等,Crit.Rev.Therapeutic DrugCarrier Systems 10:307-377(1993)。
在特定的实施方式中,本发明的多肽包含白蛋白融合蛋白氨基酸序列、G-CSF蛋白和/或人血清白蛋白氨基酸序列的片段或变体,或由其组成,其中所述片段或变体与参比氨基酸序列相比,具有1-5、5-10、5-25、5-50、10-50或50-150个氨基酸残基的添加、取代和/或缺失。在优选的实施方式中,所述氨基酸取代是保守的。本发明也包括编码这些多肽的核酸。
本发明的多肽可以由通过肽键或修饰的肽键,即肽等排物彼此连接的氨基酸组成,并可包含20种基因编码的氨基酸以外的氨基酸。所述多肽可以通过天然过程修饰,例如翻译后加工,或者通过本领域熟知的化学修饰技术修饰。这些修饰在基础文章和更具体的专著以及大量研究文献中已详尽描述。修饰可发生在多肽的任何地方,包括肽主链、氨基酸侧链和氨基或羧基末端。应当了解相同类型的修饰可以在给定多肽的多个位置以相同或不同程度出现。并且,给定多肽也可包含许多类型的修饰。多肽可以是分支的,例如因泛素化造成,它们可以是含有或不含分支的环状。环状、分支和分支环状多肽可以由翻译后的自然过程造成也可通过合成方法产生。修饰包括乙酰化、酰基化、ADP-核糖基化、酰胺化、共价结合黄素、共价结合血红素部分、共价结合核苷酸或核苷酸衍生物、共价结合脂质或脂质衍生物、共价结合磷脂酰肌醇、交联、环化、二硫键形成、去甲基化、形成共价交联、形成半胱氨酸、形成焦谷氨酸、甲酰化、γ-羧化、糖基化、GPI锚体形成、羟基化、碘化、甲基化、十四烷基化、氧化、聚乙二醇化、蛋白水解加工、磷酸化、异戊二烯化、外消旋化、硒化、硫化、转移RNA介导的蛋白质氨基酸加成如精氨酰化、以及泛素化。(参见,例如PROTEINS--STRUCTURE AND MOLECULAR PROPERTIES(蛋白质结合和分子性质),第2版,T.E.Creighton,W.H.Freeman and Company,New York(1993);POST-TRANSLATIONAL COVALENT MODIFICATION OFPROTEINS(蛋白质翻译后的共价修饰),B.C.Johnson编著,Academic Press,New York,第1-12页(1983);Seifter等,Meth.Enzymol.182:626-646(1990);Rattan等,Ann.N.Y.Acad.Sci.663:4862(1992))。
C.功能活性
“具功能活性的多肽”是指能显示与G-CSF蛋白的全长、前蛋白、和/或成熟形式相关的一种或一种以上已知功能活性的多肽。这些功能活性包括但不限于,生物学活性、抗原性[与抗多肽抗体结合(或与多肽竞争结合)的能力]、免疫原性(产生结合本发明特定多肽的抗体的能力)、与本发明的多肽形成多聚体的能力、和与多肽的受体或配体结合的能力。
“具生物学活性的多肽”是指如特定生物学实验所检测,剂量依赖或不依赖性地显示与本发明的G-CSF蛋白包括其成熟形式相似,但未必相同活性的多肽。
在优选的实施方式中,本发明的白蛋白融合蛋白具有未与白蛋白融合的G-CSF蛋白部分(或其片段或变体)相关的至少一种生物学和/或治疗活性。
可以采用本领域已知试验或对它们作常规改良以及本文所述试验以试验其本发明的白蛋白融合蛋白的功能活性(例如,生物学活性)。另外,本领域技术人员可以对白蛋白融合蛋白的G-CSF蛋白部分所对应的G-CSF蛋白片段进行常规试验。此外,本领域技术人员可以采用本领域已知试验和/或下文实施例部分所述试验对白蛋白融合蛋白的白蛋白部分所对应的白蛋白片段进行常规试验分析活性。
例如,在一个实施方式中,试验白蛋白融合蛋白结合或与G-CSF蛋白竞争结合抗G-CSF多肽抗体和/或抗白蛋白抗体的能力,可以采用本领域已知的各种免疫试验,包括但不限于,使用例如放射免疫试验、ELISA(酶联免疫吸附测定)、“夹心”免疫试验、免疫放射试验、凝胶扩散沉淀反应、免疫扩散试验、原位免疫试验(例如,采用胶体金、酶或放射性同位素标记)、Western印迹、沉淀反应、聚集试验(例如凝胶聚集试验、血凝试验)、补体固定试验、免疫荧光试验、蛋白质A试验和免疫电泳试验等技术的竞争性和非竞争性试验系统。在一种实施方式中,通过检测初次抗体上的标记测得抗体的结合。在另一个实施方式中,通过检测二次抗体或试剂与初次抗体的结合测得初次抗体。在另外一实施方式中,标记所述二次抗体。本领域已知并在本发明范围内有多种方法用于在免疫试验中检测结合。
在一个优选实施方式中,识别了G-CSF蛋白质的结合伴侣(例如,受体或配体),例如,可以通过本领域熟知的方法如还原和非还原性凝胶层析、蛋白亲和层析及亲和印迹法,分析白蛋白融合蛋白与该结合伴侣的结合,所述融合蛋白包含G-CSF蛋白作为融合体的G-CSF蛋白部分。一般参见Phizicky等,Microbiol.Rev.59:94-123(1995)。在另一实施方式中,可采用本领域已知技术常规分析白蛋白融合蛋白与融合体G-CSF蛋白部分所对应G-CSF多肽的受体结合的生理关联能力。
在另一个实施方式中,评估白蛋白融合蛋白形成多聚体的能力,可以通过本领域熟知的方法,例如还原和非还原性凝胶层析、蛋白亲和层析及亲和印迹分析多聚体与其它组分的结合。通常参见Phizicky等,同上。
可用于分析蛋白结合和交叉反应性或鉴定的免疫试验包括但不限于:使用诸如Western印迹、放射性免疫实验、ELISA(酶联免疫吸附测定)、“夹心”免疫试验、免疫沉淀试验、沉淀素反应、凝胶扩散沉淀素反应、免疫扩散试验、凝集试验、补体固定实验、免疫放射分析、荧光免疫分析、蛋白A免疫试验等技术的竞争性和非竞争性测定系统。这类实验是本领域熟知的常规实验(参见例如,Ausubel等编,1994,Current Protocols in Molecular Biology(新编分子生物学实验),第1卷,John Wiley & Sons,Inc.,New York,通过引用全文纳入本文)。
与白蛋白融合蛋白的G-CSF蛋白部分所对应的G-CSF蛋白结合的抗体也可就其对给定蛋白或抗原的结合亲和力加以描述或限定,优选其特异性结合的抗原。优选的结合亲和力包括解离常数或Kd低于5x10-2M、10-2M、5x10-3M、10-3M、5x10-4M、10-4M。更优选的结合亲和力包括解离常数或Kd低于5x10-5M、10-5M、5x10-6M、10-6M、5x10-7M、10-7M,5x10-8M或10-8M。更优选的结合亲和力包括解离常数或Kd低于5x10-9M、10-9M、5x10-10M、10-10M、5x10-11M、10-11M、5x10-12M、10-12M、5x10-13M、10-13M、5x10-14M、10-14M、5x10-15M、或10-15M。此外,本文描述的试验和本领域已知的试验可以常规应用于检测白蛋白融合蛋白及其片段、变体、衍生物引起所述白蛋白融合蛋白的G-CSF蛋白部分和/或白蛋白部分相关的生物学活性和/或G-CSF活性(体内或体外)的能力。其它方法会是本领域技术人员已知的,并在本发明范围内。
V.G-CSF和HSA的融合蛋白
重组人白蛋白-人粒细胞集落刺激因子(rHA-G-CSF)是G-CSF类似物。rHA-G-CSF的例子见美国专利第5,665,863号以及美国专利第7,041,478号的描述,两者都通过引用纳入本文。
rHA-G-CSF的另一例子是美国特华生物药物有限公司(TevaBiopharmaceuticals USA LTD.)开发的NeugraninTM(“NEUG”)。NEUG是分子量约为85kDa的融合多肽。NEUG是759个氨基酸的单链多肽,1-585位残基对应于HSA的成熟形式,586-759位残基对应于人G-CSF的成熟形式。图1显示了NEUG融合蛋白的氨基酸序列。
VI.产生融合蛋白
生产rHA-G-CSF的合成过程的示范性方法见美国专利申请第11/929,828号的描述,其全文通过引用纳入本文。在一些实施方式中,采用遗传工程改造的表达NEUG融合蛋白的酵母(例如酿酒酵母(Saccharomyces cerevisiae))宿主系统生成NEUG。采用本领域熟知的方法(例如通过一系列层析和过滤步骤,如亲和层析及离子交换层析)从酵母培养的发酵培养基中收集并纯化NEUG。
在一种非限制性的示例中,如下开发NEUG融合构建物。从英国牛津大学F.E.Baralle博士实验室的人cDNA文库中分离出全长白蛋白cDNA。该克隆作为质粒pAT153ALB送往英国诺丁汉的Delta Biotechnology Limited。此外,修饰6-氨基酸HSA前肽(RGVFRR)以促进酵母中的更有效加工(RSLDKR)。
NEUG的生产质粒,基于pSAC35改良的表达质粒,是基于野生型酿酒酵母中发现的2-μ质粒。基于pSAC35的表达载体(参见例如,专利EP 286424B、美国专利第5,637,504号)含有来自酿酒酵母的LEU2基因作为选择标记,该基因补充酿酒酵母生产宿主中的亮氨酸缺陷。该生产质粒还包含强酵母启动子PRB1,和来自质粒pUC9的可在大肠杆菌中克隆和增殖的序列。此外,一旦转化到酵母中,所述质粒消除在大肠杆菌中增殖所需的pUC9衍生序列。这通过侧翼FLP识别靶标(FRT)和进入酵母后质粒表达酵母FLP重组酶而实现。因此,用于生产NEUG的生物体中没有细菌DNA。这通过在产生主细胞库后从酵母中取出2μm质粒并测序得到证实。
如上所述,名为CID1643(pSAC35:HSA.GCSF(T31-P204))的NEUG生产质粒是从基于pSAC35的表达载体衍生得到。通过PCR扩增与人G-CSFT31-P204对应的区段,添加适当的5’和3’限制性位点以允许与HSA开放读框3’末端的无缝融合。
在马里兰州罗克维尔(Rockville,MD)的Human Genome Sciences公司用NEUG种子瓶制备cGMP主细胞库。NEUG主细胞库的测试和鉴定在美国宾夕法尼亚州马尔文的Charles River Laboratories(Malvern,PA,USA)和美国得克萨斯州休斯敦的Lark Technologies(Houston,TX,USA),遵照ICH指南Q5D(Derivation and Characterization of Cell Substrates Used for Production ofBiotechnological/Biologicals Products(用于生产生物技术/生物产品的细胞基质的产生和鉴定))进行。
随后在Charles River Laboratories产生并检测从该主细胞库衍生的cGMP工作细胞库。
生产NEUG细胞系库的所有培养基成分都是合成、生物合成或植物来源。在细胞系或细胞库制备过程中没有使用动物来源或人源的成分。
所述细胞库在<-135℃下保存在预灭菌的1.8mL带内螺口封盖Nunc聚丙烯管内的冻存培养基中。
图21显示了分离、纯化和制备药用rHSA-G-CSF融合蛋白的非限制性示范方法。用0.2μm过滤器将配制的药物物质无菌过滤入已灭菌的特富龙(Teflon)瓶中。装有药物物质的液体在约-80℃(标称值,保存温度的可接受范围为约-65℃)下冷冻保存.
为改善制剂在运输和临床地点存放的稳健性,并提供具有预期的长保存期的稳定产品,还可采用本领域已知方法冻干NEUG。
VII:白细胞减少症和嗜中性白细胞减少症的示范性原因
如上所述,白细胞减少症是循环血液白细胞计数(WBC)的减少,嗜中性白细胞减少症以血嗜中性粒细胞计数的减少为特征,常导致对细菌和真菌感染的易感性提高。下列是能使人对象面临引起白细胞减少症或嗜中性白细胞减少症风险的因素的不全面列举:药物(例如,苯妥英、氯霉素、磺胺药物和化疗);维生素B12或叶酸缺乏;饮酒过量;涉及骨髓的癌症和其它疾病(例如,再生障碍性贫血、丙种球蛋白异常血症、阵发性夜间血红蛋白尿、脊髓发育不良、脊髓发育不良综合征、骨髓纤维化、白血病、骨髓瘤、淋巴瘤或浸润并置换骨髓的转移性实体肿瘤);病毒感染(例如,流感、HIV、早期传染性单核细胞增多症、儿童病毒疾病);细菌感染(例如,结核);辐射;毒素(例如,苯和杀虫剂);骨髓衰竭(例如,Schwachman-Diamond综合征、软骨毛发发育不良、先天性角化不良、IB型糖原贮积病);脾脏紊乱、各种原因导致的脾肿大;骨髓细胞或其前体的内在缺陷;过敏性疾病;自体免疫疾病;T-γ淋巴增生性疾病(T-γLPD);血液透析或移植;毒素。
大量药物,例如多种化疗方案(如细胞毒性化疗方案)与发热性嗜中性白细胞减少症的高风险(如风险>20%)。在一些化疗方案中,没有G-CSF治疗时的发热性嗜中性白细胞减少症发病率约为40%(例如静脉内用多柔比星和多西他赛的化疗方案)。下表1提供了与发热性嗜中性白细胞减少症相关的各种癌症和治疗方案的非限制性例子。在一些实施方式中,图1所示的HSA-G-CSF融合蛋白给予患者以预防、治疗或减轻与这些药物治疗施用相关的嗜中性白细胞减少症。

小细胞肺癌的细胞毒性治疗方案,如顺铂加依托泊苷以及CAE,也与发热性嗜中性白细胞减少症有关。
已知多种嗜中性白细胞减少症,以及在一些实施方式中用图1的HSA-G-CSF融合蛋白预防、治疗或减轻一种或多种嗜中性白细胞减少症,包括但不限于,化疗引起的嗜中性白细胞减少症、原发性嗜中性白细胞减少症、急性嗜中性白细胞减少症、重度慢性嗜中性白细胞减少症(SCN)、重度先天性嗜中性白细胞减少症(Kostmann综合征)、重度婴儿遗传性粒细胞缺乏症、良性嗜中性白细胞减少症、周期性嗜中性白细胞减少症、慢性特发性嗜中性白细胞减少症、继发性嗜中性白细胞减少症、嗜中性白细胞减少症相关综合征或免疫介导的嗜中性白细胞减少症。
VIII.实验实施例
提供下列实施例以阐述本发明。但应当了解,本发明不限于这些实施例所述的特定条件和细节。本文参考的所有公开可得的文献,包括但不限于美国专利,都通过引用纳入本文。
在下列非限制性实施例中,NeugraninTM(“NEUG”)在细胞上以及小鼠和猴中进行了测试,还用于预防、治疗或减轻人对象中因乳腺癌药物治疗(如化疗)引起的嗜中性白细胞减少症和/或白细胞减少症。
NEUG是G-CSF类似物,因为活性G-CSF部分与人血清白蛋白的融合而降低了血浆清除速率。所得完全重组的蛋白质保留了G-CSF在体内的药理学活性,即它刺激嗜中性粒细胞和造血干细胞从骨髓向外周血流移动。在小鼠和猴中评估了该蛋白的活性(见下文实验实施例)。在单次给药后,观察到的嗜中性粒细胞和造血祖细胞计数增加迅速开始并持续数天。该效应为剂量依赖性,较高剂量比较低剂量引起的NEUG响应幅度更大且持续更久。NEUG的清除比Neupogen (非格司亭)慢。单次或重复给药NEUG后血液嗜中性粒细胞的动力学与用Neulasta
(非格司亭)慢。单次或重复给药NEUG后血液嗜中性粒细胞的动力学与用Neulasta (聚乙二醇化非格司亭)治疗的动物中所观察到的几乎相同。由于NEUG是Neulasta(聚乙二醇化非格司亭)4.5倍(质量),可使用更大剂量(重量)但等摩尔剂量的NEUG以获得相似的体内效应。在猴中,等摩尔剂量的NEUG与聚乙二醇化非格司亭等效,而在小鼠中1.5倍剂量的NEUG显示能获得等效的AUCANC。这些研究中,1mg Neulasta
(聚乙二醇化非格司亭)治疗的动物中所观察到的几乎相同。由于NEUG是Neulasta(聚乙二醇化非格司亭)4.5倍(质量),可使用更大剂量(重量)但等摩尔剂量的NEUG以获得相似的体内效应。在猴中,等摩尔剂量的NEUG与聚乙二醇化非格司亭等效,而在小鼠中1.5倍剂量的NEUG显示能获得等效的AUCANC。这些研究中,1mg Neulasta (聚乙二醇化非格司亭)的效果相当于4.5-7.7mg NEUG。对于上下文,NEUG在猴中显示的无不良作用水平(“NOAEL”)大于1毫克/公斤/周。在猴中1mg/kg的剂量引起的接触(Cmax和AUC)约~12倍于接受0.45mg/kg NEUG的人患者中所观察到的。因此,猴所显示的NOAEL高于NEUG临床评估所用剂量范围(见下文)。
(聚乙二醇化非格司亭)的效果相当于4.5-7.7mg NEUG。对于上下文,NEUG在猴中显示的无不良作用水平(“NOAEL”)大于1毫克/公斤/周。在猴中1mg/kg的剂量引起的接触(Cmax和AUC)约~12倍于接受0.45mg/kg NEUG的人患者中所观察到的。因此,猴所显示的NOAEL高于NEUG临床评估所用剂量范围(见下文)。
总体上,这些研究显示等摩尔剂量的NEUG提供了与Neulasta (聚乙二醇化非格司亭)相似的药理学效应,并在人体中对粒细胞群体有相似作用。
(聚乙二醇化非格司亭)相似的药理学效应,并在人体中对粒细胞群体有相似作用。
A.NEUG的体外和体内研究
下面小结了NEUG体外和体内研究的结果,并在下文的实验实施例中详细描述。
体外药理学研究显示下列结果:
1.NEUG以剂量依赖性方式引起NFS-60细胞增殖。
2.NEUG在体外的效力比Neupogen (非格司亭)低3倍。
(非格司亭)低3倍。
3.以摩尔计,NEUG与Neulasta (聚乙二醇化非格司亭)等效(1mgNeulasta(聚乙二醇化非格司亭)与4.5mg NEUG等效)。
(聚乙二醇化非格司亭)等效(1mgNeulasta(聚乙二醇化非格司亭)与4.5mg NEUG等效)。
体内药理学研究显示NEUG的下列性质:
1.NEUG在小鼠和食蟹猴中耐受良好。
2.在小鼠中,NEUG单次给药引起外周血液中的嗜中性粒细胞和造血祖细胞出现剂量依赖性、快速且持续的增加。与现有市售G-CSF产品相比,嗜中性粒细胞和祖细胞计数的升高在持续时间上比等摩尔剂量的Neupogen (非格司亭)更长,在持续时间和幅度上与等摩尔剂量的Neulasta
(非格司亭)更长,在持续时间和幅度上与等摩尔剂量的Neulasta (聚乙二醇化非格司亭)相似。
(聚乙二醇化非格司亭)相似。
3.在小鼠中,毫克剂量的NEUG与Neulasta (聚乙二醇化非格司亭)等效,且所得AUCANC是后者的7.7倍。
(聚乙二醇化非格司亭)等效,且所得AUCANC是后者的7.7倍。
4.在5-FU引发嗜中性白细胞减少症的小鼠中,单次注射等摩尔剂量的NEUG和Neulasta (聚乙二醇化非格司亭)以相似的动力学和效应幅度有效加快嗜中性粒细胞恢复。
(聚乙二醇化非格司亭)以相似的动力学和效应幅度有效加快嗜中性粒细胞恢复。
5.单次和重复(每周一次)剂量的NEUG和Neulasta (聚乙二醇化非格司亭)在猴中引起相似的外周嗜中性粒细胞计数升高。在等摩尔剂量下,猴中用Neulasta(聚乙二醇化非格司亭)所治疗动物和用NEUG所治疗动物的嗜中性粒细胞升高幅度和持续时间都相似。
(聚乙二醇化非格司亭)在猴中引起相似的外周嗜中性粒细胞计数升高。在等摩尔剂量下,猴中用Neulasta(聚乙二醇化非格司亭)所治疗动物和用NEUG所治疗动物的嗜中性粒细胞升高幅度和持续时间都相似。
6.在小鼠和猴中,IV或SC给药时,NEUG与Neupogen (非格司亭)相比清除较慢,终末半衰期较长。
(非格司亭)相比清除较慢,终末半衰期较长。
7.在食蟹猴中,SC注射后NEUG的终末半衰期(12.6小时)比Neulasta (聚乙二醇化非格司亭)(9.49小时)长约33%,且NEUG的清除与生物利用度之比(“CL/F”)约是Neulasta
(聚乙二醇化非格司亭)(9.49小时)长约33%,且NEUG的清除与生物利用度之比(“CL/F”)约是Neulasta (聚乙二醇化非格司亭)的一半。
(聚乙二醇化非格司亭)的一半。
8.NEUG和Neulasta (聚乙二醇化非格司亭)在经受5-FU引发血细胞减少症的小鼠中的清除比在正常小鼠中慢,提示这两种蛋白的受体介导清除对其清除有影响。
(聚乙二醇化非格司亭)在经受5-FU引发血细胞减少症的小鼠中的清除比在正常小鼠中慢,提示这两种蛋白的受体介导清除对其清除有影响。
9.肾脏排出(在大鼠中评估)对Neupogen (非格司亭)的清除有显著贡献,对Neulasta
(非格司亭)的清除有显著贡献,对Neulasta (聚乙二醇化非格司亭)的影响很小,对NEUG的清除没有实质影响。
(聚乙二醇化非格司亭)的影响很小,对NEUG的清除没有实质影响。
10.由人血清白蛋白和人集落刺激因子组成的人蛋白NEUG在小鼠和食蟹猴中是免疫原性的。Neulasta(聚乙二醇化非格司亭)在猴中引起相似的抗体发生率和滴度。在体外中和一些用NEUG和Neulasta (聚乙二醇化非格司亭)所治疗猴子的抗体,对于NEUG和Neulasta
(聚乙二醇化非格司亭)所治疗猴子的抗体,对于NEUG和Neulasta (聚乙二醇化非格司亭),重复接触后嗜中性粒细胞响应减少,抗体阳性动物的嗜中性粒细胞基础水平与恢复期抗体阴性动物相似。
(聚乙二醇化非格司亭),重复接触后嗜中性粒细胞响应减少,抗体阳性动物的嗜中性粒细胞基础水平与恢复期抗体阴性动物相似。
综合考虑,NEUG的体外和体内药理学性质表明,它的作用方式与Neupogen (非格司亭)和Neulasta
(非格司亭)和Neulasta (聚乙二醇化非格司亭)相似,类似地促进嗜中性粒细胞和造血细胞移动进入血流。下面提供了非限制性的体外和体内实验实施例。
(聚乙二醇化非格司亭)相似,类似地促进嗜中性粒细胞和造血细胞移动进入血流。下面提供了非限制性的体外和体内实验实施例。
实施例1:NFS-60细胞增殖
NSF-60细胞系常规用于检测G-CSF活性的生物测定。该细胞系以增殖率加快来响应G-CSF。比较了重组G-CSF(Neupogen 非格司亭)和NEUG的相对效力。
非格司亭)和NEUG的相对效力。
为检测NEUG和Neupogen (非格司亭)在刺激NFS-60细胞增殖中的有效性,在与一定浓度范围的这些类似物接触24小时后检测3H-胸苷的掺入。获得EC50值并用质量单位表达(ng/ml)。
(非格司亭)在刺激NFS-60细胞增殖中的有效性,在与一定浓度范围的这些类似物接触24小时后检测3H-胸苷的掺入。获得EC50值并用质量单位表达(ng/ml)。
简而言之,在96孔板中接种1x105NFS-60细胞/孔到终体积200μl的完全培养基中,该培养基中含有指定含量的NEUG(也称为Albugranin)或Neupogen (非格司亭)。所有样品均一式三份地进行测定。细胞在30℃下孵育24小时,在最后的4小时用0.5μCi3H胸苷/孔进行脉冲。腺苷的纳入用作增殖的度量。(图8)。
(非格司亭)。所有样品均一式三份地进行测定。细胞在30℃下孵育24小时,在最后的4小时用0.5μCi3H胸苷/孔进行脉冲。腺苷的纳入用作增殖的度量。(图8)。
NEUG和Neupogen (非格司亭)以剂量依赖性方式刺激增殖。本试验中,以质量计,Neupogen
(非格司亭)以剂量依赖性方式刺激增殖。本试验中,以质量计,Neupogen (非格司亭)的效力是NEUG的15倍。NEUG的质量是Neupogen
(非格司亭)的效力是NEUG的15倍。NEUG的质量是Neupogen (非格司亭)的~4.5倍,因此,基于摩尔表示,本试验中NEUG的体外效力比Neupogen(非格司亭)低~3倍。
(非格司亭)的~4.5倍,因此,基于摩尔表示,本试验中NEUG的体外效力比Neupogen(非格司亭)低~3倍。
Neulasta (聚乙二醇化非格司亭)根据重组G-CSF的重量装瓶,在计算剂量时未包括聚乙二醇修饰的质量。由于HSA的质量贡献,NEUG是重组G-CSF的4.5倍,因此为比较NEUG和Neulasta(聚乙二醇化非格司亭)诱导NFS-60细胞增殖的能力,比较等摩尔剂量滴度的NEUG和Neulasta
(聚乙二醇化非格司亭)根据重组G-CSF的重量装瓶,在计算剂量时未包括聚乙二醇修饰的质量。由于HSA的质量贡献,NEUG是重组G-CSF的4.5倍,因此为比较NEUG和Neulasta(聚乙二醇化非格司亭)诱导NFS-60细胞增殖的能力,比较等摩尔剂量滴度的NEUG和Neulasta (聚乙二醇化非格司亭),并且EC50以摩尔浓度表示。简而言之,在96孔板中接种1x105NFS-60细胞/孔到终体积200μl的完全培养基中,该培养基中含有指定量的NEUG(Albugranin)或Neulasta
(聚乙二醇化非格司亭),并且EC50以摩尔浓度表示。简而言之,在96孔板中接种1x105NFS-60细胞/孔到终体积200μl的完全培养基中,该培养基中含有指定量的NEUG(Albugranin)或Neulasta (聚乙二醇化非格司亭)。所有样品均一式三份地进行测定。细胞在37℃下孵育20小时,在最后的4小时用0.5μCi3H胸苷/孔进行脉冲。腺苷的纳入用作增殖的度量。结果见图9所示。
(聚乙二醇化非格司亭)。所有样品均一式三份地进行测定。细胞在37℃下孵育20小时,在最后的4小时用0.5μCi3H胸苷/孔进行脉冲。腺苷的纳入用作增殖的度量。结果见图9所示。
实施例2:在BDF-1小鼠中比较NEUG和Neupogen (非格司亭)
(非格司亭)
本研究的目的是评估BDF-1小鼠中单次皮下剂量的NEUG对外周血嗜中性粒细胞和造血干细胞的影响。BDF-1进行皮下注射(“SC”),单次给予3个剂量水平(0.25mg/kg、1.25mg/kg或5.0mg/kg)的NEUG或2个剂量水平(0.25mg/kg与1.25mg/kg)的Neupogen (非格司亭)。从第1天到第5天,每天用流式细胞术定量分析外周粒细胞(Gr.1+)和造血祖细胞(c-kit+),并与用载剂处理的动物所得的水平进行比较。
(非格司亭)。从第1天到第5天,每天用流式细胞术定量分析外周粒细胞(Gr.1+)和造血祖细胞(c-kit+),并与用载剂处理的动物所得的水平进行比较。
与载剂处理的动物相比,NEUG和Neupogen (非格司亭)都引起了外周嗜中性粒细胞计数的升高,但响应的动力学和幅度不同(图10)。在Neupogen
(非格司亭)都引起了外周嗜中性粒细胞计数的升高,但响应的动力学和幅度不同(图10)。在Neupogen (非格司亭)组,嗜中性粒细胞在第1天发生最大的3倍升高,嗜中性粒细胞到第2天回复正常水平。相反,尽管NEUG单次给药使嗜中性粒细胞计数升高到与相当剂量Neupogen(非格司亭)相似的程度,NEUG组的嗜中性粒细胞计数继续上升,达到峰值的动力学和幅度为剂量依赖性。剂量为0.25、1.25和5.0mg/kg的NEUG引起的嗜中性粒细胞计数分别是载体处理动物的5.4、10和24倍。较低的两个剂量引起的嗜中性粒细胞计数升高在第2天达到峰值,而所测试的最高剂量在第4天达到峰值。对于0.25、1.25和5.0mg/kg的NEUG剂量,嗜中性粒细胞分别在第3、4和5天回复正常水平(图10)。如图10所示,NEUG所引起的外周血嗜中性粒细胞升高与Neupogen
(非格司亭)组,嗜中性粒细胞在第1天发生最大的3倍升高,嗜中性粒细胞到第2天回复正常水平。相反,尽管NEUG单次给药使嗜中性粒细胞计数升高到与相当剂量Neupogen(非格司亭)相似的程度,NEUG组的嗜中性粒细胞计数继续上升,达到峰值的动力学和幅度为剂量依赖性。剂量为0.25、1.25和5.0mg/kg的NEUG引起的嗜中性粒细胞计数分别是载体处理动物的5.4、10和24倍。较低的两个剂量引起的嗜中性粒细胞计数升高在第2天达到峰值,而所测试的最高剂量在第4天达到峰值。对于0.25、1.25和5.0mg/kg的NEUG剂量,嗜中性粒细胞分别在第3、4和5天回复正常水平(图10)。如图10所示,NEUG所引起的外周血嗜中性粒细胞升高与Neupogen (非格司亭)所引起的相比,幅度更大且持续时间更长。
(非格司亭)所引起的相比,幅度更大且持续时间更长。
本研究中,NEUG和Neupogen(非格司亭)治疗对外周造血(c-kit+)干细胞计数的结果与外周嗜中性粒细胞所得结果非常相似(图11)。NEUG的效果是剂量依赖性,在第1天与相当剂量的Neupogen (非格司亭)相似,但在第2-4天继续上升,所有治疗组在第5天回复到载剂确定的基线。如图11所示,NEUG所引起的c-kit+细胞增加与Neupogen
(非格司亭)相似,但在第2-4天继续上升,所有治疗组在第5天回复到载剂确定的基线。如图11所示,NEUG所引起的c-kit+细胞增加与Neupogen (非格司亭)所引起的相比,幅度更大且持续时间更长。
(非格司亭)所引起的相比,幅度更大且持续时间更长。
实施例3:在BDF-1小鼠中比较NEUG和Neulasta (聚乙二醇化非格司亭)
(聚乙二醇化非格司亭)
本研究的目的是比较单次皮下(SC)注射NEUG和Neulasta (聚乙二醇化非格司亭)对BDF-1小鼠外周血液中的外周血嗜中性粒细胞和造血祖细胞的影响。这通过对BDF-1小鼠(n=5)注射单剂量的5或10mg/kg NEUG进行评估。比较NEUG的效应和单次给予等摩尔剂量聚乙二醇化非格司亭(Neulasta
(聚乙二醇化非格司亭)对BDF-1小鼠外周血液中的外周血嗜中性粒细胞和造血祖细胞的影响。这通过对BDF-1小鼠(n=5)注射单剂量的5或10mg/kg NEUG进行评估。比较NEUG的效应和单次给予等摩尔剂量聚乙二醇化非格司亭(Neulasta )(1.12mg/kg和2.24mg/kg)的效应。这两种剂量的Neulasta
)(1.12mg/kg和2.24mg/kg)的效应。这两种剂量的Neulasta (聚乙二醇化非格司亭)和NEUG大致为等摩尔。
(聚乙二醇化非格司亭)和NEUG大致为等摩尔。
结果见图12。单次给予NEUG(5和10mg/kg)或Neulasta (1.12mg/kg和2.24mg/kg)有效提高了BDF-1小鼠的外周粒细胞和造血祖细胞数量。本实验中,外周粒细胞的最大移动出现在给予5mg/kg NEUG的第3天,和给予10mg/kg NEUG的第4天。粒细胞在NEUG治疗后第6天回复正常水平。在给予单剂量Neulasta
(1.12mg/kg和2.24mg/kg)有效提高了BDF-1小鼠的外周粒细胞和造血祖细胞数量。本实验中,外周粒细胞的最大移动出现在给予5mg/kg NEUG的第3天,和给予10mg/kg NEUG的第4天。粒细胞在NEUG治疗后第6天回复正常水平。在给予单剂量Neulasta (聚乙二醇化非格司亭)的小鼠中,粒细胞最大移动发生在第4天。接受2.25mg/kg Neulasta
(聚乙二醇化非格司亭)的小鼠中,粒细胞最大移动发生在第4天。接受2.25mg/kg Neulasta (聚乙二醇化非格司亭)小鼠的ANC在给药后第6天仍显著(p=0.036)高于基线,而接受1.12mg/kg小鼠的嗜中性粒细胞绝对计数(ANC)在第6天回复正常(图12A)。
(聚乙二醇化非格司亭)小鼠的ANC在给药后第6天仍显著(p=0.036)高于基线,而接受1.12mg/kg小鼠的嗜中性粒细胞绝对计数(ANC)在第6天回复正常(图12A)。
为评估在小鼠中的相对效力,计算了PD曲线下面积(AUCANC)相对等摩尔剂量(nmol/kg)(图12B)。剂量响应线平行,提示AUCANC提供了比较该模型的适当方法。两个G-CSF类似物的AUCANC显然是剂量依赖性。本实验中,按重量计,Neulasta对NEUG的相对效力是7.7。即,1mg Neulasta (聚乙二醇化非格司亭)(临床剂量基于rhG-CSF重量)与7.7mg NEUG等效。这是NEUG和Neulasta(聚乙二醇化非格司亭)之间4.5倍分子量差异的1.5倍。
(聚乙二醇化非格司亭)(临床剂量基于rhG-CSF重量)与7.7mg NEUG等效。这是NEUG和Neulasta(聚乙二醇化非格司亭)之间4.5倍分子量差异的1.5倍。
单次给予NEUG或Neulasta (聚乙二醇化非格司亭)显著(p<0.05)提高了外周血中的造血祖细胞总数(图15)。NEUG和Neulasta
(聚乙二醇化非格司亭)显著(p<0.05)提高了外周血中的造血祖细胞总数(图15)。NEUG和Neulasta (聚乙二醇化非格司亭)治疗组中,造血祖细胞的最大移动都出现在第4天。10mg/kg NEUG和1.12与2.24mg/kg Neulasta引起了相似的c-kit+细胞增加(p<0.0001)。与HSA对照相比,5mg/kg剂量的NEUG引起了c-kit+细胞的统计学显著(p<0.0001)增加,然而,这一增加比两个Neulasta
(聚乙二醇化非格司亭)治疗组中,造血祖细胞的最大移动都出现在第4天。10mg/kg NEUG和1.12与2.24mg/kg Neulasta引起了相似的c-kit+细胞增加(p<0.0001)。与HSA对照相比,5mg/kg剂量的NEUG引起了c-kit+细胞的统计学显著(p<0.0001)增加,然而,这一增加比两个Neulasta (聚乙二醇化非格司亭)组低约50%,并且似乎是亚最大剂量,因为其2倍高剂量引起最大c-kit+细胞计数的增加(图15)。
(聚乙二醇化非格司亭)组低约50%,并且似乎是亚最大剂量,因为其2倍高剂量引起最大c-kit+细胞计数的增加(图15)。
实施例4:在5-FU引发嗜中性白细胞减少症的BDF-1小鼠中比较NEUG和Neulasta (聚乙二醇化非格司亭)
(聚乙二醇化非格司亭)
G-CSF产品在临床上用于加快骨髓抑制化疗后的嗜中性粒细胞恢复。本研究的目的是比较单次皮下(SC)注射NEUG和Neulasta (聚乙二醇化非格司亭)对模型中嗜中性粒细胞恢复的影响,该模型中由亚致死剂量的5-FU(150mg/kg)引发了嗜中性白细胞减少症。单次给予BDF1小鼠5或10mg/kg的NEUG。比较NEUG的效应和单次给予Neulasta
(聚乙二醇化非格司亭)对模型中嗜中性粒细胞恢复的影响,该模型中由亚致死剂量的5-FU(150mg/kg)引发了嗜中性白细胞减少症。单次给予BDF1小鼠5或10mg/kg的NEUG。比较NEUG的效应和单次给予Neulasta (1.12mg/kg-NEUG 5mg/kg的等摩尔剂量)的效应。两种药剂都在单剂量5-FU后第1天给予。测定第6天到第10天的外周血嗜中性粒细胞数量。接受5-FU的小鼠在此时间段的特征为,嗜中性粒细胞最低值和随后的缓慢恢复阶段。本实验设计为测定NEUG对嗜中性粒细胞恢复的时间和幅度的影响。
(1.12mg/kg-NEUG 5mg/kg的等摩尔剂量)的效应。两种药剂都在单剂量5-FU后第1天给予。测定第6天到第10天的外周血嗜中性粒细胞数量。接受5-FU的小鼠在此时间段的特征为,嗜中性粒细胞最低值和随后的缓慢恢复阶段。本实验设计为测定NEUG对嗜中性粒细胞恢复的时间和幅度的影响。
5-FU给药后第1天注射了载剂对照或HSA的小鼠到第6天达到嗜中性白细胞减少的最低值(图16)。嗜中性粒细胞水平到第10天开始恢复。相比之下,5-FU给药后第1天用NEUG或Neulasta处理小鼠的嗜中性白细胞减少症恢复被加快。与载剂对照相比,用任一种药剂在所有剂量水平下的治疗都引起嗜中性粒细胞计数在统计上的显著增加。在第9天,给予5mg/kg NEUG的效果低于等摩尔剂量的Neulasta(1.12mg/kg)(p=0.0048)。但是,到第10天,两种药剂引起外周嗜中性粒细胞总数的相似增加。
对小鼠研究数据加以总结,对正常小鼠的NEUG单次给药有效引发外周血液中嗜中性粒细胞(Gr.1+细胞)和造血祖细胞(c-kit+)的剂量依赖性、快速且持续的增加。对NEUG的响应与Neulasta (聚乙二醇化非格司亭)所引发的相似,但在本研究中,对NEUG的最大响应与Neulasta
(聚乙二醇化非格司亭)所引发的相似,但在本研究中,对NEUG的最大响应与Neulasta (聚乙二醇化非格司亭)相比稍有延迟。Neupogen
(聚乙二醇化非格司亭)相比稍有延迟。Neupogen (非格司亭)单次给药仅引起了外周血液中嗜中性粒细胞和造血祖细胞计数的短暂增加。采用临床相关的血细胞减少症小鼠模型,其中IP注射亚致死剂量的5-FU引发骨髓抑制和外周嗜中性白细胞减少症,单次给予NEUG或Neulasta
(非格司亭)单次给药仅引起了外周血液中嗜中性粒细胞和造血祖细胞计数的短暂增加。采用临床相关的血细胞减少症小鼠模型,其中IP注射亚致死剂量的5-FU引发骨髓抑制和外周嗜中性白细胞减少症,单次给予NEUG或Neulasta (聚乙二醇化非格司亭)有效促进了嗜中性粒细胞恢复。
(聚乙二醇化非格司亭)有效促进了嗜中性粒细胞恢复。
实施例5:食蟹猴中的NEUG测试
选择食蟹猴以测定重复给予NEUG的效应。进行了两次猴的研究以系列评估重复给予NEUG后的血液学:2周的药理学研究比较NEUG和Neupogen (非格司亭)的皮下剂量,以及更长(5个月)的免疫原性研究比较皮下和静脉给予NEUG与皮下给予Neulasta
(非格司亭)的皮下剂量,以及更长(5个月)的免疫原性研究比较皮下和静脉给予NEUG与皮下给予Neulasta (非格司亭)的效应。两项研究都显示,NEUG引起猴的外周血嗜中性粒细胞升高的延长,其效力和药效学情况与Neulasta
(非格司亭)的效应。两项研究都显示,NEUG引起猴的外周血嗜中性粒细胞升高的延长,其效力和药效学情况与Neulasta (聚乙二醇化非格司亭)相似。
(聚乙二醇化非格司亭)相似。
实施例6:2周的猴NEUG药理学研究
为评估NEUG在猴中的药效学,进行了2周的重复剂量研究,以血液学参数作为功效终点。20只实验用雄性和雌性原初食蟹猴随机分成5个治疗组,每组两只雄性两只雄性。在猴的肩胛中部皮下(“SC”)注射载剂、NEUG或Neupogen (非格司亭)。在研究的14天治疗期间,载剂为每4天给予(Q4D),NEUG为每4天给予(Q4D)25μg/kg,或者每4或7天给予(分别为Q4D、Q7D)100μg/kg,Neupoger
(非格司亭)。在研究的14天治疗期间,载剂为每4天给予(Q4D),NEUG为每4天给予(Q4D)25μg/kg,或者每4或7天给予(分别为Q4D、Q7D)100μg/kg,Neupoger (非格司亭)为每天给予5μg/kg。
(非格司亭)为每天给予5μg/kg。
以每4天一次的频率给予的25μg/kg或100μg/kg NEUG在食蟹猴中耐受良好,未引起不良反应。血液学变化主要由NEUG引发的外周血嗜中性粒细胞增加和较少的外周血单核细胞增加组成。嗜中性粒细胞的增加在SC给予100μg/kg后24小时达到峰值(图17)。每4天一次给予25μg/kg NEUG或每天给予5μg/kg Neupogen (非格司亭)引起嗜中性粒细胞的中度增加,与载剂相比,这一增加在给药的第二周显著起来。所有NEUG引起的血液学变化在两周的无处理恢复期内完全逆转。
(非格司亭)引起嗜中性粒细胞的中度增加,与载剂相比,这一增加在给药的第二周显著起来。所有NEUG引起的血液学变化在两周的无处理恢复期内完全逆转。
其它观察
据报道,外周单核细胞数量响应G-CSF而增加,但程度低于嗜中性粒细胞所观察到的。本研究中,只有每4天一次给予100μg/kg NEUG引起了单核细胞绝对数量的增加。外周血淋巴细胞的绝对数量未受NEUG或Neupogen(非格司亭)治疗的影响。
实施例7:猴中NEUG IV和SC与Neulasta (聚乙二醇化非格司亭)SC的比较
(聚乙二醇化非格司亭)SC的比较
在食蟹猴中进行了非GLP的NEUG重复剂量给药研究,主要目的是评估免疫原性(Covance研究第6962-129号)。评估血液学参数作为研究终点,该研究还提供了比较等摩尔剂量NEUG和Neulasta(聚乙二醇化非格司亭)的有用药理学信息。NEUG和Neulasta每周给药,持续3周。图18显示了最初3剂NEUG或Neulasta (聚乙二醇化非格司亭)每次给药后的ANC。
(聚乙二醇化非格司亭)每次给药后的ANC。
本研究中,以等摩尔剂量,经SC和IV给予的Neugranin以及SC给予的Neulasta (聚乙二醇化非格司亭)引起外周血嗜中性粒细胞的显著(<0.0001,与载剂相比)升高。在这三组中,嗜中性粒细胞响应的幅度和动力学都接近一致。
(聚乙二醇化非格司亭)引起外周血嗜中性粒细胞的显著(<0.0001,与载剂相比)升高。在这三组中,嗜中性粒细胞响应的幅度和动力学都接近一致。
实施例8:药代动力学
NEUG的药代动力学评估在正常BDF-1小鼠单次IV或SC注射后、经5-FU处理的嗜中性白细胞减少BDF-1小鼠中SC注射后、在肾切除大鼠IV注射后,以及在食蟹猴单次和多次IV和SC注射后进行。此外,对rhG-CSF(Neupogen)和聚乙二醇化rhG-CSF(Neulasta)的PK作比较。下面小结了这些研究。
在所有研究中,通过夹心ELISA用G-CSF捕获和HSA检测来测定血浆NEUG浓度。该试验形式允许完整NEUG的定量分析,没有内源G-CSF和白蛋白的干扰或交叉反应。图19中以列表形式小结了药代动力学研究。
这些研究中,当IV或SC给予BDF-1小鼠时,NEUG与Neupogen (非格司亭)相比清除较慢,终末半衰期和平均停留时间(MRT)较长。NEUG的清除比Neupogen
(非格司亭)相比清除较慢,终末半衰期和平均停留时间(MRT)较长。NEUG的清除比Neupogen (非格司亭)的清除慢约8倍,而MRT(11.2-20.7小时)约长4倍。NEUG和Neulasta
(非格司亭)的清除慢约8倍,而MRT(11.2-20.7小时)约长4倍。NEUG和Neulasta (聚乙二醇化非格司亭)在经5-FU引发血细胞减少症小鼠中的清除都比在正常小鼠中慢。这最可能是因为嗜中性粒细胞数量(在5-FU治疗后)较少,嗜中性粒细胞在G-CSF清除中发挥作用。在食蟹猴中,NEUG的MRT为17.9-27.2小时。此外,5次每周SC给药的最后一剂后的Cmax与首剂后的Cmax相比似乎降低。食蟹猴中NEUG的SC生物利用度约为22%。在食蟹猴中,SC注射后,NEUG的终末半衰期(12.6小时)比Neulasta(聚乙二醇化非格司亭)(9.49小时)约长33%。肾脏清除似乎在NEUG消除中没有显著作用(在大鼠中测定)。
(聚乙二醇化非格司亭)在经5-FU引发血细胞减少症小鼠中的清除都比在正常小鼠中慢。这最可能是因为嗜中性粒细胞数量(在5-FU治疗后)较少,嗜中性粒细胞在G-CSF清除中发挥作用。在食蟹猴中,NEUG的MRT为17.9-27.2小时。此外,5次每周SC给药的最后一剂后的Cmax与首剂后的Cmax相比似乎降低。食蟹猴中NEUG的SC生物利用度约为22%。在食蟹猴中,SC注射后,NEUG的终末半衰期(12.6小时)比Neulasta(聚乙二醇化非格司亭)(9.49小时)约长33%。肾脏清除似乎在NEUG消除中没有显著作用(在大鼠中测定)。
实施例9:非临床毒理学小结
NEUG在小鼠和猴中耐受良好。在每周一次持续4周皮下给予100、500或1000微克/公斤/剂量NEUG的猴中,未有不良发现。观察食蟹猴在多次SC或IV剂量给药后对NEUG治疗的药效学响应,发现与先前报道的G-CSF效果一致。NEUG一贯地引起显著的剂量依赖性白细胞增多和嗜中性粒细胞增多,而单核细胞、嗜酸性粒细胞和嗜碱性粒细胞的增多较不明显,并与淋巴细胞的增多不一致。GLP或非GLP研究中未鉴定出NEUG在猴中的无可观察作用水平(NOEL),因此对于皮下给药视为低于25微克/公斤/剂量。在SC NEUG治疗的猴中未观察到不良作用,因此,NEUG皮下给药在猴中的无可观察不良作用水平(NOAEL)大于1000微克/公斤/剂量。其它与NEUG药理学一致的发现包括:脾脏重量增加、骨髓增生和白细胞增多的显微迹象。
图20列表小结了非临床安全性研究。
实施例10:免疫原性
NEUG能(在理论上)引起患者中和G-CSF的免疫应答。针对HSA的抗体也是可能的,尽管考虑到HSA在血液中的极高浓度(40mg/mL)这些抗体的临床意义尚不确定。采用一系列能检测NEUG所有组分抗体的高灵敏度试验评估人体内的免疫原性。
为确定NEUG的安全性和毒理学,在多项研究中评估了免疫原性。这些研究表明,人G-CSF(Neulasta 聚乙二醇化非格司亭)、人白蛋白和NEUG在猴中都是免疫原性。在一项包括Neulasta
聚乙二醇化非格司亭)、人白蛋白和NEUG在猴中都是免疫原性。在一项包括Neulasta (聚乙二醇化非格司亭)治疗分支的免疫原性研究中,接受每周一次IV或SC给予NEUG的多数动物产生了对Neulasta
(聚乙二醇化非格司亭)治疗分支的免疫原性研究中,接受每周一次IV或SC给予NEUG的多数动物产生了对Neulasta (聚乙二醇化非格司亭)的抗体。针对NEUG(或Neulasta
(聚乙二醇化非格司亭)的抗体。针对NEUG(或Neulasta 聚乙二醇化非格司亭)的抗体最初在第22天或此后(在第3次每周给药后)测得。在很多情况中,这些抗体在体外试验中具有中和效应,尽管中和抗体的存在并不引起嗜中性白细胞减少症也不防止NEUG或Neulasta
聚乙二醇化非格司亭)的抗体最初在第22天或此后(在第3次每周给药后)测得。在很多情况中,这些抗体在体外试验中具有中和效应,尽管中和抗体的存在并不引起嗜中性白细胞减少症也不防止NEUG或Neulasta (聚乙二醇化非格司亭)在猴中的药理效应。此外,在2周和2个月的无剂量阶段后,所有组的ANC都在正常范围内,并且不论抗体状况如何ANC情况都没有显著差异。
(聚乙二醇化非格司亭)在猴中的药理效应。此外,在2周和2个月的无剂量阶段后,所有组的ANC都在正常范围内,并且不论抗体状况如何ANC情况都没有显著差异。
和多数人蛋白相似,人白蛋白在动物中是免疫原性。其它白蛋白融合蛋白的经验已经证明,在猴中的免疫原性对人体中免疫原性的发生率(或后果)不具预示性。例如,Albuferon (由人血清白蛋白和干扰素α组成的融合蛋白)在猴中是高度免疫原性(10/12的猴子在单次注射后呈抗体阳性)且免疫应答既中和接触,也受到接触的显著影响。相比之下,在最近有458名患者的Albuferon
(由人血清白蛋白和干扰素α组成的融合蛋白)在猴中是高度免疫原性(10/12的猴子在单次注射后呈抗体阳性)且免疫应答既中和接触,也受到接触的显著影响。相比之下,在最近有458名患者的Albuferon 2期研究中,出现抗体的比率很低,并且最初12周的治疗过程中,在Albuferon
2期研究中,出现抗体的比率很低,并且最初12周的治疗过程中,在Albuferon 治疗组中的这一比率(3%)显著低于聚乙二醇化干扰素治疗组(18%)。此外,抗体没有明显后果。
治疗组中的这一比率(3%)显著低于聚乙二醇化干扰素治疗组(18%)。此外,抗体没有明显后果。
在接受最多三次剂量NEUG的人患者中,未观察到针对NEUG的抗体(参见例如,下文I期5.c和II期5.e部分)。对于NEUG风险评估,可用的人和动物数据提示,G-CSF的中和不排除对药理剂量G-CSF的响应,也不排除对感染剂攻击的正常响应。在G-CSF被排除的小鼠模型中,显示仍可响应感染剂攻击而发生嗜中性粒细胞增生,提示粒细胞生成系统的冗余度。此外,有报道在费耳提综合征和系统性红斑狼疮的病例中,人体有抗G-CSF的自身抗体。这些患者产生嗜中性白细胞减少症;然而,G-CSF或GM-CSF治疗在多数患者中仍有效。
总之,NEUG的体外和体内数据以及药代动力学特征支持NEUG在进行骨髓抑制性抗癌治疗的患者中用作针对发热性嗜中性白细胞减少症的单剂量预防剂。其在单次剂量后引起外周血中高水平造血祖细胞的能力也可能有益于自体或异体造血干细胞移植的患者或供体。
B.NEUG的人体研究
在题为“I期”和“II期”的两个主要部分中提供下列实施例。每期包括两个部分,A部分和B部分。下表2小结了I期和II期的实施例。

每期分成5个部分:1)研究目的,2)患者特征,3)研究药剂,4)研究特点,以及5)A和B部分的结果。
实施例11:I期
1.研究目的
进行I期A/B、II期A/B的研究以评估接受骨髓抑制化疗(多柔比星/多西他赛)的对象中,皮下给予NeugraninTM(“NEUG”)(重组人白蛋白-人粒细胞集落刺激因子)的安全性、耐受性、免疫原性、药代动力学和药效学
I期的主要研究目的是,通过测定治疗中出现的不良事件的频率、严重程度和持续时间,并将它们与NEUG给药的时间和剂量相关联,从而评估与聚乙二醇化非格司亭相比,在可能的治疗剂量范围内皮下给予NEUG的安全性情况。
次要研究目的是评估NEUG的药代动力学和免疫原性,并在接受多柔比星/多西他赛的患者中,比较NEUG和聚乙二醇化非格司亭给药对嗜中性白细胞减少症的发病率、严重程度和持续时间的影响。
如上表2所示,I期按两个部分,A部分和B部分进行。
2.患者特征
对于I期,根据下列特征或参数筛选患者;
包括:
1.具有组织学确认的乳腺癌,已安排接受多柔比星和多西他赛的患者。
2.年龄为18岁或18岁以上。
3.血液功能合格。
4.ANC>1500/mm3
5.血小板>100,000/mm3
6.肝、肾功能合格。
7.血清肌酸酐<2.0x正常的上限
8.当地实验室测得总胆红素在正常限度内(WNL)
9.血清转氨酶(SGOT/SGPT)<1.5x正常的上限
10.碱性磷酸酶<2.5x正常的上限
11.ECOG性能状况0或1。
12.基于左心室射血分数(LVEF)在正常限度内,可以接受多柔比星。
13.能理解本研究的要求,提供已签署的知情同意书(包括同意使用并公开研究相关健康信息)并遵守研究方案步骤。
排除:
1.曾有多于1次的前期化疗方案(包括12个月内的辅助治疗);进入研究前4周内有任何化疗/免疫治疗;喜树碱累积剂量将阻止本研究中两个完全剂量的多柔比星疗程。
2.在研究化疗的6周内曾使用任何亚硝基脲(BCNU、CCNU)或丝裂霉素-C。
3.研究者的观点中,心脏记录、迹象或症状阻止使用基于喜树碱的化疗方案。
4.在化疗研究前2周内曾有手术或放射治疗。
5.骨盆或大于20%的骨髓携带面积曾受宽场辐射、或有骨髓病变。
6.曾与造血干细胞移植进行高剂量化疗。
7.在研究化疗前4周内曾使用骨髓(G-CSF或GM-CSF)生长因子。
8.在研究化疗前4周内曾使用红细胞生成素。
9.有骨髓恶性肿瘤或脊髓发育不良历史。
10.已知脑部转移,除非经充分治疗(手术或化疗),在至少3周的观察中没有发展的迹象且在不用抗惊厥药和甾体能保持神经稳定。
11.已知镰状细胞疾病。
12.诊断有成人呼吸窘迫综合征(ARDS)。
13.在当前感染中需要静脉或口服抗生素。
14.已知对酵母衍生产品有过敏史。
15.已知对大肠杆菌衍生蛋白质,聚乙二醇化非格司亭、非格司亭或聚乙二醇化非格司亭的任何其它组分有过敏性(仅针对2期)。
16.怀孕妇女或哺乳期妇女(本研究全过程中,所有女性必须采取某种可靠性大于90%的避孕方法,或者是不育或是绝经后)
17.已知HIV阳性或活动性肝炎(状态未知的患者不做测试)
18.不同意在研究全过程中和研究药剂最后给药后30天内采用有效避孕方法的男性。
对象因下列原因从后续治疗中排除:
1.疾病发展
2.尽管治疗最优,但毒性不可接受
3.研究者自由裁量的并发疾病
4.多柔比星方案-达到寿命许可累计剂量的最大值(见资格标准)
5.撤回同意
6.不合规/随访丢失
7.妊娠
若停止NEUG治疗,为了预定的安全性和PK评估,继续研究对象,在任何研究药物的最终剂量后随访至少30天。
3.研究药剂
NEUG(重组人白蛋白-人粒细胞集落刺激因子,rHSA-G-CSF)是分子量约85kDa的单链连接融合蛋白,其包含HSA成熟形式所对应的1-585位残基和人G-CSF所对应的586-759位残基。NEUG的治疗性部分是重组人DNA衍生的G-CSF。
NEUG作为一次性1型玻璃小瓶中的无菌、冻干制剂提供,并在2-8℃保存。用1.1ml注射用无菌水复溶后,各小瓶包含pH 7.2的10mM磷酸钠、200mM甘露醇、60mM无水海藻糖、0.01%(w/v)聚山梨酯80溶液中的15mg/ml(可递送15mg/小瓶)NEUG。
图13显示了I期所用的NEUG药物产品的组成。
市售的Neulasta (聚乙二醇化非格司亭)在用于皮下注射的0.6ml预装注射器中提供。各注射器包含无菌、透明、无色、无防腐剂溶液中的6mg聚乙二醇化非格司亭(基于蛋白质重量),所述溶液(pH 4.0)包含注射用水中的乙酸(0.35mg)、山梨糖醇(30.0mg),聚山梨酯20(0.02mg),钠(0.102毫克)。美国药典。
(聚乙二醇化非格司亭)在用于皮下注射的0.6ml预装注射器中提供。各注射器包含无菌、透明、无色、无防腐剂溶液中的6mg聚乙二醇化非格司亭(基于蛋白质重量),所述溶液(pH 4.0)包含注射用水中的乙酸(0.35mg)、山梨糖醇(30.0mg),聚山梨酯20(0.02mg),钠(0.102毫克)。美国药典。
通过皮下给药施用NEUG(50、150、300或450μg)或Neulasta (聚乙二醇化非格司亭)(6mg)。
(聚乙二醇化非格司亭)(6mg)。
4.研究特点
a.研究方案及持续时间
本研究是首次人体、多中心、开放标签、无对照序列剂量递增研究,继以有对照、随机临床试验,在62例患有乳腺癌计划接受多柔比星/多西他赛的对象中进行。本研究由两部分组成:为了在B部分的随机试验前评估安全性,A部分是13个对象中的剂量依序递增研究,4个剂量组(50、150、300或450μg/kg),50、150和450μg/kg为每组3个对象,在300μg/kg组有4个对象。
A部分中,对象在化疗开始前至少2周(疗程0)接受第一剂NEUG,在没有细胞毒性化疗的情况下初始评估安全性和对嗜中性粒细胞绝对计数(“ANC”)的效果。在最少两周的随访后,若未出现被认为与疗程0的NEUG有关的剂量限制不良事件,且对象继续满足所有资格标准,则对象在第1和第2疗程的化疗后接受相同剂量的NEUG。
A部分中,剂量限制毒性(DLT)定义为被认为可能、很可能或肯定与所述研究试剂2级或2级以上的临床意义不良事件,2级骨痛除外。A部分各组内,各进入试验对象的初始研究药物给药相隔至少24小时,以监控急性不良事件。
递增到下一剂量水平的决定是基于给定组所有对象的NEUG首剂给药后至少7天的安全性数据审查。若3个对象无一出现DLT,则征募3个对象继续剂量递增到下一剂量水平。若给定组3个对象中有1个出现DLT迹象,则在该剂量水平征募另3个对象,达到每组共6个对象。若6个对象中仅1个发生DLT则继续剂量递增。若6个对象中2个发生DLT,则停止剂量递增,不再给予进一步的NEUG治疗。
其余对象完成其排定的安全性、药代动力学和药效学评估。
A部分初始组证明了安全性后,进行B部分。B部分中,对象以平行方式随机分至3个治疗组之一:在研化疗约24小时后给予NEUG 300μg/kg(n=20)、NEUG 450μg/kg(n=21)、或批准剂量的6mg聚乙二醇化非格司亭(n=10)。
下表3和4小结了I期中A和B部分的对象设置。图6显示了I期研究A和B部分的化疗疗程。

b.I期A和B部分化疗的附随治疗
化疗
本试验的化疗方案由治疗第1天通过静脉输液依次给予多柔比星50mg/m2和多西他赛75mg/m2组成,最多为2个21天的疗程。
接受每个疗程的治疗前,对象的嗜中性粒细胞绝对计数(ANC)必须>1.5x109/L且血小板数必须>100x109/L。为了血液学恢复,治疗最多可以延迟2周。
据报道,联用多柔比星和多西他赛在乳腺癌患者中具有显著的临床活性。然而,所述联用是高度骨髓抑制性的,3级或4级嗜中性粒细胞缺少症的发病率比其它标准方案高。
尽管添加CSF,多柔比星和多西他赛的联用仍引起79%的患者出现4级嗜中性粒细胞缺少症,以及9-18%的发热性嗜中性粒细胞缺少症比率。这一多柔比星/多西他赛方案用于预防嗜中性粒细胞缺少症及其并发症的新药的研究。因此,多柔比星和多西他赛的联用是研究NEUG这类新试剂效力的合适化疗方案。
多柔比星
药理学数据
盐酸多柔比星是从波塞链霉菌表灰变种(streptomyces peucetius varcaesius)中获得的大环抗生素,其抑制DNA和DNA依赖性的RNA合成,以及蛋白质合成。多柔比星在细胞周期各阶段都有活性,但在S期细胞毒性最大。该药物主要通过肝排除,肾脏清除是次要的。
药学数据
该药物以10、20、50、100或200mg的小瓶市售。冻干制剂可以用无菌注射用水、注射用5%葡萄糖溶液或0.9%盐水复溶。
副作用和毒性
多柔比星的剂量限制毒性在于骨髓抑制,主要是白细胞缺少症,其最低点约在10-14天,以及心脏毒性,包括罕见的急性心包炎-心肌炎综合征和迟延的与累积剂量相关的心肌病。预期毒性有标志性脱发和中度恶心/呕吐。有报道在不慎外渗部位产生局部皮肤和组织损伤的外渗反应,口腔炎,皮肤(特别是指甲床)的色素沉着以及此前放射部位的“回忆”现象。
多西他赛
药理学数据
多西他赛是半合成的紫杉类,与游离微管蛋白结合并促进稳定微管的组装,干扰有丝分裂和细胞复制(M期细胞周期特异性)。多西他赛广泛结合蛋白,在肝中广泛代谢,在给药后7天内约有75%通过粪便排出。
药学数据
多西他赛(TaxotereTM,赛诺菲安万特公司)以80mg/2mL或20mg/0.5ml的单剂量小瓶中提供,附带稀释剂(含13%乙醇的注射用水)小瓶。每毫升Taxotere含40mg多西他赛(无水)和1080mg聚山梨酯80。
副作用和毒性
多西他赛不可用于对多西他赛或其它用聚山梨酯80配制的药物如依托泊苷和维生素E有严重过敏性反应史的患者。
不得再次刺激经历严重过敏性反应的患者。接受多西他赛的患者应当如下所述用皮质类固醇作前驱用药。
轻度到中度的肝损伤导致代谢延迟27%且系统性接触提高了38%(AUC)。多西他赛不得用于SGOT和/或SGPT>1.5倍正常限度以及碱性磷酸酶>2.5倍正常限度的患者。在III期研究中,尽管有皮质类固醇前驱用药,仍有17%(中度)和6%(重度潴留)患者出现液体潴留。观察到严重的感觉神经症状(感觉异常、感觉失常、疼痛)。
预期的副作用包括骨髓抑制,主要是白细胞缺少症,其最低点约在第9天,到第15-21天恢复。有报道脱发、指甲和皮肤变化、口腔炎、肌痛/关节痛、恶心/呕吐和低血压。
化疗剂量、给药以及剂量调整
各治疗疗程的第1天,给予化疗剂(多柔比星,然后多西他赛)。
通过IV推注给予50mg/m2多柔比星,经侧支注入静脉或中央静脉导管给药以免渗出损伤。
将75mg/m2多西他赛稀释于250mL 0.9%盐水或5%葡萄糖溶液中,用聚乙烯管输液套件静脉给药,用时约1小时。在临给药前和刚结束多西他赛输液后,获得生命体征。
接受每个疗程的化疗前,对象的嗜中性粒细胞绝对计数(ANC)必须>1500/mm3且血小板数必须>100,000/mm3。为了血液学恢复,治疗最多可以延迟2周。对于3-4级非血液学毒性,2次3-4级感染事件或4级血小板减少症,允许化疗剂量降低25%。
将经历重度过敏性反应或非血液学毒性阻止进一步化疗疗程的患者从研究治疗中除去,但完成随访.
化疗前驱用药
从多西他赛给药前一天开始连续3天口服(需要时IV)皮质类固醇(例如地塞米松8mg BID),以减少液体潴留和过敏性反应的发生率和严重程度。
由主治医生自由裁量止吐剂或其它前驱用药(例如H2拮抗剂)的使用和选择。
药物禁忌
在研究过程中以及下面指明的补充时间内对象不得接受任何下列药物或方法:
1.在启动研究试剂的30天内以及试验期间的其它在研药剂。
2.在NEUG用药后14天内不应进行后续的化疗疗程。
3.在试验期间,除非发生持续或发热性嗜中性粒细胞缺少症,不得接受细胞因子、其它造血生长因子和预防性抗生素。若对象在筛选阶段到试验第0天之间的任何时间进行了G-CSF治疗,则没有资格接受NEUG并终止其研究。
允许的药物
对象可以继续他们的基础药物。若可能,整个研究过程中各药物的每日剂量应保持一致。若出于研究者认为必须的任何理由,对象需要其它药物或改变剂量,应在CRF的适当页面记录所用药物、给药途径、及其给予所针对的适应症。
抗生素
所有对象在每次化疗过程后接受预防性口服抗生素(例如,环丙沙星)以降低感染的可能性。若发生发热性嗜中性粒细胞缺少症或持续的重度嗜中性粒细胞缺少症(持续>5天ANC<0.5x109/L),则对象视为治疗失败,从研究中除去,完成随访并接受所有标准支持治疗,包括研究者自由裁量下的生长因子支持。
也将经历重度过敏性反应或非血液学毒性阻止进一步化疗疗程的患者从研究治疗中除去,但完成随访.
c.安全性评估
通过随时间推移评价不良事件(AE)的类型、频率和严重程度,临床实验室测试(血液学和临床化学)的变化,免疫原性,身体检查,以及监控生命体征,来评估NEUG的安全性。根据国家癌症研究所常见的不良事件术语标准(National Cancer Institute Common Terminology Criteria for Adverse Events)(2003年12月12日的NCI-CTCAE 3.0版)对所有AE和实验毒性作评级。从开始研究药物给药直至任何研究药物的最终用药后的30天内,采集所有不良事件(包括严重不良事件,“SAE”)。按评估规划(Schedule of Assessments)所列获得实验室评估。若有4级嗜中性粒细胞缺少症,每天获取实验室结果直至ANC>500。若对象的下一治疗疗程被延迟(以及治疗的最后疗程后),每周至少两次获取分类的全血细胞计数(CBC)直至ANC>1500。
5.I期A和B部分的结果
a.概述
统计方法:
采用说明性统计学方法分析与安全性、药代动力学(PK)、药效学(PD)和免疫原性参数相关的数据。
对于不良事件的频率和严重程度和实验室毒性评级,给出了计数和分级。
功效分析包括4级与3-4级嗜中性粒细胞缺少症的发病率和持续时间、ANC最低值、到ANC最低值的时间、恢复的时间(到ANC>0.5x109/L和ANC>1.0x109/L)以及发热性嗜中性粒细胞缺少症的发病率。
选择本研究样本大小时未使用严格的统计强度要求。统计强度为80%的研究表明,为在5%的显著水平上证明NEUG不弱于聚乙二醇化非格司亭,计算出每个测试分支需要约37个对象。由于主要是为安全性而进行的1/2a期研究,确定效力的统计功效所要求的样本大小大于适当的样本大小。因此,评估了疗效的趋势。
设置/人口统计学:
A部分为试验的依序剂量递增部分,共招募了13个对象。B部分共招募了51个对象,随机分为NEUG 300μg/kg(n=20)、NEUG 450μg/kg(N=21)或聚乙二醇化非格司亭6mg(n=10)。
b.研究结果
在初始的剂量调查中,没有化疗的情况下,NEUG耐受良好且引起预期的ANC升高,ANC在第2-4天之间达到峰值并到第14天回复正常(图22)。
A部分中,50μg/kg NEUG剂量组的所有3个对象和450μg/kg Neugranin剂量组的1个对象经历了持续超过5天的发热性嗜中性粒细胞缺少症或重度嗜中性粒细胞缺少症。在B部分中,300μg/kg NEUG剂量组的1个对象和450μg/kg NEUG剂量组的2个对象经历持续超过5天的发热性嗜中性粒细胞缺少症或重度嗜中性粒细胞缺少症。聚乙二醇化非格司亭组中1个对象经历了持续超过5天的发热性嗜中性粒细胞缺少症或重度嗜中性粒细胞缺少症。
c.免疫原性
在各NEUG疗程的第1天用药前和治疗检查的最后(最后用药后至少15天),获取接受NEUG对象的血清样品用于针对NEUG的抗体。若在研究中的任何时间,有对象发生阳性抗NEUG抗体响应,在最终的NEUG剂量约6个月后,获取重复样品。
直到A和B部分的治疗都结束,完成所有对象的测试。所有样品都为NEUG抗体阴性。
d.不良事件
在A部分过程中,剂量限制毒性(DLT)定义为认为可能、很可能或肯定与研究药剂有关的2级或2级以上临床意义的不良事件,2级髓质骨痛除外。
A部分任何组中,在疗程0中未有DLT。仅报道了2例NEUG给药相关的不良事件:骨痛和先前存在高血压的发作,后者发生在NEUG给药后7天。两例事件都得到解决,没有后遗症。
41个NEUG治疗对象中31个经历至少1项不良事件。NEUG和聚乙二醇化非格司亭治疗对象中的AE发生率是相当的(分别为75.6%和70%)。
表5提供了B部分常见不良事件(所有对象中AE大于等于5%)的小结。

1相关=认为可能、很可能或肯定相关
2无关=认为可能不相关或者不相关
最常报道的被认为NEUG相关的不良事件是骨痛,一种与所有G-CSF产品相关的典型不良反应,有5位患者报道有骨痛,其中4位列入上表,还有一位是A部分接受450μg/kg的对象。在所有病例中,骨痛为NCI-CTCAE强度1-2级,持续时间短且解决后没有后遗症。在疗程0NEUG给药后发生了碱性磷酸酶和尿酸的1级升高;研究者认为这些事件不具临床意义,无需干预即被解决。这些是在接受G-CSF(例如,Neulasta )的患者中所预期的效应。
)的患者中所预期的效应。
化疗疗程中其它常报道的不良事件(恶心、呕吐、脱发、口腔炎)与接受多柔比星/多西他赛方案的患者中所预期的不良事件一致。
所报道的AE主要是NCI CTC严重程度1或2级。有4项AE报道为严重不良事件。有2个对象,其一接受150μg/kg,另一个接受450μg/kg,经历了导致住院的呕吐,其中一个在后续化疗疗程中经历了第二次SAE;呕吐强度为轻度但引起住院或使住院延长。第3个接受450μg/kg的对象因发热性嗜中性粒细胞缺少症住院。该事件被认为与NEUG无关。
e.药代动力学
整个研究过程中,对所有接受NEUG的对象取样分析血清NEUG浓度。采用NEUG特异性的夹心酶联免疫吸附试验(ELISA)检测该药物。用WinNonlin企业版4.1版或更高版本对血清药物浓度-时间数据作PK分析,使用非房室模型分析或基于模型的分析。
获得下列PK参数:曲线下面积(AUC0-∞)、清除(CL/F)、分布容积(Vz/F)、最大浓度(Cmax)、吸收半衰期(t1/2,abs)消除半衰期(t1/2,elim)和平均停留时间(MRT)。在方案所用剂量范围内评估药代动力学数据的线性关系。
表6小结了疗程0(预化疗)的药代动力学参数,图3显示了疗程0的PK概况。

用最大血清NEUG浓度和时间-浓度曲线下面积的剂量依赖性增加来测定药物接触。初始50μg/kg剂量组对象的血清浓度始终低于定量分析的下限(6.3ng/mL)。从150到450μg/kg的所有剂量的Tmax都在6-24小时范围内。Cmax的范围从150μg/kg剂量的72.7±59.7(均值±SD)ng/mL到450μg/kg剂量的294±351ng/mL。相应地,AUC0-∞的范围从150mcg/kg剂量的1758±1675ng/mL*hr到450μg/kg的10131±9563ng/mL*hr。第1疗程的范围相似。NEUG的平均消除半衰期范围为14-30小时。
如“研究特点”(上文第4部分)中所述,A部分的对象在开始化疗前至少2周(疗程0)接受NEUG的初始剂量,在没有细胞毒性化疗时初步评估安全性和对嗜中性粒细胞绝对计数(“ANC”)的效应。在最少2周的随访后,若未出现被认为与疗程0的NEUG有关的剂量限制不良事件,且对象继续满足所有资格标准,则对象在第1和第2疗程的化疗后接受相同剂量的NEUG。在化疗施用后24小时给予NEUG。图7显示了在第1疗程和第2疗程接受NEUG的对象的ANC和WBC。
f.药效学以及B部分剂量的确定
对研究的I期A部分数据分析得到下列观察:
1.NEUG在疗程0(化疗前)引起剂量依赖性的WBC和ANC升高(见图7A的疗程0数据)。
2.疗程0的ANC升高与等摩尔剂量聚乙二醇化非格司亭的历史数据相当
3.如所预期,化疗后WBC和ANC降低
4.从最低ANC的恢复似乎与剂量相关
5.ANC和WBC到第15天回到正常值
基于这些观察以及A部分所有剂量水平表明的安全性,B部分评估选择的剂量为300和450μg/kg。如上所述,对象随机分到NEUG 300μg/kg、NEUG 450μg/kg或聚乙二醇化非格司亭的批准固定剂量6mg。对象在多柔比星/多西他赛(用药2个疗程,相隔21天)用药后1天,接受NEUG或聚乙二醇化非格司亭。B部分的数据包括群体的第1疗程ANC情况。图2和表5为结果小结。
如下表7所示,测定了51个治疗对象中48个的3级和4级嗜中性白细胞减少症的发病率,和第1疗程中的ANC情况。应注意,在没有预防性G-CSF治疗时,多柔比星/多西他赛治疗的患者中的70-80%有平均持续时间为5天的4级嗜中性白细胞减少症。

图2给出了治疗组的平均ANC曲线。
NEUG有效治疗3级、4级和发热性嗜中性白细胞减少症。该化疗方案没有G-CSF治疗时,发热性嗜中性白细胞减少症的发病率约40%。在NEUG给药后观察到ANC的剂量相关性升高且嗜中性白细胞减少症比率低于多柔比星/多西他赛所预期。NEUG未导致超出预期的或在严重的不良事件。
在接受300μg/kg NEUG的患者中,3级和4级嗜中性白细胞减少症的发病率高于接受聚乙二醇化非格司亭(Neulasta )的患者,且在接受300μg/kgNEUG的患者中恢复到正常ANC的速度似乎也比接受聚乙二醇化非格司亭的患者慢。接受450μg/kg NEUG患者中的ANC情况与接受聚乙二醇化非格司亭的患者相似,尽管接受NEUG患者在从嗜中性白细胞减少症恢复期间的ANC一般低于接受聚乙二醇化非格司亭的患者。总之,这些剂量的NEUG看来提供了与聚乙二醇化非格司亭相似的效果。
)的患者,且在接受300μg/kgNEUG的患者中恢复到正常ANC的速度似乎也比接受聚乙二醇化非格司亭的患者慢。接受450μg/kg NEUG患者中的ANC情况与接受聚乙二醇化非格司亭的患者相似,尽管接受NEUG患者在从嗜中性白细胞减少症恢复期间的ANC一般低于接受聚乙二醇化非格司亭的患者。总之,这些剂量的NEUG看来提供了与聚乙二醇化非格司亭相似的效果。
g.I期B部分的PK/PD情况
图4显示了在乳腺癌治疗第1疗程多柔比星/多西他赛给药1天后接受450μg/kg NEUG的患者的PK/PD情况。NEUG在给药后1天内达到Cmax,并逐渐降低,到第10天降至不可检测水平。NEUG给药后,ANC到第4天升至峰值,然后如接受多柔比星/多西他赛和G-CSF治疗的患者中所预期,ANC在第8天降至最低然后在第10天恢复正常。到第12天,ANC值在正常范围内,NEUG不可检测。应注意,在未接受预防性G-CSF治疗的患者中,最低ANC的持续时间和达到ANC恢复的时间长得多(例如,5-7天)。一剂450μg/kg后,与标准剂量聚乙二醇化非格司亭报道的15-80小时相比,NEUG的清除半衰期中位数约为30小时。
h.NEUG和聚乙二醇化非格司亭之间的其它差异
在治疗第1疗程个体ANC情况的比较中,测试剂量的NEUG和聚乙二醇化非格司亭在加快嗜中性白细胞减少症恢复效果之间的更多具体差异是显著的。所有组的ANC峰值非常相似,接受300μg/kg NEUG的对象中的ANC最低值低于接受450μg/kg NEUG的对象,而接受聚乙二醇化非格司亭对象的ANC最低值平均最高。在所有治疗组中,第14天ANC都从最低值恢复到了基线,但接受300μg/kgNEUG的组比450μg/kgNEUG的组慢,接受聚乙二醇化非格司亭的对象最快。
将完成I期排定的疗程0(化疗前)的患者的NEUG PK/PD数据与相似化疗前给药的聚乙二醇化非格司亭试验的已公开数据进行比较。比较结果如下:
1.在疗程0,150μg/kg剂量NEUG的Emax(最大观测ANC)与30μg/kg剂量聚乙二醇化非格司亭匹配,该剂量此后被证明功效劣于已证实有效的100μg/kg剂量聚乙二醇化非格司亭。
2.300和450μg/kg剂量Neugranin的Emax与100μg/kg剂量聚乙二醇化非格司亭的疗程0水平更为一致。
3.300和450μg/kgNEUG的Cmax中位数和Emax中位数几乎相同,因此Cmax继续预测Emax。
4.ANC提高与等摩尔剂量聚乙二醇化非格司亭的公开数据相当。
如上所述,当按等摩尔用药时,在动物和人中的PK/PD评估与估算的NEUG和聚乙二醇化非格司亭等效剂量一致。在小鼠中,聚乙二醇化非格司亭的7.7倍剂量获得相等的AUCANC。由于白蛋白对NEUG的分子量有显著贡献,而Neulasta (聚乙二醇化非格司亭)是按rhG-CSF的重量用药(不包括聚乙二醇化非格司亭中聚乙二醇的贡献),预期4.5倍剂量的NEUG(按重量计)与等剂量的Neulasta
(聚乙二醇化非格司亭)是按rhG-CSF的重量用药(不包括聚乙二醇化非格司亭中聚乙二醇的贡献),预期4.5倍剂量的NEUG(按重量计)与等剂量的Neulasta (聚乙二醇化非格司亭)效果相当。动物中的功效数据与聚乙二醇化非格司亭的4.5-7.7倍等效(1mg聚乙二醇化非格司亭=4.5-7.7NEUG)相符。非临床安全性和功效数据与这一剂量估算相符,当与已有的临床数据一起考虑时,构成临床评估选择剂量的基础。
(聚乙二醇化非格司亭)效果相当。动物中的功效数据与聚乙二醇化非格司亭的4.5-7.7倍等效(1mg聚乙二醇化非格司亭=4.5-7.7NEUG)相符。非临床安全性和功效数据与这一剂量估算相符,当与已有的临床数据一起考虑时,构成临床评估选择剂量的基础。
i.I期结果
I期药代动力学评估的结果如下。
在疗程0和第1疗程用150μg/kg、300μg/kg和450μg/kg剂量的NEUG治疗的所有对象的血清样品中都测得NEUG。
在第1疗程,150μg/kg、300μg/kg和450μg/kg剂量组的大多数对象(50个取样对象中的45个)中,直至144小时还可测得NEUG。几乎未观察到疗程至疗程的药物积累。
各剂量组第1疗程的药物接触高于疗程0(化疗前)。第1疗程NEUG接触提高可能是由于嗜中性粒细胞数量减少,该细胞在受体介导G-CSF清除中起作用。
第1疗程中NEUG的清除半衰期中位数,300μg/kg剂量组为约36小时,450μg/kg剂量组为约30小时。据报道,非格司亭的清除半衰期为3-4小时,聚乙二醇化非格司亭视不同剂量为42-67.5小时。
观察到剂量之间达到最大血清浓度的时间(tmax)和吸收半衰期(t1/2,abs)有统计学显著差异。这两项参数都随NEUG剂量的增加而提高。未见其它剂量标准化PK参数显示剂量间的统计学显著差异。
实施例12:II期
研究的II期为对照的随机试验,在334位最多接受4个剂量多柔比星/多西他赛的乳腺癌对象中进行。所述研究在45个临床地点进行,由两个阶段组成,双向随机先导试验阶段评估皮下给予NEUG相对聚乙二醇化非格司亭的安全性和效果,后续的主试验阶段将对象随机分到聚乙二醇化非格司亭和两个耐受良好的NEUG剂量组,NEUG剂量根据先导试验选定。主试验阶段样本大小足以就主要终点,化疗第1疗程中重度(4级)嗜中性白细胞减少症(DSN)的持续时间,确立NEUG不劣于聚乙二醇化非格司亭。该研究设计的示意图如下。

1.研究目的
II期的主要目的是选择能表现与聚乙二醇化非格司亭相当效果的NEUG剂量,并评估用NEUG治疗后化疗第1疗程重度嗜中性白细胞减少症(DSN)的持续时间。次要目的是评估第2-4疗程中的DSN,评估第1-4疗程中嗜中性粒细胞绝对计数恢复的时间和发热性嗜中性白细胞减少症的比率;并评估NEUG的安全性、耐受性、药代动力学(第1疗程中)和免疫原性。
2.患者特征
对于II期,根据下列特征或参数筛选患者:
包括:
1.具有组织学确认的乳腺癌,已安排接受多柔比星60mg/m2和多西他赛75mg/m2的患者。
2.年龄为18岁或18岁以上。
3.血液功能合格:
4.ANC>1500/mm3
5.血小板>100,000/mm3
6.肝、肾功能合格:
7.血清肌酸酐<1.5x正常的上限
8.当地实验室测得总胆红素在正常限度内(WNL)
9.血清转氨酶(SGOT/SGPT)<1.5x正常的上限
10.碱性磷酸酶<2.5x正常的上限
11.美国东部肿瘤协作组(“ECOG”)性能状况0-2
12.基于左心室射血分数(LVEF)在正常限度内,可以接受多柔比星
13.能理解本研究的要求,提供已签署的知情同意书(包括同意使用并公开研究相关健康信息)并遵守研究方案步骤。
排除:
1.此前多于1次的化疗方案(包括过去12个月内给予的辅助治疗)
2.累积喜树碱剂量将阻止本研究中的4次多柔比星完全剂量疗程。
3.在研究化疗前30内有在先化疗/免疫治疗(对于亚硝基脲(BCNU,CCNU)或丝裂霉素-C,为研究化疗前6周内)
4.伴随曲妥珠单抗(Herceptin)
5.在过去30天内接受过任何在研药剂
6.在研究者的观点中,心脏记录、迹象或症状阻止使用基于喜树碱的化疗方案
7.在化疗研究前2周内曾有手术
8.在化疗研究前4周内曾有辐射治疗(针对骨髓转移瘤的定点放射除外)
9.曾与造血干细胞移植进行高剂量化疗
10.在研究化疗前4周内曾使用G-CSF、GM-CSF或红细胞生成素
11.在研究化疗72小时内接受了全身性抗生素
12.有骨髓恶性肿瘤或脊髓发育不良史。
13.已知脑部转移,除非经充分治疗(手术或化疗),在至少3周的观察中没有发展的迹象且在不用抗惊厥药和甾体能保持神经稳定。
14.已知镰状细胞疾病
15.诊断有成人呼吸窘迫综合征(ARDS)
16.已知对酵母衍生产品的过敏史
17.已知对大肠杆菌衍生的蛋白质,聚乙二醇化非格司亭、非格司亭或聚乙二醇化非格司亭的任何其它组分有过敏性
18.妊娠妇女或哺乳期妇女。(在筛选时,具有完整子宫的所有女性必须是血清妊娠测试阴性。所有非不育或非绝经后女性必须在整个研究过程中和研究药剂最后用药后30天内采取医学上接受的避孕方法。)
19.不同意在研究全过程中和研究药剂最后给药后30天内采用有效避孕方法的男性。
20.已知HIV阳性或活动性肝炎(状态未知的患者不做测试)
对象因下列原因从后续治疗中排除:
1.疾病发展
2.尽管治疗最优,但毒性不可接受
3.研究者自由裁量的并发疾病
4.多柔比星方案-达到寿命许可累计剂量的最大值(见资格标准)
5.撤回同意
6.不合规/随访丢失
7.妊娠
若停止研究药物治疗,为了预定的安全性和PK评估,继续研究对象,并在任何研究药物的最终剂量后随访至少30天。
3.研究药剂
NEUG(重组人白蛋白-人粒细胞集落刺激因子,rHSA-GCSF)是分子量约85kDa的单链连接融合蛋白,其包含HSA成熟形式所对应的1-585位残基和人G-CSF所对应的586-759位残基。NEUG的治疗性部分是重组人DNA衍生的G-CSF。
NEUG作为一次性1型玻璃小瓶中的无菌、冻干制剂提供,并在2-8℃保存。用1.0ml注射用无菌水复溶后,各小瓶包含pH 6.0的20mM磷酸钠、180mM甘露醇、60mM无水海藻糖、0.01%(w/v)聚山梨酯80溶液中的50mg/ml(可递送50mg/小瓶)NEUG。应注意,NEUG也以液体提供,在小管中或者在预装的注射器中。
图14显示了II期所用的NEUG药物产品的组成。下表8显示了I期和II期所用NEUG制剂间的区别。

I期所用制剂相当稳定,保存期至少2年。研究表明,更高离子强度和更低pH进一步稳定更高浓度(>25mg/mL)下的API(数据未显示)。为此,II期制剂pH较低(6.0相对7.2)和磷酸浓度较高(20相对10mM)。受迫降解研究表明,该制剂保护液体状态下的所述药物免于剧烈振荡、重复冻融和浓度引起的聚集。II期制剂的冻干还产生了成型良好的块。
市售的Neulasta (聚乙二醇化非格司亭)在用于皮下注射的0.6ml预装注射器中提供。各注射器包含无菌、透明、无色、无防腐剂溶液中的6mg聚乙二醇化非格司亭(基于蛋白质重量),所述溶液(pH 4.0)包含注射用水中的乙酸(0.35mg)、山梨糖醇(30.0mg),聚山梨酯20(0.02mg),钠(0.102毫克)。美国药典。
(聚乙二醇化非格司亭)在用于皮下注射的0.6ml预装注射器中提供。各注射器包含无菌、透明、无色、无防腐剂溶液中的6mg聚乙二醇化非格司亭(基于蛋白质重量),所述溶液(pH 4.0)包含注射用水中的乙酸(0.35mg)、山梨糖醇(30.0mg),聚山梨酯20(0.02mg),钠(0.102毫克)。美国药典。
通过皮下给药施用NEUG(30、40、50或60mg)或Neulasta (聚乙二醇化非格司亭)(6mg)。
(聚乙二醇化非格司亭)(6mg)。
剂量原理
I期的数据表明,300和450μg/kg的NEUG剂量是安全且耐受良好的。此外,和已批准的聚乙二醇化非格司亭固定剂量相比,NEUG的两个剂量在接受细胞毒性化疗的乳腺癌患者中产生相似的ANC情况。ANC曲线的AUC作为效果的单点度量。在这些治疗组中,就AUCANC而言没有统计学显著差异,然而,450μg/kg组的AUC略高于300μg/kg组所观察到的,并与聚乙二醇化非格司亭组所观察到的几乎一致(图23)。基于现有数据,估计300μg/kg NEUG不如聚乙二醇化非格司亭,450μg/kg大致是提供与聚乙二醇化非格司亭相当效果的最小必需剂量。
固定剂量意在确定足以为患者提供具功效和安全性的剂量,无论患者体重如何。基于I期结果,估计450μg/kg NEUG可能是提供与聚乙二醇化非格司亭相似功效的最小必需剂量,而>300μg/kg设为II期中进一步评估的最小剂量。为选择NEUG的固定剂量,模拟II期的所述患者群体(乳腺癌)。采用40-100kg的重量范围,30mg固定剂量为最重患者提供了最小剂量(300μg/kg或0.3mg/kg),而40mg的固定剂量下约75%的患者至少接受目标剂量450mg/kg。因此,II期评估所选剂量为30mg、40mg和50mg。这些剂量为平均70kg的患者分别提供0.42、0.57和0.71mg/kg剂量。
表9中提供了基于本试验所评估固定剂量的每公斤等效剂量。

NEUG的非临床安全性为这些剂量下的预期安全性提供了补充支持。在这些固定剂量下,患者的接触(AUC和Cmax)预期低于猴中的耐受良好剂量的接触。例如,猴中1mg/kg耐受良好剂量下的Cmax和AUC比0.45mg/kg下患者的接触高12倍,提示对患者中更高剂量的评估存在进一步的安全性限度,而且在猴中的重复剂量毒性研究中,高达且包括10mg/kg的剂量耐受良好。已证明,高达0.3mg/kg的聚乙二醇化非格司亭剂量在患者中是安全的。
4.研究特点
a.研究方案及持续时间
本研究是对照的随机试验,在约330位排定最多接受4个剂量多柔比星/多西他赛的乳腺癌对象中进行。本研究在45个临床地点进行,由先导试验阶段和主试验阶段这两个阶段组成。
先导试验阶段,A部分,由双向随机研究组成,以评估NEUG相对聚乙二醇化非格司亭的安全性和效果,依序使用下列剂量:NEUG 30mg(N=10)相对聚乙二醇化非格司亭(N=5);NEUG 40mg(N=20)相对聚乙二醇化非格司亭(N=10)和NEUG 50mg(N=20)相对聚乙二醇化非格司亭(N=10)。在进一步的研究中,还可测试NEUG 60mg(N=20)相对聚乙二醇化非格司亭(N=10)。在A部分先导试验阶段,对象以2∶1的NEUG与聚乙二醇化非格司亭比例随机分组,30mg组中共有10个对象,其它各组中共有20个对象。在各疗程中,化疗治疗24小时后将NEUG或聚乙二醇化非格司亭给予对象。在化疗接触和整体定位前,为平衡治疗组间的重量,采用分层随机化将对象分入治疗组(<50kg、≥50kg且<80kg、或≥80kg)。
先导试验阶段后,255个对象随机(1∶1∶1)分到聚乙二醇化非格司亭和NEUG的两个剂量组,这两个剂量在先导试验阶段(3分支、平衡的平行随机化阶段)耐受良好且效果与聚乙二醇化非格司亭更为相当。在各疗程中,化疗治疗24小时后给予NEUG或聚乙二醇化非格司亭。为平衡治疗组间的重量,采用分层随机化将对象分入治疗组(<50kg、≥50kg且<80kg、或≥80kg)。
在先导试验阶段,在研究进行的基础上审查不良事件。除非进行的数据审查提示有安全性问题,进行从30直至50mg的剂量递增。若第1疗程Neugranin40mg的ANC曲线劣于聚乙二醇化非格司亭患者中所观察到的曲线,且Neugranin 50mg是安全的,那么,可在总共30个对象的组中按2∶1的Neugranin60mg与聚乙二醇化非格司亭比例随机化补充分支。
比较NEUG各剂量水平相对聚乙二醇化非格司亭的安全性和功效。表10小结了II期A和B部分的患者分配。

安全性评估:
通过随时间推移评价AE的类型、频率和严重程度,临床实验测试(血液学和临床化学)的变化,免疫原性,身体检查,以及监控生命体征,来评估NEUG的安全性。根据国家癌症研究所常见的不良事件术语标准(2003年12月12日的NCI-CTCAE 3.0版)对所有AE和实验毒性作评级。
从开始研究药物给药直至任何研究药物的最终用药后的30天内,采集所有不良事件。从知情同意直至任何研究药物的最终用药后的30天内,采集严重不良事件(SAE)。按评估规划(Schedule of Assessments)所列获得实验室评估。
c.附随治疗
化疗
本试验的化疗方案由治疗第1天通过静脉输液依次给予多柔比星60mg/m2和多西他赛75mg/m2组成,最多为4个21天的疗程。
接受每个疗程的治疗前,要求对象的嗜中性粒细胞绝对计数(ANC)>1000/mm3且血小板数>100,000/mm3。为了血液学恢复,治疗最多可以延迟2周。对于3-4级非血液学毒性,2项3-4级感染事件或4级血小板减少症,允许化疗剂量降低25%。在参与试验期间,禁止使用预防性抗生素或其它造血生长因子。
据报道,联用多柔比星和多西他赛在乳腺癌患者中具有显著的临床活性。然而,所述联用是高度骨髓抑制性的,3级或4级嗜中性粒细胞缺少症的发病率比其它标准方案高。
尽管添加CSF,多柔比星和多西他赛的联用仍引起79%的患者出现4级嗜中性粒细胞缺少症,以及9-18%的发热性嗜中性粒细胞缺少症比率。这一多柔比星/多西他赛方案用于预防嗜中性粒细胞缺少症及其并发症的新药的研究。因此,多柔比星和多西他赛的联用是研究NEUG这类新试剂效力的合适化疗方案。
多柔比星
药理学数据
盐酸多柔比星是从波塞链霉菌表灰变种(streptomyces peucetius varcaesius)中获得的大环抗生素,与DNA碱基对直接结合(插入)并抑制DNA和DNA依赖性RNA合成以及蛋白质合成。多柔比星在细胞周期各阶段都有活性,但在S期细胞毒性最大。该药物主要通过肝排除,肾脏清除是次要的。
药学数据
该药物以10、20、50、100或200mg的小瓶市售。冻干制剂可以用无菌注射用水、注射用5%葡萄糖溶液或0.9%盐水复溶。
副作用和毒性
多柔比星的剂量限制毒性在于骨髓抑制,主要是白细胞缺少症,其最低点约在10-14天,以及心脏毒性,包括罕见的急性心包炎-心肌炎综合征和迟延的与累积剂量相关的心肌病。
预期毒性有标志性脱发和中度恶心/呕吐。有报道在不慎外渗部位产生局部皮肤和组织损伤的外渗反应,口腔炎,皮肤(特别是指甲床)的色素沉着以及此前放射部位的“回忆”现象。
多西他赛
药理学数据
多西他赛是半合成的紫杉类,与游离微管蛋白结合并促进稳定微管的组装,干扰有丝分裂和细胞复制(M期细胞周期特异性)。多西他赛广泛结合蛋白,在肝中广泛代谢,在给药后7天内约有75%通过粪便排出。
药学数据
多西他赛(TaxotereTM,赛诺菲安万特公司)在80mg/2mL或20mg/0.5ml的单剂量小瓶中提供,附带稀释剂(含13%乙醇的注射用水)小瓶。每毫升Taxotere含40mg多西他赛(无水)和1080mg聚山梨酯80。
副作用和毒性
多西他赛不可用于对多西他赛或其它用聚山梨酯80配制的药物如依托泊苷和维生素E有严重过敏性反应史的患者。
不得再次刺激经历严重过敏性反应的患者。接受多西他赛的所有患者应当如下所述用皮质类固醇作前驱用药。
轻度到中度的肝损伤导致代谢延迟27%和系统性接触提高38%(AUC)。多西他赛不得用于SGOT和/或SGPT>1.5倍正常限度以及碱性磷酸酶>2.5倍正常限度的患者。在III期研究中,尽管有皮质类固醇前驱用药,仍有17%(中度)和6%(重度潴留)患者出现液体潴留。观察到严重的感觉神经症状(感觉异常、感觉失常、疼痛)。
预期的副作用包括骨髓抑制,主要是白细胞减少症,其最低点约在第9天,到第15-21天恢复。有报道脱发、指甲和皮肤变化、口腔炎、肌痛/关节痛、恶心/呕吐和低血压。
化疗剂量、给药以及剂量调整
各治疗疗程的第1天,给予化疗剂(多柔比星,然后是多西他赛)。
通过IV推注给予60mg/m2剂量的多柔比星,经侧支注入静脉或中央静脉导管给药以免渗出损伤。
将75mg/m2多西他赛稀释于250mL 0.9%盐水或5%葡萄糖溶液中,用聚乙烯管输液套件静脉给药,用时约1小时。在临多西他赛注入前和刚结束后,获得生命体征。
将经历重度过敏性反应或非血液学毒性阻止进一步化疗疗程的患者从研究治疗中除去,但完成随访.
化疗前驱用药
从多西他赛给药前一天开始连续3天口服(需要时IV)皮质类固醇(例如地塞米松8mg BID),以减少液体潴留和过敏性反应的发生率和严重程度。
由主治医生自由裁量止吐剂或其它前驱用药(例如H2拮抗剂)的使用和选择。
药物禁忌
在研究过程中以及下面指明的补充时间内对象不得接受任何下列药物或方法:
1.在第1疗程化疗72小时内的全身性抗生素。
2.在启动研究试剂的30天内以及试验期间的其它在研药剂
3.在NEUG用药后14天内不应进行后续的化疗疗程。
4.在试验期间,除非发生持续或发热性嗜中性粒细胞缺少症,不得接受细胞因子、其它造血生长因子和预防性抗生素。若对象在筛选期间到试验第0天之间任何时间进行G-CSF治疗,则没有资格接受NEUG并终止其研究。
允许的药物
对象可以继续他们的基础药物。若可能,整个研究过程中各药物的每日剂量应保持一致。若出于研究者认为必须的任何理由,对象需要其它药物或改变剂量,应记录所用药物、给药途径、及其给予所针对的适应症。
将经历重度过敏性反应或非血液学毒性妨碍进一步化疗疗程的患者从研究治疗中除去,并完成随访.
d.药代动力学
在第1疗程中,对所有接受NEUG的对象取样分析血清NEUG浓度。采用NEUG特异性的夹心酶联免疫吸附试验(ELISA)检测该药物。用WinNonlin企业版5.0版或更高版本对血清药物浓度-时间数据作PK分析,使用非房室模型分析或基于模型的分析。测得下列PK参数:曲线下面积(AUC0-∞)、清除(CL/F)、分布容积(Vz/F)、最大浓度(Cmax)、吸收半衰期(t1/2,abs)消除半衰期(t1/2,elim)和平均停留时间(MRT)。
e.免疫原性
在各NEUG疗程的第1天用药前和治疗检查的最后(最后用药后约30天),获取接受NEUG对象的血清样品用于针对NEUG的抗体。若在研究中的任何时间,有对象发生阳性抗NEUG抗体响应,在最终NEUG剂量约6个月后,获取重复样品;若该样品为阳性,在12个月时获取样品。该方案后面修改为要求所有对象的6和12月免疫原性样品。
5.结果
a.概述
统计方法
选定本试验主阶段(B部分)每个分支的样本大小约85个对象,提供91%的强度以确立NEUG在第1疗程的重度嗜中性白细胞减少症(DSN)平均持续时间主要终点方面不劣于聚乙二醇化非格司亭,不劣于的限度为1天,对多参数测试调整(通过Hochberg方法)的总体单边显著水平为0.025。根据两个独立组的正态近似计数样本大小,估计第1疗程DSN的处理组内标准偏差为1.6天,以及第1疗程DSN主要终点不可评估的最大比例为20%。
根据3分支随机化阶段(A部分)的对象,在两个选定NEUG剂量(40mg和50mg)和聚乙二醇化非格司亭间比较功效。
次要功效分析包括2-4各化疗疗程中的DSN、1-4各疗程ANC最低值的深度、按疗程或跨疗程的FN(定义为ANC<0.5x109/L伴有偶发性口腔相等温度>38.2℃)比例、以及所有疗程中ANC恢复到>1.5x109/L的时间。
用适当的统计方法分析次要功效分析相关数据。通过描述的统计方法分析安全性、PK和免疫原性参数。
对于不良事件的频率和严重程度和实验室毒性评级,给出了计数和分级。
功效度量
在第1、3天和从第5天起每天获取全血细胞计数(“CBC”)直至在最低点后ANC>2.0x109/L,然后每周计数2次直至治疗结束。
b.II期A部分的功效
本研究的先导试验阶段征募的78个对象中,13个对象未完成本研究,3个用NEUG 30mg治疗(27.3%),3个用NEUG 40mg治疗(14.3%),3个用NEUG 50mg治疗(15.0%),还有4个用聚乙二醇化非格司亭治疗(15.4%)。提前终止的最常见原因是撤回同意(7个对象)和研究者的决定(3个对象)。一个NEUG 30mg对象因为不良事件(糖尿病足)而退出。
在各化疗疗程中,治疗组之间的重度嗜中性白细胞减少症发生率和重度嗜中性白细胞减少症平均持续时间(DSN)相似;但是,ANC恢复的时间和发热性嗜中性白细胞减少症的发生率提示,NEUG 30mg不如NEUG 40mg、NEUG 50mg或聚乙二醇化非格司亭有效。
在第1疗程,NEUG 30mg、40mg、50mg和聚乙二醇化非格司亭组中对象经历发热性嗜中性白细胞减少症的比例分别为20.0%、9.5%、10.0%和8.0%。在第2-4疗程中仅观察到3个补充对象的发热性嗜中性白细胞减少症,NEUG30mg、NEUG 40mg和聚乙二醇化非格司亭组中各1个。图5显示了接受NEUG30或聚乙二醇化非格司亭、此后出现4级嗜中性白细胞减少症的患者亚组的ANC曲线。
在第1疗程,NEUG 30mg(0.9天)、NEUG 50mg(1.1天)和聚乙二醇化非格司亭(0.9天)的平均DSN相似。尽管NEUG 40mg(1.6天)的平均DSN稍长于其它3组治疗,治疗间的差异低于1天,这是主试验阶段中认为治疗等效的标准。所有4个治疗组的DSN中位数都是0或1天。
3级或4级嗜中性白细胞减少症的发生率和持续时间的总结统计得出相似模式,即NEUG 30mg、NEUG 50mg和聚乙二醇化非格司亭组有相似结果,而NEUG 40组的3级或4级嗜中性白细胞减少症的发生率和持续时间稍高于其它治疗组。先导试验阶段对象的数量(A部分)相当小,所观察到的差异不具统计学意义。在本研究的B部分、3分支随机阶段中,选择NEUG 40mg和NEUG 50mg进行进一步评估。
c.II期B部分的功效
本研究的主试验阶段征募的256个对象中,18个对象未完成本研究;10个用NEUG 40mg治疗(11.6%),5个用NEUG 50mg治疗(6.0%),3个用聚乙二醇化非格司亭治疗(3.5%)。提前终止的最常见原因是撤回同意(7个对象)和AE(4个对象),包括2例死亡。研究者认为所有这些AE都与研究药物或化疗无关。在主试验阶段,1个(1.2%)NEUG 40mg对象在用研究药物治疗前退出。
表11小结了第1疗程中重度嗜中性白细胞减少症的发生率和持续时间。

重度嗜中性白细胞减少症的发生率从聚乙二醇化非格司亭组的58.1%到NEUG 50mg组的65.5%。治疗效果没有统计学显著性(p=0.559)。治疗组第1疗程的DSN相当,NEUG 40mg、NEUG 50mg和聚乙二醇化非格司亭组的平均值分别为1.0、1.3和1.2天。NEUG和聚乙二醇化非格司亭之间差异的95%和97.5%双侧置信区间对两个NEUG剂量都严格低于1天。这一分析确立了NEUG不劣于聚乙二醇化非格司亭。在各治疗疗程中,第2-4疗程的重度嗜中性白细胞减少症和3级或4级嗜中性白细胞减少症的发生率低于第1疗程。第2-4疗程的3级或4级嗜中性白细胞减少症的平均DSN和平均持续时间低于第1疗程。在治疗疗程内,所述治疗相似,对于任何化疗疗程内任何这些参数,治疗效果没有显著差异。
在分成重量四分位组的患者中比较DSN以确定NEUG固定剂量是否为所有体重的患者提供了充分的支持。这些结果显示,所有体重组都得到充分支持,在体重亚组间,平均DSN没有显著差异(表12)。
表13总结了所有疗程的发热性嗜中性白细胞减少症。在第1疗程中,NEUG40mg、50mg和聚乙二醇化非格司亭组中经历发热性嗜中性白细胞减少症的对象比例分别为2个对象(3.5%)、5个对象(6.0%)和2个对象(2.3%)。在第2-4疗程中,仅有3个补充对象观察到发热性嗜中性白细胞减少症,2个是NEUG 40mg组的对象且1个是聚乙二醇化非格司亭组的对象。在任何化疗疗程中,治疗效果没有统计学显著性。

第2-4疗程重度嗜中性白细胞减少症的持续时间在各治疗间没有显著差异(表14)。

Neugranin 40mg、NEUG 50mg和聚乙二醇化非格司亭组的ANC恢复(>1.5x109/L)平均时间分别为2.0、2.1和2.6天(表15)。ANC最低值的深度或达到最低值的时间在治疗组间没有显著差异。


d.II期B部分的药代动力学
采用量化下限为6.312ng/mL的已验证夹心ELISA测定血清Neugranin浓度。除用一级吸附、一级消除的一室模型确定吸收半衰期外,采用非房室模型技术计算药代动力学参数。用WinNonlin专业版(5.0.1版)进行模拟。测定II期NEUG治疗的所有对象在化疗第1疗程中的血清NEUG浓度。在II期A部分,NEUG消除半衰期中位数在30mg剂量组为33小时,40mg剂量组为46小时,50mg剂量组为18小时(表16)。在B部分,NEUG消除半衰期的中位数在40mg剂量组为40小时,50mg剂量组为39小时(表17)。A部分过程中的PK取样(给药前、3h、6h、12h、24h、第3天、第5-9天、第11天)比B部分(给药前、第3天、第5-8天)更频繁。

采用已验证的夹心ELISA测定II期聚乙二醇化非格司亭治疗的所有对象在化疗第1疗程中的血清聚乙二醇化非格司亭浓度。在A部分,聚乙二醇化非格司亭消除半衰期的中位数为约40小时。在B部分,聚乙二醇化非格司亭消除半衰期的中位数为约50小时。据报道,非格司亭的清除半衰期为3-4小时,聚乙二醇化非格司亭(取决于剂量)为42-67.5小时。
e.免疫原性
在研究参与者中,在Neugranin治疗对象中有1例证实有抗G-CSF/新表位抗体应答,在聚乙二醇化非格司亭治疗组中有1例抗G-CSF应答,或分别为0.5%和0.9%(表18)。两个案例中,所述对象都在给药前样品中有升高的非特异性结合。

NEUG治疗后,在患者中发现证实阳性抗体的水平极低,在重复剂量后应答幅度未有明显提高(数据未显示)。在聚乙二醇化非格司亭治疗的患者中,观察到异常的高度非特异性背景结合;但是,只在第2疗程治疗后发现短暂的确定抗体应答(数据未显示)。没有中和性抗体应答。
在该人群中,抗HSA抗体以低水平天然产生,对象中6.9%在给药前评估中测得HSA抗体阳性。在4个NEUG治疗对象,1.8%,中观察到治疗引起的HSA抗体(表19)。所有应答都是短暂且微弱的。3个应答在第一治疗疗程后出现,在第2、3和4疗程后检测不到。在第三次治疗后发生1个应答,但在第四次治疗后的30天随访时检测不到(数据未显示)。
f.II期B部分中治疗引起的不良事件
在II期B部分中,各治疗组中≥90%的对象经历至少一项治疗引起的不良事件(TEAE),有至少一项研究药物相关TEAE的对象百分比从聚乙二醇化非格司亭组的23.1%到Neugranin 50mg组的35.0%。在NEUG 30mg组中有至少一项SAE的对象百分比最高(30%),但在其它三个治疗组中约为15%。SAE中无一与研究药物相关。一个患者(NEUG 30mg)因糖尿病足从研究中退出,认为该糖尿病足与研究药物无关。在B部分,除8个对象(2个为NEUG40mg、3个为NEUG 50mg、3个为聚乙二醇化非格司亭)外的所有对象都有至少一项TEAE。有至少一项研究药物相关TEAE的对象百分比在NEUG 50mg组为20.2%,在NEUG 40mg组为22.4%,在接受聚乙二醇化非格司亭的对象中为22.1%。两个对象(1个为NEUG 40mg、1个为聚乙二醇化非格司亭)在研究过程中死亡,每个治疗组中有6-8个对象经历至少一项SAE。认为没有死亡或SAE与研究药物相关。
当考虑到NEUG 30mg剂量的样本大小时,A部分以及B部分中治疗组间的TEAE总数相似。在A和B部分中,NEUG和聚乙二醇化非格司亭的CTC 3级或3级以上TEAE的百分数和与研究药物相关TEAE的百分数都相似。
g.剂量反应
II期的结果表明,40和50mg的NEUG固定剂量在用骨髓毒性化疗治疗的乳腺癌对象中提供了与6mg聚乙二醇化非格司亭相当的安全性和功效。尽管40mg治疗组的平均DSN稍低于50mg组的平均DSN,这些差异在统计上不显著。考虑体重调整剂量和固定剂量组群时,都观察到AUCANC(第1疗程0-15天)的剂量反应(图24)。30mg组的AUCANC稍低于聚乙二醇化非格司亭组,表明本研究中30mg固定剂量效力较低,而40和50mg群组的AUCANC是剂量相关的,且大于(尽管不显著)聚乙二醇化非格司亭治疗对象的AUCANC。根据上述分析,当NEUG基于体重调整(mg/kg)给药时有明显的剂量反应。但是,与II期B部分第1疗程的DSN的比较显示,所有重量四分位组的患者都得到充分支持,DSN在治疗分支(40和50mg NEUG以及聚乙二醇化非格司亭)间或在重量调整剂量(mg/kg)间都没有显著改变。此外,没有迹象表明固定剂量可能引起较轻患者中安全性情况的改变,相关不良事件(特别是骨痛,数据未显示)的发生率和严重程度与每公斤体重接受的剂量没有关联,与聚乙二醇化非格司亭的情况也没有差异。
****
对本领域的技术人员显而易见的是,可以对本发明的方法和组合物进行各种修改和变动而不偏离本发明的精神或范围。因此,本发明旨在覆盖本发明的修改和变动,所述修改和变动在所附权利要求及其等同方案的范围之内。
Claims (22)
1.一种在人对象中治疗或预防嗜中性白细胞减少症的方法,包括对显示嗜中性白细胞减少症或有产生嗜中性白细胞减少症风险的人对象施用有效量的重组人白蛋白-人粒细胞集落刺激因子以治疗对象。
2.一种在人对象中治疗或预防白细胞减少症的方法,包括对显示白细胞减少症或有产生白细胞减少症风险的人对象施用有效量的重组人白蛋白-人粒细胞集落刺激因子以治疗对象。
3.如权利要求1或2所述的方法,其特征在于,所述人对象患有非骨髓恶性肿瘤并正接受至少一种骨髓抑制型抗癌药,所述药物与发热性嗜中性白细胞减少症的临床显著发病率相关。
4.一种在患有非骨髓性恶性肿瘤并在接受至少一种骨髓抑制型抗癌药的人对象中降低感染发病的方法,所述感染表现为发热性嗜中性白细胞减少症,所述抗癌药物与发热性嗜中性白细胞减少症的临床显著发病率相关,所述方法包括给予对象有效剂量的人白蛋白-人粒细胞集落刺激因子以治疗对象。
5.如权利要求1-4中任一项所述的方法,其特征在于,
(a)对象中4级嗜中性白细胞减少症消除;
(b)对象中4级嗜中性白细胞减少症减轻;
(c)对象中重度嗜中性白细胞减少症的持续时间缩短;
(d)对象中4级嗜中性白细胞减少症的持续时间低于5天;
(e)对象中3级嗜中性白细胞减少症的持续时间消除;
(f)对象中3级嗜中性白细胞减少症的持续时间减少;或
(g)其任意组合。
6.如权利要求1-5中任一项所述的方法,其特征在于,所述给予重组人白蛋白-人粒细胞集落刺激因子引起血液白细胞计数(WBC)升高。
7.如权利要求1-6中任一项所述的方法,其特征在于,
(a)对象中嗜中性粒细胞的数量增加;
(b)对象中嗜中性粒细胞数量的降低被抑制;
(c)对象中嗜中性粒细胞绝对计数(ANC)的最低值增加;
(d)对象中恢复的ANC增加;
(e)对象中ANC恢复的时间缩短;或
(f)其任意组合。
8.如权利要求1-7中任一项所述的方法,其特征在于,所述给予对象的重组人白蛋白-人粒细胞集落刺激因子的量选自下组:
(a)从约50μg/kg到约450μg/kg;
(b)约50μg/kg;
(c)约150μg/kg;
(d)约300μg/kg;
(e)约450μg/kg;
(f)从约30mg到约60mg;
(g)约30mg;
(h)约40mg;
(i)约50mg;
(j)约60mg;或
(k)其任意组合。
9.如权利要求1-8中任一项所述的方法,其特征在于,所述嗜中性白细胞减少症选自原发性嗜中性白细胞减少症、急性嗜中性白细胞减少症、重度慢性嗜中性白细胞减少症(SCN)、重度先天性嗜中性白细胞减少症(Kostmann综合征)、重症婴儿遗传性粒细胞缺乏症、良性嗜中性白细胞减少症、周期性嗜中性白细胞减少症、慢性特发性嗜中性白细胞减少症、继发性嗜中性白细胞减少症、嗜中性白细胞减少症相关综合征和免疫介导的嗜中性白细胞减少症。
10.如权利要求1-9中任一项所述的方法,其特征在于,所述嗜中性白细胞减少症是由辐射、酗酒、药物、过敏性疾病、自体免疫疾病、T-γ淋巴增生性疾病(T-γLPD)、脊髓发育不良、骨髓纤维化、丙种球蛋白异常血症、阵发性夜间血红蛋白尿、癌症、维生素B12缺乏、叶酸缺乏、病毒感染、细菌感染、脾脏疾病、血液透析、或移植、白血病、骨髓瘤、淋巴瘤、浸润并置换骨髓的转移性实体瘤、毒素、骨髓衰竭、Schwachman-Diamond综合征、软骨-毛发发育不全、先天性角化不良、IB型糖原贮积病、各种原因导致的脾肿大、骨髓细胞或其前体的内在缺陷造成或与之相关。
11.如权利要求3所述的方法,其特征在于,所述重组人白蛋白-人粒细胞集落刺激因子给药的时间选自下组:
(a)骨髓抑制性抗癌药物给药后至少12小时;
(b)骨髓抑制性抗癌药物给药后至少18小时;
(c)骨髓抑制性抗癌药物给药后至少24小时。
12.如权利要求11所述的方法,其特征在于,在骨髓抑制性抗癌药物之前给予重组人白蛋白-人粒细胞集落刺激因子引起WBC升高。
13.如权利要求11-12中任一项所述的方法,其特征在于,在化疗前给予重组人白蛋白-人粒细胞集落刺激因子引起ANC升高。
14.如权利要求3或11-13中任一项所述的方法,其特征在于,所述非骨髓恶性肿瘤包括乳腺癌。
15.如权利要求3或11-14中任一项所述的方法,其特征在于,所述骨髓抑制性抗癌药物包括多柔比星和多西他赛。
16.如权利要求15所述的方法,其特征在于,对至少一个疗程,通过静脉输液在同一天依次给予约50mg/m2多柔比星和约75mg/m2多西他赛。
17.如权利要求15所述的方法,其特征在于,对至少一个疗程,通过静脉输液在同一天依次给予约60mg/m2多柔比星和约75mg/m2多西他赛。
18.如权利要求3或11-17中任一项所述的方法,其特征在于,所述ANC和WBC在选自下组的时间段内回复正常:
(a)截止化疗后的第10天;
(b)截止化疗后的第11天;
(c)截止化疗后的第12天;
(d)截止化疗后的第13天;
(e)截止化疗后的第14天;或
(f)截止化疗后的第15天。
19.如权利要求3或11-18所述的方法,其特征在于,在化疗给药后第14天用重组人白蛋白-人粒细胞集落刺激因子治疗的患者中所述ANC的升高低于用相等剂量聚乙二醇化非格司亭治疗的患者中ANC的升高。
20.如权利要求1-19中任一项所述的方法,其特征在于,所述给予重组人白蛋白-人粒细胞集落刺激因子引起淋巴细胞、单核细胞、嗜酸性粒细胞、嗜碱性粒细胞、或其任意组合的升高。
21.如权利要求1-20中任一项所述的方法,其特征在于,所述对象中淋巴细胞、单核细胞、嗜酸性粒细胞或嗜碱性粒细胞或其任意组合的数量增加。
22.如权利要求1-21中任一项所述的方法,其特征在于,所述对象中淋巴细胞、单核细胞、嗜酸性粒细胞或嗜碱性粒细胞或其任意组合的数量降低受到抑制。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14543609P | 2009-01-16 | 2009-01-16 | |
| US14544009P | 2009-01-16 | 2009-01-16 | |
| US61/145,440 | 2009-01-16 | ||
| US61/145,436 | 2009-01-16 | ||
| PCT/US2010/021241 WO2010083439A2 (en) | 2009-01-16 | 2010-01-15 | Recombinant human albumin-human granulocyte colony stimulating factor for the prevention of neutropenia |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN102378635A true CN102378635A (zh) | 2012-03-14 |
Family
ID=42260335
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN2010800127233A Pending CN102378635A (zh) | 2009-01-16 | 2010-01-15 | 重组人白蛋白-人粒细胞集落刺激因子融合蛋白的新稳定制剂 |
| CN201080012749.8A Expired - Fee Related CN102395379B (zh) | 2009-01-16 | 2010-01-15 | 重组人白蛋白-人粒细胞集落刺激因子融合蛋白的新稳定制剂 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201080012749.8A Expired - Fee Related CN102395379B (zh) | 2009-01-16 | 2010-01-15 | 重组人白蛋白-人粒细胞集落刺激因子融合蛋白的新稳定制剂 |
Country Status (16)
| Country | Link |
|---|---|
| US (4) | US20100227818A1 (zh) |
| EP (2) | EP2387420A2 (zh) |
| JP (2) | JP5753095B2 (zh) |
| KR (2) | KR20110132327A (zh) |
| CN (2) | CN102378635A (zh) |
| AU (2) | AU2010204547B2 (zh) |
| BR (2) | BRPI1004940A2 (zh) |
| CA (2) | CA2749786A1 (zh) |
| EA (2) | EA023344B1 (zh) |
| IL (2) | IL214051A0 (zh) |
| MX (2) | MX2011007582A (zh) |
| NZ (2) | NZ594056A (zh) |
| SG (3) | SG196821A1 (zh) |
| UA (2) | UA103221C2 (zh) |
| WO (2) | WO2010083439A2 (zh) |
| ZA (1) | ZA201105171B (zh) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107206053A (zh) * | 2014-11-03 | 2017-09-26 | 华鸿新药公司 | 用于治疗血细胞减少症或减少血细胞减少症的持续时间的佛波醇酯组合物和方法 |
| CN111383745A (zh) * | 2018-12-29 | 2020-07-07 | 医渡云(北京)技术有限公司 | 计算机数据处理方法、装置、存储介质及设备 |
| CN112105363A (zh) * | 2018-02-01 | 2020-12-18 | 大连万春布林医药有限公司 | 用于通过施用普那布林和g-csf剂来减少化学疗法诱导的嗜中性白血球减少症的组合物和方法 |
| CN114364393A (zh) * | 2019-05-31 | 2022-04-15 | 光谱制药有限公司 | 使用g-csf蛋白质复合物治疗的方法 |
| CN114746104A (zh) * | 2019-12-05 | 2022-07-12 | 韩美药品株式会社 | 治疗由化疗或放疗引起的中性粒细胞减少症的方法 |
Families Citing this family (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102378635A (zh) | 2009-01-16 | 2012-03-14 | 特瓦制药工业有限公司 | 重组人白蛋白-人粒细胞集落刺激因子融合蛋白的新稳定制剂 |
| EP2384759A1 (en) | 2010-05-06 | 2011-11-09 | Suomen Punainen Risti Veripalvelu | Sulphated hyaluronic acid in combination with G-CSF for use in mobilising blood stem cells |
| SG186094A1 (en) * | 2010-06-07 | 2013-01-30 | Amgen Inc | Drug delivery device |
| CN102628869B (zh) * | 2012-04-19 | 2014-04-02 | 上海蓝怡科技有限公司 | 提高甲胎蛋白抗体冻干稳定性的制剂 |
| EP2945703A4 (en) * | 2013-01-15 | 2016-08-31 | Teva Pharma | ALBU-BCHE FORMULATIONS, PREPARATION THEREOF AND USES THEREOF |
| WO2014147489A2 (en) | 2013-03-15 | 2014-09-25 | Teva Pharmaceutical Industries Ltd. | Recombinant human albumin-human granulocyte colony stimulating factor for the prevention of neutropenia in pediatric patients |
| AU2016229294B2 (en) | 2015-03-06 | 2021-11-04 | Beyondspring Pharmaceuticals, Inc. | Method of treating cancer associated with a RAS mutation |
| BE1023343B1 (nl) * | 2015-05-20 | 2017-02-09 | Mycartis Nv | Opslagbuffer |
| JP6969848B2 (ja) | 2015-07-13 | 2021-11-24 | ビヨンドスプリング ファーマシューティカルズ,インコーポレイテッド | プリナブリン組成物 |
| US11020455B2 (en) * | 2015-07-30 | 2021-06-01 | Endor Technologies, S.L | Colony stimulating factor for use in pancreatic or colon cancer treatment |
| KR20180105685A (ko) | 2016-02-08 | 2018-09-28 | 비욘드스프링 파마수티컬스, 인코포레이티드. | 투카레솔 또는 이의 유사체를 함유하는 조성물 |
| JP7025416B2 (ja) | 2016-06-06 | 2022-02-24 | ビヨンドスプリング ファーマシューティカルズ,インコーポレイテッド | 好中球減少症を低減させるための組成物および方法 |
| CN106222221A (zh) * | 2016-08-05 | 2016-12-14 | 山东科兴生物制品有限公司 | 制备重组人粒细胞刺激因子原液的纯化方法 |
| ES2985905T3 (es) | 2016-11-22 | 2024-11-07 | Lts Device Tech Ltd | Aparato para suministrar una sustancia terapéutica |
| WO2018129381A1 (en) | 2017-01-06 | 2018-07-12 | Beyondspring Pharmaceuticals, Inc. | Tubulin binding compounds and therapeutic use thereof |
| BR112019015974A2 (pt) | 2017-02-01 | 2020-03-31 | Beyondspring Pharmaceuticals, Inc. | Método para reduzir neutropenia |
| WO2018187786A1 (en) * | 2017-04-07 | 2018-10-11 | La Jolla Institute For Allergy And Immunology | Unipotent neutrophil progenitor cells, methods of preparation, and uses thereof |
| CN109420159A (zh) * | 2017-08-23 | 2019-03-05 | 江苏泰康生物医药有限公司 | 一种重组蛋白药物的新型稳定制剂 |
| KR20200112881A (ko) | 2018-01-24 | 2020-10-05 | 비욘드스프링 파마수티컬스, 인코포레이티드. | 플리나불린의 투여를 통해 혈소판감소증을 감소시키는 조성물 및 방법 |
| US11583633B2 (en) | 2018-04-03 | 2023-02-21 | Amgen Inc. | Systems and methods for delayed drug delivery |
| EP3632487B1 (en) | 2018-10-05 | 2024-06-12 | LTS Device Technologies Ltd | Triggering sequence |
| US11684655B2 (en) | 2019-05-31 | 2023-06-27 | Spectrum Pharmaceuticals, Inc. | Methods of treating neutorpenia using G-CSF protein complex |
| CN112316120A (zh) * | 2019-08-05 | 2021-02-05 | 天津派格生物技术有限公司 | 使用低动员型g-csf有效且安全治疗粒细胞减少症的方法 |
| KR102485892B1 (ko) * | 2020-04-09 | 2023-01-09 | 주식회사 에이프릴바이오 | 고양이 과립구 집락 자극인자 및 혈청 알부민에 대한 항원 결합 단편을 포함하는 융합 단백질 및 이의 용도 |
| CN111751552B (zh) * | 2020-06-18 | 2022-09-16 | 广州市伊川生物科技有限公司 | 一种缺血修饰白蛋白测定试剂盒及其使用方法 |
| KR102375269B1 (ko) * | 2021-01-27 | 2022-03-17 | 한미약품 주식회사 | 단백질 액상 제제 및 이의 제조방법 |
| CA3215047A1 (en) | 2021-04-09 | 2022-10-13 | Lan Huang | Therapeutic compositions and methods for treating tumors |
| US11723955B1 (en) | 2022-05-13 | 2023-08-15 | Allgenesis Biotherapeutics Inc. | VEGFR fusion protein pharmaceutical composition |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5665863A (en) * | 1992-01-31 | 1997-09-09 | Rhone-Poulenc Rorer S.A. | Polypeptides having granulocyte colony stimulating activity, their preparation and pharmaceutical compositions containing them |
| CN1729018A (zh) * | 2001-07-11 | 2006-02-01 | 马克西根控股公司 | G-csf偶联物 |
Family Cites Families (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2873012B2 (ja) | 1987-04-09 | 1999-03-24 | デルタ バイオテクノロジー リミテッド | 酵母ベクター |
| JPH01215289A (ja) | 1988-02-22 | 1989-08-29 | Toa Nenryo Kogyo Kk | 遺伝子組換えによる正常ヒト血清アルブミンaの製造方法 |
| FR2649991B2 (fr) | 1988-08-05 | 1994-03-04 | Rhone Poulenc Sante | Utilisation de derives stables du plasmide pkd1 pour l'expression et la secretion de proteines heterologues dans les levures du genre kluyveromyces |
| FR2686899B1 (fr) * | 1992-01-31 | 1995-09-01 | Rhone Poulenc Rorer Sa | Nouveaux polypeptides biologiquement actifs, leur preparation et compositions pharmaceutiques les contenant. |
| DE4242863A1 (de) * | 1992-12-18 | 1994-06-23 | Boehringer Mannheim Gmbh | Stabile lyophilisierte pharmazeutische Zubereitungen von G-CSF |
| DE19549232C2 (de) * | 1995-12-20 | 1998-05-20 | Boehringer Mannheim Gmbh | Verwendung von G-CSF in Kombination mit einem Chemotherapeutikum bei der Behandlung von Erkrankungen, die eine periphere Stammzelltransplantation erfordern |
| US7321023B2 (en) * | 1997-11-07 | 2008-01-22 | Incyte Corporation | SP16 protein |
| WO1999044630A1 (en) * | 1998-03-06 | 1999-09-10 | Chugai Seiyaku Kabushiki Kaisha | Protein-free preparations |
| US6831158B2 (en) * | 2000-01-10 | 2004-12-14 | Maxygen Holdings Ltd. | G-CSF conjugates |
| EP2213743A1 (en) | 2000-04-12 | 2010-08-04 | Human Genome Sciences, Inc. | Albumin fusion proteins |
| WO2002101412A2 (en) * | 2001-06-08 | 2002-12-19 | Powderject Vaccines, Inc. | Spray freeze-dried compositions |
| AU2002364586A1 (en) | 2001-12-21 | 2003-07-30 | Delta Biotechnology Limited | Albumin fusion proteins |
| WO2005003296A2 (en) | 2003-01-22 | 2005-01-13 | Human Genome Sciences, Inc. | Albumin fusion proteins |
| AU2003210806A1 (en) * | 2002-03-05 | 2003-09-22 | Eli Lilly And Company | Heterologous g-csf fusion proteins |
| CN1241946C (zh) * | 2002-07-01 | 2006-02-15 | 美国福源集团 | 对多种细胞具刺激增生作用的人血清白蛋白重组融合蛋白 |
| WO2004082640A2 (en) * | 2003-03-19 | 2004-09-30 | New Century Pharmaceuticals, Inc. | Human serum albumin conjugates with therapeutic compounds |
| US6972332B1 (en) * | 2004-05-20 | 2005-12-06 | Acura Pharmaceuticals, Inc. | Process for the production of opiates |
| WO2007021494A2 (en) * | 2005-08-12 | 2007-02-22 | Human Genome Sciences, Inc. | Albumin fusion proteins |
| CA2584927C (en) * | 2006-04-13 | 2011-02-08 | Wix Filtration Corp. | A body for actuating a standpipe of a replaceable filter element |
| JP2008146587A (ja) * | 2006-12-13 | 2008-06-26 | Sony Corp | 表示装置、表示プログラム、表示方法、画像提供装置、画像提供プログラム、画像提供方法及び記録媒体 |
| CN102378635A (zh) | 2009-01-16 | 2012-03-14 | 特瓦制药工业有限公司 | 重组人白蛋白-人粒细胞集落刺激因子融合蛋白的新稳定制剂 |
-
2010
- 2010-01-15 CN CN2010800127233A patent/CN102378635A/zh active Pending
- 2010-01-15 KR KR1020117018162A patent/KR20110132327A/ko not_active Ceased
- 2010-01-15 BR BRPI1004940A patent/BRPI1004940A2/pt not_active IP Right Cessation
- 2010-01-15 CN CN201080012749.8A patent/CN102395379B/zh not_active Expired - Fee Related
- 2010-01-15 MX MX2011007582A patent/MX2011007582A/es active IP Right Grant
- 2010-01-15 EP EP10701082A patent/EP2387420A2/en not_active Withdrawn
- 2010-01-15 AU AU2010204547A patent/AU2010204547B2/en not_active Ceased
- 2010-01-15 WO PCT/US2010/021241 patent/WO2010083439A2/en not_active Ceased
- 2010-01-15 JP JP2011546396A patent/JP5753095B2/ja not_active Expired - Fee Related
- 2010-01-15 KR KR1020117018153A patent/KR20110132326A/ko not_active Ceased
- 2010-01-15 CA CA2749786A patent/CA2749786A1/en not_active Abandoned
- 2010-01-15 US US12/688,754 patent/US20100227818A1/en not_active Abandoned
- 2010-01-15 EA EA201190080A patent/EA023344B1/ru not_active IP Right Cessation
- 2010-01-15 WO PCT/US2010/021235 patent/WO2010083434A2/en not_active Ceased
- 2010-01-15 EP EP10700685A patent/EP2387419A2/en not_active Withdrawn
- 2010-01-15 UA UAA201109963A patent/UA103221C2/uk unknown
- 2010-01-15 BR BRPI1005159A patent/BRPI1005159A2/pt not_active IP Right Cessation
- 2010-01-15 CA CA2749802A patent/CA2749802C/en not_active Expired - Fee Related
- 2010-01-15 EA EA201190079A patent/EA201190079A1/ru unknown
- 2010-01-15 AU AU2010204552A patent/AU2010204552B2/en not_active Ceased
- 2010-01-15 MX MX2011007583A patent/MX2011007583A/es active IP Right Grant
- 2010-01-15 NZ NZ594056A patent/NZ594056A/xx not_active IP Right Cessation
- 2010-01-15 JP JP2011546399A patent/JP2012515222A/ja active Pending
- 2010-01-15 SG SG2014003073A patent/SG196821A1/en unknown
- 2010-01-15 SG SG2011050358A patent/SG172940A1/en unknown
- 2010-01-15 US US12/688,655 patent/US8323634B2/en not_active Expired - Fee Related
- 2010-01-15 UA UAA201109961A patent/UA105201C2/uk unknown
- 2010-01-15 SG SG2011050366A patent/SG172941A1/en unknown
- 2010-01-15 NZ NZ594055A patent/NZ594055A/xx not_active IP Right Cessation
-
2011
- 2011-07-12 IL IL214051A patent/IL214051A0/en unknown
- 2011-07-12 IL IL214052A patent/IL214052A0/en unknown
- 2011-07-13 ZA ZA2011/05171A patent/ZA201105171B/en unknown
-
2012
- 2012-10-25 US US13/660,915 patent/US8993519B2/en not_active Expired - Fee Related
-
2014
- 2014-12-18 US US14/574,553 patent/US20150202268A1/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5665863A (en) * | 1992-01-31 | 1997-09-09 | Rhone-Poulenc Rorer S.A. | Polypeptides having granulocyte colony stimulating activity, their preparation and pharmaceutical compositions containing them |
| CN1729018A (zh) * | 2001-07-11 | 2006-02-01 | 马克西根控股公司 | G-csf偶联物 |
Non-Patent Citations (1)
| Title |
|---|
| WENDY HALPERN: "AlbugraninTM, a Recombinant Human Granulocyte Colony Stimulating Factor (G-CSF) Genetically Fused to Recombinant Human Albumin Induces Prolonged Myelopoietic Effects in Mice and Monkeys", 《PHARMACEUTICAL RESEARCH》 * |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107206053A (zh) * | 2014-11-03 | 2017-09-26 | 华鸿新药公司 | 用于治疗血细胞减少症或减少血细胞减少症的持续时间的佛波醇酯组合物和方法 |
| CN112105363A (zh) * | 2018-02-01 | 2020-12-18 | 大连万春布林医药有限公司 | 用于通过施用普那布林和g-csf剂来减少化学疗法诱导的嗜中性白血球减少症的组合物和方法 |
| CN111383745A (zh) * | 2018-12-29 | 2020-07-07 | 医渡云(北京)技术有限公司 | 计算机数据处理方法、装置、存储介质及设备 |
| CN114364393A (zh) * | 2019-05-31 | 2022-04-15 | 光谱制药有限公司 | 使用g-csf蛋白质复合物治疗的方法 |
| CN114746104A (zh) * | 2019-12-05 | 2022-07-12 | 韩美药品株式会社 | 治疗由化疗或放疗引起的中性粒细胞减少症的方法 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2010204552B2 (en) | New stable formulations of recombinant human albumin-human granulocyte colony stimulating factor fusion protein | |
| CN105209054A (zh) | 使用白细胞介素-10治疗疾病和病症的方法 | |
| KR20150121715A (ko) | Csf1 치료제 | |
| US20220370563A1 (en) | Methods of administration of il-2 receptor agonists | |
| AU2014208224A1 (en) | New Stable Formulations of Recombinant Human Albumin-Human Granulocyte Colony Stimulating Factor Fusion Protein | |
| WO2021219940A1 (en) | Homing peptide-guided decorin conjugates for use in treating epidermolysis bullosa | |
| AU2014202190B2 (en) | New Stable Formulations of Recombinant Human Albumin-Human Granulocyte Colony Stimulating Factor Fusion Protein | |
| US20140271538A1 (en) | Recombinant Human Albumin-Human Granulocyte Colony Stimulating Factor for the Prevention of Neutropenia in Pediatric Patients | |
| AU2016371021B2 (en) | Ameliorating systemic sclerosis with death receptor agonists | |
| US20240245752A1 (en) | Methods for effective and safe treatment of neutropenia using de-mobilized g-csf | |
| WO2023097111A2 (en) | Methods and compositions for treating calcinosis associated conditions | |
| CN117683140A (zh) | 肿瘤靶向的以白介素2为活性成分的融合蛋白型药物前体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| RJ01 | Rejection of invention patent application after publication | ||
| RJ01 | Rejection of invention patent application after publication |
Application publication date: 20120314 |