WO2001060806A2 - Substituted arylpyrazines - Google Patents
Substituted arylpyrazines Download PDFInfo
- Publication number
- WO2001060806A2 WO2001060806A2 PCT/US2001/005264 US0105264W WO0160806A2 WO 2001060806 A2 WO2001060806 A2 WO 2001060806A2 US 0105264 W US0105264 W US 0105264W WO 0160806 A2 WO0160806 A2 WO 0160806A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- compound according
- named
- amine
- ethylpropyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *c(nc1*)c(*)nc1[Al] Chemical compound *c(nc1*)c(*)nc1[Al] 0.000 description 4
- RSMHAWFILXRCTO-UHFFFAOYSA-N CCC(CC)Nc(nc1)c(CC)[n+]([OH2+])c1-c(ccc(OC(F)(F)F)c1)c1OC Chemical compound CCC(CC)Nc(nc1)c(CC)[n+]([OH2+])c1-c(ccc(OC(F)(F)F)c1)c1OC RSMHAWFILXRCTO-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5355—Non-condensed oxazines and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/10—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D241/12—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/10—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D241/14—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D241/18—Oxygen or sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/10—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D241/14—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D241/20—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
Definitions
- the present invention relates to novel arylpyrazine compounds that have useful pharmacological properties in that they bind to cellular receptors, including CRF receptors. Certain of these compounds can bind to and modulate the activity of such receptors. Importantly, the compounds of the invention include compounds that bind with high selectivity and/or high affinity to CRFI receptors (Corticotropin Releasing Factor type 1 Receptors).
- This invention also relates to pharmaceutical compositions comprising such compounds and to the use of such compounds as pharmaceutical agents, e.g., in treatment of psychiatric disorders and neurological diseases, including major depression, anxiety-related disorders, post-traumatic stress disorder, supranuclear palsy and feeding disorders, as well as treatment of immunological, cardiovascular or heart-related diseases and colonic hypersensitivity associated with psychopathological disturbance and stress. Additionally this invention relates to the use such compounds as probes for the localization of cellular receptors in tissue sections.
- Corticotropin releasing factor a 41 amino acid peptide
- POMC proopiomelanocortin
- CRF has a role in psychiatric disorders and neurological diseases including depression, anxiety-related disorders and feeding disorders.
- a role for CRF has also been postulated in the etiology and pathophysiology of Alzheimer's disease, Parkinson's disease, Huntington's disease, progressive supranuclear palsy and amyotrophic lateral sclerosis as they relate to the dysfunction of CRF neurons in the central nervous system.
- CRF cerebral spinal fluid
- CSF cerebral spinal fluid
- CRF receptors are significantly decreased in the frontal cortex of suicide victims, consistent with a hypersecretion of CRF.
- ACTH blunted adrenocorticotropin
- Preclinical studies in rats and non-human primates provide additional support for the hypothesis that hypersecretion of CRF may be involved in the symptoms seen in human depression.
- tricyclic antidepressants can alter CRF levels and thus modulate the numbers of CRF receptors in brain.
- CRF has also been implicated in the etiology of anxiety-related disorders.
- CRF produces anxiogenic effects in animals and interactions between benzodiazepine / non- benzodiazepine anxiolytics and CRF have been demonstrated in a variety of behavioral anxiety models.
- Preliminary studies using the putative CRF receptor antagonist -helical ovine CRF (9-41) in a variety of behavioral paradigms demonstrate that the antagonist produces "anxiolytic-like" effects that are qualitatively similar to the benzodiazepines.
- Neurochemical, endocrine and receptor binding studies have all demonstrated interactions between CRF and benzodiazepine anxiolytics providing further evidence for the involvement of CRF in these disorders.
- Chlordiazepoxide attenuates the "anxiogenic" effects of CRF in both the conflict test and in the acoustic startle test in rats.
- the benzodiazepine receptor antagonist Ro 15-1788 which was without behavioral activity alone in the operant conflict test, reversed the effects of CRF in a dose-dependent manner, while the benzodiazepine inverse agonist FG 7142 enhanced the actions of CRF.
- CRF has also been implicated in the pathogeneisis of certain immunological, cardiovascular or heart-related diseases such as hypertension, tachycardia and congestive heart failure, stroke and osteoporosis, as well as in premature birth, psychosocial dwarfism, stress-induced fever, ulcer, diarrhea, post-operative ileus and colonic hypersensitivity associated with psychopathological disturbance and stress.
- arylpyrazine compounds including arylpyrazines that can bind with high affinity and high selectivity to CRF* receptors, including human CRF j receptors.
- the invention thus includes methods for treatment of disorders and diseases associated with RF 1 receptors, including CNS-related disorders and diseases, particularly affective disorders and diseases, and acute and chronic neurological disorders and diseases.
- Arylpyrazine compounds of the invention are preferably substituted at the 2-ring position by a carbocyclic aryl group such as phenyl, naphthyl and the like, or a heteroaromatic group, particularly a heteroaromatic having a nitrogen ring member such as pyridyl, pyrimidinyl and the like.
- Arylpyrazine compounds of the invention also are preferably substituted at the 5-ring position (para with respect to the aryl substituent) by a non-hydrogen substituent, particularly a non-aromatic group such as alkyl, alkenyl, alkynyl, heteroalkyl and the like, preferably aminoalkyl and alkoxy.
- Ar is substituted phenyl, optionally substituted napthyl, or an optionally substituted heterocyclic group having from 1 to 3 rings, and 3 to 8 ring members in each ring and 1 to about 3 hetero atoms;
- R t and R 3 are each independently hydrogen, halogen, cyano, nitro, amino, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted mono or dialkylamino, optionally substituted alkoxy, optionally substituted alkylthio, optionally substituted alkylsulfinyl, or optionally substituted alkylsulfonyl; and
- R 2 is halogen, cyano, nitro, amino, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted alkylamino, optionally substituted alkoxy, optionally substituted alkylthio, optionally substituted alkylsulfiny
- Preferred compounds of the invention also include those of the following Formula
- R j is selected from H, C M alkyl, C 2 . 4 alkenyl, C 2 . 4 alkynyl, halogen, CN, C*_ 4 haloalkyl, trifluoromethyl, trifluoromethoxy, -NH(C._ 4 alkyl), -N(C alkyl)(C M alkyl), - O(C alkyl), and S(O) n (C M alkyl);
- R 2 is selected from the group consisting of -XR A and Y, wherein -X, R A , and Y are defined below; and R 3 is selected from the group consisting of hydrogen, halogen, C*_ 4 alkyl, -O(C*_ 4 alkyl), - NH(C 1 . 4 alkyl), -N(C M alkyl)(C,. 4 alkyl), and -S(O) n (C*. 4 alkyl), haloalkyl, trifluoromethyl, trifluoromethoxy, -XR A and Y;
- R 4 is absent or an oxygen atom
- Ar is phenyl, mono-, di-, or tri-substituted with R c , or
- Ar is selected from the group consisting of: naphthyl, pyridyl, pyridonyl, pyrimidinyl, and thiophenyl, each of which is unsubstituted or mono-, di-, or tri-substituted with Rc; with the proviso that if Ar is phenyl substituted with halogen, napthyl, or naphthyl substituted with halogen, then the compounds where R 3 is hydrogen are excluded;
- R A and R B which may be the same or different, are independently selected at each occurrence from the group consisting of: hydrogen and straight, branched, or cyclic alkyl groups consisting of 1 to 8 carbon atoms, which may contain one or more double or triple bonds, each of which may be further substituted with one or more substituent(s) selected from oxo, hydroxy, halogen, -O(C*_ 4 alkyl), -NH(C*.. 4 alkyl), -N(C alky ⁇ )(C M alkyl), -NHC(O)(C,.
- Rc is independently selected at each occurrence from the group consisting of halogen, cyano, haloalkyl, trifluoromethyl, trifluoromethoxy, hydroxy, amino, and C 6 alkyl optionally substituted with 0-2 R D , C 2 . 6 alkenyl substituted with 0-2 R D , C M alkynyl substituted with 0-2 R D , C 3 . 7 cycloalkyl substituted with 0-2 R D , (C 3 .
- R D is independently selected at each occurrence the group consisting of halogen, hydroxy, cyano, C 1 . 4 alkyl, -O(C 1.4 alkyl), -NH(C 1 . 4 alkyl), -N(C M alky ⁇ )(C.. 4 alkyl), morpholino, pyrrolidino, piperidino, thiomorpholino, piperazino, 4- hydroxypiperidino, -S(O) n (C*. 4 alkyl), trifluoromethyl, trifluoromethoxy, CO(C*. 4 alkyl), CONH(C 1 . 4 alkyl), CON(C 1 . 4 alkyl)(C 1 .
- Y and Z are independently selected at each occurrence from the group consisting of: 3- to 7-membered carbocyciic and heterocyclic groups, which are saturated, unsaturated, or aromatic, which may be substituted with one or more substituents selected from halogen, haloalkyl, oxo, hydroxy, amino, C 4 alkyl, -O(C M alkyl), - NH(C M alkyl), -N(C M alkyl)(C 1.4 alkyl), and -S(O) n (C M alkyl), and said 3- to 7-membered heterocyclic groups contain one or more heteroatom(s) selected from N, O, and S, with the point of attachment being either carbon or nitrogen; and n is independently selected at each occurrence from 0, 1, and 2.
- arylpyrazines of the invention include compounds of the following Formula IB:
- Ar is selected from the group consisting of phenyl, 1- or 2-naphthyl, 2-, 3-, or 4-pyridyl, 2-, 4-, or 5-pyrimidinyl, each Ar optionally substituted with 1 to 5 with R 6 group, with the proviso that at least one of the positions ortho or para to the point of attachment of Ar to the pyrazine ring system is substituted;
- R is selected from H, C,. 4 alkyl, C 2 . 4 alkenyl, C 2 . 4 alkynyl, (C 3 . 6 alkyl, halogen, CN, C M haloalkyl, -NH(C,. 4 alkyl), -N(C M alkyl)(C,.
- R 2 is selected from the group consisting of-XR 4 and Y, wherein X, R 4 , and Y are as defined below;
- Y and Z are independently selected at each occurrence from the group consisting of: 3- to 7-membered heterocycles, saturated or unsaturated, containing one or more heteroatom(s) selected from N, O, and S, with the point of attachment being either carbon or nitrogen (where applicable), and which may be further substituted with one or more substituents selected from halogen, oxo, hydroxy, amino, C,. 4 alkyl, -0(0 ⁇ 4 alkyl),
- R 3 is selected from the group consisting of hydrogen, halogen,
- R 4 and R 5 are independently selected at each occurrence from the group consisting of: hydrogen, straight, branched, or cyclic alkyl groups consisting of 1 to 8 carbon atoms, which may contain one or more double or triple bonds, each of which may optionally be further substituted with one or more substituent(s) selected from oxo, hydroxy, - O(C M alkyl), -NH(C 1 . 4 alkyl), -N(C M alkyl)(C 1 .
- R 6 is independently selected at each occurrence from halogen, trifluoromethyl, trifluoromethoxy, hydroxy, amino, and C,-C 6 alkyl (optionally substituted with 0-2 R 7 , C 2 . 6 alkenyl substituted with 0-2 R 7 , C 1 alkynyl substituted with 0-2 R 7 , C 3 . 6 cycloalkyl substituted with 0-2 R 7 , (C 3 . 6 cycloalky ⁇ )C-- 4 alkyl substituted with 0-2 R 7 , O(C,.
- R 7 is independently selected at each occurrence the group consisting of halogen, C,.
- preferred Ar groups include 2,4- dichlorophenyl, 2,4-dimethoxyphenyl, 4-methoxy-2-methylphenyl, 2,4,6- trimethylphenyl, 4-methoxy-2,6-dimethylphenyl, 4-cyano-2,6-dimethylphenyl, 2- methoxy-4,6-dimethylphenyl, 2-methoxy-4,6-bis(trifluoromethyl)phenyl, 2,6- dichloro-4-methoxyphenyl, 2-(2-(l-morpholino)ethoxy)-4,6-dimethylphenyl, 2- (2-(4-hydroxy- 1 -piperidino)ethoxy)-4,6-dimethylphenyl, 2-methyl-4- (dimethylamino)-3-pyridyl, and 2-chloro-4-di
- preferred R* groups include methyl, ethyl, chloro, bromo, iodo, trifluoromethyl, methoxy, dimethylamino, and thiomethoxy.
- preferred R 2 groups include dipropylamino, bis(2-methoxyethyl)amino, 4-methyl-l-piperazino, 3-pentylamino, 4- heptylamino, l,3-dimethoxy-2-propylamino, l-(dimethylamino)-2-pentylamino, l-(3- pyridyl)-2-butylamino, (2-methoxy-5-pyridine)amino, 3-pentyloxy, 4-heptyloxy, 1- methoxy-2-butyloxy, and l,3-dimethoxy-2-propyloxy.
- preferred R 3 groups include methyl, ethyl, chloro, trifluoromethyl, methoxy, dimethylamino, thiomethoxy, methanesulfonyl, (l-morpholino)methyl, 2-(l-pyrrolidino)ethyl, and (2-methoxy)ethoxy.
- Preferred arylpyrazines of the invention exhibit good activity in a standard in vitro CRF receptor binding assays, specifically the assay as specified in Example 96 which follows.
- Particularly preferred arylpyrazines of the invention have an IC 50 in such a defined standard in vitro CRF receptor binding assay of about 1.5 micromolar or less, still more preferably an IC 50 of about 100 nanomolar or less, or even more preferably an IC 50 of about 10 nanomolar or less or even 1 nanomolar or less in a such a defined standard in vitro CRF receptor binding assay.
- Preferred arylpyrazines of the invention do not show activity in a standard in vitro ⁇ a channel functional assay, specifically the assay as specified in Example 99 which follows. Particularly preferred arylpyrazines of the invention do not show any statistically significant activity in a defined standard in vitro ⁇ a channel functional assay
- the invention further comprises methods of treating patients suffering from or susceptible to (i.e. prophylactic treatment) certain disorders or diseases with an effective amount of a compound of the invention.
- the patient may be a human or other mammal such as other primates.
- Treatment of humans, domesticated companion animals (pet) or livestock animals suffering from certain with an effective amount of a compound of the invention is encompassed by the invention.
- this invention relates to the use of the CRFI compounds of the invention as probes for the localization of receptors in tissue sections.
- the compounds of the present invention are useful for the treatment of CNS disorders particularly affective disorders, anxiety disorders, stress-related disorders, eating disorders and substance abuse.
- Affective disorders include all types of depression, bipolar disorder, cyclothymia, and dysthymia.
- Anxiety disorders include generalized anxiety disorder, panic, phobias and obsessive-compulsive disorder.
- Stress-related disorders include post-traumatic stress disorder, hemorrhagic stress, stress-induced psychotic episodes, psychosocial dwarfism, stress headaches, stress-induced immune systems disorders such as stress-induced fever, and stress-related sleep disorders.
- Eating disorders include anorexia nervosa, bulimia nervosa, and obesity. These conditions are herein after referred to as the primary CRF-related CNS disorders.
- Arylpyrazine compounds of the invention are also useful in the treatment of a variety of neurological disorders including supranuclear palsy, AIDS related dementias, multiinfarct dementia, neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease, and Huntington's disease, head trauma, spinal cord trauma, ischemic neuronal damage, amyotrophic lateral sclerosis, disorders of pain perception such as fibromyalgia and epilepsy. These conditions are hereinafter referred to as the secondary CRF-related C ⁇ S disorders.
- Arylpyrazine compounds of the invention are useful as modulators of the G- protein coupled receptor function. Additionally, arylpyrazine compounds of the invention are useful as modulators of the CRF receptor in the treatment of a number of gastrointestestinal, cardiovascular, hormonal, autoimmune and inflammatory conditions. Such conditions include irritable bowel syndrome, ulcers, Crohn's disease, spastic colon, diarrhea, post operative ilius and colonic hypersensitivity associated with psychopathological disturbances or stress, hypertension, tachycardia, congestive heart failure, infertility, euthyroid sick syndrome, inflammatory conditions effected by rheumatoid arthritis and osteoarthritis, pain, asthma, psoriasis and allergies. These conditions are referred to hereinafter as the non-CNS CRF- related disorders.
- Arylpyrazine compounds of the invention are also useful as modulators of the CRF, receptor in the treatment of animal conditions associated with aberrant CRF levels. These conditions include porcine stress syndrome, bovine shipping fever, equine paroxysmal fibrillation, and dysfunctions induced by confinement in chickens, sheering stress in sheep or human-animal interaction related stress in dogs, psychosocial dwarfism and hypoglycemia. These animal conditions are referred to hereinafter as the CRF- related animal disorders.
- the present invention provides pharmaceutical compositions comprising one or more aryl pyrazine compound of the invention of a pharmaceutically acceptable salt thereof, particularly for use in the treatment of any of the primary CNS disorders, secondary CNS disorders, non-CNS CRF-related disorders, or CRF-related animal disorders.
- arylpyrazine compounds of the invention are useful as standards and reagents in determining the ability of a potential pharmaceutical to bind to the CRF receptor.
- Other aspects of the invention are disclosed infra.
- the invention provides compounds of Formula I shown above.
- Preferred compound of the invention include those of the following Formula la:
- R is selected from H, C M alkyl, C 2.4 alkenyl, C 2 . 4 alkynyl, halogen, CN, C M haloalkyl, trifluoromethyl, trifluoromethoxy, -NH(C M alkyl), -N(C,_ 4 alkyl)(C,. 4 alkyl), - O C ⁇ alkyl), and S(O) n (C M alkyl);
- R 2 is selected from the group consisting of-XR A and Y, wherein -X, R A , and Y are defined below; and R 3 is selected from the group consisting of hydrogen, halogen, C M alkyl, -O(C*. 4 alkyl), - NH(C W alkyl), -N(C M alkyl)(C 1 . 4 alkyl), and -S(O) n (C M alkyl), haloalkyl, trifluoromethyl, trifluoromethoxy, -XR A and Y;
- R 4 is absent or an oxygen atom
- Ar is phenyl, mono-, di-, or tri-substituted with Rc, or Ar is selected from the group consisting of: naphthyl, pyridyl, pyridonyl, pyrimidinyl, and thiophenyl, each of which is unsubstituted or mono-, di-, or tri-substituted with R c ; with the proviso that if Ar is phenyl substituted with halogen, napthyl, or naphthyl substituted with halogen, then the compounds where R 3 is hydrogen are excluded;
- R A and R B which may be the same or different, are independently selected at each occurrence from the group consisting of: hydrogen and straight, branched, or cyclic alkyl groups consisting of 1 to 8 carbon atoms, which may contain one or more double or triple bonds, each of which may be further substituted with one or more substituent(s) selected from oxo,
- Rc is independently selected at each occurrence from the group consisting of halogen, cyano, haloalkyl, trifluoromethyl, trifluoromethoxy, hydroxy, amino, and C,_ 6 alkyl optionally substituted with 0-2 R D , C 2 . 6 alkenyl substituted with 0-2 R D , C M alkynyl substituted with 0-2 R D , C 3 . 7 cycloalkyl substituted with 0-2 R D , (C 3 .
- R D is independently selected at each occurrence the group consisting of halogen, hydroxy, cyano, Cl. 4 alkyl, -O(C,. 4 alkyl), -NH(C,. 4 alkyl), -N(C M alkyl)(C M alkyl), morpholino, pyrrolidino, piperidino, thiomorpholino, piperazino, 4- hydroxypiperidino, -S(O) admir(C*. 4 alkyl), trifluoromethyl, trifluoromethoxy, CO(C*. 4 -alkyl), CONH(C 1 . 4 alkyl), CON(C M alkyl)(C M alkyl), -XR A, and Y;
- X is independently selected at each occurrence from the group consisting of -CH 2 -, -
- 3- to 7-membered heterocyclic groups contain one or more heteroatom(s) selected from N, O, and S, with the point of attachment being either carbon or nitrogen; and n is independently selected at each occurrence from 0, 1, and 2.
- Preferred compounds of general Formula la are those where: R 4 is absent and Ar is phenyl, mono-, di-, or tri-substituted with R ⁇ , and R, and R 3 are independently selected from the group consisting of hydrogen, halogen, methyl, ethyl, ethoxy and methoxy.
- Such compounds are referred to herein as compounds of general Formula lb.
- More preferred compounds of the invention are those of general Formula la where:
- R 4 is absent and Ar is phenyl, mono-, di-, or tri-substituted with R c ; and R A and R B , which may be the same or different, are independently straight, branched, or cyclic alkyl groups having from 1 to 8 carbon atoms, which may contain one or more double or triple bonds.
- R A and R B which may be the same or different, are independently straight, branched, or cyclic alkyl groups having from 1 to 8 carbon atoms, which may contain one or more double or triple bonds.
- Such compounds are referred to as compounds of Formula Ic.
- R 4 is absent and Ar is phenyl mono-, di-, or tri-substituted with I ⁇ ;
- R A and R B which may be the same or different, are independently selected at each occurrence from the group consisting of: straight, branched, and cyclic alkyl groups having from 1 to 8 carbon atoms, which may contain one or more double or triple bonds; and
- Rj and R 3 are independently selected from the group consisting of hydrogen, halogen, methyl, ethyl, ethoxy, and methoxy.
- Such compounds are referred to herein as compounds of general Formula Id.
- A is NR * or O.
- Such compounds are referred to herein as compounds of general Formula le.
- R x and R ⁇ are the same or different and are independently selected straight, branched, or cyclic alkyl groups having from 1 to 8 carbon atoms, which may contain one or more double or triple bonds, each of which may be further substituted with one or more substituent(s) independently selected from hydroxy, halogen, -O(C*_ 4 alkyl), -NH(C M alkyl), -N(C M alkyl)(C*. 4 alkyl), and optionally substituted phenyl.
- Ar is phenyl, mono-, di-, or tri-substituted with Rc; and R, and R 3 are independently selected from the group consisting of hydrogen, halogen, methyl, ethyl, ethoxy and methoxy.
- Such compounds are referred to herein as compounds of general Formula Ila.
- R, and R 3 are independently chosen from halogen, C*_ 4 alkyl, -O(C- .4 alkyl), -NH(C,. 4 alkyl), -N(C M alkyl)(C 1 . 4 alkyl), haloalkyl, trifluoromethyl, and trifluoromethoxy; and
- Ar is phenyl, which is mono-, di-, or trisubstituted with one or more substituent(s) independently selected from: halogen, cyano, haloalkyl, trifluoromethyl, trifluoromethoxy, hydroxy, amino, and Cj. 6 alkyl, C M alkoxy, C 1 . 4 alkoxy(C M alkoxy), mono- or di(C 1 . 4 )amino(C 1 .
- Such compounds are referred to herein as compounds of general Formula lib.
- R ⁇ is chosen from the group consisting of: straight, branched, or cyclic alkyl groups having from 1 to 8 carbon atoms, which may contain one or more double or triple bonds;
- R, and R 3 are independently chosen from halogen, C M alkyl, -O(C M alkyl), -NH(C M alkyl), -N(C M alkyl)(C 1 .
- Ar is phenyl, which is mono-, di-, or trisubstituted with substituent(s) independently selected from: halogen, cyano, haloalkyl, trifluoromethyl, trifluoromethoxy, hydroxy, amino, and C,. 6 alkyl, C*_ 4 alkoxy, C,. 4 alkoxy(C,. 4 alkoxy), mono- or di(C )amino(C*.
- R x is hydrogen
- R ⁇ is chosen from the group consisting of: straight, branched, or cyclic alkyl groups having from 1 to 8 carbon atoms, which may contain one or more double or triple bonds
- R t and R 3 are independently chosen from halogen, C M alkyl, -O(C M alkyl), -NH(C 1 . 4 alkyl), -N(C, .4 alkyl)(C M alkyl), haloalkyl, trifluoromethyl, and trifluoromethoxy
- Ar is a phenyl group of the formula:
- L indicates a bond to the pyrazine ring in Formula A; and the phenyl group is substituted at one, two or three of positions 2, 4, and 6 with substituent(s) independently selected from: halogen, cyano, haloalkyl, trifluoromethyl, trifluoromethoxy, hydroxy, amino, and C-. 6 alkyl, C M alkoxy, C M alkoxy(C M alkoxy), mono- or di(C M )amino(C..
- Such compounds are referred to herein as compounds of general Formula lid.
- R ⁇ is chosen from the group consisting of: straight, branched, or cyclic alkyl groups having from 1 to 8 carbon atoms, which may contain one or more double or triple bonds;
- R t and R 3 are independently chosen from halogen, C M alkyl, -O(C M alkyl), -NH(C M alkyl), -N(C M alkyl)(C*. 4 alkyl), haloalkyl, trifluoromethyl, and trifluoromethoxy; and
- Ar is a phenyl group of the formula:
- L indicates a bond to the pyrazine ring in Formula A; and the phenyl group is substituted at positions 2 and 4 with substituents independently selected from: halogen, cyano, haloalkyl, trifluoromethyl, trifluoromethoxy, hydroxy, amino, and C,. 6 alkyl, C M alkoxy, C- ⁇ alkoxy ⁇ alkoxy), mono- or di(C 1 . 4 )amino(C ] . 4 alkoxy), and mono- or di(C M alkyl)amino.
- substituents independently selected from: halogen, cyano, haloalkyl, trifluoromethyl, trifluoromethoxy, hydroxy, amino, and C,. 6 alkyl, C M alkoxy, C- ⁇ alkoxy ⁇ alkoxy), mono- or di(C 1 . 4 )amino(C ] . 4 alkoxy), and mono- or di(C M alkyl)amino.
- R j and R 3 are independently chosen at each occurrence from halogen, C, profession 4 alkyl, -O(C M alkyl), -NH(C M alkyl), -N(C M alkyl)(C M alkyl), haloalkyl, trifluoromethyl, and trifluoromethoxy; and
- Ar is a phenyl group of the formula:
- L indicates a bond to the pyrazine ring in Formula A; and the phenyl group is substituted at one, two or three of positions 2, 4, and 6 with substituent(s) independently selected from: halogen, cyano, haloalkyl, trifluoromethyl, trifluoromethoxy, hydroxy, amino, and C, profession 6 alkyl, C M alkoxy, C 1 . 4 alkoxy(C 1 . 4 alkoxy), mono- or di(C I . 4 )amino(C 1 . 4 alkoxy), and mono- or c ⁇ C ⁇ alkyl)amino.
- R is independently selected at each occurrence from the group consisting of: hydrogen, halogen, cyano, haloalkyl, trifluoromethyl, trifluoromethoxy, hydroxy, amino,
- R. and R 3 are independently chosen from halogen, C M alkyl, -O(C ⁇ 4 alkyl), -NH(C*. 4 alkyl), -N(Cj. 4 alkylXC, ⁇ alkyl), haloalkyl, trifluoromethyl, and trifluoromethoxy.
- R x and R ⁇ are independently selected at each occurrence from the group consisting of: straight, branched, and cyclic alkyl groups having from 1 to 8 carbon atoms, which may contain one or more double or triple bonds; and R, and R 3 are independently selected from the group consisting of hydrogen, halogen, methyl, ethyl, ethoxy, and methoxy.
- Such compounds are referred to herein as compounds of general Formula Ilh.
- R x and R y are independently hydrogen or C,_ 8 alkyl; or
- NR x R Y is:
- Such compounds are referred to herein as compounds of general Formula Iii.
- R x is chosen from the group consisting of: straight, branched, or cyclic alkyl groups, including (cycloalkyl)alkyl groups, having from 1 to 8 carbon atoms, which may contain one or more double or triple bonds, each of which may be further substituted with one or more substituent(s) independently selected from
- R x is selected from straight, branched, or cyclic alkyl groups containing of 1 to 8 carbon atoms, which may contain one or more double or triple bonds;
- R t and R 3 are independently chosen from halogen, C 1 alkyl, -O(C ! . 4 alkyl), -NH(C 1 . 4 alkyl), -N(C M alkyl)(C 1 . 4 alkyl), haloalkyl, trifluoromethyl, and trifluoromethoxy; and Ar is phenyl, which is mono-, di-, or trisubstituted with one or more substituent(s) independently selected from: halogen, cyano, haloalkyl, trifluoromethyl, trifluoromethoxy, hydroxy, amino, and C 6 alkyl, C M alkoxy, C*. 4 alkoxy(C*. 4 alkoxy), mono- or di(C*_ 4 )amino(C,_ 4 alkoxy), and mono- or h ⁇ C ⁇ alkyl)amino.
- substituent(s) independently selected from: halogen, cyano, haloalkyl, triflu
- L indicates a bond to the pyrazine ring in Formula III; and the phenyl group is substituted at one, two, or three of positions 2, 4, and 6 with substituent(s)independenfiy selected from: halogen, cyano, haloalkyl, trifluoromethyl, trifluoromethoxy, hydroxy, amino, and C-*_ 6 alkyl, C M alkoxy, C 1 . 4 alkoxy(C ⁇ . 4 alkoxy), mono- or di(C w )amino(C*_ 4 alkoxy), and mono- or di(C ⁇ _ 4 alkyl)amino.
- substituent(s)independenfiy selected from: halogen, cyano, haloalkyl, trifluoromethyl, trifluoromethoxy, hydroxy, amino, and C-*_ 6 alkyl, C M alkoxy, C 1 . 4 alkoxy(C ⁇ . 4 alkoxy), mono- or di(C w )
- L indicates a bond to the pyrazine ring in Formula III; and the phenyl group is substituted at positions 2 and 4 with substitutuents independently selected from halogen, cyano, haloalkyl, trifluoromethyl, trifluoromethoxy, hydroxy, amino, C 6 alkyl, C 1 . 4 alkoxy, C 1 . 4 alkoxy(C 1 . 4 alkoxy), mono- or di(Cj_ 4 )amino(C M alkoxy), and mono- or ch C ⁇ alkyl)amino.
- substitutuents independently selected from halogen, cyano, haloalkyl, trifluoromethyl, trifluoromethoxy, hydroxy, amino, C 6 alkyl, C 1 . 4 alkoxy, C 1 . 4 alkoxy(C 1 . 4 alkoxy), mono- or di(Cj_ 4 )amino(C M alkoxy), and mono- or ch C ⁇ alkyl)
- R, and R 3 are independently chosen from halogen, C M alkyl, -O(C M alkyl), -NH(C M alkyl), -N(C M alkyl)(C 1 . 4 alkyl), haloalkyl, trifluoromethyl, and trifluoromethoxy; and
- Ar is a phenyl group of the formula: wherein L indicates a bond to the pyrazine ring in Formula B; and the phenyl group is substituted at one, two, or three of positions 2, 4, and 6 with substituent(s)independently selected from: halogen, cyano, haloalkyl, trifluoromethyl, trifluoromethoxy, hydroxy, amino, C*. 5 alkyl, C,. 4 alkoxy, C-_
- Such compounds are referred to herein as compounds of general Formula Hid.
- R, and R 3 are independently selected from the group consisting of hydrogen, halogen, methyl, ethyl, ethoxy and methoxy.
- R, and R 3 are independently selected from the group consisting of hydrogen, halogen, methyl, ethyl, ethoxy and methoxy.
- Such compounds are referred to herein as compounds of general Formula Ille.
- Another preferred compound of the invention include 2-(2-methoxy-5- triflurormethoxyphenyl)-3-ethyl-6-methylamino-5-(l-ethylpropoxy)-pyrazine. and pharmaceutically acceptable salts thereof, the compound structure is:
- the compounds herein described may have one or more asymmetric centers or planes.
- Cis and trans geometric isomers of the compounds of the present invention are described and may be isolated as a mixture of isomers or as separated isomeric forms. All chiral (enantiomeric and diastereomeric), and racemic forms, as well as all geometric isomeric forms of a structure are intended, unless the specific stereochemistry or isomeric form is specifically indicated.
- the CRF antagonist compounds provided by this invention and labeled derivatives thereof are also useful as standards and reagents in determining the ability of a potential pharmaceutical to bind to the CRF receptor.
- Labeled derivatives the CRF antagonist compounds provided by this invention are also useful as radiotracers for positron emission tomography (PET) imaging or for single photon emission computerized tomography (SPECT).
- PET positron emission tomography
- SPECT single photon emission computerized tomography
- substituted means that any one or more hydrogens on the designated atom is replaced with a selection from the indicated group, provided that the designated atom's normal valence is not exceeded, and that the substitution results in a stable compound.
- 2 hydrogens on the atom are replaced.
- Keto substituents are not present on aromatic moieties.
- the present invention is intended to include all isotopes of atoms occurring in the present compounds. Isotopes include those atoms having the same atomic number but different mass numbers, By way of general example and without limitation, isotopes of hydrogen include tritium and deuterium.
- Isotopes of carbon include n C, 13 C, and 14 C.
- its definition at each occurrence is independent of its definition at every other occurrence.
- R * R * at each occurrence is selected independently from the definition of R * .
- combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
- substituents of the various formulae are "optionally substituted", including Ar, R*, R 2 , and R 3 of Formula I, and such substituents as recited in the sub-formulae such as Formulae la and the like.
- those substituents Ar, Rute R 2 , and R 3
- those substituents may be substituted by other than hydrogen at one or more available positions, typically 1 to 3 or 4 positions, by one or more suitable groups such as those disclosed herein.
- suitable groups that may be present on a "substituted" Ar, R-, R 2 , and R 3 group or other substituent include e.g.
- alkanoyl such as a C 6 alkanoyl group such as acyl and the like; carboxamido; alkyl groups including those groups having 1 to about 12 carbon atoms, or 1, 2, 3, 4, 5, or 6 carbon atoms; alkenyl and alkynyl groups including groups having one or more unsaturated linkages and from 2 to about 12 carbon, or 2, 3, 4, 5 or 6 carbon atoms; alkoxy groups having those having one or more oxygen linkages and from 1 to about 12 carbon atoms, or 1, 2, 3, 4, 5 or 6 carbon atoms; aryloxy such as phenoxy; alkylthio groups including those moieties having one or more thioether linkages and from 1 to about 12 carbon atoms, or 1, 2, 3, 4, 5 or 6 carbon atoms; alkylsulfinyl groups including those moieties having one or more sulfinyl groups including those moieties having one or more sulfinyl groups including those moieties having one or more sulfin
- an Ar group being a substituted or unsubstituted biphenyl moiety
- aralkyl having 1 to 3 separate or fused rings and from 6 to about 18 carbon ring atoms, with benzyl being a preferred group
- aralkoxy having 1 to 3 separate or fused rings and from 6 to about 18 carbon ring atoms, with O-benzyl being a preferred group
- a heteroaromatic or heteroalicyclic group having 1 to 3 separate or fused rings with 3 to about 8 members per ring and one or more N, O or S atoms, e.g.
- the substitution position of a substituent on a phenyl ring is dependent on the point of attachment of the substitutent to the phenyl ring relative to the point of attachment of the phenyl group to the remainder of the chemical compound.
- L indicates the point of attachment of the phenyl ring to the remainder of the chemical compound.
- the numbers 2, 3, 4, 5 and 6 identify the individual ring atoms to which substituents can be attached.
- alkyl is intended to include both branched and straight-chain saturated aliphatic hydrocarbon groups, having the specified number of carbon atoms.
- alkyl include, but are not limited to, methyl, ethyl, n-propyl, i-propyl, n- butyl, s-butyl, t-butyl, n-pentyl, and s-pentyl.
- Alkyl groups typically have 1 to about 16 carbon atoms, more typically 1 to about 12 carbon atoms.
- Preferred alkyl groups are C-- C 6 alkyl groups.
- Especially preferred alkyl groups are methyl, ethyl, propyl, butyl, 3- pentyl.
- cycloalkyl is intended to include saturated ring groups, having the specified number of carbon atoms, such as cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl. Cycloalkyl groups typically will have 3 to about 8 ring members.
- (C 3 . 6 cycloalkyl)C 4 alkyl as defined above, the point of attachment is on the alkyl group. This term encompasses, but is not limited to, cyclopropylmethyl, cyclohexylmethyl, cyclohexylmethyl.
- alkenyl is intended to include hydrocarbon chains of either a straight or branched configuration and one or more unsaturated carbon-carbon bonds which may occur in any stable point along the chain, such as ethenyl and propenyl. Alkenyl groups typically will have 2 to about 12 carbon atoms, more typically 2 to about 12 carbon atoms.
- alkynyl is intended to include hydrocarbon chains of either a straight or branched configuration and one or more triple carbon-carbon bonds which may occur in any stable point along the chain, such as ethynyl and propynyl. Alkynyl groups typically will have 2 to about 16 carbon atoms, more typically 2 to about 12 carbon atoms.
- haloalkyl include, but are not limited to, trifluoromethyl, trichloromethyl, pentafluoroethyl, and pentachloroethyl.
- Typical haloalkyl groups will have 1 to about 16 carbon atoms, more typically 1 to about 12 carbon atoms.
- alkoxy represents an alkyl group as defined above with the indicated number of carbon atoms attached through an oxygen bridge.
- alkoxy include, but are not limited to, methoxy, ethoxy, n-propoxy, i-propoxy, n-butoxy, 2-butoxy, t-butoxy, n-pentoxy, 2-pentoxy, 3- pentoxy, isopentoxy, neopentoxy, n-hexoxy, 2-hexoxy, 3-hexoxy, and 3-methylpentoxy.
- Alkoxy groups typically have 1 to about 16 carbon atoms, more typically 1 to about 12 carbon atoms.
- alkylthio includes those groups having one or more thioether linkages and suitably from 1 to about 16 carbon atoms, more typically 1 to about 12 carbon atoms, still more typically 1 to about 6 or 8 carbon atoms.
- alkylsulfinyl includes those groups having one or more sulfoxide (SO) linkage groups and suitably from 1 to about 16 carbon atoms, more typically 1 to about 12 carbon atoms, still more typically 1 to about 6 or 8 carbon atoms.
- SO sulfoxide
- alkylsulfonyl includes those groups having one or more sulfonyl (SO 2 ) linkage groups and suitably from 1 to about 16 carbon atoms, more typically 1 to about 12 carbon atoms, still more typically 1 to about 6 or 8 carbon atoms.
- alkylamino includes those groups having one or more primary, secondary and/or tertiary amine groups and suitably from 1 to about 16 carbon atoms, more typically 1 to about 12 carbon atoms, still more typically 1 to about 6 or 8 carbon atoms.
- halo or halogen as used herein refers to fluoro, chloro, bromo, and iodo; and "counter-ion” is used to represent a small, negatively charged species such as chloride, bromide, hydroxide, acetate, sulfate, and the like.
- carrier or “carbocyclic residue” is intended to mean any stable 3- to 7-membered monocyclic or bicyclic or 7-to 13-membered bicyclic or tricyclic, any of which may be saturated, partially unsaturated, or aromatic.
- carbocycles include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, adamantyl, cyclooctyl, [3.3.0]bicyclooctane, [4.3.0jbicyclononane, [4.4.0]bicyclodecane, [2.2.2]bicyclooctane, fluorenyl, phenyl, naphthyl, indanyl, adamantyl, and tetrahydronaphthyl.
- heterocycle or “ heterocycloalkyl” is intended to mean a stable 5-to 7-membered monocyclic or bicyclic or 7-to 10-membered bicyclic heterocyclic ring which is saturated partially unsaturated or unsaturated (aromatic), and which consists of carbon atoms and from 1 to 4 heteroatoms independently selected from the group consisting of N, 0 and S and including any bicyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring.
- the nitrogen and sulfur heteroatoms may optionally be oxidized.
- the heterocyclic ring may be attached to its pendant group at any heteroatom or carbon atom which results in a stable structure.
- heterocyclic rings described herein may be substituted on carbon or on a nitrogen atom if the resulting compound is stable.
- a nitrogen in the heterocycle may optionally be quaternized. It is preferred that when the total number of S and 0 atoms in the heterocycle exceeds 1, then these heteroatoms are not adjacent to one another. It is preferred that the total number of S and 0 atoms in the heterocycle is not more than 1.
- aromatic heterocyclic system or similar term such as “heteroaryl” is intended to mean a stable 5-to 7-membered monocyclic or bicyclic or 7-to 10-membered bicyclic heterocyclic aromatic ring which consists of carbon atoms and from 1 to 4 heteroatoms independently selected from the group consisting of N, O and S. It is preferred that the total number of S and O atoms in the aromatic heterocycle. is not more than 1.
- heterocycles include, but are not limited to, acridinyl, azocinyl, benzimidazolyl, benzofuranyl, benzothiofuranyl, benzothiophenyl, benzoxazolyl, benzthiazolyl, benztriazolyl, benztetrazolyl, benzisoxazolyl, benzisothiazolyl, benzimidazolinyl, carbazolyl, NH-carbazolyl, carbolinyl, chromanyl, chromenyl, cinnolinyl, decahydroquinolinyl, 2H,6H-l,5,2-dithiazinyl, dihydrofuro[2,3-b]tetrahydrofuran, furanyl, furazanyl, imidazolidinyl, imidazolinyl, imidazolyl, l ⁇ -indazolyl, indolenyl, indolinyl,
- Preferred heterocycles include, but are not limited to, pyridinyl, furanyl, thienyl, pyrrolyl, pyrazolyl, pyrrohdinyl, imidazolyl, indolyl, benzimidazolyl, IH-indazolyl, oxazolidinyl, benzotriazolyl, benzisoxazolyl, oxindolyl, benzoxazolinyl, and isatinoyl. Also included are fused ring and spiro compounds containing, for example, the above heterocycles.
- carbocyclic aryl includes groups that contain 1 to 3 separate or fused rings and from 6 to about 18 ring atoms, without hetero atoms as ring members.
- Specifically preferred carbocyclic aryl groups include phenyl, and naphthyl including 1 -napthyl and 2-naphthyl.
- pharmaceutically acceptable refers to those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- pharmaceutically acceptable salts refer to derivatives of the disclosed compounds wherein the parent compound is modified by making acid or base salts thereof. Examples of pharmaceutically acceptable salts include, but are not limited to, mineral or organic acid salts of basic residues such as amines; alkali or organic salts of acidic residues such as carboxylic acids; and the like.
- the pharmaceutically acceptable salts include the conventional non-toxic salts or the quaternary ammonium salts of the parent compound formed, for example, from non-toxic inorganic or organic acids.
- such conventional non-toxic salts include those derived from inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the like; and the salts prepared from organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pamoic, malefic, hydroxymaleic, phenylacetic, glutamic, benzoic, salicylic, sulfanilic, 2-acetoxybenzoic, fumaric, toluenesulfonic, methanesulfonic, ethane disulfonic, oxalic, isethionic, HOOC-(CH 2 )n-COOH where n is 0-4, and the like.
- the pharmaceutically acceptable salts of the present invention can be synthesized from the parent compound which contains a basic or acidic moiety by conventional chemical methods.
- such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two; generally, nonaqueous media like ether, ethyl acetate, ethanol, isopropanol, or acetonitrile are preferred.
- Lists of suitable salts are found in Remington's Pharmaceutical Sciences, 17th ed., Mack Publishing Company, Easton, PA, p. 1418 (1985), the disclosure of which is hereby incorporated by reference,
- Prodrugs are intended to include any covalently bonded carriers which release the active parent drug according to formula I in vivo when such prodrug is administered to a mammalian subject.
- Prodrugs of a compound of formula I are prepared by modifying functional groups present in the compound in such a way that the modifications are cleaved, either in routine manipulation or in vivo, to the parent compound.
- Prodrugs include compounds of formula I wherein a hydroxy, amino, or sulfhydryl group is bonded to any group that, when the prodrug or compound of formula I is administered to a mammalian subject, cleaves to form a free hydroxyl, free amino, or free sulfhydryl group, respectively.
- Examples of prodrugs include, but are not limited to, acetate, formate and benzoate derivatives of alcohol and amine functional groups in the compounds of formula I, and the like.
- a stable compound or stable structure is meant to imply a compound that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and formulation into an effective therapeutic agent.
- therapeutically effective amount of a compound of this invention means an amount effective to antagonize abnormal level of CRF or treat the symptoms of affective disorder, anxiety or depression in a host.
- the compounds of general Formula I may be administered orally, topically, parenterally, by inhalation or spray or rectally in dosage unit formulations containing conventional non-toxic pharmaceutically acceptable carriers, adjuvants and vehicles.
- parenteral as used herein includes subcutaneous injections, intravenous, intramuscular, intrasternal injection or infusion techniques.
- a pharmaceutical formulation comprising a compound of general Formula I and a pharmaceutically acceptable carrier.
- One or more compounds of general Formula I may be present in association with one or more non-toxic pharmaceutically acceptable carriers and/or diluents and/or adjuvants and if desired other active ingredients.
- compositions containing compounds of general Formula I may be in a form suitable for oral use, for example, as tablets, troches, lozenges, aqueous or oily suspensions, dispersible powders or granules, emulsion, hard or soft capsules, or syrups or elixirs.
- compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide pharmaceutically elegant and palatable preparations.
- Tablets contain the active ingredient in admixture with non-toxic pharmaceutically acceptable excipients which are suitable for the manufacture of tablets.
- excipients may be for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example, corn starch, or alginic acid; binding agents, for example starch, gelatin or acacia, and lubricating agents, for example magnesium stearate, stearic acid or talc.
- the tablets may be uncoated or they may be coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period.
- a time delay material such as glyceryl monosterate or glyceryl distearate may be employed.
- Formulations for oral use may also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, for example peanut oil, liquid paraffin or olive oil.
- an inert solid diluent for example, calcium carbonate, calcium phosphate or kaolin
- water or an oil medium for example peanut oil, liquid paraffin or olive oil.
- Aqueous suspensions contain the active materials in admixture with excipients suitable for the manufacture of aqueous suspensions.
- excipients are suspending agents, for example sodium carboxymethylcellulose, methylcellulose, hydropropylmethylcellulose, sodium alginate, polyvinylpyrrolidone, gum tragacanth and gum acacia; dispersing or wetting agents may be a naturally-occurring phosphatide, for example, lecithin, or condensation products of an alkylene oxide with fatty acids, for example polyoxyethylene stearate, or condensation products of ethylene oxide with long chain aliphatic alcohols, for example heptadecaethyleneoxycetanol, or condensation products of ethylene oxide with partial esters derived from fatty acids and a hexitol such as polyoxyethylene sorbitol monooleate, or condensation products of ethylene oxide with partial esters derived from fatty acids and hexitol anhydrides, for example polyethylene sorbitan monooleate
- the aqueous suspensions may also contain one or more preservatives, for example ethyl, or n-propyl p-hydroxybenzoate, one or more coloring agents, one or more flavoring agents, and one or more sweetening agents, such as sucrose or saccharin.
- preservatives for example ethyl, or n-propyl p-hydroxybenzoate
- coloring agents for example ethyl, or n-propyl p-hydroxybenzoate
- flavoring agents for example ethyl, or n-propyl p-hydroxybenzoate
- sweetening agents such as sucrose or saccharin.
- Oily suspensions may be formulated by suspending the active ingredients in a vegetable oil, for example arachis oil, olive oil, sesame oil or coconut oil, or in a mineral oil such as liquid paraffin.
- the oily suspensions may contain a thickening agent, for example beeswax, hard paraffin or cetyl alcohol. Sweetening agents such as those set forth above, and flavoring agents may be added to provide palatable oral preparations. These compositions may be preserved by the addition of an anti-oxidant such as ascorbic acid.
- Dispersible powders and granules suitable for preparation of an aqueous suspension by the addition of water provide the active ingredient in admixture with a dispersing or wetting agent, suspending agent and one or more preservatives.
- a dispersing or wetting agent e.g., glycerol, glycerol, glycerol, glycerol, glycerol, glycerol, glycerin, glycerin, glycerin, glycerin, glycerin, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, glycerol, glycerol, glycerol, glycerol, glycerol, glycerol, glycerol, glycerol, glycerol
- compositions of the invention may also be in the form of oil-in- water emulsions.
- the oily phase may be a vegetable oil, for example olive oil or arachis oil, or a mineral oil, for example liquid paraffin or mixtures of these.
- Suitable emulsifying agents may be naturally-occurring gums, for example gum acacia or gum tragacanth, naturally-occurring phosphatides, for example soy bean, lecithin, and esters or partial esters derived from fatty acids and hexitol, anhydrides, for example sorbitan monoleate, and condensation products of the said partial esters with ethylene oxide, for example polyoxyethylene sorbitan monoleate.
- the emulsions may also contain sweetening and flavoring agents.
- Syrups and elixirs may be formulated with sweetening agents, for example glycerol, propylene glycol, sorbitol or sucrose. Such formulations may also contain a demulcent, a preservative and flavoring and coloring agents.
- the pharmaceutical compositions may be in the form of a sterile injectable aqueous or oleaginous suspension. This suspension may be formulated according to the known art using those suitable dispersing or wetting agents and suspending agents which have been mentioned above.
- the sterile injectable preparation may also be sterile injectable solution or suspension in a non-toxic parentally acceptable diluent or solvent, for example as a solution in 1,3- butanediol.
- Suitable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil may be employed including synthetic mono-or diglycerides.
- fatty acids such as oleic acid find use in the preparation of injectables.
- the compounds of general Formula I may also be administered in the form of suppositories for rectal administration of the drug.
- These compositions can be prepared by mixing the drug with a suitable non-irritating excipient which is solid at ordinary temperatures but liquid at the rectal temperature and will therefore melt in the rectum to release the drug.
- suitable non-irritating excipient which is solid at ordinary temperatures but liquid at the rectal temperature and will therefore melt in the rectum to release the drug.
- Such materials are cocoa butter and polyethylene glycols.
- Compounds of general Formula I may be administered parenterally in a sterile medium.
- the drug depending on the vehicle and concentration used, can either be suspended or dissolved in the vehicle.
- adjuvants such as local anesthetics, preservatives and buffering agents can be dissolved in the vehicle.
- Typical subjects to which compounds of the invention may be administered will be mammals, particularly primates, especially humans.
- mammals particularly primates, especially humans.
- livestock such as cattle, sheep, goats, cows, swine and the like; poultry such as chickens, ducks, geese, turkeys, and the like; and domesticated animals particularly pets such as dogs and cats.
- rodents e.g. mice, rats, hamsters
- rabbits primates, and swine such as inbred pigs and the like.
- body fluids and cell samples of the above subjects will be suitable for use such as mammalian, particularly primate such as human, blood, urine or tissue samples, or blood urine or tissue samples of the animals mentioned for veterinary applications.
- Dosage levels of the order of from about 0.1 mg to about 140 mg of a compound of the invention per kilogram of body weigh per day are useful in the treatment of the above-indicated conditions (about 0.5 mg to about 7 g per patient per day).
- the amount of active ingredient that may be combined with the carrier materials to produce a single dosage form will vary depending upon the host treated and the particular mode of administration.
- Dosage unit forms will generally contain between from about 1 mg to about 500 mg of an active ingredient.
- Frequency of dosage may also vary depending on the compound used and the particular disease treated. However, for treatment of most CNS disorders, a dosage regimen of 4 times daily or less is preferred. For the treatment of stress and depression a dosage regimen of 1 or 2 times daily is particularly preferred.
- the specific dose level for any particular ' patient will depend upon a variety of factors including the activity of the specific compound employed, the age, body weight, general health, sex, diet, time of administration, route of administration, and rate of excretion, drug combination and the severity of the particular disease undergoing therapy,
- Preferred compounds of the invention will have certain pharmacological properties. Such properties include, but are not limited to oral bioavailability, low toxicity, low serum protein binding and desirable in vitro and in vivo half-lifes. Penetration of the blood brain barrier for compounds used to treat CNS disorders is necessary, while low brain levels of compounds used to treat periphereal disorders are often preferred. Assays may be used to predict these desirable pharmacological properties. Assays used to predict bioavailability include transport across human intestinal cell monolayers, including Caco-2 cell monolayers. Toxicity to cultured hepatocyctes may be used to predict compound toxicity. Penetration of the blood brain barrier of a compound in humans may be predicted from the brain levels of the compound in laboratory animals given the compound intravenously.
- Serum protein binding may be predicted from albumin binding assays. Such assays are described in a review by Oravcova, et al. (Journal of Chromatography B (1996) volume 677, pages 1-27).
- Compound half-life is inversely proportional to the frequency of dosage of a compound.
- In vitro half-lives of compounds may be predicted from assays of microsomal half-life as described by Kuhnz and Gieschen (Drug Metabolism and
- compound half-life may be predicted from an in vitro microsomal assay such as the assay given in example 99b.
- the present invention also pertains to methods for altering the activity of CRF receptors, said method comprising exposing cells expressing such receptors to an effective amount of a compound of the invention, wherein the compound is present in the solution at a concentration sufficient to specifically alter the signal transduction activity in response to CRF in cells expressing high levels of CRFI receptors in vitro.
- This method includes altering the signal transduction activity of CRF receptors in vivo, e.g., in a patient given an amount of a compound of Formula I that would be sufficient to alter the signal transduction activity in response to CRF in cells expressing high levels of CRFI in vitro.
- the amount of a compound that would be sufficient to alter the signal transduction activity in response to CRF receptors may be determined via an assay of CRF receptor mediated signal transduction, such as an assay wherein the binding of CRF to a cell surface CRF receptor effects a changes in reporter gene expression.
- the present invention also pertains to packaged pharmaceutical compositions for. treating disorders responsive to C5a receptor modulation, e.g., eating disorders, depression or stress.
- the packaged pharmaceutical compositions include a container holding a therapeutically effective amount of at least one CRFI receptor modulator as described supra and instructions for using the treating disorder responsive to CRFI receptor modulation in the patient.
- the compounds of the present invention can be prepared in a number of ways well known to one skilled in the art of organic synthesis.
- the compounds of the present invention can be synthesized using the methods described below, together with synthetic methods known in the art of synthetic organic chemistry, or variations thereon as appreciated by those skilled in the art. Preferred methods include but are not limited to those methods described below.
- Each of the references cited below are hereby incorporated herein by reference.
- Preferred methods for the preparation of compounds of the present invention include, but are not limited to, those described in Scheme I, Scheme II, and Scheme III. Those who are skilled in the art will recognize that the starting materials may be varied and additional steps employed to produce compounds encompassed by the present invention. All references cited herein are hereby incorporated in their entirety herein by reference. The following abbreviations are used herein: AcOH acetic acid
- Ri and R3 are as defined for formula I and Hal represents a halogen atom, suitably chloride or bromide
- the halide in VI can be displaced by an amine or (thio)alkoxide nucleophile.
- aminopyrazine can be prepared from VI and an amine in the presence of a suitable transition metal catalyst such as but not limited to palladium(II) acetate or tris(dibenzylideneacetone)dipalladium(0), a ligand such as but not limited to l,l'-bis(diphenylphosphine)ferrocene, 2,2'- bis(diphenylphosphine)- 1 , 1 '-binaphthyl, dicyclohexyl(2-biphenyl)phosphine, tricyclohexylphosphine, or tri-tert-butylphosphine, and a base such as sodium or potassium tert-butoxide in inert solvents such as but not limited to toluene, ethyleneglycol dimethyl ether, diglyme, DMF, or N-methylpyrrolidinone at temperatures ranging from ambient to 100°C.
- a suitable transition metal catalyst such as but not limited
- (Thio)alkoxypyrazines can be prepared by treating NI with a sodium or potassium salt of an alcohol or thiol in an inert solvent such as THF, DMF, ⁇ -methylpyrrolidinone, or methyl sulfoxide at ambient temperature or at elevated temperature up to the boiling point of the solvent employed.
- an inert solvent such as THF, DMF, ⁇ -methylpyrrolidinone, or methyl sulfoxide
- Halogenation may be accomplished by a variety of methods known in the art, including treatment with ⁇ - chlorosuccinimide, bromine, ⁇ -bromosuccinimide, pyridiniurn tribromide, triphenylphosphine dibromide, iodine, and ⁇ -iodosuccinimide in solvents such as but not limited to dichloromethane, acetic acid, or methyl sulfoxide.
- the bromopyrazine can be converted to arylpyrazine Nil by a transition metal-catalyzed coupling reaction with a metalloaryl reagent (Ar-[MJ).
- More commonly employed reagent/catalyst pairs include aryl boronic acidVpalladium(O) (Suzuki reaction; ⁇ . Miyaura and A. Suzuki, Chemical Review 1995, 95, 2457), aryl trialkylstannane/palladium(0) (Stille reaction; T. ⁇ . Mitchell, Synthesis 1992, 803), arylzinc/palladium(0) and aryl Grignard/nickel(II).
- Palladium(O) represents a catalytic system made of a various combination of metal/ligand pair which includes, but not limited to, tetrakis(triphenylphosphine)palladium(0), palladium(II) acetate/tri(o-tolyl)phosphine, tris(dibenzylideneacetone)dipalladium(0)/tri- tert-butylphosphine and dichloro[l,l '-bis(diphenylphosphine)ferrocene]palladium(0).
- metal/ligand pair includes, but not limited to, tetrakis(triphenylphosphine)palladium(0), palladium(II) acetate/tri(o-tolyl)phosphine, tris(dibenzylideneacetone)dipalladium(0)/tri- tert-butylphosphine and dichloro[l,l '-bis(diphenylphosphine
- Nickel(II) represents a nickel-containing catalyst such as [1,2- bis(diphenylphosphino)ethane]dichloronickel(II) and [1,3- bis(diphenylphosphino)propane]dichloronickel(II).
- the arylpyrazine VII when X is NH, may be further transformed to NIII by ⁇ -alkylation.
- the ⁇ -H group is deprotonated by a strong base such as but not limited to alkali metal hydride, alkali metal amide, or alkali metal alkoxide in inert solvents such as but not limited to THF, DMF, or methyl sulfoxide.
- Alkylation may be conducted using alkyl halide, suitably bromide or iodide, at temperatures ranging from 0°C to 100°C.
- compounds of Formula VII can be prepared as outlined in Scheme II.
- Transition metal-catalyzed coupling of the halo pyrazine NI as described in the Method A can provide the intermediate NIII.
- Oxidation of sterically less hindered nitrogen can be effected by using a variety of oxidizing agents known in the art, which includes m-chloroperoxybenzoic acid, trifluoroperacetic acid, hydrogen peroxide, and monoperoxyphthalic acid.
- the ⁇ -oxide can undergo rearrangement to give chloropyrazine IX upon the action of phosphorus oxychloride at temperatures ranging from ambient to 100°C.
- Displacement of the chloride with a nitrogen, oxygen, or sulfur nucleophile as described in the Method A can furnish the compounds of Formula Nil.
- X may react with an amine in solvents such as but not limited to dichloromethane, acetonitrile, THF, DMF, ⁇ - methylpyrrolidinone, methyl sulfoxide, methanol, ethanol, and isopropanol at temperatures ranging from 0°C to the boiling point of the solvent.
- solvents such as but not limited to dichloromethane, acetonitrile, THF, DMF, ⁇ - methylpyrrolidinone, methyl sulfoxide, methanol, ethanol, and isopropanol at temperatures ranging from 0°C to the boiling point of the solvent.
- X may react with a sodium or potassium (thio)alkoxide in inert solvents such as but not limited to THF, DMF, N-methylpyrrolidinone, or methyl sulfoxide at temperatures ranging from 0°C to ambient temperature.
- a sodium or potassium (thio)alkoxide inert solvents such as but not limited to THF, DMF, N-methylpyrrolidinone, or methyl sulfoxide at temperatures ranging from 0°C to ambient temperature.
- Resulting monochloropyrazine XI can be halogenated by using the conditions described in the Method A to give a mixture of regioisomeric bromides Xlla and Xllb.
- the aforementioned nucleophiles may include sodium or potassium
- (thio)alkoxide, alkylamine, and organometallic reagent such as but not limited to alkyl Grignard reagents, alkylboronic acids or its ester, or alkylstannanes.
- the aforementioned transition metal catalyst may represent palladium or nickel catalysts described in the Method A.
- the other regioisomeric bromide Xllb can also be converted to VII by changing the order of the transformation sequence. Those who are skilled in the art will also recognize that one can further change the order of transformations to prepare the compounds of Formula VII by way of the intermediate XIII.
- preferred arylpyrazines of the invention exhibit good activity in standard in vitro CRF receptor binding assays, specifically the assay as specified in Example 96, which follows.
- References herein to "standard in vitro receptor binding assay" are intended to refer to that protocol as defined in Example 96 which follows.
- preferred compounds preferred arylpyrimidines of the invention have an IC 50 of about 10 micromolar or less, still more preferably and IC 50 of about 100 nanomolar or less even more preferably an IC 50 of about 10 nanomolar or less or even 1 nanomolar or less in such a defined standard in vitro CRF receptor binding assay as exemplified by Example 96 which follows.
- the Examples 2-20a in the Table I may be prepared following the methods described in Example 1.
- the Examples 24-48 in the Table 13 may be prepared following the general procedure described in Example 23.
- Examples 50 - 53 may be prepared according to the general procedure described in Example 49.
- Examples 55-62 in Table El may be prepared following the general procedure described in Examples 54 and 63.
- Examples 64-74 in Table IN may be prepared following the general procedure described in Example 63.
- Example 75 The bromopyrazine obtained in Example 75 is converted to the desired product by using allylboronic acid following the same procedure as in Example IC: ⁇ NMR (CDC1 3 , 400 MHz) ⁇ 0.93 (t, 6H), 1.50 - 1.71 (m, 4H), 3.50 (d, 2H), 4.05 (m, IH), 4.55 (d, IH), 5.22 (m, 2H), 5.92 (m, IH), 7.34 (m, 2H), 7.48 (m, IH); MS(CI) 384.
- Example 77 NMR (CDC1 3 , 400 MHz) ⁇ 0.93 (t, 6H), 1.50 - 1.71 (m, 4H), 3.50 (d, 2H), 4.05 (m, IH), 4.55 (d, IH), 5.22 (m, 2H), 5.92 (m, IH), 7.34 (m, 2H), 7.48 (m, IH); MS(CI) 384.
- Example 77
- Example 75 A 6-chloro-[N-(l-ethyl)propyl]pyrazine-2-amine obtained in Example 75 A reacts with ethylmagnesium bromide as in Example 21 to give 6-ethyl-[N-(l- ethyl)propyl]pyrazine-2-amine.
- the product of Example 78 is brominated as in Example 75B to give the title compound: ⁇ NMR (CDC1 3 , 400 MHz) ⁇ 0.97 (t, 6H), 1.14 (t, 3H), 1.56 (m, 2H), 1.65 (m, 2H), 2.45 (m, 2H), 4.02 (m, IH), 5.00 (d, IH), 7.25 (d, IH), 7.30 (dd, IH), 7.46 (d, IH); MS (CI) 416.
- Examples 80-82 in Table N may be prepared following the general procedure described in Example 79 using the corresponding halogenating agent indicated in the table.
- methylmagnesium bromide (3.0M in diethyl ether, 15.7 mL) is added slowly dropwise at 0 °C. The mixture is stirred at room temperature for 1 hr. The resulting dark solution is poured into aqueous ammonium chloride and extracted twice with ether. Combined extracts are dried (sodium sulfate), filtered, concentrated, and chromatographed to give the desired product as a light brown oil(98%).
- Example 75C 6-Chloro-5-(2,4-dichlorophenyl)-[N-(l -ethyl)propyl]pyrazine-2-amine obtained in Example 75C is converted to 5-(2,4-dichlorophenyl)-6-methoxy-[N-(l- ethyl)propyl]pyrazine-2-amine according to the same procedure used in Example 83, which is further converted to the title compound according to the same procedure used in Example 79: ⁇ NMR (CDC1 3 , 400 MHz) ⁇ 0.97 (t, 6H), 1.60 (m, 2H), 1.70 (m, 2H), 3.89 (s, 3H), 3.92 (m, IH), 4.98 (d, IH), 7.27 (dd, IH), 7.34 (d, IH), 7.44 (d, IH); MS (CI) 418.
- Examples 86C-86D in Table NI may be prepared following the general procedure described in Example 86b or Example 86e using the corresponding phenylboronic acids
- step 3 (l-ethyl-propyl)-amine.
- the aminopyrazine obtained in step 3 (370 mg, 0.97 mmol) was dissolved in dichloromethane (15 mL) and treated with solid m-chloroperoxybenzoic acid
- reaction mixture was diluted with ethyl ether (100 mL) and washed with ⁇ aOH (2M, 50 mL) and brine (3 x 50 mL), dried (MgSO4), filtered, and the solvent evaporated under reduced pressure to afford the desired chloropyrazine (40 mg, 96%>).
- a solution in DMF (2 mL) of 3-bromo-6-chloro-[N-(l-ethyl)propyl]pyrazine- 2-amine (0.50 g; 1.8 mmol) obtained as a minor product in Example 75B is added into a solution of sodium methoxide (freshly prepared from 90 mg of sodium metal in methanol; 3.9 mmol) in DMF (3 mL). The mixture is stirred at room temperature overnight, then poured into aqueous ammonium chloride solution and extracted twice with hexane-ethyl ether.
- Example 79 The above material is brominated according to the same procedure used in Example 79 and further converted to the title compound according to the same Suzuki coupling procedure used in Example IC: ⁇ NMR (CDC1 3 , 400 MHz) ⁇ 0.95 (t, 6H), 1.5 - 1.7 (m, 4H), 2.15 (s, 3H), 3.8 (s, 3H), 3.85 (s, 3H), 3.95 (s, 3H), 4.0 (m, IH), 4.8 (br d, IH), 6.55 (s, IH), 6.55 (d, IH), 7.3 (d, IH).
- Example 87A 6-chloro-3-methoxy-[N-(l-ethyl)propyl]pyrazine-2-amine obtained in Example 87A is converted to 6-methyl derivative according to the same procedure used in Example 85 and further brominated according to the same procedure used in Example 79.
- Example 93 a 2-Sec-butylsulfanyl-5-(2,4-dichloro-phenyl)-3,6-diethyl-pyrazine.
- 2-Bromo-5-(2-methoxy-5-trifluoromethoxyphenyl)-6-ethylpyrazine can be prepared by the halogenation method of Example 91, step H by substituting (5-(2- methoxy-5-trifluoromethoxyphenyl)-6-ethylpyrazin-2-yl)amine for ⁇ 3-ethyl-6-methoxy- 5-[2-methoxy-4-(trifluoromethoxy)phenyl]pyrazin-2-yl ⁇ amine.
- 2-(3-Pentoxy)-(5-(2-methoxy-5-trifluoromethoxyphenyl)-6-ethylpyrazine can be prepared by the method of Example 91, step I by substituting 2-bromo-(5-(2-methoxy- 5-trifluoromethoxyphenyl)-6-ethylpyrazin-2-yl)[(4-methoxyphenyl)methyl]amine for [4- (5-bromo-6-ethyl-3-methoxypyrazin-2-yl)-3-methoxyphenoxy]trifluoromethane.
- step I by substituting 2-bromo-(5-(2-methoxy- 5-trifluoromethoxyphenyl)-6-ethylpyrazin-2-yl)[(4-methoxyphenyl)methyl]amine for [4- (5-bromo-6-ethyl-3-methoxypyrazin-2-yl)-3-methoxyphenoxy]trifluoromethane.
- 2-(3-Pentoxy)-3-bromo-(5-(2-methoxy-5-trifluoromethoxyphenyl)-6- ethylpyrazine can be prepared by the method of step A of the present Example by substituting 2-(3-Pentoxy)-3-bromo-(5-(2-methoxy-5-trifluoromethoxyphenyl)-6- ethylpyrazine for (6-chloropyrazin-2-yl)[(4-methoxyphenyl)methyl]amine.
- the following assay is defined herein as a standard in vitro CRF receptor binding assay.
- the pharmaceutical utility of compounds of this invention is indicated by the following assay for CRFI receptor activity.
- the CRF receptor binding is performed using a modified version of the assay described by Grigoriadis and De Souza (Methods in Neurosciences, Vol. 5, 1991).
- IMR-32 human neuroblastoma cells, a cell-line that naturally expresses the CRFI receptor, are grown to confluency in DMEM containing
- receptor containing membranes cells are homogenized in wash buffer (50 mM Tris HC1, 10 mM MgCl 2 , 2 mM EGTA, pH 7.4) and centrifuged at 48,000 x g for 10 minutes at 4°C. The pellet is re-suspended in wash buffer and the homogenization and centrifugation steps are performed two additional times.
- Membrane pellets containing CRF receptors are re-suspended in 50 mM Tris buffer pH 7.7 containing 10 mM MgCl 2 and 2 mM EDTA and centrifuged for 10 minutes at 48000g.
- Membranes are washed again and brought to a final concentration of 1500 mg/ml in binding buffer (Tris buffer above with 0.1 % BSA, 15 mM bacitracin and 0.01 mg/ml aprotinin.).
- binding buffer Tris buffer above with 0.1 % BSA, 15 mM bacitracin and 0.01 mg/ml aprotinin.
- 100 ml of the membrane preparation are added to 96 well microtube plates containing 100 ml of l2 T-CRF (SA 2200 Ci/mmol, final concentration of 100 pM) and 50 ml of test compound. Binding is carried out at room temperature for 2 hours. Plates are then harvested on a Brandel 96 well cell harvester and filters are counted for gamma emissions on a Wallac 1205 Betaplate liquid scintillation counter.
- Non specific binding is defined by 1 mM cold CRF.
- IC 50 values are calculated with the non-linear curve fitting program RS/1 (BBN Software Products Corp., Cambridge, MA).
- the binding affinity for the compounds of Formula I expressed as IC 50 value generally ranges from about 0.5 nanomolar to about 10 micromolar.
- Preferred compounds of Formula I exhibit IC 50 values of less than or equal to 1.5 micromolar, more preferred compounds of Formula I exhibit IC 50 values of less than 500 nanomolar, still more preferred compounds of Formula I exhibit IC 50 values of less than 100 nanomolar, and most preferred compound of Formula I exhibit IC 50 values of less than 10 nanomolar.
- the compounds shown in Examples 1-95 and Examples 100-396 have been tested in this assay and found to exhibit IC 50 values of less than or equal to 1.5 micromolar.
- the compounds of the invention are prepared as radiolabeled probes by carrying out their synthesis using precursors comprising at least one atom that is a radioisotope.
- the radioisotope is preferably selected from of at least one of carbon (preferably 14 C), hydrogen (preferably 3 H), sulfur (preferably 35 S), or iodine (preferably 125 I).
- Such radiolabeled probes are conveniently synthesized by a radioisotope supplier specializing in custom synthesis of radiolabeled probe compounds. Such suppliers include Amersham Corporation, Arlington Heights, IL; Cambridge Isotope Laboratories, Inc.
- Tritium labeled probe compounds are also conveniently prepared catalytically via platinum-catalyzed exchange in tritiated acetic acid, acid-catalyzed exchange in tritiated trifluoroacetic acid, or heterogeneous-catalyzed exchange with tritium gas. Such preparations are also conveniently carried out as a custom radiolabeling by any of the suppliers listed in the preceding paragraph using the compound of the invention as substrate. In addition, certain precursors may be subjected to tritium-halogen exchange with tritium gas, tritium gas reduction of unsaturated bonds, or reduction using sodium borotritide, as appropriate.
- Receptor autoradiography Receptor mapping
- Receptor mapping Receptor mapping
- the most preferred compounds of the invention are suitable for pharmaceutical use in treating human patients. Accordingly, such preferred compounds are non-toxic. They do not exhibit single or multiple dose acute or long-term toxicity, mutagenicity (e.g., as determined in a bacterial reverse mutation assay such as an Ames test), teratogenicity, tumorogenicity, or the like, and rarely trigger adverse effects (side effects) when administered at therapeutically effective dosages.
- administering does not result in prolongation of heart QT intervals (i.e., as determined by electrocardiography, e.g., in guinea pigs, minipigs or dogs).
- such doses of such preferred compounds When administered daily for 5 or preferably ten days, such doses of such preferred compounds also do not cause liver enlargement resulting in an increase of liver to body weight ratio of more than 100%), preferably not more than 75% and more preferably not more than 50%) over matched controls in laboratory rodents (e.g., mice or rats). In another aspect such doses of such preferred compounds also preferably do not cause liver enlargement resulting in an increase of liver to body weight ratio of more than 50%>, preferably preferably not more than 25%, and more preferably not more than 10% over matched untreated controls in dogs or other non-rodent animals.
- such doses of such preferred compounds also preferably do not promote the release of liver enzymes (e.g., ALT, LDH, or AST) from hepatocytes in vivo.
- liver enzymes e.g., ALT, LDH, or AST
- such doses do not elevate such enzymes by more than 100%, preferably not by more than 75% and more preferably not by more than 50% over matched untreated controls in laboratory rodents.
- concentrations (in culture media or other such solutions that are contacted and incubated with cells in vitro) equivalent to two, fold, preferably five-fold, and most preferably ten-fold the minimum in vivo therapeutic concentration do not cause release of any of such liver enzymes from hepatocytes in vitro.
- preferred compounds of the invention exert their receptor-modulatory effects with high selectivity. This means that they do not bind to certain other receptors (i.e., other than CRF receptors) with high affinity, but rather only bind to, activate, or inhibit the activity of such other receptors with affinity constants of greater than 100 nanomolar, preferably greater than 1 micromolar, more preferably greater than 10 micromolar and most preferably greater than 100 micromolar.
- Such receptors preferably are selected from the group including ion channel receptors, including sodium ion channel receptors, neurotransmitter receptors such as alpha- and beta-adrenergic receptors, muscarinic receptors (particularly ml, m2, and m3 receptors), dopamine receptors, and metabotropic glutamate receptors; and also include histamine receptors and cytokine receptors, e.g., interleukin receptors, particularly IL-8 receptors.
- ion channel receptors including sodium ion channel receptors, neurotransmitter receptors such as alpha- and beta-adrenergic receptors, muscarinic receptors (particularly ml, m2, and m3 receptors), dopamine receptors, and metabotropic glutamate receptors; and also include histamine receptors and cytokine receptors, e.g., interleukin receptors, particularly IL-8 receptors.
- the group of other receptors to which preferred compounds do not bind with high affinity also includes GABA A receptors, bioactive peptide receptors (including NPY and NIP receptors), neurokinin receptors, bradykinin receptors (e.g., BK1 receptors and BK2 receptors), and hormone receptors (including thyrotropin releasing hormone receptors and melanocyte-concentrating hormone receptors).
- GABA A receptors include GABA A receptors, bioactive peptide receptors (including NPY and NIP receptors), neurokinin receptors, bradykinin receptors (e.g., BK1 receptors and BK2 receptors), and hormone receptors (including thyrotropin releasing hormone receptors and melanocyte-concentrating hormone receptors).
- Sodium channel activity may be measured a standard in vitro sodium channel binding assays such as the assay given by Brown et al. (J. ⁇ eurosci. (1986) 265: 17995-18004).
- Preferred compounds of the invention exhibit less than 15 percent inhibition, and more preferably less than 10 percent inhibition, of sodium channel specific ligand binding when present at a concentration of 4 uM.
- the sodium ion channel specific ligand used may be labeled batrachotoxinin, tetrodotoxin, or saxitoxin.
- Such assays including the assay of Brown referred to above, are performed as a commercial service by CEREP, INC., Redmond, WA.
- sodium ion channel activity may be measured in vivo in an assay of anti-epileptic activity.
- Anti-epileptic activity of compounds may be measured by the ability of the compounds to inhibit hind limb extension in the supra maximal electro shock model.
- Male Han Wistar rats (150-200mg) are dosed i.p. with a suspension of 1 to 20 mg of test compound in 0.25% methylcellulose 2 hr. prior to test. A visual observation is carried out just prior to testing for the presence of ataxia. Using auricular electrodes a current of 200 mA, duration 200 millisec, is applied and the presence or absence of hind limb extension is noted.
- Preferred compounds of the invention do not exhibit significant anti-epileptic activity at the p ⁇ 0.1 level of significance or more preferably at the p ⁇ 0.05 level of significance as measured using a standard parametric assay of statistical significance such as a student's T test.
- Example 99b
- Compound half-life values may be determined via the following standard liver microsomal half-life assay. Liver microsomes obtained from pooled liver samples and prepared so that the P-450 enzyme content is approximately 0.5 nmol/ mg protein. Reactions are preformed in a 5ml well deep-well plate as follows: Phosphate buffer: 19 mL 0.1 M NaH 2 PO 4 , 81 mL 0.1 Na ⁇ PO,, pH 7.4 with H 3 PO 4 . CoFactor Mixture: 16.2 mg NADP, 45.4 mg Glucose-6-phosphate in 4 mL 100 mM MgCl 2 . Glucose-6-phosphate dehydrogenase: 214.3 ul glucose-6-phosphate dehydrogenase, 1285.7 ul distilled water
- Starting Reaction Mixture 3 mL CoFactor Mixture, 1.2 mL Glucose-6-phosphate dehydrogenase 6 identical samples wells each containing 25 ul microsomes, 5 ul of test compound (from a 100 uM stock), and 399 ul 0.1 M phosphate buffer, pH 7.4, are prepared.
- a seventh well containing 25 ul microsomes, 399 ul 0.1 M phosphate buffer, pH 7.4, and 5 ul (from a 100 uM stock) of a compound, e.g. DIAZEPAM, CLOZEPINE, with known metabolic properties is used as a positive control. Reactions are preincubated at 39 °C for 10 minutes.
- 71 ul Starting Reaction Mixture is added to 5 of the 6 reaction wells and to the positive control well, 71 ul 100 mM MgCl 2 is added to the sixth reaction well, which is used as a negative control.
- 75 ul reaction is pipetted into a 96-well deep-well plate reaction well containing 75 ul ice-cold acetonitrile.
- Samples are vortexed and centrifuged 10 minutes at 6000 rpm (Sorval T 6000D rotor).
- Supernatant, 75 ul from each reaction well is transferred to a 96-well plate containing 150 ul internal standard per well.
- the remaining test compound is quantitated via LCMS and Compound concentration vs time is plotted and commercially available statistical software is used to extrapolate to the t ⁇ n value of the test compound.
- Preferred compounds of the invention exhibit in vitro t 1 2 values of greater than 10 minutes and less than 4 hours. Most preferred compounds of the invention exhibit in vitro t 1/2 values of between 30 minutes and 1 hour in human liver microsomes.
- Toxicity of a test compound may be assessed by measuring the effect of the compound on ATP production by MDCK cells.
- MDCK cells product no. CCL-34, are purchased from ATCC, Manassas, NA and maintained in sterile conditions by the methods described in the supplier's production information sheet.
- the PACKARD BIOSCIE ⁇ CE, (Groningen, The Netherlands) ATP- LITE-M Luminescent ATP decection kit, product no. 6016941, may be used to monitor ATP production in MDCK cells. Prior to assay 1 ul of test compound or control sample is pipetted into PACKARD
- Test compounds and control samples are diluted in DMSO to give final concentration in the assay of 10 micromolar, 100 micromolar, or 200 micromolar.
- Control samples are drug compounds having known toxicity properties.
- Confluent MDCK cells are trypsinized, harvested, and diluted to a concentration of 0.1 x 10 6 cells/ ml with warm ATCC Eagle's Minimum Essential Medium (catalog # 30-2003). Warm Eagle's MEM without cells (lOOul) is pipetted in five wells a 96-well plate. These wells are used to determine the standard curve. Cells in Eagle's MEM (lOOul or 10,000 cells) are pipetted into the remaining wells of the 96-well plates.
- lyophilized ATP standard solution is reconstituted in deionized water to give a 10 mM stock. Standard (10 ul), diluted so that a 10 ul aliquot yields a final concentration of 200 nM, 100 nM, 50 iiM, 25 nM, or 12.5 nM is added to each of the five standard curve wells, which do not contain cells. Substrate solution (50 ul) is added to all wells. Wells are covered with Packard topseal stickers, and plates are shaken at approximately 700 rpm on a microtiter shaker for 2 minutes.
- a white Packard sticker is attached to the bottom of each plates and samples are dark adapted by wrapping plates in foil and placing in the dark for 10 minutes. Luminescence is then quantitated at 22 °C using a luminescence counter, e.g. Packard TopCount Microplate Scintillation and Luminescense Counter or Tecan Spectrafluor plus.
- a luminescence counter e.g. Packard TopCount Microplate Scintillation and Luminescense Counter or Tecan Spectrafluor plus.
- Luminscence values at each concentration are compared to the values computed from the standard curve for that concentration.
- Preferred test compounds exhibit luminescence values 80 % ⁇ or more of the standard, or preferably 90 % or more of the standard, when 10 micromolar concentration of the test compound is used.
- preferred test compounds exhibit luminescence values 50 % or more of the standard, or more preferably 80 % or more of the standard.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Psychiatry (AREA)
- Rheumatology (AREA)
- Vascular Medicine (AREA)
- Child & Adolescent Psychology (AREA)
- Pain & Pain Management (AREA)
- Endocrinology (AREA)
- Reproductive Health (AREA)
- Hematology (AREA)
- Diabetes (AREA)
- Obesity (AREA)
- Urology & Nephrology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Photoreceptors In Electrophotography (AREA)
- Pyridine Compounds (AREA)
Abstract
Description




















































































































Claims




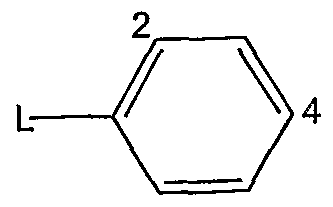











Priority Applications (22)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DZ013293A DZ3293A1 (en) | 2000-02-16 | 2001-02-16 | Substituted arylpyrazines |
| UA2002076329A UA75586C2 (en) | 2000-02-16 | 2001-02-16 | Substituted arylpyrazines, pharmaceutical composition based thereof, method for modulation of crf receptors function, method for treatment of cns-related disorders and diseases, method for localization of crf receptors in tissue specimens, method for inhibition of cfr receptor binding with crf1 receptor, method for changing activity of transduction cfr1 receptor signal, method for modulation of function g protein-coupled receptor |
| AT01910939T ATE307121T1 (en) | 2000-02-16 | 2001-02-16 | SUBSTITUTED ARYLPYRAZINE |
| SK1154-2002A SK11542002A3 (en) | 2000-02-16 | 2001-02-16 | Substituted arylpyrazines |
| NZ520484A NZ520484A (en) | 2000-02-16 | 2001-02-16 | Substituted arylpyrazines that are modulators of CRF (corticotropin releasing factor) receptor and methods of treatment using them |
| BR0108363-5A BR0108363A (en) | 2000-02-16 | 2001-02-16 | Compound, methods for modulating g protein-coupled crf receptor function, for treating a snc disorder or disease, for locating crf receptors in tissue section samples, for inhibiting the binding of crf to a crf¹ receptor and for alter signal transduction activity of a crf¹ receptor and pharmaceutical composition |
| AU38494/01A AU783915B2 (en) | 2000-02-16 | 2001-02-16 | Substituted arylpyrazines |
| HU0301573A HUP0301573A3 (en) | 2000-02-16 | 2001-02-16 | Substituted arylpyrazines, pharmaceutical compositions containing them and their use |
| CA002398937A CA2398937A1 (en) | 2000-02-16 | 2001-02-16 | Substituted arylpyrazines |
| JP2001560191A JP2004500383A (en) | 2000-02-16 | 2001-02-16 | Substituted arylpyrazine |
| DE60114153T DE60114153T2 (en) | 2000-02-16 | 2001-02-16 | SUBSTITUTED ARYLPYRAZINE |
| MXPA02007868A MXPA02007868A (en) | 2000-02-16 | 2001-02-16 | Substituted arylpyrazines. |
| HK03103353.2A HK1051191B (en) | 2000-02-16 | 2001-02-16 | Substituted arylpyrazines |
| EEP200200453A EE200200453A (en) | 2000-02-16 | 2001-02-16 | Substituted arylpyrazines |
| HRP20020747 HRP20020747A2 (en) | 2000-02-16 | 2001-02-16 | Substituted arylpyrazines |
| APAP/P/2002/002620A AP2002002620A0 (en) | 2000-02-16 | 2001-02-16 | Substituted arylpyrazines |
| EP01910939A EP1255740B1 (en) | 2000-02-16 | 2001-02-16 | Substituted arylpyrazines |
| EA200200766A EA006938B1 (en) | 2000-02-16 | 2001-02-16 | Substituted arylpyrazines |
| IL15102001A IL151020A0 (en) | 2000-02-16 | 2001-02-16 | Substituted arylpyrazines |
| IS6488A IS2192B (en) | 2000-02-16 | 2002-07-29 | Substituted arylpyrazine |
| BG106968A BG106968A (en) | 2000-02-16 | 2002-07-31 | Substituted arylpyrazines |
| NO20023869A NO324473B1 (en) | 2000-02-16 | 2002-08-15 | Substituted arylpyrazines, use thereof for the preparation of pharmaceutical preparations for the treatment of disease, method for locating CRF receptors in tissue sections based on such compounds, use of the compounds to inhibit, alter or modulate receptor functions, pharmaceutical preparations and packaged pharmaceutical preparations compounds. |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US18293400P | 2000-02-16 | 2000-02-16 | |
| US60/182,934 | 2000-02-16 | ||
| US20645500P | 2000-05-22 | 2000-05-22 | |
| US60/206,455 | 2000-05-22 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2001060806A2 true WO2001060806A2 (en) | 2001-08-23 |
| WO2001060806A3 WO2001060806A3 (en) | 2002-02-07 |
Family
ID=26878562
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2001/005264 Ceased WO2001060806A2 (en) | 2000-02-16 | 2001-02-16 | Substituted arylpyrazines |
Country Status (33)
| Country | Link |
|---|---|
| US (3) | US6995161B2 (en) |
| EP (1) | EP1255740B1 (en) |
| JP (1) | JP2004500383A (en) |
| KR (2) | KR20080021603A (en) |
| CN (1) | CN1231473C (en) |
| AP (1) | AP2002002620A0 (en) |
| AT (1) | ATE307121T1 (en) |
| AU (1) | AU783915B2 (en) |
| BG (2) | BG106968A (en) |
| BR (1) | BR0108363A (en) |
| CA (2) | CA2398937A1 (en) |
| CR (1) | CR6735A (en) |
| CZ (1) | CZ20022739A3 (en) |
| DE (1) | DE60114153T2 (en) |
| DK (1) | DK1255740T3 (en) |
| DZ (1) | DZ3293A1 (en) |
| EA (1) | EA006938B1 (en) |
| EE (1) | EE200200453A (en) |
| ES (1) | ES2247070T3 (en) |
| HR (1) | HRP20020747A2 (en) |
| HU (1) | HUP0301573A3 (en) |
| IL (1) | IL151020A0 (en) |
| IS (1) | IS2192B (en) |
| MA (1) | MA26874A1 (en) |
| MX (1) | MXPA02007868A (en) |
| NO (1) | NO324473B1 (en) |
| NZ (1) | NZ520484A (en) |
| OA (1) | OA12178A (en) |
| PL (1) | PL365238A1 (en) |
| SK (1) | SK11542002A3 (en) |
| TW (1) | TWI232215B (en) |
| WO (1) | WO2001060806A2 (en) |
| YU (1) | YU61002A (en) |
Cited By (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002024681A3 (en) * | 2000-09-20 | 2002-06-20 | Ortho Mcneil Pharm Inc | Pyrazine derivatives as modulators of tyrosine kinases |
| WO2002096421A1 (en) * | 2001-05-22 | 2002-12-05 | Neurogen Corporation | 5-substituted-2-arylpyridines as crf1 modulators |
| WO2002100838A1 (en) * | 2001-06-12 | 2002-12-19 | Neurogen Corporation | 2,5-diarylpyrazines, 2,5-diarylpyridines and 2,5-diarylpyrimidines as crf1 receptor modulators |
| WO2003004472A1 (en) * | 2001-07-05 | 2003-01-16 | Astrazeneca Ab | Arylamines for the treatment of conditions associated with gsk-3 |
| WO2003004475A1 (en) * | 2001-07-05 | 2003-01-16 | Astrazeneca Ab | Heterocyclic amines for the treatment of conditions associated with gsk-3 |
| WO2003045924A1 (en) * | 2001-11-21 | 2003-06-05 | Pharmacia & Upjohn Company | Substituted aryl 1,4-pyrazine derivatives |
| WO2003091225A1 (en) * | 2002-04-26 | 2003-11-06 | Pharmacia & Upjohn Company | Substituted pyrazine derivatives |
| WO2004018437A1 (en) * | 2002-08-20 | 2004-03-04 | Neurogen Corporation | 5-substituted-2-arylpyrazines as modulators of crf receptors |
| WO2004024719A1 (en) * | 2002-09-12 | 2004-03-25 | Pharmacia & Upjohn Company Llc | Substituted 1,4-pyrazine derivatives |
| WO2004046136A1 (en) * | 2002-11-21 | 2004-06-03 | Pharmacia & Upjohn Company Llc | Pyrazine compounds as crf modulators |
| WO2004055005A1 (en) * | 2002-12-17 | 2004-07-01 | Astrazeneca Ab | Novel compounds having selective inhibiting effect at gsk3 |
| WO2004055009A1 (en) * | 2002-12-17 | 2004-07-01 | Astrazeneca Ab | Novel compounds having selective inhibiting efect at gsk3 |
| WO2004099201A1 (en) * | 2003-05-09 | 2004-11-18 | Pharmacia & Upjohn Company Llc | Compounds as crf1 receptor antagonists |
| WO2005007644A1 (en) * | 2003-06-27 | 2005-01-27 | Banyu Pharmaceutical Co., Ltd | Heteroaryloxy nitrogenous saturated heterocyclic derivative |
| WO2005040151A1 (en) * | 2003-10-27 | 2005-05-06 | Astellas Pharma Inc. | Pyrazine derivatives and pharmaceutical use thereof |
| US6992087B2 (en) | 2001-11-21 | 2006-01-31 | Pfizer Inc | Substituted aryl 1,4-pyrazine derivatives |
| US7015227B2 (en) | 2002-06-21 | 2006-03-21 | Cgi Pharmaceuticals, Inc. | Certain amino-substituted monocycles as kinase modulators |
| US7087617B2 (en) | 2003-05-09 | 2006-08-08 | Pfizer Inc. | Substituted pyrimidine derivatives |
| WO2006114666A1 (en) * | 2005-03-17 | 2006-11-02 | Pfizer Products Inc. | SUBSTITUTED ARYL 1,4-PYRAZlNE DERIVATIVES |
| US7230098B2 (en) | 2003-02-26 | 2007-06-12 | Sugen, Inc. | Aminoheteroaryl compounds as protein kinase inhibitors |
| US10000468B2 (en) | 2014-02-14 | 2018-06-19 | Takeda Pharmaceutical Company Limited | Pyrazines as modulators of GPR6 |
| WO2021214494A1 (en) * | 2020-04-24 | 2021-10-28 | University Of Greenwich | Interleukin inhibitors |
| US11261186B2 (en) | 2014-12-24 | 2022-03-01 | Lg Chem. Ltd. | Biaryl derivative as GPR120 agonist |
| US20220340546A1 (en) * | 2019-06-14 | 2022-10-27 | Disarm Therapeutics, Inc. | Inhibitors of sarm1 |
| US11661401B2 (en) | 2016-07-12 | 2023-05-30 | Revolution Medicines, Inc. | 2,5-disubstituted 3-methyl pyrazines and 2,5,6-trisubstituted 3-methyl pyrazines as allosteric SHP2 inhibitors |
| US11673901B2 (en) | 2017-12-15 | 2023-06-13 | Revolution Medicines, Inc. | Polycyclic compounds as allosteric SHP2 inhibitors |
| US11673896B2 (en) | 2017-01-23 | 2023-06-13 | Revolution Medicines, Inc. | Pyridine compounds as allosteric SHP2 inhibitors |
| US11702411B2 (en) | 2017-10-12 | 2023-07-18 | Revolution Medicines, Inc. | Pyridine, pyrazine, and triazine compounds as allosteric SHP2 inhibitors |
| US11739093B2 (en) | 2017-01-23 | 2023-08-29 | Revolution Medicines, Inc. | Substituted pyrazolopyrazines, imidazopyrazines and [1,2,4]triazolopyrazines as allosteric SHP2 inhibitors |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ITMI20011726A1 (en) | 2001-08-06 | 2003-02-06 | Recordati Ind Chimica E Farma | POLYMORPHIC FORMS OF LERCANIDIPINE HYDROCHLORIDE |
| US6852737B2 (en) * | 2001-08-06 | 2005-02-08 | Recordati Ireland Limited | Crude and crystalline forms of lercanidipine hydrochloride |
| TW200716594A (en) * | 2005-04-18 | 2007-05-01 | Neurogen Corp | Substituted heteroaryl CB1 antagonists |
| PL1959955T3 (en) * | 2005-12-05 | 2011-04-29 | Pfizer Prod Inc | Method of treating abnormal cell growth |
| DK1963302T3 (en) * | 2005-12-05 | 2013-04-02 | Pfizer Prod Inc | Polymorphs of a C-MET / HGFR Inhibitor |
| SG157264A1 (en) * | 2008-06-02 | 2009-12-29 | Inzign Pte Ltd | A golf tee and method of producing a golf tee |
| US7932256B2 (en) * | 2008-07-31 | 2011-04-26 | Bristol-Myers Squibb Company | (S)-4-(1-cyclopropyl-2-methoxyethyl)-6-(6-(difluoromethoxy)-2,5-dimethylpyridin-3-ylamino)-5-oxo-4,5-dihydropyrazine-2-carbonitrile: a pyrazinone modulator of corticotropin-releasing factor receptor activity |
| RU2672470C1 (en) * | 2017-07-10 | 2018-11-15 | Федеральное государственное бюджетное учреждение науки Институт нефтехимии и катализа Российской академии наук | Production process of 2,3,5,6-tetrafenyl(benzyl)pyrasine |
| US11708376B2 (en) * | 2018-04-20 | 2023-07-25 | Virginia Tech Intellectual Properties, Inc. | Substituted imidazo[4,5-b]pyridines, imidazo[4,5-b]pyrazines, and oxazolo[4,5- b]pyrazines as mitochondrial uncouplers |
Family Cites Families (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2425355A1 (en) | 1974-05-25 | 1975-12-04 | Basf Ag | METHOD FOR MANUFACTURING PYRAZINES |
| EG12387A (en) * | 1975-04-21 | 1978-12-31 | Merck & Co Inc | Process for preparing of piperazinyl pyrazines |
| FR2398738A1 (en) * | 1977-06-22 | 1979-02-23 | Lilly Co Eli | PREPARATION OF BENZOYLUREES |
| US4293552A (en) | 1978-02-27 | 1981-10-06 | Eli Lilly And Company | Novel 1-(mono-o-substituted benzoyl)-3-(substituted pyrazinyl) ureas |
| US4211870A (en) * | 1979-04-06 | 1980-07-08 | Eli Lilly And Company | Preparation of substituted 2-aminopyrazines |
| JPS59144772A (en) * | 1983-02-08 | 1984-08-18 | Ube Ind Ltd | Liquid crystalline pyrazine derivative and its production method |
| JPS625970A (en) | 1985-03-15 | 1987-01-12 | Terumo Corp | Pyrazine derivative and platelet coagulation inhibitor containing same |
| JPH0739398B2 (en) * | 1986-10-04 | 1995-05-01 | 宇部興産株式会社 | Liquid crystalline pyrazine derivative |
| NZ227684A (en) * | 1988-01-30 | 1991-07-26 | Merck Sharp & Dohme | Pyrazine, pyridazine, and pyrimidine derivatives and pharmaceutical compositions |
| JPH0348666A (en) | 1989-07-17 | 1991-03-01 | Rasa Kogyo Kk | Production of aromatic diamine compound |
| DE3940477A1 (en) * | 1989-12-07 | 1991-06-13 | Bayer Ag | HETARYL-SUBSTITUTED PYRIDINYLPYRIMIDINE DERIVATIVES |
| GB2272216A (en) * | 1992-11-04 | 1994-05-11 | Merck Patent Gmbh | Liquid crystalline di-, tri- and tetra-fluorinated phenylpyrazines |
| WO1995010506A1 (en) * | 1993-10-12 | 1995-04-20 | The Du Pont Merck Pharmaceutical Company | 1n-alkyl-n-arylpyrimidinamines and derivatives thereof |
| WO1997011943A1 (en) * | 1995-09-26 | 1997-04-03 | Nippon Soda Co., Ltd. | Pyrazole compounds, process for preparing the same, and agrohorticultural bactericide |
| US6159980A (en) * | 1996-09-16 | 2000-12-12 | Dupont Pharmaceuticals Company | Pyrazinones and triazinones and their derivatives thereof |
| MA26473A1 (en) * | 1997-03-01 | 2004-12-20 | Glaxo Group Ltd | PHARMACOLOGICALLY ACTIVE COMPOUNDS. |
| BE1011077A3 (en) | 1997-03-28 | 1999-04-06 | Univ Catholique Louvain | Pharmaceutical composition, cosmetic and / or food to properties antioxidant. |
| ID24959A (en) | 1998-01-28 | 2000-08-31 | Shionogi & Co | NEW TRISICLIC COMPOUNDS |
| GB9818881D0 (en) | 1998-08-28 | 1998-10-21 | Glaxo Group Ltd | Compounds |
| WO2000045800A2 (en) | 1999-02-02 | 2000-08-10 | K.U. Leuven Research & Development | Immunosurpressive effects of pteridine derivatives |
-
2001
- 2001-02-16 PL PL01365238A patent/PL365238A1/en not_active Application Discontinuation
- 2001-02-16 AP APAP/P/2002/002620A patent/AP2002002620A0/en unknown
- 2001-02-16 TW TW090103566A patent/TWI232215B/en not_active IP Right Cessation
- 2001-02-16 MX MXPA02007868A patent/MXPA02007868A/en active IP Right Grant
- 2001-02-16 ES ES01910939T patent/ES2247070T3/en not_active Expired - Lifetime
- 2001-02-16 NZ NZ520484A patent/NZ520484A/en unknown
- 2001-02-16 AT AT01910939T patent/ATE307121T1/en not_active IP Right Cessation
- 2001-02-16 AU AU38494/01A patent/AU783915B2/en not_active Ceased
- 2001-02-16 CN CNB018051316A patent/CN1231473C/en not_active Expired - Fee Related
- 2001-02-16 US US09/788,315 patent/US6995161B2/en not_active Expired - Lifetime
- 2001-02-16 DZ DZ013293A patent/DZ3293A1/en active
- 2001-02-16 EP EP01910939A patent/EP1255740B1/en not_active Expired - Lifetime
- 2001-02-16 HR HRP20020747 patent/HRP20020747A2/en not_active Application Discontinuation
- 2001-02-16 EE EEP200200453A patent/EE200200453A/en unknown
- 2001-02-16 BR BR0108363-5A patent/BR0108363A/en not_active IP Right Cessation
- 2001-02-16 CZ CZ20022739A patent/CZ20022739A3/en unknown
- 2001-02-16 DK DK01910939T patent/DK1255740T3/en active
- 2001-02-16 EA EA200200766A patent/EA006938B1/en unknown
- 2001-02-16 CA CA002398937A patent/CA2398937A1/en not_active Abandoned
- 2001-02-16 KR KR1020077027017A patent/KR20080021603A/en not_active Ceased
- 2001-02-16 CA CA002651825A patent/CA2651825A1/en not_active Abandoned
- 2001-02-16 JP JP2001560191A patent/JP2004500383A/en active Pending
- 2001-02-16 KR KR1020027010403A patent/KR20030031886A/en not_active Ceased
- 2001-02-16 DE DE60114153T patent/DE60114153T2/en not_active Expired - Lifetime
- 2001-02-16 YU YU61002A patent/YU61002A/en unknown
- 2001-02-16 WO PCT/US2001/005264 patent/WO2001060806A2/en not_active Ceased
- 2001-02-16 IL IL15102001A patent/IL151020A0/en unknown
- 2001-02-16 HU HU0301573A patent/HUP0301573A3/en unknown
- 2001-02-16 OA OA1200200251A patent/OA12178A/en unknown
- 2001-02-16 SK SK1154-2002A patent/SK11542002A3/en not_active Application Discontinuation
-
2002
- 2002-07-29 IS IS6488A patent/IS2192B/en unknown
- 2002-07-31 BG BG106968A patent/BG106968A/en unknown
- 2002-07-31 BG BG110174A patent/BG110174A/en unknown
- 2002-08-13 MA MA26774A patent/MA26874A1/en unknown
- 2002-08-15 NO NO20023869A patent/NO324473B1/en not_active IP Right Cessation
- 2002-08-23 CR CR6735A patent/CR6735A/en not_active Application Discontinuation
-
2005
- 2005-04-15 US US11/107,148 patent/US7202250B2/en not_active Expired - Fee Related
-
2007
- 2007-02-16 US US11/675,648 patent/US20070225287A1/en not_active Abandoned
Cited By (47)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6710048B2 (en) | 2000-09-20 | 2004-03-23 | Ortho-Mcneil Pharmaceutical, Inc. | Pyrazine derivatives as modulators of tyrosine kinases |
| WO2002024681A3 (en) * | 2000-09-20 | 2002-06-20 | Ortho Mcneil Pharm Inc | Pyrazine derivatives as modulators of tyrosine kinases |
| WO2002096421A1 (en) * | 2001-05-22 | 2002-12-05 | Neurogen Corporation | 5-substituted-2-arylpyridines as crf1 modulators |
| US7223778B2 (en) | 2001-05-22 | 2007-05-29 | Neurogen Corporation | 5-substituted-2-arylpyridines |
| WO2002100838A1 (en) * | 2001-06-12 | 2002-12-19 | Neurogen Corporation | 2,5-diarylpyrazines, 2,5-diarylpyridines and 2,5-diarylpyrimidines as crf1 receptor modulators |
| WO2003004472A1 (en) * | 2001-07-05 | 2003-01-16 | Astrazeneca Ab | Arylamines for the treatment of conditions associated with gsk-3 |
| WO2003004475A1 (en) * | 2001-07-05 | 2003-01-16 | Astrazeneca Ab | Heterocyclic amines for the treatment of conditions associated with gsk-3 |
| WO2003045924A1 (en) * | 2001-11-21 | 2003-06-05 | Pharmacia & Upjohn Company | Substituted aryl 1,4-pyrazine derivatives |
| US6992087B2 (en) | 2001-11-21 | 2006-01-31 | Pfizer Inc | Substituted aryl 1,4-pyrazine derivatives |
| US6964965B2 (en) | 2002-04-26 | 2005-11-15 | Pharmacia & Upjohn | Substituted pyrazine derivatives |
| WO2003091225A1 (en) * | 2002-04-26 | 2003-11-06 | Pharmacia & Upjohn Company | Substituted pyrazine derivatives |
| US7015227B2 (en) | 2002-06-21 | 2006-03-21 | Cgi Pharmaceuticals, Inc. | Certain amino-substituted monocycles as kinase modulators |
| WO2004018437A1 (en) * | 2002-08-20 | 2004-03-04 | Neurogen Corporation | 5-substituted-2-arylpyrazines as modulators of crf receptors |
| US7179807B2 (en) | 2002-08-20 | 2007-02-20 | Neurogen Corporation | 5-substituted-2-arylpyrazines |
| WO2004024719A1 (en) * | 2002-09-12 | 2004-03-25 | Pharmacia & Upjohn Company Llc | Substituted 1,4-pyrazine derivatives |
| WO2004046136A1 (en) * | 2002-11-21 | 2004-06-03 | Pharmacia & Upjohn Company Llc | Pyrazine compounds as crf modulators |
| JP2006514628A (en) * | 2002-11-21 | 2006-05-11 | ファルマシア・アンド・アップジョン・カンパニー・エルエルシー | Pyrazine compounds as CRF modulators |
| EP2161265A3 (en) * | 2002-12-17 | 2010-06-02 | Astra Zeneca AB | Compounds having selective inhibiting effect at GSK3 |
| WO2004055009A1 (en) * | 2002-12-17 | 2004-07-01 | Astrazeneca Ab | Novel compounds having selective inhibiting efect at gsk3 |
| WO2004055005A1 (en) * | 2002-12-17 | 2004-07-01 | Astrazeneca Ab | Novel compounds having selective inhibiting effect at gsk3 |
| US7585853B2 (en) | 2002-12-17 | 2009-09-08 | Astrazeneca Ab | Compounds having selective inhibiting effect at GSK3 |
| US7595321B2 (en) | 2002-12-17 | 2009-09-29 | Astrazeneca Ab | Compounds having selective inhibiting effect at GSK3 |
| US8106197B2 (en) | 2003-02-26 | 2012-01-31 | Pfizer Inc. | Aminoheteroaryl compounds as protein kinase inhibitors |
| US7230098B2 (en) | 2003-02-26 | 2007-06-12 | Sugen, Inc. | Aminoheteroaryl compounds as protein kinase inhibitors |
| US7087617B2 (en) | 2003-05-09 | 2006-08-08 | Pfizer Inc. | Substituted pyrimidine derivatives |
| US7250418B2 (en) | 2003-05-09 | 2007-07-31 | Pfizer Inc | Compounds as CRF1 receptor antagonists |
| WO2004099201A1 (en) * | 2003-05-09 | 2004-11-18 | Pharmacia & Upjohn Company Llc | Compounds as crf1 receptor antagonists |
| WO2005007644A1 (en) * | 2003-06-27 | 2005-01-27 | Banyu Pharmaceutical Co., Ltd | Heteroaryloxy nitrogenous saturated heterocyclic derivative |
| JPWO2005007644A1 (en) * | 2003-06-27 | 2006-08-31 | 萬有製薬株式会社 | Heteroaryloxy nitrogen-containing saturated heterocyclic derivatives |
| JP2007510620A (en) * | 2003-10-27 | 2007-04-26 | アステラス製薬株式会社 | Pyrazine derivatives and their pharmaceutical use |
| WO2005040151A1 (en) * | 2003-10-27 | 2005-05-06 | Astellas Pharma Inc. | Pyrazine derivatives and pharmaceutical use thereof |
| WO2006114666A1 (en) * | 2005-03-17 | 2006-11-02 | Pfizer Products Inc. | SUBSTITUTED ARYL 1,4-PYRAZlNE DERIVATIVES |
| EA012874B1 (en) * | 2005-03-17 | 2009-12-30 | Пфайзер Продактс Инк. | Substituted aryl 1.4-pyrazine derivatives |
| US10000468B2 (en) | 2014-02-14 | 2018-06-19 | Takeda Pharmaceutical Company Limited | Pyrazines as modulators of GPR6 |
| US10273225B2 (en) | 2014-02-14 | 2019-04-30 | Takeda Pharmaceutical Company Limited | Pyrazines as modulators of GPR6 |
| US11261186B2 (en) | 2014-12-24 | 2022-03-01 | Lg Chem. Ltd. | Biaryl derivative as GPR120 agonist |
| US12565476B2 (en) | 2016-07-12 | 2026-03-03 | Revolution Medicines, Inc. | 2,5-disubstituted 3-methyl pyrazines and 2,5,6-trisubstituted 3-methyl pyrazines as allosteric SHP2 inhibitors |
| US11661401B2 (en) | 2016-07-12 | 2023-05-30 | Revolution Medicines, Inc. | 2,5-disubstituted 3-methyl pyrazines and 2,5,6-trisubstituted 3-methyl pyrazines as allosteric SHP2 inhibitors |
| US12365688B2 (en) | 2017-01-23 | 2025-07-22 | Revolution Medicines, Inc. | Substituted pyrazolopyrazines, imidazopyrazines and [1,2,4]triazolopyrazines as allosteric SHP2 inhibitors |
| US11739093B2 (en) | 2017-01-23 | 2023-08-29 | Revolution Medicines, Inc. | Substituted pyrazolopyrazines, imidazopyrazines and [1,2,4]triazolopyrazines as allosteric SHP2 inhibitors |
| US11673896B2 (en) | 2017-01-23 | 2023-06-13 | Revolution Medicines, Inc. | Pyridine compounds as allosteric SHP2 inhibitors |
| US11702411B2 (en) | 2017-10-12 | 2023-07-18 | Revolution Medicines, Inc. | Pyridine, pyrazine, and triazine compounds as allosteric SHP2 inhibitors |
| US11673901B2 (en) | 2017-12-15 | 2023-06-13 | Revolution Medicines, Inc. | Polycyclic compounds as allosteric SHP2 inhibitors |
| EP3982949A4 (en) * | 2019-06-14 | 2023-10-11 | Disarm Therapeutics, Inc. | Inhibitors of sarm1 |
| US20220340546A1 (en) * | 2019-06-14 | 2022-10-27 | Disarm Therapeutics, Inc. | Inhibitors of sarm1 |
| US12595252B2 (en) * | 2019-06-14 | 2026-04-07 | Disarm Therapeutics, Inc. | Inhibitors of SARM1 |
| WO2021214494A1 (en) * | 2020-04-24 | 2021-10-28 | University Of Greenwich | Interleukin inhibitors |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1255740B1 (en) | Substituted arylpyrazines | |
| RS61002B1 (en) | Rna formulation for immunotherapy | |
| US6887875B2 (en) | 2,5-diarypyrimidine compounds | |
| US6943173B2 (en) | 5-substituted 2-aryl-4-pyrimidinones | |
| US7179807B2 (en) | 5-substituted-2-arylpyrazines | |
| US20020072521A1 (en) | 5-substituted arylpyrimidines | |
| EP1500653A1 (en) | Substituted arylpyrazines | |
| ZA200206103B (en) | Substituted arylpyrazines. | |
| AU2002310117A1 (en) | 2,5-diarylpyrazines, 2,5-diarylpyridines and 2,5-diarylpyrimidines as CRF1 receptor modulators |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: P-610/02 Country of ref document: YU |
|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2398937 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2001 106968 Country of ref document: BG Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002/06103 Country of ref document: ZA Ref document number: 151020 Country of ref document: IL Ref document number: 200206103 Country of ref document: ZA Ref document number: 520484 Country of ref document: NZ Ref document number: 38494/01 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: IN/PCT/2002/01065/MU Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11542002 Country of ref document: SK Ref document number: PV2002-2739 Country of ref document: CZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020027010403 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2002/007868 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200200766 Country of ref document: EA |
|
| ENP | Entry into the national phase |
Ref document number: 2001 560191 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 018051316 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 88.312.2002 Country of ref document: BZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2001910939 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P20020747A Country of ref document: HR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 88.312.2002 Country of ref document: BZ |
|
| WWP | Wipo information: published in national office |
Ref document number: 88.312.2002 Country of ref document: BZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: AP/P/2002/002620 Country of ref document: AP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1200200843 Country of ref document: VN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2001910939 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: PV2002-2739 Country of ref document: CZ |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020027010403 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 520484 Country of ref document: NZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 520484 Country of ref document: NZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2001910939 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 38494/01 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 11017401 Country of ref document: BG Kind code of ref document: A |